
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAerobic oxidation of 1,6-hexanediol to adipic acid over Au-based catalysts: the role of basic supports
Noemi
Capece
ab,
Achraf
Sadier
a,
Camila
Palombo Ferraz
a,
Joëlle
Thuriot-Roukos
a,
Mariusz
Pietrowski
 c,
Michał
Zieliński
c,
Sébastien
Paul
a,
Fabrizio
Cavani
b and
Robert
Wojcieszak
*a
c,
Michał
Zieliński
c,
Sébastien
Paul
a,
Fabrizio
Cavani
b and
Robert
Wojcieszak
*a
aUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: Robert.wojcieszak@univ-lille.fr
bDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
cFaculty of Chemistry, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
First published on 30th March 2020
Abstract
1,6-Hexanediol is one of the most relevant building blocks originating from biomass and it transforms into adipic acid for polymer synthesis. Herein, we examine the selective oxidation of 1,6-hexanediol to adipic acid over Au-based catalysts, in the aqueous phase, under base-free conditions. Particularly, the absence of a base allows the neutralization step at the end of the reaction to be avoided. The influences of various supports (MgO, BaO, NiO, and TiO2) and substrate/gold molar ratios were studied. Under the conditions used, the leaching of Mg in the mixture of MgF2 and MgO was limited by diluting the basic sites in the support. The highest selectivity to adipic acid (43%) was achieved at 110 °C under 15 bar of air in the presence of 2 wt% Au/0.6MgF2–0.4MgO.
Introduction
Adipic acid (AA) is an important aliphatic dicarboxylic acid with the highest world production and is mostly used as a starting material in the synthesis of polyamide (nylon-6,6), fibers, adipic esters as plasticizers in PVC, polyurethanes, lubricants, food additives and many other applications.1,2 In 2014, 2839 kton was the global production and it is predicted to further grow with about 2% increase each year.3 It is due also to the retreat of the nylon sector over non-nylon applications.4 AA is generally synthesized by a two-step oxidation process. In the first step, the oxidation of cyclohexane to a mixture of cyclohexanone/cyclohexanol (one/Ol, KA oil) is performed in air and in the second step the oxidation of KA oil to AA with an excess of nitric acid HNO3, which is seriously harmful to the environment leading to a huge impact on global warming. Indeed, the nitrous oxide (N2O) produced in this process is an unavoidable stoichiometric waste that is commonly considered as a major contributor to global warming and ozone depletion.5 Moreover, AA can be obtained via catalytic petrochemical routes including the oxidation of cyclohexane with molecular O2,6,7 of cyclohexanol/one with air8 or hydrogen peroxide,9 of cyclohexene with hydrogen peroxide,10,11 and of n-hexane,12 or through methoxycarbonylation of 1,3-butadiene followed by hydrolysis.13 On the other hand, the bio-based production of AA has become an alternative candidate to the petrochemical process.3,14,15 In an alternative approach, Boussie et al.16 presented a two-step catalytic process for the synthesis of AA. 99% yield of AA was obtained by C–O bond hydrogenolysis of tetrahydrofuran-2,5-dicarboxylic acid (obtained by the hydrogenation of 2,5-furandicarboxylic acid, yield of 88%).16 In parallel, Boussie et al.17,18 discovered a new heterogeneous catalyst to produce AA in two steps from glucose: first, 66% of glucaric acid was obtained by oxidation of an aqueous solution of glucose in the presence of a Pt/SiO2 catalyst, followed by selective catalytic hydrodeoxygenation of glucaric acid to AA (89% yield) over a PdRh/SiO2 catalyst and a halogen source. Another route is a two-step reaction starting with the acid catalyzed ring opening of γ-valerolactone to pentenoic acid in the presence of a silica/alumina catalyst, and its subsequent conversion to AA on a palladium acetate catalyst in the presence of CO and water.19 Recently, scientific attention has been paid toward another bio-based route to produce AA by the oxidation of bio-HDO and oxidative cleavage of trans-1,2-cyclohexanediol.20 The synthesis of bio-HDO from renewable feedstocks has been disclosed using many catalytic reaction pathways: i) selective C–O bond hydrogenolysis of 2,5-tetrahydrofurandimethanol,21,22 ii) selective ring opening of the intermediate tetrahydropyran-2-methanol,21,23,24 or its sequential dehydration/hydration/hydrogenation,25 iii) hydrogenolysis of sorbitol,26,27 and (iv) hydrogenation of levoglucosenone.28 However, the HDO yield was still moderate (25–40%). The biological oxidation of HDO to AA was previously reported.29 Nevertheless, the catalytic oxidation of aldehydes or alcohols over a noble metal based catalyst in the presence of molecular oxygen is also probable.30–33 A 2.69 wt% Pt/C catalyst yielded 85% AA in 0.35 M acetic acid at 100% conversion after 24 h of reaction at 70 °C under 10 bar O2, using a molar substrate/metal ratio of 100.31 On the other hand, the oxidation of an aqueous solution of HDO (0.1 M) using 1 M NaOH at 70 °C under 10 bar of oxygen yielded 97% sodium adipate after 4 h in the presence of a 1 wt% Au/C catalyst.34 Au-Based materials present a variety of advantages over other noble metals, including high catalytic activity and stability which enable a higher selectivity to be obtained in selective oxidation processes of organic compounds in water compared with Pt- and Pd-based catalysts. These advantages appear because Au exhibits much better resistance to molecular O2 and water.35,36Furthermore, a number of previous research studies demonstrated that alloying Pd or Pt with Au enhanced significantly the catalytic activity and stability.36–41 Supported bimetallic catalysts (2wt% Au–0.1wt% Pd and 2wt% Au–0.1wt% Pt) were reported to oxidize the aqueous solution of 1,6-HDO (0.1 M) to AA with yields up to 99% under 6 bar of oxygen and 34 bar of nitrogen at 140–160 °C for 2 h.42 An Au–Pt/ZrO2 catalyst (nAu/nPt = 1 prepared by the co-wet impregnation method) was investigated in the aqueous phase oxidation of HDO (2.77 g HDO + 150 mL H2O). After 48 h at 70 °C under 40 bar of air, full conversion of HDO was achieved with a maximum yield of AA of ca. 96%.43,44 They hypothesized that the reaction pathway of HDO went through sequential oxidation (Scheme 1): first, HDO is oxidized to an intermediate 6-hydroxyhexanal (HH) which is a structural isomer of ε-caprolactol; the intermediate is subsequently oxidized to 6-hydroxyhexanoic acid (HA) which is a structural isomer of ε-caprolactone. Finally, AA was obtained by subsequent oxidation of HA via 6-oxohexanoic acid (OH).
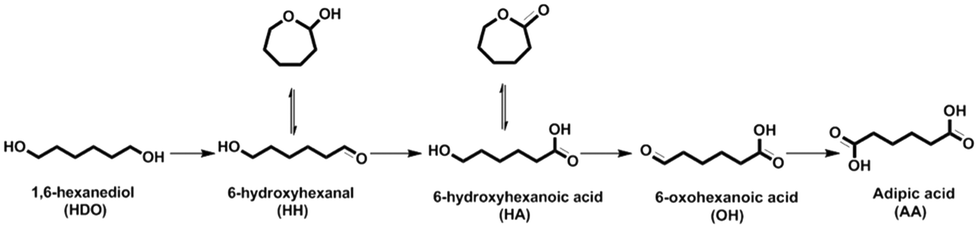 | ||
| Scheme 1 Oxidation of 1,6-hexanediol to adipic acid. | ||
The objective of this paper is to study the effect of different metal oxide supports for base-free HDO oxidation. We thus devoted our efforts to the development of an environmentally friendly process based on a base-free oxidation reaction using heterogeneous catalysts. Generally, reactions performed with base-free conditions (MgO) suffer from the leaching of Mg2+.45 Herein, we develop a new Au-based catalyst supported on MgF2–MgO mixed solid phases, in which the catalytic performances are compared with those of various catalysts supported on different metal oxides.
Results and discussion
The XRD patterns of the calcined samples at 500 °C are shown in Fig. 1. They confirmed the presence of crystallized MgF2 and MgO phases. After calcination at 500 °C, MgF2 and MgO phases were identified in the studied supports. Magnesium fluoride after calcination at 500 °C shows a typical MgF2 X-ray diffraction pattern (Sellait, PDF 41-1443) and has a tetragonal structure (P42/mnm). No other diffraction peaks are detected showing the high purity of the phase. According to the JCPDS card (JCPDS card, No. 45-0946), the phase of the powders obtained from the magnesium precursor can be indexed to a cubic MgO structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, periclase, PDF 45-496), which is consistent with the results reported in the literature.50,51 Furthermore, it is worth nothing that the XRD patterns of the Au-based catalysts (not shown here) exhibited no diffraction peaks other than those associated with the supports suggesting that a small particle size of Au is achieved after sol immobilization.
m, periclase, PDF 45-496), which is consistent with the results reported in the literature.50,51 Furthermore, it is worth nothing that the XRD patterns of the Au-based catalysts (not shown here) exhibited no diffraction peaks other than those associated with the supports suggesting that a small particle size of Au is achieved after sol immobilization.
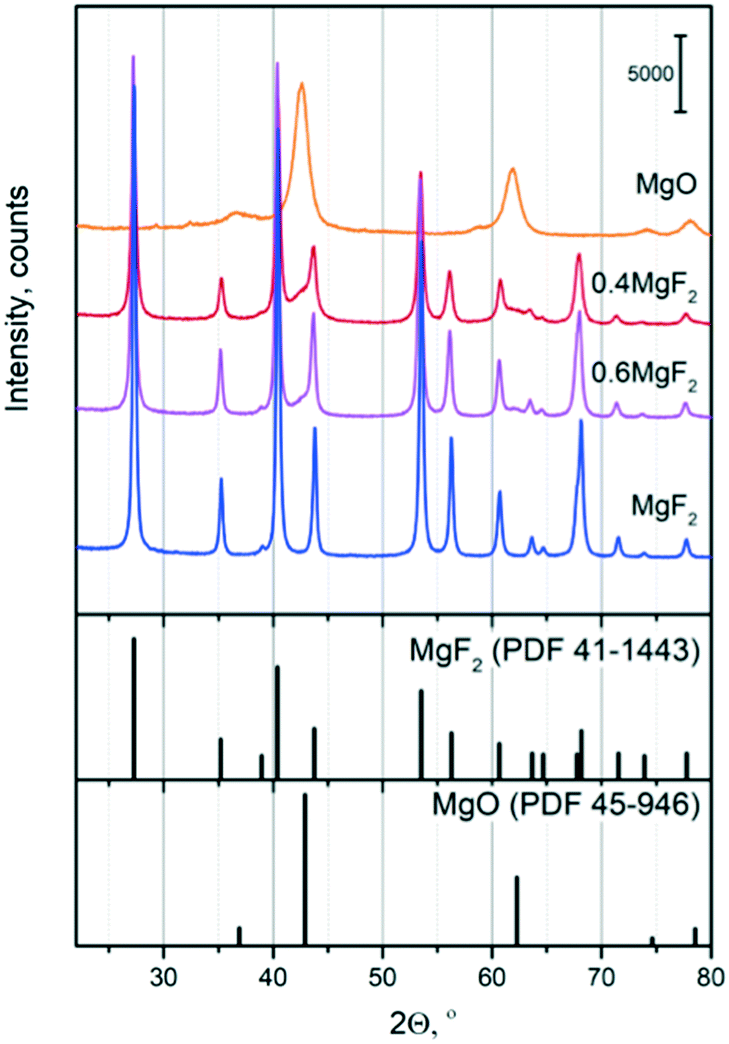 | ||
| Fig. 1 XRD patterns of the MgO, MgF2 and MgF2–MgO supports after calcination at 500 °C (top). MgF2 and MgO PDF patterns (bottom). | ||
To determine the effect of MgO in MgF2–MgO mixed solid samples on the nature of the basic sites, temperature programmed desorption of CO2 (TPD-CO2) was performed – Fig. 2.
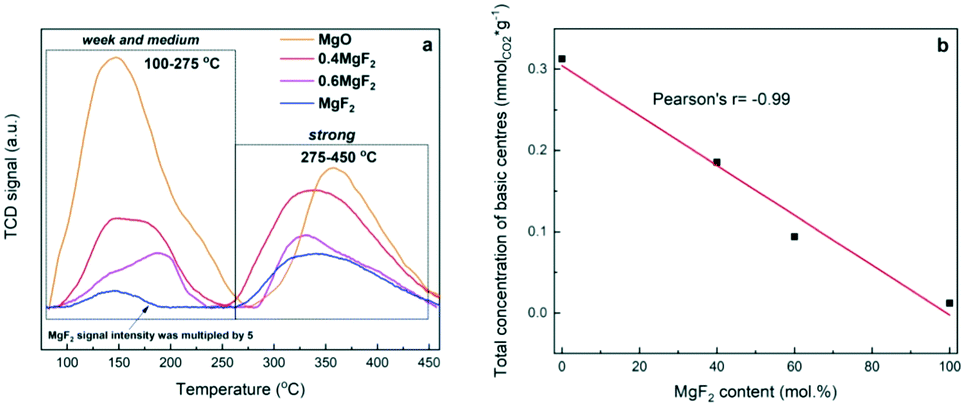 | ||
| Fig. 2 a) TPD-CO2 profiles of MgF2–MgO, MgO and MgF2 supports; b) concentration of basic centres as a function of MgF2 content in the sample. | ||
The curves recorded (Fig. 2a) for the mixed systems MgF2–MgO and those for pure MgF2 and MgO reveal different types of desorption signals, indicating the presence of basic sites of different natures and strengths:52 i) weak sites – desorption signals below 150 °C – correspond to weak sites assigned to CO2 linearly bound to OH− or correspond to bicarbonate adsorbed at weakly basic OH groups; ii) medium strong sites – desorption signals in the range 150–350 °C – are assigned to CO2 bridged with a magnesium cation and an oxygen anion; iii) strong sites – desorption signals above 350 °C – correspond to strong sites assigned to CO2 linearly bound to O2−. As reported by Aramendia et al.,52 the basic sites correspond to three desorption temperature ranges, viz. 100–145 °C (weak sites), 170–225 °C (medium sites) and 240–345 °C (strong sites). The desorption peak appearing at the lowest temperature corresponds to bicarbonate adsorbed at weakly basic OH groups. The bidentate carbonate adsorbed at Mg–O pairs shows the desorption peak in the intermediate temperature range. Finally, the peak at the highest temperature corresponds to the unidentate carbonate bound to strongly basic O2− sites. According to ref. 52, the shape of the TPD-CO2 profiles (Fig. 2a) clearly indicates the presence of weak and medium basic sites, which are responsible for the desorption peaks at about 100–275 °C, and strong sites from which CO2 is desorbed at 275–450 °C.
For pure magnesium fluoride, two desorption peaks (at 144 and 334 °C) were recorded, which indicates the presence of a small number of weak and strong basic sites. It is known from our earlier results53 that the surface area of MgF2 contains weak acidic Lewis sites (coordinatively unsaturated magnesium ions) and basic sites (OH groups). The surface of MgO contains predominantly basic centres with the maximum of CO2 desorption at 144 (weak sites) and 358 °C (strong sites), respectively. As reported by Wang et al., on the basis of CO2 adsorption measurements,54 it has been shown that the basic centres on the surface of MgO are of two types (probably O2− ions and OH− groups). As reported by Prescott et al.,55 the introduction of fluorine to MgO reduces the basic strength of O2− (OH−). Also, Wuttke et al. reported that by increasing the fluorine content of magnesium oxide fluoride, the Lewis acidity increases whereas the basicity decreases.56 This is clearly visible in the case of our systems, with increasing MgF2 content, the total concentration of basic centers decreases in the sample – Fig. 2b and Table 1. The ratio of strong to weak and medium basic centers also changes. With the increase of MgF2 in the support, the proportion of strong basic centers increases – Table 1.
| Sample | Ratio of peak surfacesa | Number of basic sites, mmolCO2 g−1 | ||
|---|---|---|---|---|
| Total | 100–275 °C, weak and medium | 275–450 °C, strong | ||
| a Ratio of the peak area at 275–450 °C to the peak area at 100–275 °C. | ||||
| MgO | 0.5 | 0.3126 | 0.2074 | 0.1052 |
| 0.4MgF2 | 1.8 | 0.1854 | 0.0663 | 0.1191 |
| 0.6MgF2 | 1.7 | 0.0940 | 0.0350 | 0.0590 |
| MgF2 | 7.4 | 0.0120 | 0.0014 | 0.0106 |
The BET surface area decreased as the content of MgO in the MgF2–MgO material decreased. The lowest average pore diameter was recorded for the bare MgO support with the highest surface area (Table 2).
| Sample | BET surface area, m2 g−1 | Total pore volume, cm3 g−1 | Average pore diameter, nm |
|---|---|---|---|
| MgO | 100 | 0.2 | 5.5 |
| 0.4MgF2 | 81 | 0.4 | 16.8 |
| 0.6MgF2 | 51 | 0.3 | 23.8 |
| MgF2 | 29 | 0.2 | 22.4 |
The TEM images of the Au-based catalysts are shown in Fig. 3. For each sample, approximately 300 particles were used to estimate the average particle size. The average particle size of PVA-stabilized Au NPs was 3 nm (not shown). The average particle sizes of Au nanoparticles were conserved after immobilization on different supports (Fig. 3). Indeed, the Au NPs deposited on the supports were monodispersed with an average size of 3.6 nm ± 0.8 nm and no significant agglomeration of gold nanoparticles was detected.
 | ||
| Fig. 3 TEM images of the (a) Au/MgO, (b) Au/NiO, (c) Au/TiO2 and (d) supported Au/0.6MgF2–0.4MgO catalysts. | ||
As mentioned in the introduction, several works have shown that the heterogeneous catalysts based on noble metals were efficient for catalytic oxidation. The effect of noble metals on HDO conversion and product selectivity is presented in Table 3. The carbon balance was always over ca. 90%. The non-complete carbon balance can be attributed to the adsorption of the intermediate aldehyde (HA), or the formation of gaseous products by decarbonylation of aldehydes, decarboxylation of acid or di-acid products, or over oxidation.43,44
| Catalyst | X HDO (%) | S HA (%) | S AA (%) | CB (%) |
|---|---|---|---|---|
| Au/MgO | 58 | 58 | 23 | 90 |
| Pd/MgO | 27 | 58 | 15 | 93 |
At 100 °C under 6 bar of O2, and for the same reaction time, Au NPs showed a higher catalytic activity (58%) toward HDO conversion compared to Pd NPs (27%). Whatever the noble metal, the main product of the reaction was the intermediate HA (SHA = 58%). This result suggests that both catalysts preferentially oxidize HDO and the HA desorption is the rate-determining step of the reaction. Moreover, AA formation was favored in the presence of the Au/MgO catalyst with a selectivity of 23%. For that reason, Au-based catalysts were chosen in the selective oxidation of HDO. Au/MgO, Au/BaO, Au/NiO and Au/TiO2 catalysts were investigated in the catalytic oxidation of HDO. The effect of the support on the catalytic activity and selectivity to reaction products is presented in Table 4. First, it is worth mentioning that the carbon balance at the defined conversions was always higher than 90%. Au/MgO showed a higher catalytic activity (XHDO = 58%) compared to the other Au-supported catalysts in the present work with 23% selectivity to AA. At 100 °C under 6 bar of O2, and for 3 h reaction time, the catalytic activity for HDO conversion and selectivity to AA followed the order: MgO > BaO > NiO > TiO2, which indicates that the active sites of the support play an important role in the oxidation reactions.57 Indeed, this was in agreement with Davis et al.58 They said that the number of hydroxyl groups on the surface of the catalyst will increase by using basic supports which thus trigger the oxidation of HDO.
| Catalyst | X HDO (%) | S HA (%) | S AA (%) | CB (%) | pHfinal |
|---|---|---|---|---|---|
| Au/TiO2 | 16 | 26 | 5 | 90 | 5.0 |
| Au/NiO | 23 | 39 | 6 | 91 | 5.9 |
| Au/BaO | 35 | 60 | 15 | 91 | 8.2 |
| Au/MgO | 58 | 58 | 23 | 90 | 9.4 |
| Au/0.6MgF2–0.4MgO | 42 | 41 | 33 | 89 | 5.6 |
The basic oxide support (MgO) reacts with water to form a weak base inorganic compound (Mg(OH)2) resulting in the increase of pH of the reaction mixture.45 In contrast, the pH of the acidic mixture will not change in the presence of much less basic TiO2.59 This result is in good agreement with the report of Ebitani et al., who investigated the oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid. They reported that catalysts using neutral or acidic supports exhibited a lower catalytic activity than those with basic supports [SiO2 < activated carbon (AC) < Al2O3 < MgO < hydrotalcite (HT)].60 Although the catalytic activity depends on the basic sites on the surface of the catalyst, by using basic oxides (NiO, BaO, MgO), hydroxide ions in aqueous solutions will be formed, dissolving part of the support. The leaching of Mg2+ from HT was detected during the oxidation of HMF in the presence of a Au/HT catalyst.61 The pH values obtained before (pHinitial = 7.6) and after catalytic tests (pHfinal = 9.4, Table 4) and ICP-OES measurements (110 ppm for Au/MgO) confirmed the dissolution of the supports; according to the literature,59 the acid formed during the reaction lowered the pH of the reaction medium, due to the acid–base reaction. Then, the formation of acidic products (AA and HA) will intensify the support dissolution resulting in lower pH values at the end of the reaction. This means that the catalysts with basic supports are very active, but they are not stable, due to the leaching of the support. Only the solution corresponding to the Au/TiO2 catalyst showed an acidic pH value with negligible leaching (15 ppm). Finally, whatever the support, HA was the main product formed. The highest selectivity (SHA = 60%) was recorded in the presence of Au/BaO.
Unfortunately, during the catalytic tests, leaching issues at the end of the reaction have been noted. Nevertheless, apparently the basic properties of the supports contribute significantly to the catalytic activity of the final materials. Their catalytic performances strongly depend on the basicity of the support. For that reason, the basicity effect was studied by changing the amount of MgO and MgF2 in MgO–MgF2 mixed phases. The effect of MgO addition to Au/MgF2 catalysts on conversion and selectivity to reaction products is shown in Fig. 4.
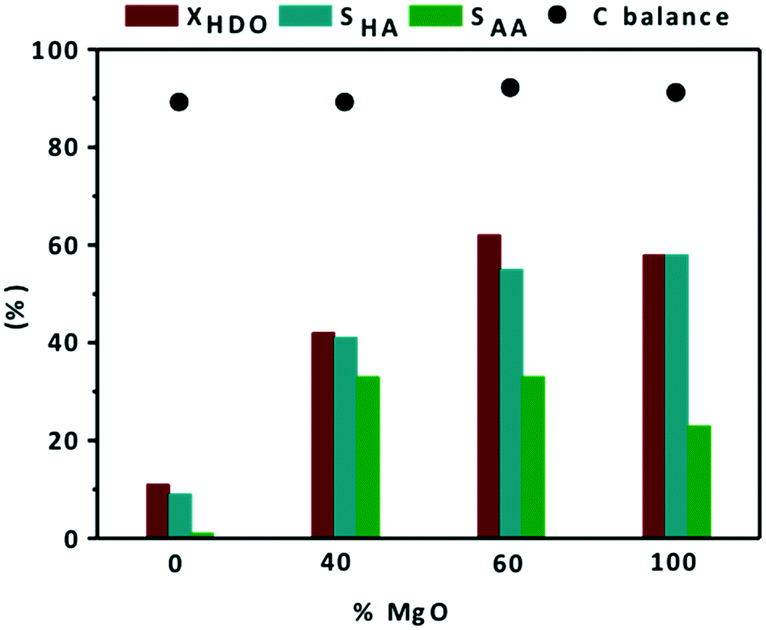 | ||
| Fig. 4 Effect of MgO addition to Au/MgF2 catalysts on conversion and selectivity to reaction products during catalytic oxidation of HDO. Reaction conditions: HDO 14 mmol L−1, 20 mL H2O, 100 °C, 6 bar O2, 600 rpm, 3 h, nHDO/nAu = 100. (■) Conversion of 1,6-hexandiol, (■) selectivity to 6-hydroxyhexanoic acid, (■) selectivity to adipic acid, and (●) carbon balance. | ||
First, it should be noted that the carbon balance at the defined conversions was always close to 90%. Au/MgF2 exhibited low catalytic activity, similar to the Au/TiO2 catalyst (11% HDO conversion) with negligible formation of AA (SAA = 1%). Over the Au/MgF2 catalyst, there was no change in the pH value at the beginning and end of the reaction suggesting the absence of leaching. Therefore, the lower catalytic performance can be assigned to the acidic reaction medium which does not favor the Au-based solid catalytic functions. Increasing the content of MgO to an certain level in the mixed phase support (MgO–MgF2) enhanced the catalytic performance (Fig. 5 and 2b).
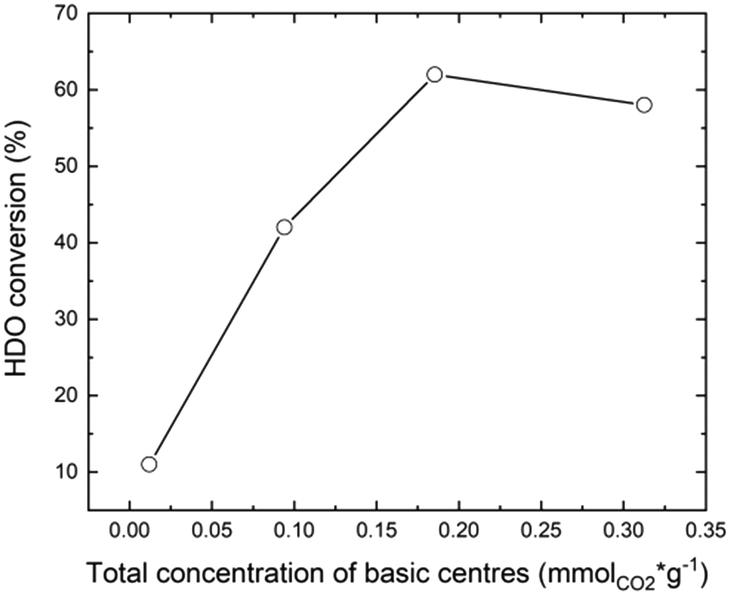 | ||
| Fig. 5 HDO conversion as a function of strong basic site quantity. | ||
The XHDO and SAA increased from 11 and 1% to 62 and 33%, respectively, when the content of MgO was increased from 0 to 60%. Surprisingly, Au/0.4MgF2−0.6MgO and Au/MgO showed similar catalytic activities (62% and 59%, respectively). However, the effect was mainly on the AA selectivity (33% vs. 23%, Fig. 4). These corroborate the synergic effect between MgF2 and MgO for the activity and selectivity to AA. These data are consistent with observations during catalytic oxidation of furfural to furoic acid at 110 °C, under 26 bar of O2, for 2 h over the Au/MgF2–MgO catalyst; they showed that upon increasing the MgO content from 0 to 60%, the conversion of furfural and yield of furoic acid increased from 20 and 5% to 95 and 99%, respectively. Under the same conditions, they also found that 60 mol% of MgO in the mixed phase material (MgF2–MgO) was already enough to provide the same catalytic activity as that of pure MgO.47,57 At the end, ICP-OES measurements showed much lower Mg leaching (82 ppm) in the case of mixed phases than in the case of MgO (110 ppm). This confirms that the leaching of Mg in the mixture of MgF2 and MgO was limited by diluting the basic sites in the support.
The effect of the HDO/Au ratio was studied using a screening pressure reactor (SPR) at 110 °C under 15 bar of air for 4 h in the presence of the Au/0.6MgF2−0.4MgO catalyst. The effect of the HDO/Au ratio on catalytic conversion and product selectivity is shown in Fig. 6. In these experiments, the total volume of water was fixed and the number of moles of HDO was adjusted by varying the amount of HDO added.
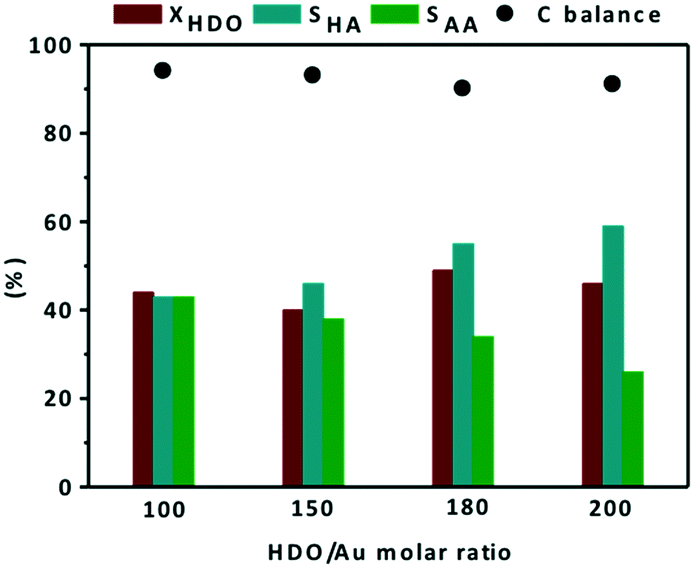 | ||
| Fig. 6 Effect of the HDO/Au molar ratio on conversion and selectivity to reaction products during catalytic oxidation of HDO. Reaction conditions: 2 mL aqueous solution of HDO (14 mmol L−1), 110 °C, 15 bar of air, 600 rpm, 4 h, 10 mg Au/0.6MgF2–0.4MgO. (■) conversion of 1,6-hexandiol, (■) selectivity to 6-hydroxyhexanoic acid, (■) selectivity to adipic acid, and (●) carbon balance. | ||
At 110 °C and under 15 bar of air, whatever the HDO/Au molar ratio, the catalytic conversion of HDO remained roughly constant (40–49%). Under these conditions, the selectivity to the monoacid compound (HA) resulting from the first oxidation of HDO increased from 43 to 59%, whereas the selectivity to the diacid compound (AA) decreased from 43 to 26%, as the HDO/Au molar ratio increased from 100 to 200. This tendency agrees with the literature dealing with the selective oxidation of 5-hydromethylfurfural (HMF) to 2,5-furandicarboxylic (FDCA) at 110 °C under 26 bar of air over an Au/MgO catalyst.47,57 In this study, they reported that the yield of the intermediate monacids [5-hydroxymethyl-2-furancarboxylic monoacid (HMFCA) and 5-formyl-2-furancaboxylic acid (FFCA)] increased from 5 to 70%, whereas the selectivity to diacid molecules [2,5-furandicarboxylic (FDCA)] decreased from 95 to 30% as the HMF/Au molar ratio increased from 100 to 400. It is worth mentioning that the highest selectivity to adipic acid (43%) at 49% conversion over the monometallic catalyst (Au/0.6MgF2−0.4MgO) is comparable with the literature when using the bimetallic Au–Pt/ZrO2 catalyst under similar reaction conditions (40% selectivity at 50% conversion).43 Finally, the mass balance in the range of HDO/Au molar ratios was >90%.
Experimental
Supports
NiO, MgO, and TiO2 were purchased from Sigma Aldrich®. The reaction between 4MgCO3·Mg(OH)2·5H2O powder (POCH – Polish Chemicals Reagents) and controlled amounts of a 40 wt% aqueous solution of HF (POCH – Polish Chemicals Reagents) formed the supports (MgF2 and MgF2–MgO) with different MgF2/MgO ratios.46,47 40, 60 and 100 mol% of MgF2 in the support were obtained by modifying the amount of HF. The resulting dense gels of MgF2-unreacted magnesium carbonate were subjected to ageing for 40 h at room temperature then dried at 80 °C for 24 h before being calcined under air flow (50 mL min−1) at 500 °C for 4 h. The MgF2–MgO samples were labeled as 0.4MgF2–0.6MgO and 0.6MgF2–0.4MgO. BaO was obtained by calcination of 1 g of BaNO3 (Sigma-Aldrich) at 600 °C for 6 h (20 °C min−1).Catalyst preparation
The supported Au catalysts were prepared by using the sol-immobilization method.48,49 2 wt% polyvinyl alcohol solution (PVA, Sigma-Aldrich, MW 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was dropped to the HAuCl4 (99.9%, Sigma-Aldrich) water solution (0.5 mM) under magnetic stirring [PVA/Au (w/w) = 1.2]. To reduce the gold particles, a solution of NaBH4 (98%, Sigma-Aldrich, 0.1 M, NaBH4/Au (mol/mol) = 5] was added to the PVA/Au solution. A light-red color was obtained indicating the reduction of gold. After 30 minutes of sol generation, the Au nanoparticles were immobilized by adding different supports (TiO2, NiO, BaO, MgO, 0.4MgF2–0.6MgO and 0.6MgF2–0.4MgO) under vigorous stirring. Then, the solid was filtered and washed with ethanol and hot water. Finally, the catalyst was dried at 100 °C for 1 h. The final loading of gold was ∼2 wt% for all the Au-containing samples, as determined by ICP analysis.
000 g mol−1) was dropped to the HAuCl4 (99.9%, Sigma-Aldrich) water solution (0.5 mM) under magnetic stirring [PVA/Au (w/w) = 1.2]. To reduce the gold particles, a solution of NaBH4 (98%, Sigma-Aldrich, 0.1 M, NaBH4/Au (mol/mol) = 5] was added to the PVA/Au solution. A light-red color was obtained indicating the reduction of gold. After 30 minutes of sol generation, the Au nanoparticles were immobilized by adding different supports (TiO2, NiO, BaO, MgO, 0.4MgF2–0.6MgO and 0.6MgF2–0.4MgO) under vigorous stirring. Then, the solid was filtered and washed with ethanol and hot water. Finally, the catalyst was dried at 100 °C for 1 h. The final loading of gold was ∼2 wt% for all the Au-containing samples, as determined by ICP analysis.
Characterization of catalysts
The powder X-ray diffraction patterns (XRD) of the samples were recorded using a Bruker AXS D8 Advance diffractometer equipped with a nickel filter, a copper tube (λKα (Cu) = 1.54184 Å) and a multi-channel fast detector. The samples were scanned at 0.014° s−1 over the range 20 ≤ 2θ ≤ 80°. The XRD data for the well-crystallized phases of MgF2 and MgO calcined at 900 °C for 4 h were used to determine the composition of the MgF2–MgO supports. Then, the content of MgO and MgF2 could be determined by comparing the intensities of the diffraction peaks of the studied samples with those of the mixture of MgF2 and MgO calcined at 900 °C, containing 85, 70 or 30 mol% of MgF2. The DQuant program was used for the calculations, taking into consideration the XRD patterns of MgO and MgF2 which exhibited diffraction peaks at 2θ = 36.92°, 42.92°, and 62.29° (MgO) and at 2θ = 35.21°, 53.48° and 60.60° (MgF2).50 The (BET) specific surface areas of the catalysts were determined by N2 adsorption at −196 °C using a Micromeritics ASAP 2010 sorptometer. The Barrett–Joyner–Halenda (BJH) method was used to determine the total pore volume and average pore radius. TEM images were obtained by using an FEI Tecnai microscope. A drop of the particles dispersed in ethanol was placed on a carbon-coated copper-grid. Elemental analysis of the Au-based catalysts was carried out by ICP-OES (inductively coupled plasma optical emission spectrometry) analysis by using Agilent 720-ES ICP-OES equipment combined with a Vulcan 42S automated digestion system. The pH value was measured for each sample using a pH meter (FE20 FiveEasy). TPD-CO2 was performed on a Micromeritics ChemiSorb 2705. Prior to CO2 adsorption, the samples were pre-treated in situ in He at 480 °C for 30 min to remove the molecules adsorbed on the surface. Carbon dioxide was adsorbed at 50 °C for 1 h. TPD-CO2 analysis was carried out in the temperature range of 50–480 °C using a ramp rate of 10 °C min−1. The desorption of CO2 was monitored with a thermal conductivity detector (TCD) and the signal intensity was normalized to 1 g for each sample studied.HDO oxidation catalytic tests
The catalytic oxidations of HDO were carried out using two catalytic systems: first, the reactions were carried out in a 30 mL TOPIndustry autoclave reactor equipped with a high precision heating system and a mechanical stirrer. In a typical experiment, an aqueous solution (20 mL) of HDO and the appropriate amount of catalyst that corresponds to the HDO/metal molar ratio of 100 were introduced into the reactor. After this, the reactor was closed and purged with a flow of oxygen. The reaction was carried out under 6 bar of O2, at 100 °C, 600 rpm for 3 h. The time necessary to reach the desired temperature was not considered in the reaction time; indeed the 3 hours of reaction started just when the system reached the right temperature. The second system is a multi-reactor system: the screening pressure reactor (SPR). It consists of 24 parallel batch reactors with a final volume of 6 mL, in which each reactor was loaded with an aqueous HDO solution (2 mL, 14 mmol L−1) and the considered Au-based catalyst (ca. 10 mg). The reaction was carried out under 15 bar of air, at 110 °C, 600 rpm for 4 h. At the end of the reactions, the autoclave was cooled, the pressure was released, and the suspension was collected and filtered with a nylon membrane filter (0.2 μm). AA, HDO, and HA were analyzed using a high performance liquid chromatograph (HPLC, Waters 2410 RJ) equipped with refractive index (RI) and UV detectors and a Rezex ROA–organic acid H+ column (∅ 7.8 mm × 300 mm). Dilute H2SO4 (5 mM, 0.5 mL min−1) was used as a mobile phase. The response factor was determined experimentally for the commercial compounds. It was therefore possible to calculate conversion (X), selectivity (S) and carbon balance (CB), according to the following formulas: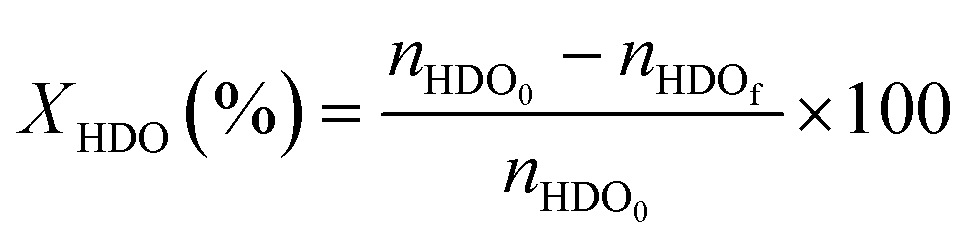
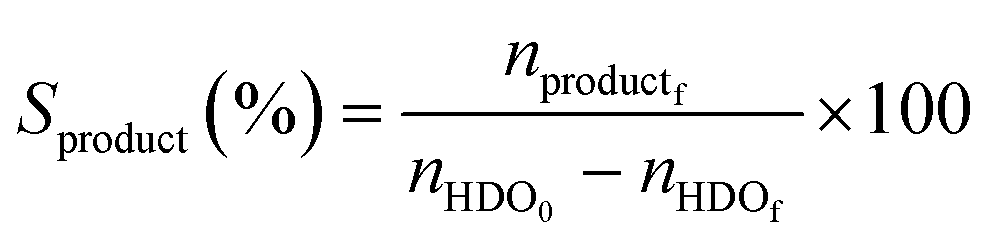
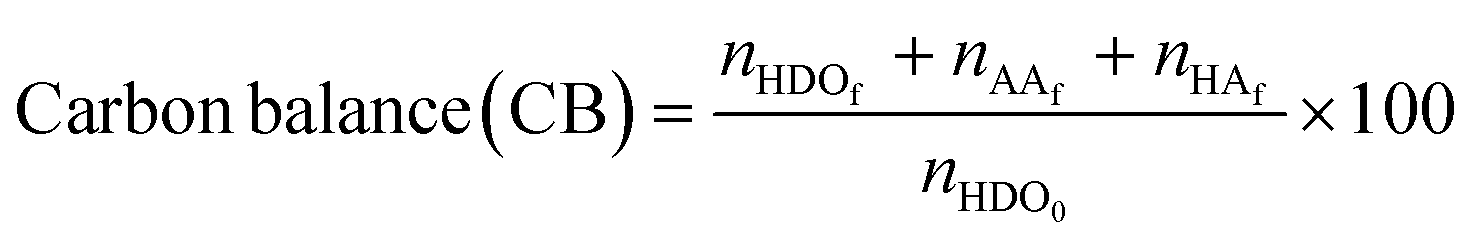 where nHDOi is the initial number of moles of HDO, nHDOf is the final number of moles of HDO, nproductf is the final number of moles of products, nAAf is the final number of moles of adipic acid, and nHAf is the final number of moles of HA.
where nHDOi is the initial number of moles of HDO, nHDOf is the final number of moles of HDO, nproductf is the final number of moles of products, nAAf is the final number of moles of adipic acid, and nHAf is the final number of moles of HA.
Conclusions
The selective aqueous phase oxidation of 1,6-hexanediol to adipic acid was investigated under base-free conditions in the presence of Au-basic oxide materials. The formation of AA followed multiple sequential oxidations of HDO via the aldehyde. The catalytic performance of Au-based catalysts prepared by the sol immobilization method showed that the conversions and selectivity to AA highly depend on the basicity of the support. Au/MgO was the most efficient catalyst among the other base oxides. However, the main drawback was the leaching of Mg. Subsequently, new Au/MgF2–MgO-based catalysts were developed in which the active MgO phase was diluted within an inactive MgF2 phase. This development limited the leaching of Mg. The highest selectivity to adipic acid (43%) was achieved at 110 °C under 15 bar of air in the presence of 2 wt% Au/0.6MgF2–0.4MgO with 100 as the HDO/Au molar ratio.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The REALCAT platform benefits from a state subsidy administrated by the French National Research Agency (ANR) within the frame of the ‘Investments for the Future’ program (PIA), with the contractual reference ‘ANR-11-EQPX-0037’. The European Union, through the FEDER funding administered by the Hauts-de-France Region, has co-financed the platform. Centrale Lille, CNRS, and the University of Lille as well as the Centrale Initiatives Foundation are thanked for their financial contributions to the acquisition and implementation of the equipment of the REALCAT platform. Chevreul Institute (FR 2638), Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation, Hauts-de-France Region and FEDER are acknowledged for supporting and partially funding this work.Notes and references
- J. P. Oppenheim and G. L. Dickerson, Kirk-Othmer Encycl. Chem. Technol., 2000, 553–582 Search PubMed.
- S. Van de Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465–1479 RSC.
- R. Beerthuis, G. Rothenberg and N. Raveendran Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- A. Mazzi, S. Paul, F. Cavani and R. Wojcieszak, ChemCatChem, 2018, 10, 3680–3682 CrossRef CAS.
- A. Rahman, M. Mupa and C. Mahamadi, Catal. Lett., 2016, 146, 788–799 CrossRef CAS.
- L.-X. Xu, C.-H. He, M.-Q. Zhu and S. Fang, Catal. Lett., 2007, 114, 202–205 CrossRef CAS.
- B. P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270, 16–25 CrossRef CAS.
- A. Dutta, M. Pramanik, A. K. Patra, M. Nandi, H. Uyama and A. Bhaumik, Chem. Commun., 2012, 48, 6738–6740 RSC.
- M. Moudjahed, L. Dermeche, S. Benadji, T. Mazari and C. Rabia, J. Mol. Catal. A: Chem., 2016, 414, 72–77 CrossRef CAS.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS PubMed.
- S.-O. Lee, R. Raja, K. D. M. Harris, J. M. Thomas, B. F. G. Johnson and G. Sankar, Angew. Chem., Int. Ed., 2003, 42, 1520–1523 CrossRef CAS PubMed.
- F. Cavani, G. Centi, S. Perathoner and F. Trifirò, Sustainable Industrial Processes, Wiley-VCH, Weinheim, 2007 Search PubMed.
- M. Röper, in Stud. Surf. Sci. Catal., Elsevier, 1991, pp. 381–429 Search PubMed.
- J. Han, Energy Convers. Manage., 2016, 129, 75–80 CrossRef CAS.
- S. Gunukula and R. P. Anex, Biofuels, Bioprod. Biorefin., 2017, 11, 897–907 CrossRef CAS.
- T. R. Boussie, E. L. Dias, Z. M. Fresco and V. J. Murphy, Production of Adipic Acid and Derivatives from Carbohydrate-Containing Materials, US Pat., 2010/0317822 to Rennovia, 2010 Search PubMed.
- G. M. Diamond, V. Murphy and T. R. Boussie, Mod. Appl. High Throughput RD Heterog. Catal., ed. A. Hagemeyer and F. Volpe, Bentham Sci. Publ., 2014, pp. 288–309 Search PubMed.
- T. R. Boussie, E. L. Dias, Z. M. Fresco, V. J. Murphy, J. Shoemaker, R. Archer and H. Jiang, Production of Adipic Acid and Derivatives from Carbohydrate-Containing Materials, US Pat., 2014/8669397 to Rennovia, 2014 Search PubMed.
- J. Q. Bond, D. M. Alonso, R. M. West and J. A. Dumesic, Langmuir, 2010, 26, 16291–16298 CrossRef CAS PubMed.
- (a) S. Solmi, E. Rozhko, A. Malmusi, T. Tabanelli, S. Albonetti, F. Basile, S. Agnoli and F. Cavani, Appl. Catal., A, 2018, 557, 89–98 CrossRef CAS; (b) E. Amadio, J. Gonzalez-Fabra, D. Carraro, W. Denis, B. Gjoka, C. Zonta, K. Bartik, F. Cavani, S. Solmi, C. Bo and G. Licini, Adv. Synth. Catal., 2018, 360, 3286–3296 CrossRef CAS.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Top. Catal., 2012, 55, 612–619 CrossRef CAS.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- K. Chen, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, ChemCatChem, 2010, 2, 547–555 CrossRef CAS.
- M. Chia, Y. J. Pagán-Torres, D. Hibbitts, Q. Tan, H. N. Pham, A. K. Datye, M. Neurock, R. J. Davis and J. A. Dumesic, J. Am. Chem. Soc., 2011, 133, 12675–12689 CrossRef CAS PubMed.
- S. P. Burt, K. J. Barnett, D. J. McClelland, P. Wolf, J. A. Dumesic, G. W. Huber and I. Hermans, Green Chem., 2017, 19, 1390–1398 RSC.
- A. Said, D. Da Silva Perez, N. Perret, C. Pinel and M. Besson, ChemCatChem, 2017, 9, 2768–2783 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- S. H. Krishna, D. J. McClelland, Q. A. Rashke, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1278–1285 RSC.
- M. Faber, Process for Producing Adipic Acid from Biomass, US Pat., 1983/4400468 to Hydrocarbon Research, 1983 Search PubMed.
- J. Xie, B. Huang, K. Yin, H. N. Pham, R. R. Unocic, A. K. Datye and R. J. Davis, ACS Catal., 2016, 6, 4206–4217 CrossRef CAS.
- M. S. Ide, D. D. Falcone and R. J. Davis, J. Catal., 2014, 311, 295–305 CrossRef CAS.
- M. S. Ide and R. J. Davis, J. Catal., 2013, 308, 50–59 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- T. Wang, M. S. Ide, M. R. Nolan, R. J. Davis and B. H. Shanks, Energy Environ. Focus, 2016, 5, 13–17 CrossRef.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CrossRef CAS.
- (a) A. Lolli, S. Albonetti, L. Utili, R. Amadori, F. Ospitali, C. Lucarelli and F. Cavani, Appl. Catal., A, 2015, 504, 408–419 CrossRef CAS; (b) S. Solmi, C. Morreale, F. Ospitali, S. Agnoli and F. Cavani, ChemCatChem, 2017, 9, 2797–2806 CrossRef CAS.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102–103, 203–212 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- A. Villa, D. Wang, D. Su, G. M. Veith and L. Prati, Phys. Chem. Chem. Phys., 2010, 12, 2183–2189 RSC.
- W. Hou, N. Dehm and R. Scott, J. Catal., 2008, 253, 22–27 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20–44 RSC.
- E. L. Dias, V. J. Murphy and J. A. W. Shoemaker, Process for Production of Adipic Acid from 1,6-Hexanediol, US Pat., 2013/0331606 to Rennovia, 2015 Search PubMed.
- M. Mounguengui-Diallo, F. Vermersch, N. Perret, C. Pinel and M. Besson, Appl. Catal., A, 2018, 551, 88–97 CrossRef CAS.
- M. Mounguengui-Diallo, A. Sadier, D. Da Silva Perez, C. Nikitine, L. Puchot, Y. Habibi, C. Pinel, N. Perret and M. Besson, New J. Chem., 2019, 43, 9873–9885 RSC.
- M. Douthwaite, X. Huang, S. Iqbal, P. J. Miedziak, G. L. Brett, S. A. Kondrat, J. K. Edwards, M. Sankar, D. W. Knight, D. Bethell and G. Hutchings, Catal. Sci. Technol., 2017, 7, 5284–5293 RSC.
- M. Zieliński, A. Kiderys, M. Pietrowski, I. Tomska-Foralewska and M. Wojciechowska, Catal. Commun., 2016, 76, 54–57 CrossRef.
- M. Zieliński, Appl. Catal., A, 2012, 449, 15–22 CrossRef.
- C. Ferraz, M. Zielinski, M. Pietrowski, S. Heyte, F. Dumeignil, L. Marcia Rossi and R. Wojcieszak, ACS Sustainable Chem. Eng., 2018, 6, 16332–16340 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. Carley, L. Prati and G. J. Hutchings, Catal. Today, 2007, 122, 317–324 CrossRef CAS.
- K. Chhor, J. F. Bocquet and C. Pommier, Mater. Chem. Phys., 1995, 40, 63–68 CrossRef CAS.
- E. Alvarado, L. M. Torres-Martinez, A. F. Fuentes and P. Quintana, Polyhedron, 2000, 19, 2345–2351 CrossRef CAS.
- (a) Z. Liu, J. A. Cortés-Concepcion, M. Mustian and M. D. Amiridis, Appl. Catal., A, 2006, 302, 232–236 CrossRef CAS; (b) M. A. Aramendia, V. Borau, C. Jimenez, J. M. Marinas, J. R. Ruiz and F. J. Urbano, Appl. Catal., A, 2003, 244, 207–215 CrossRef CAS.
- M. Wojciechowska, B. Czajka, M. Pietrowski and M. Zieliński, Catal. Lett., 2000, 66, 147–153 CrossRef CAS.
- J. A. Wang, O. Novaro and X. Bokhimi, J. Phys. Chem. B, 1997, 101, 7448–7451 CrossRef CAS.
- H. A. Prescott, Z.-J. Li, E. Kemnitz, J. Deutsch and H. Lieske, J. Mater. Chem., 2005, 15, 4616–4628 RSC.
- S. Wuttke, S. M. Coman, J. Kröhnert, F. C. Jentoft and E. Kemnitz, Catal. Today, 2010, 152, 2–10 CrossRef CAS.
- C. P. Ferraz, M. A. S. Garcia, É. Teixeira-Neto and L. M. Rossi, RSC Adv., 2016, 6, 25279–25285 RSC.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 70–74 CrossRef PubMed.
- J. A. Duffy, J. Chem. Educ., 1996, 73, 1138–1142 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- B. N. Zope, S. E. Davis and R. J. Davis, Top. Catal., 2012, 55, 24–32 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |