
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono- and bimetallic metal catalysts based on Ni and Ru supported on alumina-coated monoliths for CO2 methanation†
Ainhoa
Bustinza
a,
Marina
Frías
a,
Yuefeng
Liu
 b and
Enrique
García-Bordejé
*a
b and
Enrique
García-Bordejé
*a
aInstituto de Carboquímica (ICB-CSIC), Miguel Luesma Castán 4, E-50018 Zaragoza, Spain. E-mail: jegarcia@icb.csic.es
bDalian National Laboratory for Clean Energy (DNL), Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
First published on 19th May 2020
Abstract
Structured reactors such as monoliths have some favorable properties over fixed bed reactors such as low pressure drop, ease in handling and fast heat and mass transfer. Some of these properties make them advantageous reactors for the catalytic reduction of CO2 contained in post-combustion gases. Mono- and bimetallic Ru and Ni catalysts on alumina-washcoated cordierite monoliths have been scrutinized for CO2 methanation. The methodology for the preparation of NiRu bimetallic catalysts has been optimized to obtain an outperforming monolithic catalyst. The catalyst providing the highest CH4 productivity corresponds to a bimetallic NiRu catalyst with a small amount of Ru, prepared by consecutive impregnation of Ni and Ru precursors with an intermediate reduction step. This catalyst consists of 2–4 nm Ni nanoparticles interspersed with atomic Ru homogeneously distributed on the alumina coating. This nanostructure endows the catalyst with the highest density of basic sites of medium strength and the highest degree of Ni oxidation in the passivated catalyst compared to the other tested catalysts. The bimetallic monolithic catalyst afforded a stable CO2 methanation activity at high space velocities and with a low pressure drop, reducing energy consumption.
Introduction
CO2 hydrogenation to CH4 using H2 from renewable energy sources is proposed as a promising way for storing intermittent renewable energy in the stable and transportable form of synthetic natural gas.1 This technology, usually known as “power to gas”, combines the reduction of CO2 emission and the storage of renewable energy to overcome the mismatch between renewable energy generation and demand.For the feasibility of the reaction between CO2 and H2 yielding CH4, the use of a catalyst and high temperature are required. A crucial aspect for the industrial deployment of this technology is the design of the catalytic reactor, which should implement a heat evacuation system because the reaction is highly exothermic ( = −165 kJ mol−1). In fact, conventional fixed bed catalytic reactors are not suitable because the heat released by the reaction leads to temperature rises, resulting in a decrease of CH4 conversion due to thermodynamic constraints and shortening of catalyst lifetime due to sintering. Different solutions have been applied to overcome temperature rises such as fluidized beds, multi-tubular reactors externally cooled or staged reactors with intermediate cooling by heat exchangers.2,3 Another less explored option is washcoating a catalytic layer on the surface of heat exchangers, hence integrating both reaction and cooling. In this context, catalyst washcoating on monoliths, widely studied for automotive catalysts, has also been proposed as a model of catalytic coating for heat exchangers in CO2 methanation.4–6 On the other hand, mesostructured reactors and, in particular, honeycomb monoliths are ideal reactors to convert flue gases emitted at nearly atmospheric pressure such as post-combustion gases. Post-combustion gases are generated in heaters and industrial utility boilers, representing the major part of the current fossil-fuel-based electricity generation. Since reaction equilibrium is favoured by higher pressures, it could be necessary to pressurize. In contrast to fixed beds, the monoliths oppose a much lower pressure drop, reducing the energy consumption for pressurization.
= −165 kJ mol−1). In fact, conventional fixed bed catalytic reactors are not suitable because the heat released by the reaction leads to temperature rises, resulting in a decrease of CH4 conversion due to thermodynamic constraints and shortening of catalyst lifetime due to sintering. Different solutions have been applied to overcome temperature rises such as fluidized beds, multi-tubular reactors externally cooled or staged reactors with intermediate cooling by heat exchangers.2,3 Another less explored option is washcoating a catalytic layer on the surface of heat exchangers, hence integrating both reaction and cooling. In this context, catalyst washcoating on monoliths, widely studied for automotive catalysts, has also been proposed as a model of catalytic coating for heat exchangers in CO2 methanation.4–6 On the other hand, mesostructured reactors and, in particular, honeycomb monoliths are ideal reactors to convert flue gases emitted at nearly atmospheric pressure such as post-combustion gases. Post-combustion gases are generated in heaters and industrial utility boilers, representing the major part of the current fossil-fuel-based electricity generation. Since reaction equilibrium is favoured by higher pressures, it could be necessary to pressurize. In contrast to fixed beds, the monoliths oppose a much lower pressure drop, reducing the energy consumption for pressurization.
Regarding the catalytic active phase, the state-of-the-art catalyst for this reaction is usually based on monometallic metal (Ni or Ru) nanoparticles supported on alumina or titania.7–10 Ru exhibits intrinsic activity higher than that of Ni but it is significantly more expensive.11,12 Different alternatives are currently being explored to reduce the cost of the catalyst. Recently, the benefits of bimetallic catalysts based on several metal combinations have been reported. Bimetallic ensembles of metal sites combining a transition metal and a noble metal may enhance the catalytic performance due to geometric, electronic or cooperative effects. Moreover, the combination of a noble metal and a transition metal has the potential to decrease the noble metal content, leading to cost reduction.13 Under these premises, the combination of Ni and a noble metal (Pt, Pd) has been explored for CO2 hydrogenation.14 Elsewhere, iron catalysts have proven the ability to promote the water gas-shift reaction15 and the oligomerization to C2+.16 The addition of Fe to a Ni catalyst has been found to promote the CO2 methanation reaction.12,16,17 Bimetallic Ni–Fe alloy nanoparticles have been prepared from a hydrotalcite precursor and the bimetallic catalyst exhibited higher activities and stabilities than a monometallic catalyst in CO2 hydrogenation.18 Nevertheless, the best catalyst was that with the lowest amount of Fe. Another aspect to consider when designing a catalyst for CO2 methanation is that the reaction requires two catalytic functions operating cooperatively: one function for the dissociative chemisorption of H2, which is usually carried out mainly by a noble metal or nickel, and another function to chemisorb and activate CO2. Since CO2 is mildly acidic, this function is carried out by basic sites such as transition metals or the hydroxyl groups of alumina support.
Herein, we have tackled the preparation of a catalytic coating on honeycomb monoliths for the methanation of CO2 flue gas at atmospheric pressure, as encountered in post-combustion gases. Monometallic and bimetallic catalyst nanoparticles have been dispersed homogeneously on the walls of alumina-washcoated monoliths. For the preparation of the bimetallic catalyst, the consecutive adsorption method has been employed. The monolithic catalysts have been tested in isothermal CO2 hydrogenation and subsequently have been characterized by TEM, XPS, ICP-OES and CO2-TPD. The order of metal addition and the treatment after the first metal addition have a profound impact on the bimetallic metal nanostructure, on the amount and strength of basic sites and thus on the catalytic performance. The addition of small amounts of Ru to reduced Ni/Al2O3 monolith catalysts improves the performance remarkably with respect to the rest of the catalysts. The resulting monolithic catalysis afforded a highly efficient CO2 methanation with stable performance and negligible pressure drop at a space velocity as high as 5 × 105 h−1.
Experimental details
Catalyst preparation
Cordierite monoliths (from Corning, 10 mm diameter, 60 mm length, 400 cpsi, ca. 2.2 g) were washcoated with alumina using a dip-coating method as described previously.19 In this method, a sol is prepared from pseudoboehmite (AlOOH, Pural from Sasol), urea and 0.3 M nitric acid with a weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5. After stirring for 24 h, the monolith was dip-coated with the sol. To this end, the dried monolith is dipped in this sol for a few minutes until all air bubbles inside the channels are removed. Subsequently, the monolith is withdrawn from the sol and the liquid inside the channels is removed by flushing thoroughly with pressurized air. Then the monolith is dried at room temperature for 24 h while continuously being rotated around its axis. Finally, the monolith is calcined in air at a rate of 1 K min−1 up to 600 °C, and this temperature is kept for 2 h to obtain the γ-alumina washcoating. The alumina content in the monolith amounted to ca. 5 wt% of the monolith weight.
:1:5. After stirring for 24 h, the monolith was dip-coated with the sol. To this end, the dried monolith is dipped in this sol for a few minutes until all air bubbles inside the channels are removed. Subsequently, the monolith is withdrawn from the sol and the liquid inside the channels is removed by flushing thoroughly with pressurized air. Then the monolith is dried at room temperature for 24 h while continuously being rotated around its axis. Finally, the monolith is calcined in air at a rate of 1 K min−1 up to 600 °C, and this temperature is kept for 2 h to obtain the γ-alumina washcoating. The alumina content in the monolith amounted to ca. 5 wt% of the monolith weight.
On the γ-alumina-washcoated monolith, the metal was impregnated. Table 1 lists the different prepared catalysts along with the actual metal loading and the preparation protocol. Due to the different price of the two metals, Ni and Ru were impregnated using different methodologies. Ni was impregnated by equilibrium adsorption with a solution containing excess Ni. In brief, for Ni impregnation, 29 g of Ni(NO3)2·6H2O (pure, Sigma-Aldrich), a buffer solution of 80 g of NH4NO3 (pure, Sigma-Aldrich) and 4 mL of ammonia solution (25%) were mixed in a 1 L bottle. 100 ml of this solution is allowed to flow overnight through the monolith channels until equilibration. The equilibrium adsorption method led to 12.3–12.7% Ni with respect to the weight of alumina on the monolith. For the Ru precursor impregnation, a smaller and defined amount of precursor was used to ensure that the entire Ru precursor was adsorbed on the alumina-washcoated monolith. To prepare the solution, a certain weight of Ru(NO)(NO3)3 precursor was diluted in 4 ml water. The weight of the precursor was calculated to correspond to 5 wt% Ru with respect to the weight of alumina for the Ru and RuNi catalyst and 1 wt% Ru for the other bimetallic catalysts. The employed method is illustrated in Scheme S1 of the ESI.† In brief, the monolith was placed inside a test tube fitted to the walls using a Teflon strip. The test tube with the monolith was filled with 4 ml of the impregnating metal solution inside and closed with a plug. The test tube with the monolith and the impregnating solution was rotated continuously overnight (Fig. 1A). Likewise, the liquid is forced to flow through the channels overnight. Then, the Ru solution became transparent and no Ru was detected by inductively coupled plasma-optical emission spectroscopy (ICP-OES), indicating that all the Ru precursor has been adsorbed on the alumina-coated monolith. Then the monoliths were rinsed thoroughly with deionized water, followed by drying first at room temperature overnight and later at 383 K for 1 h. The colour of the monolith changed to homogeneous green, indicative of the adsorption of the precursor. Subsequently, the monoliths were calcined under flowing nitrogen (1 K min−1) up to 873 K followed by a 2 h dwell time (Scheme S1b, ESI†) and subsequently the same heating treatment was performed under a H2 atmosphere (Scheme S1c, ESI†) except for sample NiRu(cal), for which the latter treatment was omitted. After these preparation steps, the monolith exhibited a homogenous grey colour, indicating that the metal has been impregnated uniformly along the width and length of the monolith (Scheme S1d, ESI†).
| Sample short name | Precursor adsorption steps | Loadinga wt% on Al2O3 | Gas treatment between the two metal impregnations | |
|---|---|---|---|---|
| 1st metal | 2nd metal | |||
| a Measured by ICP-OES. | ||||
| Ru | Ru | 4.8 | — | — |
| Ni | Ni | 12.5 | — | — |
| NiRu(cal) | Ni + Ru | 12.3 | 1.0 | N2 at 873 K |
| NiRu | Ni + Ru | 12.7 | 0.9 | N2 + H2, 873 K |
| RuNi | Ru + Ni | 4.4 | 11.1 | N2 + H2, 873 K |
| NiNi | Ni + Ni | 12.5 | 4.3 | N2 + H2, 873 K |
| NiFe | Ni + Fe | 12.2 | 5 | N2 + H2, 873 K |
 | ||
| Fig. 1 (A) CO2 conversion; (B) selectivity to CH4 and (C) Arrhenius plot at low conversions for the different monolithic catalysts. (D) Stability test during 2 days at 350 °C for the NiRu catalyst in the form of a monolith and in the form of a packed bed after crushing the monolith. Feed composition: 5 vol% CO2, 15 vol% H2, Ar to balance. | ||
Catalyst testing
Catalytic testing was carried out in a continuous-flow 15 mm inner diameter quartz reactor inside a vertical furnace equipped with a temperature controller (Eurotherm). The monolith wrapped with a quartz fiber strip was tightly fitted to the walls of the quartz reactor. The reaction temperature was controlled using a thermocouple in contact with the inlet of the monolith. The flow rates of the gases were fixed using Bronkhorst mass flow controllers. Prior to catalytic tests, the catalyst was heated to 500 °C in a N2 flow at a heating rate of 10 K min−1 and it was reduced with H2 at 500 °C for 1 h. The reaction conversion and selectivities were recorded at steady state using a 60 mL min−1 reaction mixture consisting of 5% CO2, 20% H2 and Ar to balance. This flow rate corresponds to a space velocity of 1.04 × 105 h−1 calculated as flow rate divided by the volume of catalytic washcoating. Stability tests were conducted under the same conditions but leaving the reaction overnight. The outlet reaction gases were analysed using an Agilent Micro GC 3000A instrument. H2 and CO were analysed in a molsieve column and CO2 in a Plot-Q column. The GC signal was calibrated using certified gas cylinders. To ensure repeatability, 2–3 separate GC samples were taken and averaged for each experimental data point. The carbon balance deviated only a ±3% of deviation. The conversion was calculated taking into account the variations in the flow due to the mole variation in the reaction. After discharging the monolith, it was slowly passivated with 1% O2 overnight.The Weisz–Prater criterion was applied to assess the absence of internal and external mass transfer limitations according to the following equations:
 | (1) |
 | (2) |
Catalyst characterization
XPS spectra were recorded using an ESCAPlus Omnicrom system equipped with an Al Kα radiation source to excite the sample. Calibration of the instrument was done with the Ag 3d5/2 line at 368.27 eV. All measurements were performed under ultra-high vacuum, higher than 10−10 torr. Internal referencing of spectrometer energies was made using the dominant Al 2p peak at 74.4 eV of Al2O3 support. The program used to do curve fitting of the spectra was CASA XPS using a Shirley baseline.Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was performed using a Spectroblue de Ametek apparatus.
Scanning transmission electron microscopy in high-angle annular dark field mode (STEM-HAADF) was carried out on a Titan Themis ETEM G3 electron microscope (Thermo Fisher) at 300 kV. Atomic resolution STEM-HAADF images and energy dispersive X-ray (EDX) spectroscopy mapping were conducted using a JEOL JEM ARM-200F microscope with the probe corrector at 200 kV.
Temperature-programmed desorption of pre-adsorbed CO2 (CO2-TPD) experiments were conducted in the same set-up as catalytic testing. The catalyst was heated to 500 °C at a heating rate of 10 °C min−1 in inert gas. At this temperature, the catalyst was reduced with a 100 mL min−1 H2/N2 mixture for 1 h. Subsequently, the temperature was set at 300 °C and CO2 was flushed for 1 h. The gas was switched to 100 mL min−1 Ar and the reactor was allowed to cool to room temperature. At this temperature, Ar flow was maintained during 2 h to remove all weakly physisorbed CO2. Then the gas was adjusted to 60 mL min−1 Ar for CO2-TPD experiments. When the signal of the mass spectrometer was stable, the temperature was increased to 500 °C at a rate of 10 K per minute while monitoring the desorbed gases. Gas analysis was performed using a Pfeiffer vacuum mass spectrometer. The main m/z signals used for each gas were 2 (H2), 16 (CH4), 18 (H2O), 28 (CO), 40 (Ar) and 44 (CO2). The signals of the gases were calibrated taking into account the baseline of Ar and the fragmentation pattern of each mass. The concentration of CO was calculated by subtracting the contribution of CO2 from m/z = 28. The concentration of CH4 was calculated by subtracting the contribution of CO2 and CO from m/z = 16. The correct calibration of the mass spectrometer was double-checked by analysing the gases using a calibrated Agilent Micro GC 3000A instrument.
Results and discussion
Catalytic performance
Fig. 1 shows the steady-state catalytic performance in CO2 hydrogenation for the different monometallic and bimetallic catalysts. The CO2 conversion at steady state increases as the reaction temperature rises (Fig. 1A) until reaching a maximum at the thermodynamic equilibrium. Beyond the temperature of the maximum, the conversion decreases because the equilibrium of the exothermic reaction is shifted to the reactants. Therefore, it is essential to develop a catalyst with a high activity at the lowest temperature as possible to maximize the attainable equilibrium conversion. The selectivity to CH4 for all the Ni- and Ru-containing catalysts is close to 100% below 400 °C (Fig. 1B). When the temperature surpasses 400 °C, the selectivity to CH4 decays because the formation of CO is favored due to the reverse water-gas-shift reaction (rWGS). In contrast, the selectivity to CH4 for the NiFe catalyst is lower than for the rest of the catalysts, ca. 80%, in all the temperature ranges. This is consistent with fact that Fe is known to catalyze the rWGS reaction.16,20The kinetic rate constants (k) were calculated from the results in Fig. 1A. To calculate the reaction rate, first we used the equation of a differential reactor (eqn (3))21 which is applicable only for low conversions:
 | (3) |
| Kinetic rate constant (k) at 553 K | Apparent activation energy | Apparent pre-exponential factor | |
|---|---|---|---|
| mmolCH4 min−1 gcat−1 | kJ mol−1 | s−1 | |
| Ru | 0.0110 | 51 | 38.4 |
| Ni | 0.0066 | 74 | 3380 |
| NiRu(cal) | 0.0112 | 32 | 0.67 |
| NiRu | 0.0280 | 60 | 626 |
| RuNi | 0.0124 | 67 | 1248 |
| NiNi | 0.0176 | 63 | 638 |
| NiFe | 0.0042 | 58 | 6.6 |
Comparing NiRu vs. RuNi activity, it is evident that the order of metal addition is important, being favorable adding first Ni and second Ru. When Ni is added in the second impregnation step over the Ru monometallic catalyst, it does not supply a higher activity than the monometallic Ru catalyst, making the second step redundant. Comparison of NiRu and NiRu(cal) activity reveals that an intermediate reduction step under H2 after the first Ni deposition (NiRu) is beneficial for the catalyst performance. Poorer catalytic activity was found if only the calcination step was performed after Ni impregnation, i.e. for the catalyst NiRu(cal).
Stability tests were performed using the best performing catalyst (NiRu) for 48 hours (Fig. 1D). The catalyst is stable, showing negligible deactivation in this time span. Additionally, a stability test was also performed using the monolith crushed to slabs of sieve fraction <500 μm and subsequently tested as a packed bed. The crushed catalyst in the fixed bed provided a stable performance but lower conversion probably due to the easier heat removal for the monolith, preventing hot spots. This local rise of temperature would decrease thermodynamic conversion due to the exothermicity of the reaction. The monolith has enhanced heat transfer compared to the packed bed. In the packed bed the main mechanism for radial heat transfer is by gas convection, while heat conduction in the thermally connected solid monolith matrix becomes the dominant heat transport mechanism.23 Preferential gas passages can likely occur in the packed bed while the monolith affords enhanced accessibility of process gas to the active catalyst, which provide higher conversion and enhanced heat removal by gas convection.24
It is noticeable that the monolith worked at atmospheric pressure while the packed bed had a pressure drop of 0.8 bar. In fact, when the flow rate was increased 5 times (520000 h−1) the monolith still exhibited a negligible pressure drop, while the pressure in the packed bed increased so much that the connections between the O-rings and the quartz reactor of our experimental set-up are not adapted to work at this high pressure. Therefore, the washcoated monoliths are more suitable than packed beds because of the higher conversion, the lower energy consumption to pressurize the gas and enhanced heat management, which could be optimized further if the washcoating is applied on a monolithic heat exchanger. Accordingly, the catalytic monoliths are ideal reactors for converting CO2 from post-combustion gases, which are emitted at atmospheric pressure. Post-combustion gases are those emitted by conventional process heaters and industrial utility boilers, which represent most of the current fossil-fuel-based electricity generation.
To calculate the apparent activation energy, the linearization of the Arrhenius equation was applied (eqn (4)):
 | (4) |
k vs. 1/T is linear. This occurs for low temperatures such as those depicted in Fig. 1C, that is, before entering the diffusion limited regime. The calculated apparent activation energies are displayed in Table 2.
The activation energy for the Ni catalyst is similar to that reported in the literature for alumina-supported nickel catalysts (74 kJ mol−1),20,25–27 while that for the Ru monolith is slightly lower than that reported (51 vs. 73 kJ mol−1). The lower activation energy for the Ru catalyst suggests that the reaction is more favored over the Ru catalyst. The bimetallic catalysts have values of apparent activation energy between those of monometallic Ru and Ni catalysts, except NiRu(cal), in which Ni was not reduced before Ru adsorption. The apparent activation energy of NiRu(cal) is extremely low for the chemical reaction, which may be attributed to the occurrence of significant diffusional limitations. The rest of the bimetallic catalysts exhibited similar activation energies, pointing towards similar reaction mechanisms. In the literature, different activation energies are reported for different supports, suggesting that the metal–support interaction affects the mechanism of reaction.27 In the present case, the support is the same for all the catalysts but the interaction between the two metals in bimetallic catalysts may vary depending on the preparation methodology, leading to different geometric, electronic and/or cooperative effects. To gain insight into the differences between the catalysts, they were characterized after reaction by ex situ TEM, XPS and CO2-TPD. TEM and XPS techniques give insight about the interaction between the two metals and between metal and support, while the latter technique characterizes the basicity of the catalyst surface.
Morphology and microstructure analyses of monolith catalysts
Monometallic catalysts consist of very well dispersed metal nanoparticles with an average metal size of 2.5 nm for the Ni catalyst and less than 0.8 nm for the Ru catalyst (Fig. 2 and S1, ESI†). Comparing bimetallic catalysts, the metal nanoparticles exhibited a more uniform and smaller size for the NiRu catalyst than for the rest of the catalysts (Fig. 2). For NiRu, there are no particles larger than 10 nm, while for the other catalysts a significant number of particles between 15 and 40 nm were found. Since NiRu exhibited the highest dispersion and catalytic activity, this catalyst was characterized in detail by aberration-corrected HRTEM. Fig. 3a–c shows representative HRTEM images of the NiRu catalyst, which contain very small metal nanoparticles (1.5–3 nm). The HRTEM image in Fig. 3a reveals the clear lattice fringes of cubic NiO crystals with different orientation. The gaps between lattice fringes were approximately 0.15 and 0.24 nm, which correspond to the interspace of the (220) and (111) planes, respectively.28,29 Other images (Fig. 3b) show lattice fringe distances of 0.17 and 0.20 nm corresponding to the (200) and (111) lattice planes, respectively, of fcc Ni.30Fig. 3c shows a 2.8 nm Ni nanoparticle and some small bright dots which should correspond to Ru atoms or a few atom clusters as inferred from the EDS analysis. The EDS mapping (Fig. 3d) indicates that both Ni and Ru are uniformly distributed on the alumina support. The larger nanoparticles (1.5–4 nm) found in the STEM image correspond to Ni, while Ru is not present as nanoparticles but dispersed as atoms or small clusters. | ||
| Fig. 2 Representative STEM-HAADF images of the different mono- and bimetallic catalysts based on Ni and Ru: (a) Ni catalyst, (b) Ru catalyst, (c) NiRu(cal), (d) NiRu and (e) RuNi. | ||
 | ||
| Fig. 3 (a–c) Representative STEM images corrected by bright field (BF) and HAADF models and EDS elemental mapping of the NiRu catalyst. The atomic resolution STEM-HAADF image (c) clearly shows that Ru is atomically dispersed, which is also confirmed by the EDS elemental mapping image (d). | ||
Ex situ XPS measurements
Table 3 compiles the parameters determined after fitting of XPS Ni and Ru peaks (Fig. 4A and B). The quantification of the ratios between different elements (Ni/Al, Ru/Al and Ru/Ni) must be handled with caution because there are several factors that may deviate this ratio from the actual ratio such as the non-uniform depth profile distribution and the particle size. XPS is a surface-sensitive technique and probes only about ca. 10 nm of the outer surface while the alumina coating has a few hundreds of nanometers. The Ni/Al and Ru/Al weight percentage for the monometallic catalysts is much lower than the actual loading (Table 1), suggesting a well-dispersed metal through the alumina layer depth and/or very small nanoparticles. The Ni/Al and Ru/Al ratios increase even above the actual loading for the bimetallic catalysts which can be caused by an increased metal coverage of the alumina surface after adsorption of the second metal, decreasing alumina surface exposure. Thus, XPS detects less alumina signal than the actual composition. The Ru/Ni loading is larger than the actual ratio and is larger when Ru is added in the second step, suggesting that Ru is dispersed predominantly on the surface.| Ni/Al wt% | Ru/Al wt% | Ru/Ni wt% | Ru0/RuTOT | Ni0/NiTOT | |
|---|---|---|---|---|---|
| Ru | — | 2.8 | — | 73.5 | — |
| Ni | 2.1 | — | — | — | 66.4 |
| NiRu(cal) | 3.4 | 6.7 | 1.9 | 75.2 | 35.4 |
| NiRu | 7.8 | 11.2 | 1.4 | 63.2 | 14.5 |
| RuNi | 6.9 | 6.5 | 0.9 | 79.1 | 11.2 |
| NiNi | 2.2 | — | — | — | 34.7 |
| NiFe | 1.9 | — | — | — | 40.9 |
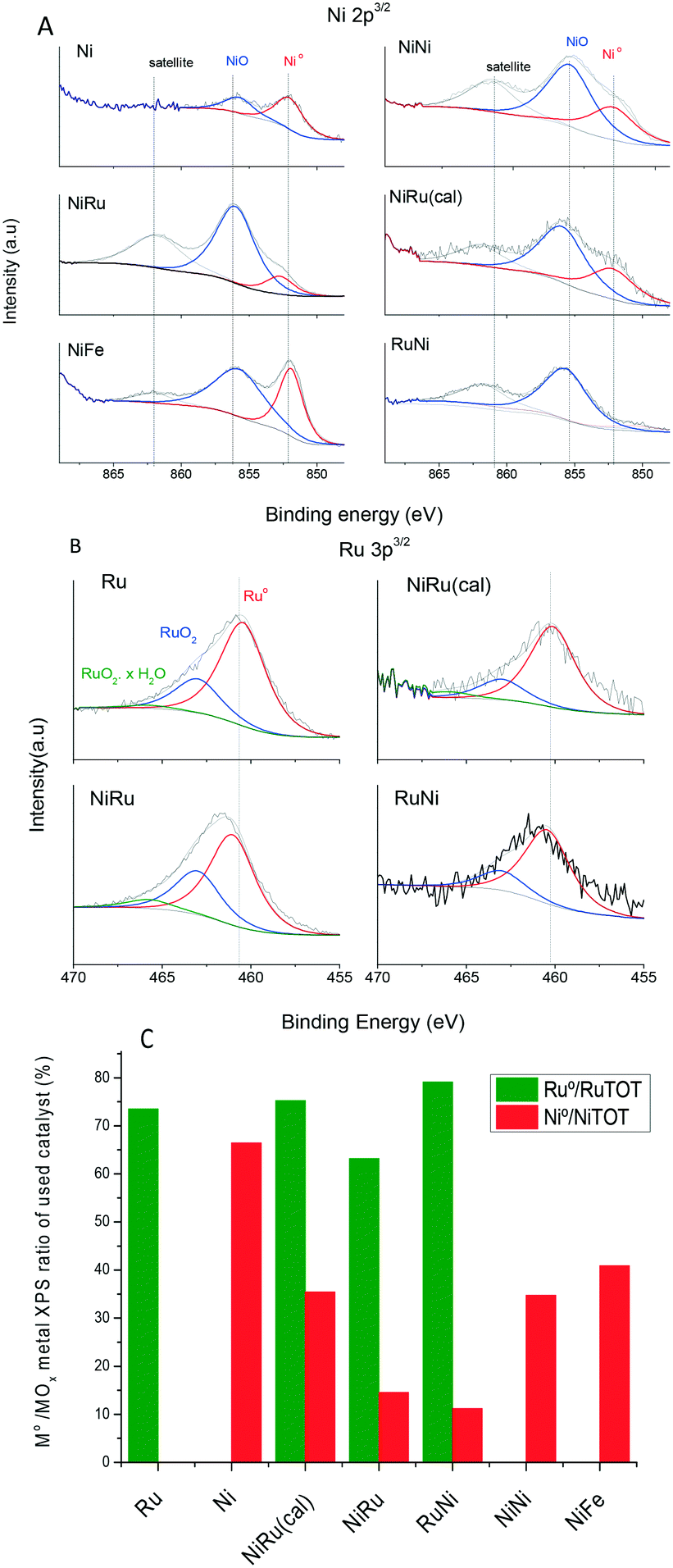 | ||
| Fig. 4 XPS core-level spectra fitting of Ni 3p3/2 (A) and Ru 3p3/2 (B) and quantification of the atomic percentage of reduced metal (C) for catalysts after reaction. | ||
XPS peak fitting for the monometallic and bimetallic catalysts after reaction (Fig. 4A and B, respectively, and Table 3) reveals information about the oxidation state of Ni and Ru in the passivated catalyst. The fitting of XPS peaks corresponding to Ni 2p3/2 core electrons exhibited three peaks, namely, a main peak at around 856.1–856.4 eV, which is attributed to Ni2+ and Ni3+ in nickel oxides such as NiO, Ni2O3 and Ni(OH)2,31 another peak at 852.7 eV attributed to reduced nickel (Ni0) and a peak at 862 eV, which is a satellite.31Table 3 and Fig. 4C compile the quantification of the atomic ratio of Ni0 to total Ni (Ni0 + Ni oxide). A lower Ni0/NiTOT ratio indicates that the catalyst is more active to oxidation by dissociation of atmospheric O2 or CO2 upon air exposure at room temperature after reaction. It is reported that reduced Ni nanoparticles exposed to air undergo surface oxidation, becoming covered with a NiO layer.32,33 This agrees with the TEM observation of NiO lattice fringes for the passivated catalyst (Fig. 3a). The higher oxidation ability suggests a weaker interaction with the support and/or a stronger interaction between the two metals in the bimetallic catalyst. The highest percentage of Ni0 (66%) is found in monolith Ni. For monolith NiNi, the percentage of Ni0 decreases to half that of the Ni catalyst. A higher degree of Ni oxidation to NiO has been reported for increasing Ni loadings,34,35 which may be attributed to a larger metal particle size or weaker metal–support interaction. The catalysts with the lower Ni0/NiTOT ratio are bimetallic Ru and Ni monoliths, suggesting that the close interaction between Ni and Ru favors Ni reoxidation. The interaction between Ni and Ru is apparently weaker in NiRu(cal) as indicated by its larger Ni0/NiTOT ratio. Therefore, the reduction of Ni previously to Ru deposition (as in the NiRu catalyst) is essential to enhance the interaction between the two metals and hence the reoxidation ability. On the other hand, the presence of Fe in the NiFe catalyst hinders the oxidation of Ni, remaining in a high degree of reduction. Accordingly, it is apparent that the extent of Ni reoxidation, afforded by a cooperative effect between Ru and Ni, is a figure of merit for the CO2 methanation reaction. However, this is not the only factor affecting the activity because the high Ni oxidation extent for the RuNi monolith does not supply a high activity.
The oxidation state of Ru post-reaction was also studied by ex situ XPS (Fig. 4B and C and Table 3). Because the binding energy of Ru 3d overlaps with the 1s region of carbon (284.6 eV), the region Ru 3p3/2 (458–468 eV) was chosen for further analysis. The XPS Ru 3p3/2 core level peak was fitted to three peaks at 461.2–461.6 eV, 463.4–463.8 eV, and 465.7–466.1 eV which are attributed to Ru0, RuO2, and hydrated RuO2 or oxyhydroxide, respectively.36,37 The peak ascribed to hydrated RuO2·xH2O at 466 eV is very weak or even absent for all the samples, suggesting that there is a strong metal–support interaction. In fact, Ru on other supports holding a weaker metal–support interaction, such as carbon nanofibers, exhibited a significant contribution of the RuO2·xH2O peak at 466 eV.38 The Ru0/RuTOT atomic ratios (Table 3 and Fig. 4C) are very similar for all the catalysts and larger than the Ni0/NiTOT ratios, indicating than Ru is more inert to oxidation than Ni. The most reactive to oxidation, i.e. lower Ru0/RuTOT atomic ratio, is the NiRu catalyst which confirms again a stronger and mutual Ni–Ru interaction, explaining its highest activity. The highest Ru0/RuTOT ratio value found for RuNi can be attributed to the fact that ruthenium was impregnated first and it established more interaction with the support than the other bimetallic catalysts.
Analysis of basic sites by CO2-TPD
An important parameter for CO2 hydrogenation is the number and strength of basic sites, which chemisorb CO2 in the form of reactive intermediates.39 The alumina support is also reported to play a role in CO2 chemisorption by storing reactive intermediates.22,40,41 CO2 is dissociated on metal and it spills over to the alumina support where it is chemisorbed. The chemisorbed CO2 enhances the catalytic activity since may reverse-spill over to the Ni nanoparticles, where it reacts with chemisorbed H2.42 To analyze and quantify the basic groups chemisorbing CO2 or CO2-decomposition intermediates, a convenient technique is temperature-programmed desorption of pre-adsorbed CO2 (CO2-TPD). To apply this technique, first CO2 was chemisorbed at 300 °C until saturation and later cooled in inert gas. Subsequently, a temperature ramp was applied under flowing Ar while CO2 desorbs from basic sites of increasing strength as the temperature of desorption rises. The CO2-TPD curves attained for the different catalysts are displayed in Fig. 5A and B. According to the temperature of desorption, the basic groups have been classified according to their strength. Likewise, weak basic sites are considered as those releasing physisorbed or weakly chemisorbed CO2 at temperatures below 150 °C, medium basic sites are those desorbing CO2 between 150 and 400 °C and strong basic sites are those releasing CO2 at temperatures above 400 °C. The quantification is shown in Table 4 and Fig. 5C. Among monometallic catalysts, the Ru catalyst has a significantly higher density of medium basic sites than the Ni catalyst, while the latter has a higher density of weak basic sites. The catalysts featuring the highest density of basic sites are Ru and NiRu, the latter exhibiting the highest activity among all the catalysts. Moreover, these catalysts have the lowest number of weak basic sites and the highest number of medium basic sites. To have a more complete overview, Fig. 5D plots together the Ni0/NiTOT ratio and the amount of medium basic sites as a function of the activity. In general, the activity increases roughly as the Ni0/NiTOT ratio decreases and as the density of medium basic sites increases. The highest activity, garnered by the NiRu monolith, correlates to the highest number of basic sites and the lowest Ni0/NiTOT ratio indicative of a strong interaction between Ru and Ni. Accordingly, medium basic sites seem to be a good descriptor of the catalyst activity, but other intrinsic parameters of the metallic phase such as the Ni0/NiTOT or uniform dispersion of the two metal, i.e. Ru (Fig. 2 and 3), also have to be taken into account. The correlation of activity with the medium basic sites makes sense because the highest activity of the catalyst occurs in the temperature range whereby CO2 is desorbed from medium basic sites.43,44 The basic sites that adsorb CO2 either too weakly (desorption temperature <150 °C) or too strongly (desorption temperature >400 °C) are not useful for the reaction, in agreement with the universal principle of volcano dependence of the activity on the adsorption of reactants.11 In fact, Ni and NiFe have some of the highest numbers of weak basic sites and exhibited the lowest activities. According to the literature,45 basic sites of different strengths chemisorb CO2 in different chemical forms such as bicarbonate, bidentate and monodentate carbonates, etc. These species have different chemical stabilities that give rise to the different features in the TPD profile. Monodentate carbonates are reported to adsorb on medium-strength basic sites and this species is reported to be the fastest to undergo hydrogenation.44 The characterization of these sites by DRIFTS FTIR spectroscopy is an interesting point for future research.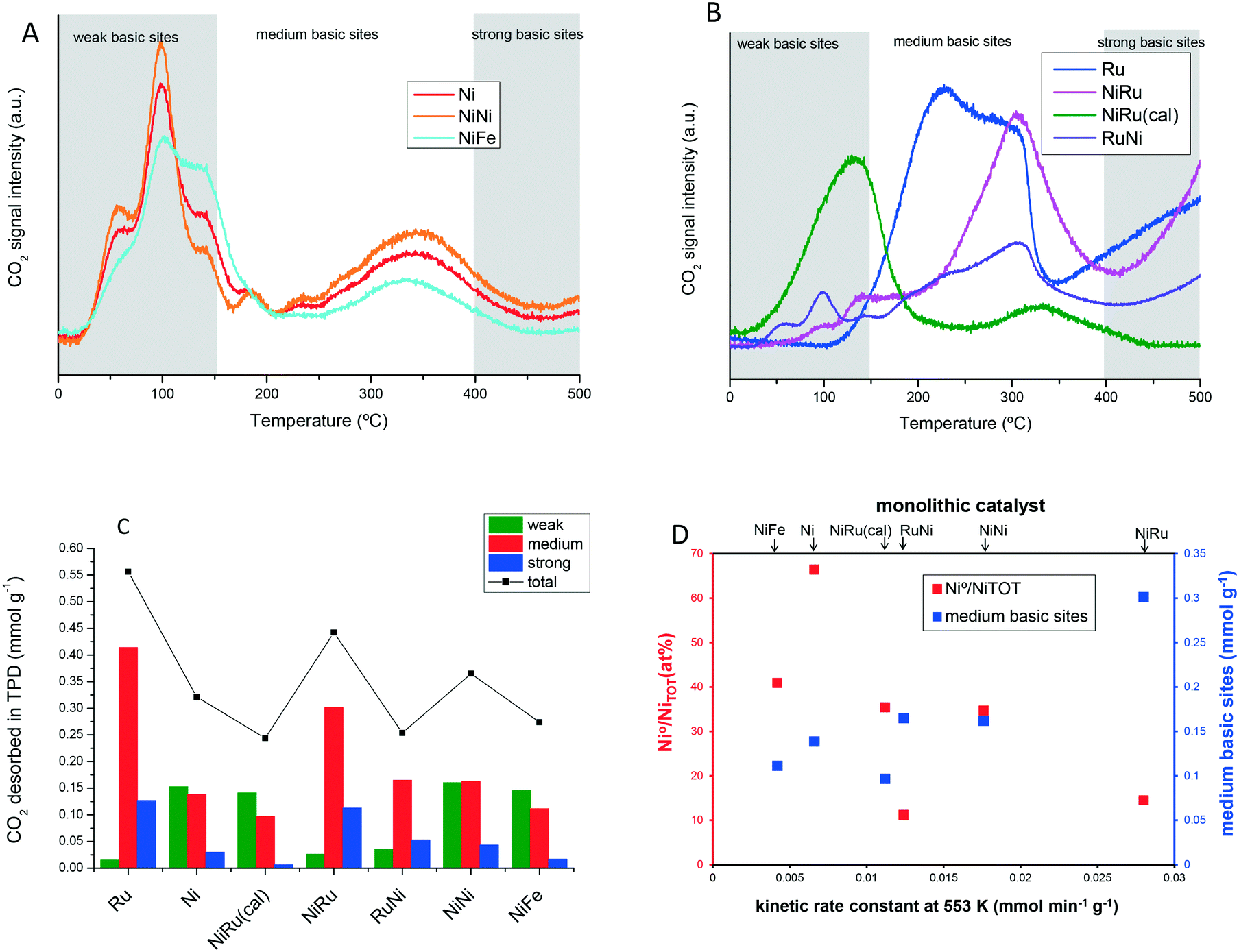 | ||
| Fig. 5 Characterization of basic sites by temperature-programmed desorption of pre-adsorbed CO2 for (A) catalysts based on Ni without Ru and (B) catalysts containing Ru. (C) Quantification of weak, medium and strong basic sites. (D) Plot of percentage of reduced Ni (left Y-axis) and basic sites of medium strength (right Y-axis) as a function of the catalyst kinetic rate constant at 553 K. | ||
| Weak basicity | Medium basicity | Strong basicity | Total basicity | |
|---|---|---|---|---|
| <150 °C | 150–400 °C | 400–500 °C | ||
| mmol g−1 | mmol g−1 | mmol g−1 | mmol g−1 | |
| Ru | 0.015 | 0.414 | 0.127 | 0.556 |
| Ni | 0.153 | 0.138 | 0.030 | 0.321 |
| NiRu(cal) | 0.141 | 0.096 | 0.006 | 0.244 |
| NiRu | 0.026 | 0.301 | 0.113 | 0.442 |
| RuNi | 0.036 | 0.165 | 0.053 | 0.253 |
| NiNi | 0.160 | 0.162 | 0.043 | 0.365 |
| NiFe | 0.146 | 0.111 | 0.016 | 0.274 |
Conclusions
γ-Al2O3-washcoated monoliths as catalyst support meet some special engineering properties such as low pressure drop, thin catalytic layer to favor heat and mass diffusion, ease in handling and robustness, among others. Due to these reasons, a washcoated monolithic reactor is an advantageous contender against a fixed bed reactor for its implementation in a process of power-to-gas using CO2 from postcombustion flue gases, which are emitted at nearly atmospheric pressure. Here, it is demonstrated that the bimetallic NiRu catalyst on γ-Al2O3-washcoated monoliths provides a stable CO2 conversion during 48 h of testing, affording high space velocities (up to 5 × 105 h−1) with a low pressure drop. The monolithic catalyst outperformed the fixed bed operation in terms of both conversion and pressure drop. Among the different tested monometallic catalysts, the Ru catalyst is intrinsically more active than the Ni catalyst. The preparation methodology of the bimetallic (Ni,Ru) catalyst by successive adsorption affected profoundly the methanation performance. The order of adsorption first Ni and second Ru was found to be more favorable than the reverse order. Performing not only a calcination but also a reduction step between Ni and Ru impregnation was also critical for catalyst activity. This optimum preparation methodology led to atomic Ru and small Ni nanoparticles homogenously interspersed on the alumina coating. Some figures of merit contributing to yield a high activity are the number of basic sites of medium strength as determined by CO2-TPD and the degree of Ni oxidation for the passivated catalyst measured by XPS, which denotes a cooperative effect between the two metals (Ni and Ru). The bimetallic monolithic catalyst based on Ni on Al2O3 with a small amount of Ru prepared according to the optimized protocol is an efficient and cost-effective catalyst for the CO2 hydrogenation to CH4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Financial support from Spanish Ministry MINECO (project ENE2016-79282-C5-1-R), Gobierno de Aragón (Grupo Reconocido DGA T03_17R), and associated EU Regional Development Funds are gratefully acknowledged. The DICP team acknowledges the National Natural Science Foundation of China (21872144 and 21972140), Liaoning Revitalization Talents Program (XLYC1907053) and CAS Youth Innovation Promotion Association (2018220) for financial support. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- R. C. Armstrong, C. Wolfram, K. P. de Jong, R. Gross, N. S. Lewis, B. Boardman, A. J. Ragauskas, K. Ehrhardt-Martinez, G. Crabtree and M. V. Ramana, Nat. Energy, 2016, 1, 15020 CrossRef.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- T. Schaaf, J. Grünig, M. R. Schuster, T. Rothenfluh and A. Orth, Energy Sustain Soc., 2014, 4, 2 CrossRef.
- C. Janke, M. S. Duyar, M. Hoskins and R. Farrauto, Appl. Catal., B, 2014, 152–153, 184–191 CrossRef CAS.
- A. Vita, C. Italiano, L. Pino, P. Frontera, M. Ferraro and V. Antonucci, Appl. Catal., B, 2018, 226, 384–395 CrossRef CAS.
- D. Schlereth, P. J. Donaubauer and O. Hinrichsen, Chem. Eng. Technol., 2015, 38, 1845–1852 CrossRef CAS.
- G. Garbarino, D. Bellotti, P. Riani, L. Magistri and G. Busca, Int. J. Hydrogen Energy, 2015, 40, 9171–9182 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 11, 2449–2455 CrossRef.
- D. Li, N. Ichikuni, S. Shimazu and T. Uematsu, Appl. Catal., A, 1999, 180, 227–235 CrossRef CAS.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 RSC.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS.
- J. Sehested, K. Larsen, A. Kustov, A. Frey, T. Johannessen, T. Bligaard, M. Andersson, J. Nørskov and C. Christensen, Top. Catal., 2007, 45, 9–13 CrossRef CAS.
- A. V. Boix, M.-ü. A. Ulla and J. O. Petunchi, J. Catal., 1996, 162, 239–249 CrossRef CAS.
- M. D. Porosoff and J. G. Chen, J. Catal., 2013, 301, 30–37 CrossRef CAS.
- T. Riedel, M. Claeys, H. Schulz, G. Schaub, S.-S. Nam, K.-W. Jun, M.-J. Choi, G. Kishan and K.-W. Lee, Appl. Catal., A, 1999, 186, 201–213 CrossRef CAS.
- A. V. Puga and A. Corma, Top. Catal., 2018, 61, 1810–1819 CrossRef CAS.
- B. Mutz, M. Belimov, W. Wang, P. Sprenger, M.-A. Serrer, D. Wang, P. Pfeifer, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2017, 7, 6802–6814 CrossRef CAS.
- C. Mebrahtu, F. Krebs, S. Perathoner, S. Abate, G. Centi and R. Palkovits, Catal. Sci. Technol., 2018, 8, 1016–1027 RSC.
- E. García-Bordejé, I. Kvande, D. Chen and M. Rønning, Adv. Mater., 2006, 18, 1589–1592 CrossRef.
- G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1984, 87, 352–362 CrossRef CAS.
- O. Levenspiel, Chemical reaction engineering, John Wiley and Sons, New York, 1962, p. 578 Search PubMed.
- X. Wang, Y. Hong, H. Shi and J. Szanyi, J. Catal., 2016, 343, 185–195 CrossRef CAS.
- G. Groppi, E. Tronconi, C. Cortelli and R. Leanza, Ind. Eng. Chem. Res., 2012, 51, 7590–7596 CrossRef CAS.
- L. Baharudin and M. J. Watson, Rev. Chem. Eng., 2018, 34, 481–501 CAS.
- R. Mutschler, E. Moioli, W. Luo, N. Gallandat and A. Züttel, J. Catal., 2018, 366, 139–149 CrossRef CAS.
- L. Falbo, M. Martinelli, C. G. Visconti, L. Lietti, C. Bassano and P. Deiana, Appl. Catal., B, 2018, 225, 354–363 CrossRef CAS.
- C. K. Vance and C. H. Bartholomew, Appl. Catal., 1983, 7, 169–177 CrossRef CAS.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- K. Nguyen, N. D. Hoa, C. M. Hung, D. T. Thanh Le, N. Van Duy and N. Van Hieu, RSC Adv., 2018, 8, 19449–19455 RSC.
- M. K. Singh, A. Agarwal, R. Gopal, R. K. Swarnkar and R. K. Kotnala, J. Mater. Chem., 2011, 21, 11074–11079 RSC.
- B. W. Hoffer, A. Dick van Langeveld, J.-P. Janssens, R. L. C. Bonné, C. M. Lok and J. A. Moulijn, J. Catal., 2000, 192, 432–440 CrossRef CAS.
- C. Plana, S. Armenise, A. Monzón and E. García-Bordejé, J. Catal., 2010, 275, 228–235 CrossRef CAS.
- J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, Inorg. Chem., 2009, 48, 7389–7393 CrossRef CAS PubMed.
- S. Hwang, J. Lee, U. G. Hong, J. G. Seo, J. C. Jung, D. J. Koh, H. Lim, C. Byun and I. K. Song, J. Ind. Eng. Chem., 2011, 17, 154–157 CrossRef CAS.
- J. G. Seo, M. H. Youn, I. Nam, S. Hwang, J. S. Chung and I. K. Song, Catal. Lett., 2009, 130, 410–416 CrossRef CAS.
- R. Chetty, W. Xia, S. Kundu, M. Bron, T. Reinecke, W. Schuhmann and M. Muhler, Langmuir, 2009, 25, 3853–3860 CrossRef CAS PubMed.
- R. K. Raman, A. K. Shukla, A. Gayen, M. S. Hegde, K. R. Priolkar, P. R. Sarode and S. Emura, J. Power Sources, 2006, 157, 45–55 CrossRef CAS.
- S. Armenise, L. Roldán, Y. Marco, A. Monzón and E. García-Bordejé, J. Phys. Chem. C, 2012, 116, 26385–26395 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 9, 2094–2100 CrossRef.
- L. Roldán, Y. Marco and E. García-Bordejé, ChemCatChem, 2015, 7, 1347–1356 CrossRef.
- L. Roldán, Y. Marco and E. García-Bordejé, ChemSusChem, 2017, 10, 1139–1144 CrossRef PubMed.
- A. E. Aksoylu, A. N. Akin, Z. İ. Önsan and D. L. Trimm, Appl. Catal., A, 1996, 145, 185–193 CrossRef CAS.
- T. Burger, F. Koschany, A. Wenng, O. Thomys, K. Köhler and O. Hinrichsen, Catal. Sci. Technol., 2018, 8, 5920–5932 RSC.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS.
- X. Wang, M. Shen, L. Song, Y. Su and J. Wang, Phys. Chem. Chem. Phys., 2011, 13, 15589–15596 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy00639d |
| This journal is © The Royal Society of Chemistry 2020 |