
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong wavelength visible light-responsive SrTiO3 photocatalysts doped with valence-controlled Ru for sacrificial H2 and O2 evolution†
Sho
Suzuki
a,
Akihide
Iwase
 ab and
Akihiko
Kudo
*ab
ab and
Akihiko
Kudo
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.tus.ac.jp
bPhotocatalysis International Research Center, Research Institute for Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda-shi, Chiba 278-8510, Japan
First published on 15th May 2020
Abstract
SrTiO3 doped with Ru, H2-reduced SrTiO3 doped with Ru and SrTiO3 codoped with Ru and Sb were developed as active photocatalysts for sacrificial H2 and O2 evolution under visible light irradiation. H2-Reduced SrTiO3:Ru showed the highest activity responding up to 750 nm, almost the whole range of visible light.
Photocatalytic water splitting is a promising chemical reaction to convert solar energy into storable chemical energy, so-called artificial photosynthesis.1–6 Utilization of visible light is a key issue to achieve highly efficient solar energy conversion. Accordingly, development of photocatalysts with responses to a wide range of visible light is an important research topic.
Doping of metal ions is a useful technique to make materials responsive to light with longer wavelengths.2,7 The doped metal ions form impurity levels in the forbidden band of the host material, and hence new energy gaps appear in addition to the band gap of the host material. For example, Rh-doped SrTiO3 is an established photocatalyst which is highly active for sacrificial H2 evolution under visible light irradiation.8 In addition to Rh ions, Cr, Ir, and Ni ions are known as effective dopants.9–13 We have preliminarily reported that Ru-doped SrTiO3 shows photocatalytic activities for sacrificial H2 and O2 evolution under visible light irradiation.8 However, the details of the photocatalytic properties and the band structure have not been clarified yet. It is important to improve the Ru-doped SrTiO3 by some modifications.
Codoping with a second dopant can control the oxidation number of the main dopant. For example, Rh ions are mainly doped as Rh4+ at Ti4+ sites when only Rh is doped into SrTiO3.14 In contrast, the oxidation number of the rhodium species is controlled from Rh4+ to Rh3+ by codoping of Sb5+ into SrTiO3 to maintain the charge balance, according to 2Ti4+ = Rh3+ + Sb5+.15,16 As a result, Rh,Sb-codoped SrTiO3 shows activity for water splitting under visible light irradiation,16 being different from Rh-doped SrTiO3. Thus, the oxidation number of the doped metal ions drastically affects the photocatalytic properties.
In the present study, we investigated the photocatalytic properties of Ru-doped SrTiO3 to develop and improve a photocatalyst responding to long wavelength visible light. Sb-Codoping and H2-reduction were applied to Ru-doped SrTiO3 to control the oxidation number of doped Ru. The band structures of Ru-doped SrTiO3, Ru,Sb-codoped SrTiO3, and Ru-doped SrTiO3 after H2 reduction were also discussed.
Metal ion-doped SrTiO3 was prepared by a solid-state reaction. The starting materials SrCO3 (Kanto Chemical, 99.9%), TiO2 (Soekawa Chemical, 99.9%), RuO2 (Rare Metallic, 99.9%), Sb2O5 (Nakarai Tesque, 98%), Nb2O5 (Kojundo Chemical, 99.99%) and Ta2O5 (Rare Metallic, 99.99%) were mixed in atomic ratios of Sr/Ti/Ru = 1.015![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.997:0.003 for Ru(0.3%)-doped SrTiO3, Sr/Ti/Ru/Sb = 1.015:0.9925:0.003:0.0045 for Ru(0.3%),Sb(0.45%)-codoped SrTiO3, and Sr/Ti/Ru/M = 1.015:0.991 − x:0.003:0.006 for Ru(0.3%),Nb(0.6%)- and Ru(0.3%),Ta(0.6%)-codoped SrTiO3. The mixture was calcined in an alumina crucible at 1273 K for 10 h. H2-Reduced SrTiO3 doped with Ru was prepared by reduction in one atmosphere of H2 at 473 K or 673 K for 2 h. The crystal phase of the prepared powder was analyzed on an X-ray diffractometer (Rigaku, MiniFlex) using CuKα radiation. Diffuse reflectance spectra were obtained using a UV-vis-NIR spectrometer (JASCO, Ubest-570) equipped with an integrating sphere and were converted from reflection to K–M function by the Kubelka–Munk method. Electron spin resonance (ESR) spectra were recorded at 77 K on an ESR spectrometer (JEOL, JES-FA200).
:0.997:0.003 for Ru(0.3%)-doped SrTiO3, Sr/Ti/Ru/Sb = 1.015:0.9925:0.003:0.0045 for Ru(0.3%),Sb(0.45%)-codoped SrTiO3, and Sr/Ti/Ru/M = 1.015:0.991 − x:0.003:0.006 for Ru(0.3%),Nb(0.6%)- and Ru(0.3%),Ta(0.6%)-codoped SrTiO3. The mixture was calcined in an alumina crucible at 1273 K for 10 h. H2-Reduced SrTiO3 doped with Ru was prepared by reduction in one atmosphere of H2 at 473 K or 673 K for 2 h. The crystal phase of the prepared powder was analyzed on an X-ray diffractometer (Rigaku, MiniFlex) using CuKα radiation. Diffuse reflectance spectra were obtained using a UV-vis-NIR spectrometer (JASCO, Ubest-570) equipped with an integrating sphere and were converted from reflection to K–M function by the Kubelka–Munk method. Electron spin resonance (ESR) spectra were recorded at 77 K on an ESR spectrometer (JEOL, JES-FA200).
Photocatalytic reactions of sacrificial H2 and O2 evolution were carried out using a gas-tight circulation system with a top-irradiation cell with a Pyrex window. Photocatalyst powder (0.2 g) was dispersed in an aqueous solution (120 mL) containing 10 vol% methanol as a hole scavenger and a certain amount of H2PtCl6 as a source of a Pt cocatalyst for sacrificial H2 evolution. Photocatalyst powder (0.2 g) was dispersed in an aqueous solution (120 mL) containing 20 mmol L−1 AgNO3 as an electron scavenger for sacrificial O2 evolution. The suspension was irradiated with visible light using a 300 W Xe lamp (PerkinElmer, Cermax PE300BF) with a long-pass filter (HOYA L42). The amounts of evolved gases were determined using an online gas chromatograph (Shimadzu, GC-8A, MS-5A column, TCD, Ar carrier). The apparent quantum yield (AQY) for the sacrificial O2 evolution was estimated using the following equation.
| [AQY%] = 100 × [the number of reacted holes]/[the number of incident photons] = 100 × [the number of evolved O2 molecules] × 4/[the number of incident photons] |
XRD measurements revealed that Ru(0.3%)-doped SrTiO3 (SrTiO3:Ru) was obtained without noticeable impurities (Fig. S1†), indicating that Ru ions were doped into the SrTiO3 lattice. Judging from the ionic radii of Ru3+ (68 pm, 6 coordination) and Ru4+ (62 pm, 6 coordination) compared to that of Ti4+ (60.5 pm, 6 coordination),17 the Ru ions should be doped at Ti4+ sites. In the ESR measurements, no signal was observed for non-doped SrTiO3, while Ru-doped SrTiO3 gave a small signal, as shown in Fig. 1. The intensity of the signal increased by H2-reduction and Sb-codoping, indicating that the observed ESR signal was from either Ti3+ or Ru3+. Upon considering the stability of SrTiO3, Ti3+ may not be formed by H2 reduction at 673 K. Thus, we can conclude that Ru was mainly doped as Ru4+ which is ESR inactive and was reduced to Ru3+ by H2-reduction and Sb-codoping.
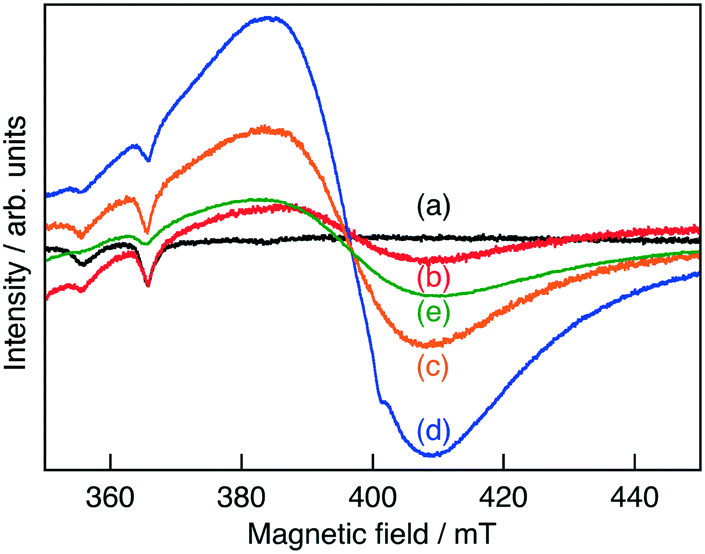 | ||
| Fig. 1 ESR spectra for Ru3+ in (a) non-doped SrTiO3, (b) SrTiO3:Ru(0.3%), SrTiO3:Ru(0.3%) after H2-reduction at (c) 473 K and (d) 673 K and (e) SrTiO3:Ru(0.3%),Sb(0.45%). | ||
The change in the oxidation number of a dopant, especially a transition metal cation with dn configuration (n = 1–9), usually affects the photoabsorption properties of the material, as observed for Rh-doped SrTiO3 and Ir-doped SrTiO3.13–16 SrTiO3:Ru possessed a wide absorption band in the visible light region in addition to the band gap absorption of the SrTiO3 host (Fig. 2b). Upon reduction with H2, the absorption at around 500–700 nm increased, while the absorption at around 400–450 nm decreased (Fig. 2c and d). On the basis of the change in the absorption profile, the absorption bands at around 500–700 nm and 400–450 nm were assigned to Ru3+- and Ru4+-related transitions, respectively. This behavior corresponded to the change in ESR signals.
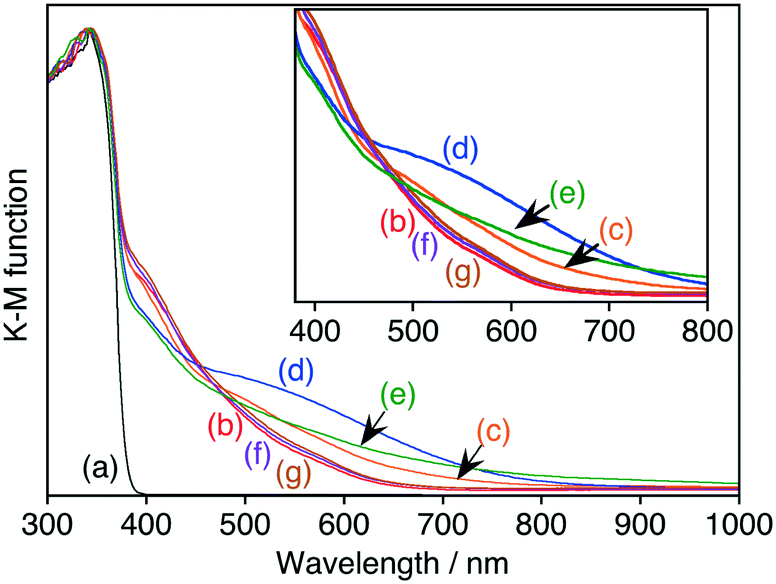 | ||
| Fig. 2 Diffuse reflectance spectra of (a) non-doped SrTiO3, (b) SrTiO3:Ru(0.3%), SrTiO3:Ru(0.3%) after H2-reduction at (c) 473 K and (d) 673 K, (e) SrTiO3:Ru(0.3%),Sb(0.45%), (f) SrTiO3:Ru(0.3%),Nb(0.6%), and (g) SrTiO3:Ru(0.3%),Ta(0.6%). | ||
Sb, Nb and Ta ions were codoped with Ru into SrTiO3 to control the Ru to be trivalent. The XRD patterns of Ru,Sb-, Ru,Nb- and Ru,Ta-codoped SrTiO3 were the same as that of SrTiO3:Ru (Fig. S1†), indicating the successful doping of Sb, Nb and Ta. Upon codoping with Sb ions, the absorption in the diffuse reflectance spectrum at around 500–800 nm increased, whereas the absorption at around 400–450 nm decreased (Fig. 2e). This is because the doped Ru ions were controlled to be trivalent by codoping with Sb ions. In more detail, two Ti4+ ions were substituted with Ru3+ and Sb5+ ions to maintain the charge balance, according to 2Ti4+ = Ru3+ + Sb5+. Actually, the intensity of the ESR signal also increased by codoping with Sb ions (Fig. 1e). However, the intensities of the absorption at around 500–800 nm in the diffuse reflectance spectra and the ESR signal were lower than those of the sample after H2-reduction. These lower intensities indicate that Ru4+ ions still existed even in Ru,Sb-codoped SrTiO3. The profile of the diffuse reflectance spectrum of SrTiO3:Ru did not change upon codoping of either Nb or Ta ions (Fig. 2f and g). This indicates that the Nb and Ta ions do not contribute to control of the oxidation number of the doped Ru ions, being different from Sb ions. The codopants should locate close to the Ru ions to maintain the charge balance. Both Ru3+ (68 pm, 6 coordination) of the dopant and Nb5+ or Ta5+ (64 pm, 6 coordination) of the codopants possess larger ionic radii than Ti4+ (60.5 pm, 6 coordination). Therefore, it is unfavourable that Ru3+ and Nb5+ or Ta5+ are closely located to each other. In contrast, Sb5+ (60 pm, 6 coordination) possesses a slightly smaller ionic radius than Ti4+ (60.5 pm, 6 coordination). This suggests that Sb5+ can locate closely to Ru ions resulting in control of the oxidation number of the doped Ru ions compared to Nb and Ta ions.
Table 1 shows the photocatalytic activities for sacrificial H2 and O2 evolution over SrTiO3:Ru, H2-reduced SrTiO3:Ru and codoped SrTiO3:Ru under visible light irradiation. SrTiO3:Ru showed activities for both sacrificial H2 evolution and O2 evolution, as previously reported.8 H2-Reduced SrTiO3:Ru showed a higher activity for the sacrificial O2 evolution than the pristine SrTiO3:Ru and the activity increased with increasing temperature of H2 reduction.
| Photocatalysta | Activity/μmol h−1 | |
|---|---|---|
| H2b | O2c | |
| Photocatalyst: 0.2 g; reactant solution: 120 mL; light source: 300 W Xe lamp with a long-pass filter (λ > 420 nm, L42).a Prepared at 1273 K for 10 h by a solid-state reaction with 1.5 at% excess Sr.b Pt(0.3 wt%)-Cocatalyst; 10 vol% MeOH aq.c 20 mmol L−1 AgNO3 aq. | ||
| SrTiO3:Ru(0.3%) | 4.0 | 4.4 |
| SrTiO3:Ru(0.3%) with H2-red. (473 K) | 1.7 | 8.0 |
| SrTiO3:Ru(0.3%) with H2-red. (673 K) | 1.8 | 16.1 |
| SrTiO3:Ru(0.3%),Sb(0.45%) | 0.2 | 7.3 |
| SrTiO3:Ru(0.3%),Nb(0.6%) | 3.3 | 3.3 |
| SrTiO3:Ru(0.3%),Ta(0.6%) | 2.4 | 4.3 |
In contrast, the activity for the sacrificial H2 evolution was decreased by H2 reduction. The H2-reduced SrTiO3:Ru continuously produced O2 under visible light irradiation, as shown in Fig. 3. The turnover number which is the ratio of the number of reacted holes to the number of doped Ru ions is calculated to be 45 using the amount of evolved O2 (37 μmol for 4 h) and doped Ru ions (3.3 μmol in 0.2 g of SrTiO3:Ru). The activity for sacrificial O2 evolution over Ru,Sb-codoped SrTiO3 was also higher than that over SrTiO3:Ru, while the activity for sacrificial H2 evolution was lower. This trade-off between the sacrificial H2 evolution and O2 evolution for the codoped photocatalyst was similar to that for Rh,Sb-codoped SrTiO3.15,16 A doped photocatalyst with impurity levels formed by a dopant with an oxidation number stabilized by H2-reduction and Sb-codoping is sometimes not suitable for H2 evolution, as observed for Rh,Sb-codoped SrTiO3.15,16 Ru,Nb- and Ru,Ta-codoped SrTiO3 showed similar activities for sacrificial H2 and O2 evolution to that of SrTiO3:Ru. Thus, the activity for sacrificial O2 evolution increased upon increasing the rate of doped Ru3+ by H2 reduction and Sb-codoping.
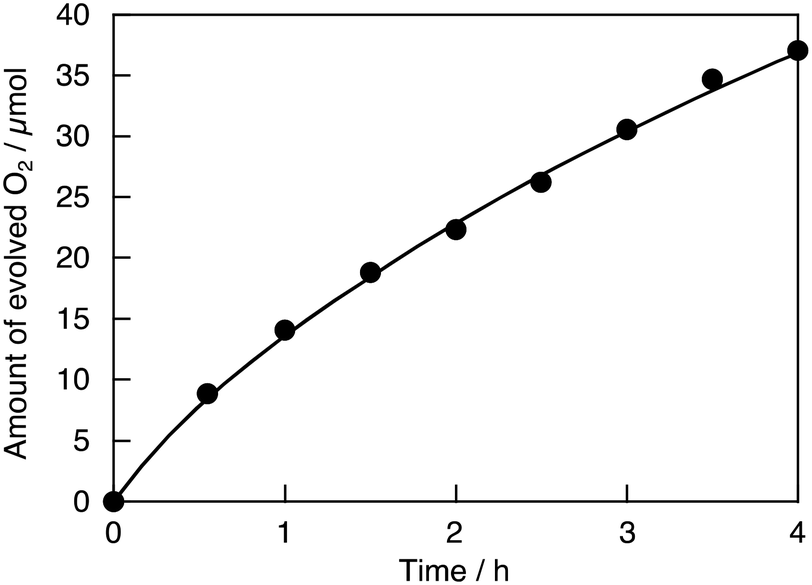 | ||
| Fig. 3 Photocatalytic O2 evolution over SrTiO3:Ru(0.3%) after H2-reduction at 673 K from an aqueous AgNO3 solution under visible light irradiation. Photocatalyst: 0.2 g; reactant solution: 20 mmol L−1 AgNO3 aq., 120 mL; light source: 300 W Xe lamp with a long-pass filter (λ > 420 nm, L42). | ||
To further understand the relationship between the doped Ru3+ ions and the activity for O2 evolution, action spectra were measured, as shown in Fig. 4. SrTiO3:Ru, H2-reduced SrTiO3:Ru and Ru,Sb-codoped SrTiO3 (SrTiO3:Ru,Sb) showed activity for sacrificial O2 evolution using light up to 660 nm (1.88 eV), 750 nm (1.65 eV) and 670 nm (1.85 eV), respectively.
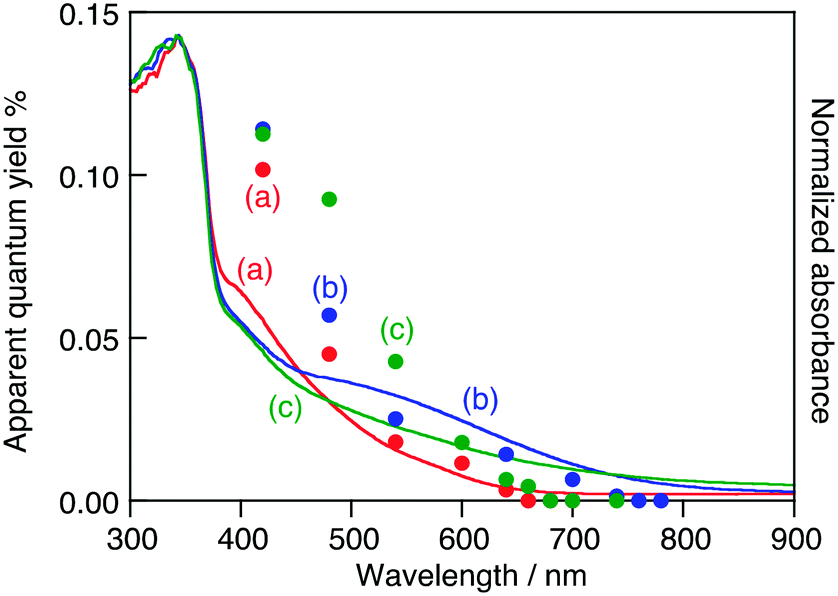 | ||
| Fig. 4 Action spectra for photocatalytic O2 evolution from an aqueous AgNO3 solution (closed circles) and diffuse reflectance spectra (solid lines) of (a) SrTiO3:Ru(0.3%), (b) SrTiO3:Ru(0.3%) after H2-reduction at 673 K and (c) SrTiO3:Ru(0.3%),Sb(0.45%). Photocatalyst: 0.2 g; reactant solution: 20 mmol L−1 AgNO3 aq., 120 mL; light source: 300 W Xe lamp with a band-pass filter; cell: top-irradiation cell with a Pyrex window. | ||
The onset of the action spectrum for the O2 evolution over the SrTiO3:Ru in which doped Ru was mainly tetravalent was similar to that of the photoanodic current of an RuO2-doped SrTiO3 photoelectrode, though the condition of the doped Ru was not clear for the photoelectrode.18 As discussed above, the corresponding absorption bands were assigned to Ru3+-related transitions. The possible band structures of SrTiO3:Ru, H2-reduced SrTiO3:Ru and SrTiO3:Ru,Sb judging from the action spectra are summarized in Fig. 5. The valence band maximum consisting of the O2p orbitals of metal oxides is generally located at around +3.0 V vs. NHE at pH 0,19 and the band levels of metal oxides shift with −0.059 V pH −1. Accordingly, the conduction band minimum and the valence band maximum of SrTiO3 with a band gap of 3.2 eV are estimated to be at −0.61 V and +2.59 V vs. NHE at pH 7, respectively. When the Ru3+-related transition is the excitation of electrons from impurity levels formed by Ru3+ to the conduction band formed by Ti3d, the potential of impurity levels formed by Ru3+ are estimated to be +1.04 V–+1.27 V vs. NHE at pH 7. The redox potential of Ag+/Ag and H2O/O2 are +0.80 V and +0.82 V vs. NHE at pH 7, respectively. Thus, photogenerated electrons in the conduction band and holes at the impurity levels possess thermodynamically enough potential to reduce Ag+ to Ag and oxidize H2O to O2, respectively. In contrast to this, assuming that the Ru3+-related transition is the excitation of electrons from the valence band formed by O2p to the impurity levels formed by Ru3+, the potential of the impurity levels are estimated to be +0.71–+0.94 V vs. NHE at pH 7.
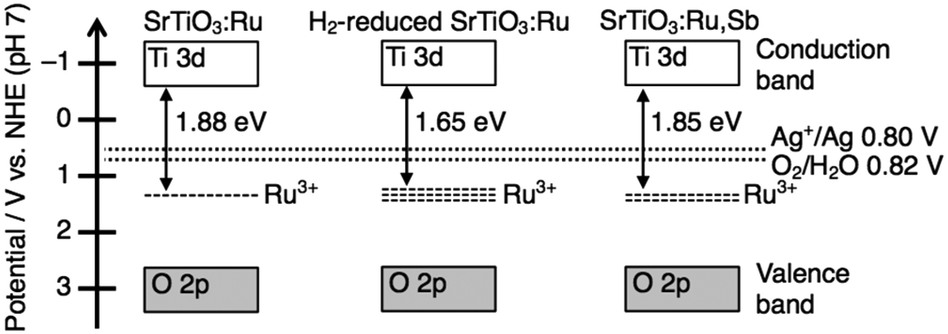 | ||
| Fig. 5 Proposed band structures of SrTiO3:Ru, H2-reduced SrTiO3:Ru and SrTiO3:Ru,Sb. | ||
These potentials are insufficient to reduce Ag+ to Ag (+0.80 V vs. NHE). Thus, we can conclude that the sacrificial O2 evolution from an aqueous AgNO3 solution proceeded by the excitation from the impurity levels formed by Ru3+ to the conduction band of SrTiO3. Although the impurity levels are formed by Ru4+, the Ru4+-related absorption does not contribute to the photocatalytic reactions, judging from the action spectra shown in Fig. 4. Sb5+ does not form impurity levels in the forbidden band of SrTiO3. Thus, the impurity levels formed by Ru4+ and Sb5+ are not described in Fig. 4 to simplify the band structure relating the photocatalytic reactions.
SrTiO3:Ru,Sb showed higher apparent quantum yields than H2-reduced SrTiO3:Ru under light irradiation at around 500 nm, while the former showed lower apparent quantum yields than the latter under light irradiation at around 650 nm, as shown in Fig. 4. The doped Ru was controlled to become Ru3+ by both H2-reduction and Sb-codoping. The advantage of the Sb-codoping at Ti4+ sites is the self-charge compensation due to the formation of Sb3+ with an excess amount of Sb2O5 in the starting material.9 The excessively doped antimony was doped as Sb3+ and Sb5+ at Ti4+ to maintain the charge balance, according to 2Ti4+ = Sb3+ + Sb5+. In contrast, oxygen vacancies were formed by H2-reduction. Therefore, SrTiO3:Ru,Sb should basically show higher apparent quantum yields than H2-reduced SrTiO3:Ru because of less defects. However, the H2-reduced SrTiO3:Ru possessed a narrower energy gap than SrTiO3:Ru,Sb resulting in absorption of more photons. Therefore, the H2-reduced SrTiO3:Ru showed higher apparent quantum yields than SrTiO3:Ru,Sb at wavelengths close to the absorption edge of H2-reduced SrTiO3:Ru at around 650 nm.
There is a negative correlation between the order of the energy gap (SrTiO3:Ru > SrTiO3:Ru,Sb > H2-reduced SrTiO3:Ru) and the order of the rate of Ru3+ ions (SrTiO3:Ru < SrTiO3:Ru,Sb < H2-reduced SrTiO3:Ru) judging from the ESR spectra (Fig. 1). When the rate of Ru3+ increases, the impurity levels formed by Ru3+ become wide due to the increased density of states. Accordingly, the energy gap between the conduction band and the impurity levels formed by Ru3+ becomes narrow, as shown in Fig. 5. The widened impurity levels are also considered to be favorable for migration of photogenerated holes. Thus, the possible reasons why H2-reduced SrTiO3:Ru showed higher activity for the sacrificial O2 evolution than SrTiO3:Ru and SrTiO3:Ru,Sb are the longest response wavelength and favorable impurity levels for hole migration.
Conclusions
In conclusion, the SrTiO3:Ru,Sb and H2-reduced SrTiO3:Ru photocatalysts as well as SrTiO3:Ru showed activities for sacrificial H2 and O2 evolution under visible light irradiation. Ru ions were mainly doped as tetravalent Ru in SrTiO3:Ru and the Ru4+ ions became Ru3+ ions by Sb-codoping and H2-reduction. The H2 evolution activity decreased by controlling Ru to become trivalent, while the O2 evolution activity increased. Photocatalytic reactions over SrTiO3:Ru, SrTiO3:Ru,Sb and H2-reduced SrTiO3:Ru proceeded by the excitation from the impurity levels formed by Ru3+ to the conduction band of SrTiO3. Among them, H2-reduced SrTiO3:Ru especially showed the highest O2 evolution activity and the longest response wavelength up to 750 nm, because of widening of the impurity level formed by Ru3+. Thus, we successfully developed metal oxide photocatalysts with a response to long wavelength visible light (near infrared) by Ru3+ doping. This responsive wavelength is almost the longest among those of photocatalysts which are active for O2 evolution. Thus, Ru-doping will be one strategy to develop metal oxide photocatalysts responding to a wide range of visible light. Moreover, these photocatalysts can be employed as O2-evolving photocatalysts in a Z-scheme photocatalyst system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 17H06433 and 17H06440 in Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)”, and 17H01217.References
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179 CrossRef CAS.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237 CrossRef CAS.
- T. Yamada and K. Domen, ChemEngineering, 2018, 2, 36 CrossRef.
- S. Chen, Y. Qi, C. Li, K. Domen and F. Zhang, Joule, 2018, 2, 2260 CrossRef CAS.
- A. Kudo, H. Kato and I. Tsuji, Chem. Lett., 2004, 33, 1534 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 5029 CrossRef CAS.
- T. Ishii, H. Kato and A. Kudo, J. Photochem. Photobiol., A, 2004, 163, 181 CrossRef CAS.
- R. Niishiro, H. Kato and A. Kudo, Phys. Chem. Chem. Phys., 2005, 7, 2241 RSC.
- A. Iwase and A. Kudo, Chem. Commun., 2017, 53, 6156 RSC.
- S. Suzuki, H. Matsumoto, A. Iwase and A. Kudo, Chem. Commun., 2018, 54, 10606 RSC.
- S. Kawasaki, K. Akagi, K. Nakatuji, S. Yamamoto, I. Matsuda, Y. Harada, J. Yoshinobu, F. Komori, R. Takahashi, M. Lippmaa, C. Sakai, H. Niwa, M. Oshima, K. Iwashina and A. Kudo, J. Phys. Chem. C, 2012, 116, 24445 CrossRef CAS.
- R. Niishiro, S. Tanaka and A. Kudo, Appl. Catal., B, 2014, 150, 187 CrossRef.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- M. Matsumura, M. Hiramoto and H. Tsubomura, J. Electrochem. Soc., 1983, 130, 326 CrossRef CAS.
- D. E. Scaife, Sol. Energy, 1980, 25, 41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns of the photocatalysts. See DOI: 10.1039/d0cy00600a |
| This journal is © The Royal Society of Chemistry 2020 |