
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEthanol-to-butadiene: the reaction and its catalysts
Guillaume
Pomalaza
,
Paola
Arango Ponton
,
Mickaël
Capron
and
Franck
Dumeignil
 *
*
Univ. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: franck.dumeignil@univ-lille.fr
First published on 2nd June 2020
Abstract
Butadiene is a crucial chemical, notably for the manufacturing of car tires. The ethanol-to-butadiene reaction has emerged as a promising solution to both environmental concerns and the disruption caused by the emergence of shale gas associated with butadiene production. As a result, it has gathered significant scientific interest and private companies are investigating its application. Although there has been much insight into the reaction and its catalysts in recent years, several issues need to be resolved before becoming financially viable. This review aims to provide up-to-date insight into the ethanol-to-butadiene reaction, including topics that are seldom addressed. It can serve as a starting point for any new research projects on the subject of ethanol conversion to butadiene.
Introduction
Biorefineries convert renewable resources to chemicals and fuels.1 The carbon content of biomass makes it an interesting substitute for fossil-based feedstocks on which the petrochemical industry currently relies.2 A more sustainable economy could potentially be achieved by replacing petrorefineries with biorefineries. However, the low return on investment of bio-based processes hinders their development. The integration of a biorefinery with the production of a single platform molecule from which many value-added chemicals can be derived has been proposed as a strategy to overcome this obstacle.3 The “drop-in” concept is another tactic for improving the financial viability of biomass valorization;4 platform molecules are used to produce existing fossil-based intermediates, thereby benefiting from preexisting existing value chains and infrastructure. Note that, in that case, direct competition with petro-sourced molecules becomes in turn an issue due to the higher price of the raw material.For instance, ethylene and propylene—olefins highly important to the manufacturing of plastics—can be obtained by dehydration of ethanol on solid acid catalysts, a process often compared to the methanol-to-olefin reaction.5 The topic of this review concerns another reaction that generates olefins from ethanol: the catalytic conversion of ethanol to butadiene (Scheme 1). The latter is also a crucial commodity, as it is the main feedstock for the production of synthetic rubber, a key product in the automotive industry.6
 | ||
| Scheme 1 Global equation of conversion of ethanol to butadiene. | ||
Two ethanol-to-butadiene processes following the same mechanism exist: the Lebedev (or one-step) process consists of direct conversion of gaseous ethanol to butadiene over multifunctional catalysts;7 the Ostromislensky (or two-step) process divides the conversion in two reactors—a first one for partial dehydrogenation of ethanol to acetaldehyde and a subsequent one for the conversion of the as-obtained ethanol–acetaldehyde mixtures to butadiene.8 The aim of this first step is to feed the second reactor with ethanol–acetaldehyde mixtures. In practice, most authors have preferred substituting it by simply co-feeding acetaldehyde with ethanol in a single reactor dedicated to butadiene formation. After all, the relative simplicity of optimizing an ethanol dehydrogenation process to feed a subsequent reactor with the desired ethanol–acetaldehyde ratio9,10 gives merit to this shortcut, despite the fact the hydrogen produced may participate in the reaction. Consequently, references to the two-step or the Ostromislensky process are to be understood as the conversion of acetaldehyde–ethanol mixtures as well as experiments with two reactors.
Interestingly, the Lebedev and Ostromislensky processes were important sources of butadiene from the 1930s to 1970s, before being supplanted by petroleum-based routes.11–13 However, environmental and economic concerns have spurred a recent interest in this reaction, both in industry and academia.14 Much effort has been dedicated to improving catalytic performances with the ultimate aim of turning the Lebedev and Ostromislensky processes into technologies capable of competing financially with current butadiene production methods.11,14 However, achieving this goal has been hindered by the complexity of the reaction, which is only partly understood, and the collectively uncoordinated approach used to study the activity of catalysts. A myriad of catalytic systems have been studied under various conditions with different methods in either of the two processes. Consequently, a wide range of sometimes contradictory observations have been made at the expense of a comprehensive understanding of the reaction, as well as the relationship between the properties of catalysts and their activity. Without this knowledge, the rational design of better performing catalysts is hindered.
This bibliographic part aims at detailing the recent advances in the understanding of the reaction and in the design of improved catalytic systems. A brief summary of the rich historical and economic context of the ethanol-to-butadiene reaction will also be provided. In addition, we hope to build on previous reviews11–16 by discussing topics not yet reviewed, such as the deactivation mechanism.
Historical & economic context
Brief history
Butadiene was first isolated from the pyrolysis of acetylene and ethylene by Caventou in 1863. Its structure was elucidated in 1895 by Ciamician and Magnaghi.17 The interest in butadiene began in 1910 when Lebedev found that it could be converted to rubber-like polymers one year after the discovery of isoprene.17–20During the Second Industrial Revolution (1870–1914), the vulcanization of natural rubber was the sole means of producing the elastomers used to manufacture the tires demanded by the ongoing industrialization.19–22 As a result, synthetic rubber greatly interested countries that lacked colonial empires spanning into tropical zones, the only geographic regions where the rubber plant Hevea Brasiliensis actually grows.
Although Armstrong and Miller detected butadiene among the products of petroleum cracking in 1886,17 ethanol (ethanol) remained the main feedstock of butadiene production for the early decades of polybutadiene rubber manufacturing. In 1901, Nef first reported the conversion of gaseous ethanol to butadiene using zinc and pumice.16,23 Two years later, Ipatiev detected traces of butadiene after passing ethanol on powdered aluminum at high temperatures.16 In 1915, Ostromislensky reported obtaining practical yields of butadiene by co-feeding ethanol with acetaldehyde on alumina–clay mixtures.24 Finally, Lebedev reached appreciable butadiene production from pure ethanol on ZnO–Al2O3 catalysts, first reported in a 1928 patent.16 Both catalytic reactions became large-scale industrial processes. In the United States, the Union Carbide and Carbon Chemical Corporation commercialized a process akin to that developed by Ostromislensky. In the Soviet Union, Lebedev's work was industrialized so that by the eve of the Second World War, 62% of the 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of synthetic rubber were derived from ethanol.16
000 tons of synthetic rubber were derived from ethanol.16
The First World War demonstrated the importance of rubber to modern warfare when shortages forced the Central Powers to use the inferior rubber-like material derived from thermal polymerization of isoprene for the production of tires.20 The Interwar period saw not only a further mechanization of war, but also of logistics: armies now relied on fleets of lorries to supply fuel and other goods. The Second World War became a conflict in which natural and industrial resources were crucial. When the dazzling expansion of the Japanese Empire into Southeast Asia drew the United States into the War, it also deprived the allies of their rubber plantations in British Malaya and Dutch Indonesia.25 Consequently, a special American war committee concluded that—of all strategic goods crucial to Ally victory—rubber shortages posed the most immediate threat, more than steel, aluminum or gasoline. In response, the U.S. invested heavily in emergency synthetic rubber research.7,20,26–30
In 1939, the American government launched a synthetic rubber program in partnership with industry and academe with the aim of expanding the U.S. synthetic rubber industry.20,30,31 It also sponsored research on the catalytic conversion of ethanol to butadiene to meet the demand for the monomer.7,26 In this context, scientists at the Carbide and Carbon Chemicals Corporation and the Mellon Institute conducted seminal works on the reaction, including high-throughput catalyst screening,7,32 process optimization26,28 and mechanistic studies.33,34 Soon, the U.S. government was able to commission three plants with a combined yearly production capacity of 220000 tons of butadiene from ethanol.16 By the end of the War, the Allies dwarfed the Axis in every strategic resource reserve, including butadiene and synthetic rubber.
With the advent of the inexpensive butadiene from petroleum cracking following the Second World War, the ethanol route fell into obsolescence. Industrially, very few plants remain in operation and only in countries where a specific economic situation makes them profitable.11
Scientific interest declined simultaneously: only a few papers on the subject were published from the 1960s to the 2000s.14 However, recent years have seen a renewed interest in the ethanol-to-butadiene reaction due to economic and environmental factors, which Weckhuysen et al. described as a renaissance of the subject. Although a subjective choice, we find that the 2011 publication by Jones et al.35 is an ideal starting point for this renaissance, which has been marked by new insight into the reaction mechanism and more productive catalysts, as discussed below. Key events in the discovery of the ethanol-to-butadiene reaction are depicted in Fig. 1.
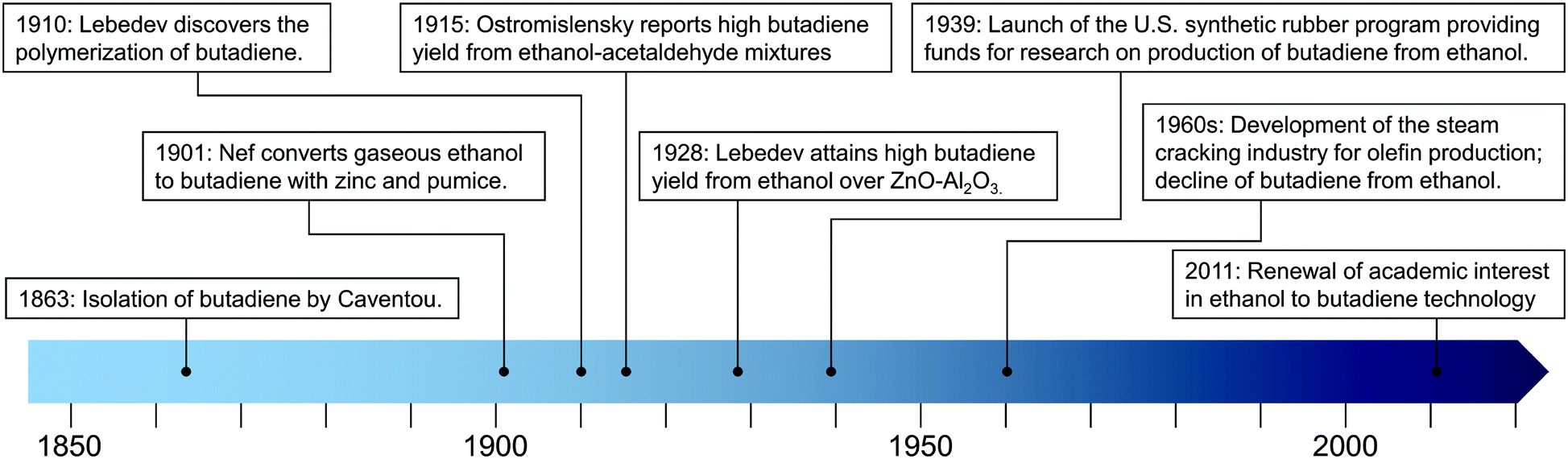 | ||
| Fig. 1 Timeline of key events in the development of the butadiene-from-ethanol process. | ||
Butadiene today
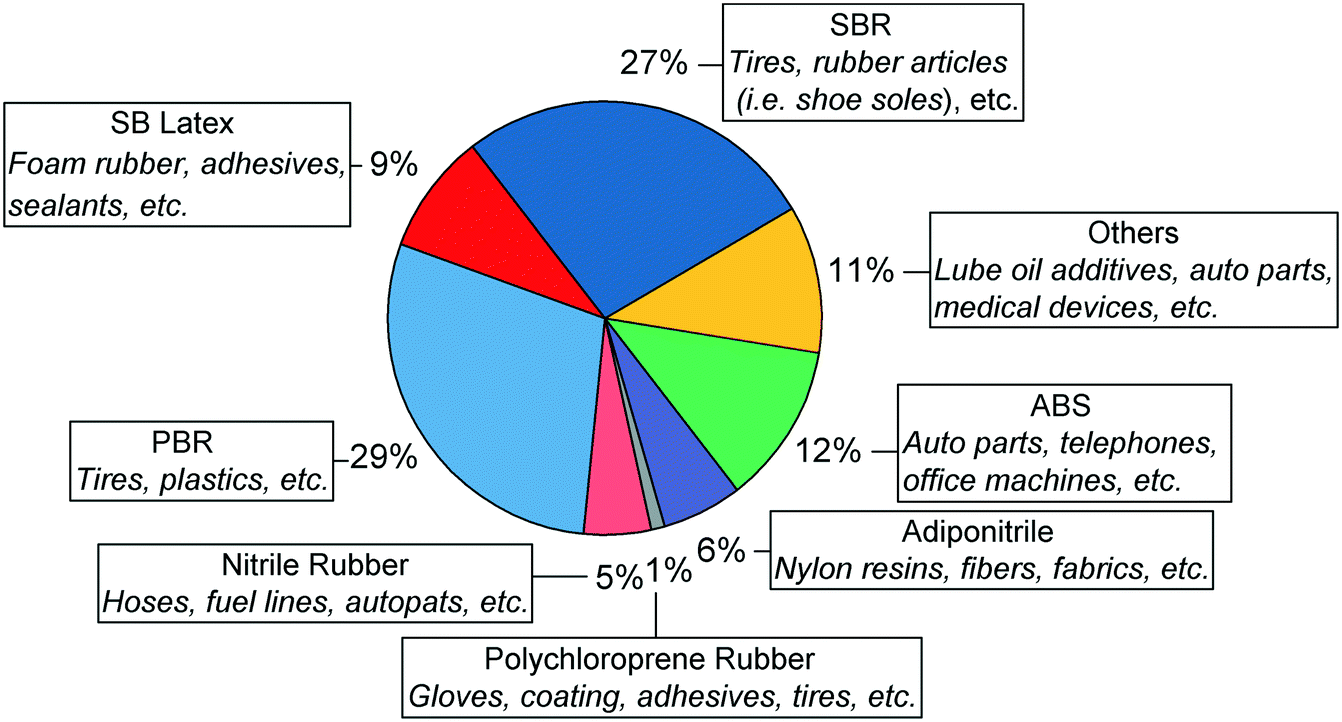 | ||
| Fig. 2 Butadiene demand and its end uses in 2015.36,40 | ||
Valued at around 30–40 billion USD, the global butadiene market is predicted to gradually increase in the coming years. While stagnating in the West, Asian countries are expected drive this growth, owing to the rising appetite for automobiles of their burgeoning middle classes. Dargay et al. forecasted that personal car ownership would reach 2 billion units in 2030, twice the ownership of 2010.37 China alone could reach 330 million cars and match the projected ownership of the US by that date.38 A paradigm shift in tire manufacturing technology withstanding, butadiene production must keep pace with the increasing global vehicle stock, an issue with which current processes may be ill-equipped for.
Current butadiene production
Nowadays, butadiene is primarily derived from ethylene production via steam cracking of naphtha, a C6–11 petroleum distillate. The process is a highly endothermic pyrolysis conducted in the presence of steam.17,18 A vaporized hydrocarbon feed is heated to more than 1073 K at low pressure in a pyrolysis chamber. Under these conditions, carbon–carbon and carbon–hydrogen bonds can break down, resulting in a mixture of olefins, aromatics, tar and gases. Cracking lasts less than one second to prevent product degradation through secondary reactions. The addition of steam reduces the partial pressure of hydrocarbons, inhibiting the problematic formation of coke.After cooling, these products are fractionated into different cuts: C1, C2, C3, C4, etc. Due to the shared boiling range, simple distillation cannot extract butadiene from the C4 fraction. Refineries generally address this issue by using extractive distillation, which operates using selective organic solvents to decrease the volatility of target compounds, thereby enabling their separation by distillation. The purity of butadiene is important because polymerization catalysts cannot operate properly in the presence of impurities.17,39 Modern plants use N-methylpyrrolidone or dimethylformamide, and reach butadiene recovery rates of 99–100%. Other C4 species include butane, n-butenes and harmful acetylenes. The C4 products' distribution depends on the cracking conditions and the nature of the raw material. Generally, heavier hydrocarbons such as naphtha generate more butadiene. However, the main economic incentive of steam cracking remains the production of ethylene which is the most important olefin in the world. Consequently, crackers may seek to maximize ethylene output at the expense of butadiene and other byproducts by using lighter raw materials. Fig. 3 illustrates the influence of the feedstock on the production ratio between butadiene and ethylene: lighter hydrocarbons generate significantly less butadiene for the same amount of ethylene.17
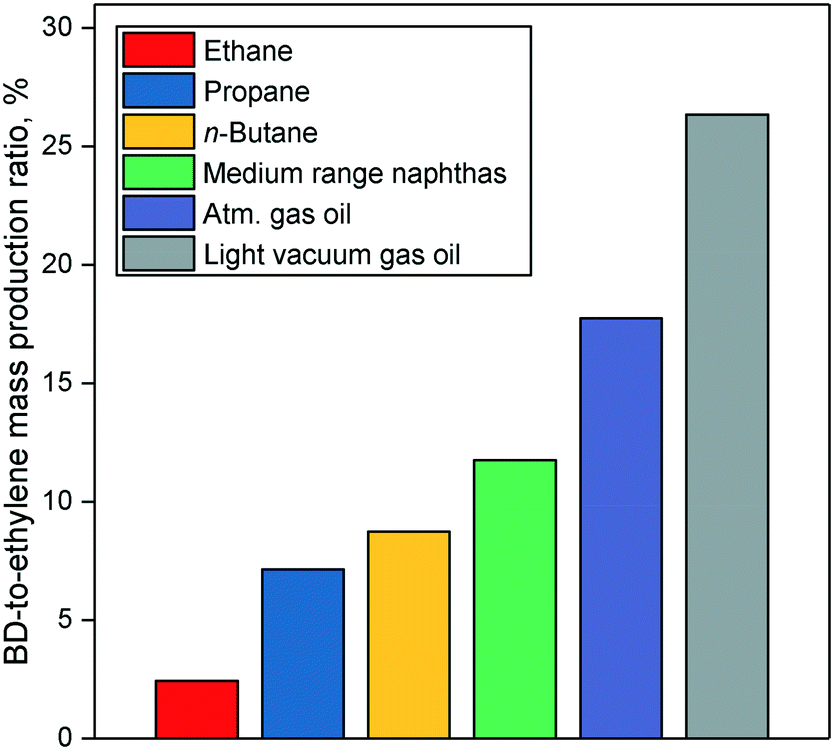 | ||
| Fig. 3 Projected figures for the yield of ethylene and butadiene from North American steam crackers relative to 2000 production levels.44 | ||
Due to its ties with ethylene production, recent trends in the petrochemical industry threaten the supply of butadiene. Ethylene-producing steam cracking plants are moving toward lighter feedstocks, yielding less butadiene for the same amount of ethylene.40–43 Most notably, the emergence of inexpensive butane from shale gas as a cracking feedstock has led to a decrease in the production of butadiene in North America.42,43 As Fig. 4 illustrates, North American steam crackers are projected to generate less butadiene.44 European crackers are also switching from naphtha to liquefied petroleum gas, further reducing global butadiene production.40 China—the world's primary supplier of naphtha-derived butadiene—is commercializing the coal-to-olefin process: by converting coal to methanol via syngas, this technology can synthesize ethylene through the well-established methanol-to-olefin reaction.40,41,45 However, these plants do not generate butadiene. If this these trends continue, butadiene shortages and the ensuing price increase can be expected. Filling the gap caused by the issues resulting from production may require new technologies capable of producing butadiene on purpose.42
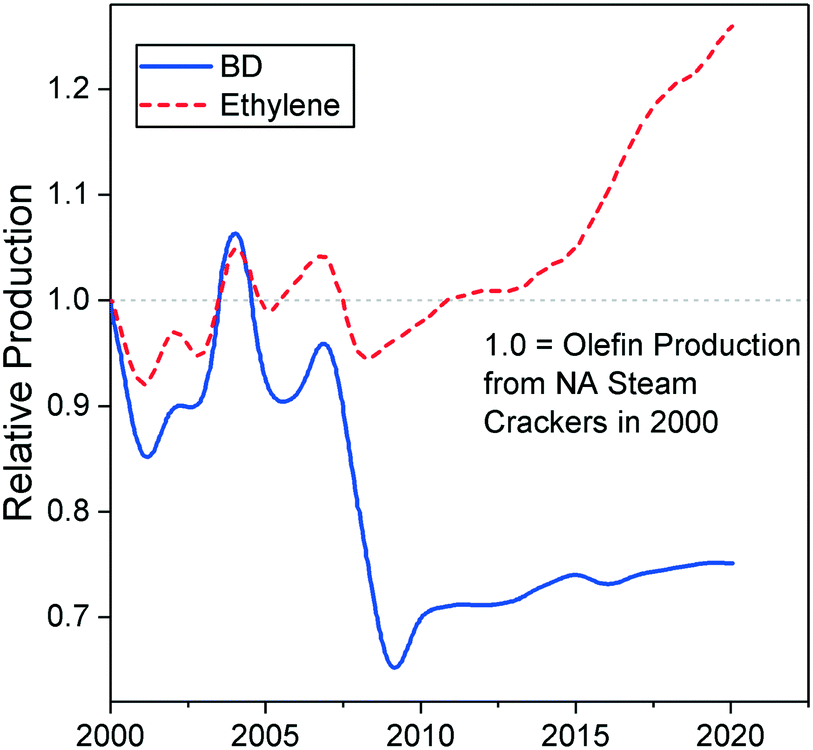 | ||
| Fig. 4 Mass production ratio between butadiene and ethylene in steam crackers according to the feed used.17 | ||
In addition, current butadiene production is unsustainable. Due to the highly endothermic pyrolysis step, steam cracking is the most energy-consuming process used by the chemical industry.46 Because of its prevalence, the process generates yearly 180–200 million tons of CO2, which is the most significant long-lived greenhouse gas (GHG).46–48 Therefore, the naphtha cracking route to butadiene contributes significantly to climate change. With the adoption of the historic global climate accord, nations have agreed to limit global warming to below 2 °C by reducing their GHG emissions.49 Achieving this goal requires emissions to be cut by 40–70% by 2050. However, lowering the CO2 production of an industrial process is highly complex.48 Optimizing naphtha cracking using state-of-the-art technologies would reduce CO2 emissions by 30% at best, according to the open literature.46,47 Ultimately, considering that petroleum reserves are finite, it is unlikely that current butadiene production methods will ever be environmentally sustainable.
In summary, butadiene is predominantly obtained by the steam cracking of naphtha, which is primarily used to produce ethylene. However, the emergence of lighter alternative feedstocks threatens the supply of butadiene by favoring ethylene yield at expense of the former. As a result, meeting the butadiene demand, which is expected to grow with the increase of car ownership, may not be achieved with current technologies. Furthermore, steam cracking emits large quantities of CO2, which is harmful to the environment. This situation is a good opportunity to develop and implement a sustainable and on-purpose process to produce butadiene.11,50–52
Alternative butadiene production method
Oxidative dehydrogenation of butene, i.e., the oxo-D-type process, offers an additional route to butadiene.17,56 The oxidation step significantly favors the reaction, resulting in butadiene yields comparatively higher than those with the Catadiene process.17 Furthermore, the formation of steam reduces coking. However, butanes are detrimental to this process, instead forming undesirable by-products.17 Consequently, it cannot directly operate from a C4 fraction without prior purification. In any case, butanes are predominantly obtained from the steam cracking of naphtha,53 defeating the purpose of detaching butadiene production from ethylene. Integrating oxidative dehydrogenation with a Catofin process producing propylene and butenes from LPG may be a practical way to obtain butadiene from hydrocarbon feedstocks other than naphtha. Nevertheless, their reliance on finite fossil resources does not address the sustainability issues of butadiene production.
Sustainable butadiene
In recent years, new technologies have been developed to produce butadiene from renewable resources. In 2015, Genomatica, in collaboration with Braskem, announced the lab scale conversion of sugars to butadiene with bioengineered microorganisms.57 More recently, Genomatica with Versalis favored a different approach to renewable butadiene: the conversion of butanediol (BDO), further identifying butanediol as the best suited precursor for this process.58 Butanediol isomers can be produced from sugar fermentation,59–62 from industrial waste or from biomass-sourced syngas.63,64 These can be subsequently converted to butadiene by the double dehydrogenation of the alcohol groups. In their review, Sato et al. highlighted how the butadiene yield depends on the type of BDO and catalyst used, as well as the reaction conditions.65 Generally, BDO dehydrogenation reactions generate significant amounts of the corresponding butenols, formed by partial dehydrogenation of the substrates, but their recovery and further dehydration can ensure a high butadiene yield.65BDO is well suited to be a precursor of butadiene: high selectivity towards butadiene are achieved at temperatures below 573 K using acid metal oxides, i.e., Al2O3, ZrO2, TiO2, and Al2O3–SiO2, and liquid 3-buten-1-ol (3B1OL) is easily recycled so that 90% yield is possible.65,66 Our group reported how a 60% butadiene yield could be obtained at 473 K using Al-SBA-15 catalysts, demonstrating the ease with which BDO undergoes conversion.67
1,4-BDO has the advantages of being a crucial commodity traded in millions of tons via the Reppe process62,68,69 and of notably being an intermediate of the obsolete acetylene-to-butadiene process.17,18,68–70 Double dehydration of 1,4-BDO generates butadiene, but high selectivity towards tetrahydrofuran (THF) when using acidic and amphoteric oxide catalysts at high temperatures reduces the overall yield.11,71,72 Further research is needed to improve butadiene selectivity.65
In addition to butadiene, 2,3-BDO dehydration produces large amounts of methyl ethyl ketone (MEK) via a Pinacol rearrangement.73–80 Although a potentially desirable product, MEK cannot be further converted to butadiene. As a result, butadiene production maximization necessitates specific catalytic systems and appropriate reaction conditions, namely high temperature. For instance, using a Sc2O3/Al2O3 double-bed, Sato et al. achieved 94% selectivity towards butadiene.65 However, it was the only rare-earth oxide screened out of 17 not to predominantly yield MEK, highlighting the unfavourability of butadiene formation. It should be noted that MEK synthesis uses a new process in which 2,3-BDO is esterified with a carboxylic acid, where butadiene is obtained from the steam pyrolysis of the resulting ester.81
Many technologies are being developed for producing sustainable butadiene. Most advances have focused on the catalytic conversion of butanediols, obtainable from the fermentation of bio-derived feedstocks (Scheme 2). Selectivity issues have hindered butadiene productivity, but these are being addressed by recent breakthroughs. However, butanediol production levels remain too low to significantly replace current production methods.50
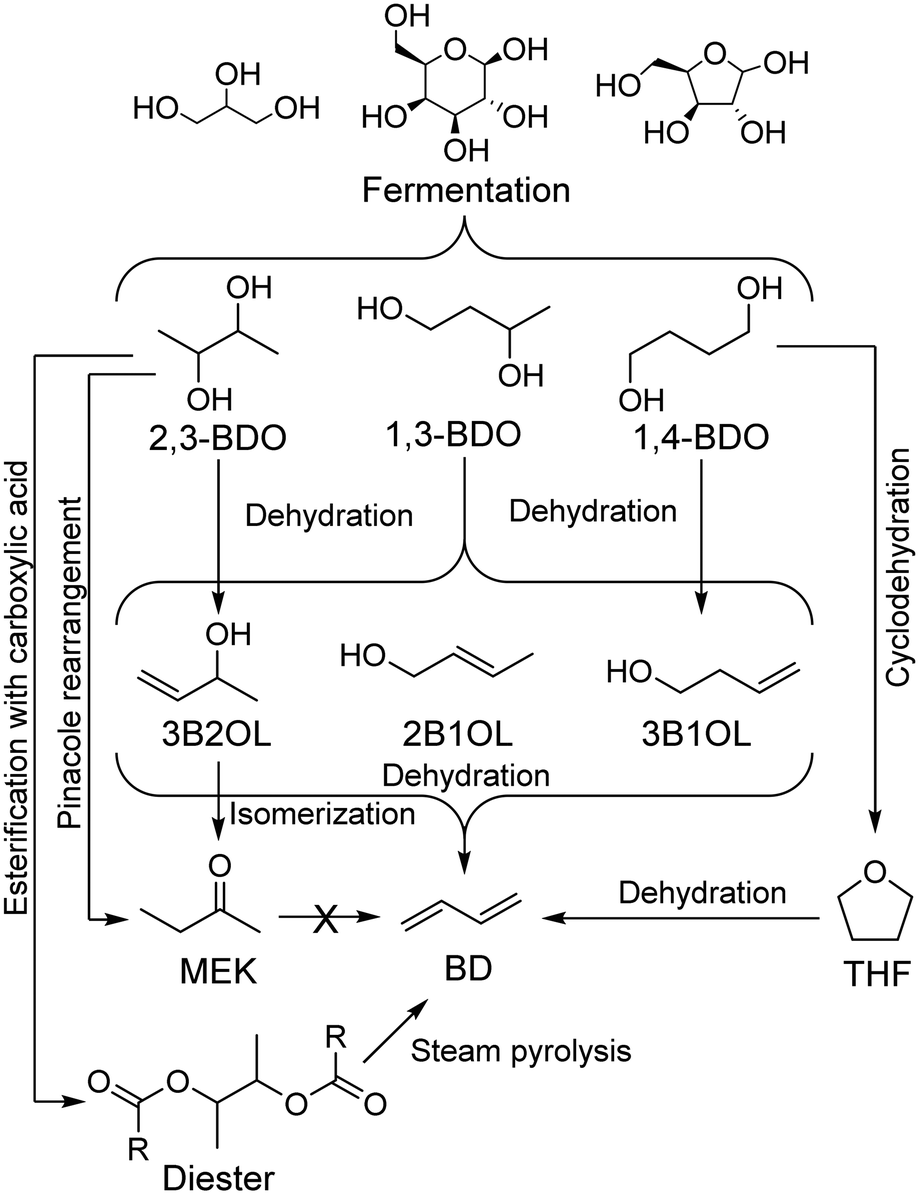 | ||
| Scheme 2 Main pathways from sugars to butadiene via butanediols.65 | ||
Ethanol-sourced butadiene
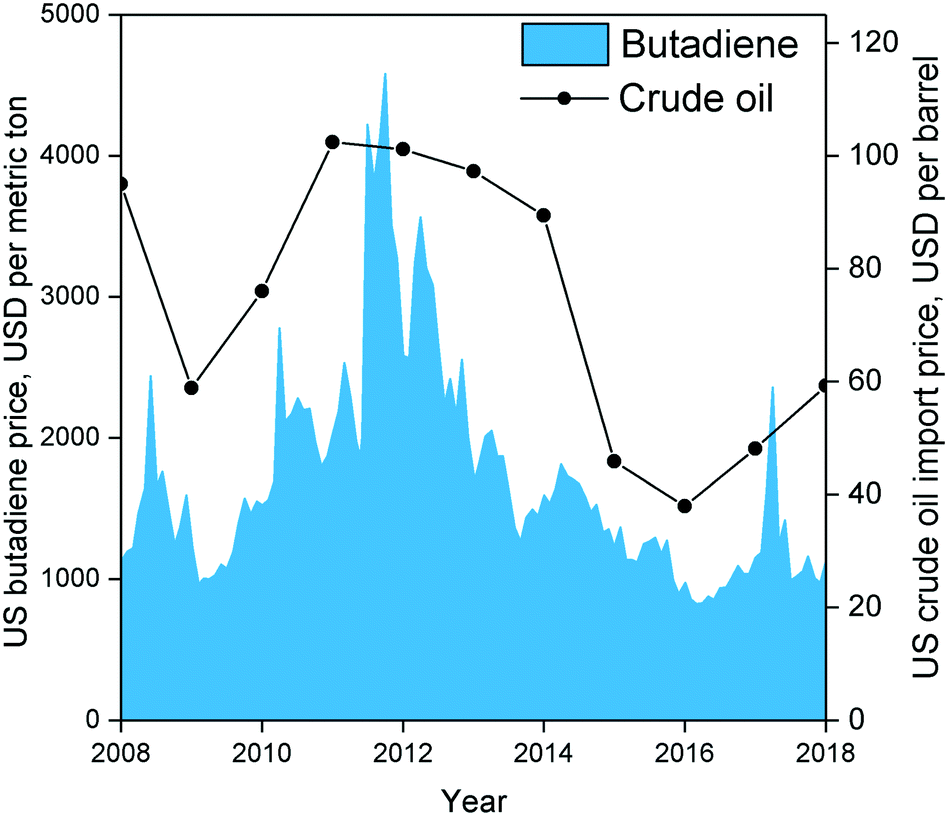
 | ||
| Fig. 5 Historical American butadiene price in USD per metric ton from Intratec Solutions LLC (www.intratec.us)89versus historical US crude oil import prices.90 | ||
More specific to the practical production of butadiene from ethanol is the question of water content. Biomass-derived ethanol must be purified with water before being used in combustion engines,85,86 which increases its cost, notably because of the ethanol–water azeotrope (95.5 wt% ethanol). However, ethanol-to-butadiene processes can operate in the presence of water, albeit not without changes in catalytic performances compared to anhydrous ethanol.4 Nevertheless, under optimized conditions, it would be beneficial both financially and environmentally to use water-containing ethanol as a feedstock for producing butadiene as it would it would not require more expensive drying steps.
Techno-economic analyses of Brazilian and American scenarios have provided further insight into the profitability of ethanol-to-butadiene processes.87,88 Using 2012 prices, Burla et al.87 determined a US-based plant employing the two-step process to be a highly promising venture. However, butadiene prices, which are partly influenced by that of oil, peaked in 2012 and have fluctuated significantly in the last 10 years (Fig. 6).89,90 Considering the average price of butadiene between 2008 and 2018, the scenario described by Burla et al. would not be profitable. Incidentally, using a 5 year price average (2007–2011), Farzad et al.88 found a Brazil-located plant employing the two-step process to have a 0% chance at profitability. Nevertheless, they found that integrating the butadiene production plant into the ethanol production process increased the profitability by reducing infrastructure. It should be noted that both simulations were conducted using reaction parameters dating back to the 1950s; recent catalytic systems boast superior performances, albeit at the laboratory scale. To the best of our knowledge, no techno-economic analysis of the Lebedev process has been performed, ostensibly due to the lack of kinetic data. Incidentally, Michelin, in partnership with French energy enterprises, announced in 2013 the launch of a project for the development of a bio-sourced Lebedev process for the purpose of developing renewable synthetic rubber.91 Accurately assessing the economic viability of ethanol-based butadiene would necessitate modelling the reaction kinetics of modern catalytic systems. Whereas the prices of ethanol and butadiene are subject to the complexity of our modern economy, scholars have pointed out that improving the catalytic performances of the process would enhance the viability of the ethanol-to-butadiene process.11,92
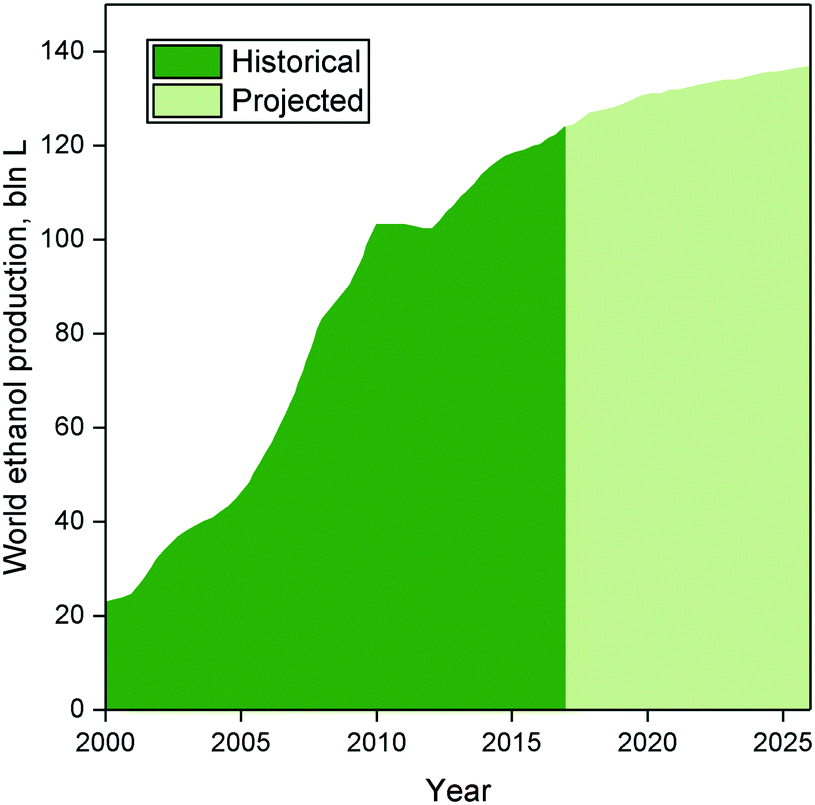
 | ||
| Fig. 6 Historical and projected ethanol production.83 | ||
Farzad et al.88 simulated a process integrating a Brazilian sugar cane mill with on-site ethanol production and conversion to butadiene via the two-step process. The reduction of transport needs and the omission of ethanol purification beyond distillation prior to the ethanol-to-butadiene process both reduced the energy needs. The combustion of butadiene and ethanol production residues also enabled energy self-sufficiency, further reducing GHG emissions. Overall, the integrated process presented significantly less harm to the environment, including human toxicity, ecotoxicity and climate change, than naphtha-derived butadiene. By situating a simulated ethanol-to-butadiene plant in existing American petrochemical facilities, Shylesh et al. argued that CO2 sequestration can be increased through hydrogen exports, thereby offsetting the use of natural gas in petroleum refineries.93 However, this strategy could not outweigh the significant amounts of GHG emitted by corn grain-based ethanol; only by using Brazilian ethanol, imported through the port of Houston, and corn stub-derived ethanol was the process found to combat climate change. Under the right circumstances, ethanol-to-butadiene processes have great potential for producing sustainable butadiene. While the geographic location largely influences the amount of harm to the environment, several strategies can be used to further improve the sustainability of the process.
The ethanol-to-butadiene reaction
Toussaint–Kagan pathway
Fully elucidating the mechanism of a reaction not only enables rational design of catalysts with tailored performances,94 but also assists in kinetic modelling, which is important for process design.95 Although alternatives have recently been proposed,50,96–98 the Lebedev and Ostromislensky processes are now generally recognized to follow the same pathway (Scheme 3).99–101 It consists of five steps: dehydrogenation of ethanol to acetaldehyde (Scheme 3(a)); aldol condensation or aldolization of acetaldehyde to 3-hydroxybutanal, commonly known as acetaldol (Scheme 3(b)); dehydration of acetaldol to crotonaldehyde (Scheme 3(c)); reduction of crotonaldehyde to crotyl alcohol (Scheme 3(d)); dehydration of crotyl alcohol to butadiene (Scheme 3(e)).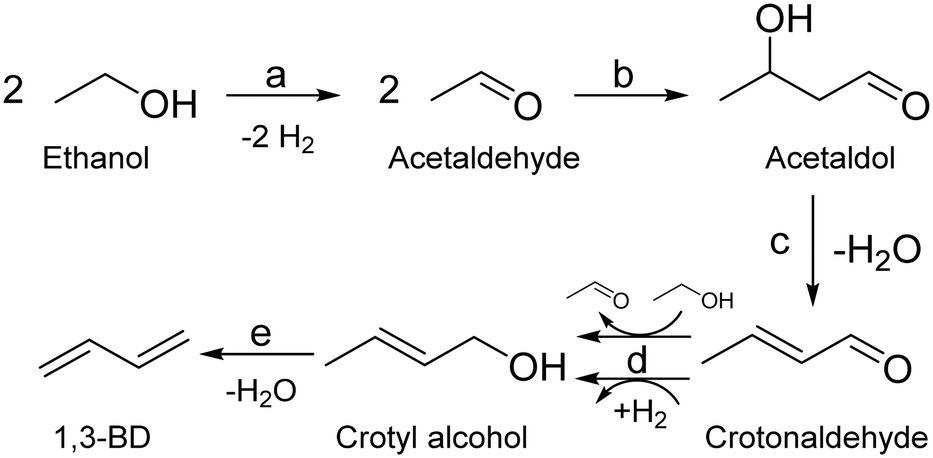 | ||
| Scheme 3 The generally accepted pathway from ethanol to butadiene in the Lebedev and Ostromislensky processes. The five steps are: (a) dehydrogenation of ethanol to acetaldehyde; (b) aldol condensation or aldolization of acetaldehyde to 3-hydroxybutanal, commonly known as acetaldol; (c) dehydration of acetaldol to crotonaldehyde; (d) reduction of crotonaldehyde to crotyl alcohol; (e) dehydration of crotyl alcohol to butadiene. | ||
However, the consensus remains limited to the overall pathway: the molecular-level mechanism leading to butadiene remains under debate. Part of the issue can be attributed to the relative difficulty of observing intermediate species at the surface of heterogeneous catalysts.102 Our current understanding of the reaction mechanism comes predominantly from fragmentary evidence gathered over the span of several decades by different research teams. Consequently, attempts at elucidating the mechanism have relied on various techniques, which have led to some discrepancies. This situation is further aggravated using diverse catalytic systems and reactions, possibly leading to different yet valid mechanisms. In our opinion, not enough evidence has been gathered to conclude whether the generally accepted pathway follows a single mechanism or is dependent on the experimental conditions. Often denominated as the Toussaint12,28,33,100 or Kagan13,103–105 pathway or mechanism, we will refer to the pathway illustrated in Scheme 3 as the Toussaint–Kagan mechanism to highlight the pioneering work of both teams.
Ethanol dehydrogenation
The Lebedev process begins with the non-oxidative dehydrogenation of ethanol, forming acetaldehyde and hydrogen (Scheme 3(a)). In an Ostromislensky-type process, it takes place in a separate reactor so that the butadiene-producing reactor operates with an ethanol–acetaldehyde feed. This step is important because it generates acetaldehyde which participates in the formation of C4 precursors leading to butadiene.Ostromislensky was the first to propose acetaldehyde as a key intermediate based on the high yields obtained with his two-step process.24 By applying Wojciechowski's criteria,106,107 which involves plotting the product yield as a function of the reactant conversion to deduce the kinetic sequence of reaction products, Ivanova et al.108 and Cabello González et al.109 determined that acetaldehyde was a primary product of ethanol conversion which was involved in subsequent reaction steps leading to butadiene (Fig. 7). Isotopic tracer experiments co-feeding ethanol with deuterated and 14C-labelled acetaldehyde demonstrated the reversibility of ethanol dehydrogenation110 and confirmed that acetaldehyde participated in the formation of butadiene.35,104
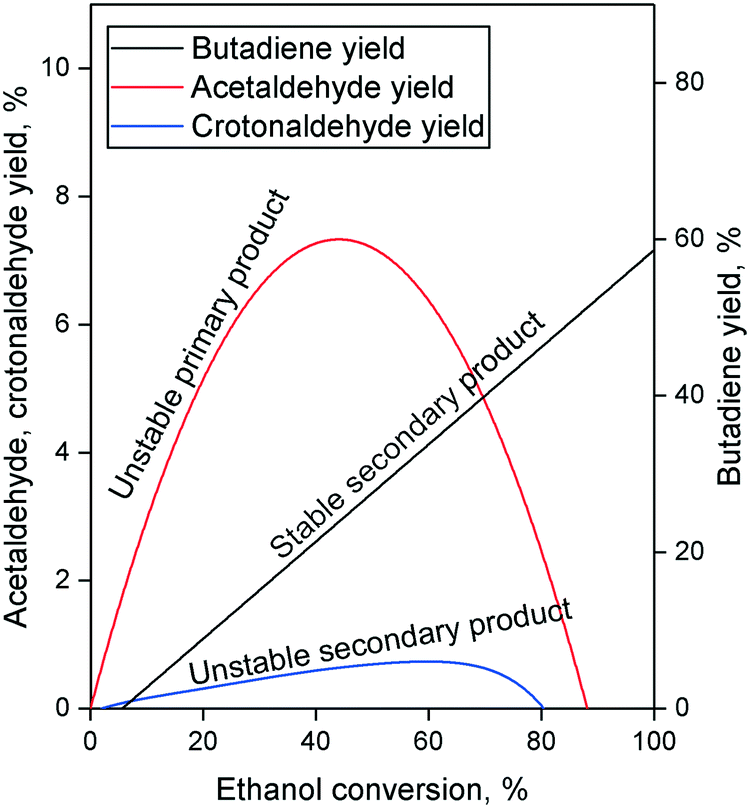 | ||
| Fig. 7 Typical Wojciechowski plots of the Lebedev process.108,109 | ||
Much insight on the mechanism of ethanol dehydrogenation in the context of the Lebedev process has been acquired using recent temperature-programmed surface reaction (TPSR) studies. By coupling this technique with in situ direct reflectance infrared spectroscopy and mass spectrometry, Taifan et al. identified ethoxy species formed by the dissociation of ethanol on the surface of MgO–SiO2 and correlated their disappearance at increased temperature with the emergence of acetaldehyde.111
Similar observations were made on pure MgO.96,112 It has been proposed that during the Lebedev process, ethanol dehydrogenation proceeds via surface ethoxide intermediates.96,111,112 On metal oxides with acid–base pairs, the literature suggests that acetaldehyde is formed by a sequential mechanism that begins with the dissociation of ethanol into a surface ethoxy intermediate followed by the E2 or E1cb-elimination of a proton (Scheme 4(a)).100,113–116 According to Sykes et al., acetaldehyde formation occurs similarly on defective Cu,117 suggesting that this mechanism is not limited to metal oxides. Surface ethoxy species were also detected on other transition metal oxide catalysts during IR-TPSR experiments with ethanol, leading to similar conclusions.118–120 It should be noted that studies of the sort have observed that surface ethoxy species are also intermediates in the formation of ethylene from ethanol.121 Therefore, ethoxy species must be correlated with the formation of acetaldehyde alone to confirm their involvement in mechanisms found in Scheme 4.
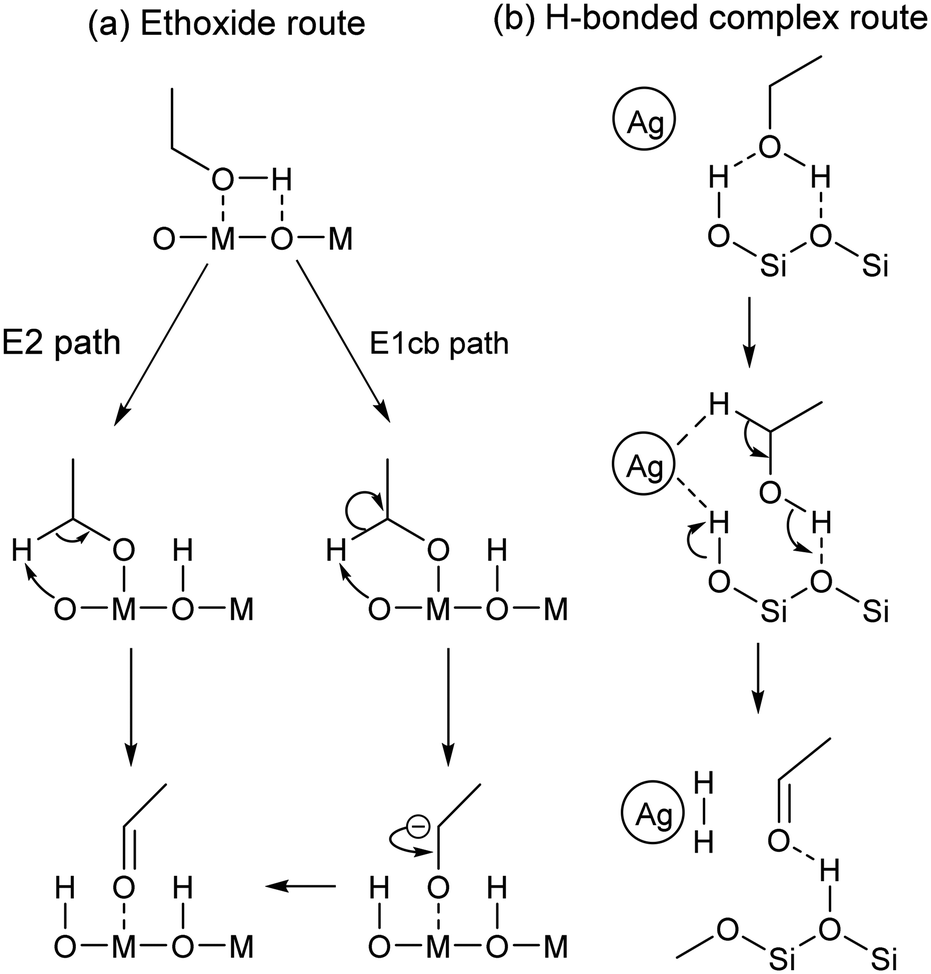 | ||
| Scheme 4 Proposed mechanisms for the dehydrogenation of ethanol to acetaldehyde via (a) an ethoxide intermediate116 or (b) a H-bonded complex.118 | ||
Ivanova et al. did not detect surface ethoxy species during the dehydrogenation of ethanol on SiO2-supported silver using in situ IR spectroscopy.115 The authors explained this discrepancy by a weak interaction between the substrate and the catalyst carrier. Instead of ethoxy intermediates, density functional theory (DFT) calculations and spectroscopic evidence suggested that ethanol forms a H-bonded complex with silanol groups prior to a simultaneous proton abstraction by metallic silver nanoparticles that forms hydrogen and acetaldehyde (Scheme 4(b)).122 Additionally, kinetic isotope effect (KIE) studies with various ethanol isotopomers on Ag/ZrO2/SiO2 indicated that both Cα–H bond cleavage and proton abstraction occur simultaneously during the Lebedev process, disproving the involvement of surface ethoxy species.99
Ethanol dehydrogenation to acetaldehyde and hydrogen is well-established as the first step of the Lebedev process according to the Toussaint–Kagan pathway. Studies of the surface intermediates suggested that the reaction mechanism depends on the type of catalyst. Over metal oxides on metallic copper, a sequential mechanism involving the dissociation of ethanol to ethoxy species and the subsequent elimination of a proton to form acetaldehyde has been proposed. On silica-supported silver, the reaction has instead been proposed to proceed via the simultaneous proton abstraction of a H-bonded complex formed between ethanol and surface silanol groups. However, it should be noted that in the broad context of light alcohol dehydrogenation, other mechanisms have been proposed.123 Further studies should seek to confirm the nature of surface intermediates on the various catalysts active in the Lebedev process.
Aldol condensation of acetaldehyde
Synthesizing butadiene from C2 compounds necessarily involves the formation of new C–C bonds. Although the aldol condensation/aldolization of acetaldehyde to crotonaldehyde (Scheme 3(b)) is now recognized as the bond-forming reaction, this topic has been the subject of much debate.11,50,96 A summarized by Quattlebaum et al.33 the involvement of crotonaldehyde was proposed following the observation that: (1) crotonaldehyde readily forms when passing acetaldehyde over Ostromislensky active catalysts; (2) crotonaldehyde is not present in significant amounts when converting ethanol–acetaldehyde mixtures over the same catalyst; (3) crotonaldehyde converts to butadiene when co-fed with ethanol under the same conditions.33,34A glaring issue with this theory has been the repeated absence of acetaldol, the immediate product of acetaldehyde coupling, in the output stream of catalytic tests. Several authors attributed the absence of acetaldol to its rapid dehydration to crotonaldehyde, which subsequently reacted with ethanol.100,108,110,125 Nonetheless, Corson et al. failed to observe any improvement in the yield of butadiene when co-feeding ethanol with acetaldol on a commercial Ta/SiO2 catalyst, concluding instead that the latter reverted to acetaldehyde.34,50,96 Cavani et al. confirmed this reversibility of acetaldol when testing the substrate over MgO catalysts.96 However, Taifan et al. argued that the reverse reaction of acetaldol was promoted by the absence of ethanol to react with crotonaldehyde, thereby shifting the equilibrium towards acetaldehyde instead of butadiene.126 Cavani et al. further suggested that crotonaldehyde may instead be a byproduct of ethanol conversion to butadiene, not an intermediate, and have conceived an alternative mechanism (vide infra).
Recent publications have provided further evidence for the intermittent presence of acetaldol during the conversion of ethanol to butadiene. For instance, Gao et al. studied the Lebedev process using the pulse reaction technique,127 which is better suited than continuous flow heterogeneous catalytic reactions for observing intermediates.128 Using mass-spectrometry to monitor the products formed, ethanol pulsed over MgO–SiO2 indicated the sequential formation of acetaldehyde, acetaldol, crotonaldehyde and butadiene. Surprisingly, crotyl alcohol, another key intermediate discussed further below, was undetected, possibly because of its rapid dehydration. Furthermore, Taifan et al. observed with DRIFTS a band at 1273 cm−1 previously assigned to acetaldol when reacting ethanol over MgO–SiO2 during TPSR experiments.111 It should be noted that the authors recognized that the conditions used were not optimized for the observation of acetaldol.
Whether or not the elusive acetaldol plays a role in the ethanol-to-butadiene process, the validity of the aldol condensation pathway is also dependent on the involvement of crotonaldehyde. New publications further suggest crotonaldehyde to be a key intermediate. Gao et al. found butadiene to be kinetically subsequent to crotonaldehyde in their pulse study of the reaction on MgO–SiO2.127 TPSR experiments with EtOH on MgO–SiO2,111 Ag/ZrO2 (ref. 118) and ZnY/SiBEA118 reported the appearance of IR bands associated with crotonaldehyde with increasing temperature. In the latter case, these also coincided with the detection of butadiene in the gas phase by mass spectrometry.119 Müller et al. conducted complex modulated co-feeding experiments monitored by operando DRIFTS-MS.101 The mechanistic implication of their observation was that acetaldehyde formed crotonaldehyde which was consumed by ethanol to form butadiene but would accumulate when switching the feed of ethanol off and the surface species were depleted. Furthermore, an unidentified surface intermediate could form only in the presence of crotonaldehyde and ethanol was detected and was proposed to play an important role in the formation of butadiene. Ivanova et al.108 and Cabello González et al.109 additionally found crotonaldehyde to be a secondary unstable product of the Lebedev process using Wojciechowski's criteria (Fig. 7), suggesting that: (1) crotonaldehyde is formed from a primary product, e.g., acetaldehyde; (2) crotonaldehyde is consumed during the reaction.
Although most authors now recognize that the conversion of ethanol to butadiene comprises the aldol condensation of acetaldehyde, there remain disagreements concerning the molecular-level mechanism of this reaction step. In its simplest form, the prevalent rationalization involves the enolization of an acetaldehyde molecule on acid–base pair sites; the resulting activated intermediate reacts with a neighboring acetaldehyde molecule to create a new C–C bond. Dehydration to crotonaldehyde is assumed to occur readily upon formation of the aldol. However, different molecular-level mechanisms have been proposed to take place depending on the type of catalyst used. On predominantly basic oxides, i.e., earth-alkaline oxides such as MgO and CaO, the strong basic sites of acid–base pairs are believed to fully abstract the α-proton of acetaldehyde, resulting in an enolate that is stabilized by the Lewis acid moiety of the acid–base pair. Because the d orbital of pre-transition metal oxides is not accessible, the role of their Lewis acidic cations is limited to stabilizing via electrostatic interactions the electron-rich carbonyl group of both the enolate and the second acetaldehyde molecule. Once in proximity, the β-carbon of the enolate reacts with the α-carbon of the acetaldehyde molecule to form the new C–C bond. Acetaldol is finally formed by the back-transfer of a proton from the surface of the catalyst. Scheme 5(b) depicts the sequential steps involved in this mechanism. In agreement with this mechanism, Taifan et al. reported observing surface enolates during the TPSR experiments with ethanol and acetaldehyde on MgO–SiO2 using DRIFTS.111
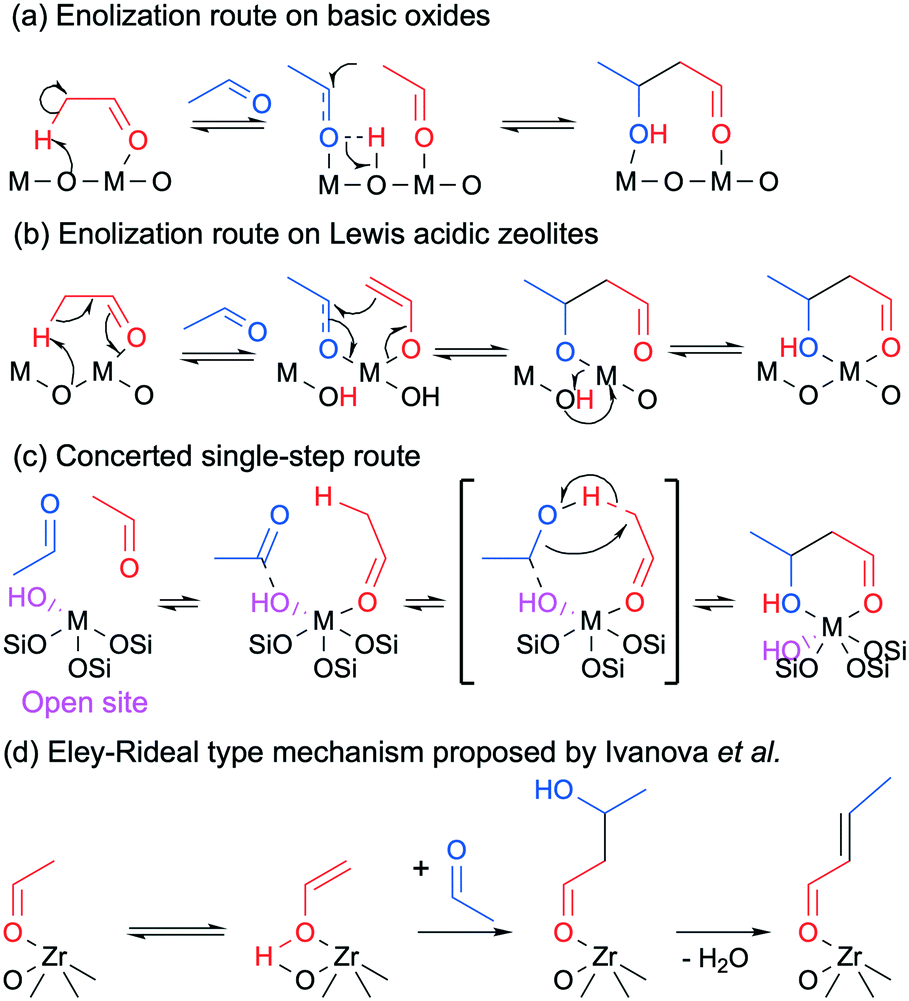 | ||
| Scheme 5 Proposed mechanisms for the aldolization of acetaldehyde taking place during the conversion of ethanol to butadiene where ‘M’ is a metal atom.124 | ||
Over transition metal oxides possessing strong Lewis acid characteristics, the metal cation is believed to polarize the carbonyl group of the first acetaldehyde molecule, acidifying the α-proton.129 Oxygen from the catalyst framework acts as a base and abstracts the proton, resulting in cleavage of Si–O–M bonds in the case of silica-supported catalysts.124 In contrast to pre-transition metal oxides, the partially filled d orbital of transition metal oxides is accessible for bonding. It can form enolate or enol intermediates coordinated on cation sites depending on the type of metal used (Scheme 5(b)).124 The metal center can accommodate a second acetaldehyde molecule, which is followed by a similar C–C bond formation and proton back-transfer previously described for basic oxides. The unstable presence of surface enolates was observed by Li et al. using DRIFTS on Zn–Y/SiBEA catalysts during TPSR experiments following the adsorption of ethanol and acetaldehyde.
However, Ivanova et al. recently proposed an alternative mechanism in which the coupling of both acetaldehyde molecules on transition metal cations does not involve the formation of an enolate intermediate, proceeding instead via a single-step concerted mechanism (Scheme 5(c)).124 This theory comes from the failure to observe the in situ formation of enolates with IR spectroscopy during the condensation of acetaldehyde on Zr-BEA and Ti-BEA. Hydrogen–deuterium exchange activity studies with heavy water further suggested that the catalysts did not stabilize enolate species, as evidenced by the low degree of deuterium incorporation in acetaldehyde. In the case of Zr-BEA, DFT calculations indicated that the two acetaldehyde molecules preferably formed a H-bonded complex stabilized by the metal cation and OH group of so-called Lewis open sites—isolated atoms in the tetrahedral positions of a zeolite structure connected to three –O–Si linkages and one OH group.130 C–C coupling would occur in a single concerted step involving an α-proton transfer from one aldehyde molecule to the carbonyl group of the second. Having failed to observe enolate species with in situ IR spectroscopy during the two-step process, Müller et al. advocated a similar one-step mechanism for the same condensation reaction over Ta-BEA.101 However, rather than an adsorption on the metal sites, the authors suggested that acetaldehyde reacts with OH groups—either coordinated to Ta(V) or from neighboring silanol groups.
Ivanova et al. later revisited the aldol condensation mechanism by combining kinetic measurements, steady-state isotopic transient kinetic analysis (SSITKA) and deuterium-tracing techniques for the conversion of ethanol to butadiene over Ag/ZrO2/SiO2.99 Since a kinetic effect was observed with β-deuterated ethanol, the authors dismissed the direct aldol condensation pathway previously proposed (Scheme 5(c)), which would have otherwise involved the α-proton of acetaldehyde (the β-proton of ethanol), in favor of a stepwise mechanism involving enolization. Furthermore, the rapid growth and decay of labeled and unlabeled butadiene response curves following the switch from the isotope-labelled to unlabeled feed during SSTIKA experiments suggested that the coupling reaction occurred between an acetaldehyde molecule strongly adsorbed on the catalyst surface and an acetaldehyde molecule in the gas phase. Consequently, a new mechanism combining both observations was proposed for the aldolization of acetaldehyde: it proceeds via a stepwise mechanism involving enolization, followed by C–C bond formation between the enolate intermediate and a gas phase acetaldehyde molecule, e.g., an Eley–Rideal mechanism (Scheme 5(d)). This proposal differs from most mechanistic interpretations of acetaldehyde coupling in the context of the ethanol-to-butadiene reaction100,101,111,119,126 or the closely related Guerbet coupling,114,131 which assumes a Langmuir–Hinshelwood type reaction, although Eley–Rideal mechanisms were proposed for the condensation of other short-chain aldehydes.132,133
While the formation of crotonaldehyde via the aldol condensation of acetaldehyde is generally accepted as a key-step of the Toussaint–Kagan pathway from ethanol to butadiene, our understanding of the molecular-level phenomena taking place remains limited. Several mechanisms have been proposed, but the direct observation of surface intermediates and their correlation to activity has been limited, ostensibly due to their high instability.111,119 Clarifying these issues would benefit catalyst design—are open Lewis acid sites necessary for one-step aldol condensation?—and kinetic modelling by identifying intermediate steps and answering whether an Eley–Rideal or a Langmuir–Hinshelwood type reaction is involved. However, DFT calculations have also shown the favorability of different mechanisms to depend on the nature of the catalytic systems used.100,124,134–136 This may limit the general validity of observations made and limit them to the system under study, meaning Ag/ZrO2/SiO2.
Crotonaldehyde conversion to butadiene
Obtaining butadiene from a crotonaldehyde feed on catalysts for the Ostromislensky process has only proceeded when co-feeding ethanol.33,34 This observation highlights the importance of ethanol to reaction beyond its role as a source of acetaldehyde. It also explains why the Ostromislensky process requires ethanol–acetaldehyde mixtures to operate. Early mechanistic theories considered the direct catalytic deoxygenation of crotonaldehyde to butadiene with ethanol as a hydrogen source to be the reaction step.33 Having noted that: (1) crotonaldehyde was reduced to crotyl alcohol under H2 and (2) crotyl alcohol readily dehydrated to butadiene under typical reaction conditions, Kagan et al. proposed that crotonaldehyde was first reduced to crotyl alcohol, which dehydrated to form butadiene.11,16,105Although sometimes absent from the output stream of reactors during catalytic testing due to its rapid dehydration, the participation of crotyl alcohol is now generally recognized and supported by several experimental observations. For instance, ethanol conversion over magnesia at a very short contact time enabled the detection of crotyl alcohol and identified it as a kinetic precursor to butadiene.96 In DRIFTS-monitored TPSR experiments, the disappearance of chemisorbed crotyl alcohol coincided with the detection of signals attributed to vapor-phase butadiene.111 The MS-monitored operando modulated co-feeding experiments for the Lebedev and Ostromislensky processes further provided insight on this reaction step.101,119 As the signal patterns of crotyl alcohol and butadiene behaved similarly upon feed switching, i.e., from ethanol–acetaldehyde to pure acetaldehyde, the authors concluded that both shared the same precursor, e.g., crotonaldehyde. The detection of deuterated crotyl alcohol when reacting labelled ethanol with crotonaldehyde, and its replacement by unlabeled crotyl alcohol upon switching to a non-isotopic feed further confirmed crotyl alcohol to be the intermediate between crotonaldehyde and butadiene, and also that ethanol was involved in its formation.
How crotonaldehyde is reduced to crotyl alcohol has raised further questions. Undoubtedly, ethanol enables the conversion of crotonaldehyde to butadiene, but so does hydrogen,137 propanol,34,138 which yields C3 and C5 byproducts, or even crotyl alcohol itself.34 Therefore, its role has been the subject of debate. Some scholars have argued that ethanol is the reducing agent (Scheme 6(a) and (b)),99–101,136,138 others that it acts as an in situ source of surface hydrogen species responsible for reducing the carboxyl group of crotyl alcohol (Scheme 6(c)),139 or both.119 Old137,140 and new11,12 thermodynamic studies have shown the reduction of crotyl alcohol with hydrogen to be less favorable than that with ethanol. As a result, publications often disregard the role of hydrogen,100 focusing instead on crotonaldehyde reduction with ethanol, which Niiyama et al. proposed to proceed via an intermolecular transfer involving acid–base sites.11,140 This reaction is believed to follow a Meerwein–Ponndorf–Verley reduction mechanism, also referred to as the Meerwein–Ponndorf–Verley–Oppenauer (MPVO) reaction to account for the oxidation of ethanol to acetaldehyde.
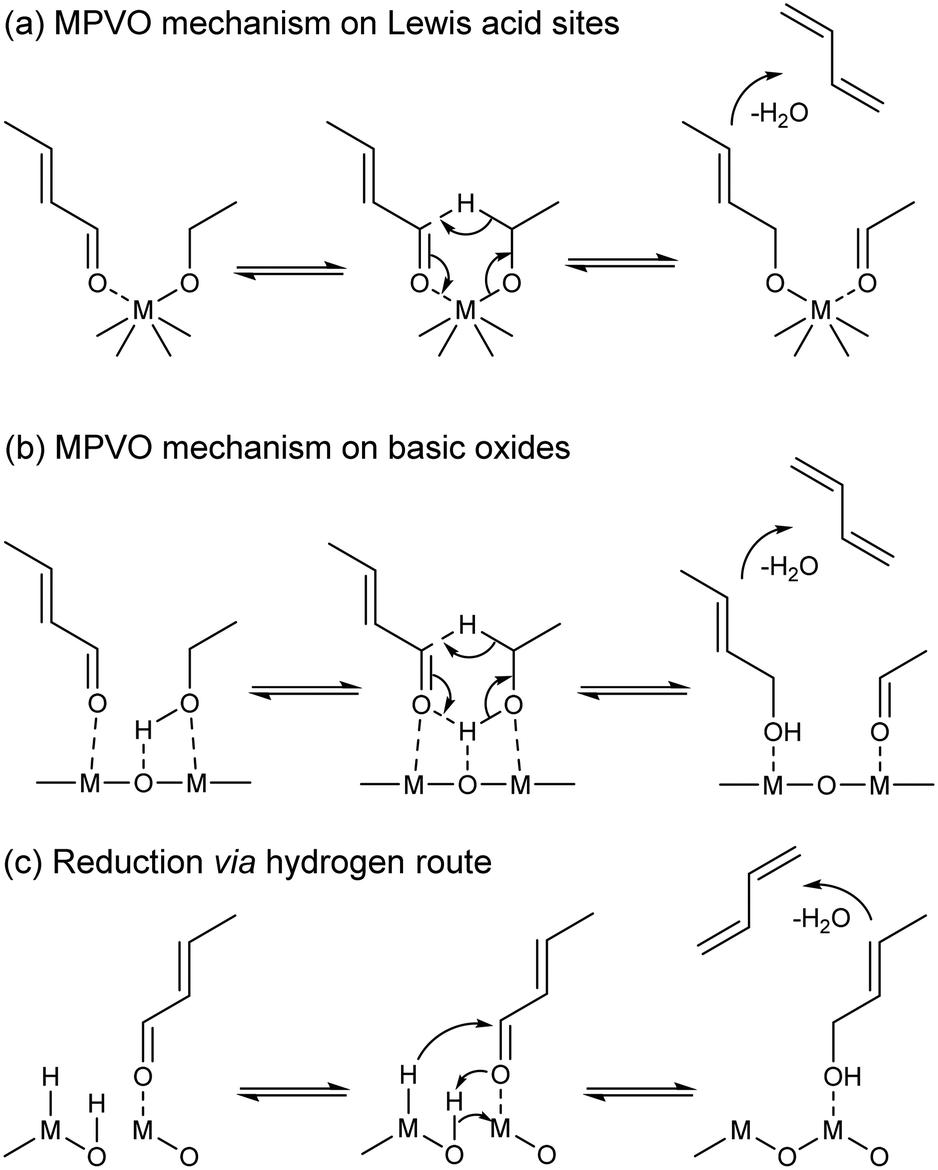 | ||
| Scheme 6 Mechanisms of crotonaldehyde reduction to crotyl alcohol and its dehydration to butadiene on different catalysts.99,116,141 (a) MPVO mechanism on Lewis acid sites; (b) MPVO mechanism on basic oxides; (c) reduction via hydrogen route. | ||
The MPVO reaction mechanism consists of the stabilization of ethanol and crotonaldehyde on Lewis acid sites—the same sites for transition metals such as Zr (ref. 99) and Ta (ref. 101) (Scheme 6(a)) and neighboring sites on pre-transition metal oxides116 (Scheme 6(b))—followed by the formation of a six-membered transition state; the reaction then proceeds by a hydride transfer from the alcohol to the carbonyl group, which forms acetaldehyde and crotyl alcohol. Relatively few studies have focused on studying this mechanism in the context of the ethanol-to-butadiene reaction. Ivanova et at. observed a reactivity trend typical of Langmuir–Hinshelwood type mechanisms when changing the partial pressure of ethanol co-fed with crotonaldehyde to form butadiene over Ag/ZrO2/SiO2.99 More interestingly, their catalytic tests with ethanol deuterated in the α-carbon position resulted in the formation of CD2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CD–CH2, a butadiene isotopomer that is only obtainable by the D-shift taking place with this six-membered transition state.99 MPVO reactions between other carbonyl and reductive alcohols on heterogeneous catalysts substantiate the validity of this mechanism in the conversion of crotonaldehyde to crotyl alcohol.143–145
CD–CH2, a butadiene isotopomer that is only obtainable by the D-shift taking place with this six-membered transition state.99 MPVO reactions between other carbonyl and reductive alcohols on heterogeneous catalysts substantiate the validity of this mechanism in the conversion of crotonaldehyde to crotyl alcohol.143–145
Recently, Baba et al. reiterated that ethanol is not the sole reducing agent of crotonaldehyde in the ethanol-to-butadiene reaction. This claim stems from the fact that the reaction can proceed on MgO,96,139,142 but not CaO, despite both being alkaline-earth metal oxides with basic properties. According to the authors, contrary to MgO, CaO cannot perform the heterolytic dissociation of hydrogen. Therefore, the hydride and hydroxide species formed at the surface of MgO were attributed a role to explain the discrepant activity of the two oxides. Baba et al. suggested that chemisorbed hydrogen could reduce crotonaldehyde to crotyl alcohol during the conversion of ethanol to butadiene (Scheme 7). Incidentally, ZrO2,146 Ag (ref. 147) and ZnO,148,149 well-established components of ethanol-to-butadiene catalysts, can also promote the heterolytic dissociation of hydrogen. Bhattacharyya and Ganguly compared the yields of H2 and acetaldehyde following the conversion of ethanol–crotonaldehyde mixtures on ZnO–Al2O3 to investigate the reduction step.137 According to these authors, the relative excess of acetaldehyde compared to hydrogen suggests that crotonaldehyde reduction by ethanol preferably takes place. However, the opposite conclusion—that the observed lower quantity of H2 demonstrates its consumption in the reduction step—may also be drawn from the same results. Clarifying the degree of participation of dissociated hydrogen in the conversion of ethanol to butadiene is crucial for proper kinetic modelling which would consist of rate laws depending on the concentration of reactants involved. Accurately predicting the output of hydrogen is also crucial considering that selling it has been considered a strategy for reducing the environmental impact of the Lebedev process.93
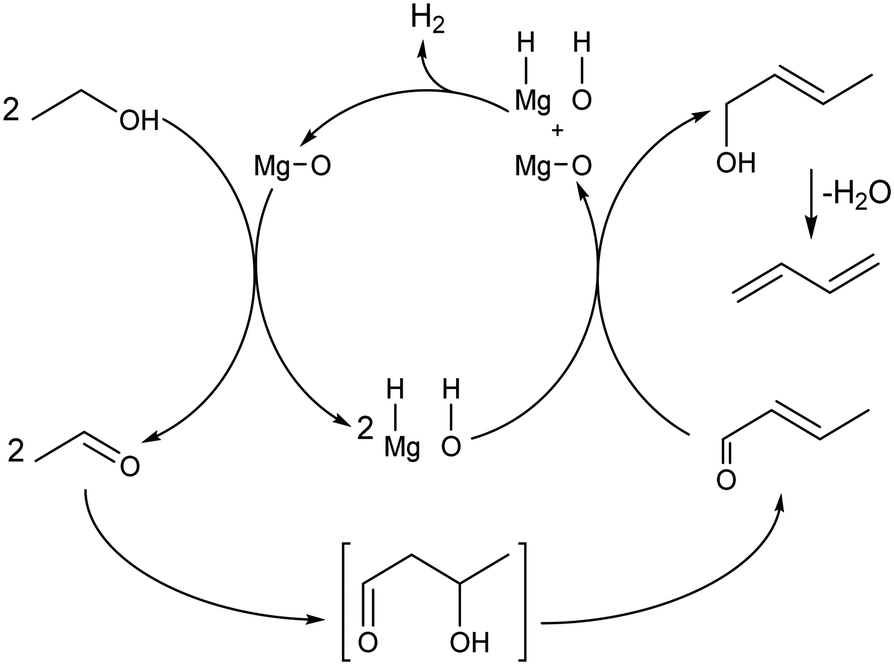 | ||
| Scheme 7 Ethanol-to-butadiene pathway on MgO with dissociated hydrogen as the reductive agent for crotonaldehyde conversion.96,139,142 | ||
Dehydration of crotyl alcohol to butadiene and water (Scheme 6) has long been known to occur readily on catalysts active for the Ostromislensky process.34,105 Recently, Cabello González et al. confirmed that it also occurred on catalysts for the Lebedev process.109 Studies found the dehydration of crotyl alcohol to be highly favored thermodynamically.11,12,137,140 Consequently, this reaction step has not attracted significant attention: it is assumed to readily take place during the conversion to butadiene upon crotyl alcohol formation.
All-inclusive Toussaint–Kagan mechanism proposals
In 2017, Ivanova et al. presented the first fully-fledged molecular-level mechanism of the ethanol-to-butadiene reaction.99 Their proposal stems from results obtained with kinetic measurements, SSITKA and deuterium tracing techniques performed using Ag/ZrO2/SiO2. The authors interpreted the reaction as two distinct catalytic cycles (Scheme 8): cycle I, the dehydrogenation of ethanol, and cycle II, the condensation–reduction–dehydration pathway of acetaldehyde to butadiene; two distinct active sites were considered. Cycle II essentially describes the reactions taking place during the Ostromislensky process on catalysts lacking dehydrogenating properties. At present, the mechanistic interpretation of Ivanova et al. is the most detailed in the literature. Müller et al. further elaborated on catalytic cycle II with Ta-BEA, namely by distinguishing two activated surface intermediates for the aldol condensation and MPVO reaction steps.101 Li et al. proposed a similar reaction mechanism over a bifunctional Zn–Y/SiBEA catalyst, albeit in the form of a single cycle containing both active sites.119 The authors chose to represent crotonaldehyde reduction with dissociated hydrogen rather than ethanol, but recognized that both pathways may take place. Comprehensive mechanistic elucidations of the mechanism on MgO and ZrO2 were also performed using DFT calculations by Taifan et al.100 and Zhang et al.136 However, the mechanisms proposed in the literature disagree on certain aspects, notably at the molecular level. We have highlighted some points of contention that could be addressed in future works: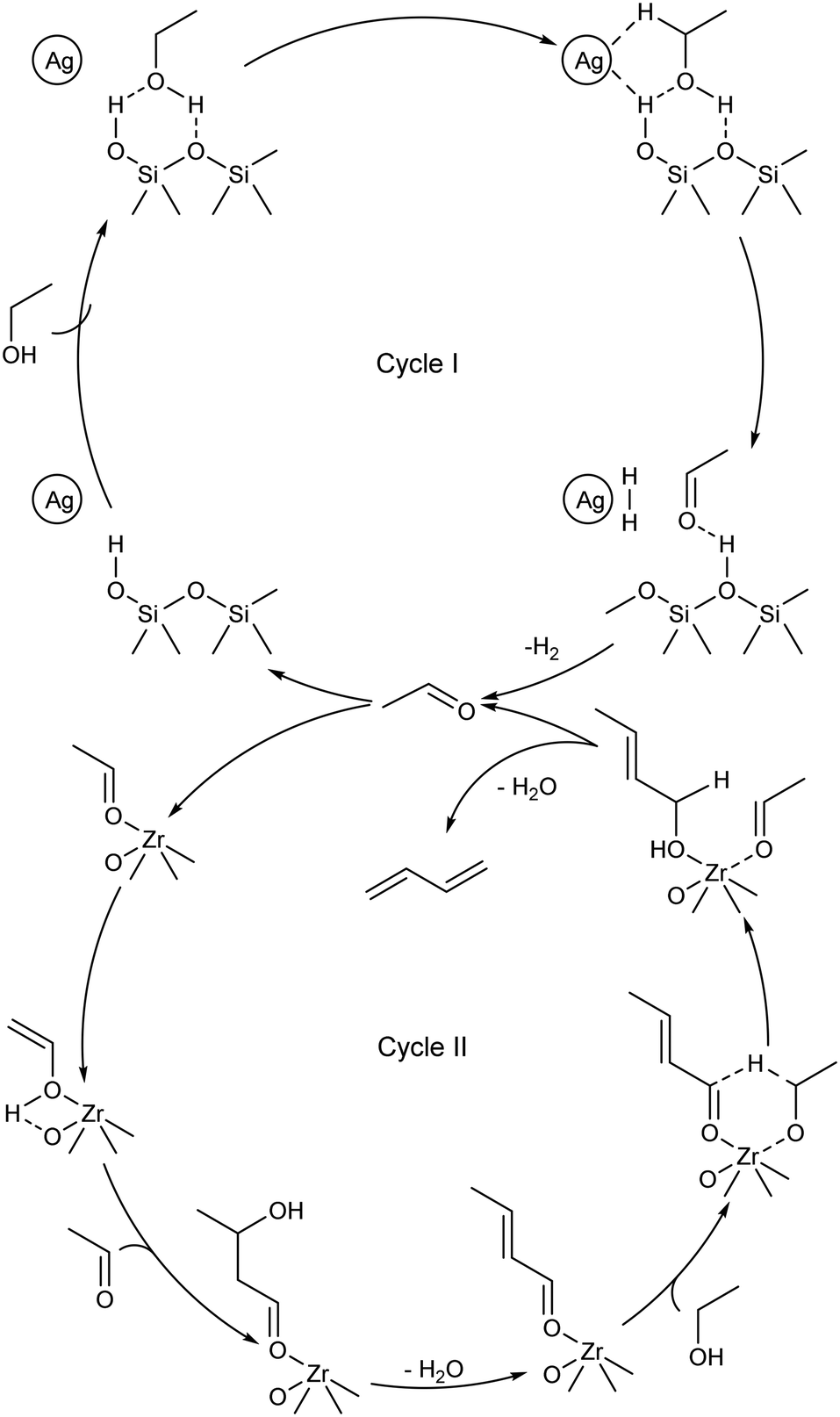 | ||
| Scheme 8 Dual cycle molecular-level mechanism for the ethanol-to-butadiene reaction proposed by Ivanova et al.99 | ||
• Verifying whether the choice of catalyst causes significant mechanistic differences at the molecular level, and whether these differences are relevant to catalyst design and kinetic modelling. At present, the evidence gathered suggests this to be the case;
• Verifying the involvement of surface intermediates, such as ethoxy species for the dehydrogenation of ethanol, and enolate species for the aldolization of acetaldehyde;
• Clarifying the mechanism forming crotonaldehyde from acetaldehyde. Notably whether it involves a direct coupling reaction,124 or a step-wise coupling with enolate intermediates, and whether the latter follows an Eley–Rideal102 or Langmuir–Hinshelwood mechanism;150
• Measuring the contribution of hydrogen and ethanol to the reduction of crotonaldehyde to crotyl alcohol. Is the participation of hydrogen dismissible?
Alternative pathways to butadiene
More than variants of the Toussaint–Kagan pathway, alternative pathways with different reaction steps have been considered for the conversion of ethanol to butadiene. The main point of divergence with the common only accepted pathway concerns the formation of the C–C bond. Unfortunately, only a few scholars have taken account of these alternative mechanisms in their own study of the mechanism.Consequently, it is difficult to assess their validity; they may co-exist as complementary pathways to butadiene, may be wrong, but also may be the main pathway under specific conditions.
Ostromislensky24 and Balandin151 proposed that ethanol reacted with acetaldehyde to form 1-ethoxyethanol. The latter would undergo a hemiacetal rearrangement to form butanediol, which would dehydrate to form butadiene (Scheme 9(a)). Quattlebaum et al. later dismissed this mechanism, arguing that the rearrangement reaction did not have experimental support.33 Furthermore, butanediol yielded significantly less butadiene than ethanol–acetaldehyde mixtures when reacted on a Ta2O5/SiO2 catalyst active in the Ostromislensky process.34 Recently, Taifan et al. studied the viability of this pathway on MgO using DFT calculations.100 They found the thermodynamic stability of 1-ethoxyethanol to prevent further reaction via a hemiacetal rearrangement.
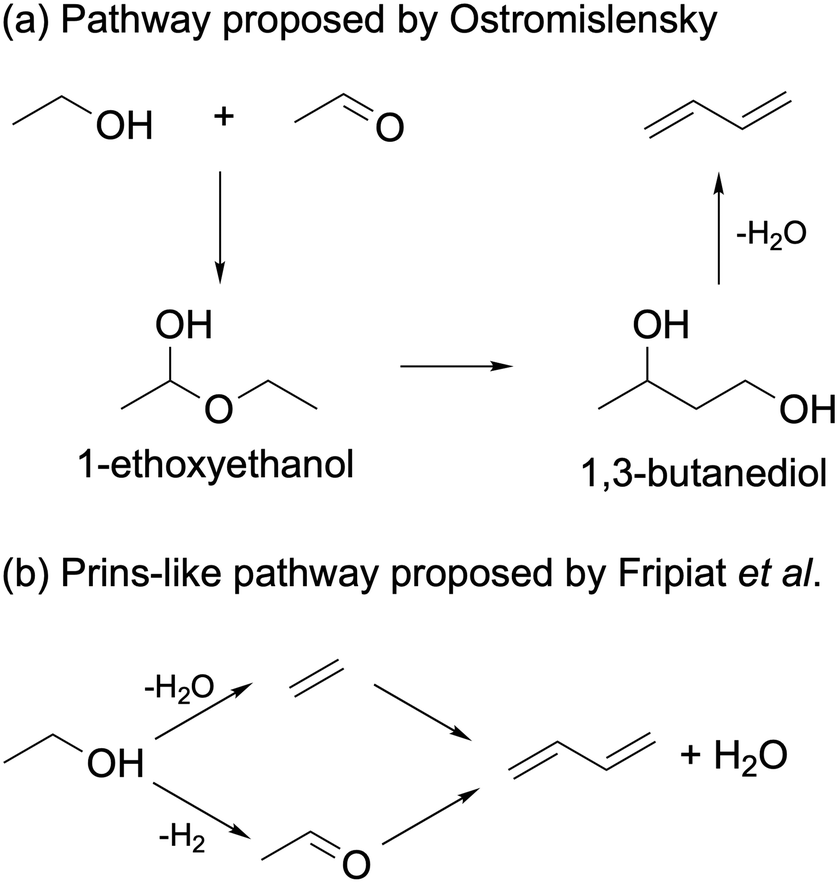 | ||
| Scheme 9 Ethanol-to-butadiene pathways proposed by (a) Ostromislensky and Balandin and (b) Fripiat et al.152 | ||
Fripiat et al. studied the ethanol-to-butadiene reaction on silver-exchanged aluminated sepiolite.152 In plotting product selectivity versus ethanol conversion, the authors found a linear relation between ethylene and butadiene yields. On this basis, they argued that ethylene reacted with acetaldehyde via a Prins-like mechanism to form butadiene (Scheme 9(b)). Thermodynamic equilibrium calculations of the proposed mechanism found it to be favorable, albeit slightly less than the Toussaint–Kagan mechanism.12 However, these calculations omitted the formation of the intermediate 3-buten-2-ol, the expected product of a Prins reaction between acetaldehyde and ethylene. Our own calculations, made using the Aspen Plus® software with mixtures of pure components (vide infra), indicated that the intermediate product formation step is highly endergonic. Although they also proposed a Prins-like mechanism, Natta and Rigamonti rejected it after the addition of 20% ethylene to the reactant feed failed to improve the butadiene yield. Taifan et al.100 and Zhang et al.136 investigated the Prins-like mechanism in their DFT calculations on the conversion of ethanol, finding the formation of the new C–C bond to have a higher energy barrier than the aldol condensation on MgO and ZrO2, respectively. Taifan et al. added that the aldolization pathway must be kinetically favored, as the Prins-like mechanism was more exergonic, suggesting that the latter occurs under specific conditions.
Instead of acetaldehyde self-coupling, Cavani et al. proposed that the new C–C bonds stemmed from the reaction between C2-oxygenated species and a surface carbanion formed from ethanol.96 The authors reached this conclusion after studying the reactivity of ethanol over basic oxides, e.g., MgO and MgO-containing catalysts. Certain observations were considered incompatible with the Toussaint–Kagan pathway. Catalytic tests performed at a very short contact time showed crotonaldehyde kinetically consecutive to crotyl alcohol, which was recognized as the precursor to butadiene. Furthermore, Cavani et al. questioned the role of acetaldol, the presumed transient intermediate of the aldol coupling of acetaldehyde to crotonaldehyde. Alone or co-fed with ethanol, the compound failed to produce butadiene on MgO. In FTIR-TPSR experiments using MgO with pre-adsorbed ethanol, peaks assigned to crotyl alcohol also emerged at lower temperature than those attributed to crotonaldehyde. Consequently, the aldolization step of the Toussaint–Kagan pathway was rejected on the grounds that C4 alcohols appeared to kinetically precede their alleged precursor. Instead, the authors proposed that a surface carbanion formed by the methyl deprotonation of ethanol participated in the coupling reaction (Scheme 10). DFT and spectroscopic studies served as the basis for this argument. In the aforementioned FTIR-TPSR experiments, the increase of the band at v = 1143 cm−1 above 573 K coinciding with the formation of crotyl alcohol, but distinct to it, was observed, suggesting a relationship between the two species. This unidentified signal matched with one IR band of a C2 carbanion on MgO simulated by DFT calculations, to which it was tentatively assigned. However, the experimental results lacked the other peaks of the simulated IR spectra, a discrepancy that admittedly remained unclear.
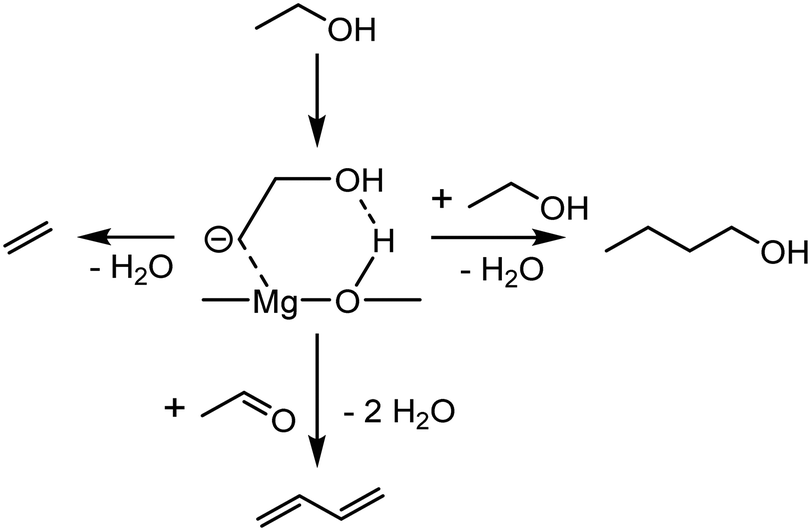 | ||
| Scheme 10 Pathway to butadiene, ethylene and butanol proposed by Cavani et al.96 | ||
With DFT calculations, a pathway involving the reaction between the theorized surface C2 carbanion and acetaldehyde was found to rival the Toussaint–Kagan pathway energy-wise (Scheme 10). First, ethanol underwent proton abstraction from its β-carbon to form the carbanion, which was stabilized by the catalyst surface. This intermediate could then follow three different pathways. Its hydroxyl group may react with the previously dissociated proton to form water and ethylene. It may also attack the oxygen-bound carbon atom, a neighboring adsorbed ethanol or an acetaldehyde molecule. In the first case, butanol and water were produced. With acetaldehyde, the reaction generated water and either crotyl alcohol or 3-buten-1-ol. These alkenols were proposed to dehydrate into butadiene. Cavani et al. further made similar observations on MgO–SiO2, a more conventional catalyst for the Lebedev process than pure MgO.97 The carbanion mechanism was thus suggested to take place on MgO-containing catalysts, explaining the presence of ethylene, butanol and butadiene amongst the products. So far, no other scholars have corroborated some key observations made by the authors. DFT calculations made by Taifan et al. found the carbanion to be unstable on MgO, preferably forming ethylene rather than following the pathway leading to butadiene.100 Furthermore, Taifan et al. did not report the IR bands attributed to the carbanion species over MgO–SiO2.111 Admittedly, Cavani et al. noted that a Mg:Si ratio of 15 was necessary to observe it distinctively, whereas the catalyst used by Taifan et al. had a ratio of 1. It may be the case that this pathway occurs preferably on specific catalysts, such as pure MgO and MgO–SiO2 with high magnesia content.
Recently, Dussol et al. proposed a new reaction pathway operating in parallel with the Toussaint–Kagan pathway.98 Dubbed the Inoue route after the work of Inoue et al. on butadiene from 4-oxy-2-butanone and ethanol,153 it explains the presence of side-products that the authors detected when conducting the Ostromislensky process over a Ta2O5–SiO2 catalyst. At the core of the Inoue route is an intramolecular MPVO reaction of acetaldol that would produce 4-hydroxy-butan-2-one (Scheme 11, (b)). From this point on, the reaction steps are identical to the Toussaint–Kagan pathway: dehydration into methyl vinyl ketone (MVK), MPVO reaction forming 3-buten-2-ol and dehydration to butadiene (Scheme 11, (c)–(e), respectively). Not only does this pathway explain the presence of MVK and 3B2O, but also of methyl ethyl ketone formed via 3B2O, and 2-butanol by its dehydration. Dussol et al. demonstrated the validity of their proposal with a kinetic study, which is discussed below.
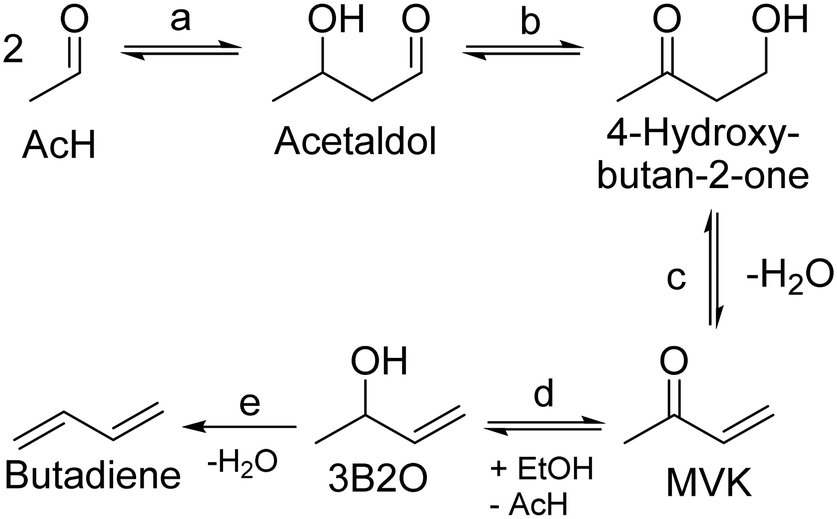 | ||
| Scheme 11 Inoue route to butadiene proposed by Dussol et al.98 MVK: methyl vinyl ketone. 3B2O: 3-buten-2-ol. (a) Aldol condensation; (b) intramolecular MPVO reaction; (c) dehydration to methyl vinyl ketone; (d) MPVO reaction; (e) dehydration to butadiene. | ||
Alternative pathways should be considered when studying the ethanol-to-butadiene reaction. Although the Toussaint–Kagan pathway is now generally accepted, the possibility that these different pathways may occur in the conversion of ethanol remains to be disproved. Notably, the Prins-like mechanism proposed by Fripiat et al. was shown to be thermodynamically favorable with DFT calculations. In addition, the observations suggesting the involvement of a carbanion on MgO should be investigated further. The Inoue route, which has been proposed to complement the Toussaint–Kagan route during the Ostromislensky process, is insightful in explaining the formation of unexpected products seemingly unrelated to the formation of butadiene; research on this topic could focus on determining its validity on other catalytic systems, its occurrence during the Lebedev process and the extent to which it contributes to butadiene formation.
Thermodynamic considerations
Several thermodynamic studies of the Toussaint–Kagan pathway and the Prins-like mechanism for the conversion of ethanol to butadiene have been performed. Natta and Rigamontti first calculated the Gibbs free energy change of potential reaction steps at 673–703 K, concluding that what we now refer to as the Toussaint–Kagan pathway was the most likely route to butadiene.140 Bhattacharyya and Ganguly, although neglecting the reduction of crotonaldehyde by combining it with the subsequent dehydration of crotyl alcohol, found it to be more endergonic with hydrogen rather than ethanol as a reductive agent.137 Recently, Weckhuysen et al. performed thermodynamic calculations with the HSC7 software package, confirming the favorability of the ethanol-induced reduction pathway at 673 K. Their results further indicate that the reaction becomes favorable above 420 K. The authors also determined the conversion of ethanol to butadiene to be endothermic by 102–109 kJ mol−1 from 473 to 773 K. In their review of the ethanol-to-butadiene reaction, Sels et al. reported the most comprehensive thermodynamic study of the reaction, which was performed with the Aspen Plus® software using pure compounds in their gaseous states for simulating the various reaction steps at different temperatures. We performed thermodynamic simulations of our own using the same approach as Sels et al. with the Aspen Plus® software to further investigate the theoretical influence of pressure on the feasibility of the reaction. Since our results coincide mostly with those of Sels et al., we consider this section complimentary to their work and refer the reader to their article for further details.As depicted in Fig. 8(a) step i, ethanol conversion to butadiene becomes thermodynamically favorable beyond 418–420 K.11,12 However, ethanol dehydrogenation to acetaldehyde (Fig. 8(a), step ii) only becomes exergonic at higher temperature: 550 K according to our calculations and 585 K according to Sels et al.11 Acetaldol formation from acetaldehyde (Fig. 8(a), step iii) is the least thermodynamically favored step of the Toussaint–Kagan mechanism.11 At 1 atm, it is always endergonic and becomes increasingly so at greater temperature. In contrast, thermodynamics favor acetaldol dehydration to crotonaldehyde significantly (Fig. 8(a), step iv), substantiating the claims of its rapid disappearance and absence from the output of catalytic tests. Contrary to Weckuysen et al. the Aspen® simulation showed the MPVO reaction between crotonaldehyde and ethanol (Fig. 8(a), step v) to have comparatively low, but steady ΔG values over the entire temperature range studied.12 Sels et al. also found this step to be endergonic, but with a Gibbs free energy change almost close to zero. Although thermodynamically favored overall, the Toussaint–Kagan pathway possesses a single highly endergonic step: the acetaldehyde condensation to acetaldol.11,12 According to gas-phase thermodynamic calculations for the pure components, all other steps have either lower ΔG values or are highly exergonic. Fig. 8(b) illustrates the influence of pressure on the Gibbs free energy change of the overall reaction: it becomes exergonic at 400 K temperature when reducing the pressure to 0.5 atm.
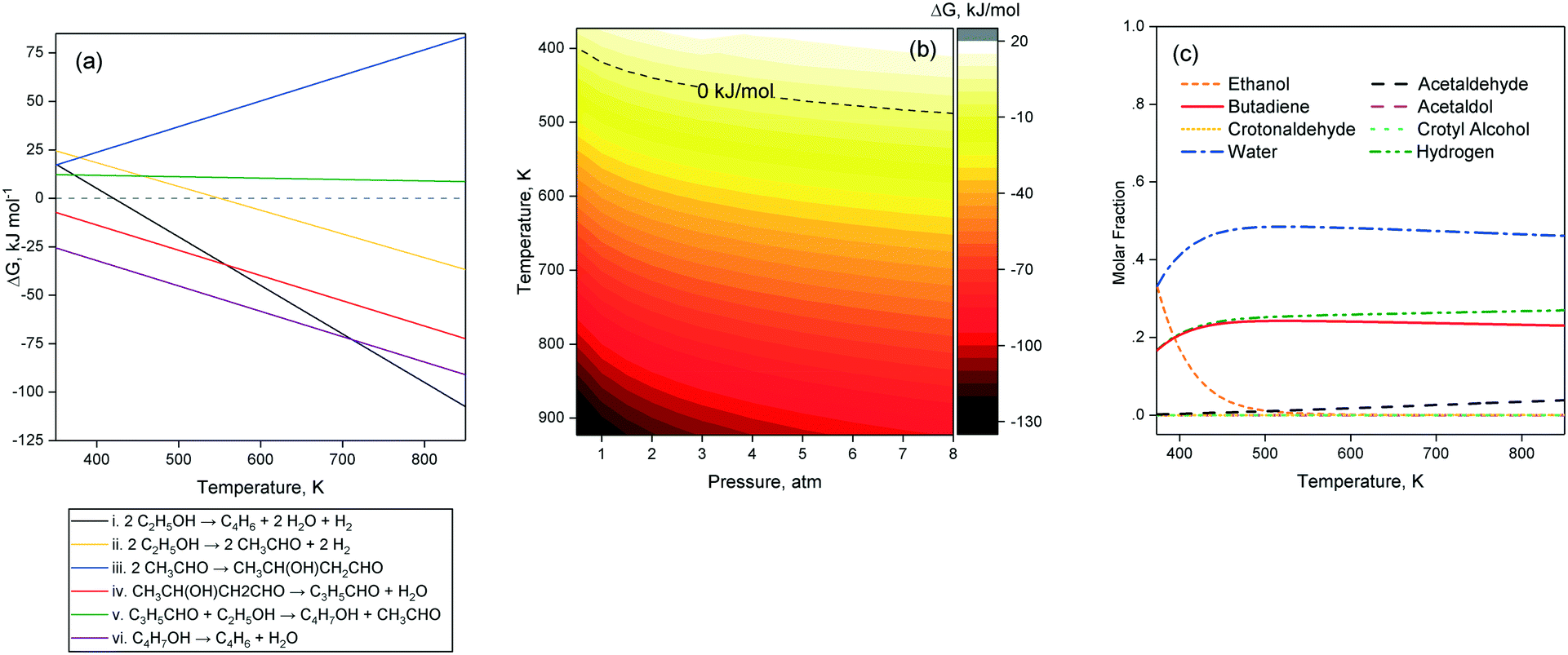 | ||
| Fig. 8 Thermodynamic modelling of the ethanol-to-butadiene conversion considering the Toussaint–Kagan mechanism. Calculations were performed using the Aspen Plus® software for mixtures of pure components at given temperatures and pressures. (a) Ellingham-type of Gibbs free energy change of the reaction steps in the Toussaint–Kagan pathway at 1 atm. (b) Gibbs free energy change of the overall reaction at different pressures. (c) Molar composition at equilibrium for the overall reaction at different temperatures and atmospheric pressure. | ||
According to Sels et al., the direct conversion of ethanol to butadiene at equilibrium was complete above 400 K.11 However, modelling the equilibrium composition of the reaction including every reaction intermediate showed a slight decrease in butadiene above 613 K due to unconverted acetaldehyde and crotonaldehyde. Our calculations confirm this assessment (Fig. 8(c)). According to this thermodynamic model, the maximum butadiene yield at a typical temperature of 673 K is 90%. At 1 atm, Sels et al. further found that the individual reactions steps were limited by the thermodynamic equilibrium and could not reach full conversion, except for ethanol dehydrogenation above 800 K and crotyl alcohol dehydration at every temperature tested. The authors concluded their thermodynamic study by stating that the preferable reaction temperature lay between 602 K and 703 K. Furthermore, within this temperature yield, butadiene yield and selectivity were determined by reaction kinetics, highlighting the importance of the catalyst choice.
Kinetics & reaction conditions
| No. | Reaction scheme | Rate law of the limiting steps, s−1 | Rate parameters, s−1 |
|---|---|---|---|
| NB: Xi refers to the output molar fraction of compound I, Kn is the equilibrium adsorption constant for active sites A, B or C, 500, 100, 550, respectively. | |||
| 1 | C2H5OH → C2H4 + H2O |
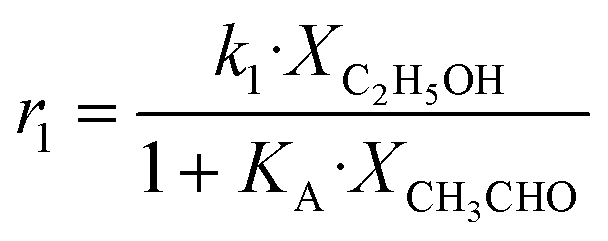
|
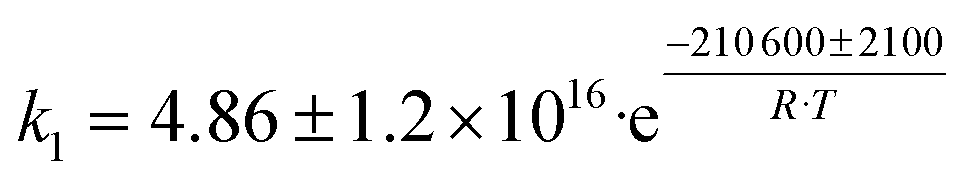
|
| 2 | C2H5OH → CH3CHO + H2 |
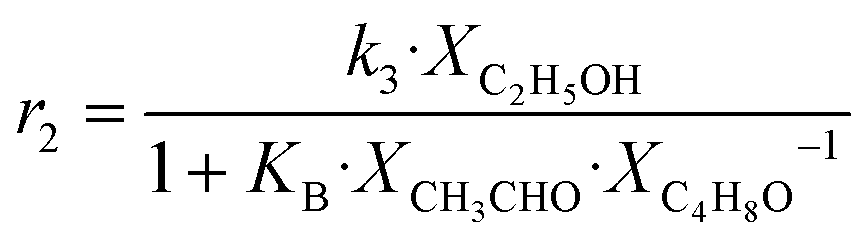
|
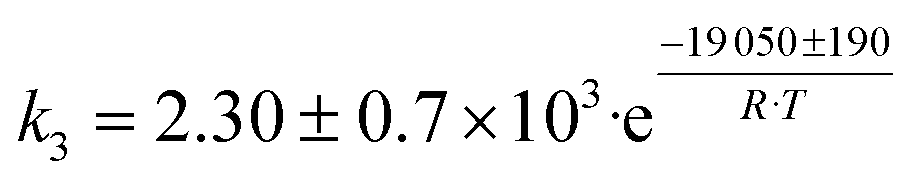
|
| 3 | CH3CHO + C2H4 → C4H6 + H2O |
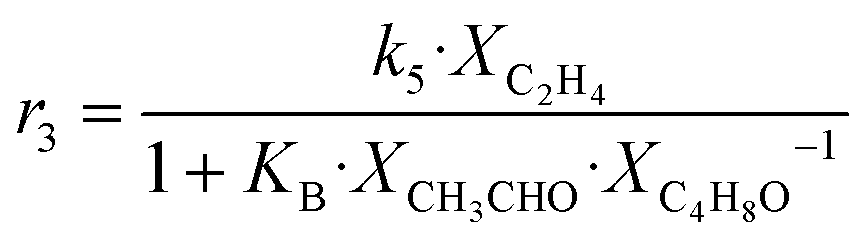
|
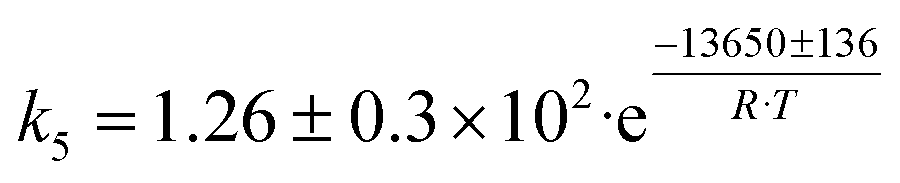
|
| 4 | CH3CHO + C2H4 → C4H8O |
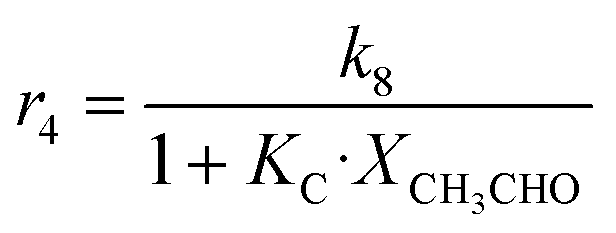
|
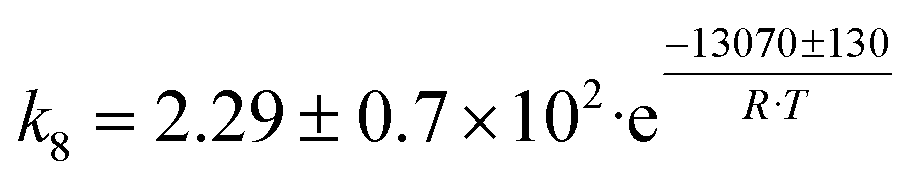
|
| 5 | 2 CH3CHO + H2 → C4H6 + 2 H2O |
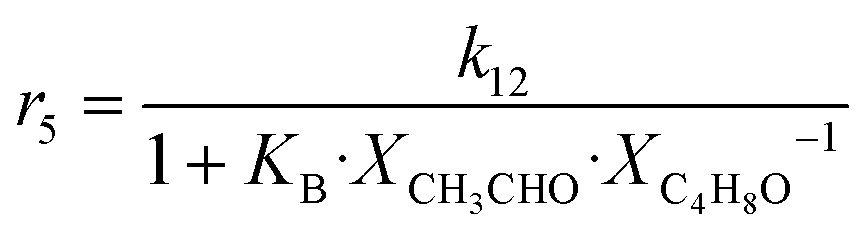
|
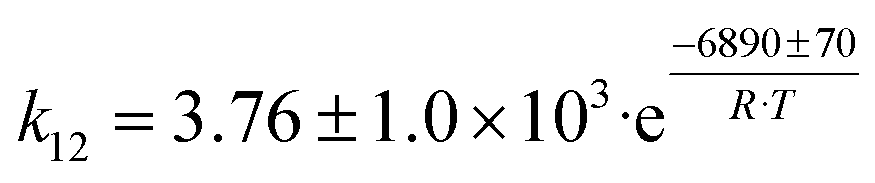
|
| 6 | 2 C2H4 → C4H8 |
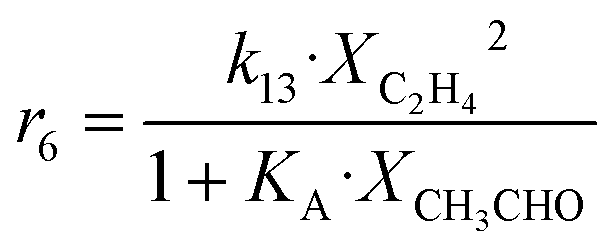
|
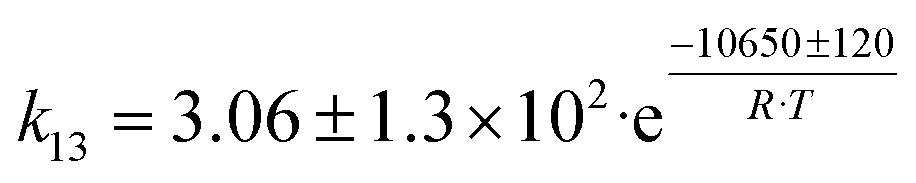
|
| 7 | C4H8 → C4H6 + H2 |
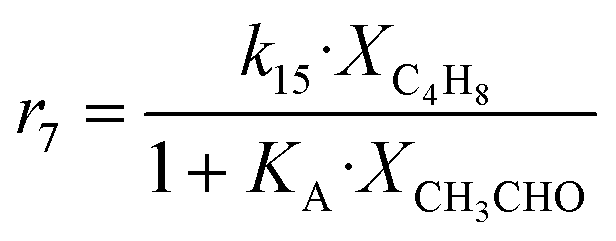
|
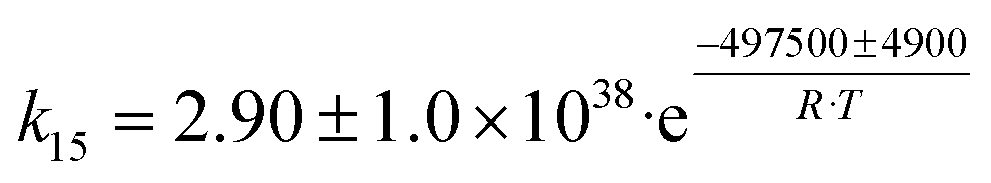
|
Although the latter reaction is feasible,17,18 Sels et al. have shown it to be thermodynamically unfavored due to the stability of 1-butene.11 The corresponding set of rate expressions (Table 1) was used to solve the mass balance of an integral reactor and fit the model. The kinetic parameters obtained showed butadiene formation from aldol condensation (Table 1(5)) to have the lowest activation energy barrier. Other notable features of this kinetic model are: the apparent zero order of the butadiene formation steps, the competition between the adsorption of acetaldehyde and butanal on the main active site, and the Eley–Rideal mechanism of ethylene dimerization. Unfortunately, no statistical information was provided to assess the validity of the model (Table 2). However, it remains the only formal kinetic model of the Lebedev process, which may require further research on the subject.
| No | Reaction scheme | Reaction type | k° × 103 | E a (kJ mol−1) |
|---|---|---|---|---|
| ↔: reversible reaction. →: irreversible reaction. k° at 613 K.a m3 kgcata−1 s−1.b (m3)2 kgcata−1 mol−1 s−1.c (m3)3 kgcata−1 mol−2 s−1. | ||||
| 1 | 2 AcH ↔ Crotonaldehyde + H2O | Aldol reaction | 0.3b | 40 |
| 2 | 2 AcH ↔ MVK + H2O | Aldol reaction + intra-MPV | 6.4 × 10–2b | 40 |
| 3 | Crotonaldehyde + EtOH ↔ 2-buten-1-ol + AcH | MPV reaction | 36b | 45 |
| MVK + EtOH ↔ 3-buten-2-ol + AcH | ||||
| Butanal + EtOH ↔ 1-butanol + AcH | ||||
| MEK + EtOH ↔ 2-butanol + AcH | ||||
| 4 | 2-Buten-1-ol → BD + H2O | Dehydration | 9.4a | 55 |
| 3-Buten-2-ol → BD + H2O | ||||
| 1-Butanol → butene + H2O | ||||
| 2-Butanol → butene + H2O | ||||
| 5 | 2-Buten-1-ol ↔ butanal | Isomerisation | 0.14a | |
| 3-Buten-2-ol ↔ MEK | ||||
| 6 | 2 AcH + EtOH ↔ EtOAc + H2O + EtOH | Ester formation | 3.6 × 10–4b | 41 |
| 7 | 2 AcH ↔ acetone + formaldehyde | Aldol addition + intra-MPV + retro-aldol | 4.5 × 10–3b | |
| 8 | Formaldehyde + EtOH → C3 + 2 H2O | C3 pathway from acetone | 0.65b | |
| 9 | Acetone + EtOH → C3 + AcH + H2O | C3 pathway from formaldehyde | 5.4 × 10–3c | |
| 10 | Acetone + EtOH → C5 + 2 H2O | C5 pathway | 0.54b | |
| 11 | 2 AcH + EtOH → hexatriene + 3 H2O | C6 pathway | 5.0 × 10–4c | |
| 12 | EtOH → ethylene + H2O | EtOH dehydration | 8.0 × 10–3a | 157 |
| 13 | 2 EtOH ↔ DEE + H2O | DEE formation | 4.4 × 10–4b | 103 |
A kinetic study of the Ostromislensky process using Ta2O5–SiO2 has recently been conducted by Dussol et al.98 As discussed previously, the authors proposed a parallel pathway to butadiene from ethanol and acetaldehyde mixtures—the Inoue route—characterized by the intramolecular MVPO reaction of acetaldol to form 4-hydroxybutan-2-one. The latter subsequently transforms into butadiene via the same reaction sequence as the Toussaint–Kagan pathway (Scheme 11) which would take place simultaneously.
Effects of the ethanol flow rate and temperature
In lieu of formal kinetic modelling, statistical modelling has been used to assess the effect of reaction conditions on catalytic performances. Da Ros et al.157 and Cabello González et al.4 each developed empirical mathematical models to predict the activity of K2O–ZrO2–ZnO/MgO–SiO2 and hemimorphite-HfO2/SiO2 catalysts, respectively, in the Lebedev process as a function of the temperature and ethanol flow rate. The effect of water, which Cabello González et al. studied, will be discussed later. Both models were constructed by fitting polynomial equations to experimental results, choosing selectivity, yield, molar fraction or productivity as a response. Although the accuracy of empirical models is limited to the range of operation conditions studied, the activity trends observed are comparable to results obtained by other scholars in their studies of the ethanol-to-butadiene reaction. Consequently, it is ideal for visualizing the general impact of operation conditions.As depicted in Fig. 9(a), temperature showed a positive effect on ethanol conversion, whereas WHSVEtOH displayed a negative influence. Although similar results were found on other catalyst systems,109,158 the linear relationship between conversion and temperature depicted was not observed.109 Ethanol–acetaldehyde mixtures also displayed comparable conversion trends.5 Due to the endergonic nature of the reaction, a temperature increase is expectedly beneficial to conversion. However, Bhattacharyya and Avashti noted a decrease in ethanol conversion between 673 and 723 K on ZnO–Al2O3,159 which may be explained by the thermodynamic unfavourability of aldol condensation at higher temperatures.
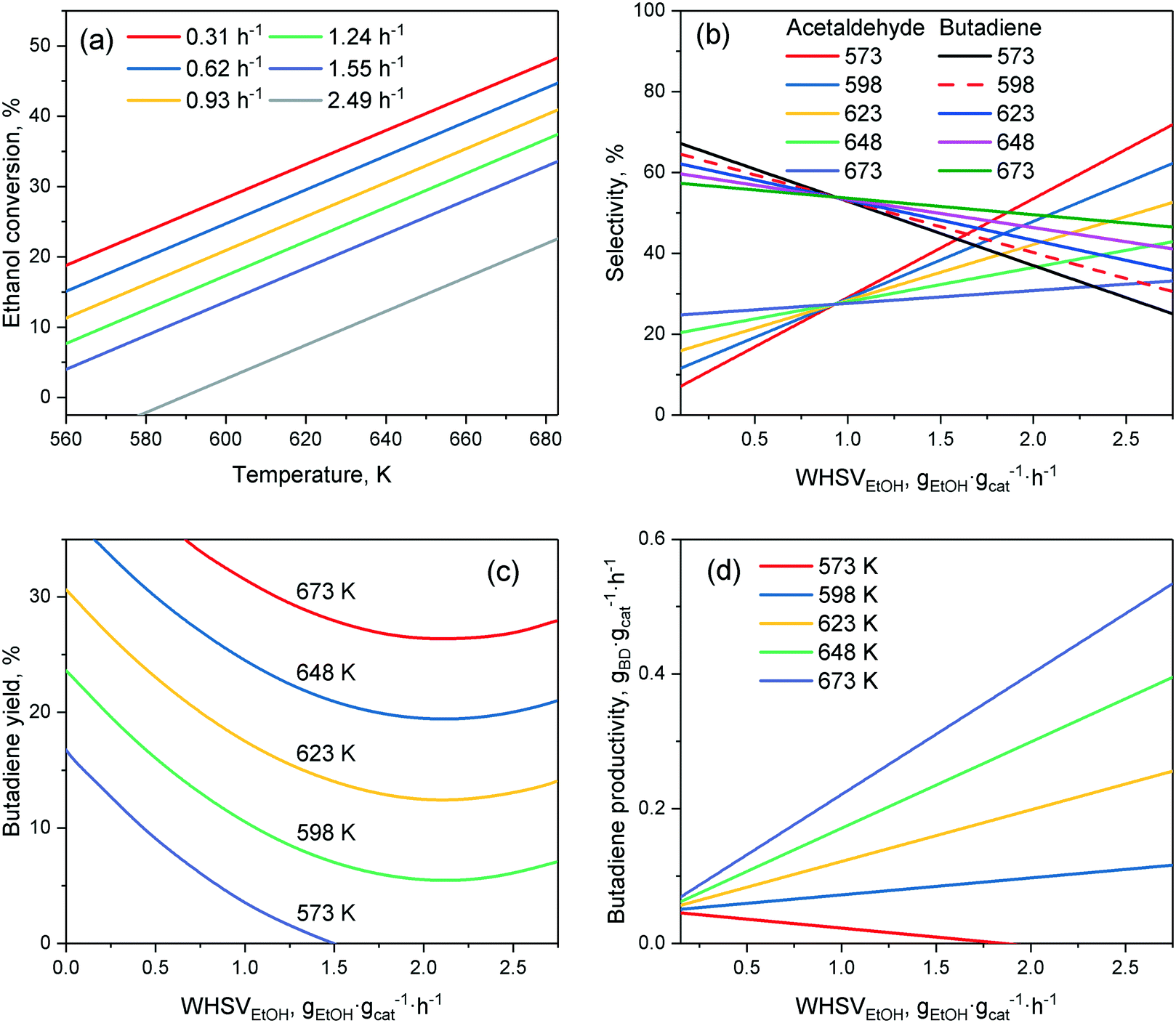 | ||
| Fig. 9 Empirical model by Pinto et al. predicting the effect of the temperature and ethanol flow rate on selected performance metrics: (a) ethanol conversion; (b) acetaldehyde and butadiene selectivity; (c) butadiene yield; (d) butadiene productivity. Catalyst used: K2O–ZrO2–ZnO/MgO–SiO2. Reproduced from ref. 157. Copyright 2017 with permission from Elsevier. | ||
Fig. 9(b) depicts the change in selectivity occurring when tuning the operation conditions. When the ethanol flow was increased, the selectivity towards butadiene decreased in favor of acetaldehyde, as well as of other byproducts (not shown). In the Lebedev process, the accumulation of acetaldehyde with reduced contact time is often observed, allowing many authors to conclude that aldol condensation is the rate-limiting step.28,33,108,160 Product selectivity was also highly dependent on temperature. Increasing it had the opposite effect, favoring butadiene selectivity significantly. Different results were obtained on other catalytic systems: on Cu–Ta/SiBEA, higher temperature (>573 K) significantly reduced butadiene selectivity to the benefit of ethylene;161 on Au/MgO–SiO2, butadiene selectivity increased, before falling when the temperature rose above 573 K.93 Interestingly, butadiene selectivity was almost unaffected by temperature on MgO–SiO2 when conversion was maintained at 40% by adjusting the ethanol flow rate, whereas byproduct selectivity changed significantly.139 Explaining the discrepancies reported is difficult, due to the drastically different catalysts and reactor set-ups used. However, it is noteworthy that the product selectivity does not have a clear relationship with temperature.
The butadiene yield (Fig. 9(c)), important for the practical application of the reaction, was favored in a non-linear fashion by high temperatures and low ethanol flow rates, which are also beneficial to conversion.97,137,159–161 Increasing WHSVEtOH suppressed the butadiene yield non-linearly, in agreement with observations made on other catalytic systems.109 This is highly relevant due to the fact that a high ethanol flow rate is required for attaining the high butadiene productivity needed to meet industrial standards.92 Although the model of Da Ros et al. indicated a linear relationship between productivity and WHSVEtOH (Fig. 9(d)), it is unlikely that its validity extends beyond the range of studied reaction conditions due to productivity plateaus appearing at high WHSVEtOH, either because of diminishing conversion, or reduced selectivity to butadiene. For instance, Kyriienko et al. Found that WHSVEtOH in excess of 1 h−1 lowered the butadiene productivity on Cu–Ta/SiBEA as a sharp decrease in butadiene yield occurred.161
The relationship between activity and temperature or WHSVEtOH appears to vary greatly depending on the catalytic system used. The empirical model established by Da Ros et al. is only valid for the specific operation conditions studied. Generally, reducing the ethanol flow rate during the Lebedev process improved the catalytic butadiene selectivity and yield. However, the resulting lower butadiene productivity is not very interesting for industrial application. High WHSVEtOH increases productivity, but only to some extent, as it suppressed butadiene formation, possibly due to kinetic or diffusion limitations. In the range found ideal by Sels et al.11 (602–703 K), increasing the temperature improves ethanol conversion. However, the unclear relationship between butadiene selectivity, temperature and the catalytic system used means that the benefits to the butadiene yield are not straightforward; ideal temperatures are still found on a case-by-case basis.
Da Ros et al. also performed a microkinetic analysis of the ethanol-to-butadiene reaction on MgO–SiO2 which highlighted this phenomenon.168 The authors established that the experimental fluctuations of the molar fraction of distinct species in the output stream of their catalytic tests were not independent of one another. Local microkinetic information was extracted from the covariance matrix of experimental fluctuations. A negative correlation coefficient between the molar fractions of two given products signaled a fluctuation in opposite direction, indicative of a reactant–product relationship. Contrarily, a positive correlation coefficient could either be interpreted a co-reactant or co-product relationship, or a reactant–product connection with no bearing on the reaction rate. Da Ros et al. reported a change in mechanism with increasing temperature on the basis of a change in correlation coefficient. At 723 K, the reactant–product relationship between ethanol and acetaldehyde becomes unclear. Simultaneously, the 723 K mark sees the emergence of a reactant–product relationship between acetaldehyde and butadiene, when there were none at lower temperatures. Da Ros et al. interpreted these observations as a change in the reaction kinetics. Between 573 K and 673 K, the rate-determining step was the aldol condensation. At 723 K, the availability of acetaldehyde became kinetically relevant, e.g., its formation by ethanol dehydrogenation determined the reaction rate. Interestingly, the correlation coefficient of H2 remained positive, indicating that it was not involved in the reduction of crotonaldehyde.
Effects of the ethanol–acetaldehyde ratio on the two-step process
In the Ostromislensky process, the ratio of ethanol to acetaldehyde in the reactant feed is a crucial reaction parameter, usually ranging between 2 and 4,9,14,162 and between 0.7 (ref. 10) and 9 (ref. 5) in extreme cases. Although catalysts for the Ostromislensky process generally lack a dehydrogenation function, the best ethanol–acetaldehyde ratios reported are often above 1, indicating that the reaction regenerates acetaldehyde by the MPVO reaction. Several scholars have sought to improve their process by tuning the ethanol–acetaldehyde ratio, such as Zhu et al.5 (Fig. 10) who studied the influence of this parameter with the MgO–SiO2 catalyst.10,34,162 However, there is no recognized optimal value. This phenomenon is ostensibly due to the different properties of each catalytic system and the reaction conditions used.5,10 Consequently, the ethanol–acetaldehyde ratio adds another dimension to the reaction conditions that must be optimized to maximize butadiene formation. To the best of our knowledge, no modelling of this parameter has been reported.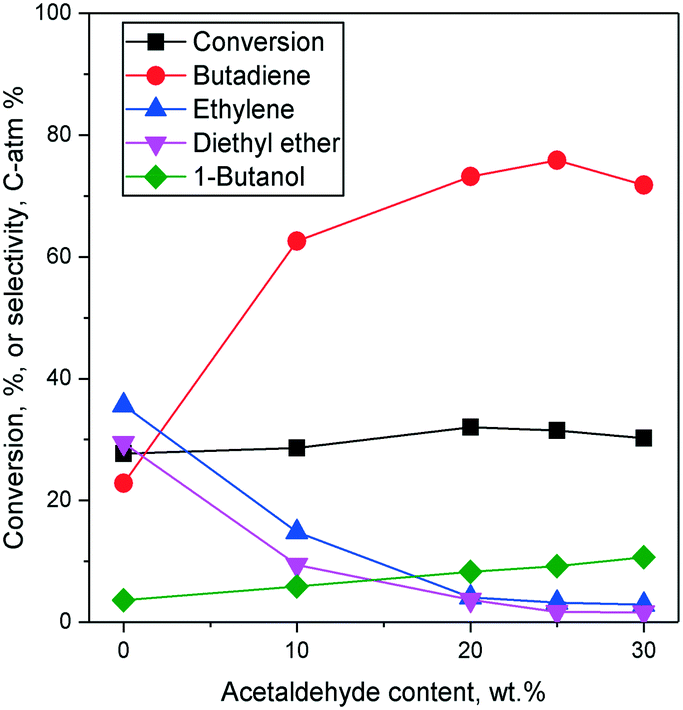 | ||
| Fig. 10 Effect of acetaldehyde content in the ethanol–acetaldehyde mixture used for the Ostromislensky process on MgO–SiO2.5 | ||
Effects of the water in the feed
As previously mentioned, using water-containing ethanol would be economically and environmentally beneficial. In this regard, the influence of co-feeding water with ethanol or ethanol–acetaldehyde mixtures is a crucial reaction parameter for the practical application of the ethanol-to-butadiene reaction and what degree of water, if any, must be removed. Furthermore, since water is generated in situ, processes designed to recycle unconverted ethanol would inevitably operate with water in the feed due to their azeotrope, unless incorporating additional purification steps.4 Unfortunately, the effect of water is an understudied subject, with most researchers preferring to use anhydrous ethanol when performing catalytic tests.Recent studies have provided insight on the subject, but some discrepancies remained to be answered. In the Lebedev process, three major effects have been observed. First, co-feeding water suppresses ethanol conversion.4,97,159 Cabello González et al., who modeled the impact of water, the ethanol flow rate and temperature on the reaction, found that increasing the water content of the feed progressively reduced conversion (Fig. 11(a)).4 Second, product selectivity was altered by the presence of water, generally reducing butadiene selectivity in favor of ethylene and acetaldehyde,4,97 although this effect also depended on the other reaction conditions (Fig. 11(b)). Third, water decreased the rate of catalytic deactivation.4,97,159 The poisoning effect was attributed to the adsorption of water molecules on active sites, notably the Lewis acid sites responsible for acetaldehyde condensation. This also explained the reduced deactivation observed, as the condensation of aldehydes into heavier carbonaceous species has been identified as a source of catalyst poisoning.109,163,164 IR spectroscopy identified the in situ formation of Brønsted acid sites upon addition of water, which are known to catalyze the dehydration of ethanol, explaining the increased ethylene selectivity.4,97 Ultimately, process design and economic analysis will decide whether an optimal water content—where the drawbacks of using water-containing ethanol for the Lebedev process mainly include the loss of butadiene yield and the increase of byproducts—is outweighed by the economic benefits of reduced separation cost, as well as the increased catalyst lifespan.
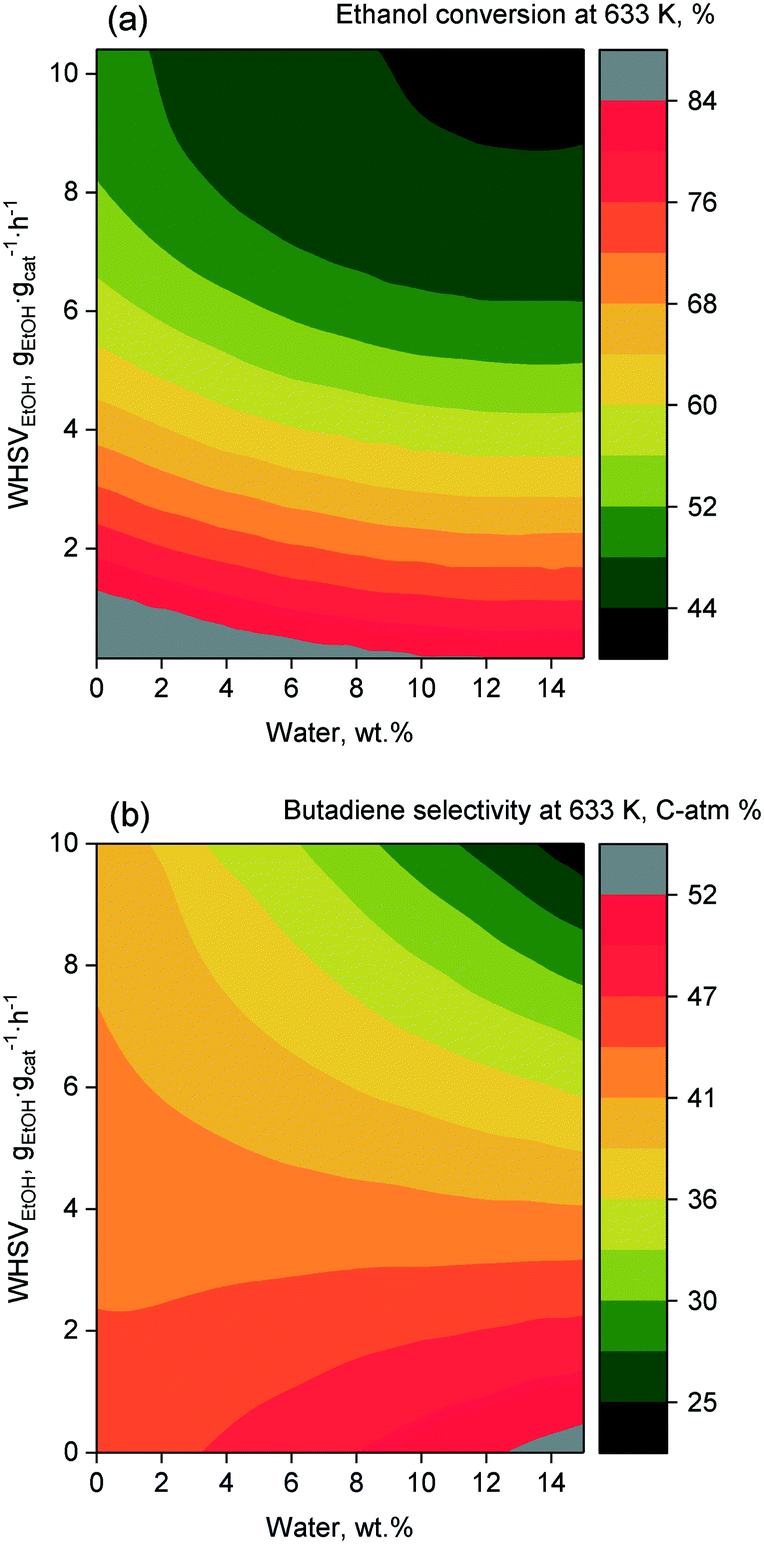 | ||
| Fig. 11 Empirical model for the effect of water content in the Lebedev process over hemimorphite-HfO2/SiO2. Reproduced from ref. 90. Copyright 2019, with permission from Elsevier. (a) Ethanol conversion at 633 K; (b) butadiene selectivity at 633 K. | ||
In the case of the Ostromislensky process, the suppression of heavy carbon species formation by co-feeding water was also observed.5,164 Although ethanol–acetaldehyde conversion was slightly reduced, Zhang et al. and Zhu et al. have found butadiene selectivity over ZnO–ZrO2 and MgO–SiO2 to be mostly unaffected by the addition of up to 50 wt% of water to the feed.5,164 Toussaint et al. found 10 wt% water to decrease the rate of butadiene formation on Ta2O5/SiO2, while also reporting an enhance catalyst lifetime.28 Whether this contrast in activity with the Lebedev process is owed to the difference in reaction conditions or is inherent to the Ostromislensky process remains to be answered.
Catalyst deactivation
Sels et al. identified catalytic stability to be a factor in the ethanol-to-butadiene route's ability to compete with existing fossil-based technologies.11 Despite this, research has focused on strategies to improve stability, rather than on understanding deactivation itself, which is a field of study of its own.165,166 Carbon species formed during ethanol conversion to butadiene are generally recognized to result in catalyst decay.137,163,164 But details on the exact nature and mechanism of deactivation are lacking. Deactivation is reported in almost every instance of catalytic testing. The time-scale of deactivation can be in a matter of hours, comparable to the ethanol-to-propylene process with ZSM-5, another pathway to renewable olefins,167–169 or extend to hundreds of hours depending to the reaction conditions. Most published catalytic reactions did not go beyond 10–20 hours on stream, with the longest instances of stability testing lasting 143 hours (ref. 170) and 175 hours.28Numerous factors influence the observed catalyst decay. Like selectivity, deactivation rates vary greatly depending on the catalyst. The choice of metal demonstrably affects stability of materials in otherwise identical catalytic tests.171–173 In turn, the resulting chemical surface properties correlate with the ability of a sample to resist deactivation: strong acidic and basic sites are believed to accelerate carbonaceous compound formation.160,174 The catalyst support and its morphology unambiguously influence the speed of catalytic decay.35,160,170,175 Toussaint et al. reported that lower temperature reduces deactivation.28,33 They also noted that the ethanol–acetaldehyde ratio is a factor affecting the stability of catalysts in the Ostromislensky process.
Based on a review of the literature, Corson et al. stated that the Ostromislensky process suffers from a slower decay than the Lebedev process.7 However, the disparity may be owed to the different natures of catalysts employed at the time, e.g., Ta2O5/SiO2 compared with ZnO–Al2O3 in the two- and one-step processes, respectively. Many recent two-step processes show remarkable stability albeit at relatively low WHSVEtOH,10,164,170,171 possibly a contributing factor. An accurate comparison would require catalytic testing using similar materials and conditions. In one such instance, the normalized activity of Ta/SiBEA in the two-step process remained slightly more stable than that of Cu-Ta/SiBEA in converting pure ethanol (Fig. 12(a)).161,171 When comparing deactivation rates on silica-supported zirconia at a longer time-on-stream, the Ostromislensky process again showed a superior stability (Fig. 12(b)).10,176 However, the different reaction conditions make this comparison less conclusive. So far, the conclusion of Corson et al. appears to be correct.
 | ||
| Fig. 12 Stability comparison between two similar catalytic systems in the one- and two-step processes. (a) Cu-1% %Ta/SiBEA (T = 598 K, WHSVEtOH = 0.5 h−1);164 1% Ta/SiBEA (T = 598 K, WHSVEtOH = 0.792 h−1, EtOH:AcH = 3.2).171 (b) Ag-2.95% Zr/SiO2 (T = 598 K, WHSVEtO0 h = 0.23 h−1);176 2.7% Zr/SiO2 (T = 673 K, WHSVEtOH = 1.5 h−1, EtOH:AcH = 1.16–1.60).17 Normalized activity was defined as the specific activity at any time-on-stream divided by the initial activity obtained by extrapolation to TOS = 0 h.165 | ||
Deactivation during alcohol catalytic conversions generally takes place via two mechanisms:177–179 (i) active site poisoning by carbonaceous molecules and/or (ii) pore obstruction or blockage by large species. In the first case, adsorbed molecules sterically prevent the access of reactants to the active sites.165 In the second case, pore obstruction decreases reactant mass transfer rates by reducing accessibility to the pore network; complete blockage may also occur, further hindering the access to active sites.165 The contribution of each mechanism to deactivation depends on several factors, such reaction conditions, i.e., temperature and contact time, the nature of reactants and the catalytic system. In the ethanol-to-butadiene reaction, pore blockage by carbonaceous species has been speculated as a significant deactivation mechanism. Notably, mesoporous catalyst carriers with larger pores have remained relatively more stable, suggesting that pore size played a role in deactivation.10,14,35,170 BET analysis of deactivated catalysts also demonstrated the loss of pore volume, pore size and specific surface area.164 The fact that calcination under air has repeatedly been used to regenerate spent catalysts10,170,180,181 is a strong indicator that such structural changes are owed to the deposition of organic species and not framework collapse. A feature of the blockage mechanism is the accumulation of large amounts of heavy carbonaceous molecules.165 Thermogravimetric analysis (TGA) of different mesoporous catalysts showed significant accumulation of such species, identifiable by high-temperature weight loss.10,164,170 For instance, Kim et al. reported nearly 25 wt% heavy carbon content in the spent Ta/SBA-100 catalyst after a TOS of 40 h.170 Large carbonaceous species are formed by the polymerization of lighter hydrocarbons, which can take place on the catalytic sites active for condensation reactions. In their study of deactivation on ZnO doped ZrO2–SiO2,164 Zhang et al. characterized the surface of spent catalysts using X-ray photoemission spectroscopy (XPS). They found a large proportion of the C 1s signal to be attributable to graphitized carbon. Consequently, Zhang et al. argued that pore blockage by large polymerized aromatic species contributes to deactivation. As noted above, co-feeding water consistently suppressed the formation of carbon species and extended the catalyst lifetime, ostensibly due to the inhibition of its activity in condensation reactions.4 Interestingly, this suppression effect shifted the nature of retained carbonaceous species from heavy carbon to lighter carbon molecules.164
Li et al. investigated the deactivation mechanism during the Lebedev process on a microporous Zn–Y/SiBEA catalyst.163 Contrary to mesoporous materials, the TGA of spent catalysts revealed more light carbonaceous species and fewer heavy compounds—the coking rate was lower than those with other catalytic systems.164,170 In addition, organic extracts from HF-dissolved samples contained no polycyclic aromatics indicative of large amounts of polymerized coke, but many carbonyl-possessing unsaturated five- or six-membered rings. In situ DRIFTS and UV-vis spectroscopy corroborated not only the presence of these compounds, but also the formation of C5+ carbonyl molecules. Accordingly, Li et al. proposed that, rather than pore blockage, deactivation on the zeolite-based catalyst results from the gradual coverage of Zn and Y sites by the deposition of large unsaturated cyclic compounds. Aldolization of acetaldehyde and acetone formed C5+ carbonyl intermediates, which were also observed on Zr-containing materials,108,175 subsequently undergoing cyclization, resulting in the deactivating species (Fig. 13). At present, it is difficult to judge whether the alternative deactivation mechanism reported by Li et al. can be attributed to the chemical or morphological properties of the catalyst—which is microporous, contrary to the mesoporous samples of Kim et al.—or different reaction conditions. Incidentally, Cabello González et al. similarly found the deposition of oxygenated aromatic species on active sites to be a source of deactivation for the Lebedev process on hemimorphite-HfO2/SiO2, suggesting that this mechanism is not limited to microporous catalysts.109
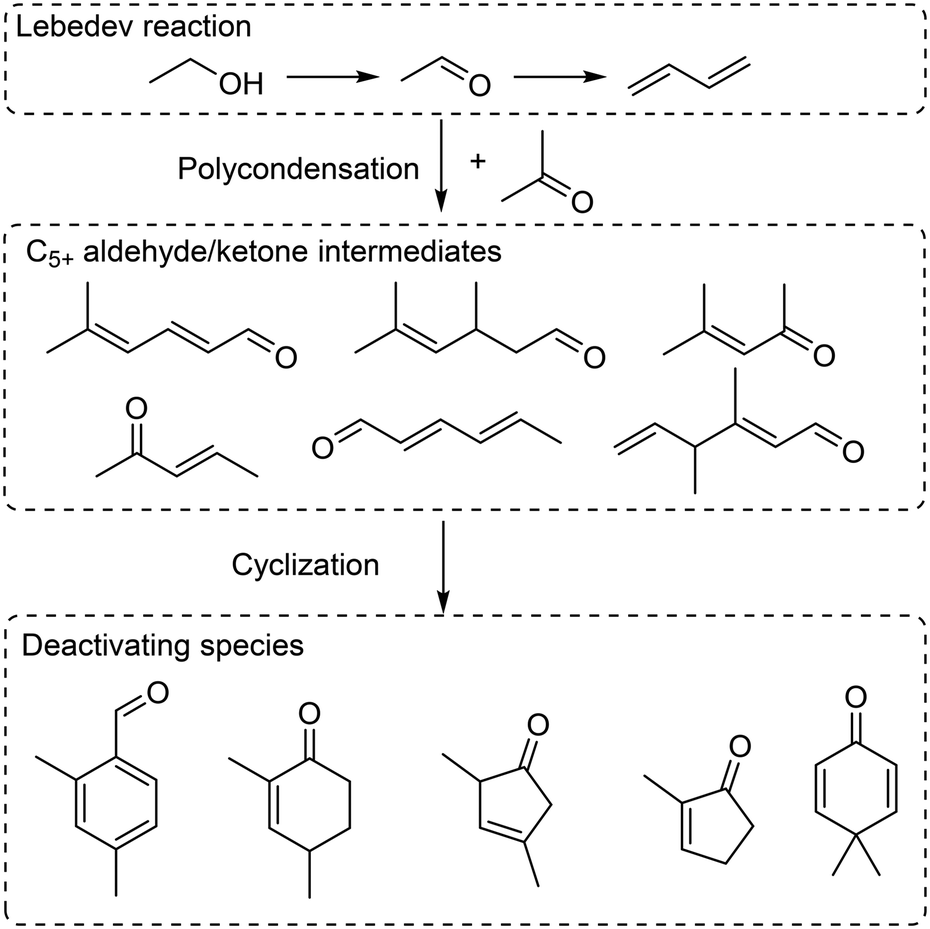 | ||
| Fig. 13 Deactivation mechanism on Zn-Y/SiBEA proposed by Li et al.163 The list of intermediate and deactivating species is not exhaustive. | ||
In studying the deactivation of MgO–SiO2 in the Ostromislensky process, Zhang et al. demonstrated that the nature of deposited carbonaceous species differed depending on their location of the surface with a multi-level extraction method.182 CCl4 was used for the outer surface, CHCl2 for middle layers and CHCl2 following catalyst dissolution in HF for the inner layers of the catalyst. The authors determined by GC-MS that chained alkanes were predominant on the outer surface and aromatic species within the pores of the spent catalyst. Considering the results of Li et al., it can also be concluded that the type of deactivating species depends on the nature of catalyst used.
For the Lebedev process, both conversion and selectivity towards butadiene tend to decrease with time-on-stream, which also results in greater acetaldehyde selectivity (Fig. 14(a)).106,122,186 Contrarily, during the Ostromislensky process, although ethanol conversion also declines, butadiene selectivity is generally unaffected (Fig. 14(b)).10,164,170,171 A possible explanation for this discrepancy may be the selective poisoning of active sites. If sites responsible for the aldol condensation also form the heavier carbonaceous species ostensibly responsible for deactivation, they are more likely to be blocked by carbon depositions. On multi-functional catalysts for the Lebedev process, this phenomenon could lead to an excess of dehydrogenating sites, explaining the greater acetaldehyde selectivity observed. In the Ostromislensky process, catalysts generally lack a dehydrogenating function. As a result, poisoning would proceed uniformly by only targeting the sites responsible for condensation, lowering the negative impact on selectivity.
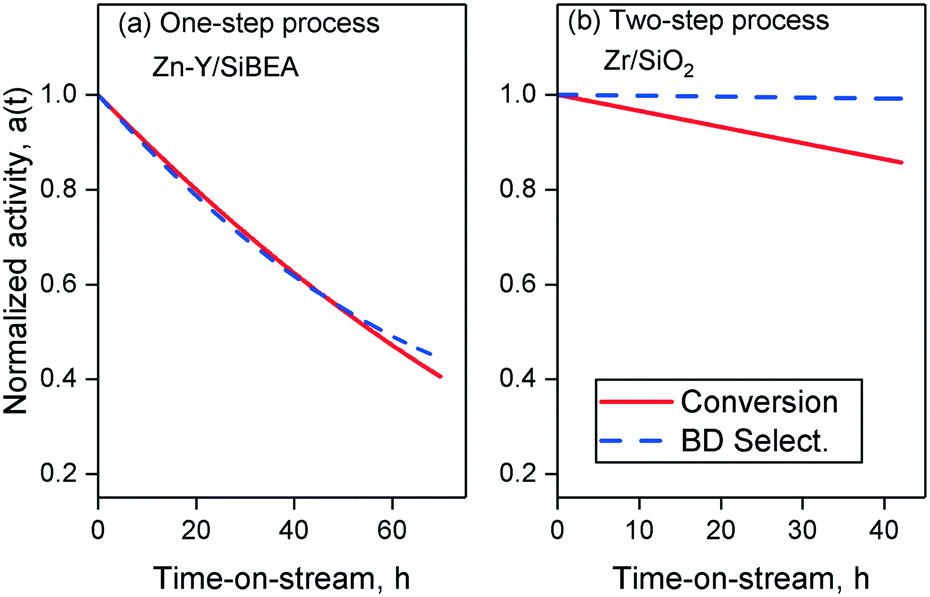 | ||
| Fig. 14 Effect of time-on-stream on conversion and selectivity for catalysts in the one- and two-step processes. (a) ZnY/SiBEA (T = 673 K, WHSVEtOH = 1.0 h−1).163 (b) Zr/SiO2 (T = 673 K, WHSVEtOH = 1.5 h−1).10 | ||
Deactivation processes unrelated to carbon deposition have also been proposed for specific catalytic systems. For instance, Taifan et al., who studied the Lebedev process on CuO and ZnO-modified MgO–SiO2, distinguished two deactivation mechanisms depending on the choice of promoter.181 Using operando X-ray analysis, the authors observed the disappearance of Cu–O bonds and the emergence of Cu–Cu pairs at 673 K after several hours on stream, whereas zinc bonds were resilient under the same operating conditions. They proposed that deactivation on Cu/MgO–SiO2 resulted in part from Cu reduction and sintering. Carbon deposition was judged by Taifan et al. to be the more likely cause of deactivation on the ZnO-containing catalyst. Cabello González et al. tested a HfO2/SiO2 catalyst modified with the zinc silicate hemimorphite in the Lebedev process.109 XPS analysis indicated that Zn(II) was reduced in the spent catalyst. The authors proposed that part of the deactivation could be attributed to this phenomenon, as the Zn(II) sites of hemimorphite were considered the sites responsible for ethanol dehydrogenation. Ostensibly, this phenomenon is reserved to hemimorphite, and other scholars analyzing the Zn 2p peak of ZnO-containing catalysts observed no change in the oxidation state of Zn after the reaction.163
Understanding and preventing catalyst deactivation during the conversion of ethanol to butadiene has progressed in recent years. Pore blockage appears to be the predominant deactivation mechanism at high coking rates, whereas active site poisoning by bulky oxygenated species has been reported at low coking rates on a zeolite catalyst.163 Whether catalyst properties or reaction conditions favor one mechanism over the other is not yet understood. Due to the repeated observation of bulky oxygenated carbonaceous species,108,109,163 both mechanisms are possibly initiated by the condensation of carboxylic species on Lewis acid sites. It is unlikely that the olefin condensation mechanism leading to the formation of deactivating polyaromatic coke that takes place in other ethanol-to-olefin reactions183 occurs in the conversion of ethanol to butadiene. Indeed, catalysts active in the Lebedev and Ostromislensky processes predominantly possess Lewis acidity,5,126,161 whereas the condensation of olefins produced by the dehydration of ethanol has been found to preferably take place on Brønsted acid sites.183,184,185 Incidentally, the co-feeding of water, which has been found to not only poison Lewis acid sites, but also to generate new Brønsted acid sites,52,109 alleviated the formation of heavy coke species and extended the catalyst lifetime. Consequently, one approach to consider for reducing catalyst deactivation is the passivation of acid sites to prevent the over-condensation of carboxylic intermediates. As discussed below, alkali-doping in one strategy to achieve this. Deactivation mechanisms unrelated to carbon formation should be considered, as Taifan et al. observed that particle sintering can take place during the reaction181 and Cabello González et al. found metal oxide active sites to be reduced in situ,109 both hindering the catalytic activity. However, such mechanisms appear to be limited to specific catalytic systems and should therefore be dealt with on a case-by-case basis.
Byproducts
Ethanol conversion to butadiene generates many byproducts. Their presence increases separation costs, reducing the ability of ethanol-to-butadiene processes to compete with petroleum-based routes. Consequently, suppressing the formation of undesired compounds coincides with the aim of improving butadiene selectivity. Doing so requires an understanding of byproduct formation. Butadiene itself does not convert to other compounds, as evidenced by its stability under catalytic test conditions.109 Side-reactions occurring along the pathway to the final product cause the emergence of such undesired species. However, their amount and nature depend on the reaction conditions and the catalytic system employed. We review in this section the theorized or demonstrated routes to the many undesired compounds formed during butadiene production regardless of reaction parameters. Fig. 15 illustrates the reaction network of the main byproducts.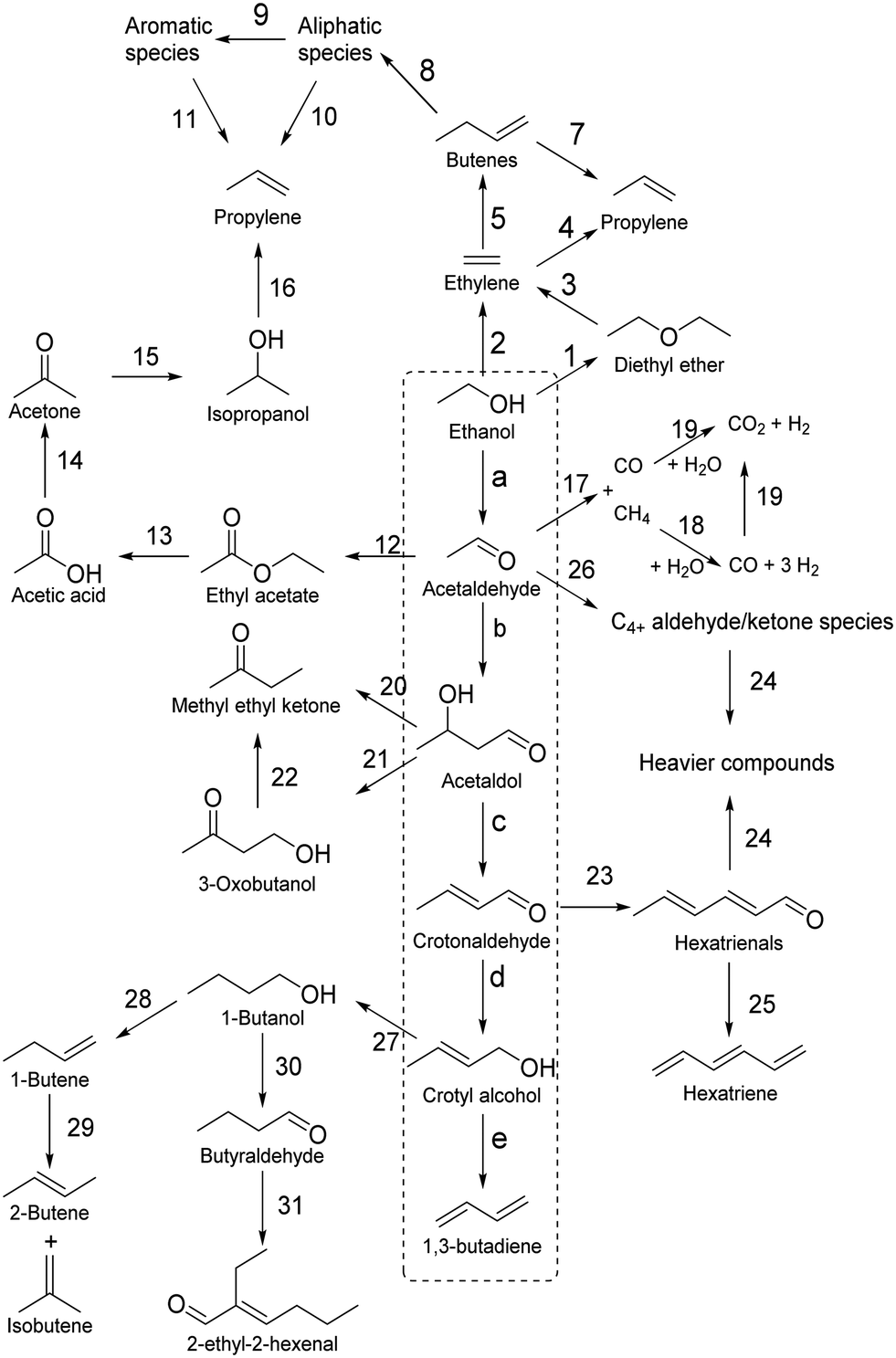 | ||
| Fig. 15 Main byproducts' pathway believed to take place during the ethanol-to-butadiene reaction. In the dashed boxed: the main pathway to butadiene from ethanol. The list is not exhaustive, as other byproducts may be detected. | ||
Ethylene and diethyl ether are the principal undesired byproducts of butadiene synthesis. Both species result from the dehydration of ethanol, potentially taking place on acidic186 or basic sites.96 Diethyl ether forms from an intermolecular dehydration of two ethanol molecules (Fig. 15(1)). Ethylene may not only result from the direct intramolecular dehydration of ethanol (Fig. 15(2)), but also from a subsequent conversion of diethyl ether (Fig. 15(3)), alternatively described as a dehydration186 or cracking187 reaction. The triangular scheme describes the parallel series of reactions ostensibly happening. The reaction conditions and the nature of the catalyst dictate which route dominates the production of ethylene.188–190 Generally, the diethyl ether pathway to ethylene takes place at lower temperature (i.e., below 543 K),190 whereas the direct dehydration route predominates at higher temperatures. With the kinetic curves obtained with a Zn and Hf-containing catalytic tests at 633 K—a temperature more typical of the Lebedev process—Cabello González et al. concluded that ethylene was a primary stable product, not a secondary one derived from diethyl ether.109 Compared to acetaldehyde, ethylene is thermodynamically the favored product of ethanol conversion.11 Furthermore, DFT calculations on MgO showed that the direct dehydration of ethanol possessed a lower energy barrier than its dehydrogenation.100 Consequently, ethylene formation is in direct competition with the ethanol-to-butadiene pathway. High ethylene yields plague the process so much so that catalyst design strategies have been adopted to specifically limit ethanol dehydration, namely the use of alkaline dopants to suppress the responsible acid sites.8,172,191
Ethylene is also an intermediate of various ethanol-to-hydrocarbon reactions. Propylene can be formed from the reaction between surface carbene species and ethylene (Fig. 15(4)),192 from the cracking of bigger aliphatic species (Fig. 15(7) and (10)),193 or via the hydrocarbon pool mechanism involving aromatic intermediates (Fig. 15(11)).194 The dimerization of ethylene (Fig. 15(5))195 can also lead to the formation of larger aliphatic species (Fig. 15(8)) and their cyclisation to aromatics (Fig. 15(9)).196
Several less important byproducts may form during the subsequent steps of the ethanol-to-butadiene reaction. Besides condensing to crotonaldehyde, the highly active acetaldehyde may undergo the Lewis acid-driven Tischenko reaction to yield ethyl acetate (Fig. 15(12)).11,108,109,111,175 Hydrolysis of ethyl acetate forms acetic acid (Fig. 15(13)), which produces acetone through decarboxylation (Fig. 15(14)).11,108,109,111,175 Propylene can ultimately be obtained by the reduction of acetone to isopropanol (Fig. 15(15)) and its subsequent dehydration (Fig. 15(16)).108,109,175 Ethanol reforming,197–199 which involves acetaldehyde decarbonylation (Fig. 15(17)), steam reforming of methane (Fig. 15(18)) and the water-gas shift of carbon monoxide to carbon dioxide (Fig. 15(19)), may explain the traces of these compounds often detected amongst the product distribution.92,139,160 Methyl ethyl ketone formation33 can be explained by various mechanisms: the rearrangement of deoxygenated acetaldol (Fig. 15(20))11,200 or the dehydration of 3-oxobutanol formed via the intramolecular H transfer of acetaldol (Fig. 15(21) and (22)).197 Other possibilities include the dehydration of butanediol (not shown) or crotyl alcohol isomerization (not shown).11
Many scholars identified crotonaldehyde as a precursor to heavier,108,109,175 potentially poisoning163 compounds. It may undergo aldol coupling with other carboxyl-containing molecules, such as acetaldehyde and acetone to form larger C6+ compounds. For instance, cross-coupling between acetaldehyde and crotonaldehyde leads to hexadienal (Fig. 15(23)), which may convert to hexatriene (Fig. 15(25)) or couple with aldehydes into heavier compounds (Fig. 15(24)). Besides crotonaldehyde, a host of aldehyde and ketone molecules may form from the aldol condensation of acetaldehyde.163 Li et al. argued that the cyclization of such heavy compounds initiates the coking process responsible for catalytic deactivation (Fig. 15(26)).163
The Guerbet reaction, believed to follow a pathway akin to that of the ethanol-to-butadiene reaction, likely forms the majority of C4 byproducts.96,114,131 1-Butanol, ostensibly obtained from hydrogenation of crotonaldehyde via crotyl alcohol (Fig. 15(27)),114,131 can undergo dehydration to 1-butene (Fig. 15(28)) which subsequently isomerizes to isobutene and 2-butene (Fig. 15(29)).11,108,109,175 C4 olefins are problematic since their separation, required to achieve high-purity butadiene, is an expensive process.18 The small presence of butyraldehyde139,201 may be owed to the dehydrogenation of 1-butanol (Fig. 15(30))109 or the partial hydrogenation of crotonaldehyde (not shown).200,201 Self-coupling of butyraldehyde may also lead to bulky oxygenated carbonaceous compounds (Fig. 15(31)).109
The high reactivity of oxygenated hydrocarbons and olefins causes the ethanol-to-butadiene reaction to generate several unwanted species. Reaction intermediates such as acetaldehyde may be recycled, but these byproducts impede the economic viability of the ethanol-to-butadiene reaction. Catalyst design should aim to suppress their formation, notably that of ethylene, which forms in large amounts, and butenes, due to their difficult separation from butadiene.
Catalytic systems
Many catalysts have been tested and reported in the literature since research first began on the conversion of ethanol to butadiene. Notably, screening studies by Corson et al.7 and Bhattacharyya et al.202,203 have generated over 600 different materials. This section reviews the two predominant catalytic systems: (i) group 4 and 5 transition metals and (ii) magnesia–silica mixed oxides. These two categories have been the subject of several studies, affording precious insight on their activity and the different methods used to prepare them. In addition, rare-earth metal oxide catalysts, a recent topic of interest, are discussed. Furthermore, the use of alkali and alkaline-earth dopants, a design strategy used to tune the chemical properties of any catalytic system, is addressed.Other catalytic systems, notably mixed oxides of other transition metals, have not been the subject of recent investigation. As a result, there is a comparative lack of characterization using modern techniques available. The reader is referred to previous reviews, which have addressed the omitted catalytic systems in depth.11,12,16
Group 4 and 5 transition metals
Catalysts containing transition metals belonging to groups 4 and 5 of the periodic table have shown remarkable activity in converting ethanol to butadiene. Initially reported in the 1940s by scientists from the Carbide and Carbon Chemicals Corporation,28,33,204 these catalytic systems have been carefully studied throughout the years. As detailed in the original patent, silicates of zirconium, tantalum or niobium oxide were first used to convert ethanol–acetaldehyde mixtures into butadiene due to their condensation ability.204 Furthermore, the wartime screening of over 500 catalysts by Corson et al. found silica-supported titanium and hafnium oxides capable of high butadiene yield in the Ostromislensky process.7,27,34 It also established that dehydrogenation promoters, i.e., MgO or CuO, could be incorporated to make these catalysts active in the Lebedev process. As vanadium oxide performed poorly,7 research then and now has generally concerned Zr, Nb, Hf and Ta—Ti has mostly been ignored, ostensibly due to its lower activity. Since scholars have argued that these metals share a similar catalytic activity in the ethanol-to-butadiene reaction,101,125 this section will discuss them as closely related catalytic systems, despite Zr and Ta clearly being the most studied of all group 4 and 5 transition metals. The important parameters of concern from preparing catalysts active in the one- and two-step processes are discussed by providing examples from the literature.Acidity and activity
Due to the their high activity in the Ostromislensky process,7,171,205 but poor performances in the Lebedev process when lacking dehydrogenation promoters,170,171 Zr, Nb, Hf and Ta oxides have long been assumed to catalyze the aldol condensation and MPVO steps of the ethanol-to-butadiene reaction.34 Through spectroscopic studies of these reactions steps, Ivanova et al. evidenced that the Lewis acid character of Zr(IV) was the source of catalytic activity.206,207 As Nb, Hf and Ta-containing catalysts also display Lewis acidity208–211 and show comparable activity in various organic reactions,215 it is generally assumed that the involvement of Lewis acid sites is valid for all four transition metals.101,109,125,205Solid Lewis acids consisting of transition and post-transition metal oxides, often supported on zeolites and other silicates, have emerged as adaptable catalysts for organic chemistry due to their capacity to activate and convert oxygen-containing molecules. For metal sites incorporated within a silica framework or in bulk metal oxides, Lewis acidity, e.g., the ability to accept electron pairs, is owed to their partial positive charge resulting from the formation of covalent bonds with adjacent oxygen atoms, which become Lewis bases.212 Due to their partial positive charge, metal sites can stabilize oxygenated organic molecules via electrostatic interaction or activate them by accepting electrons from their electron-rich groups. With their basic character, neighboring oxygen atoms are also available for reacting with organic molecules. These acid–base pairs can work in tandem to catalyze a variety of organic reactions, notably the aldol condensation129 and MPVO reaction.213,214
In the Zr, Nb, Hf and Ta catalysts for the ethanol-to-butadiene reaction, the relationship between the catalytic activity and the properties of these Lewis acid sites is not fully understood, in part due to the difficulty of defining and quantifying Lewis acidity.215 For instance, Corson et al. observed a trend with regard to the activity of silica-supported metal oxides in the Ostromislensky process that is generally valid: Sn < Nb < Ti < Zr ≈ Hf < Ta (Table 3, entries 1 to 6).7,125,171,205 However, when compared to their Lewis acid strength when incorporated inside a zeolite, as determined by Román-Leshkov using Mulliken electronegativities and pyridine adsorption energies as descriptors (Fig. 16(a)),6 there appears to be little correlation with activity, as expressed by the butadiene yield (Fig. 16(b)).7 Admittedly, the difference in preparation methods and catalyst carrier between both studies may explain this discrepancy. However, even for other organic reactions, there is generally only a loose correlation between Lewis acid strength and activity;212 as the distinct electronic properties of each type of metal site also influence catalytic performances.215 How these factors impact the activity of group 4 and 5 transition metals in the ethanol-to-butadiene reaction remains to be elucidated.
| Entry | Catalyst | E:A | WHSV | TOS | T | X | BD S. | C2=S. | BD Y. | P BD | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| h−1 | h | K | % | % | % | % | |||||
| a LHSV = 0.4. b Pressure = 1.5 atm. X, S., Y. and P. are conversion, selectivity, yield and productivity. BD and C2= are butadiene and ethylene, respectively. Productivity is expressed in terms of gBD gcat−1 h−1. Active phases are expressed in terms of wt%. | |||||||||||
| 1 | 1.2%Ta2O5/SiO2a | 2.75 | 0.3 | 8 | 623 | — | — | — | 69 | — | 7 |
| 2 | 2.8%ZrO2/SiO2a | 2.75 | 0.3 | 8 | 573 | — | — | — | 64 | — | 7 |
| 3 | 2%HfO2/SiO2a | 2.75 | 0.3 | 8 | 573 | — | — | — | 64 | — | 7 |
| 4 | 1%TiO2/SiO2a | 2.75 | 0.3 | 8 | 623 | — | — | — | 52 | — | 7 |
| 5 | 1%Nb2O5/SiO2a | 2.75 | 0.3 | 8 | 623 | — | — | — | 39 | — | 7 |
| 6 | 2%SnO2/SiO2a | 2.75 | 0.3 | 8 | 673 | — | — | — | 20 | — | 7 |
| 7 | 1.7%Ta2O5/1.8%Zr/SiO2a | 2.75 | 0.3 | 48 | 623 | — | — | — | 64 | — | 7 |
| 8 | 0.5%Ta–0.5%Nb/SiO2b | 2.6 | 9.5 | — | 623 | 25 | 71 | — | 18 | 1.02 | 228 |
| 9 | 0.5%Ta/SiO2 + 0.5%Nb/SiO2b | 2.6 | 4.4 | — | 623 | 25 | 61 | — | 15 | 0.36 | 228 |
| 10 | 0.7%Nb/SiBEA | 2.7 | 0.8 | 4 | 623 | 43 | 55 | 36 | 24 | 0.11 | 205 |
| 11 | 2%Nb/SiBEA | 2.7 | 0.8 | 4 | 623 | 26 | 53 | 30 | 14 | 0.06 | 205 |
| 12 | 1%Ta/SiBEA | 3.2 | 0.8 | 4 | 623 | 45 | 78 | 14 | 36 | 0.17 | 171 |
| 13 | 3%Ta/SiBEA | 3.2 | 0.8 | 4 | 623 | 59 | 73 | 21 | 43 | 0.20 | 171 |
| 14 | 3%Ta/SiBEA | 2.2 | 0.8 | 4 | 598 | 31 | 90 | 0 | 28 | 0.13 | 171 |
| 15 | 2.7%Zr/MCF | 1.38 | 3.7 | 1 | 673 | 95 | 70 | 6 | 67 | 1.4 | 10 |
| 16 | 2%ZrO2–SiO2 | 3.5 | 1.8 | 3 | 593 | 45 | 70 | 16 | 32 | 0.33 | 162 |
| 17 | 2%Ta/SBA-15 | 2.5 | 2.1 | 10 | 623 | 31 | 73 | 9 | 23 | 0.28 | 170 |
| 18 | 2%Ta/SBA-15 | 2.5 | 2.1 | 10 | 623 | 47 | 79 | 5 | 37 | 0.46 | 170 |
| 19 | 0.5%ZnO–ZrO2–SiO2 | 3.5 | 1.8 | 10 | 593 | 37 | 84 | 5 | 31 | 0.32 | 232 |
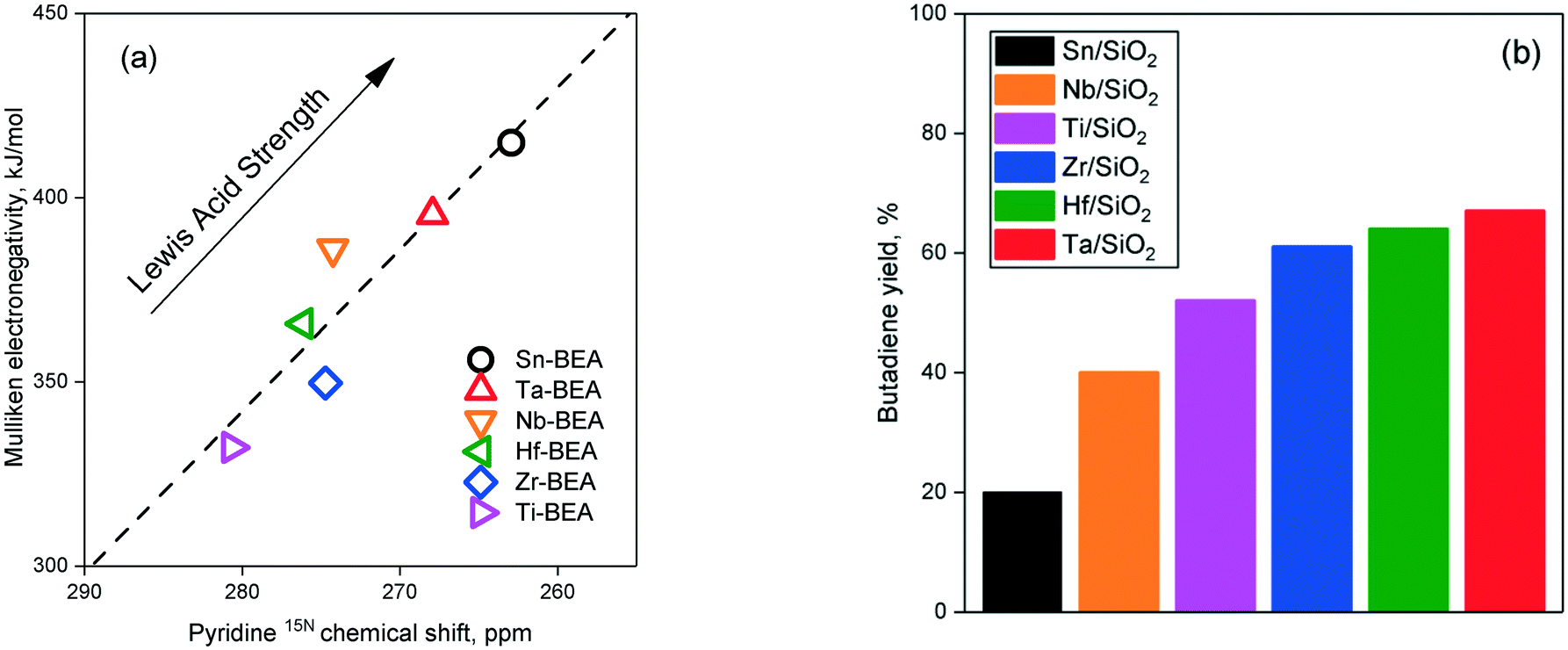 | ||
| Fig. 16 (a) Experimental Mulliken electronegativity versus pyridine 15 N magic angle spinning nuclear magnetic resonance chemical shift. Reproduced from ref. 6. Copyright 2016, with permission from American Chemical Society. (b) Butadiene yield in the two-step process obtained at 573–623 K, LHSVEtOH of 0.4 h−1, 2.75 ethanol–acetaldehyde ratio.7 | ||
Another important structural property of silicate-supported catalysts related to activity was identified by Ivanova et al. In their study of Ag/Zr-BEA, the authors found a direct correlation between the relative amount of tetrahedral Zr(IV) “open” Lewis acid sites (Fig. 18(a)) present in the zeolite matrix and the initial rate of butadiene formation.130,216,217 As Fig. 18(b) illustrates, metals incorporated into a silicate carrier can be fully coordinated with the silica framework, e.g., “closed” sites, or have “open” sites due to the hydrolysis of Si–O–M bonds. Ivanova et al. argued that these configurations influence the reactivity of a metal site: “open” sites are known to require less energy to adopt the geometric distortion required by the transition state of a reaction mechanism; the metal hydroxide and its adjacent silanol group may also participate in the reaction.130,212,218 Accordingly, “open” Lewis metal sites are believed to be more active in the aldol condensation of acetaldehyde, the rate-limiting step on supported metal oxides of periodic group 4 and 5, due to their acid strength and steric accessibility.99,101,130,171 Tetrahedral Lewis acid sites were also detected on ZrO2, ZrO2/SiO2 and ZrO2/MCM-41 using FTIR with CO adsorption, but were found to be less active in the MPVO reaction.207 Tetrahedral hafnium, niobium and tantalum oxides sites are also known to form when dispersed on catalysts carriers, but can adopt different structures when in bulk.219–224
Linear correlations between the acidity and catalytic activity with different catalysts were observed by other scholars: Kyriienko et al. reported a correlation between the relative concentration of Lewis acid sites in Zr-MTW catalysts determined by FTIR spectroscopy of adsorbed CD3CN and butadiene productivity;225 the present authors reported a direct correlation between the number of acid sites probed in Zn–Ta catalysts by NH3 and the selectivity towards butadiene.103 Contrarily, Dagle et al., who quantified the Lewis acid sites of Ag/ZrO2/SiO2 catalysts with FTIR spectroscopy of pyridine, found that excessive Lewis acid site concentration caused by modifying the ZrO2 loading lowered both butadiene selectivity and productivity in favor of ethanol dehydration products (Fig. 17).176 The authors argued that a small number of acid sites was preferable to avoid site reactions. Nevertheless, a scientific explanation as to why excessive Lewis acid site concentration favors undesirable reactions has yet to be provided.
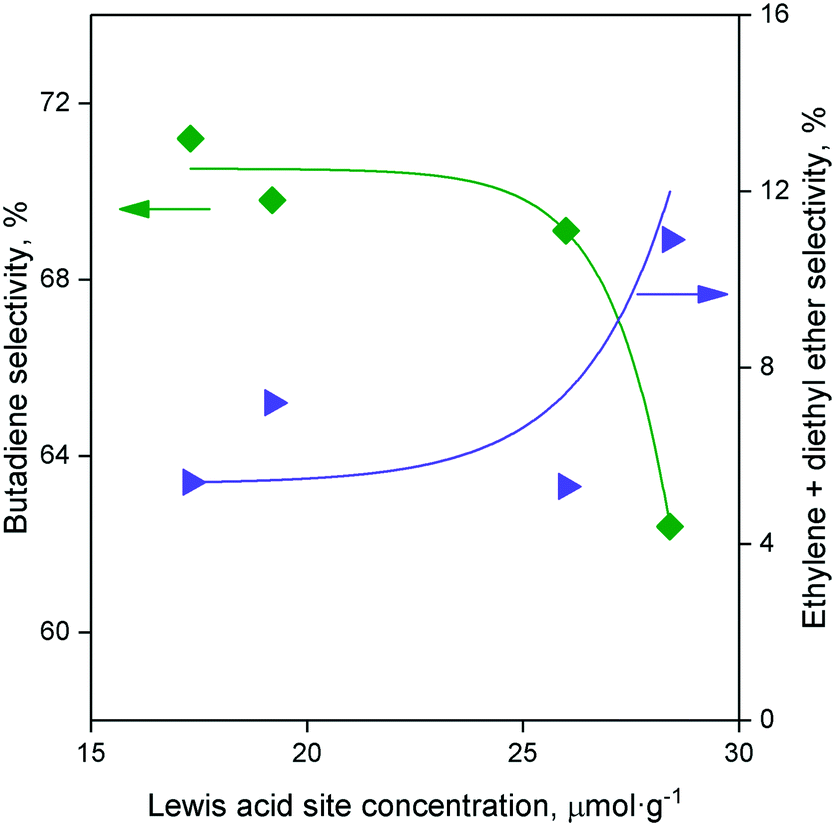 | ||
| Fig. 17 Evolution of selectivity in the Lebedev process on Ag/ZrO2/SiO2versus the concentration of Lewis sites determined by FTIR of pyridine.182T = 598 K, WHSVEtOH = 0.45 h−1. | ||
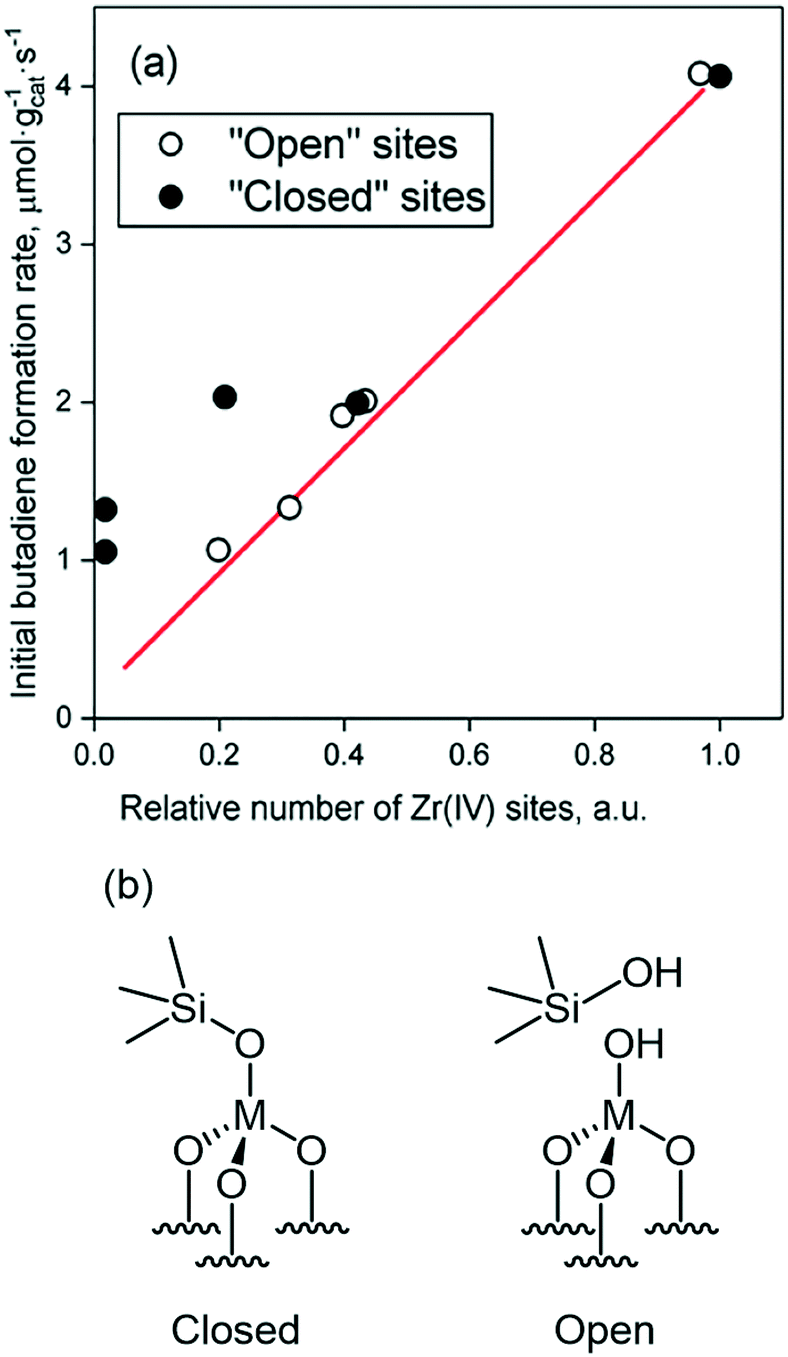 | ||
| Fig. 18 (a) Correlation between the relative number of open and closed Lewis sites determined by FTIR spectroscopy of adsorbed CO and the initial rates of butadiene formation on Ag/Zr-BEA.130 (b) Closed and open Lewis acid sites. | ||
Spectroscopic studies of supported Zr, Nb, Hf and Ta showed that the most active materials not only predominantly possess Lewis acid sites, but also small amounts of Brønsted acid sites.125,161,205,217 The latter may be so weak that they risk being undetected with pyridine or CDCN3, requiring the use of alternative probes, such as the strong organic base 2,6-di-tert-butylpyridine or CO.109,161 Brønsted acids undermine selectivity towards butadiene by catalyzing the dehydration of ethanol. It should be noted that due to the Lewis acid–base pair nature of these metal oxides, basic properties can also be detected,2,221,226 although amphoteric catalysts highly active in the conversion of ethanol to butadiene are primarily acidic.103,171,206
On the basis of observations made with Zr by Ivanova et al.,130,206,207,217 catalysts containing group 4 and 5 metals possess the Lewis acid characteristics required to catalyze the aldol condensation of acetaldehyde to crotonaldehyde and its subsequent conversion to butadiene via an MPVO reaction (reaction mechanisms are detailed above).101,109,125,161,205 Nonetheless, there exists a complex relationship between the properties of Lewis acid sites and their activity in the conversion of ethanol to butadiene. An explanation for the trend observed when comparing the performances of silica-supported group 4 and 5 transition metals in the Ostromislensky process has yet to be found, as no correlation was found with intrinsic acid strength, one possibility being the difference in electronic properties of each metal. Direct correlations between the number of Lewis acid sites and the butadiene formation rate have been reported;103,130,161,217,227 however excessive Lewis acid site concentration appears to be detrimental by favoring side reactions.176 Furthermore, not all Lewis acid sites perform equally: Ivanova et al. identified the ideal active sites as “open” isolated metal atoms in tetrahedral positions of the zeolite crystalline structure due to their enhanced steric accessibility and greater acid strength.124 Other Lewis acid sites are believed to be comparatively less active.207
Catalyst design and activity
Supported metal oxides are the predominant types of catalysts containing Zr, Nb, Hf or Ta. Designing materials highly active in the conversion of ethanol to butadiene has generally involved the following considerations: (i) the choice of metal; (ii) the metal loading method; (iii) the choice of catalyst carrier; (iv) the choice of dopant if used in the Lebedev process. Comparison between the performances of the various catalysts discussed in this section and those found in the literature is discussed below as summarized in Tables 3 and 4. The reader is also invited to consult them for details on the reaction conditions used in each test.| Entry | Catalyst | WHSV | TOS | T | X | BD S. | AcH S. | C2=S. | BD Y. | P BD | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| h−1 | H | K | % | % | % | % | % | ||||
| a Prepared with Zr(NO3)2. b Prepared with Zr(NO3)2. c Zn was in the form of hemimorphite. d Pressure was 1.4 atm. e Si–Zr ratio = 200. f SiO2 was Davasil 636. g NS = nanosheet, M = microporous. h Includes diethyl ether selectivity. i Zr–Zn ratio = 10. j CZA refers to Cu/ZnO/Al2O3; tetragonal ZrO2 was used. WHSV: weighted hourly space velocity of ethanol. BD, AcH, C2= are butadiene, acetaldehyde and ethylene, respectively. X, S., Y. and P. are conversion, selectivity, yield and productivity, respectively. Productivity is expressed in terms of gBD gcat−1 h−1. Active phases are expressed in terms of wt%. | |||||||||||
| 1 | 1%Cu–1%Zr–0.5Zn%/SiO2a | 0.21 | 0.5 | 633 | 98 | 61 | 6 | 16 | 60 | 0.07 | 125 |
| 2 | 1%Cu–1%Zr–0.5Zn%/SiO2b | 0.21 | 0.5 | 633 | 96 | 65 | 8 | 10 | 63 | 0.08 | 125 |
| 3 | 1%Cu–3%–Hf-0.5%Zn/SiO2 | 0.21 | 0.5 | 633 | 99 | 72 | 3 | 6 | 71 | 0.09 | 125 |
| 4 | 3%Hf–9.3%Zn/SiO2 | 0.64 | 0.5 | 633 | 99 | 70 | 5 | 10 | 69 | 0.26 | 125 |
| 5 | 10%Zn–1%Ta–0.25%Nb/SiO2c | 0.7 | — | 648 | 55 | 64 | — | — | 35 | 0.18 | 228 |
| 6 | 5%Zn–1%Ta/SiO2 + 5%Zn–0.25%Nb/SiO2d | 0.8 | — | 648 | 55 | 59 | — | — | 32 | 0.14 | 228 |
| 7 | 1%Ag–3.5%Zr/SiBEA | 1.2–15 | 3 | 593 | 15 | 59 | — | 10 | 9 | 0.58 | 217 |
| 8 | 1%Ag/Zr-BEAe | 0.32 | 3 | 593 | 31 | 66 | — | 5 | 20 | 0.04 | 175 |
| 9 | 1%/Zr-MCM-41e | 0.32 | 3 | 593 | 30 | 66 | — | 7 | 20 | 0.04 | 175 |
| 10 | 1%Ag–ZrO2/SiO2e | 0.32 | 3 | 593 | 12 | 67 | — | 3 | 8 | 0.01 | 175 |
| 11 | 0.3%Ag–4%ZrO2/SiO2 | 0.3 | 5 | 593 | 30 | 74 | — | 3 | 22 | 0.04 | 108 |
| 12 | 0.3%Cu–4%ZrO2/SiO2 | 0.3 | 5 | 593 | 27 | 74 | — | 3 | 20 | 0.03 | 108 |
| 13 | 0.3%Ni–4%ZrO2/SiO2 | 0.3 | 5 | 593 | 10 | 68 | — | 6 | 7 | 0.01 | 108 |
| 14 | 6.1%Zn–3.4%Ta-TUD-1 | 8 | 3 | 673 | 82 | 63 | 27 | 9 | 52 | 2.45 | 103 |
| 15 | 4%Ag–4%ZrO2/SiO2f | 0.45 | — | 598 | 91 | 67 | 7 | 11 | 61 | 0.16 | 176 |
| 16 | 1.5%Zn–8.9%Hf/MFI-NSg | 0.47 | 3 | 693 | 67 | 53 | 0 | 15 | 36 | 0.10 | 3 |
| 17 | 1.5%Zn–8.9%Hf/MFI-Mg | 0.47 | 3 | 693 | 64 | 43 | 1 | 11 | 27 | 0.08 | 3 |
| 18 | 1%Ir–4%ZrO2SiO2f | 0.35 | — | 598 | 85 | 63 | 9 | 2 | 54 | 0.11 | 176 |
| 19 | 1%Ag–1%Ta/SiBEA | 0.5 | 3.5 | 598 | 83 | 63 | 24 | 8 | 52 | 0.15 | 161 |
| 20 | 1%Cu–1%Ta/SiBEA | 0.5 | 3.5 | 598 | 88 | 73 | 15 | 2 | 64 | 0.19 | 161 |
| 21 | 1%Zn–1%Ta/SiBEA | 0.5 | 3.5 | 598 | 52 | 43 | 22 | 17 | 22 | 0.06 | 161 |
| 22 | 2%Cu/2%Zr–MTW | 0.5 | — | 648 | 81 | 68 | 8 | 18h | 55 | 0.06 | 225 |
| 23 | 1%Cu–1.5%–Zr0.5%Zn/SiO2 | — | 3 | 648 | 45 | 67 | 5 | 21 | 30 | — | 35 |
| 24 | 3%Hf–9.3%Zn/SiO2 | 1.12 | — | 633 | 87 | 43 | 7 | 4 | 38 | 0.25 | 109 |
| 25 | 3%Hf–9.3%Zn/SiO2 | 11.2 | — | 633 | 50 | 29 | 43 | 2 | 15 | 0.96 | 109 |
| 26 | ZnZrOxi | 0.8 | — | 623 | 98 | 26 | 51 | 11 | 26 | 0.06 | 8 |
| 27 | 02.5CZA + ZrO2j | — | 6 | 673 | 76 | 54 | 12 | 16 | 42 | 0.73 | 118 |
| 28 | 5%ZnO–5%ZrO2/KIT-6 | 9.47 | 12 | 698 | 95 | 52 | 28 | 10 | 49 | 2.49 | 242 |
Choice of metal
As highlighted in section 4.1.1. there appears to be a hierarchy in the catalytic activity of group 4 and 5 metal tested in the Ostromislensky process when in the form of silica-supported metal oxides: Nb < Ti < Zr ≈ Hf < Ta (Table 3, entries 1 to 6).7 Due to their relatively poor performances, Nb and Ti are seldom used.108,205 Instead, research has predominantly focused on Zr and Ta. Hf was found slightly superior to Zr as it lowered the selectivity towards dehydration products while maintaining a similar activity; this observation has been attributed to the softer nature of Hf according to the hard–soft acid–base theory.125 We believe that all three metals are suitable for preparing highly active catalysts for the conversion of ethanol to butadiene. Likely, the availability of raw material and price will be important factors in choosing the right active phase.Two group 4 and 5 metals are rarely combined in a single supported catalyst. Corson et al. tested Ta2O5 and ZrO2 on silica, which was highly active in the two-step process, reaching a butadiene yield of 64%, but showed no obvious advantage over using each metal oxide alone other than their price difference (Table 3, entry 7).7 Cadran and Chaumonnot reported a synergetic effect between tantalum and niobium oxide supported on silica.228 The combination of both metals improved butadiene productivity and selectivity in the Ostromislensky process and Lebedev process when doped with zinc oxide when compared to summed performances of monometallic catalysts with equivalent metal content (Table 3, entries 8 and 9).
Metal loading methods
Generally, the group 4 and 5 metal loading of highly active supported catalysts ranges between 0.1 and 10 wt%, preferably between 0.5 and 5 wt% on a mass basis.11,14 Generally, activity does not proportionally scale with metal loading. For instance, Kyriienko et al. observed that increasing from 0.7–1.0 wt% to 2.0–3.0 wt% the content of Nb and Ta in zeolite catalysts did not proportionally increase the butadiene yield in the Ostromislensky process, instead resulting in moderate improvements in selectivity (Table 3, entries 10 to 13).171,205 This phenomenon was attributed to the formation of less-active extra-framework metal oxide particles by sintering, which was evidenced by solid UV-vis spectroscopy. Furthermore, Ivanova et al. found that the low Zr content in Zr-BEA catalysts prepared by hydrothermal synthesis favored the formation of “open” Lewis acid sites.130 Metal content can be considered to affect the morphological and chemical properties of a catalyst and should therefore be adjusted to favor the dispersion of the active phase for enhanced catalytic activity.Various means of metal incorporation have been used to prepare catalysts with highly dispersed active phases. Wet and dry impregnation can be found extensively in the literature concerning the ethanol-to-butadiene conversion.7,108,109,125,161,171,217,229,230 Several parameters influence the properties of the final material: the type and amount of precursor, solvent and carrier, as well as experimental conditions such as pH, temperature, contact time, etc.231 For instance, De Baerdemaeker et al. noted that using ZrCl4 instead of Zr(NO)3 in the impregnation slurry improved the stability and butadiene selectivity of a Cu–Zr–Zn/SiO2 catalyst (Table 4, entries 1 and 2).125 Ivanova et al. developed an impregnation method to induce the formation of monoatomic “open” metal sites at higher metal loadings in their Ag/Zr/SiBEA catalyst.217 By impregnating dealuminated commercial β zeolite with ZrOCl2 in DMSO rather than in alcohol, they prevented the formation of “closed” Lewis acid sites. The authors explained that Zr cations in DMSO—either through diffusion limitations, steric hindrance of the nested silanol groups or energetically unfavorable formation of Zr(OSi)4—grafted onto the terminal silanol groups formed by the dealumination process, but not on silanol nests within the carrier, thereby exclusively forming “open” Zr(IV) sites (Fig. 20). A catalyst with 3.5 wt% of Zr and a high concentration of “open” Lewis acid sites was achieved, resulting in a high butadiene formation rate and a butadiene selectivity of ∼60% (Table 4, entry 7). Unfortunately, the dealumination of commercial zeolite could not remove all traces of Brønsted acidity, which caused a high selectivity towards dehydration products. Introduction of metal precursors in the synthesis gel of aluminum-free β zeolite enabled the synthesis of solid Lewis acids without the need for post-synthesis modifications.175 However, the necessity for a hydrofluoric medium and long preparation time may discourage this approach.
Cheong et al. reported a highly active Zr catalyst on a mesocellular siliceous foam (MCF).10 The authors used the urea hydrolysis method, which consists of a controlled precipitation step that avoids formation of concentration gradients of precipitants in the solution, to achieve a high degree of dispersion, as evidenced by TEM images depicted in Fig. 19(a) and (b). Accordingly, the Zr/MCF catalyst performed exceptionally in the Ostromislensky process, reaching a butadiene selectivity of 73% and unprecedented butadiene productivity (Table 3, entry 15).
 | ||
| Fig. 19 (a) HAADF-STEM of Zr/MCF; (b) EDX mapping of Zr/MCF showing the high degree of dispersion obtained by urea hydrolysis precipitation. Reproduced from ref. 10. Copyright 2016, with permission from American Chemical Society. | ||
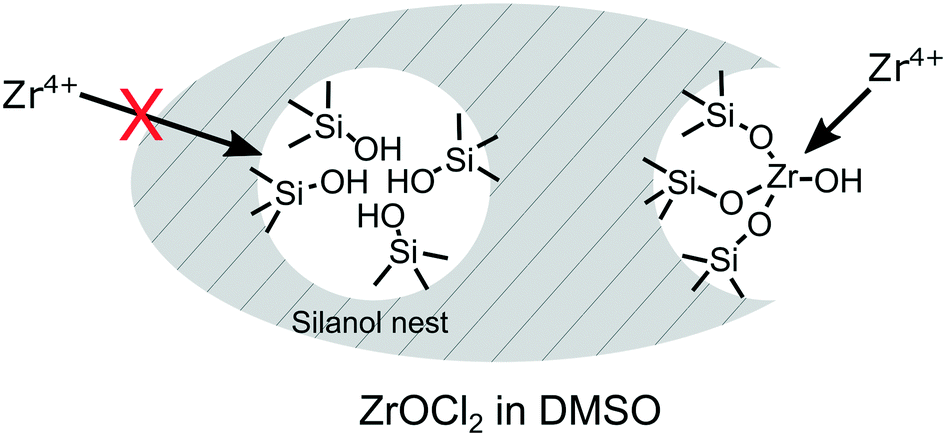 | ||
| Fig. 20 Schematic representation of Zr grafting to generate “open” Zr(IV) sites by treatment of dealuminated β zeolite with ZrOCl2 in DMSO. Reproduced from ref. 217. Copyright 2017, with permission from John Wiley & Sons, Inc. | ||
Foam-like mesoporous silica was also used by the present authors as a carrier for a highly active Zn–Ta catalyst for the Lebedev process.103,227 Using the TUD-1 methodology with tetraethylene glycol as both the chelating and structuring agent, metal incorporation was included during the sol–gel step of the silica synthesis. The Zn–Ta-TUD-1 catalyst achieved a butadiene selectivity of 63% in spite of a high ethanol flow rate (8 h−1) resulting in the highest butadiene productivity reported for the Lebedev process (Table 4, entry 14), which was attributed to the morphological properties of the catalyst and the high dispersion of the active phase. Sol–gel synthesis was used by Zhang et al. to synthesize a ZrO2–SiO2 catalyst with highly dispersed ZrO2 that achieved remarkable butadiene selectivity in the Ostromislensky process.232 Importantly, the concentration and strength of predominant Lewis acid sites could be tuned by adjusting the metal loading, as evidenced by NH3-TPD and pyridine-FTIR. 2 wt% of ZrO2 leading to a moderate amount of relatively weak acid sites was found ideal to maximize butadiene formation (Table 3, entry 16). A non-hydrolytic sol–gel process was also used by Dochain et al. to produce Ta-containing mesoporous silica, which was subsequently doped with silver or copper to promote activity in the Lebedev process.233 A large surface area and monomeric species of TaOx were obtained with this procedure.
Choice of catalyst Carrier
Three broad types of supports have been used to prepare such catalysts for the ethanol-to-butadiene reaction: microporous and mesoporous molecular sieves, and amorphous silica. These materials predominantly differ in their morphology, i.e., pore size, porous volume and specific surface area, with repercussions on the activity of the end-material. The structure of microporous molecular sieves causes steric hindrance, which may result in a potentially beneficial shape selectivity of the products.234 By providing confinement effects, small pores may enhance the stability of reaction transition states of a reaction, thus improving catalytic activity.150 However, the size similarity between the micropore diameter and reacting molecules may restrict molecular transportation, resulting in intraparticle diffusion limitations curtailing catalytic activity.235 Mesoporous carriers generally lack the benefits of confinement effects and shape selectivity but are less prone to mass transfer issues. Due to their porosity, both types of materials usually possess a large specific surface area, increasing the active phase accessible to reactants per volume of catalyst and facilitating the dispersion of metal oxide phases during the preparation process.231 In fact, Da Ros et al. reported a direct correlation between the specific surface area of K/ZnO–ZrO2/MgO–SiO2 catalysts and their butadiene yield (Table 9, entry 3), highlighting the importance of this parameter.191 Compared to other oxide carriers, i.e., ZrO2, Al2O3 or TiO2, the relative inertness of amorphous silica can be advantageous—it is a simple and convenient catalyst support. Corson et al. tested a variety of metal oxide supports in the one- and two-step processes, finding SiO2 to be a more suitable carrier for highly active Ta2O5, HfO2 and ZrO2.7Ivanova et al. compared the activity of all three categories of support in the Lebedev process with equimolar Ag and Zr as the active phase; β zeolite (BEA), MCM-41 and commercial silica were respectively used for the three types of carriers. The authors found performances to scale with the content of Lewis acid sites, highlighting the influence of the chosen support on the chemical properties of the catalyst. As illustrated in Fig. 21 the activity trend is as follows: Ag/ZrO2/SiO2 < Ag/Zr-MCM-41 < Ag/Zr-BEA (Table 4, entries 8 to 10). The authors argued that only the microporous material generated an ideal tetrahedral Zn(IV) isolated site. As a result, subsequent efforts by the team have focused on β zeolite. The latter has been extensively used to create highly active, hydrophobic, single-site Lewis acids via the incorporation of transition metals. This hydrophobic nature enhances the hydrothermal stability of the material. As a result, catalysts are less susceptible to framework collapse and deactivation by the effect of steam produced via the dehydration of alcohol groups in the ethanol-to-butadiene reaction.236 At low group 4 and 5 metal loading, mononuclear Lewis acids can be generated in β zeolite catalysts by impregnation. Consequently, many excellent catalysts capable of achieving butadiene selectivity greater than 60% in the one- and two-step processes have been reported with Ta and Zr in the literature. In its aluminosilicate form, unmodified β zeolite performs poorly in the one- and two-step processes due to its strong Brønsted acid sites which favor dehydration reactions.9,35 Fortunately, aluminum-free synthesis of β zeolite237,238 or post-synthesis removal of aluminum,210,239,240 which also generates new mesopores, is easily accomplished.
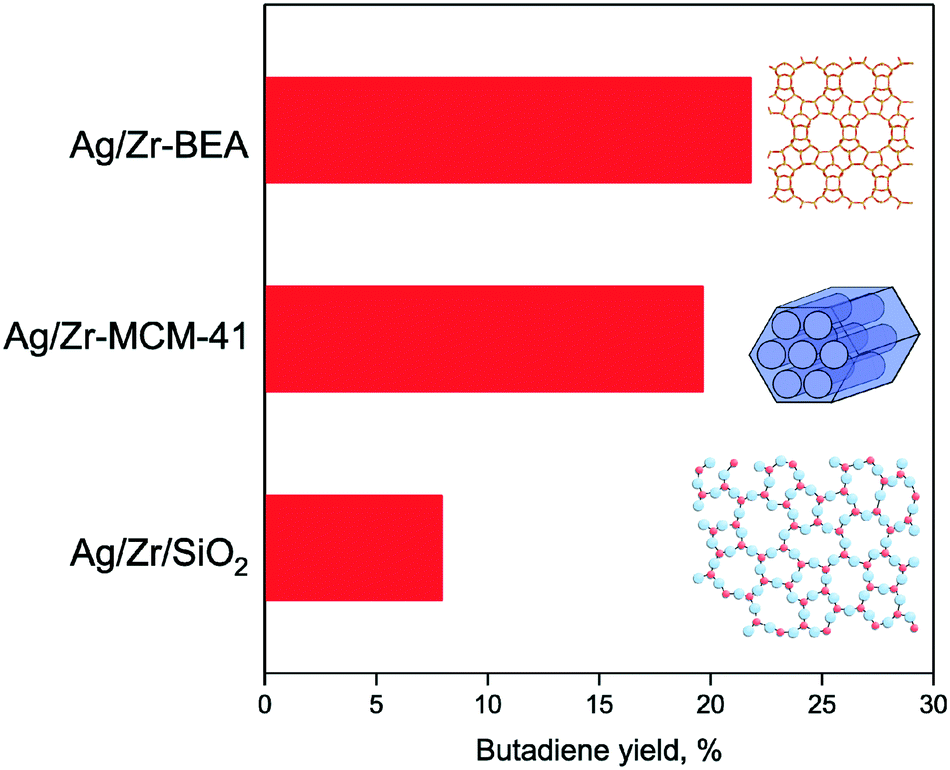 | ||
| Fig. 21 Effect of the silicate carrier on the butadiene yield of Ag–Zr catalysts in the Lebedev process.175T = 593 K, WHSVEtOH = 0.32, TOS = 3 h. | ||
Klein et al. observed that dealuminated zeolite catalysts used in the Ostromislensky process benefited from a hierarchical micro–meso porous structure resulting in an increase in ethanol conversion compared to microporous materials.173 The introduction of mesopores into a microporous zeolite is an established strategy to address diffusion limitations.235 Two instances of successful designs of hierarchical catalysts for the Lebedev process have been reported. Zhang et al. synthesized a hierarchical catalyst by impregnating alumina-free MFI zeolite nanosheets with Zn and Hf.3 The microporosity–mesoporosity provided by the three-dimensionally intergrown zeolite nanosheets increased the butadiene yield from 27.3% to 40.8% when compared to a similar Zn/Hf-MFI catalyst prepared with microporous MFI zeolite (Table 4, entries 16 and 17). These results coincided with a greater concentration of Lewis acid sites. Kyriienko et al. prepared a hierarchical copper-doped Zr-MTW zeolite as a catalyst for the Lebedev process.225 It was found that the nature and concentration of fluoride-containing mineralizing agents used during synthesis influenced the morphological characteristics, acid–base properties and catalytic activity in the Lebedev process. Better catalytic performances were obtained with HF and a Si:F ratio of 4, affording a butadiene selectivity of 68% and an ethanol conversion of 81% (Table 4, entry 22).
Although Ivanova et al. first found microporous β zeolite to outperform mesoporous MCM-41, Zr and Ta-containing mesoporous catalysts have since demonstrated exceptional activity in the one- and two-step processes. The present authors conducted a similar study comparing the activity of Zn–Ta catalysts supported on TUD-1 mesoporous silica, dealuminated β zeolite and amorphous silica.14,103 Contrary to Ivanova et al. mesoporous silica-supported samples outperformed Zn–Ta/SiBEA, suggesting that microporous materials are not necessarily the best suited carriers for this reaction. In another comparison of the catalyst carrier influence on the performances of Zn–Y, Li et al. agreed with Ivanova et al. that dealuminated zeolites were superior to MCM-41 and commercial silica.180 A possible explanation could be that MCM-41, with its two-dimensional pore structure, was unsuited for the ethanol-to-butadiene reaction, which was found to benefit from a three-dimensional mesoporous structure. At present, concluding whether one carrier morphology is superior to the other is difficult considering that these studies were conducted under different reaction conditions with distinct catalytic systems. Nevertheless, both materials can be used to prepare highly active catalysts.
The IUPAC defines mesoporous materials as solids possessing pores with diameters between 2 and 50 nm.241 A wide range of mesoporous silicates exist, many of which have been tested as catalyst carriers in the ethanol-to-butadiene reaction. Although ostensibly lacking the morphology required to induce confinement effects or size selectivity, several scholars highlight the influence of mesopore size on catalytic activity. Using a Zn–Zr active phase supported on mesoporous silica for the Lebedev process, Jones et al. observed a progressive improvement in catalytic performances by increasing the average pore diameter from 4 nm to 15 nm (Fig. 23, green data points).35 Although the ethanol conversion was not significantly affected, the butadiene selectivity rose by 20%, resulting in an increase of 8% in butadiene yield. Similarly, Kim et al. reported a 15% improvement in butadiene yield during the Ostromislensky process over ordered mesoporous Ta/SBA-15 (Table 3, entries 17 and 18) after increasing the average pore diameter from 6 nm to 13 nm (Fig. 23, red data points).170 The authors attributed this phenomenon to a better active site accessibility of the reactants and products, noting that pore and crystal sizes were more important than the topology of the ordered silica. Additionally, Zr and Ta-containing mesoporous catalysts displayed notable catalytic activity and stability under industrially relevant conditions, namely a high hourly space velocity, for both types of ethanol-to-butadiene processes.10,14,103,242 Li et al. partly attributed such performances to the three-dimensional pore structure enabling greater reactant accessibility and improved mass transfer within the pore channels, thereby preventing coke formation and pore blockage.10
This theory was explored by Bharadwaj et al. with a multi-scale modelling of the Lebedev process with Ag/ZrO2/SBA-16 catalysts.243 Their molecular dynamics and finite element method simulations demonstrated that the intraparticle effective diffusivity was positively correlated with the effective pore diameter of the support. According to their model, which was fitted to experimental data, the catalytic activity and lifetime improved with greater pore size by improving reactant access to active sites. The benefits of mesoporous carriers are supported by experimental and theoretical studies.
One downside of the mesoporous silica support is its relatively poorer hydrothermal stability,244 which may be an issue for post-synthesis modifications involving water. For instance, Mg-containing MCM-41 collapsed after aqueous impregnation introduction of the oxide phase, rendering the active sites inaccessible, resulting in poor catalytic performances.165 Thicker pore walls and more micropores can increase the hydrothermal stability of mesoporous silicates.244
Dehydrogenation promoters
Supported group Zr, Nb, Hf and Ta catalysts are inadequate to perform the Lebedev process due to their poor ability to dehydrogenate ethanol into acetaldehyde. The addition of metals or metal oxides with dehydrogenation capabilities has been used to address this limitation. The use of a promoter provides the missing chemical properties needed to perform every step that leads to conversion of ethanol to butadiene. However, it adds a new dimension to catalyst design; the nature, amount and structure of promoters, and the influence of these parameters on catalyst performances have to be also considered.Ivanova et al. demonstrated that the performances of Ag-modified ZrO2/SiO2 could be improved by increasing the promoter content from 0.3 wt% and 2 wt% (Table 4 entry 10 and Fig. 22(a)).108 However, more than 1 wt% of Ag reduced the catalytic stability over time on stream—a sign that promoter loading had more effect than simply introducing new active sites. This phenomenon was attributed to an excess of aldehyde, as evidenced by the increase in C6+ compounds resulting from undesired aldehyde coupling, which were believed to act as precursors to deactivating carbonaceous species. Dagle et al. further found that the activity of Lebedev catalysts depended on promoter dispersion (Fig. 22(b)).176 With equimolar amounts of Ag-modified ZrO2 dispersed on various silicates, the authors established a correlation between Ag nanoparticle size and ethanol conversion, 1 nm being the preferred diameter to enhance ethanol conversion (Table 4, entry 17). Recent computational and experimental techniques have highlighted the relationship between the dehydration–dehydrogenation activity of silver in Ag/ZrO2/SiO2 catalysts and particle size.245 Poorly dispersed metallic silver clusters were found to promote ethanol dehydration at the expense of acetaldehyde formation, resulting in lower butadiene selectivity. Rousseau et al. concluded that high dispersion resulted in the partial charge of silver particles, which was identified as a crucial property for ethanol dehydrogenation. In general, alcohol dehydrogenation with metals such as Au and Ag benefits from nanosized particles; appropriate synthesis methods must therefore be employed when preparing promoted catalysts for the Lebedev process.123
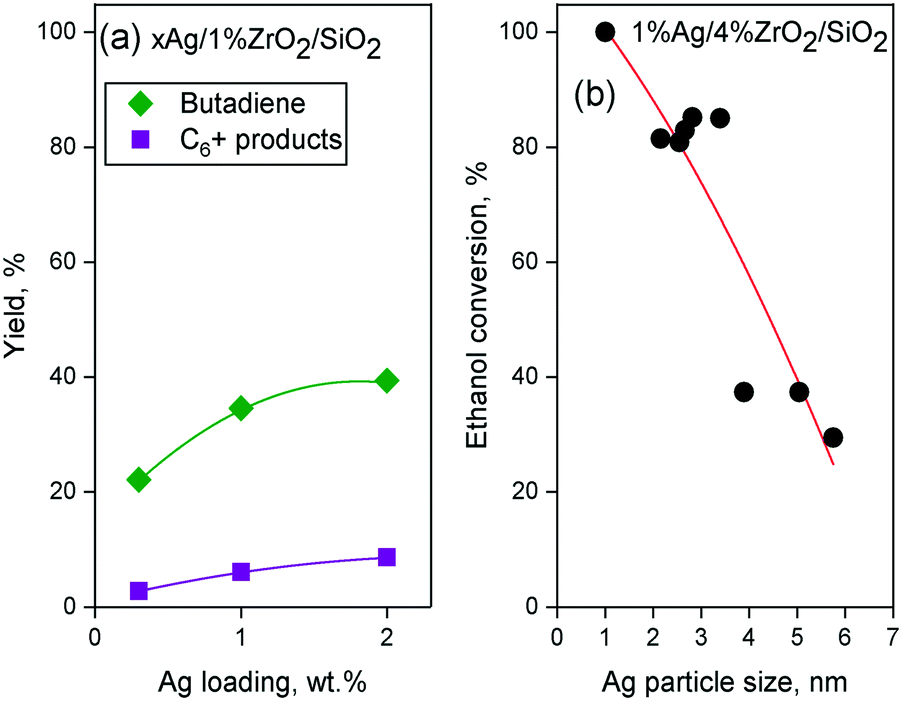 | ||
| Fig. 22 (a) Effect of Ag loading on the performances of Ag/ZrO2/SiO2 in the Lebedev process. T = 593 K, WHSVEtOH = 0.31 h−1.109 (b) Effect of particle size on the activity of Ag/ZrO2/SiO2 in the Lebedev process at equimolar metal loading. T = 598 K, WHSVEtOH = 0.45 h−1.182 | ||
 | ||
| Fig. 23 Effect of pore size on the butadiene yield during the one- and two-step processes over mesoporous catalysts.35,170 | ||
The type of promoter used is another crucial aspect of catalyst design when preparing group 4 and 5 materials active in the Lebedev process. Kyriienko et al. compared the performances of Ta–SiBEA catalysts modified with different metal dopants.161 Impregnating these catalysts with Ag, Cu or Zn ion solutions modified their acid–base properties, resulting in a change in catalytic performances. Studied by FTIR spectroscopy of chemical probes, the promoters generated new Lewis acid sites, weak Brønsted acid sites and weak basic sites. Of the three resulting catalysts, performances followed the trend: Zn–Ta/SiBEA < Ag–Ta/SiBEA < Cu–Ta/SiBEA, with the latter being one of the most butadiene selective catalysts in the literature (Table 4, entries 19 to 21 and Fig. 24).
 | ||
| Fig. 24 Influence of dehydrogenation promoters on the catalytic performances of Ta/SiBEA.161T = 598 K, WHSVEtOH = 0.5 h−1. | ||
This hierarchy reflected some observations made for each promoter dispersed on pure silica for the dehydrogenation of ethanol: Cu/SiO2 had previously been found to be more selective towards acetaldehyde and more stable than Ag/SiO2,9 whereas Zn/SiO2, although active, was reported to produce significant amounts of ethylene along with acetaldehyde.230 However, this promoter hierarchy is not valid for every catalytic system. For instance, Ivanova et al., who compared 0.3 wt% of Ag, Cu, and Ni as promoters for ZrO2/SiO2, found Ag to be the superior dopant, whereas Ni gave very poor performances (Table 4, entries 11 to 13).108 This suggests that the performance enhancement of dehydrogenation promoters also depends on the properties of the unmodified material.
The loading of a dehydrogenation promoter in relation to the active phase also influences the selectivity towards the desired product. Dochain et al. recently highlighted this by tuning the Ag:Ta ratio of a Ag–Ta–SiO2 catalyst prepared by a non-hydrolytic sol–gel procedure using silica and a tantalum precursor, followed by impregnation.233
Both Ag and Cu can enable high butadiene formation under the right conditions. The same can be said for zinc, which has successfully been combined with group 4 and 5 transition metals to prepare catalysts highly active in the Lebedev process.103,109,125,227
The zinc silicate hemimorphite is one such promoter: it was used by De Baerdemaeker et al.125 and Cabello González et al.4,109 to make HfO2/SiO2 highly active, selective and stable in the Lebedev process, performing better than Cu-containing catalysts (Table 4, entries 3, 4, 22 and 23). The authors noted that the addition of hemimorphite, in addition to increasing the ethanol dehydrogenation activity, passivated Brønsted acid sites, reducing selectivity towards dehydration products and improving stability. Baylon et al. reported a similar effect with ZnO–ZrO2 mixed metal oxide. As illustrated in Fig. 25, increasing the Zr:Zn ratio reduced the selectivity towards dehydration products (Table 4, entry 24), which was correlated with a reduction in the number of strong acid sites.8 Even in the Ostromislensky process, where dehydrogenation activity is not required, Xu et al. used ZnO to suppress ethylene formation on a ZrO2–SiO2 catalyst (Table 4, entry 19); it decreased the number of acid sites without reducing their strength.232
 | ||
| Fig. 25 Effect of Zn content in ZnZrOx on the selectivity of the Lebedev process.197T = 623 K, WHSVEtOH = 0.789 h−1. | ||
With zinc oxide being an amphoteric material, Zn promoters are generally believed to introduce basic sites capable of ethanol dehydrogenation. However, it is unclear why some instances of zinc modification have resulted in highly active catalysts and, in other cases, an important formation of dehydration products.161,230,246 Research on bulk zinc oxide suggests that oxygen vacancies in the oxide phase are active sites for ethanol dehydrogenation, and that the synthesis method used can significantly influence whether ZnO becomes primarily a dehydration or dehydrogenation promoter. Recent findings concerning Zn-modified ZrO2 catalysts used in the Lebedev process lead to the same conclusion, that zinc-dopants also introduce oxygen vacancies, and that the latter act as the basic sites needed for acetaldehyde formation.247
The addition of ZnO or hemimorphite has also been shown to introduce new Lewis acid sites in catalysts.109,125,227,230,248 Some authors have attributed the higher selectivity towards dehydration products compared to Ag or Cu to these acid properties.161,230 Others have found the Lewis acid properties of Zn(II) to enhance butadiene formation, proposing that Zn(II) possessed a condensation activity of its own, contributing to that of the active phase.119,227
Although Ag was ultimately found to be the best performing promoter of their study, Dagle et al. achieved a high butadiene yield when using iridium to enhance the dehydrogenation abilities of ZrO2/SiO2 (Table 4, entry 18).176 Interestingly, when compared to Ag, Ir suppressed ethylene formation, but generated significantly more butenes. From an industrial standpoint, it is unlikely that Ir will be used due to its restrictive cost compared to silver and the fact that undesired C4 compounds drive up the cost of butadiene purification.
Cu and Zn were combined with HfO2/SiO2 and ZrO2/SiO2 to prepare catalysts highly active and selective towards butadiene in the Lebedev process (Table 4, entries 1 to 3).35,125 With both group 4 metals, bimetallic dehydrogenation promoters resulted in higher butadiene selectivity compared to Cu and Zn alone. It is difficult to judge whether this improvement resulted from a synergy between both promoters and a simple increase in the number of dehydrogenation sites because monometallic catalysts with equimolar promoter loadings were not tested. Concerning the deactivation of Cu–Zn promoted catalysts, Jones et al. and De Vos et al. observed contradictory results.35,125 Cu/ZnO/Al2O3 (CZA), a catalyst active in the steam reforming of short-chain alcohols, was combined with tetragonal ZrO2 to prepare a new material active in the Lebedev process (Table 4, entry 27). CZA suppressed ethanol dehydration and greatly increased selectivity towards butadiene, outperforming Ag-doped tetragonal ZrO2.118 However, the study lacked Cu and Zn-promoted materials for adequate comparison. Consequently, these results show that the use of Cu–Zn promoters, although promising, has yet to demonstrate a clear advantage over their monometallic counterparts.
Many other transition and noble metals have been tested as dehydrogenation promoters for Zr-containing catalysts but failed to give satisfactory performances in the Lebedev process. When introduced to ZrO2 over SiO2, 1 wt% of Co, Mn and Ce were each unable to achieve butadiene selectivity greater than 29%, instead favoring ethanol dehydration products.35 Despite being capable of non-oxidative dehydrogenation of light alcohols,123 Pt gave remarkably poor performances when added to ZrO2/SiO2, yielding little butadiene to the benefit of methane, CO and CO2.176 As previously mentioned, 0.3% Ni/4% ZrO2/SiO2 was also not very active with a butadiene yield below 8%.108 Ostensibly, these metals and metal oxides are not active enough in the non-oxidative dehydrogenation of ethanol to acetaldehyde for the consecutive steps of the Lebedev process to take place.
The chemistry behind the activity of dehydrogenation promoters, exemplified by the cases of Ag and Zn, is complex. In many cases, the selected promoters have increased the selectivity to undesired byproducts, notably ethylene. As a result, preparing catalysts for the Lebedev process is challenging because it requires an understanding of two structure–activity relationships, whereas synthesis of catalysts for the Ostromislensky process only considers the condensation activity of the catalyst.
In summary, catalysts consisting of metal oxides belonging to group 4 and 5 elements (Zr, Nb, Hf and Ta) are active in the Ostromislensky process and can perform in the Lebedev process by introducing metal or metal oxide promoters with dehydrogenation capabilities. The majority of these catalytic systems consist of silicate-supported materials, although mixed metal oxides have also been reported. Their condensation ability, which has been correlated with Lewis acid sites, is the key characteristic for achieving good catalytic performances, as the aldol condensation of acetaldehyde to crotonaldehyde is recognized to be the rate limiting step.
A primary concern of catalyst design has been to obtain isolated Lewis acid sites, which have been identified as the most active and selective in the aldol condensation.130 Impregnation of a support with metal precursor salts appears to be the predominant method of choice. However, it is susceptible to particle sintering during the synthesis process, which may result in large oxide particles with inferior catalytic activity. Consequently, a variety of synthesis methods have been employed to achieve highly dispersed active phases. In most cases, metal loading was shown to be an important parameter, often necessitating to be optimized in order to obtain the ideal acid characteristics that lead to good performances.
Besides the metal introduction method, the catalyst carrier was highly influential on catalytic performances. Al-free zeolites, mesoporous silicates and amorphous silica are generally used to disperse metal oxides belong to groups 4 and 5 of the periodic table. Several authors have reported enhanced activity and selectivity by tuning the morphological properties of catalyst carriers, notably with the introduction of mesopores, which are believed to reduce mass transfer limitations.35,103,170,173 Cheong et al. suggested preparing catalysts possessing: (i) a highly dispersed active phase and (ii) a three-dimensional pore structure. A high surface area was also shown to improve catalytic performances.191 However, other scholars have instead found that microporous zeolite supports afforded better catalysts than their mesoporous equivalents.175,180 Some instances have shown that hierarchical materials possessing both micro and mesoporous morphologies were also very active in the ethanol-to-butadiene reaction.3,225
Zr, Nb, Hf and Ta catalysts can become active in the Lebedev process by introducing metal or metal oxide promoters with dehydrogenation capabilities. So far, Ag, Cu and Zn have been used to prepare catalysts demonstrating remarkable performances. However, their usage adds new dimensions to catalyst design that must be taken into consideration. Not only are the promoter effects dependent on their nature and properties, but these dopants can also affect the properties of the unmodified catalyst, notably by altering their acid characteristics.161,176 Consequently, preparing highly active and selective materials for the Lebedev process requires tuning both the condensation component provided by group 4 and 5 transition metals and the dehydrogenation component, as well as to consider interactions between the two. As the cases of Ag and Zn have demonstrated, the structure–activity relationship of dehydrogenation promoters can also be complex and must be taken into consideration.
Catalytic performance data
Tables 3 and 4 list the catalysts consisting of group 4 and 5 transition metals discussed in this section, and their performances in the one- and two-step processes, respectively. Accurate comparison between the catalysts is impossible due to the different reaction conditions used, namely temperature, contact time and conversion (Tables 3 and 4), as well as reactant concentration (not shown).109,249 In the absence of such data, butadiene productivity and selectivity are useful metrics for comparison, as the former is important to the industrialization of the ethanol-to-butadiene reaction,92 and the latter is necessary to reduce separation costs. To some extent, high acetaldehyde selectivity can be considered beneficial, as it can be recycled into the reactant stream,191 whereas ethylene selectivity should be kept as low as possible.In the Lebedev process, the most productive catalysts consisted of tantalum oxide (Table 4, entry 14)103 and zirconium oxide (Table 4, entry 28),242 both doped with Zn and highly dispersed on mesoporous silica. Productivities of 2.45 and 2.49 gBD gcat−1 h−1 were achieved by maintaining relatively high ethanol conversion and selectivity towards butadiene despite elevated ethanol flows. Pomalaza et al. used the TUD-1 preparation method to incorporate the Zn–Ta active phase into amorphous mesoporous silica. Ahn et al. found KIT-6—an ordered mesoporous catalyst—to be the best carrier for the Zn–Zr active phase introduced via impregnation. These remarkable performances were attributed to the three-dimensional mesopore structure, high specific surface area, high active phase dispersion and high Lewis acid site concentration.
A similar conclusion was reached for explaining the highest productivity obtained in the Ostromislensky (1.4 gBD gcat−1 h−1) process with Zr supported on meso-cellular foam (Table 3, entry 15).10 The best selectivity reported was achieved on Ta/SiBEA for both the Ostromislensky process and the Lebedev process, with copper being the dehydrogenation promoter (Tables 3, entry 14 and 4, entry 20).161,171 This high activity was attributed to the formation of “open” Lewis Ta(V) sites in the tetrahedral position of the dealuminated zeolite framework.210
As discussed previously, catalyst deactivation is one issue that plagues the ethanol-to-butadiene reaction. Few studies have investigated this phenomenon in group 4 and 5 transition metal catalysts. Yet, the primary cause of deactivation has been attributed to coke deposition.109,164,170 According to Cheong et al., three-dimensional pore structures help to reduce coking by facilitating mass transport within the catalyst.10 Although direct evidence remains to be found, it should be noted that the two most productive mesoporous catalysts exhibited remarkable resistance to deactivation.10,103,227
Magnesia–silica catalysts
Catalysts consisting of mixed magnesia and silica can perform the one-step conversion of ethanol to butadiene without the need for dehydrogenation promoters. This catalytic system dates back at least to 1944,250 but some scholars251 suggested MgO and SiO2 to be the components of an undisclosed catalyst in Lebedev's earlier patents on the one-step process.252,253 MgO–SiO2 has since become one of the most studied catalytic systems. Research predominantly focuses on elucidating the relationship between properties of MgO–SiO2 and its catalytic activity—a prerequisite of rational catalyst design. This section reviews the literature on the structure–activity relationship of MgO–SiO2 catalysts, synthesis methods and parameters and presents examples of MgO–SiO2 catalysts with remarkable performances.Acidity, basicity & activity
At present, the relationship between the reactivity of MgO–SiO2 and its properties is not fully understood. Scholars agree that combining magnesia and silica produces materials with acidic and basic properties that enable the conversion of ethanol; a subtle balance between these properties is believed to be crucial for maximizing butadiene synthesis.51 Still, only a limited consensus exists regarding the exact nature of these active sites, or what role each play in the reaction. Notably, the contribution of silica to the catalytic activity of MgO–SiO2 is a contested topic. Disagreements appear to result from conflicting experimental observations reported in the literature. Likely, the diversity of methods to prepare and study MgO–SiO2 catalysts, and also the limits of analytic techniques often employed are partly responsible for these discrepancies. The wide variety of characterization and preparation methods used has made comparison between different materials described in the literature difficult.Kvisle et al. observed that active MgO–SiO2 possesses structural defects in the magnesia phase and magnesium silicate domains, but did not establish a direct correlation with them and catalytic activity.1 Defects in magnesia are known to possess specific chemical properties and are often catalytically active sites.2 The authors questioned whether silica, through the mixing process, merely induced catalytically active defects in MgO, or whether it was an essential component needed for the formation of catalytically active Mg–O–Si linkages. Generally, pure MgO predominantly converts ethanol to 1-butanol via the Guerbet reaction.96,131,254 However, Baba et al. reported that MgO, when subjected to hydrothermal treatment, became highly selective towards butadiene.139,142 This suggests that silica is not an essential component of the catalytic system and MgO structural defects, which are formed by the hydrothermal treatment, are the active sites of the ethanol-to-butadiene reaction. However, the two answers to the question above are not mutually exclusive: silica mixing may not only simultaneously induce defects in MgO, but also generate active magnesium silicates. In fact, the latter have often been associated with superior catalytic activity. As a result, SiO2 and magnesium silicates, though not a prerequisite for enabling the activity of MgO in the Lebedev process, should not be dismissed when discussing this catalytic system.
Weckhuysen et al. studied the influence that three groups of magnesium silicates—anhydrous crystalline magnesium silicates, amorphous hydrous magnesium silicates and layered hydrous crystalline magnesium silicates—had on the catalytic activity of MgO–SiO2.255 By relatively quantifying each group within various MgO–SiO2 samples using 1H–29Si CP/MAS-NMR, the authors were able to correlate specific structures with catalytic activity. The butadiene yield directly correlated with the relative amount of layered hydrous magnesium silicates, i.e., talc, stevensite, and lizardite (Fig. 26(a)). Incidentally, Zn-modified talc has proved to be one of the most active catalysts in the conversion of ethanol to butadiene, and pure talc was found to be highly active in the aldol condensation of acetaldehyde.139,142 Recently, Zhang et al. demonstrated that pure magnesium silicates were active in the two-step process, reaching butadiene selectivity comparable, albeit inferior, to MgO–SiO2, confirming their coupling activity.182
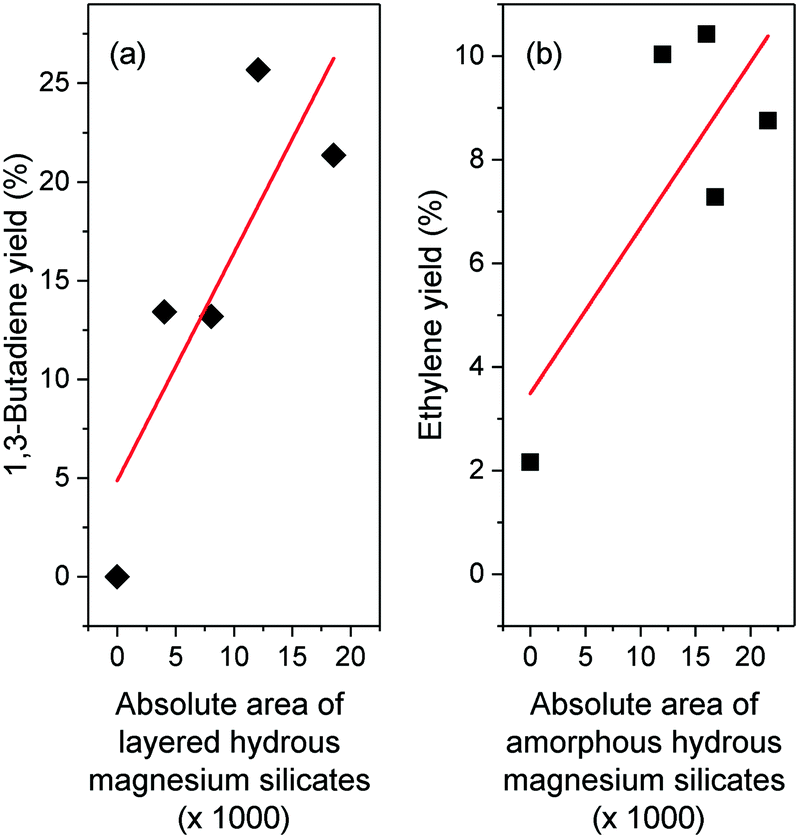 | ||
| Fig. 26 (a) Butadiene and (b) ethylene yield versus absolute area detected for layered hydrous magnesium silicates and amorphous hydrous magnesium silicates, respectively, according to Weckhuysen et al. Reproduced from ref. 255. Copyright 2016, American Chemical Society. | ||
Weckhuysen et al. further established a correlation between the ethylene yield and the relative amount of amorphous hydrous magnesium silicate phase (Fig. 26(b)). Contrarily, Zhu et al. attributed the high activity of their MgO–SiO2 catalyst in the Ostromislensky process to the presence of amorphous magnesium silicates, which XPS analysis confirmed to have a low binding energy, possibly related to their disordered configuration.5
In other instances, scholars have noted the negative effect of anhydrous magnesium silicate forsterite (Mg2SiO2) sometimes detected in MgO–SiO2 catalysts, the presence of which coincided with greater selectivity towards dehydration products. Forsterite formation occurred when using high calcination temperature during prepration5,256 or using the sol–gel method with a low Mg to Si ratio.97 Unfortunately, the chemical properties and catalytic activity of these magnesium silicates have not been properly evaluated.255 As a result, their role in the ethanol-to-butadiene reaction has yet to be confirmed.
Weckhuysen et al. determined through a Hammett indicator study that well-performing MgO–SiO2 catalysts were predominantly basic.174 Nevertheless, these catalysts also possessed significant amounts of acid sites, as evidenced by IR spectroscopy with chemical probes. Consequently, MgO–SiO2 can be considered an amphoteric catalyst. Identifying the nature of these sites and their role has been an important research topic.
Catalyst poisoning studies with propionic acid demonstrated that weaker basic sites were active for the dehydrogenation of ethanol, whereas stronger basic sites catalyzed the aldol condensation and MPVO reactions.93,126 Temperature-programmed and spectroscopic studies with probe molecules confirmed the presence of weak, medium and strong basic sites on MgO–SiO2.93,97,126,160 These studies, including in situ DRIFT spectroscopy, further identified surface Mg–OH groups as the weak basic sites responsible for ethanol dehydrogenation, as evidenced by their consumption during the reaction.126
The stronger basic sites responsible for the aldol condensation and MVPO reaction are believed to belong to Lewis acid–base pairs.93,126,160 In basic metal oxides, such sites are well-known for operating in tandem to promote alcohol coupling, aldolization and MPVO reactions.113,116 In Mg–O ion pairs, Mg2+ and O2− exhibit Lewis acidic and basic characters, respectively. The higher the strength of each moiety, the lower their coordination number, owing to electron deficiency and lowered stability, respectively.257 Consequently, pairs with varying degrees of coordination (Mg3C2+O4C2−, Mg3C2+O3C2− and Mg4C2+O4C2−, see Fig. 27 (ref. 258)) have been proposed as active sites and found to be active in the ethanol-to-butadiene reaction through DFT computational studies.96,100,134,259 Acid–base pairs with vicinal Si4+ ions have also been suggested as active sites.97 Sels et al. argued that the stronger basic sites, ostensibly tricoordinated oxygen anions found in corner defects, rapidly deactivated under the poisoning effect of polycondensation products formed by their larger strength.160,206 Many authors have noted that excessive basicity, both in terms of site quantity and strength, correlated with poorer catalytic performances.5,160,174 Consequently, acid–base pairs of moderate basic strength are believed to participate in the aldol condensation and MPVO reactions.93,160 DFT computational calculations further confirmed that Mg3C2+O4C2, as opposed to tricoordinated oxygen anions O3C2−, was the most active site for the condensation step.134
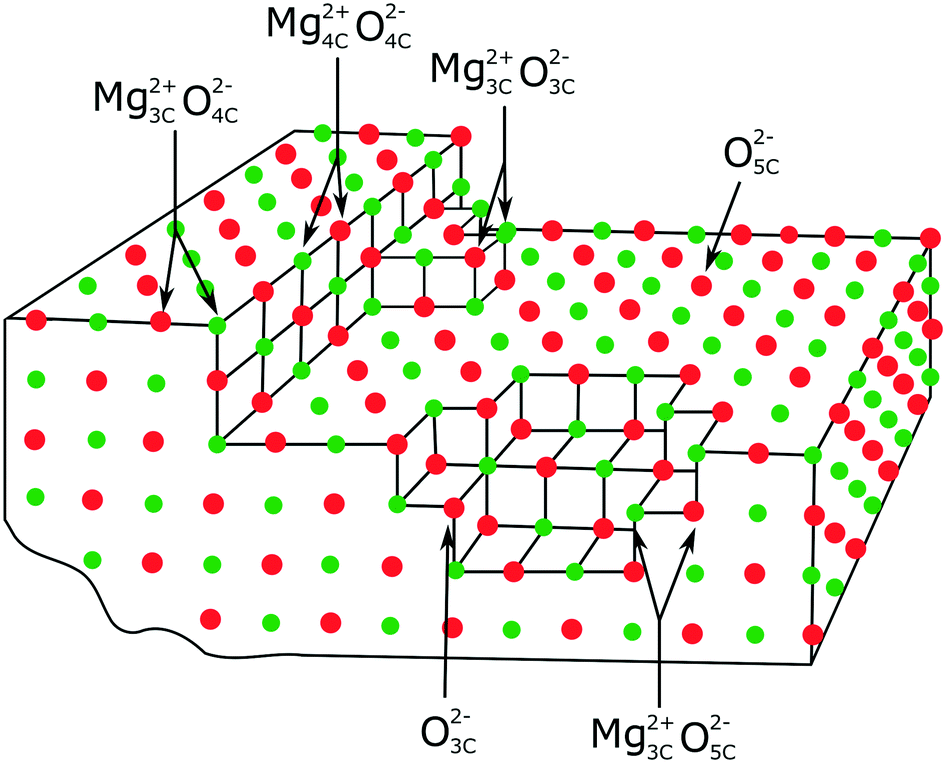 | ||
| Fig. 27 Model of structural defects on the surface of MgO.258 | ||
Interestingly, the XPS spectra of O 1s for hydrothermally treated MgO prepared by Baba et al. showed the presence of a noticeable shoulder peak at 532.1 eV which was less pronounced in calcined MgO (Fig. 28).139 The higher binding energy indicated that the oxygen atoms contributing to this peak were comparatively weaker basic sites.230 The authors suggested that these sites participated in the formation of butadiene, as only the hydrothermally treated MgO showed a significant butadiene formation rate. This conclusion agrees with the aforementioned theory that reducing the basic strength of MgO is beneficial to its performances in the Lebedev process. Although Taifan et al. assigned this peak to uncoordinated oxygen anions,126 the literature suggests that it could belong to the oxygen in Mg(OH)2 (ref. 260–262) or MgCO3.263
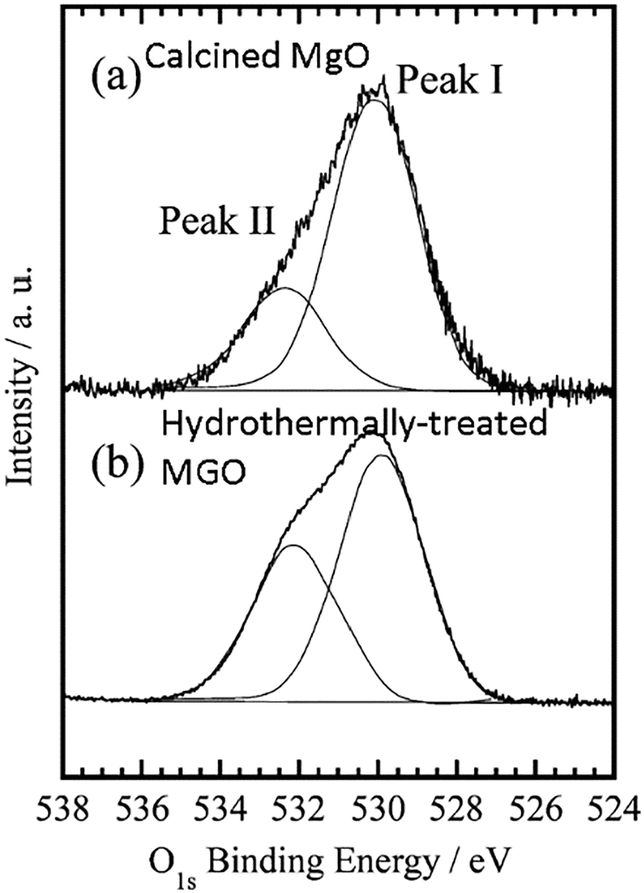 | ||
| Fig. 28 X-ray photoelectron spectra of O 1s for (a) calcined MgO and (b) hydrothermally-treated MgO, reproduced from Baba et al. Reproduced from ref. 139. Copyright 2016, American Chemical Society. | ||
Generally, introducing Si into MgO generates new acid sites.174 However, the contribution of surface acidity to the catalytic activity of MgO–SiO2 in the ethanol-to-butadiene reaction is not fully understood. Correlating the nature of acid sites (Brønsted or Lewis) or their strength to any specific reaction step has proven difficult, in part due to the limitations of spectroscopic techniques used to characterize the surface acidity of MgO–SiO2.
In theory, the Lewis acid moiety of Mg–O acid–base pairs is limited to electrostatic interactions with electron-rich intermediate species such as alkoxides, carbonyls and alcohols.257 Unlike its transition metal counterparts, the d orbital of the Mg cation is not accessible for bonding. However, its partial positive charge can stabilize the electron-rich function of transition state species in organic reactions catalyzed by the vicinal basic oxygen anion. According to DFT calculations, this ability—which increases with Lewis acid strength—is believed to play a crucial role in the ethanol-to-butadiene reaction.134 Acid–base pairs are active not only in aldol condensation113,116 and MPVO reactions,264 but also in alcohol dehydration.265 The Mg3C2+O4C2− pair was shown to be active in each catalytic step of the Lebedev mechanism by a DFT study.100
In practice, the relationship between acid sites and activity is not obvious. Most scholars agree that the acid sites of MgO–SiO2 are not only responsible for the dehydration of acetaldol and crotyl alcohol, but also of ethanol. However, the dehydration of ethanol and C4 alcohols is believed to occur on different sites. NH3 poisoning experiments of MgO–SiO2 in the one-step process revealed that weak acid sites dehydrated ethanol to ethylene and strong acid sites participated in the formation of butadiene.126 The authors proposed that the latter were responsible for the dehydration of acetaldol and crotyl alcohol as NH3 poisoning irreversibly suppressed the butadiene formation rate. Contrarily, acid quantification techniques with NH3 showed that MgO–SiO2 catalysts with excessive numbers of strong acid sites generated more dehydration products—an observation which led the authors to conclude that weak and medium strength acid sites were instead responsible for the dehydration of C4 alcohols which are necessary to the formation of butadiene. Additionally, MgO–SiO2 possessing forsterite phases showed possessed stronger acidity and higher selectivity towards dehydration products.97 The possibility that the mildly acidic silanol group on the silica phase—too weak to be probed by pyridine—is responsible for the dehydration of crotyl alcohol, a reaction thermodynamically favorable, was also proposed.11
Having observed with IR spectroscopy that Brønsted acids formed after co-adsorbing water and pyridine on the surface of MgO–SiO2, Cavani et al. proposed a different role for Lewis acid sites. The authors suggested that Mg–O–Si Lewis acid sites become Brønsted acid sites in the presence of water which forms by the dehydration of ethanol.97 These sites would possess the right acid strength for dehydrating the alkenol intermediates of the ethanol-to-butadiene reaction due to the influence of neighboring Si groups. Such phenomena may result in acid properties characterized ex situ that do not reflect those of MgO–SiO2 as the ethanol-to-butadiene reaction takes place. Contrarily, Taifan et al., using in situ DRIFT spectroscopy with pyridine to assess the surface acidity of spent catalyst, did not observe new Brønsted acid sites,126 suggesting that the chemical properties of different MgO–SiO2 are not influenced by water in the same fashion.
The conflicting theories regarding the role of acid sites may also partly be explained by experimental limitations. For instance, several research teams have observed an absence of Brønsted acid sites on the surface of MgO–SiO2 when using pyridine as a chemical probe with IR spectroscopy.5,12,160,266 On this basis, several authors described highly active catalysts as possessing the right balance of Lewis acid sites and basic sites. However, Taifan et al. recently identified Brønsted acid sites on the surface of wet-kneaded MgO–SiO2 from the deconvoluted DRIFT spectra of chemisorbed ammonia.126 According to the authors, the bulkiness of pyridine presumably prevented it from reaching isolated, less-accessible Brønsted acid sites of unspecified strength. Consequently, theories regarding the participation of acid sites in the Lebedev process may have been formulated with an inaccurate assessment of the acidic character of MgO–SiO2 catalysts.
The role of MgO–SiO2 surface acidity in the ethanol-to-butadiene reaction has not yet been fully understood. Ostensibly, Lewis acid sites—found on highly active catalysts—participate as part of the acid–base pair catalyzing condensation and MPVO reactions. Their in situ transformation into Brønsted acid sites active in the dehydration of alkenol intermediates requires further investigation. Of unspecified nature, weak acid sites dehydrated ethanol. The role of stronger acid sites in the different alcohol dehydration reactions remains under debate. In comparison, scholars generally agree on the contribution of basic sites: weak basic sites, likely hydroxyl groups on the surface of MgO, dehydrogenate ethanol; Mg–O pairs of medium-strength basic sites enable aldol condensation and MPVO reaction; strongly basic tri-coordinated oxygen anions deactivate quickly due to their poisoning by condensation products. How these chemical properties related to the structural properties of MgO–SiO2, notably of different magnesium silicates, is another topic of research that requires further investigation. Fig. 29 summarizes the active sites on MgO–SiO2 evidenced or theorized to participate in the Lebedev process.
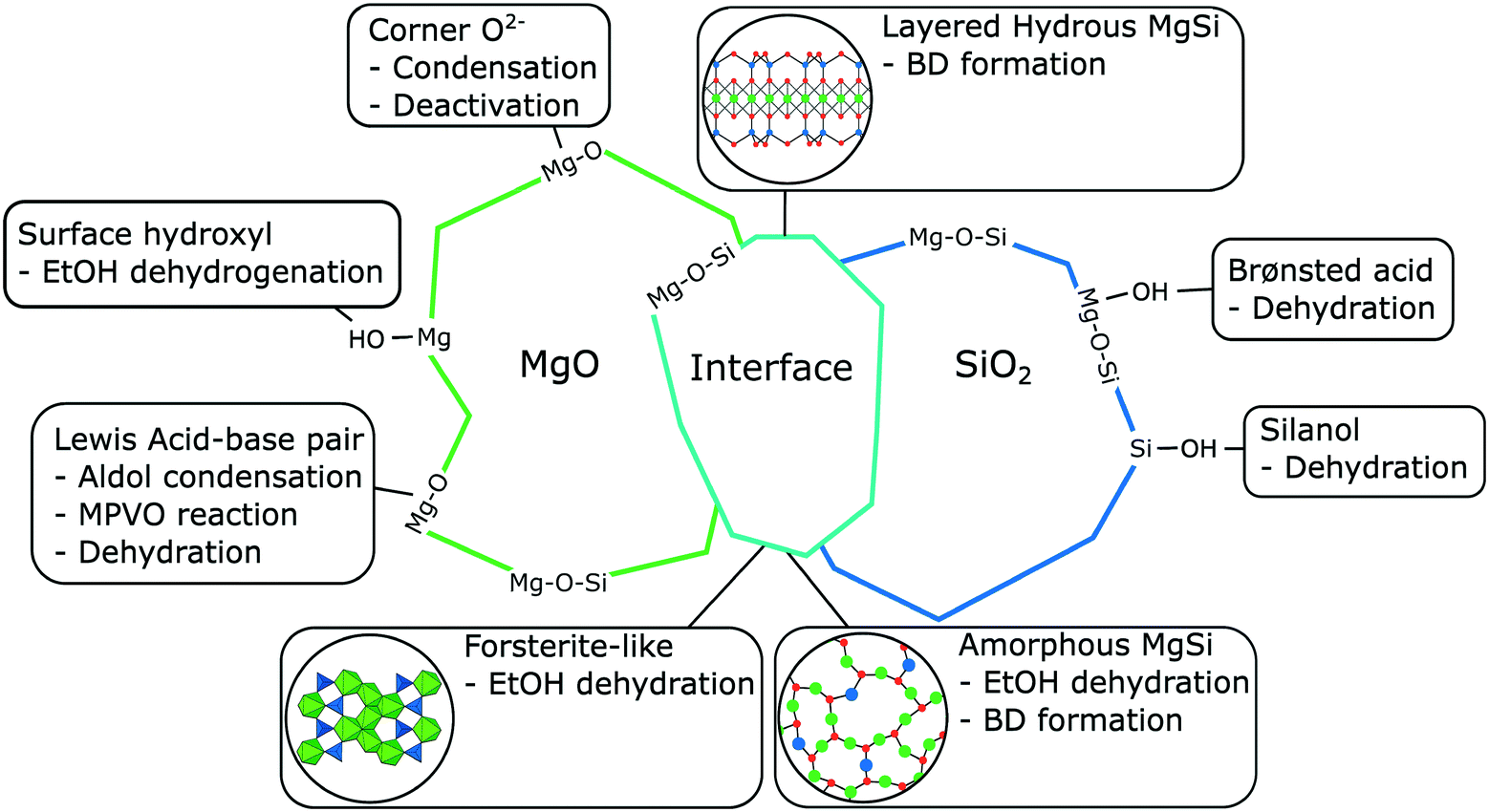 | ||
| Fig. 29 Visual representation of the different active sites on MgO–SiO2 catalysts theorized to participate in the ethanol-to-butadiene reaction. Expanded on the work of Taifan et al.126 | ||
Catalyst preparation
Despite uncertainty concerning the exact nature and role of active sites, there is a consensus that maximizing the catalytic performances of MgO–SiO2 catalysts requires a balance between the acidic and basic properties, although precisely what that balance is remains under debate. Many authors agree with the recommendation of Weckhuysen et al.: the presence of small amounts of strong basic sites in the proximity of intermediate amounts of moderate strength acid sites is the optimal balance for maximizing butadiene yield and suppressing byproduct formation.5,97,174 In light of the findings described above, the presence of weak basic sites Mg–OH may be added to this recommendation. Men et al. provided an experimental value for the ideal acid–base balance based on the results of TPD quantification techniques using NH3 and CO2 with various MgO–SiO2 catalysts.267 A surface total basicity/total acidity between 0.24 and 0.3 with an optimal strong acidity/total acidity between 0.46 and 0.5—small amounts of strong acid sites—maximized the butadiene yield. However, Weckhuysen et al. demonstrated that CO2 did not probe all the basic sites of MgO–SiO2.174 Consequently, this limitation must be taken into consideration when interpreting the results provided by the approach of Men et al. Likely, the trends observed provide more insight into the desirable chemical properties than do the precise ratios calculated from chemical surface analysis techniques.In practice, optimizing the acidic and basic properties of MgO–SiO2 to maximize butadiene formation often proceeds by tuning the Mg-to-Si ratio. Since MgO is a strong basic oxide and the introduction of Si generally produces new acid sites, the Mg-to-Si ratio loosely reflects the basic-to-acid site ratio. However, the properties and activity of MgO–SiO2 catalysts also depend on the level of interaction between each phase, which depends on the synthesis method used. As several preparation techniques are available to prepare MgO–SiO2 materials, different optimal Mg-to-Si ratios have been reported.143,157,255,257 Consequently, the Mg-to-Si ratio is a parameter better suited for comparing series of catalysts prepared by the same method. As exemplified in Fig. 30, a crucial performance metric such as butadiene selectivity can be improved by tuning the Mg-to-Si ratio with the optimal value dependent on the synthesis method.97,255
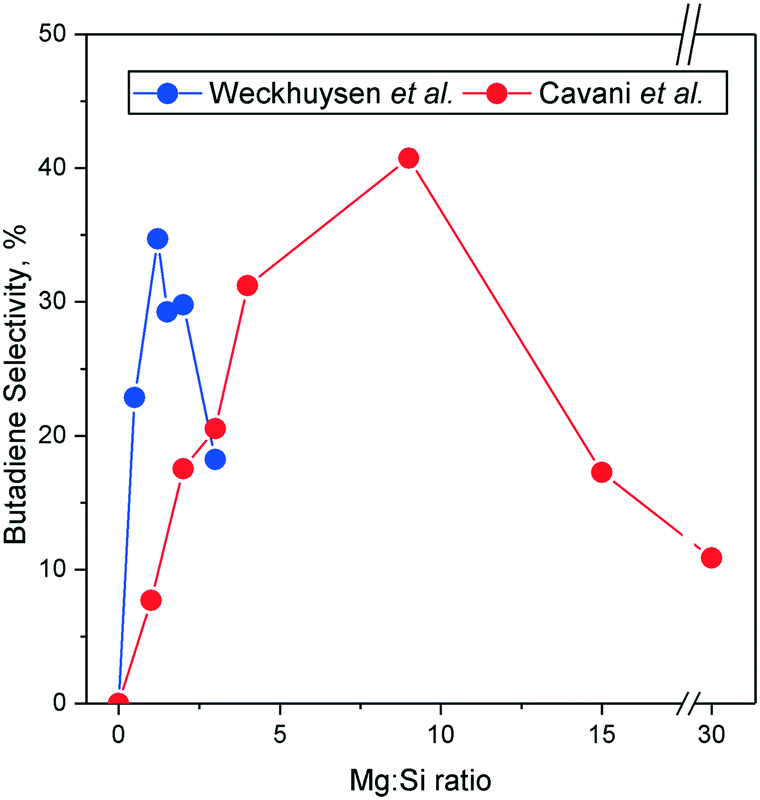 | ||
| Fig. 30 Influence of the molar Mg:Si ratio on the selectivity of MgO–SiO2 prepared by wet-kneading (blue)255 and the sol–gel technique (red).97 | ||
Several procedures are used to prepare MgO–SiO2 materials. Wet-kneading—the process of combining two or more solid precursor materials (mechanically or magnetically) in a liquid medium255—is a very common method for preparing the MgO–SiO2 catalyst in the ethanol-to-butadiene reaction; with water as a solvent, it has produced the most active catalyst of its kind267 and a majority of studies used wet-kneaded samples.1,51,92,111,126,127,174,230,251,255,266–269 The high butadiene yield provided by wet-kneaded MgO–SiO2 has been attributed to the controlled mixing it provides. TEM and EDX-STEM have indicated that wet-kneaded materials exist in two bulk phases—crystalline MgO sheets and amorphous SiO2 particles—with limited contact with one another.1,255 LEIS revealed that MgO and SiO2 are only intimately mixed at the surface of the two bulk oxide phases.126 Weckhuysen et al. explained that, during the preparation, water dissolved MgO into Mg(OH), which provides alkali conditions that dissolve further silica; each ion is free to redeposit on the surface of the two oxides due to the use of water.257 Wet-kneading conditions are known to influence the properties and activity of MgO–SiO2.51,251 Notably, the choice of oxide precursor is important for achieving high activity. Weckhuysen et al. found that using nanosized Mg(OH) enhanced the degree of mixing between both phases, resulting in better performances.255 Men et al. prepared the most productive MgO–SiO2 catalyst (Mg-to-Si = 1.86) by using magnesium acetate, which resulted in a hierarchical flower-like MgO phase.267 Similarly, the MgO precursor possessed a large surface area, making it suitable to maximize the interaction between the two oxide phases.
Other synthesis methods include: the sol–gel technique,97 co-precipitation,51,157,174,191,270 incipient wetness impregnation,5,9,172,269 dry milling1,160 and mechanochemical mixing.266 Co-precipitated metal oxides resulted in excessive phase mixing; more Mg–O–Si linkages were detected compared to wet-kneaded MgO–SiO2. These materials also possessed high amounts of acid sites and strong basic sites, resulting in poor butadiene selectivity. Catalysts prepared by the sol–gel method were highly sensitive to the Mg-to-Si ratio used.97 At a ratio below 9, forsterite and silica domains formed, giving rise to high ethylene selectivity. Optimal activity was achieved with a Mg-to-Si ratio of 15 (Fig. 30), which resulted in highly dispersed Mg–O–Si linkages within a predominant magnesia phase. Impregnation of silica with ethanol-dissolved Mg precursors showed that Mg–O–Si linkages can be obtained in the absence of water, forming amorphous magnesium silicates in addition to crystalline MgO and silica phases.5 With balanced acid–base properties, this MgO–SiO2 catalyst proved highly active in the Ostromislensky process. Generally, dry-milling proved inferior to wet-kneading due to the lesser degree of mixing generated.1 Mechanochemical mixing—heating during a dry-milling process—provided the energy to generate chemical interaction between magnesia and silica, resulting in a material comparable to wet-kneaded MgO–SiO2 in terms of activity.266
Hydrothermal treatment was shown to turn calcined MgO highly active in the ethanol-to-butadiene reaction. The origin of this enhancement is unclear, but the treatment induced changes in the morphological and surface chemical properties of this oxide.139,271 Kovařík et al. discovered that post-synthesis hydrothermal treatment of MgO–SiO2 led to an increase in both its surface area and pore volume, improving activity and butadiene selectivity.272 Men et al. prepared their highly active MgO–SiO2 by wet-kneading hydrothermally synthesized MgO with silica; its catalytic performances were attributed in part to the morphological properties of the magnesia phase, which consisted of hierarchical flow-like inter-grown nanosheets.267 Hydrothermal treatment was also suggested as an alternative regeneration procedure for deactivated catalysts.11,272
Calcination conditions affect the chemical and structural properties of MgO–SiO2 catalysts. Generating new basic sites on MgO requires the removal of deactivating surface contaminants—notably acidic CO2—which generally proceeds by heating.2 Furthermore, thermal processing provides the energy to create new bonds between the magnesia and silica phases forming new crystalline magnesium silicates.5,251 Consequently, calcination conditions can influence the activity of MgO–SiO2 catalysts by modifying their acid–base and structural properties. Zhu et al. studied the relationship between the calcination temperature under air of MgO–SiO2 and its chemical properties;5 their results are summarized in Fig. 31. As illustrated, the ratio between the number of Lewis acid sites and basic sites was significantly affected by the calcination conditions. Calcination between 773 and 873 K generated a balanced number of Lewis acid and basic sites, resulting in higher butadiene selectivity. However, higher temperature generated too many basic sites, favoring undesirable side reactions. Weckhuysen et al. also found the thermal treatment conditions to influence the properties of CuO/MgO–SiO2, including the nature of the atmosphere, noting that stagnant air was preferable to a N2 atmosphere.51
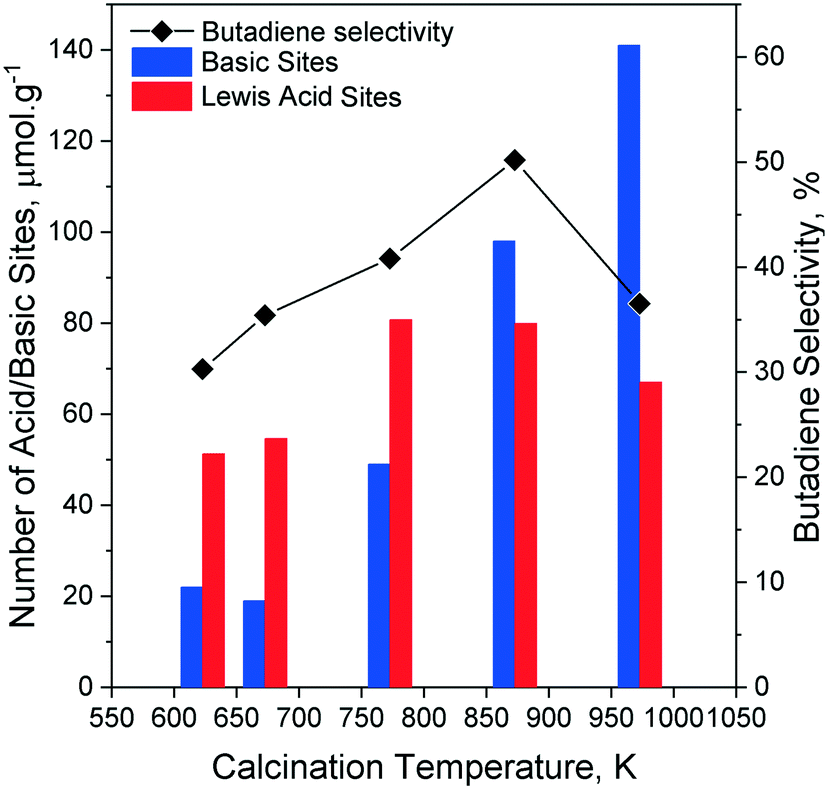 | ||
| Fig. 31 Effect of calcination temperature on the acid and basic properties of MgO–SiO2 catalysts versus butadiene selectivity in the Ostromislensky process.5 Acid sites quantified with pyridine chemisorption; basic sites quantified with CO2 chemisorption. | ||
Over bare MgO–SiO2 catalysts, temperature-programmed surface reactions, catalytic tests, DFT calculations and poisoning studies suggest that the conversion of ethanol to acetaldehyde limits the reaction.100,111,126,138,160,259 Consequently, several dehydrogenation promoters have successfully been used to improve catalytic performances. Taifan et al.181 and Weckhuysen et al.51,268 studied the promoting role of copper. It was found to increase the butadiene yield due to its dehydrogenation abilities improving the acetaldehyde formation rate, but also by poisoning acid sites, ostensibly suppressing undesired side-reactions. However, contrary to bare MgO–SiO2, Cu-modified samples deactivated faster. Both the sintering of metallic copper particles and blockage by coke have been proposed as deactivation mechanisms. Ag, Au and Pd have also been used to promote the activity of MgO–SiO2.93,158,160 Transition and noble metal nanoparticles, well established for promoting alcohol dehydrogenation reactions, are believed to provide redox properties to the catalytic system. Shylesh et al. investigated the potential of all three elements and copper: the catalytic test results are listed in Table 5.93
| Catalyst | Selectivity at 25% conversion, % | |||||
|---|---|---|---|---|---|---|
| Butadiene | Acetaldehyde | Ethylene | Butenes | Butanol | Methane | |
| Conditions: T = 523 K, WHSVEtOH = 1.1 h−1. | ||||||
| Au/MgO–SiO2 | 61 | 30 | 5 | 3 | 1 | — |
| Ag/MgO–SiO2 | 54 | 43 | 2 | 1 | — | — |
| Cu/MgO–SiO2 | 37 | 55 | 2 | 1 | 5 | — |
| Pd/MgO–SiO2 | 2 | 12 | 1 | — | — | 85 |
A clear hierarchy can be seen in terms of selectivity at iso-conversion: Au > Ag > Cu ≫ Pd, but its origin has yet to be determined. Several parameters can influence the nonoxidative dehydrogenation ability of noble and transition metals, such as particle size, pretreatment conditions and metal–support interactions.
Sels et al. used Ag to enhance the activity of dry-milled MgO–SiO2 after conducting a screening of promoters which included Zn, Cu, Ni, Co, Fe, Mn and Cr.92,160 The introduction of Ag by aqueous impregnation improved acetaldehyde formation and generated new Lewis acid sites attributable to silver cations. Additionally, water itself increases the mixing between the oxide by dissolving Mg cations. Tripathi et al. reported a Cu–Ag/MgO–SiO2 catalyst with high activity and stability compared to catalysts with monometallic promoters, a phenomenon which was attributed to a synergism between Cu and Ag.158
Zinc oxide and zirconium oxide dispersed on MgO–SiO2 were also used to enhance its catalytic performances. Zinc oxide contributed to the dehydrogenation reactivity of MgO–SiO2. Zn-Modified MgO–SiO2 was not only found to yield more butadiene and acetaldehyde, but also to be more stable than a Cu-modified equivalent, possibly due to the lower reducibility of Zn(II).181 Both Zn and Zr oxides introduced new Lewis acid sites believed not only to promote the aldol condensation reaction (see above), but also to increase the rate of ethanol dehydration.230,266 As a result, another dimension is added to catalyst design: the ZnO–ZrO2 must also be balanced to maximize ethanol conversion to butadiene.191 One may consider that the contribution of zinc and zirconium goes beyond that of a promoter. In fact, Kyriienko et al. found ZnO–ZrO2/SiO2 to be more active than ZnO–ZrO2/MgO–SiO2.266
Examples of MgO–SiO2 catalysts and performance data
Men et al. reported the most productive MgO–SiO2 catalyst in the literature: at a high WHSVEtOH of 4.1 h−1 and a temperature of 723 K, it exhibited an initial conversion of 95% with a butadiene selectivity of 77%, resulting in a productivity of 1.76 gBD gcat−1 h−1.267,273 These performances were attributed to the morphological and chemical properties of the catalyst resulting from the synthesis method used. The authors prepared MgO using an ethylene glycol mediated self-assembly method reported by Cui et al.274 Magnesium acetate and polyvinylpyrrolidone—a structure-directing polymer—are dissolved in ethylene glycol and hydrothermally-treated in a Teflon-lined autoclave before calcination of the MgO precursors at 773 K. This procedure afforded flower-like hierarchical oxide with a larger surface area and pore size than commercial MgO. It was combined with SiO2 by a wet-kneading process.The acidic and basic properties were optimized by tuning the Mg-to-Si ratio between 1 and 19, which also affected the textural properties of the mixed oxides; butadiene selectivity ranged between 50 and 80%. Ultimately, the authors obtained an optimal ratio of 1.87, arguing that it provided the material with the right balance of acid and basic sites, notably by limiting the number of strong acid sites. They also attributed the impressive catalytic performances to the high surface area and better access to catalytic sites provided by the hierarchical morphology. A stability test indicated that the catalyst deactivated slowly over a period of 42 hours: ethanol conversion and butadiene selectivity decreased from 95% and 77% to 51% and 49%, respectively (Fig. 32). The authors attributed the loss of activity to gradual covering of active sites by coke deposition. This catalyst was further put to test by comparing it with materials possessing different morphologies.273 Flower-like MgO–SiO2 outperformed catalysts with nanodisk and nanosheet morphologies. The authors identified the greater presence of Mg–O–Si linkages and a better balance of acid–base properties in the former catalyst to be the cause of its superiority.
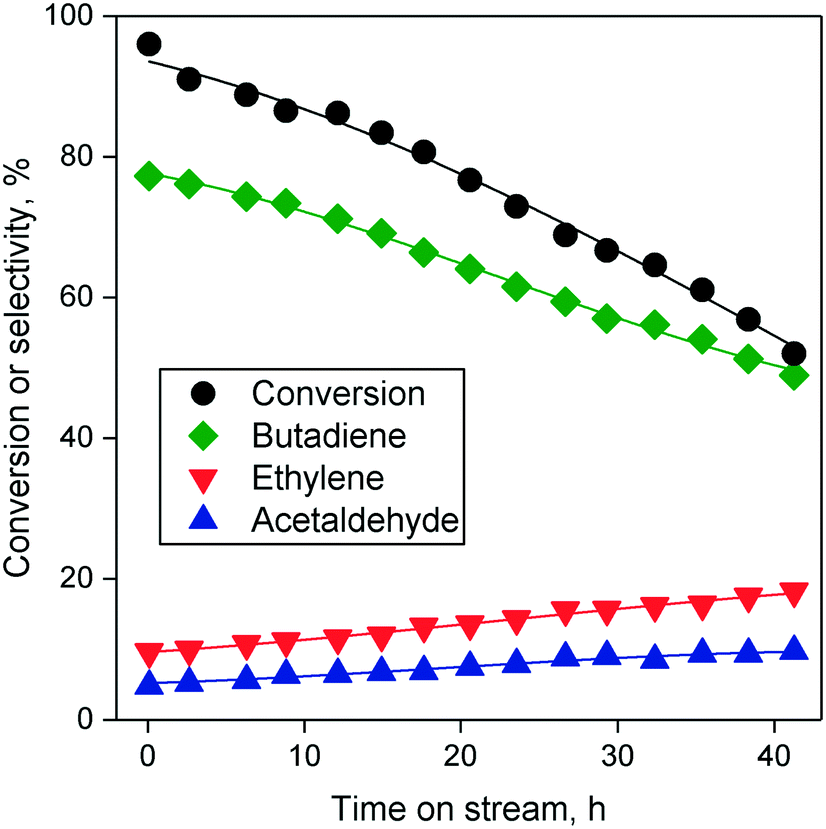 | ||
| Fig. 32 Stability test of a highly active hierarchical MgO–SiO2 catalyst prepared using magnesium acetate and polyvinylpyrrolidone. T = 723 K, WHSVEtOH = 4.1 h−1. Reproduced from ref. 267 with permission from The Royal Society of Chemistry. | ||
As previously mentioned, Shylesh et al. used metallic dopants to enhance the catalytic activity of MgO–SiO2.93 Their work illustrates the beneficial effects of promoters. Prepared by incipient wetness impregnation of commercial silica using a magnesium nitrate solution, the bare initial catalyst with a Mg-to-Si ratio of 2.6 performed poorly. With an ethanol conversion below 10% at 573 K and a WHSVEtOH of 4.1, it mainly yielded ethylene. Au was added using the deposition–precipitation method with urea, well-known for introducing highly dispersed nanoparticles. As illustrated in Fig. 33, ethanol conversion underwent a seven-fold increase; the addition of Au further suppressed the ethylene selectivity, increasing the selectivity towards butadiene to near 60%. The authors attributed the performance enhancement observed to the redox properties provided by Au which participates in the dehydrogenation of ethanol. Characterization further indicated a close, but limited mixing of the magnesia and silica phases, a property that Weckhuysen et al. had associated with superior activity. The contribution of the deposition–precipitation method to the catalytic activity was not explored. It may be possible that a basic pH combined with the mixing required replicated conditions akin to those of wet-kneading, further enhancing the textural and chemical properties of the catalyst. The work of Shylesh et al. illustrates how post-synthesis modifications can drastically improve the catalytic performances of MgO–SiO2.
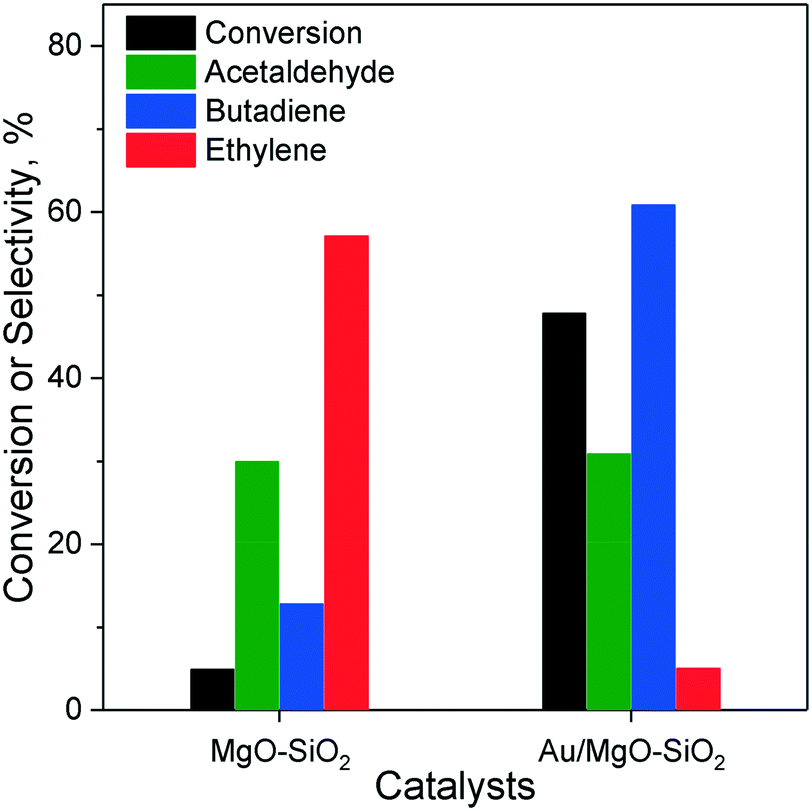 | ||
| Fig. 33 Effect of 3 wt% Au on the performance of MgO–SiO2.93T = 573 K, WHSVEtOH = 1.1 h−1. | ||
While Weckhuysen et al. correlated butadiene yield with the relative quantity of layered hydrous magnesium silicates,255 Baba et al. studied the catalytic activity of talc, a layered hydrous magnesium silicate, in the ethanol-to-butadiene reaction.139,142,271 Pure synthetic talc produced very little butadiene from ethanol but showed >75% selectivity towards dehydration products at 38.8% conversion. The addition of zinc oxide to the hydrothermal synthesis process produced talc catalysts with Zn(II) substituted within the layered structure and highly selective towards butadiene. By tuning the Zn concentration, it reached 51.8% at a WHSVEtOH of 8.4 h−1 and a temperature of 673 K, amounting to a productivity of 1.1 gBD gcath−1—one of the highest in the literature. Catalytic tests with acetaldehyde alone demonstrated that Zn not only suppressed ethanol dehydration, but also crotonaldehyde formation, indicating that pure talc was more active in the aldol condensation. Experimental and computational studies were conducted to explain the promoter effect of Zn. XPS revealed that the growing incorporation of Zn increased the binding energy of the O 1s level, indicating a progressive lowering of the basic character of talc. These results not only support the theory that limiting the amount of strong basic sites is beneficial to butadiene yield, but also indicate that strong basic sites are more active in the aldol condensation. The promoter effect of Zn was attributed to its contribution to ethanol dehydrogenation. Based on DFT calculations and the hard–soft acid–base theory, the authors argued that Zn cations, acting as softer Lewis acid–base pairs, favored dehydrogenation compared to purely Mg-containing catalysts. Zinc also increased the lifetime of the catalyst. Pure talc deactivated after 1 hour due to severe coking, whereas ethanol decreased by around half over a period of 15 hours with Zn–talc. However, the selectivity towards acetaldehyde progressively increased at the expense of butadiene, indicating that the stronger basic sites responsible for aldol condensation were progressively poisoned.
MgO–SiO2 has proved to be one of the best catalytic systems for the Lebedev process. Table 6 provides the performance details of many catalysts directly or indirectly addressed in this section. Again, accurately comparing the different materials reported in the literature is hindered by the different reaction conditions employed. Nonetheless, from an industrial standpoint, it is worth highlighting that high butadiene selectivity, low selectivity towards dehydration products and high butadiene productivity were achieved, despite the challenges associated with MgO–SiO2 catalyst design. Part of the issue can be attributed to unknowns regarding the identity of active sites. For instance, the extent to which SiO2 is necessary to the formation of the active phase has yet to be identified.1 Nevertheless, it is understood that a balance between acidic and basic properties is required to maximize catalytic activity. The prevalent theory on catalyst design suggests that weak and medium basic sites found on the MgO phase are required to catalyze the ethanol dehydrogenation and aldol condensation, respectively, with strong basic sites being detrimental to activity. However, the role of acid sites remains under debate: although moderate numbers of Lewis acid sites have been correlated with superior activity, there is no consensus on their identity. Identifying their ideal acid strength and the influence of neighboring Si atoms requires further investigation. Finally, the role of different magnesium silicates formed by the mixing of MgO and SiO2 must be clarified.255
| Entry | Catalyst | Mg:Si |
WHSV | TOS | T | X | BD S. | AcOH S. | C2=S. | BD Y. | P BD | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h−1 | h | K | % | % | % | % | % | |||||
| a Wet-kneading with hierarchical MgO. b Wet-kneading with nano-sized MgO. c Mechano-chemical synthesis. d Hydrogen was co-fed. WHSV: weighted hourly space velocity of ethanol. X: ethanol conversion. BD, AcH, C2= are butadiene, acetaldehyde and ethylene, respectively. S., Y. and P. are selectivity, yield and productivity, respectively. Productivity is expressed in terms of gBD gcat−1 h−1. Active phases are expressed in terms of wt%. | ||||||||||||
| 1 | MgO–SiO2a | 1.86 | 4.1 | — | 723 | 95 | 77 | 2 | 13 | 73 | 1.76 | 267 |
| 2 | 3%Au/MgO–SiO2 | 2.6 | 1.1 | 3.3 | 573 | 45 | 60 | 28 | 7 | 27 | 0.14 | 93 |
| 3 | Zn–talc | — | 8.4 | 7 | 673 | 42 | 52 | 22 | 8 | 22 | 1.06 | 139 |
| 4 | MgO–SiO2b | 1 | 0.275 | 4 | 748 | 93 | 41 | — | — | 38 | 0.06 | 275 |
| 5 | MgO–SiO2c | 1 | 1 | — | 673 | 41 | 57 | 5 | 34 | 24 | 0.14 | 266 |
| 6 | 1%Ag/MgO–SiO2 | 2 | 1.2 | 3.3 | 753 | 84 | 50 | 6 | 10 | 42 | 0.29 | 160 |
| 7 | 1%CuO/MgO–SiO2 | 1 | 1.1 | 0.5 | 698 | 74 | 48 | 7 | 5 | 38 | 0.25 | 51 |
| 8 | 6.3%Zr–1.4%Zn/MgO–SiO2 | 3 | 0.62 | 3 | 648 | 40 | 36 | 8 | 32 | 30 | 0.13 | 191 |
| 9 | 4%ZnO/MgO–SiO2 | 1 | 1 | 3 | 648 | 56 | 62 | 22 | 10 | 35 | 0.20 | 230 |
| 10 | 2.5%Cu–2.5%Ag/MgO–SiO2 | 2 | — | — | 573 | 64 | 72 | 15 | 9 | 46 | — | 158 |
| 11 | 0.5%Ag/MgO–SiO2d | 1 | 0.2 | 6 | 598 | 85 | 76 | 6 | 3 | 64 | 0.02 | 276 |
| 12 | MgO–SiO2 | 0.63 | 0.03 | 11 | 623 | 53 | 30 | 10 | 53 | 16 | 0.003 | 1 |
Even with the knowledge of the active phase, achieving the ideal balance of acidic and basic properties would be challenging. No systematic understanding of the numerous synthesis parameters susceptible to affect the acid–base character of MgO–SiO2 (Mg-to-Si ratio, calcination temperature, mixing of the oxide phases, solvents, etc.) has been formulated. An overview of the best performing catalysts suggests that a limited mixing at the interface of MgO and SiO2 phases, resulting in layered hydrous amorphous silicate, is preferable. Ostensibly, the remaining MgO phases provides the weak and medium basic sites required, while the new magnesium silicate phase introduces new Lewis acid sites required for catalytic activity. Excessive mixing between the two oxides leads to amorphous magnesium silicates responsible for ethanol dehydration.
The most successful preparation method appears to be wet-kneading MgO and SiO2 precursors with morphological properties (nanosized, hierarchical) intended to maximize the limited interaction between them. The optimal Mg-to-Si ratio has varied depending on the synthesis method used. Contrarily, the ideal calcination temperature was found to vary between 773 K and 873 K.5
Because the rate limiting step of the ethanol-to-butadiene conversion over MgO–SiO2 is believed to be the formation of acetaldehyde, the catalytic activity can be further improved by introducing transition and noble metals active in the dehydrogenation of ethanol. According to the results of Shylesh et al. Au is the most promising promoter for MgO–SiO2.93
As illustrated in Fig. 32, MgO–SiO2 is susceptible to deactivation, ostensibly by coke deposition believed to be formed by strong basic sites. Yet, few studies have addressed the improvement of catalytic stability, or even the regeneration of the spent catalyst.272 It may be worth investigating the deactivation mechanism of MgO–SiO2.
Rare earth elements
Rare earth metal oxides have found numerous uses in heterogeneous catalysis.277,278 Recently, some authors have investigated the potential application of rare earth elements as catalysts for the conversion of ethanol to butadiene. Li et al. studied the activity of various rare earth (Y, La, Ce, Pr, Nd) oxide catalysts supported on dealuminated zeolite. Although zeolite-supported monometallic catalysts primarily dehydrated ethanol to ethylene and diethyl ether, Y, La and Ce were found to be capable of converting acetaldehyde to crotonaldehyde, indicating their condensation ability. By combining these elements with metal oxides capable of ethanol dehydrogenation, catalysts highly active in the ethanol-to-butadiene reaction were recently reported in the literature. However, due to their novelty, these catalytic systems have not been investigated as other catalytic systems detailed above. Consequently, the understanding of their activity is limited.Owing to the work of Li et al., yttrium-containing catalysts have been the most studied of all rare earth-based ones for the ethanol-to-butadiene reaction.119,180 Supported on dealuminated β zeolite, Y lead primarily to ethanol dehydration products. Promotion with copper marginally increased the butadiene yield, yet diethyl ether and other carbonaceous species remained the principal products. Significant activity was obtained by combining Zn and Y. Li et al. tuned the Zn-to-Y ratio, ultimately obtaining 2%–Zn8%Y/SiBEA as the most active catalyst. Butadiene selectivity as high as 81% was reported. This catalyst achieved an exceptional productivity of 2.33 gBD gcat−1 h−1 at 673 K and a WHSVEtOH of 7.9 h−1, making it one of the best for the Lebedev process found in the literature. The authors attributed these remarkable performances to a synergic effect between both Zn(II) and Y(III), which was enhanced by their proximity within the zeolite pores. Owing to the confinement effect of zeolites, Zn–Y clusters were suggested to form within the zeolite cages, as evidenced by the fact that MCM-41-supported Zn–Y did not perform as well. Furthermore, XPS analysis found a stronger interaction between Zn(II) and Y(III) unique to the zeolite supported catalyst. The deactivation of 2%Zn8%Y/BEA was discussed previously. Li et al. determined that pore blockage did not play a significant role in the loss of activity.131 Rather, cyclic unsaturated aldehydes and ketones gradually cover the Zn and Y sites of the catalyst. Under conditions enabling high productivity, the butadiene yield dropped by half in the first 10 hours of catalyst testing (Fig. 34), which is more severe than other highly productive catalysts recently reported, such as hierarchical MgO–SiO2 and Zn–Ta–TUD-1.103,267 Nevertheless, ZnY/SiBEA recovered its high productivity after calcination under air for 4 hours.
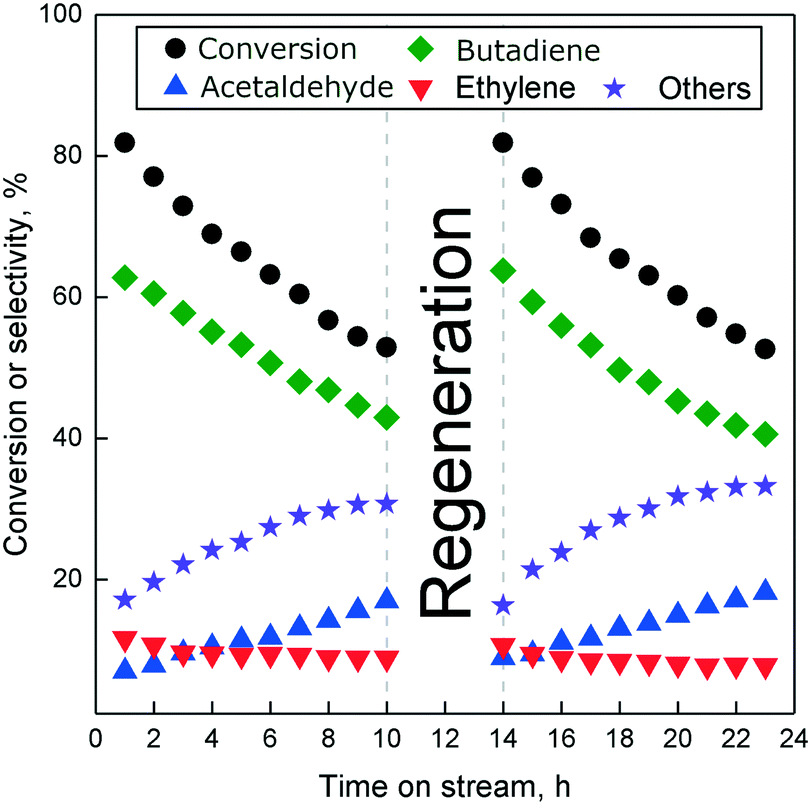 | ||
| Fig. 34 Stability and regeneration test of 2%Zn–8%Y/SiBEA. Regeneration under air flow at 673 K for 4 hours. T = 673 K, WHSVEtOH = 7.9 h−1. Reproduced from ref. 180. Copyright 2017, American Chemical Society. | ||
Li et al. have proposed a model of activity and deactivation of their 2%Zn–8%Y/SiBEA catalyst.163 Since Zn/SiBEA produced not only acetaldehyde from ethanol, but also some butadiene, the authors argued that Zn(II) primarily provided the catalyst with redox properties active in the dehydrogenation reaction. Since each component taken as a stand-alone catalytic system was found by MAS-NMR coupled with NH3 and acetone-2-13C adsorption experiments to introduce Lewis acidity to SiBEA, the authors argued that both metals contributed to the condensation activity, although Y(III) as found to be more active. Li et al. concluded that not only the synergistic effect between Zn(II) and Y(III), but also the proximity of the two phases enables the high selectivity observed, as it increased the chance of the intermediates to react with each other on these sites during the aldol condensation.
Zhu et al. focused on understanding the structure–activity relationship of Y/SiO2 catalysts in the two-step process, providing insight on the coupling activity of yttrium-containing catalysts.279 Y/SiO2 catalysts with varying Y:Si ratios were prepared via wet impregnation of fumed silica. Characterization revealed that the amount and coordination of Y–O–Si linkages depended on the elementary composition of the catalyst. This parameter had repercussions on the acid–base properties of the materials and, in turn, on their catalytic activity. The authors used chemical probes to quantify and characterize the acidic and basic sites of their Y/SiO2 samples. In an approach reminiscent of works on MgO–SiO2,255,267 they were able to correlate the chemical properties with the product distribution. Accordingly, materials possessing a high concentration of strong Lewis acid sites and intermediate basicity displayed higher selectivity towards butadiene; such properties were obtained with moderate Y loading. High loadings resulted in bulk Y2O3 formation on the surface of fumed silica. Such samples displayed stronger basic sites monitored with CO2-TPD, which favored butanol formation. Therefore, as with MgO–SiO2, Y-containing catalysts are subject to a careful balance between acidic and basic properties.
Kyriienko et al. mixed lanthanum oxide, which the literature identifies as silica-supported La2O3, a multifunctional catalyst,280 with silica, zirconia and zinc oxide to prepare a catalyst highly active in the Lebedev process.230 The preparation process combined incipient wetness impregnation and wet-kneading; several samples with different compositions were synthesized to study the role of each component in catalytic tests at 748 K and a WHSVEtOH of 1 h−1. La2O3–SiO2 proved poorly active, yielding primarily dehydration products. The addition of zinc oxide successfully suppressed ethanol dehydration—a phenomenon observed in many catalytic systems for the ethanol-to-butadiene reaction.127,197,247 Still, the butadiene yield remained below 20%. Only with the addition of ZrO2 could a yield of 52.5% be achieved, marked by a sharp consumption of acetaldehyde compared to other samples. Ultimately, the best catalyst was 2%ZnO–7%La2O3–1%ZrO2–SiO2, which reached a butadiene yield of 60.2% at 743 K. The authors suggested that a synergy between each component explained the catalytic performances of the mixed catalyst. ZnO promoted ethanol dehydrogenation, whereas both ZrO2 and La2O3 catalyzed the aldol condensation and MPVO reactions. In fact, La2O3 been shown to be active in aldol condensation180,281 and MPVO reactions.282 The condensation activity of La2O3 can be attributed to its basic properties, confirmed by Kyriienko et al. with pyrrole-FTIR, but also well-known in the literature on La2O3.229 Potential active sites are La–O acid–base pairs and isolated O2−.
Zhao et al. reported a Zn–Ce catalyst encapsulated within mesoporous SBA-15 active in the Lebedev process.283 10%Zn–5%Ce/SBA-15 catalysts were prepared using the solid-state grinding method to mix SBA-15 samples with metal precursors. By mixing the as-prepared SBA-15 sample containing the organic templates, a higher degree of active phase dispersion was obtained. This dispersion increased the concentration of Lewis acid sites compared to calcined SBA-15, which resulted in higher ethanol conversion and butadiene selectivity in the Lebedev process. At 648 K and a WHSVEtOH of 1.62 h−1, the butadiene yield reached 36% after 5 hours on stream. These performances were ostensibly attributed to a better condensation activity provided by the greater number of Lewis acid sites.
Development of rare earth catalysts for the ethanol-to-butadiene reaction is a recent subject. Characterization studies together with the literature available for other organic reactions suggest that parallels may be drawn with other catalytic systems, as Y, La and Ce oxides possess chemical properties comparable to those of the previously discussed systems: basic sites and Lewis acid–base pairs. Correlating these properties with specific catalytic activity remains to be fully elucidated before proceeding to rational design. As demonstrated by Li et al.163 rare earth metal oxides are active in the aldol condensation and MPVO reaction, implying that dehydrogenation promoters must be introduced to make them active in the Lebedev process. So far, Zn has been the preferred promoter, which some authors have suggested to be in synergy with Y.180 The performances of the most active catalysts reviewed are listed in Table 7.
| Entry | Catalyst | WHSV | TOS | T | X | BD S. | AcOH S. | C2=S. | BD Y. | P BD | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| h−1 | h | K | % | % | % | % | % | ||||
| WHSV: weighted hourly space velocity of ethanol. X: ethanol conversion. BD, AcH, C2= are butadiene, acetaldehyde and ethylene, respectively. S., Y. and P. are selectivity, yield and productivity, respectively. Productivity is expressed in terms of gBD gcat−1 h−1. Active phases are expressed in terms of wt%. | |||||||||||
| 1 | 2%Zn–8%Y/SiBEA | 7.9 | 1 | 673 | 82 | 63 | 7 | 2 | 52 | 2.33 | 180 |
| 2 | 2%ZnO–7%La2O3–1%ZrO2–SiO2 | 2 | — | 673 | 100 | 60 | — | — | 60 | 0.71 | 229 |
| 3 | 10%Zn–5%Ce/SBA-15 | 1.62 | 5 | 648 | 79 | 45 | 22 | 22 | 36 | 0.339 | 283 |
Alkali & alkaline earth dopants
The balance of acidic and basic properties influences the activity of catalysts in the ethanol-to-butadiene reaction. A simple, but efficient strategy has been used to modulate them with alkali and alkaline-earth dopants,3,8,9,118,172,191,284 an approach employed when dealing with solid acids.285,286 The main benefit is the suppression of ethanol dehydration, sometimes at the expense of the overall activity.16,198 Although successfully put in practice, the fundamental aspects of this approach are not fully understood and no established method for using these dopants has been formulated.The suppression of side reactions is attributed to the modulation of acid sites resulting from their interaction with alkali and alkaline-earth cations, which can be introduced into catalysts via impregnation.3,8,9,118,172,191,284 In theory, these cations undergo proton-exchange with Brønsted acid sites, thereby deactivating them. The suppression of ethylene and diethyl ether formation are attributed to this poisoning effect, as Brønsted acid sites are known dehydration active sites. In practice, alkali and alkaline-earth cations have been shown to not only weaken or poison acid sites, but also to act as Lewis acid sites themselves, owing to their positive charge.287 The influence of alkali and alkaline-earth dopants depends on both their nature and the amount added to the parent material. Baylon et al. demonstrated that the principal effect of Na doping was to weaken the acid strength of a ZnO–ZrO2 catalyst without significantly lowering the total number of sites (Fig. 35).8 As a result, acetaldehyde and butadiene formation became favored over ethanol dehydration (Table 6, entries 1 and 2). However, the enhancement of butadiene yield plateaued when the acid strength decreased. Similarly, Da Ros et al. progressively increasing the amount of Na from 0 to 2 wt% on ZnO–ZrO2/MgO–SiO2 which lowered the butadiene yield (Table 9, entry 3).191 These results suggest that, although these dopants can alleviate unwanted side-reactions by tuning the acid properties of a catalyst through poisoning, a minimum number of acid sites with moderate strength is required to enable to conversion of ethanol to butadiene.
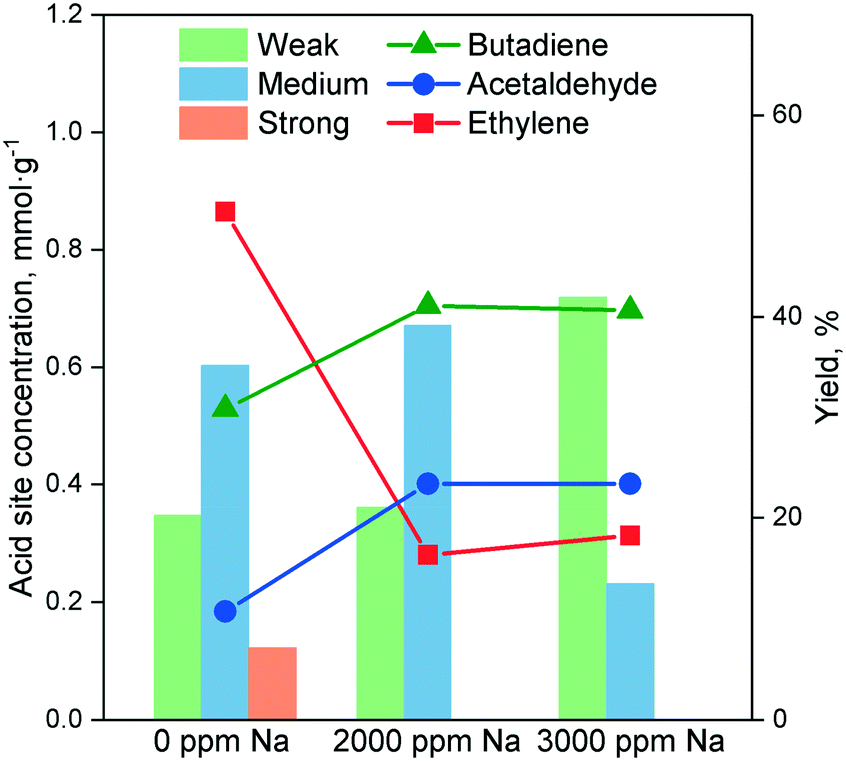 | ||
| Fig. 35 Influence of alkali and alkaline-earth dopants on the acid (NH3-TPD)/base (CO2-TPD) site concentration of the reference 1%ZnO–5%ZrO2/SiO2versus butadiene yield.172 | ||
Beyond their role as dehydration suppressors, Patil et al.172 have proposed that alkali and alkaline-earth oxides could also contribute to butadiene production since they are known to possess basic properties.230 After all, the basic alkaline-earth metal oxide MgO is active in the ethanol-to-butadiene reaction. In comparing the performance enhancement of various promoters (Li, Na, K, Cs), the authors found that—although every dopant increased the number of basic sites and suppressed ethylene selectivity—only Cs increased the butadiene yield compared to the ZnO–ZrO2 reference material (Fig. 36). Notably, this promoter effect was verified at equimolar amounts of alkali and alkaline-earth dopants. The authors argued that Cs alone possessed the basic strength necessary to promote the aldol condensation step, thereby increasing the butadiene yield (Table 6, entry 3).
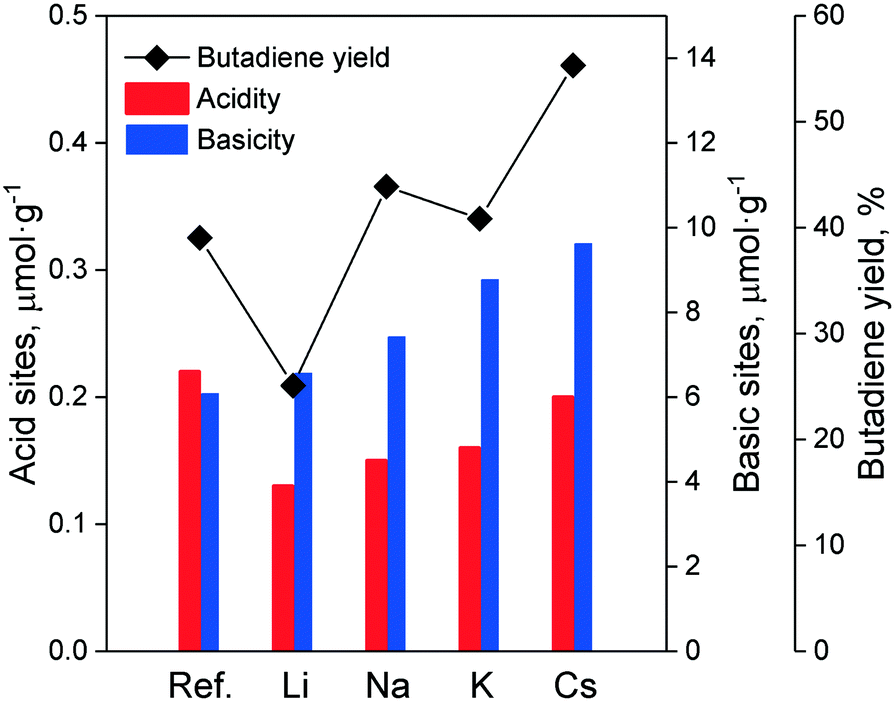 | ||
| Fig. 36 Product yield for the Lebedev process and the concentration of weak, medium and strong acid sites on ZnZrOx (Zr:Zn = 10) versus the concentration of the Na dopant.8 | ||
However, Klein et al. possibly contradicted this conclusion. Catalytic tests of alkali and alkaline-earth (Cs, K, Ca, Mg) doped β zeolites during the Ostromislensky process indicated that Cs/BEA did not catalyze the aldol condensation, reaching only 5% conversion; magnesium-doped samples alone showed significant activity. This suggests that magnesium oxide is better suited for providing the condensation activity required in the ethanol-to-butadiene reaction.139,142
The only instance of an alkaline-earth oxide other than MgO being used as the catalyst component providing condensation activity was reported by La-Salvia et al.288 The authors used barium oxide to promote the condensation of acetaldehyde on the acidic mesoporous aluminosilicate Al-MCM-41, with chromium oxide being used as a dopant for ethanol dehydrogenation. With barium oxide and the aluminosilicate alone, ethylene was the predominant species, indicating that the alkaline-earth oxide did not perform adequately in the dehydrogenation of ethanol. Only by adding chromium oxide and barium oxide could selectivity towards butadiene attain a maximum of 28% (Table 8, entry 5). Compared to other doped catalysts reviewed in this section, the ethylene selectivity remained very high, a phenomenon ostensibly due to Brønsted acids formed by the presence of Al cations within the silicate framework.
| Catalyst | Conversion | Selectivity | ||
|---|---|---|---|---|
| Butadiene | Acetaldehyde | Ethylene | ||
| NS = nanosheets. M = microporous. | ||||
| 1.5%Zn–8.9%Hf/MFI (M) | 63.6 | 43.0 | 1.1 | 11.1 |
| 1.5%Zn8.9%Hf/MFI (NS) | 76.2 | 53.3 | 0 | 14.6 |
| 1.7%Li–1.5%–Zn–8.9%Hf/MFI (M) | 21.3 | 36.4 | 10.3 | 3.8 |
| 1.7%Li–1.5%–Zn8.9%Hf/MFI (NS) | 64.6 | 73.0 | 0 | 5.3 |
| 1.7%Na–1.5%Zn–8.9%Hf/(NS) | 54.2 | 45.2 | 3.2 | 2.3 |
| 1.7%K–1.5%Zn–8.9%Hf/MFI (NS) | 35.6 | 32.1 | 5.6 | 1.2 |
Wang et al. recently demonstrated how the morphology of a catalyst plays a significant role in the promoter effect of alkali and alkaline-earth cations.3 Two zinc–hafnium oxide catalysts were compared: one supported on microporous MFI zeolite, another on nanosheets of the same molecular sieve. As listed in Table 8, the latter performed better, ostensibly due to the hierarchical morphology of the carrier. Interestingly, introducing 1.5 wt% Li led to very different changes in activity depending on the support. In the microporous material, the alkali cation reduced ethanol conversion by two-thirds, also suppressing the butadiene selectivity. Contrarily, adding Li to the nanosheet-supported catalyst significantly enhanced the butadiene selectivity while lowering ethylene formation at minimal loss in ethanol conversion. The reason for the discrepancy is unclear, but it demonstrated that the properties of the catalyst support influenced the dopant effect of alkali and alkaline-earth dopants. Furthermore, the authors compared the promoter effect of other cations (Na, K), concluding that Li was a better dopant (Table 8).
Post-synthesis introduction of alkali and alkaline-earth cations in various catalytic systems—mixed oxides, supported metal oxides, zeolites—suppresses side-reactions, most notably ethanol dehydration. Generally, this is accompanied with a decrease in ethanol conversion. Alkali and alkaline-earth modifications also influence acetaldehyde and butadiene selectivity. The extent to which the catalytic performances are modified depends on the type and amount of dopant, as well as the nature of the catalytic system. Various studies indicated that these oxides lower the strength and/or number of acid sites.
Generally, an excess of dopant results in a clear decrease in butadiene yield, to the benefit of acetaldehyde yield, a phenomenon attributed to the poisoning of acid sites required to form butadiene. It is still unclear whether alkali and alkali-earth oxides also promote condensation activity via their basic properties, as discrepant results have been published.9,172Table 9 lists the performances of notable instances in which alkali and alkaline-earth dopants were used to enhance the catalytic activity of a parent material. Judging from the studies reviewed, it is difficult to conclude whether one dopant is inherently superior in their ability to enhance catalytic performances, a problem exacerbated by the different conditions used. For instance, Ohishi et al. reported one of the best catalytic performances by adding Na2O to MgO–SiO2 (Table 9, entry 7). However, many authors have pointed out that product distribution was recorded at a very short time on stream, thereby ignoring any contribution from deactivation.11,12 Nevertheless, alkali and alkaline-earth doping offer a facile approach to modify the crucial acid–base properties of a given catalytic system. And, as Da Ros et al. pointed out, the loss of butadiene yield sometimes observed can be compensated for by the reduction of undesired byproducts considering that acetaldehyde can be recycled to take part in a two-step process.191
| Entry | Catalyst | WHSV | TOS | T | X | BD S. | AchOH S. | C2=S. | BD Y. | PBD | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| h−1 | h | K | % | % | % | % | % | ||||
|
a Zr:Zn ratio = 10.
|
|||||||||||
| 1 | 2000 ppm Na/ZnZrOxa | 6.2 | — | 623 | 54 | 26 | 37 | 28 | 14 | 0.49 | 8 |
| 2 | 2000 ppm Na/ZnZrOxa | 0.2 | — | 623 | 97 | 47 | 11 | 47 | 46 | 0.06 | 8 |
| 3 | 1.2%K–1.5%Zr–0.5%Zn/MgO–SiO2 | 0.62 | 3 | 648 | 26 | 55 | 17 | 12 | 27 | 0.12 | 190 |
| 4 | 0.5%Cs2O–17%ZnO–5%ZrO2/SiO2 | 1 | — | 673 | 98 | 56 | 16 | 15 | 55 | 0.32 | 172 |
| 5 | 1.4%Cr–16%Ba/Al-MCM-41 | 0.07 | 10 | 723 | 90 | 28 | — | — | 25 | 0.01 | 288 |
| 6 | 1.7%Li–1.5%Zn8.9%Hf/MFI (NS) | 0.47 | 3 | 593 | 65 | 73 | 0 | 5 | 47 | 0.13 | 3 |
| 7 | 0.1%Na2O/MgO–SiO2 | 0.18 | 0.17 | 623 | 100 | 87 | — | — | 87 | 0.09 | 284 |
Conclusions
After decades of being considered an obsolete technology, ethanol conversion to butadiene has begun to gather attention again. The primary cause of interest is the necessity of finding alternatives to the steam cracking of naphtha, which suffers from sustainability and supply issues. Under the right circumstances, notably the geographical location and the type of biomass feedstock, butadiene from bio-ethanol is more sustainable than fossil-based routes. Financially, ethanol-to-butadiene processes cannot yet compete with current production methods, in part due to low petroleum cost, as well as due to insufficient catalyst performances.In the last 8 years, significant progress has been made in gaining insight into several aspects of the ethanol-to-butadiene reaction, namely its mechanism and the design of materials exhibiting optimal catalytic activity. The mechanism of the ethanol-to-butadiene reaction is known to be complicated, partly because several of the intermediate species believed to be involved are seldom detected. Nevertheless, recent development owing to the use of techniques such as infrared spectroscopy, isotope tracing experiments and DFT studies has provided new evidence. The general pathway formulated by various authors more than fifty years ago has now come to be recognized as valid: ethanol first dehydrogenates to acetaldehyde, which condensates to crotonaldehyde; the latter reacts with another ethanol molecule to form crotyl alcohol and acetaldehyde via an MPVO reaction; crotyl alcohol dehydrates to butadiene.
At the molecular level, there remains differences amongst the conclusions of the various teams, particularly concerning the aldol condensation mechanism, which may consist of an Eley–Rideal or a Langmuir–Hinshelwood-type mechanism. These discrepancies are not inconsequential, as kinetic modelling greatly benefits from our understanding of the reaction. Incidentally, the ethanol-to-butadiene reaction lacks adequate kinetic studies that can be used in process simulations. Consequently, studies on the viability of this technology often rely on industrial data—only for the Ostromislensky process—obtained after the Second World War with catalysts that have now be outclassed by recent findings. Another topic important to the implementation of ethanol-to-butadiene processes is the deactivation mechanism. The major cause of deactivation on almost all catalytic systems has been identified as the deposition of coke, with calcination under air being a viable method of catalyst regeneration. However, some studies have found alternative sources of deactivation, such as the poisoning of active sites by bulky oxygenated species that are not coke, the reduction of metal oxide active phases and particle sintering.
A wide variety of catalysts have been reported throughout the years. Two primary categories of catalytic systems have been reviewed: group 4 and 5 transition metals, and MgO–SiO2. In the first case, the catalytic activity has been attributed to the Lewis acid characteristics of these metals when in their oxide form, generally supported over silicates. Lewis acid sites are known to catalyze the aldol condensation, making Ta, Hf and Zr catalysts some of the most active found in the literature. The condensation ability of these catalysts is maximized by achieving monoatomic dispersion, preferably in the form of tetrahedral “open” sites within the framework of silicate carriers. To this end, several metal dispersion methods and catalyst carriers have been investigated, with exceptional performances obtained using mesoporous silica supports. The Lebedev process requires the addition of dehydrogenation promoters, with Ag, Cu and Zn being the most successful. Recent research has found rare-earth metal oxides to be also active in the ethanol-to-butadiene reaction. Notably, zeolite-supported yttrium oxide catalysts have shown remarkable performances when combined with zinc oxide. Ostensibly, the Lewis acid properties of Y play a role similar to Ta, Hf, Zr and Nb catalysts, which is to say to provide the material with condensation abilities.
MgO–SiO2 catalysts are inherently active in the Lebedev process and have also shown remarkable performances. The main issue regarding this catalytic system concerns the uncertainty regarding the nature of the active sites. Weak and medium-strength basic sites on the MgO phase have been shown to enable the dehydrogenation of ethanol and the aldol condensation. However, the role and character of the acid sites formed by the mixing between MgO and SiO2 remain under debate. To maximize the activity of MgO–SiO2 catalysts, a subtle balance between the acid and base properties must be attained. However, the effect that many synthesis parameters have on this balance makes the design of catalysts challenging. The activity can be further improved by the use of dehydrogenation promoters, as the formation of acetaldehyde has been identified as the rate-limiting step on MgO–SiO2.
Modifying catalysts with alkali and alkaline-earth ions was shown to be a practical strategy to tune the acid–base properties of catalysts. These can poison stronger acid sites associated with undesired side-reactions, enhancing the selectivity towards butadiene. To some extent, alkali and alkaline-earth ions can also introduce basic sites, although the remains no clear evidence that these new sites significantly participate in the reaction.
Although much insight has been acquired in recent years, there remains room for improving our understanding of the ethanol-to-butadiene reaction. A full elucidation of the molecular-level mechanism has been proposed, but its validity on different catalysts remains to be confirmed. Ostensibly, the subtle differences between the conclusions reached by various research groups could be explained by the different properties of each active phase. With such elucidation, kinetic modelling and accurate process simulation will be facilitated. Such modelling should include the effect of co-feeding water and deactivation. Improving the reactivity of catalysts—all systems included—will not only require further investigation of the structure–activity relationship, but also the development of synthesis methods better suited to generate the most desirable active sites.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the French National Research Agency (ANR-15-CE07-0018-01). Chevreul Institute (FR 2638), Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation, Région Hauts-de-France and FEDER are acknowledged for support and partial funding of this work. We thank Dr. Lee and Dr. Cheong for providing us with high-resolution TEM images.Notes and references
- S. Kvisle, A. Aguero and R. P. A. Sneeden, Appl. Catal., 1988, 43, 117–131 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- C. Wang, M. Zheng, X. Li, X. Li and T. Zhang, Green Chem., 2019, 21, 1006–1010 RSC.
- G. M. Cabello González, P. Concepción, A. L. Villanueva Perales, A. Martínez, M. Campoy and F. Vidal-Barrero, Fuel Process. Technol., 2019, 193, 263–272 CrossRef.
- Q. Zhu, B. Wang and T. Tan, ACS Sustainable Chem. Eng., 2017, 5, 722–733 CrossRef CAS.
- W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Román-Leshkov, J. Phys. Chem. C, 2016, 120, 28533–28544 CrossRef CAS PubMed.
- B. B. Corson, H. E. Jones, C. E. Welling, J. A. Hinckley and E. E. Stahly, Ind. Eng. Chem., 1950, 42, 359–373 CrossRef CAS.
- R. A. L. Baylon, J. Sun and Y. Wang, Catal. Today, 2014, 259, 446–452 CrossRef.
- A. Klein, K. Keisers and R. Palkovits, Appl. Catal., A, 2016, 514, 192–202 CrossRef CAS.
- J. L. Cheong, Y. Shao, S. J. R. Tan, X. Li, Y. Zhang and S. S. Lee, ACS Sustainable Chem. Eng., 2016, 4, 4887–4894 CrossRef CAS.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2013, 6, 1595–1614 CrossRef CAS PubMed.
- G. O. Ezinkwo, V. P. Tretyakov, A. Aliyu and A. M. Ilolov, ChemBioEng Rev., 2014, 1, 194–203 CrossRef CAS.
- G. Pomalaza, M. Capron, V. Ordomsky and F. Dumeignil, Catalysts, 2016, 6, 203 CrossRef.
- M. Jones, Chem. Cent. J., 2014, 8, 53 CrossRef PubMed.
- G. Egloff and G. Hulla, Chem. Rev., 1945, 36, 63–141 CrossRef CAS.
- M. Dahlmann, J. Grub and E. Löser, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, vol. 100 C, pp. 1–24 Search PubMed.
- H. N. Sun and J. P. Wristers, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, NJ, USA, 2002, vol. 4s Search PubMed.
- D. Threadingham, W. Obrecht, W. Wieder, G. Wachholz and R. Engehausen, Ullmann's Encycl. Ind. Chem., 2011, pp. 597–622 Search PubMed.
- M. Morton, J. Macromol. Sci., Part A: Pure Appl. Chem., 1981, 15, 1289–1302 CrossRef.
- G. S. Whitby and M. Katz, Ind. Eng. Chem., 1933, 25, 1204–1211 CrossRef CAS.
- H.-H. Greve, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, vol. 9, pp. 245–260 Search PubMed.
- J. U. Nef, Justus Liebigs Ann. Chem., 1901, 318, 137–230 CrossRef CAS.
- J. Ostromislenskiy, Zh. Russ. Fiz.-Khim. O-va., 1915, 1472–1506 Search PubMed.
- J. Toland, The Rising Sun: The Decline and Fall of the Japanese Empire, 1936–1945, Modern Library, 2014 Search PubMed.
- P. M. Kampmeyer and E. E. Stahly, Ind. Eng. Chem., 1949, 41, 550–555 CrossRef CAS.
- B. B. Corson, E. E. Stahly, H. E. Jones and H. D. Bishop, Ind. Eng. Chem., 1949, 41, 1012–1017 CrossRef CAS.
- W. J. Toussaint, J. T. Dunn and D. R. Jackson, Ind. Eng. Chem., 1947, 39, 120–125 CrossRef CAS.
- P. Wendt, South. Econ. J., 1947, 13, 203 CrossRef.
- American Chemical Society National Historic Chemical Landmarks, U.S. Synthetic Rubber Program, http://www.acs.org/content/acs/en/education/whatischemistry/landmarks/syntheticrubber.html (accessed December 20, 2018). Search PubMed.
- W. M. Tuttle, Technol. Cult., 2006, 22, 35 CrossRef.
- M. H. Whitlock, G. J. Haddad and E. E. Stahly, Anal. Chem., 1947, 19, 767–770 CrossRef CAS.
- W. M. Quattlebaum, W. J. Toussaint and J. T. Dunn, J. Am. Chem. Soc., 1947, 1491, 593–599 CrossRef.
- H. E. Jones, E. E. Stahly and B. B. Corson, J. Am. Chem. Soc., 1949, 71, 1822–1828 CrossRef CAS.
- M. Jones, C. Keir, C. Iulio, R. Robertson, C. Williams and D. Apperley, Catal. Sci. Technol., 2011, 1, 267 RSC.
- W. C. White, Chem.-Biol. Interact., 2007, 166, 10–14 CrossRef CAS PubMed.
- J. Dargay, D. Gately and M. Sommer, Energy J., 2007, 28, 143–170 Search PubMed.
- H. Huo, M. Wang, L. Johnson and D. He, Transp. Res. Rec.: J. Transp. Res. Board, 2007, 2038, 69–77 CrossRef.
- T. Ouhadi, S. Abdou-Sabet, H.-G. Wussow, L. M. Ryan, L. Plummer, F. E. Baumann, J. Lohmar, H. F. Vermeire and F. L. G. Malet, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 1–41 Search PubMed.
- J. S. Plotkin, The Continuing Quest for Butadiene, 2016, http://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/thecontinuing-quest-for-butadiene.html (Accessed on December 20, 2018) Search PubMed.
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed.
- S. E. Derosa and D. T. Allen, ACS Sustainable Chem. Eng., 2015, 3, 451–459 CrossRef CAS.
- H. Yoshimoto, presented at the Petrochemical Conclave, Gurgaon, 2013.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- T. Ren, M. K. Patel and K. Blok, Energy, 2008, 33, 817–833 CAS.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- D. J. Gielen, Y. Moriguchi and H. Yagita, J. Cleaner Prod., 2002, 10, 589–604 CrossRef.
- J. Tollefson and K. R. Weiss, Nature, 2015, 528, 315–316 CrossRef CAS PubMed.
- F. Cavani, S. Albonetti, F. Basile and A. Gandini, Chemicals and fuels from bio-based building blocks, John Wiley & Sons, 2016 Search PubMed.
- C. Angelici, M. E. Z. Velthoen, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2014, 7, 2505–2515 CrossRef CAS PubMed.
- D. Cespi, F. Passarini, I. Vassura and F. Cavani, Green Chem., 2016, 18, 1625–1638 RSC.
- M. M. Bhasin, J. H. McCain, B. V. Vora, T. Imai and P. R. Pujadó, Appl. Catal., A, 2001, 221, 397–419 CrossRef CAS.
- Y. Stallwood, Summary Statement October 18, Tokyo, 2017 Search PubMed.
- F. Jing, B. Katryniok, M. Araque, R. Wojcieszak, M. Capron, S. Paul, M. Daturi, J.-M. Clacens, F. De Campo, A. Liebens, F. Dumeignil and M. Pera-Titus, Catal. Sci. Technol., 2016, 6, 5830–5840 RSC.
- A. Al-Douri, D. Sengupta and M. M. El-Halwagi, J. Nat. Gas Sci. Eng., 2017, 45, 436–455 CrossRef CAS.
- Genomatica and Braskem produce single step butadiene through biological process, https://www.chemicals-technology.com/news/newsgenomatica-and-braskem-produce-single-step-butadiene-through-biological-process-4735614/, (accessed 28 February 2020) Search PubMed.
- Versalis and Genomatica develop bio-rubber with bio-butadiene made from sugars, https://www.chemicals-technology.com/news/newsversalis-and-genomatica-develop-bio-rubber-with-bio-butadiene-made-from-sugars-4813346/, (accessed 28 February 2020) Search PubMed.
- N. Kataoka, A. S. Vangnai, T. Tajima, Y. Nakashimada and J. Kato, J. Biosci. Bioeng., 2013, 115, 475–480 CrossRef CAS PubMed.
- H. Yim, R. Haselbeck, W. Niu, C. Pujol-Baxley, A. Burgard, J. Boldt, J. Khandurina, J. D. Trawick, R. E. Osterhout, R. Stephen, J. Estadilla, S. Teisan, H. B. Schreyer, S. Andrae, T. H. Yang, S. Y. Lee, M. J. Burk and S. Van Dien, Nat. Chem. Biol., 2011, 7, 445–452 CrossRef CAS PubMed.
- X. J. Ji, H. Huang and P. K. Ouyang, Biotechnol. Adv., 2011, 29, 351–364 CrossRef CAS PubMed.
- J. S. Plotkin, The Many Lives of BDO, https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/the-many-lives-of-bdo.html, (accessed 6 March 2019) Search PubMed.
- M. Köpke, C. Mihalcea, F. M. Liew, J. H. Tizard, M. S. Ali, J. J. Conolly, B. Al-Sinawi and S. D. Simpson, Appl. Environ. Microbiol., 2011, 77, 5467–5475 CrossRef PubMed.
- J. Daniell, M. Köpke and S. D. Simpson, Commercial biomass syngas fermentation, 2012, vol. 5 Search PubMed.
- H. Duan, Y. Yamada and S. Sato, Chem. Lett., 2016, 45, 1036–1047 CrossRef CAS.
- N. Ichikawa, S. Sato, R. Takahashi and T. Sodesawa, J. Mol. Catal. A: Chem., 2006, 256, 106–112 CrossRef CAS.
- F. Jing, B. Katryniok, S. Paul, L. Fang, A. Liebens, M. Shen, B. Hu, F. Dumeignil and M. Pera-Titus, ChemCatChem, 2017, 9, 258–262 CrossRef CAS.
- E. V. Hort and P. Taylor, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003, vol. 1 Search PubMed.
- R. E. Gannon, R. M. Manyik, C. M. Dietz, H. B. Sargent, R. O. Thribolet and R. P. Schaffer, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003, vol. 1 Search PubMed.
- A. S. Travis and E. Center, Bull. Hist. Chem., 2007, 32, 27 CAS.
- S. Sato, R. Takahashi, T. Sodesawa and N. Yamamoto, Catal. Commun., 2004, 5, 397–400 CrossRef CAS.
- S. Sato, R. Takahashi, T. Sodesawa and N. Honda, J. Mol. Catal. A: Chem., 2004, 221, 177–183 CrossRef CAS.
- H. Duan, D. Sun, Y. Yamada and S. Sato, Catal. Commun., 2014, 48, 1–4 CrossRef CAS.
- D. Tsukamoto, S. Sakami, M. Ito, K. Yamada, M. Ito and T. Yonehara, Chem. Lett., 2016, 45, 831–833 CrossRef CAS.
- L. Zhang, R. K. Singh, S. D. Z. Guo, J. Li, F. Chen, Y. He, X. Guan, Y. C. Kang and J.-K. Lee, Artificial synthetic pathway for acetoin, 2,3-butanediol, and 2-butanol production from ethanol using cell free multi-enzyme catalysis, 2017 Search PubMed.
- W. Zhang, D. Yu, X. Ji and H. Huang, Green Chem., 2012, 14, 3441 RSC.
- W. Kim, W. Shin, K. J. Lee, H. Song, H. S. Kim, D. Seung and I. N. Filimonov, Appl. Catal., A, 2016, 511, 156–167 CrossRef CAS.
- D. Song, Ind. Eng. Chem. Res., 2016, 55, 11664–11671 CrossRef CAS.
- A. Igarashi, N. Ichikawa, S. Sato, R. Takahashi and T. Sodesawa, Appl. Catal., A, 2006, 300, 50–57 CrossRef CAS.
- H. Duan, Y. Yamada and S. Sato, Appl. Catal., A, 2015, 491, 163–169 CrossRef CAS.
- M. Jacquin, N. Pacheco and C. Fauvarque-nuytten, WO2017198503A1, 2019.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- OECD-FAO Agricultural Outlook 2017–2026, OECD, 2017 Search PubMed.
- J. Baeyens, Q. Kang, L. Appels, R. Dewil, Y. Lv and T. Tan, Prog. Energy Combust. Sci., 2015, 47, 60–88 CrossRef.
- N. Kosaric, Z. Duvnjak, A. Farkas, H. Sahm, S. Bringer-Meyer, O. Goebel and D. Mayer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, pp. 1–72 Search PubMed.
- J. E. Logsdon, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2004 Search PubMed.
- J. Burla, R. Fehnel, P. Louie and P. Terpeluk, Two-step Production of 1,3-Butadiene from Ethanol, 2012 Search PubMed.
- S. Farzad, M. A. Mandegari and J. F. Görgens, Bioresour. Technol., 2017, 239, 37–48 CrossRef CAS PubMed.
- Butadiene Current Prices | Petrochemicals Prices Database, https://www.intratec.us/chemical-markets/butadiene-price, (accessed 1 January 2018) Search PubMed.
- Energy - Crude oil import prices - OECD Data, https://data.oecd.org/energy/crude-oil-import-prices.htm, (accessed 2 March 2020) Search PubMed.
- Michelin, IFPEN et Axens donnent ensemble une nouvelle dimension au projet BioButterfly | IFPEN, https://www.ifpenergiesnouvelles.fr/article/michelin-ifpen-et-axens-donnent-ensemble-nouvelle-dimension-au-projet-biobutterfly, (accessed 2 March 2020) Search PubMed.
- E. V. Makshina, W. Janssens, B. F. Sels and P. A. Jacobs, Catal. Today, 2012, 198, 338–344 CrossRef CAS.
- S. Shylesh, A. A. Gokhale, C. D. Scown, D. Kim, C. R. Ho and A. T. Bell, ChemSusChem, 2016, 9, 1462–1472 CrossRef CAS PubMed.
- V. Van Speybroeck, K. De Wispelaere, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet and M. Waroquier, Chem. Soc. Rev., 2014, 43, 7326–7357 RSC.
- F. Kapteijn, R. J. Berger and J. A. Moulijn, Handb. Heterog. Catal., 2008, 1, 1693–1714 Search PubMed.
- A. Chieregato, J. Velasquez Ochoa, C. Bandinelli, G. Fornasari, F. Cavani and M. Mella, ChemSusChem, 2015, 8, 377–388 CrossRef CAS PubMed.
- J. V. Ochoa, C. Bandinelli, O. Vozniuk, A. Chieregato, A. Malmusi, C. Recchi and F. Cavani, Green Chem., 2016, 18, 1653–1663 RSC.
- D. Dussol, N. Cadran, N. Laloue, L. Renaudot and J. M. Schweitzer, Chem. Eng. J., 2019, 123586 Search PubMed.
- V. L. Sushkevich and I. I. Ivanova, Appl. Catal., B, 2017, 215, 36–49 CrossRef CAS.
- W. E. Taifan, T. Bučko and J. Baltrusaitis, J. Catal., 2017, 346, 78–91 CrossRef CAS.
- P. Müller, S. P. Burt, A. M. Love, W. P. McDermott, P. Wolf and I. Hermans, ACS Catal., 2016, 6, 6823–6832 CrossRef.
- S. Akiyama, A. Miyaji, Y. Hayashi, M. Hiza, Y. Sekiguchi, T. Ru Koyama, A. Shiga and T. Baba, J. Catal., 2018, 359, 184–197 CrossRef CAS.
- G. Pomalaza, G. Vofo, M. Capron and F. Dumeignil, Green Chem., 2018, 20, 3203–3209 RSC.
- A. N. Kolmogorov, V. M. Tikhomirov, O. M. Vinogradova, N. P. Keier and S. Z. Roginskii, in Dokl. Akad. Nauk SSSR, 1957, vol. 112, pp. 1075–1078 Search PubMed.
- O. M. Kagan, M. Y. Lyubarskii, G. D. Podurovskaya, M. Y. Kagan, G. D. Lyubarskii and O. M. Podurovskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1947, 173–181 Search PubMed.
- J. H. Miller, L. Bui and A. Bhan, React. Chem. Eng., 2019, 4, 784–805 RSC.
- A. N. Ko and B. W. Wojciechowski, Prog. React. Kinet., 1983, 12(4), 201–262 Search PubMed.
- V. L. Sushkevich, I. I. Ivanova, V. V. Ordomsky and E. Taarning, ChemSusChem, 2014, 2527–2536 CrossRef CAS PubMed.
- G. M. Cabello González, R. Murciano, A. L. Villanueva Perales, A. Martínez, F. Vidal-Barrero and M. Campoy, Appl. Catal., A, 2019, 570, 96–106 CrossRef.
- M. Lewandowski, G. S. Babu, M. Vezzoli, M. D. Jones, R. E. Owen, D. Mattia, P. Plucinski, E. Mikolajska, A. Ochenduszko and D. C. Apperley, Catal. Commun., 2014, 49, 25–28 CrossRef CAS.
- W. Taifan, G. X. Yan and J. Baltrusaitis, Catal. Sci. Technol., 2017, 7, 4648–4668 RSC.
- N. Takezawa, C. Hanamaki and H. Kobayashi, J. Catal., 1975, 38, 101–109 CrossRef.
- J. I. Di Cosimo, V. K. Díez and C. R. Apesteguía, Appl. Catal., A, 1996, 137, 149–166 CrossRef CAS.
- C. R. Ho, S. Shylesh and A. T. Bell, ACS Catal., 2016, 6, 939–948 CrossRef CAS.
- V. L. Sushkevich, I. I. Ivanova and E. Taarning, ChemCatChem, 2013, 5, 2367–2373 CrossRef CAS.
- J. I. Di Cosimo, V. K. Díez, M. Xu, E. Iglesia and C. R. Apesteguía, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- Z. T. Wang, Y. Xu, M. El-Soda, F. R. Lucci, R. J. Madix, C. M. Friend and E. C. H. Sykes, J. Phys. Chem. C, 2017, 121, 12800–12806 CrossRef CAS.
- L. H. Chagas, C. R. V. Matheus, P. C. Zonetti and L. G. Appel, Mol. Catal., 2018, 458, 272–279 CrossRef CAS.
- T. Yan, W. Dai, G. Wu, S. Lang, M. Hunger, N. Guan and L. Li, ACS Catal., 2018, 8, 2760–2773 CrossRef CAS.
- P. Müller, S.-C. Wang, S. P. Burt and I. Hermans, ChemCatChem, 2017, 9, 3572–3582 CrossRef.
- J. N. Kondo, K. Ito, E. Yoda, F. Wakabayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 10969–10972 CrossRef CAS PubMed.
- M. A. Natal-Santiago and J. A. Dumesic, J. Catal., 1998, 175, 252–268 CrossRef CAS.
- K. I. Shimizu, K. Sugino, K. Sawabe and A. Satsuma, Chem. – Eur. J., 2009, 15, 2341–2351 CrossRef CAS PubMed.
- D. Palagin, V. L. Sushkevich and I. I. Ivanova, J. Phys. Chem. C, 2016, 120, 23566–23575 CrossRef CAS.
- T. De Baerdemaeker, M. Feyen, U. Müller, B. Yilmaz, F. S. Xiao, W. Zhang, T. Yokoi, X. Bao, H. Gies and D. E. De Vos, ACS Catal., 2015, 5, 3393–3397 CrossRef CAS.
- W. E. Taifan and J. Baltrusaitis, J. Phys. Chem. C, 2018, 122, 20894–20906 CrossRef CAS.
- M. Gao, Z. Liu, M. Zhang and L. Tong, Catal. Lett., 2014, 144, 2071–2079 CrossRef CAS.
- T. Hattori and Y. Murakami, J. Catal., 1968, 10, 114–122 CrossRef CAS.
- J. D. Lewis, S. Van De Vyver and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2015, 54, 9835–9838 CrossRef CAS PubMed.
- V. L. Sushkevich, D. Palagin and I. I. Ivanova, ACS Catal., 2015, 5, 4833–4836 CrossRef CAS.
- Z. D. Young, S. Hanspal and R. J. Davis, ACS Catal., 2016, 6, 3193–3202 CrossRef CAS.
- A. M. Hernández-Giménez, J. Ruiz-Martínez, B. Puértolas, J. Pérez-Ramírez, P. C. A. Bruijnincx and B. M. Weckhuysen, Top. Catal., 2017, 60, 1522–1536 CrossRef.
- B. Puértolas, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Appl. Catal., B, 2016, 184, 77–86 CrossRef.
- D. Fan, X. Dong, Y. Yu and M. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 25671–25682 RSC.
- X. Dong, J. Lu, Y. Yu and M. Zhang, Phys. Chem. Chem. Phys., 2018, 20, 12970–12978 RSC.
- M. Zhang, J. Zhuang and Y. Yu, Appl. Surf. Sci., 2018, 458, 1026–1034 CrossRef CAS.
- S. K. Bhattacharyya and S. K. Sanyal, J. Catal., 1967, 7, 152–158 CrossRef CAS.
- H. Niiyama, S. Morii and E. Echigoya, Bull. Chem. Soc. Jpn., 1972, 45, 655–659 CrossRef CAS.
- Y. Hayashi, S. Akiyama, A. Miyaji, Y. Sekiguchi, Y. Sakamoto, A. Shiga, T. Koyama, K. Motokura and T. Baba, Phys. Chem. Chem. Phys., 2016, 18, 25191–25209 RSC.
- G. Natta and R. Rigamonti, Chim. Ind., 1947, 29, 195–200 CAS.
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, ACS Catal., 2016, 6, 3600–3609 CrossRef CAS.
- A. Miyaji, M. Hiza, Y. Sekiguchi, S. Akiyama, A. Shiga and T. Baba, J. Jpn. Pet. Inst., 2018, 61, 171–181 CrossRef CAS.
- A. Corma, M. E. Domine and S. Valencia, J. Catal., 2003, 215, 294–304 CrossRef CAS.
- E. Plessers, G. Fu, C. Tan, D. De Vos and M. Roeffaers, Catalysts, 2016, 6, 104 CrossRef.
- C. Jiménez-Sanchidrián and J. R. Ruiz, Appl. Catal., A, 2014, 469, 367–372 CrossRef.
- B. Centers and A. Property, in Studies in Surface Science and Catalysis, 1989, vol. 51, pp. 27–213 Search PubMed.
- Y. Ono and T. Baba, Phys. Chem. Chem. Phys., 2015, 17, 15637–15654 RSC.
- J. Kiss, A. Witt, B. Meyer and D. Marx, J. Chem. Phys., 2009, 130(18), 184706 CrossRef PubMed.
- H. A. Aleksandrov, G. N. Vayssilov and N. Rösch, J. Mol. Catal. A: Chem., 2006, 256, 149–155 CrossRef CAS.
- M. Boronat, P. Concepción, A. Corma, M. T. Navarro, M. Renz and S. Valencia, Phys. Chem. Chem. Phys., 2009, 11, 2876 RSC.
- A. A. Balandin, Zh. Fiz. Khim., 1935, 6, 357 Search PubMed.
- V. Gruver, A. Sun and J. J. Fripiat, Catal. Lett., 1995, 34, 359–364 CrossRef CAS.
- R. Inoue, A. Ichikawa and K. Furukawa, Nippon Kagaku Kaishi, 1958, 61, 566–569 CAS.
- F. Kapteijn and J. A. Moulijn, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008, p. 2019 Search PubMed.
- G. O. Ezinkwo, V. F. Tretjakov, R. M. Talyshinky, A. M. Ilolov and T. A. Mutombo, Catal. Commun., 2014, 43, 207–212 CrossRef CAS.
- V. F. Tret'yakov, R. M. Talyshinskii, A. M. Ilolov, A. L. Maksimov and S. N. Khadzhiev, Pet. Chem., 2014, 54, 195–206 CrossRef.
- S. Da Ros, M. D. Jones, D. Mattia, M. Schwaab, F. B. Noronha and J. C. Pinto, Appl. Catal., A, 2017, 530, 37–47 CrossRef CAS.
- A. Tripathi, K. Faungnawakij, A. Laobuthee, S. Assabumrungrat and N. Laosiripojna, Int. J. Chem. React. Eng., 2016, 14, 945–954 CAS.
- S. K. Bhattacharyya and B. N. Avasthi, Ind. Eng. Chem. Process Des. Dev., 1963, 2, 45–51 CrossRef CAS.
- W. Janssens, E. V. Makshina, P. Vanelderen, F. De Clippel, K. Houthoofd, S. Kerkhofs, J. A. Martens, P. A. Jacobs and B. F. Sels, ChemSusChem, 2015, 8, 994–1008 CrossRef CAS.
- P. I. Kyriienko, O. V. Larina, S. O. Soloviev, S. M. Orlyk, C. Calers and S. Dzwigaj, ACS Sustainable Chem. Eng., 2017, 5, 2075–2083 CrossRef CAS.
- Z. Han, X. Li, M. Zhang, Z. Liu and M. Gao, RSC Adv., 2015, 5, 103982–103988 RSC.
- T. Yan, L. Yang, W. Dai, C. Wang, G. Wu, N. Guan, M. Hunger and L. Li, J. Catal., 2018, 367, 7–15 CrossRef CAS.
- M. Zhang, X. Tan, T. Zhang, Z. Han and H. Jiang, RSC Adv., 2018, 8, 34069–34077 RSC.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- J. A. Moulijn, A. E. van Diepen and F. Kapteijn, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- N. R. C. F. MacHado, V. Calsavara, N. G. C. Astrath, C. K. Matsuda, A. Paesano and M. L. Baesso, Fuel, 2005, 84, 2064–2070 CrossRef CAS.
- R. Johansson, S. L. Hruby, J. Rass-Hansen and C. H. Christensen, Catal. Lett., 2009, 127, 1–6 CrossRef CAS.
- K. Van der Borght, R. Batchu, V. V. Galvita, K. Alexopoulos, M.-F. Reyniers, J. W. Thybaut and G. B. Marin, Angew. Chem., Int. Ed., 2016, 55, 12817–12821 CrossRef CAS PubMed.
- T. W. Kim, J. W. Kim, S. Y. Kim, H. J. Chae, J. R. Kim, S. Y. Jeong and C. U. Kim, Chem. Eng. J., 2014, 278, 217–223 CrossRef.
- P. I. Kyriienko, O. V. Larina, S. O. Soloviev, S. M. Orlyk and S. Dzwigaj, Catal. Commun., 2016, 77, 123–126 CrossRef CAS.
- P. T. Patil, D. Liu, Y. Liu, J. Chang and A. Borgna, Appl. Catal., A, 2017, 543, 67–74 CrossRef CAS.
- A. Klein and R. Palkovits, Catal. Commun., 2016, 91, 72–75 CrossRef.
- C. Angelici, M. E. Z. Z. Velthoen, B. M. Weckhuysen and P. C. A. A. Bruijnincx, Catal. Sci. Technol., 2015, 5, 2869–2879 RSC.
- V. L. Sushkevich, I. I. Ivanova and E. Taarning, Green Chem., 2015, 17, 2552–2559 RSC.
- V. L. Dagle, M. D. Flake, T. L. Lemmon, J. S. Lopez, L. Kovarik and R. A. Dagle, Appl. Catal., B, 2018, 236, 576–587 CrossRef CAS.
- D. Chen, K. Moljord, T. Fuglerud and A. Holmen, Microporous Mesoporous Mater., 1999, 29, 191–203 CrossRef CAS.
- T. V. W. Janssens, J. Catal., 2009, 264, 130–137 CrossRef CAS.
- S. Müller, Y. Liu, M. Vishnuvarthan, X. Sun, A. C. Van Veen, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, J. Catal., 2015, 325, 48–59 CrossRef.
- W. Dai, S. Zhang, Z. Yu, T. Yan, G. Wu, N. Guan and L. Li, ACS Catal., 2017, 7, 3703–3706 CrossRef CAS.
- W. E. Taifan, Y. Li, J. P. Baltrus, L. Zhang, A. I. Frenkel and J. Baltrusaitis, ACS Catal., 2019, 9, 269–285 CrossRef CAS.
- M. Zhang, Y. Qin, X. Tan, L. Wang, Y. Yu and H. Jiang, Catal. Lett., 2020, 150, 1462–1470 CrossRef CAS.
- Q. Qian, J. Ruiz-martínez, M. Mokhtar, A. M. Asiri, S. A. Al-thabaiti, S. N. Basahel and B. M. Weckhuysen, Catal. Today, 2014, 226, 14–24 CrossRef CAS.
- F. Ferreira Madeira, K. Ben Tayeb, L. Pinard, H. Vezin, S. Maury and N. Cadran, Appl. Catal., A, 2012, 443–444, 171–180 CrossRef CAS.
- F. F. Madeira, N. S. Gnep, P. Magnoux, S. Maury and N. Cadran, Appl. Catal., A, 2009, 367, 39–46 CrossRef CAS.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- T. K. Phung and G. Busca, Chem. Eng. J., 2015, 272, 92–101 CrossRef CAS.
- C. B. Phillips and R. Datta, Ind. Eng. Chem. Res., 1997, 36, 4466–4475 CrossRef CAS.
- J. Ouyang, F. Kong, G. Su, Y. Hu and Q. Song, Catal. Lett., 2009, 132, 64–74 CrossRef CAS.
- R. Le Van Mao, T. M. Nguyen and J. Yao, Appl. Catal., 1990, 61, 161–173 CrossRef CAS.
- S. Da Ros, M. D. Jones, D. Mattia, J. C. Pinto, M. Schwaab, F. B. Noronha, S. A. Kondrat, T. C. Clarke and S. H. Taylor, ChemCatChem, 2016, 8, 2376–2386 CrossRef CAS.
- A. Takahashi, W. Xia, Q. Wu, T. Furukawa, I. Nakamura, H. Shimada and T. Fujitani, Appl. Catal., A, 2013, 467, 380–385 CrossRef CAS.
- M. Iwamoto, Catal. Today, 2015, 242, 243–248 CrossRef CAS.
- A. K. Talukdar, K. G. Bhattacharyya and S. Sivasanker, Appl. Catal., A, 1997, 148, 357–371 CrossRef CAS.
- S. Muthukumaru Pillai, M. Ravindranathan and S. Sivaram, Chem. Rev., 1986, 86, 353–399 CrossRef.
- V. R. Choudhary, P. Devadas, S. Banerjee and A. K. Kinage, Microporous Mesoporous Mater., 2001, 47, 253–267 CrossRef CAS.
- M. Benito, J. L. Sanz, R. Isabel, R. Padilla, R. Arjona and L. Daza, J. Power Sources, 2005, 151, 11–17 CrossRef CAS.
- L. V. Mattos, G. Jacobs, B. H. Davis and F. B. Noronha, Chem. Rev., 2012, 112, 4094–4123 CrossRef CAS.
- S. Cavallaro, Energy Fuels, 2000, 14, 1195–1199 CrossRef CAS.
- M. J. L. Gines and E. Iglesia, J. Catal., 1998, 176, 155–172 CrossRef CAS.
- L. Silvester, J.-F. Lamonier, J. Faye, M. Capron, R.-N. Vannier, C. Lamonier, J.-L. Dubois, J.-L. Couturier, C. Calais and F. Dumeignil, Catal. Sci. Technol., 2015, 5, 2994–3006 RSC.
- S. K. Bhattacharyya and N. D. Ganguly, J. Appl. Chem., 1962, 12, 97–104 CrossRef CAS.
- S. K. Bhattacharyya, N. D. Ganguly, B. S. K. Bhattacharyya, N. D. Ganguly and B. Ganguly-fixed-bed, J. Appl. Chem., 1962, 12, 105–110 CrossRef CAS.
- W. J. Toussaint and J. T. Dunn, United States, US2421361, 1947.
- P. I. Kyriienko, O. V. Larina, N. O. Popovych, S. O. Soloviev, Y. Millot and S. Dzwigaj, J. Mol. Catal. A: Chem., 2016, 424, 27–36 CrossRef CAS.
- V. V. Ordomsky, V. L. Sushkevich and I. I. Ivanova, J. Mol. Catal. A: Chem., 2010, 333, 85–93 CrossRef CAS.
- V. L. Sushkevich, I. I. Ivanova, S. Tolborg and E. Taarning, J. Catal., 2014, 316, 121–129 CrossRef CAS.
- M. Trejda, A. Wojtaszek, A. Floch, R. Wojcieszak, E. M. Gaigneaux and M. Ziolek, Catal. Today, 2010, 158, 170–177 CrossRef CAS.
- Y. Wang, J. D. Lewis and Y. Romn-Leshkov, ACS Catal., 2016, 6, 2739–2744 CrossRef CAS.
- S. Dzwigaj, Y. Millot and M. Che, Catal. Lett., 2010, 135, 169–174 CrossRef CAS.
- T. Ushikubo, Catal. Today, 2000, 57, 331–338 CrossRef CAS.
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef CAS.
- M. Boronat, A. Corma and M. Renz, J. Phys. Chem. B, 2006, 110, 21168–21174 CrossRef CAS PubMed.
- A. Corma, F. X. Llabrés i Xamena, C. Prestipino, M. Renz and S. Valencia, J. Phys. Chem. C, 2009, 113, 11306–11315 CrossRef CAS.
- M. Boronat, A. Corma, M. Renz and P. M. Viruela, Chem. – Eur. J., 2006, 12, 7067–7077 CrossRef CAS PubMed.
- V. L. Sushkevich, A. Vimont, A. Travert and I. I. Ivanova, J. Phys. Chem. C, 2015, 119, 17633–17639 CrossRef CAS.
- V. L. Sushkevich and I. I. Ivanova, ChemSusChem, 2016, 9, 2216–2225 CrossRef CAS PubMed.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro, R. Gounder, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- J.-M. Jehng and I. E. Wachs, J. Mol. Catal., 1991, 67, 369–387 CrossRef CAS.
- V. S. Braga, J. A. Dias, S. C. L. Dias and J. L. De Macedo, Chem. Mater., 2005, 17, 690–695 CrossRef CAS.
- M. Baltes, A. Kytökivi, B. M. Weckhuysen, R. A. Schoonheydt, P. Van Der Voort and E. F. Vansant, J. Phys. Chem. B, 2001, 105, 6211–6220 CrossRef CAS.
- P. A. Burke and E. I. Ko, J. Catal., 1991, 129, 38–46 CrossRef CAS.
- K. Tanabe and S. Okazaki, Appl. Catal., A, 1995, 133, 191–218 CrossRef CAS.
- L. Li, D. Cani and P. P. Pescarmona, Inorg. Chim. Acta, 2015, 431, 289–296 CrossRef CAS.
- M. M. Kurmach, O. V. Larina, P. I. Kyriienko, P. S. Yaremov, V. V. Trachevsky, O. V. Shvets and S. O. Soloviev, ChemistrySelect, 2018, 3, 8539–8546 CrossRef CAS.
- Y. Chen, J. L. G. Fierro, T. Tanaka and I. E. Wachs, J. Phys. Chem. B, 2003, 107, 5243–5250 CrossRef CAS.
- G. Pomalaza, P. Simon, A. Addad, M. Capron and F. Dumeignil, Green Chem., 2020, 22(8), 2558–2574 RSC.
- N. Cadran and A. Chaumonnot, WO2017009108A1, 2017.
- O. V. Larina, P. I. Kyriienko and S. O. Soloviev, Theor. Exp. Chem., 2016, 52, 51–56 CrossRef CAS.
- O. V. Larina, P. I. Kyriienko and S. O. Soloviev, Catal. Lett., 2015, 145, 1162–1168 CrossRef CAS.
- E. Marceau, X. Carrier, M. Che, O. Clause and C. Marcilly, Handb. Heterog. Catal., 2008, 1, 467–484 CAS.
- Y. Xu, Z. Liu, Z. Han and M. Zhang, RSC Adv., 2017, 7, 7140–7149 RSC.
- D. D. Dochain, A. Stýskalík and D. P. Debecker, Catalysts, 2019, 9, 1–14 CrossRef.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51(24), 5810–5831 CrossRef CAS PubMed.
- S. Higgins, W. DeSisto and D. Ruthven, Microporous Mesoporous Mater., 2009, 117, 268–277 CrossRef CAS.
- G. M. Lari, P. Y. Dapsens, D. Scholz, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2016, 18, 1249–1260 RSC.
- O. Larlus and V. P. Valtchev, Chem. Mater., 2005, 17, 881–886 CrossRef CAS.
- O. Larlus and V. Valtchev, Microporous Mesoporous Mater., 2006, 93, 55–61 CrossRef CAS.
- E. Bourgeat-Lami, F. Fajula, D. Anglerot and T. Couriers, Microporous Mesoporous Mater., 1993, 1, 237–245 CrossRef.
- S. Dzwigaj, M. J. Peltre, P. Massiani, A. Davidson, M. Che, T. Sen, S. Sivasanker and S. Mas, Chem. Commun., 1998, 87–88 RSC.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CAS.
- C. Il Ahn, C. Kim, J. W. Bae, J. Jeon, H. S. Jung, Y. B. Kim, S. Lee, J. Lee and K. S. Ha, Fuel Process. Technol., 2020, 200, 106317 CrossRef.
- V. S. Bharadwaj, M. B. Pecha, L. Bu, V. L. Dagle, R. A. Dagle and P. N. Ciesielski, Catal. Today, 2019, 338, 141–151 CrossRef CAS.
- D. Zhao, F. Zhang, B. Tu, H. Yang, C. Yu, Y. Yan and Y. Meng, J. Phys. Chem. B, 2005, 109, 8723–8732 CrossRef.
- S. A. Akhade, A. Winkelman, V. Lebarbier Dagle, L. Kovarik, S. F. Yuk, M. S. Lee, J. Zhang, A. B. Padmaperuma, R. A. Dagle, V. A. Glezakou, Y. Wang and R. Rousseau, J. Catal., 2020, 386, 30–38 CrossRef CAS.
- O. V. Larina, P. I. Kyriienko and S. O. Soloviev, Theor. Exp. Chem., 2015, 51, 244–249 CrossRef.
- L. H. Chagas, P. C. Zonetti, C. R. V. Matheus, C. R. K. Rabello, O. C. Alves and L. G. Appel, ChemCatChem, 2019, 11, 5625–5632 CrossRef CAS.
- J. Goldwasser, J. Catal., 1981, 71, 53–63 CrossRef CAS.
- F. H. Ribeiro, A. E. S. Von Wittenau, C. H. Bartholomew and G. A. Somorjai, Catal. Rev.: Sci. Eng., 1997, 39, 49–76 CrossRef CAS.
- B. Przedpelski and T. Kuzniarz, US2357855, 1944.
- G. Natta and R. Rigamonti, Chim. Ind., 1947, 29, 239–243 CAS.
- S. V. Lebedev, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010, p. FR665917 Search PubMed.
- S. V. Lebedev, GB331482, 1930.
- S. Hanspal, Z. D. Young, H. Shou and R. J. Davis, ACS Catal., 2015, 5, 1737–1746 CrossRef CAS.
- S.-H. Chung, C. Angelici, S. O. M. Hinterding, M. Weingarth, M. Baldus, K. Houben, B. M. Weckhuysen and P. C. A. Bruijnincx, ACS Catal., 2016, 6, 4034–4045 CrossRef CAS.
- T. López, R. Gomez, M. Llanos and E. López-Salinas, Mater. Lett., 1999, 38, 283–288 CrossRef.
- C. C. Chusuei, D. C. Meier and D. Wayne Goodman, in The Chemical Physics of Solid Surfaces, 2001, pp. 373–408 Search PubMed.
- N. Nilius, M. Sterrer and M. Heyde, Defects at Oxide Surfaces, 2015, vol. 58 Search PubMed.
- M. Zhang, M. Gao, J. Chen and Y. Yu, RSC Adv., 2015, 5, 25959–25966 RSC.
- Y. V. Larichev, B. L. Moroz, V. I. Zaikovskii, S. M. Yunusov, E. S. Kalyuzhnaya, V. B. Shur and V. I. Bukhtiyarov, J. Phys. Chem. C, 2007, 111, 9427–9436 CrossRef CAS.
- T. Karasuda and K. Aika, Bull. Chem. Soc. Jpn., 1998, 71, 1999–2003 CrossRef CAS.
- S. Ardizzone, C. L. Bianchi, M. Fadoni and B. Vercelli, Appl. Surf. Sci., 1997, 119, 253–259 CrossRef CAS.
- F. Khairallah and A. Glisenti, Surf. Sci. Spectra, 2007, 13, 58–71 CrossRef.
- J. R. Ruiz, C. Jiménez-Sanchidrián, J. M. Hidalgo and J. M. Marinas, J. Mol. Catal. A: Chem., 2006, 246, 190–194 CrossRef CAS.
- V. Díez, C. R. Apesteguía and J. I. Di Cosimo, Catal. Today, 2000, 63, 53–62 CrossRef.
- O. V. Larina, P. I. Kyriienko, V. V. Trachevskii, N. V. Vlasenko and S. O. Soloviev, Theor. Exp. Chem., 2016, 51, 387–393 CrossRef CAS.
- X. Huang, Y. Men, J. Wang, W. An and Y. Wang, Catal. Sci. Technol., 2017, 7, 168–180 RSC.
- C. Angelici, F. Meirer, A. M. J. van der Eerden, H. L. Schaink, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, B. M. Weckhuysen and P. C. A. Bruijnincx, ACS Catal., 2015, 5, 6005–6015 CrossRef CAS.
- R. He, Y. Men, X. Huang, J. Wang, S. Li and X. Wang, Chem. Lett., 2018, cl.180474 Search PubMed.
- S. Da Ros, M. D. Jones, D. Mattia, M. Schwaab, E. Barbosa-Coutinho, R. C. Rabelo-Neto, F. Bellot Noronha and J. Carlos Pinto, Chem. Eng. J., 2016, 308, 988–1000 CrossRef.
- Y. Sekiguchi, S. Akiyama, W. Urakawa, T. R. Koyama, A. Miyaji, K. Motokura and T. Baba, Catal. Commun., 2015, 68, 20–24 CrossRef CAS.
- B. Kovařík, Collect. Czech. Chem. Commun., 1959, 24, 1260–1267 CrossRef.
- S. Li, Y. Men, J. Wang, S. Liu, X. Wang, F. Ji, S. Chai and Q. Song, Appl. Catal., A, 2019, 577, 1–9 CrossRef CAS.
- Z. M. Cui, Z. Chen, C. Y. Cao, W. G. Song and L. Jiang, Chem. Commun., 2013, 49, 6093–6095 RSC.
- C. Angelici, PhD, Utrecht University, 2015 Search PubMed.
- D. Cai, Q. Zhu, C. Chen, S. Hu, P. Qin, B. Wang and T. Tan, J. Taiwan Inst. Chem. Eng., 2018, 82, 137–143 CrossRef CAS.
- L. Chen, Z. Si, X. Wu, D. Weng, R. Ran and J. Yu, J. Rare Earths, 2014, 32, 907–917 CrossRef CAS.
- G. A. M. Hussein, J. Anal. Appl. Pyrolysis, 1996, 37, 111–149 CrossRef CAS.
- Q. Zhu, L. Yin, K. Ji, C. Li, B. Wang and T. Tan, ACS Sustainable Chem. Eng., 2020, 8, 1555–1565 CrossRef CAS.
- G. A. H. Mekhemer, Phys. Chem. Chem. Phys., 2002, 4, 5400–5405 RSC.
- A. M. Frey, S. K. Karmee, K. P. de Jong, J. H. Bitter and U. Hanefeld, ChemCatChem, 2013, 5, 594–600 CrossRef CAS.
- Z. Boukha, L. Fitian, M. López-Haro, M. Mora, J. R. Ruiz, C. Jiménez-Sanchidrián, G. Blanco, J. J. Calvino, G. A. Cifredo, S. Trasobares and S. Bernal, J. Catal., 2010, 272, 121–130 CrossRef CAS.
- Y. Zhao, S. Li, Z. Wang, S. S. Wang, S. S. Wang and X. Ma, Chin. Chem. Lett., 2019, 3–6 Search PubMed.
- R. Ohnishi, T. Akimoto and K. Tanabe, J. Chem. Soc., Chem. Commun., 1985, 1613–1614 RSC.
- R. Otomo, R. Kosugi, Y. Kamiya, T. Tatsumi and T. Yokoi, Catal. Sci. Technol., 2016, 6, 2787–2795 RSC.
- A. Martínez and E. Peris, Appl. Catal., A, 2016, 515, 32–44 CrossRef.
- T. Yashima, H. Suzuki and N. Hara, J. Catal., 1974, 33, 486–492 CrossRef CAS.
- N. La-Salvia, J. J. Lovón-Quintana and G. P. Valença, Braz. J. Chem. Eng., 2015, 32, 489–500 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |