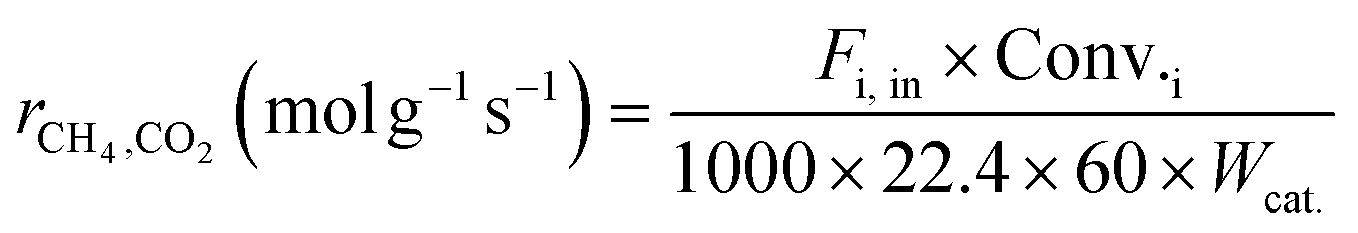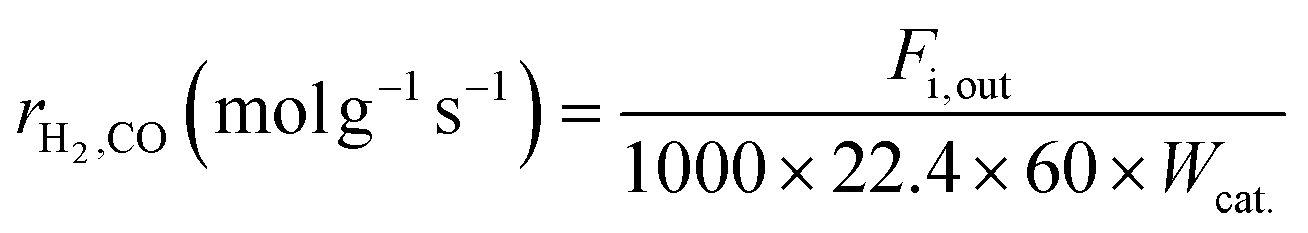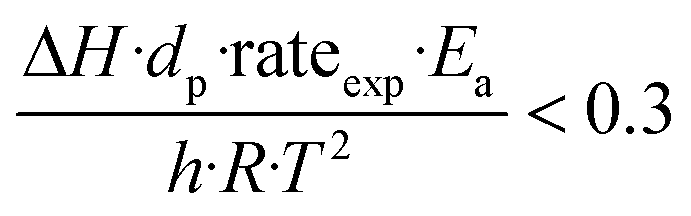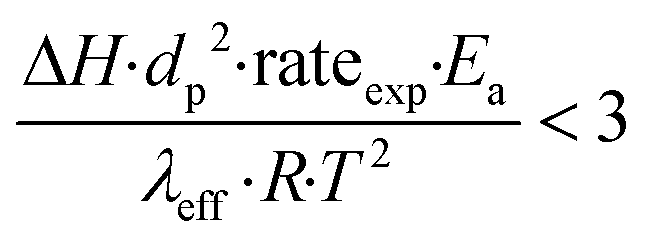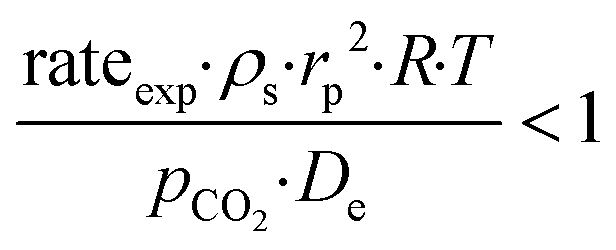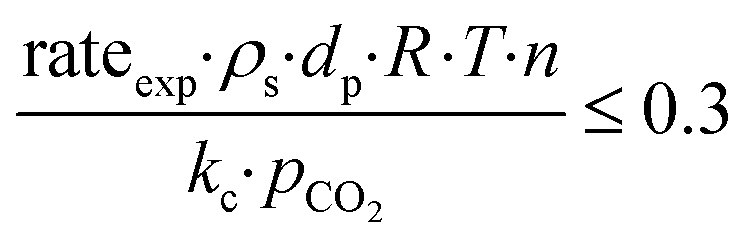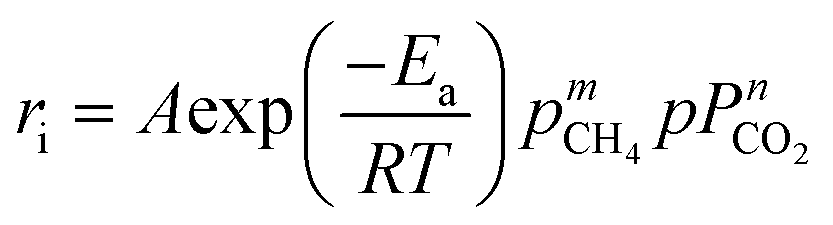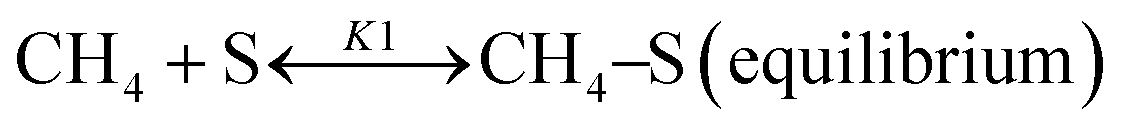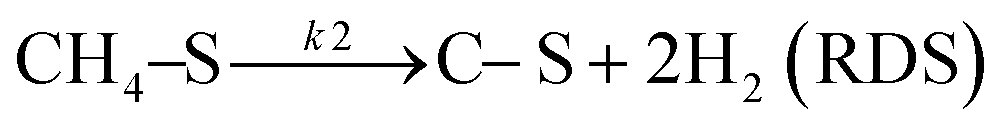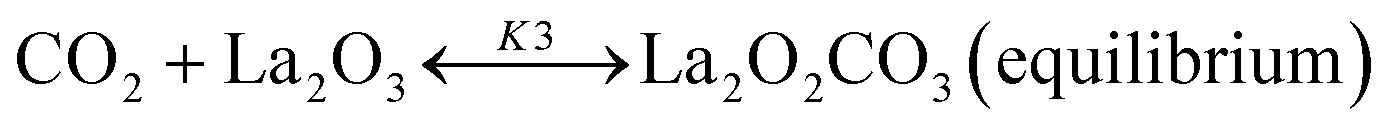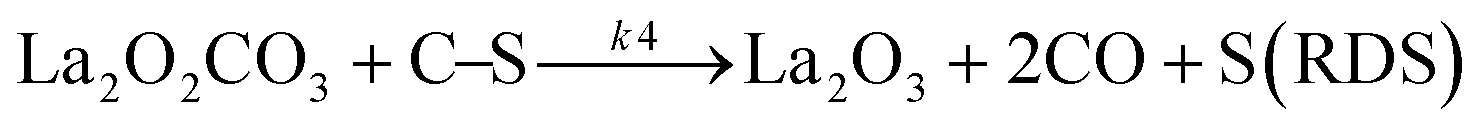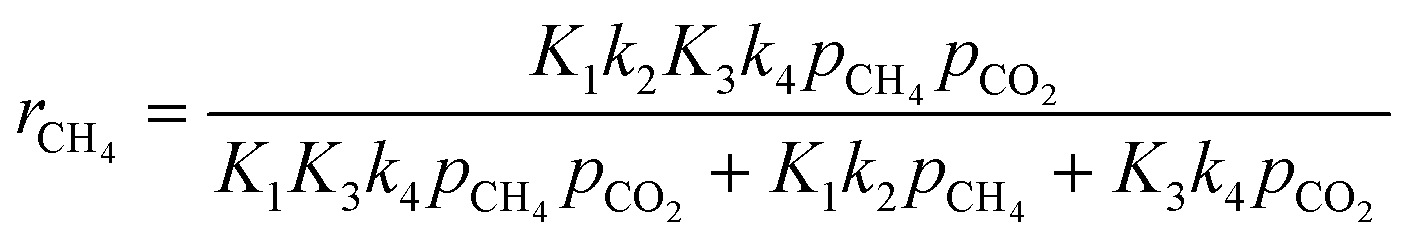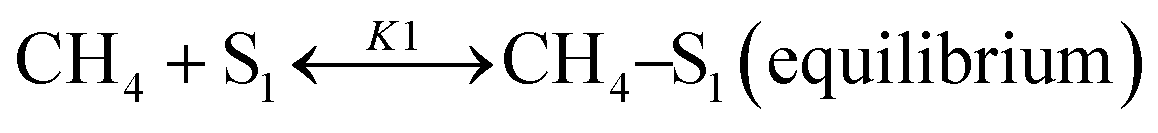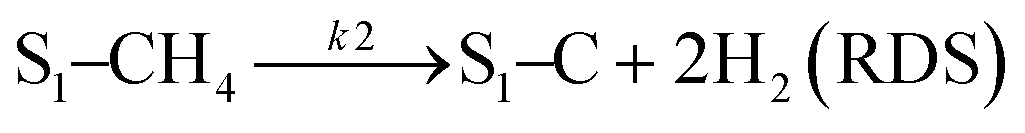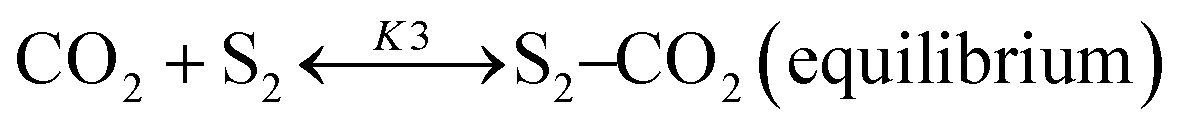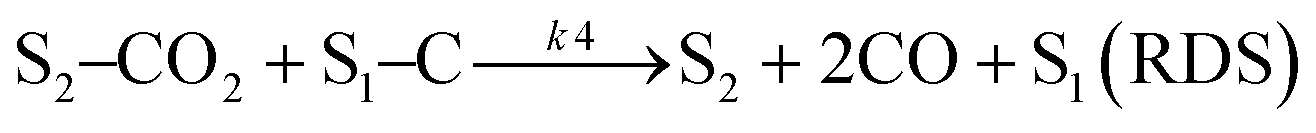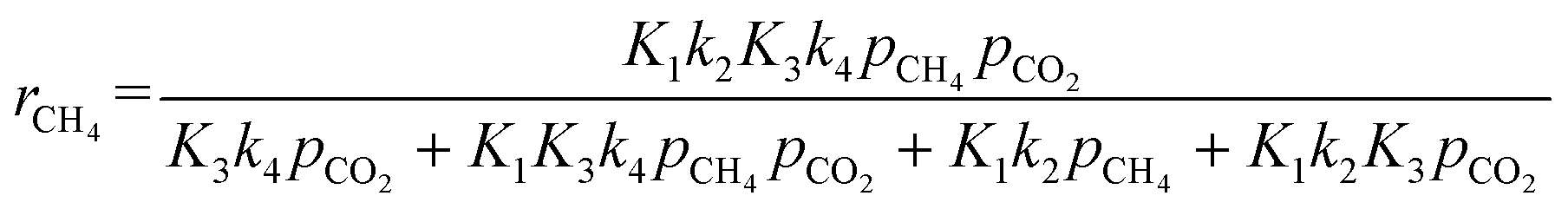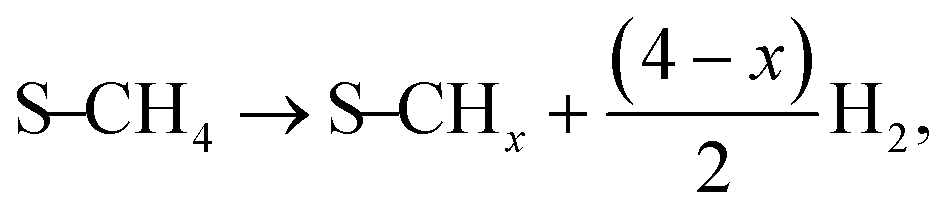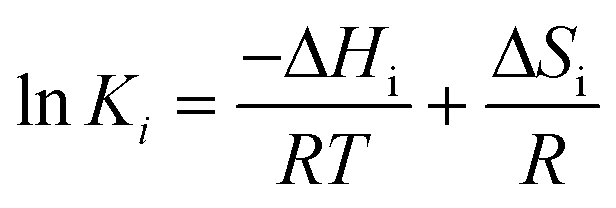Kinetic study of the methane dry (CO2) reforming reaction over the Ce0.70La0.20Ni0.10O2−δ catalyst
Received
30th October 2019
, Accepted 18th March 2020
First published on 18th March 2020
Abstract
The kinetic behaviour of the Ce0.70La0.20Ni0.10O2−δ catalyst during the methane dry reforming reaction was investigated in a fixed bed reactor in the temperature range of 923–1023 K with the partial pressure of CH4 and CO2 ranging between 5 and 50 kPa. The experimental data were fitted using the empirical power-law rate equation and Langmuir–Hinshelwood kinetic models proposed in literature for the Ni–La2O3 catalytic system and based on two-step single- and dual-site mechanisms. The obtained fitting results, after statistical and thermodynamic discrimination, showed that the mechanism of the dry reforming reaction over the Ce0.70La0.20Ni0.10O2−δ catalyst could be successfully described by the two-step dual-site mechanism. The activation energies for the CH4 consumption from the power law and Langmuir–Hinshelwood models were estimated to be 91.5 and 136.9 kJ mol−1, respectively. The lower activation energies for the CO2 consumption (70.2 and 100.6 kJ mol−1 from both models) suggested that the CO2 activation should faster than CH4. The basic nature of the Ce–La–O sites catalyzed the CO2 conversion to oxy-carbonate, decreasing the CO2 activation energy compared to that of CH4.
1. Introduction
The “Paris Agreement” signed by 195 countries under the United Nations Framework Convention on Climate Change (UNFCC) aims to reduce greenhouse gas emissions, limiting the earth to a 2 °C temperature rise.1 This ambitious perspective involves, as an energetic strategy, the conversion of the current energetic vector from petroleum to an H2-based energetic vector, which is widely considered an efficient and clean energy source.2,3 Indeed, as an energy carrier, hydrogen allows decarbonisation in many sectors like transport, industries, space heating and storage for intermittent renewable energy.4 In this context, methane dry reforming (CH4 + CO2 → 2H2 + 2CO, ΔH298 = 260.5 kJ mol−1) can be considered as a dual global warming addressing technology as it consumes and/or mitigates two greenhouse gases (CO2 and CH4), producing synthesis gas (CO and H2) with an H2/CO molar ratio closer to unity. This is adequate for the production of valuable synthetic liquid fuels and oxygenated chemicals via oxo- and Fischer–Tropsch synthesis processes.5,6
Natural gas with a high CO2 content yet very abundant in nature7,8 and biogas produced by the anaerobic digestion of biomass9,10 can represent significant sources of CO2 and CH4. Despite these advantages, methane dry reforming is not yet regarded as an industrially mature process. The main limitation lies in catalyst deactivation occurring as a result of the large amount of carbon deposition due to the high reaction temperature.11,12
Generally, two types of supported catalysts have been applied for the dry reforming reaction: noble metal (Pt, Pd, Rh, Ru, Ir, etc.)-based and transition metal (Ni, Co, Fe, etc.)-based catalysts. Although noble metals have been found to be less sensitive to coke deposition,13–16 Ni-based catalysts have been largely studied because of its abundance and low cost. However, a propensity for deactivation can be observed that normally takes place due to the sintering, cooking and phase transformation of the active metal during the reaction.17,18
Different strategies have been adopted to increase the resistance to coke deposition in Ni-based catalysts:19 modification by promoters (structural and chemical);20 selection of suitable supports (CeO2, MgO, La2O3, CeO2–ZrO2, TiO2) able to promote coke gasification by CO2 activation;21,22 the investigation of novel Ni-structures such as core–shell, core-tube and mesoporous framework.23–25 In addition, it has been evidenced that a fine-tuning and intensification of the interactions between Ni and support can induce better anti-coke and anti-sintering performance of the catalysts.26,27
The effect of interactions between the active metal and support, as a result of the formation of a solid solution, on the catalysts' performance during dry reforming reaction has been widely evaluated in literature. Excellent activity and stability have been reported by Hu for the NiO–MgO (or CoO–MgO) solid solution;28 the carbon deposition is inhibited by the isolation effect of small nickel particles formed on the catalytic surface coupled with the surface basicity of MgO. Sadykov et al. showed that a strong interaction of Ni with Ce–Zr–O oxide, as a support in the reduced state, induces a high activity and coking stability of the developed catalyst.29 Jang et al. attributed the excellent activity of the MgO-promoted Ni/Ce0.8Zr0.2O2 sample (equilibrium CH4 and CO2 conversion for 200 h without carbon deposition) to the high strength of the interactions between the components.30 Makri et al. pointed out that the metal oxide interface in the Ni/Ce0.8Pr0.2O2 catalyst plays an important role in reducing coke formation under dry reforming conditions.31
The catalytic Ni/La–Ce–Ox mixed oxide system has been widely studied by our research group and successfully applied to reforming reactions.32,33 In particular, the selected Ce0.70La0.20Ni0.10O2−δ catalyst under appropriate pre-treatment (reduction at 1023 K) exhibited promising activity and stability during the dry reforming reaction.34 The absence of carbon deposition and stable performance (CH4,conversion = 73%; CO2,conversion = 83%) for 50 h on stream at a gas hourly space velocity (GHSV) of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 h−1, were observed. In addition, a negligible amount (0.12–0.20%) was deposited at increasing GHSV (52800–105600 h−1) after H2 pre-reduction of the catalyst at 1023 K. From the characterizations of the catalyst, a complex structure has been highlighted that includes: i) a strong metal support interaction (SMSI) evidenced by the suppression of CO and H2 chemisorption; ii) the presence of oxygen vacancy defects induced by the partial incorporation of Ni2+ in the Ce–La–Ox support with partial exsolution of Ni2+ and/or La3+ ions from bulk to surface; iii) the bi-functional role of the support, where CeO2 coupled with La2O3 are involved in CO2 adsorption and coke gasification. All of these features seemed to be responsible for the observed catalytic performance.
400 h−1, were observed. In addition, a negligible amount (0.12–0.20%) was deposited at increasing GHSV (52800–105600 h−1) after H2 pre-reduction of the catalyst at 1023 K. From the characterizations of the catalyst, a complex structure has been highlighted that includes: i) a strong metal support interaction (SMSI) evidenced by the suppression of CO and H2 chemisorption; ii) the presence of oxygen vacancy defects induced by the partial incorporation of Ni2+ in the Ce–La–Ox support with partial exsolution of Ni2+ and/or La3+ ions from bulk to surface; iii) the bi-functional role of the support, where CeO2 coupled with La2O3 are involved in CO2 adsorption and coke gasification. All of these features seemed to be responsible for the observed catalytic performance.
Kinetics and mechanistic studies over Ni catalysts have been widely reported in literature, as summarized in some recent reviews.35,36 In the presence of La2O3 as a support, the proposed mechanisms include the presence of a single- or dual-active site, where the dissociation of methane on Ni particles and carbon gasification by adsorbed CO2 on the support sites are the rate-determining steps.37,38
In the current study, kinetic measurements over the Ce0.70La0.20Ni0.10O2−δ catalyst were performed. The dependency of the reactant consumption rate and product formation rate on the temperature (923–1023 K) and the reactant partial pressure (5–50 kPa) were evaluated. The results were fitted into a power-law model and the cited Langmuir–Hinshelwood models proposed in literature for the two-step single- and dual-site adsorption mechanisms.
To the best of our knowledge, the intrinsic kinetics of the CeO2–La2O3 mixed oxide-supported Ni catalyst for the dry reforming reaction have not been investigated in detail. The purpose of this study is to combine the characterizations and performance test results from previous investigations with the present kinetics data to propose a plausible kinetics model for the reaction.34
2. Experimental
2.1 Catalyst preparation
The catalyst, Ce0.70La0.20Ni0.10O2−δ (3.66 wt% Ni), was prepared by combustion synthesis as previously reported.33,34 The metal precursors of cerium ammonium nitrate ((NH4)2Ce(NO3)6·6H2O), nickel nitrate (Ni(NO3)2·6H2O), and lanthanum nitrate (La(NO3)3·6H2O) with oxalyldihydrazide as fuel (C2H6N4O2, all reagents from Aldrich) were dissolved in a minimum amount of H2O. The mixture was then transferred into an oven furnace heated at 623 K to allow water evaporation, promoting the combustion reaction. A huge amount of fumes were produced and a few seconds after ignition, a fragile foam was formed that easily crumbled into powder. The obtained sample, heated at 873 K in air to burn off the carbonaceous residues, was tableted, ground and sieved to obtain particles with the required size.
2.2 Kinetic evaluations
Kinetic studies under different conditions were carried out in a continuous-flow quartz reactor (i.d. = 6 mm) placed in an electric oven. The selection of experimental conditions (reported below) was such that the effect of the external/internal mass and heat transfer could be considered negligible. Weighted amounts of the catalyst (0.011–0.044 g) diluted with quartz of the same size (dilution 1:5) were loaded in the middle part of the reactor and supported on quartz wool. The reaction temperature was measured/controlled by two k-thermocouples. One of the thermocouples was placed inside a thermowell and centered in the catalyst bed; the other was located at the outlet of the catalytic bed. The catalyst activation before the catalytic tests was carried out in situ by treatment with a 25% H2/N2 flow (80 ml min−1) at 1023 K for 2 h, and then the temperature was maintained and/or dropped to the desired reaction temperature. The feed was controlled by mass flow controllers, and the products were analyzed with the help of a gas chromatograph (Agilent 6890 Plus) equipped with FID and TCD detectors. Kinetic measurements were performed in the temperature range of 923–1023 K, employing dilute CH4:CO2:N2 = 1:1:3 feeds. The partial pressure dependencies were evaluated by maintaining the pressure of one reactant at 20 kPa and varying the pressure of the other reactant between 5 to 50 kPa. N2, as the balance gas, was adjusted to maintain the overall pressure constant (101.32 kPa). The gas hourly space velocity was controlled at 1.2 × 106 h−1 using 0.022 g of the catalyst and appropriate feed flows to obtain significantly lower conversion (<15%) than those defined by thermodynamic equilibrium, and the reaction was controlled by kinetics. For each reactant stream and reaction temperature, the kinetic data were collected after the catalyst was stabilized for about 2 h. The reaction rates, based on the reactant consumption and product formation, were calculated by the following equations:| | 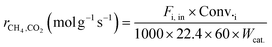 | (1) |
| | 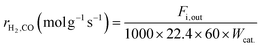 | (2) |
where Fi,in (mol min−1) is the volume flow rate of the reagent gas, Fi,out (mol min−1) is the volume flow rate of the component i in the outflow gas, Conv.i is the reagent conversion and Wcat. (g) is the weight of the catalyst.39
2.3 Diagnostic tests for mass and heat transfer limitations
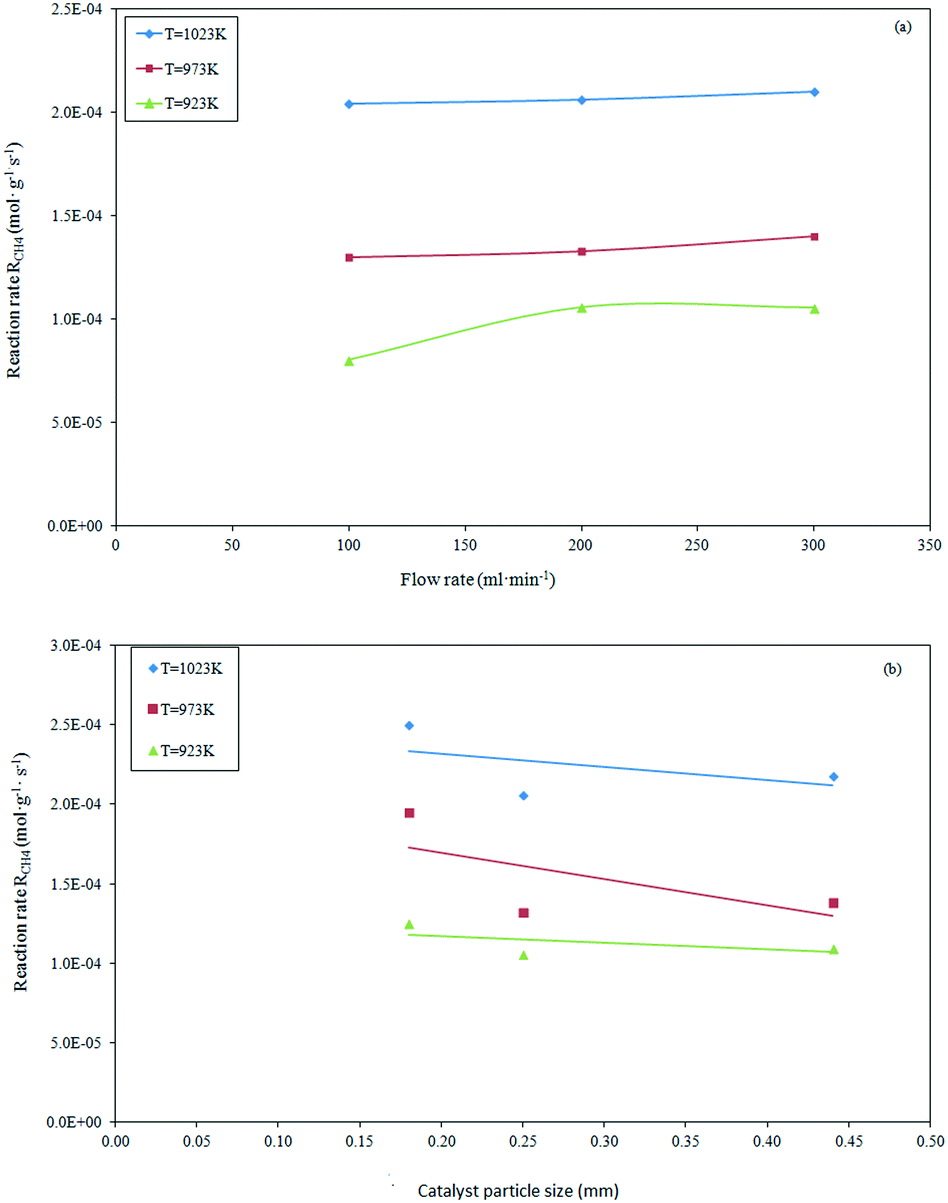
In order to evaluate the appropriate experimental conditions under which the effect of the external and internal mass transfer limitations on the methane dry reforming reaction can be considered negligible, preliminary reforming tests were performed by changing the total flow rate and catalyst particle size. The inter-particle limitation tests were performed at a GHSV of 1.2 × 106 h−1 (reaction conditions: feed composition 20/20/60 vol%; reaction temperature = 923–1023 K, pressure = 101.32 kPa), and showed an invariant rate for flow rates above 200 ml min−1. This indicated that no external limitations existed under those conditions, as shown in Fig. 1a.
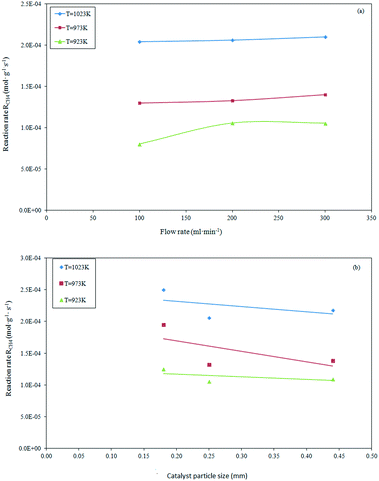 |
| | Fig. 1 Effect of volumetric feed flow rate (a) and catalyst particle size (b) on the methane consumption during dry reforming at constant GHSV = 1.2 × 106 h−1. | |
At a selected flow rate of 200 ml min−1, the absence of the intra-particle limitations was verified by using different average sizes of the catalyst particles (0.180–0.444 mm). As evidenced in Fig. 1b, the reaction rate results were almost invariant with particle sizes ranging between 0.254 to 0.444 mm. This suggests that the internal mass transfer limitations can be considered negligible. In summary, the mass transfer limitations and diffusion rate-controlling regime were fully excluded for the conditions selected to perform the catalytic tests, i.e., a flow rate of 200 ml min−1 and average particle size of 0.254 mm.
To further investigate the heat and mass transfer limitations for the reaction over the studied catalyst, suitable diagnostic criteria have been also applied. The effect of intra-/interphase heat transfer limitations on the intrinsic kinetics of the reaction was ruled out using the Mears and Anderson criteria, respectively, represented by the following notations:40,41
| | 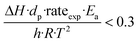 | (3) |
| | 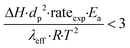 | (4) |
where Δ
H is the reaction enthalpy (260 kJ mol
−1), rate
exp is the highest reaction rate (
RCO2 = 2.07 × 10
−4 mol g
−1 s
−1),
dp is the catalyst particle diameter (254 × 10
−6 m),
Ea is the experimentally derived activation energy (70.17 kJ mol
−1),
h is the heat transfer coefficient between the gas phase and the catalyst surface (908 W m
2 K),
λeff is the catalyst thermal conductivity (11.71 W m K),
R is the universal gas constant and
T is the reaction temperature (1023 K). The derived values of 2.4 × 10
−2 and 4.6 × 10
−4 suggest that the diagnostic criteria are satisfied; thus, the heat transfer effect was not significant. In addition, the effects of the external mass transfer and intra-phase diffusion were checked by application of the Mears and Weisz–Prater criteria, respectively, that can be defined as follows:
42| | 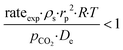 | (5) |
| | 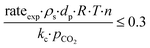 | (6) |
where
ρs is the catalyst particle density (assumed spherical, 2500 kg m
−3), n is the reaction order (0.31),
pCO2 is the partial pressure of CO
2 at the external surface (20 kPa),
kc is the mass transfer coefficient (0.206 m s
−1). Also, in this case, the derived values of 0.18 and 8.4 × 10
−2, respectively, confirm a negligible influence of the mass transfer on the reaction rate.
3. Results
3.1 Kinetic evaluations
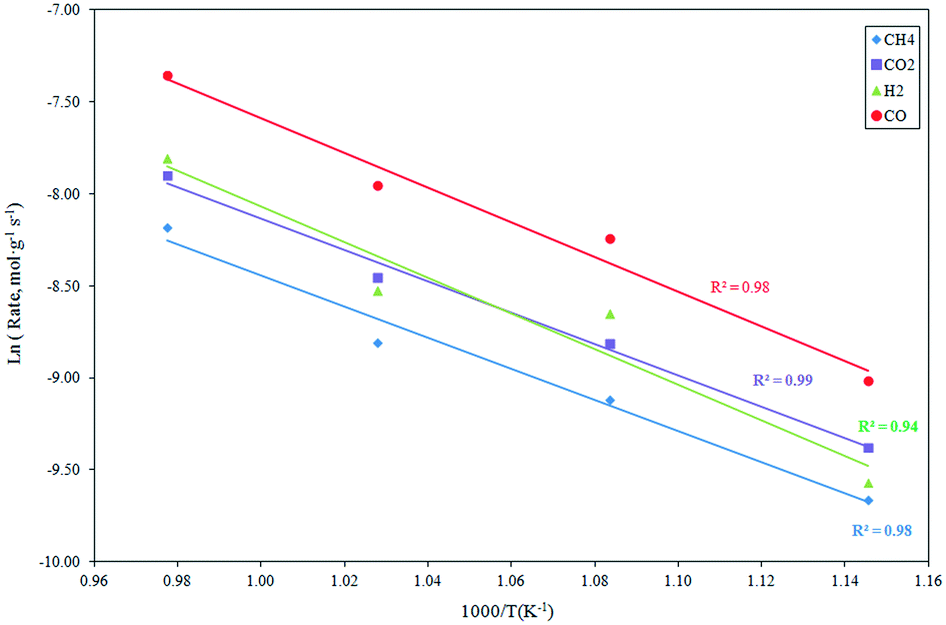
Apparent activation energy.
The effect of the temperature on the reaction rate was investigated in the temperature range 873–1023 K, employing a feed mixture consisting of CH4:CO2:N2 = 20:20:60 vol% under a pressure of 101.32 kPa. The results, in the form of an Arrhenius plot, are depicted in Fig. 2. The apparent activation energies estimated for the CH4 and CO2 consumption, as well as H2 and CO formation, are presented in Table 1 and compared with analogous data from Ni-based catalysts derived from literature. As evident, the apparent activation energy can reach very different values over the different catalytic systems applied to the reaction. The support of the Ni catalysts can significantly influence the activation energy by altering the rate-controlling step in the reaction sequence.45,49 Bradford and Vannice pointed out that the activation energies for the reforming reaction, carried out with Ni catalysts supported on different supports, were typically included in the range 30–100 kJ mol−1. Conversely, high values of up to 160 kJ mol−1 have been obtained for some classes of catalysts.50
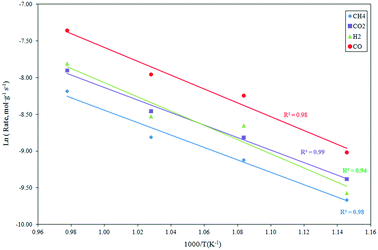 |
| | Fig. 2 Influence of the reaction temperature on the CH4 and CO2 consumption rate, as well as the H2 and CO formation rate derived at 101.32 kPa, CH4:CO2:N2 = 1:1:3 and GHSV = 1.2 × 106 h−1. | |
Table 1 Comparison of apparent activation energy (Ea, kJ mol−1) over Ni-based catalysts
| Catalyst |
Temperature (K) |
Reagent compositions (kPa) |
E
a (CH4) |
E
a (CO2) |
E
a (CO) |
E
a (H2) |
Ref. |
| CH4 |
CO2 |
N2 |
| Ce0.70La0.20Ni0.10O2−δ |
873–1023 |
20 |
20 |
60 |
70.5 |
71.0 |
78.3 |
80.4 |
This work |
| Ni/La2O3 |
773–973 |
|
|
|
68.0 |
77.0 |
|
|
38
|
| Ni° from 20 LaNi-SBA(10) |
813–893 |
50 |
50 |
— |
64.7 |
57.8 |
63.7 |
82.7 |
47
|
| Ni/CaO–Al2O3 |
893–963 |
50 |
50 |
|
106.7 |
98.8 |
103.0 |
147.4 |
44
|
| Ni15CeMgAl |
773–873 |
20 |
20 |
60 |
65.5 |
62.2 |
40.8 |
61.3 |
39
|
| 13.5Ni–2K/10CeO2–Al2O3 |
873–1073 |
20 |
20 |
60 |
46.1 |
46.2 |
47.4 |
54.0 |
45
|
| 1Ce5Ni3Mg/Al |
613–673 |
25 |
25 |
50 |
21.7 |
23.9 |
|
|
48
|
| Ni–Co/Al–Mg–O |
923–1023 |
62 |
62 |
186 |
69.4 |
25.9 |
61.8 |
85.1 |
46
|
| Ni/γAl2O3 |
773–973 |
25 |
25 |
50 |
50.9 |
56.1 |
80.5 |
|
43
|
In the current investigation with the Ce0.70La0.20Ni0.10O2−δ catalyst, the activation energies for CH4 and CO2 consumption were very close and comparable with analogous data reported by Sierra-Gallego et al. for the Ni/La2O3 catalytic system.38 The apparent activation energies for hydrogen and CO were greater than those for CH4 and CO2, suggesting that the rate-determining step for CO and H2 formation are different from those for CH4 and CO2 consumption. In addition, the activation energy for H2 production was higher than that for the formation of CO. This could be related to the parallel occurrence of the reverse water gas shift reaction (RWSR) that influences the reaction mechanism.45
Effect of reactant partial pressures on dry reforming rate.
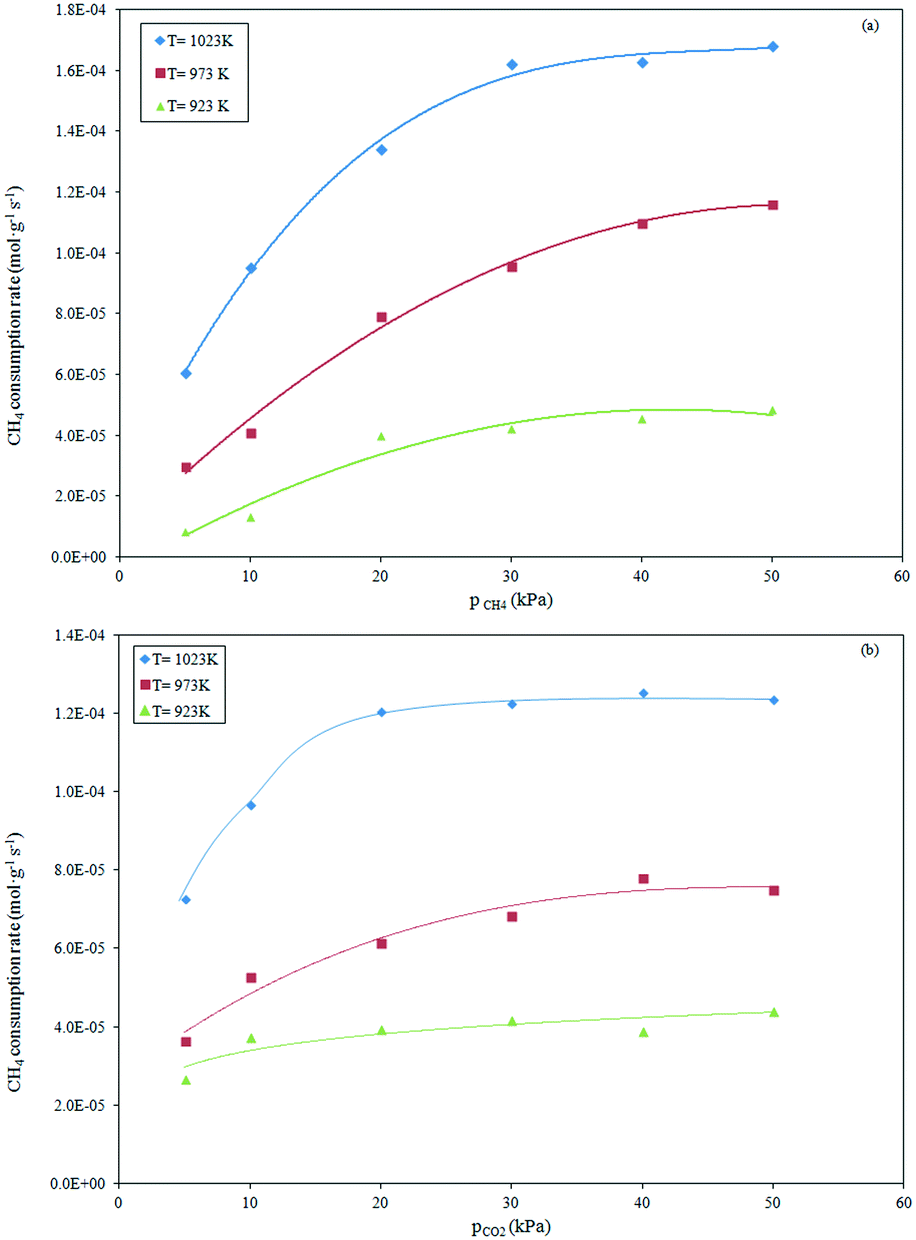
The influence of the partial pressure of CH4 and CO2 on the reforming rate was evaluated at a pressure of 101.32 kPa in the temperature range of 923–1023 K. The effect of the CH4 partial pressure on the consumption rate of CH4 was evaluated at a constant CO2 partial pressure of 20 kPa as the CH4 partial pressure was increased from 5 to 50 kPa. The results evaluated at the different temperatures are depicted in Fig. 3a.
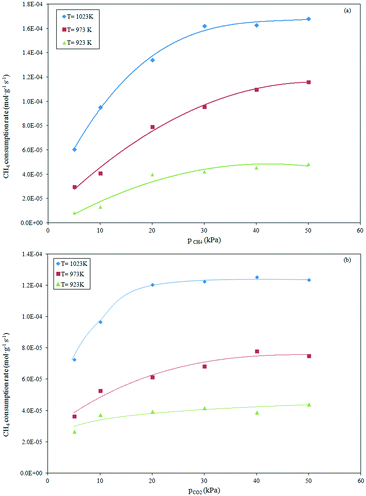 |
| | Fig. 3 Effect of variation of partial pressure of CH4 (a) and CO2 (b) at constant pCO2 and pCH4, respectively, on the reaction rate over the Ce0.70La0.30Ni0.10O2−δ catalyst in the temperature range of 923–1023 K. | |
As evident, the rate of methane consumption was noticeably influenced by the CH4 partial pressure over the whole experimental range of 5–30 kPa. A further increase of the CH4 partial pressure up to 30 kPa induced a less significant effect in the consumption rate. This behavior was observed at high reaction temperatures (973–1023 K). At 923 K, the reforming rate was sensitively affected by the low partial pressures of CH4 (≤20 kPa), remaining almost unchanged with further increase of the CH4 partial pressure.
The effect of the variation of the CO2 partial pressure on the rate of the methane consumption is depicted in Fig. 3b. It can be see that the methane consumption rate increased with increasing CO2 partial pressure in the range of 5–20 kPa, remaining almost constant at higher partial pressures (20–50 kPa). The relatively constant reaction rate, recorded at all investigated reaction temperatures, can be ascribed to a limitation of the thermodynamic equilibrium of the limiting reactant CH4 (pCH4 = 20 kPa).
In addition, the effect of the CO2 partial pressure appeared less significant in the tests carried out at a lower temperature (923 K) in relation to analogous tests at higher temperatures (973–1023 K).
A comparison of Fig. 3a and b suggests that the CH4 consumption rate was more sensitive to the CO2 partial pressure than the CH4 partial pressure at low CH4 and CO2 partial pressures, respectively. From these results, a stronger CO2 adsorption on the catalyst surface can be derived than CH4 adsorption due to the strong interaction of the CO2 molecule with the basic surface of the mixed Ni–La–CeOx oxides, as previous envisaged.32 The presence of the Ce2O3 support from the pre-reduction step of the catalyst can enhance CO2 adsorption by forming the strong ionic oxide Ce2O2CO3. The sequential dissociation of the carbonate species into CO2 and further to CO and O generates oxygen species able to react with carbon deposits at the Ni–Ce2O2CO3 interface.51 Similarly, CO2 can adsorb onto La2O3 (present on the surface) to form La–oxycarbonates, which in turn react with the carbon formed from CH4 activation at the metal–La interface, cleaning the surface of the catalyst from carbon accumulation. This scavenging effect of the carbonate species on carbon from Ni has been proven to be responsible for the stability evidenced by the Ni/La2O3, Rh/La2O3 and Rh/La2O3–SiO2 catalysts.37,44,52–54 Finally, the presence of oxygen vacancies on CeO2–La2O3 in solid solution can promote the dissociation of adsorbed CO2, generating lattice oxygen that reacts with the carbon atom. This can play a fundamental role in the stability of the doped Ni–CeO2 catalytic system.31 Results of the previous investigation with the Ce0.70La0.20Ni0.10O2−δ catalyst, under analogous experimental conditions after 50 h of reaction, showed the absence of carbon deposition.34
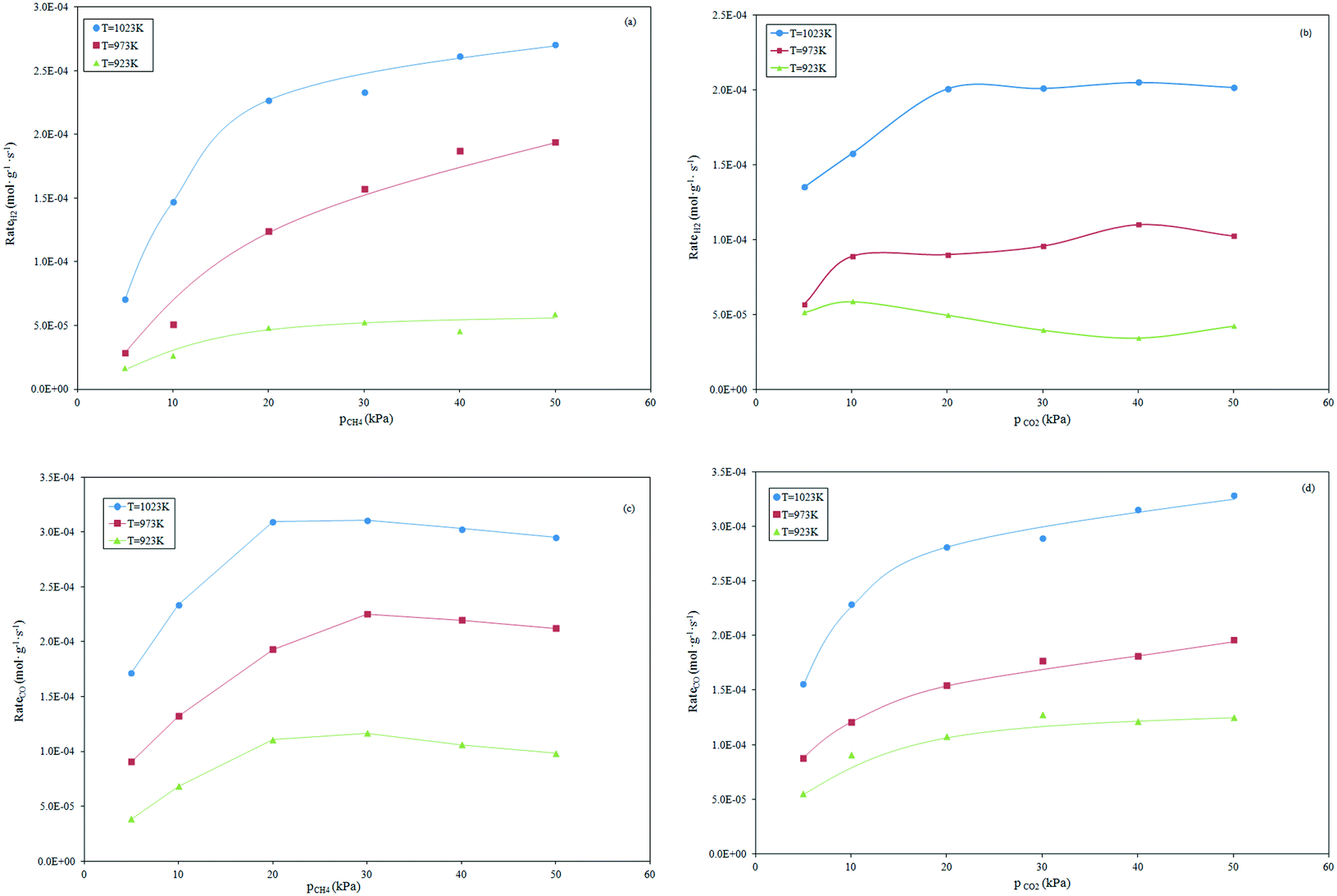
Effect of reactant partial pressures on product formation rates.
The formation rates of H2 and CO as a function of the partial pressure of the reactants are depicted in Fig. 4. The rate of H2 formation for constant CO2 partial pressure increases linearly by increasing the CH4 partial pressure in the range 5–20 kPa. A more limited increase at higher CH4 partial pressure was recorded, as shown in Fig. 4a. This suggests that the dry reforming and RWGS reactions rates become lower in the presence of a limited CO2 amount. This is supported by the experiments conducted at constant CH4 partial pressure. Depicted in Fig. 4b, the increase in the CO2 partial pressure up to the stoichiometric value (20 kPa) involves a noticeable increase in the H2 formation rates that become invariant at higher partial pressures of CO2. The equilibrium that is established between H2 produced by reforming and consumed by RWGS results responsible of the observed trend. The dependence of the CO formation rate on the partial pressures of CH4 and CO2, is depicted in Fig. 4c and d, respectively.
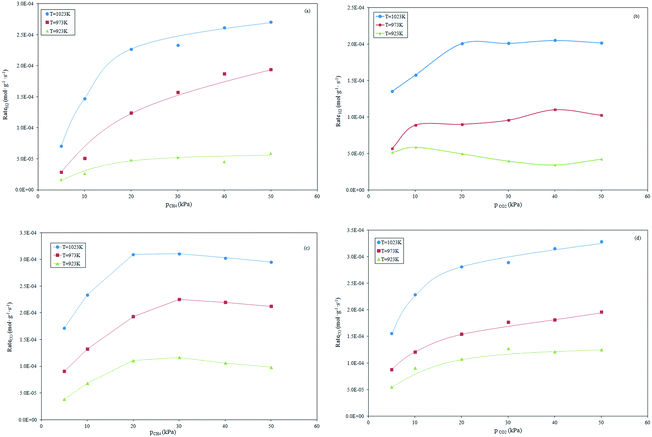 |
| | Fig. 4 Effect of the alteration of CH4 partial pressure (a and c) and CO2 (b and d) at constant pCO2 and pCH4, respectively, on the H2 and CO formation rates. | |
A quasi-linear dependence of the CO formation rate on the CH4 partial pressure ranging between 5 to 20 kPa can be observed. Conversely, a light decrease in the CO formation rate at a high partial pressure of CH4 (Fig. 4c) was observed. This could be due to the effect of side reactions such as RWGS, that limit the CO formation when CO2 is the limiting reagent. This is supported by the observation of Fig. 4d: the CO formation increases progressively by increasing the partial pressure of CO2. The observed trend agrees well with those reported by other authors such as Osazuwa et al. with the SmCoO3 perovskite catalyst,55 Nandini et al. who evaluated the catalytic activity of a Ni–K/CeO2–Al2O3 sample,45 and Fan et al. who applied a bimetallic Ni–Co/MgO–ZrO2 catalyst to the dry reforming reaction.20
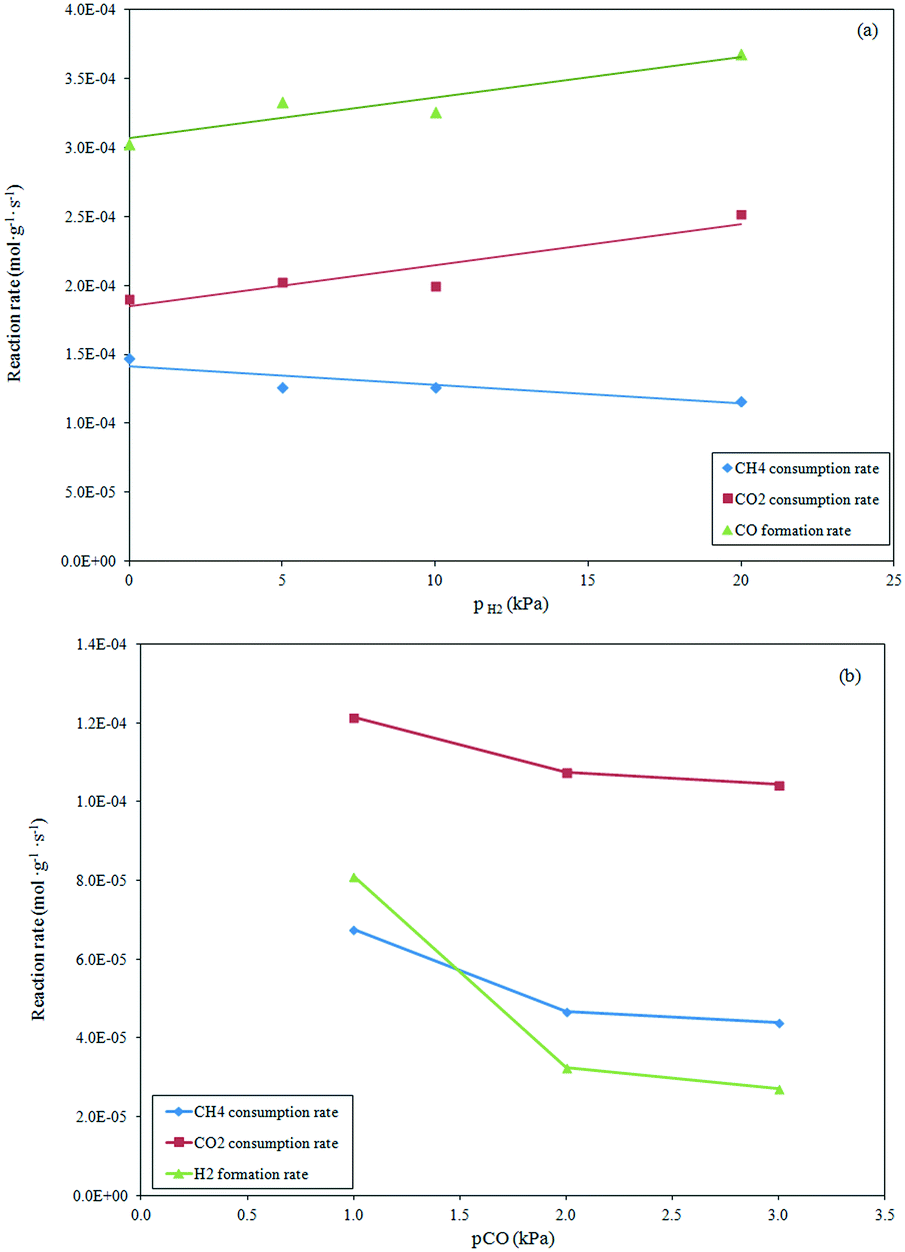
Effect of product partial pressures on dry formation rates.
The effect of H2 in the feed on the dry reforming rate was investigated in the range of 5–20 kPa at a constant CH4 and CO2 partial pressure of 10 kPa and reaction temperature of 1023 K. The results depicted in Fig. 5a show that the increase in the H2 partial pressure induced a light inhibition of the methane consumption rate, while exerting a positive effect on the CO2 consumption rate and CO production rate. This behavior, attributable to the occurrence of the reverse water gas shift reaction, shows that the Ni–H produced on the surface can reach an equilibrium state with the hydrogen product from the dry reforming reaction.44
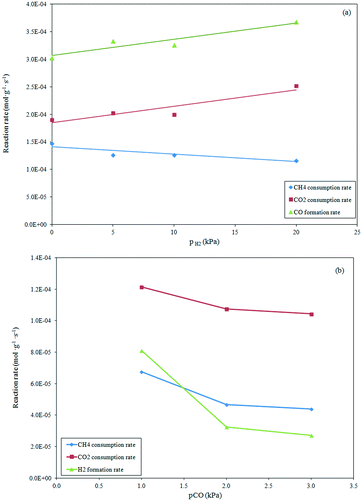 |
| | Fig. 5 Influence of H2 (a) and CO addition (b) to the feed mixture (T = 1023 K, pCH4 = pCO2 = 10 kPa) on the reaction rates. | |
The effect of the increased CO partial pressure from 1 to 3 kPa in the feed stream on the reaction rates is evidenced in Fig. 5b. The reforming rates were slightly influenced by the CO partial pressure. A decrease in the reforming rate was observed for pCO (up to 2 kPa) that remained almost constant with further increases in the CO partial pressure. Taking in consideration that the CH4 dissociation and CO adsorption occurred on the Ni active sites, the inhibition effect of CO on the reaction rate was due to the competitive adsorption of CO on active sites that limited the adsorption/dissociation of CH4 for which the reforming rate decreased. The invariant CH4 consumption rate observed at high CO partial pressure can suggest a limited CO adsorption on the Ni active sites.
3.2 Kinetic modeling
Power law model.
The obtained kinetic data, including the reaction rates and partial pressures of the reactants (CH4 and CO2), were first fitted into the empirical power law model. This provides a rough estimation of the reaction parameters even though it does not account for the reaction mechanisms. The empirical power law model for the dry reforming reaction is given in the following equation:| | 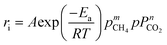 | (7) |
where ri represents the consumption rates of CH4 and CO2, as well as the formation rates of H2 and CO; A represents the pre-exponential factor, Ea is the activation energy; R is the universal gas constant; pCH4 and pCO2 are the partial pressures of CH4 and CO2, respectively, with m and n as the related reaction orders; and T is the absolute reaction temperature.
The SOLVER function, available in Excel spreadsheets, was employed to evaluate the kinetic parameters from the nonlinear power law model.56,57 The goodness of fit was determined by the coefficient of determination (R2). The estimated values of the kinetic parameters, as well as the corresponding R2, Rmsd (residual sum of square) and σ (standard deviation) values are summarized in Table 2. It can be seen that the reaction rates for the reactants and products were satisfactorily predicted with R2 ≥ 0.95.
Table 2 Estimation of kinetics parameters from the power law model
| Reaction species |
Kinetics parameters |
|
m
|
n
|
A (mol g−1 s−1) kPa−(m+n) |
E
a (kJ mol−1) |
R
2
|
Rmsd (mol g−1 s−1) |
σ (mol g−1 s−1) |
|
Rmsd: root mean square deviation; σ: standard deviation.
|
| CH4 |
0.50 |
0.20 |
0.66 |
91.50 |
0.97 |
8.6 × 10−6 |
4.7 × 10−5 |
| CO2 |
0.29 |
0.31 |
0.10 |
70.17 |
0.97 |
9.9 × 10−6 |
6.0 × 10−5 |
| H2 |
0.54 |
0.15 |
5.62 |
105.41 |
0.95 |
1.7 × 10−5 |
1.0 × 10−4 |
| CO |
0.26 |
0.28 |
0.53 |
78.52 |
0.96 |
1.8 × 10−5 |
7.8 × 10−5 |
Activation energies of approximately 91.5 kJ mol−1 and 70.17 kJ mol−1 were derived for CH4 and CO2 consumption, respectively, as evidenced in Table 2. The activation energy of the CH4 consumption was higher than that of the CO2 consumption. This indicates that the CH4 activation is more temperature-sensitive than that of CO2.
The lower CO2 activation energy related to the methane activation energy implied furthermore that the rate of CO2 consumption was faster than that for CH4. Thus, the CO2 conversion was higher than the CH4 conversion.58
The reaction orders obtained for the CH4 consumption (m = 0.50; n = 0.20) indicate that the CH4 partial pressure significantly influences the methane reforming. In addition, the CO2 consumption is influenced by the CO2 partial pressure (m = 0.29; n = 0.31).
The activation energies of H2 (105.41 kJ mol−1) and CO (78.52 kJ mol−1) are comparable to that of CH4 and CO2, respectively. Therefore, it can be derived that the H2 formation was predominantly influenced by CH4, while the CO formation was primarily influenced by CO2. Furthermore, both values were higher than those for the CH4 and CO2 consumption. This suggests, as previously observed, that the rate-determining steps for the H2 and CO formation are different from those for the reagent consumption.
The activation energy for H2 production, noticeably higher than that CO, is a clear indication of the influence of the RWGS reaction on the H2 formation. A comparison of the experimental rates for CH4 and CO2 consumption, as well as for the H2 and CO formation with those predicted by the power law model, are given in Fig. 6. A good agreement between the predicted and experimental values emerged.
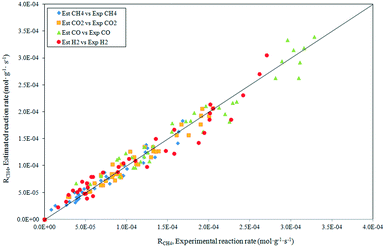 |
| | Fig. 6 Statistical fit of the postulated power law model showing the comparison of the rate predicted by the model to the experimental rate. | |
The derived kinetic parameters are very similar to the reported values by Bradford and Vannice49 and Ayodele et al. for the reaction carried out with Ni/MgO and Co/La2O3,60 respectively, as evidenced in Table 3. Its results are noticeably higher than the activation energy reported by Takano et al. over a traditional Ni/Al2O3 (cordierite) catalyst.59 Although the different supports have different influences on the activity of the metallic phase, this could be ascribed to the high interaction occurring between Ni and the La–CeOx support.
Table 3 Activation energy from the power law reported in the literature
| Catalyst |
Temperature (K) |
Partial pressure (kPa) |
E
CH4 (kJ mol−1) |
E
CO2 (kJ mol−1) |
E
CO (kJ mol−1) |
E
H2 (kJ mol−1) |
Ref. |
| 10.1Ni/MgO |
673–823 |
5–53 |
92.10 |
87.9 |
87.90 |
146.51 |
49
|
| 1.22Ni/TiO2 |
673–823 |
" |
108.8 |
87.9 |
96.28 |
133.95 |
" |
| Co/La2O3 |
923–1023 |
5–50 |
96.44 |
72.22 |
69.71 |
101.50 |
60
|
| Ni/Al2O3 |
850–1000 |
17–51 |
76.0 |
|
|
|
59
|
| 5Ni/CeO2–SiO2-c |
703–733 |
20–40 |
154.7 |
|
|
|
61
|
| 5Ni/CeO2–SiO2-p |
" |
" |
97.3 |
|
|
|
" |
| 4.7Ni/ZnO |
863–883 |
4.5–11 |
110.7 |
118.4 |
|
|
62
|
| 4.8Ni–4.3MgO/ZnO |
743–763 |
" |
97.4 |
106.3 |
|
|
" |
Indeed, Bradford and Vannice pointed out that with the Ni–TiO2 catalyst, the SMSI induced a noticeable value of the activation energy (108 kJ mol−1) that increased to 171.6 kJ mol−1 after 18 h of stream.49 In this case, the migration of TiOx on the Ni surface may increase the activation barrier for methane dissociation. Recently, Yan et al. showed that the close metal–support interface between Ni and CeO2 in the Ni–CeO2–SiO2 catalyst led to a substantial decrease in the activation energy from 154.7 kJ mol−1 (Ni–CeO2–SiO2 sample C) to 97.3 kJ mol−1 with the sample in the absence of interactions (Ni–CeO2–SiO2 sample P).61 Analogous evidence emerged from the study reported by Singha et al., where the methane activation energy of the Ni/ZnO catalyst decreased significantly from 110.7 kJ mol−1 to 97.4 kJ mol−1 by the addition of MgO in the catalyst formulation, inducing high Ni dispersion and metal–support interactions.62 Therefore, other models may be more appropriate than the power law. In the following section, the Langmuir–Hinshelwood mechanism models proposed in the literature for the Ni–La2O3 catalytic system (and as previously mentioned) were applied to evaluate the kinetic mechanism of the current sample.
Langmuir–Hinshelwood models.
Many studies related to the kinetics of methane reforming with CO2 are generally based on mechanistic steps that include: i) the dissociation/activation of CH4 and CO2; ii) adsorption of elemental and intermediate species on active sites; iii) formation of products via surface reactions; iv) desorption of product species.63 The fundamental study carried out by L. M. Aparicio showed that there is not a single rate-determining step (RDS) in the reaction.64 Therefore, in most cases, the CH4 and CO2 dissociation or activation are considered to be the RDS.
Based on DRIFT (diffuse reflectance infrared Fourier transform) and SSITKA (steady-state isotopic transient kinetic analysis) studies, Verykios and co-workers proposed a mechanistic sequence for the Ni/La2O3 catalytic system where CH4 cracking is a slow step, while the CH4 adsorption is at equilibrium.65–67 The strong interaction occurring between CO2 and La2O3 that leads to the formation of the La2O2CO3 species, can be considered a fast step at equilibrium. Conversely, the reaction between this formed species with Ni particles at the Ni/La2O2CO3 interface oxygenated species becomes a slow step.37
This mechanistic step can be synthetically represented by the following reactions:
| | 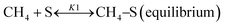 | (8) |
| | 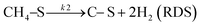 | (9) |
| | 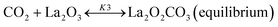 | (10) |
| | 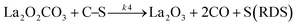 | (11) |
| | | H2 + 2–S ↔ 2H–S(equilibrium) | (12) |
In addition, the simultaneous occurrence of RGSR includes the sequence of the following reaction steps:
| | | CO2 + S ↔ S–CO2(equilibrium) | (13) |
| | | S–CO2 + H–S → S–CO + OH–S(slow) | (14) |
| | | S–OH + H–S ↔ 2S–H2O(equilibrium). | (15) |
On this basis, the authors derived the following rate expression for the methane consumption:
| | 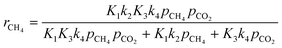 | (Model\kern2 1) |
where
K1 is the adsorption equilibrium constant for CH
4;
k2 is the rate constant of methane cracking on the metallic surface;
K3 is the adsorption equilibrium constant of the reaction between CO
2 and La
2O
3;
k4 is the rate constant of the reaction between the oxycarbonate species and carbon deposited on the Ni surface.
Sierra-Gallego et al. studied the kinetic behaviour of the Ni/La2O3 catalyst obtained from the reduction of LaNiO3 perovskite structure.38 From XPS (X-ray photoelectron spectroscopy) after the catalytic tests, the authors derived that the Ni particles were partially covered by the La2O2CO3 species due to the migration of La2O3 over the metal particles. From this evidence, the authors developed a rate reaction equation including, for the first time, the presence of two active sites on the catalyst surface: metallic nickel particles and the La2O3 metal support. Assuming a negligible H2 and CO surface coverage, the proposed dual active-site mechanism involves the following steps:
| | 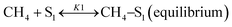 | (16) |
| | 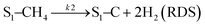 | (17) |
| | 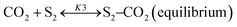 | (18) |
| | 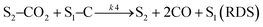 | (19) |
where
S1 represents the metallic active site (Ni°) and S
2 is the La
2O
3 support as the active site. The related rate model assumes the following form:
| | 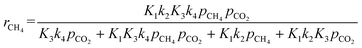 | (Model\kern2 2) |
The current catalytic system can be envisaged as a Ni-solid solution with ceria–lanthana in which the presence of anionic vacancies can help the abstraction of H species from CH
4. The sequential decomposition of the CH
3 intermediate into C, that could substitute steps 9/17 in previous models and is described as:
| | 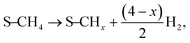 | (9a, 17a) |
occurs very fast. The reversible CO
2 adsorption on the support surface can happen on both La
2O
3 and Ce
2O
3, generating carbonate species at the boundary of Ni particles. The subsequent reaction with adsorbed carbon generates CO, restoring the active sites.
The presence of oxygen vacancies (□) on the ceria–lanthana support contributes to the CO2 adsorption/dissociation step:
Diffusion of the generated lattice oxygen towards the Ni surface exerts a promoting effect in the cleaning steps (carbon oxidation to CO) of the surface sites envisaged in the reaction sequences.
On this basis, the kinetic models proposed by Verykios's group (model 1) and Sierra-Gallego et al. (model 2) were applied to fit the experimental data obtained from this study over the Ce0.70La 0.20Ni0.10O2−δ catalyst. The parameters of both models were estimated using the nonlinear regression method with the ANEMONA.XLS tools, an Excel template previously developed and successfully tested with some kinetic models applied in literature for the dry reforming reaction.68
The summary of the kinetic parameters estimated from the two Langmuir–Hinshelwood rate expressions are summarized in Table 4. As evident, both models gave fitting regression coefficients (R2) greater than 0.90, which can be considered to be a good fitting. Thus, the models are statistically significant.
Table 4 Langmuir–Hinshelwood kinetic models parameters
| Model no. |
Temperature (K) |
Kinetic constant |
|
K
1 (kPa−1) |
k
2 (mol g−1 s−1) |
K
3 (kPa−1) |
k
4 (mol g−1 s−1) |
R
2
|
Rmsd (mol g−1 s−1) |
σ (mol g−1 s−1) |
|
Rmsd: root mean square deviation. σ: standard deviation, (values in parenthesis are the standard deviation for the kinetic constants).
|
| 1 |
1023 |
0.0323 (1.8 × 10−2) |
0.00036 (1.5 × 10−4) |
0.0037 (3.5 × 10−1) |
0.00815 (7.0 × 10−2) |
0.97 |
2.3 × 10−5 |
2.6 × 10−3 |
| 973 |
0.0250 (1.3 × 10−2) |
0.00025 (9.4 × 10−5) |
0.0080 (1.0 × 10−1) |
0.00267 (3.4 × 10−1) |
0.96 |
3.0 × 10−6 |
7.5 × 10−6 |
| 923 |
0.0811 (5.4 × 10−2) |
0.00005 (1.8 × 10−5) |
0.0100 (3.0 × 10−2) |
0.00299 (8.1 × 10−3) |
0.91 |
7.9 × 10−6 |
1.8 × 10−5 |
| 2 |
1023 |
0.0285 (2.7 × 10−2) |
0.00040 (3.2 × 10−5) |
0.0069 (2.5 × 10−2) |
0.0043 (1.4 × 10−3) |
0.97 |
1.5 × 10−5 |
8.7 × 10−4 |
| 973 |
0.0466 (5.4 × 10−2) |
0.00019 (1.8 × 10−4) |
0.0100 (9.0 × 10−2) |
0.0024 (1.1 × 10−3) |
0.93 |
1.4 × 10−5 |
2.8 × 10−4 |
| 923 |
0.0679 (7.7 × 10−2) |
0.00007 (5.6 × 10−5) |
0.0181 (3.2 × 10−1) |
0.0012 (8.2 × 10−4) |
0.92 |
2.4 × 10−6 |
4.8 × 10−6 |
In order to evaluate the related thermodynamic relevance, the models were further screened by application of the criteria designed to evaluate the adsorption equilibrium constants obtained in a Langmuirian rate expression, defined by the following equation:69
| | | 10 ≤ −ΔS ≤ 12.2 − 0.0014ΔH | (20) |
where Δ
S and Δ
H are the changes in entropy and enthalpy, respectively. However, the change in the entropies and enthalpies have been estimated using the Van't Hoff expression:
| | 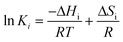 | (21) |
where
Ki is the adsorption constant (
K1 for CH
4 or K
3 for CO
2). The derived thermodynamic parameters for CH
4 and CO
2 and the related evaluations are synthesized in
Table 5. As evident,
model 2 emerges as the best kinetic model that represents the mechanism of the reaction: it satisfies the thermodynamic criterion, the
R2 values (0.98 and 0.99) are considerably higher than the corresponding values derived for
model 1 (0.58 and 0.89) that failed to satisfy the thermodynamic criteria.
Table 5 Models screening using change in entropies and enthalpies for CH4 and CO2
| Reagent |
Model no. |
ΔH (kJ mol−1) |
ΔS (kJ mol−1 K) |
R
2
|
Criteria (eqn (20)) |
| CH4 |
1 |
−74.04 |
−102.92 |
0.58 |
10 ≤ 102.92 ≤ 115.85 (yes) |
| 2 |
−68.84 |
−97.87 |
0.98 |
10 ≤ 97.87 ≤ 108.58 (yes) |
| CO2 |
1 |
−77.25 |
−121.20 |
0.89 |
10 ≤ 121.20 ≤ 120.35 (no) |
| 2 |
−76.49 |
−117.21 |
0.99 |
10 ≤ 117.21 ≤ 119.30 (yes) |
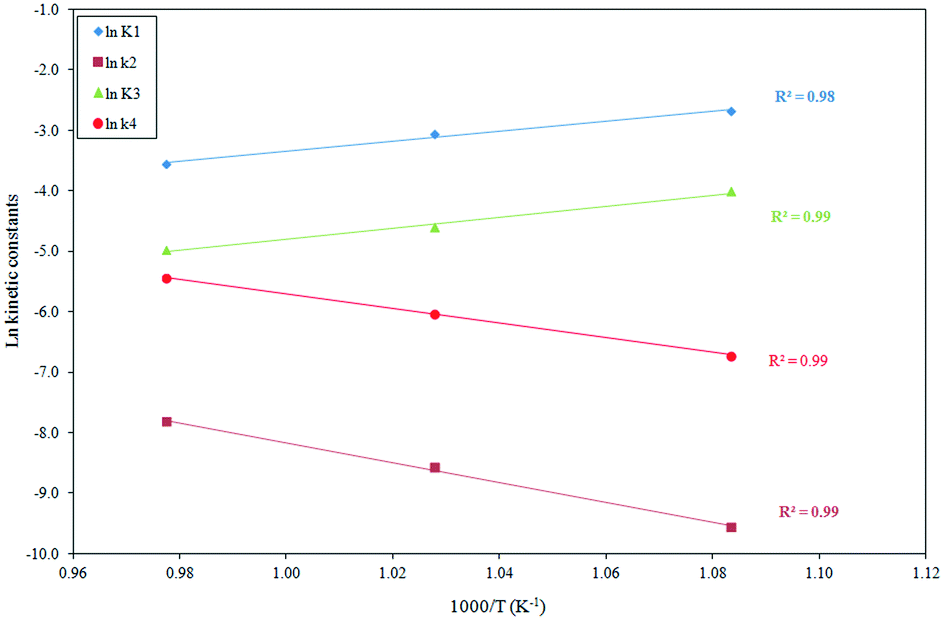
The temperature dependence of the reaction rate parameters derived from model 2 is shown in the following equations obtained from the Arrhenius plots (depicted in Fig. 7) of the K1, k2, K3 and k4 constants:
| | | K1 = 7.82 × 10−6 exp(8280/T) | (22) |
| | | k2 = 4.80 × 103 exp(−16470/T) | (23) |
| | | K3 = 7.54 × 10−7 exp(9200/T) | (24) |
| | | k4 = 6.32 × 102 exp(−12110/T). | (25) |
The adsorption equilibrium constants
K1 and
K3 increase with decreasing reaction temperature, as expected since the CH
4 and CO
2 adsorptions are exothermal processes. The reaction rate constants
k2 and
k4 follow an opposite trend. It is interesting to note that the
k4 values are noticeably higher than the
k2 rate constants related to the methane cracking. From this, it is possible to derive that the catalysts kinetically inhibited the carbon deposition in accordance with analogous observations reported by other authors.
20,39
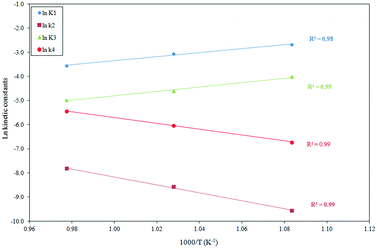 |
| | Fig. 7 Effect of the temperature on the adsorption (K1 and K3) and reaction constants (k2 and k4) for the Langmuir–Hinshelwood kinetic model (model 2). | |
The absence of the carbon deposition observed in all experimental tests suggests that the catalyst stabilizes CHx decomposition and CO disproportion that is likely the mechanism for carbon deposition during the dry reforming reaction.49 The ability to activate the C–H bond cleavage requires the electron pair donation from CH4 to the unfilled d-orbital of nickel. The high d-electron density of the Ni atoms, as previously revealed by XPS studies, inhibits this process.34 Similarly, the evidenced suppression of CO chemisorptions shown by this catalytic system may increase the activation barrier for CO dissociation.
The activation energies related to CH4 and CO2 were 136.9 and 100.6 kJ mol−1, respectively, which are higher than the corresponding values derived with the power law rate expression. This is reasonable considering that the Langmuir–Hinshelwood rate expression takes into account the adsorption/desorption enthalpies. Thus, the predicted values are expected to be higher than the power law expression.70
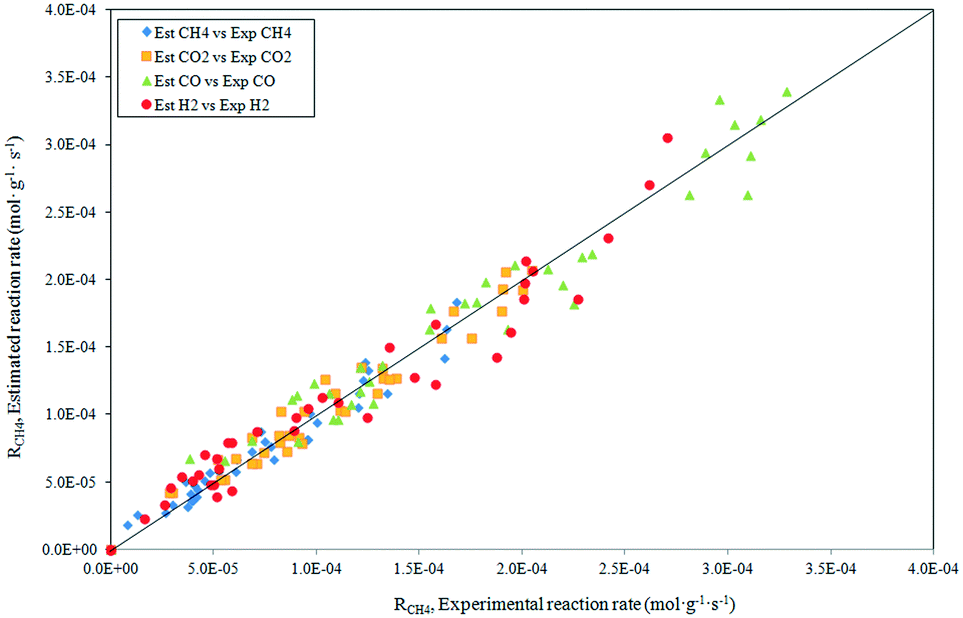
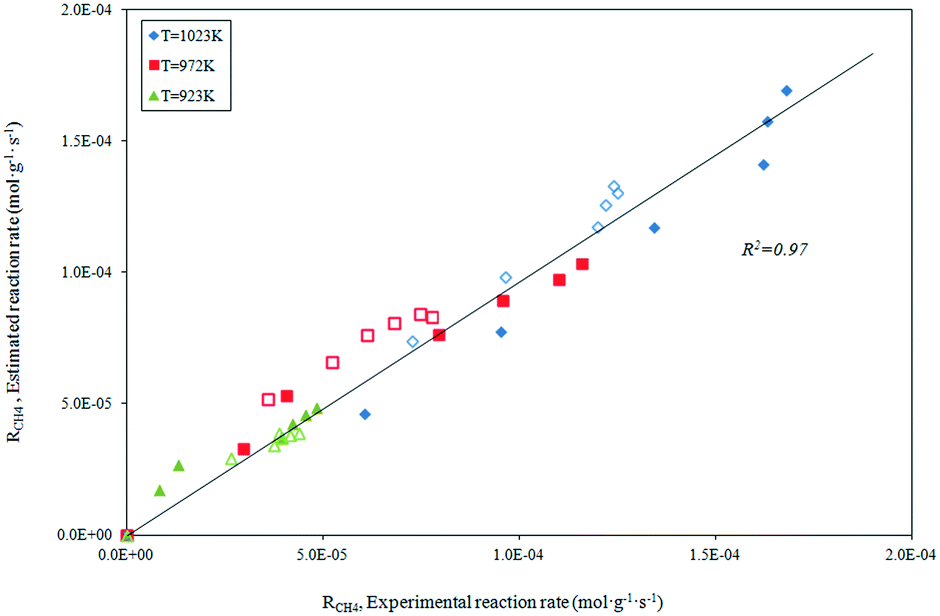
The parity plot showing the comparison between the experimental and predicted CH4 reaction rates is shown in Fig. 8. The kinetic model shows a good fit with the experimental data with a correlation coefficient of R2 = 0.97.
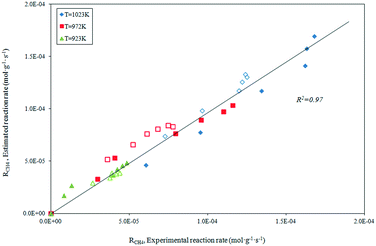 |
| | Fig. 8 Statistical fit of the postulated model 2 comparing the rate predicted by the model to the experimental rate. | |
Conclusions
The kinetics of methane dry reforming over the Ce0.70La0.20Ni0.10O2-δ catalyst has been evaluated at CH4 and CO2 partial pressures ranging between 5 to 50 kPa at 923, 973 and 1023 K. The reaction mechanism was proposed on the basis of literature and experimental observations.
The experimental data were fitted into the power law and Langmuir–Hinshelwood kinetics expression considering a dual-step with single-(Ni°) and dual-active sites (La2O3 support), respectively, for the reaction. The discrimination between the two models, statistically and thermodynamically, was evidenced as the reaction mechanism of the dry reforming reaction over the Ce0.70La0.20Ni0.10O2−δ catalyst. It can be described by a dual molecular adsorption of both CH4 and CO2 on metallic nickel and support (La2O3), respectively, with surface reactions as the rate-determining steps. The reaction between CO2 and support generates the La2O2CO3 species, and is able to react with carbon at the metal–carbon interface to produce CO and clean up the metallic surface.
The high value of the kinetic constant k4 (reaction between carbonate species and carbon deposited) compared with k2 (methane cracking) suggests that the catalyst kinetically inhibits the excess of carbon deposition.
Conflicts of interest
There are no conflicts to declare.
References
- United Nations Framework Convention on Climate Change (UNFCC), Adoption of the Paris Agreement, Report N. FCCC/CP/2015, http://Unounfccc.int/resource/docs/2015/cop21/eng/l09r01.pdf
 .
.
- J. D. Fonseca, M. Camargo, J.-M. Commenge, L. Falk and I. D. Gil, Int. J. Hydrogen Energy, 2019, 44, 9486–9504 CrossRef CAS .
- G. Sdanghi, G. Maranzana, A. Celzard and V. Fierro, Renewable Sustainable Energy Rev., 2019, 102, 150–170 CrossRef CAS .
- A. Chapman, K. Itaoka, K. Hirose, F. T. Davidson, K. Nagasawa, A. C. Lloyd, M. E. Webber, Z. Kurban, S. Managi, T. Tamaki, M. C. Lewis, R. E. Hebner and Y. Fujii, Int. J. Hydrogen Energy, 2019, 44, 6371–6382 CrossRef CAS .
- K. Nagaoka, K. Takanabe and A. Ken-Ichi, Appl. Catal., A, 2003, 255, 13–21 CrossRef CAS .
- S. A. Chattanathan, S. Adhikari and S. Taylor, Int. J. Hydrogen Energy, 2012, 37, 18031–18039 CrossRef CAS .
- R. Nehring, Philos. Trans. R. Soc., B, 2009, 364, 3067–3079 CrossRef PubMed .
- J.-M. Lavoie, Front. Chem., 2014, 2, B1 Search PubMed .
- Y. Gao, J. Jianga, Y. Meng, F. Yan and A. Aihemaiti, Energy Convers. Manage., 2018, 171, 133–155 CrossRef CAS .
- V. Nallathamb Gunaseelan, Biomass Bioenergy, 1997, 13, 83–114 CrossRef .
- S. Gaur, D. J. Haynes and J. J. Spivey, Appl. Catal., A, 2011, 403, 142–151 CAS .
- A. Fouskas, M. Kollia, A. Kambolis, C. Papadopoulou and H. Matralis, Appl. Catal., A, 2013, 474, 125–134 CrossRef .
- J. H. Bitter, K. Sehan and J. A. Lercher, J. Catal., 1991, 183, 336–343 CrossRef .
- F. Menegazzo, M. Signoretto, F. Pinna, P. Canton and N. Pernicone, Appl. Catal., A, 2012, 439–440, 80–87 CrossRef CAS .
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC .
- F. Wang, L. Xu, W. Shi, J. Zhang, K. Wu, Y. Zhao, H. Li, H. X. Li, G. Q. Xu and W. Chen, Nano Res., 2017, 10, 364–380 CrossRef CAS .
- M. Usman, W. M. A. Wan Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2015, 45, 710–744 CrossRef CAS .
- G. Zhang, J. Liu, Y. Xu and Y. Sun, Int. J. Hydrogen Energy, 2018, 43, 15030–15054 CrossRef CAS .
- S. Arora and R. Prasad, RSC Adv., 2016, 6, 108668–108688 RSC .
- M.-S. Fan, A. Z. Abdullah and S. Bhatia, ChemCatChem, 2009, 1, 192–208 CrossRef CAS .
- W.-J. Jang, J.-O. Shim, H.-M. Kim, S.-Y. Yoo and H.-S. Roh, Catal. Today, 2019, 324, 15–26 CrossRef CAS .
- I. Luisetto, S. Tuti, C. Romano, M. Boaro, E. Di Bartolomeo, J. Kopula Kesavan, S. M. Senthil Kumar and K. Selvakumar, J. CO2 Util., 2019, 30, 63–78 CrossRef CAS .
- S. Das, J. Ashok, Z. Bian, N. Dewangan, M. H. Wai, Y. Du, A. Borgna, K. Hidajat and S. Kawi, Appl. Catal., B, 2018, 230, 220–236 CrossRef CAS .
- Y. Lu, D. Guo, Y. Ruan, Y. Zhao, S. Wang and X. Ma, J. CO2 Util., 2018, 24, 191–199 Search PubMed .
- Z. Li, M. Li, Z. Bian, Y. Kathiraser and S. Kawi, Appl. Catal., B, 2016, 188, 324–341 CrossRef CAS .
- M. Steib, Y. Lou, A. Jentys and J. A. Lercher, ChemCatChem, 2017, 9, 3809–3813 CrossRef CAS .
- M. Shah, S. Das, A. K. Nayak, P. Mondal and A. Bordoloi, Appl. Catal., A, 2018, 556, 137–154 CrossRef CAS .
- Y. H. Hu, Catal. Today, 2009, 148, 206–211 CrossRef CAS .
- V. A. Sadykov, M. N. Simonov, N. V. Mezentseva and S. N. Pavlova,
et al.
, Open Chem., 2016, 14, 363–376 CAS .
- W.-J. Jang, D.-W. Jeong, J.-O. Shim, H.-M. Kim, W.-B. Han, J. W. Bae and H.-S. Roh, Renewable Energy, 2015, 79, 91–95 CrossRef CAS .
- M. M. Makri, M. A. Vasiliades, K. C. Petallidou and A. M. Efstathiou, Catal. Today, 2015, 259, 150–164 CrossRef .
- L. Pino, A. Vita, F. Cipiti, M. Laganà and V. Recupero, Appl. Catal., B, 2011, 104, 64–73 CrossRef CAS .
- L. Pino, A. Vita, M. Laganà and V. Recupero, Appl. Catal., B, 2014, 148–149, 91–105 CrossRef CAS .
- L. Pino, C. Italiano, A. Vita, M. Laganà and V. Recupero, Appl. Catal., B, 2017, 218, 779–792 CrossRef CAS .
- Y. Kathiraser, U. Oemar, E. T. Saw, Z. Li and S. Kawi, Chem. Eng. J., 2015, 278, 62–78 CrossRef CAS .
- G. Zhang, J. Liu, Y. Xu and Y. Sun, Int. J. Hydrogen Energy, 2018, 42, 15030–15054 CrossRef .
- V. A. Tsipouriari and X. E. Verykios, Catal. Today, 2001, 64, 83–90 CrossRef CAS .
- G. Sierra Gallego, C. Batiot-Dupeyrat, J. Barrault and F. Mondrago, Ind. Eng. Chem. Res., 2008, 47, 9272–9278 CrossRef CAS .
- Z. Bao, Y. Lu and F. Yu, AIChE J., 2017, 63, 2019–2029 CrossRef CAS .
- D. E. Mears, Ind. Eng. Chem. Process Des. Dev., 1971, 10, 541–547 CrossRef CAS .
-
M. E. Davis and R. J. Davis, Fundamental of chemical reaction engineering, McGraw-Hill, New York, 2003, pp. 184–239 Search PubMed .
- P. B. Weisz and C. D. Prater, Adv. Catal., 1954, 6, 143–196 CAS .
- S. Wang and G. Q. Lu, Ind. Eng. Chem. Res., 1999, 38, 2615–2625 CrossRef CAS .
- A. A. Lemonidou and I. A. Vasalos, Appl. Catal., A, 2002, 228, 227–235 CrossRef CAS .
- A. Nandini, K. K. Pant and S. C. Dhingra, Appl. Catal., A, 2006, 308, 119–127 CrossRef CAS .
- J. Zhang, H. Wang and A. K. Dalai, Ind. Eng. Chem. Res., 2009, 48, 677–684 CrossRef CAS .
- J. Dacquin, D. Sellam, C. Batiot-Dupeyrat, A. Tougerti, D. Duprez and S. Royer, ChemSusChem, 2014, 7, 631–637 CrossRef CAS PubMed .
- S. Das, S. Thakur, A. Bag, M. S. Gupta, P. Mondal and A. Bordoloi, J. Catal., 2015, 330, 46–60 CrossRef CAS .
- M. C. J. Bradford and M. A. Vannice, Appl. Catal., A, 1996, 142, 97–122 CrossRef CAS .
- M. C. J. Bradford and M. A. Vannice, Catal. Rev.: Sci. Eng., 1991, 41, 1–42 CrossRef .
- F. A. J. Al-Doghachi, U. Rashid, Z. Zainal, M. I. Saiman and Y. H. T. Yap, RSC Adv., 2015, 5, 81739–81752 RSC .
- X. E. Verikyos, Int. J. Hydrogen Energy, 2003, 28, 1045–1063 Search PubMed .
- J. F. Munera, S. Irusta, L. M. Cornaglia, E. A. Lombardo, D. V. Cesar and M. Schmal, J. Catal., 2007, 245, 25–34 CrossRef CAS .
- J. F. Munera, L. Cornaglia, D. V. Cesar, M. Schmal and E. A. Lombardo, Ind. Eng. Chem. Res., 2007, 46, 7543–7549 CrossRef CAS .
- O. U. Osazuwa, H. D. Setiabudi, S. Abdullah and C. K. Cheng, Int. J. Hydrogen Energy, 2017, 42, 9707–9721 CrossRef CAS .
- A. M. Brown, Comput. Methods Programs Biomed., 2001, 65, 191–200 CrossRef CAS PubMed .
- M. D. A. Hossain, H. H. Ngo and W. Guo, J. Water Sustainability, 2013, 3, 223–237 CAS .
-
V. C. H. Kroll, G. J. Tjatjopoulos and C. Mirodatos, Natural Gas Conversion V. (Stud. Surf. Sci. Catal.), 1998, vol. 119, pp. 753–758 Search PubMed .
- A. Takano, T. Tagawa and S. Goto, J. Chem. Eng. Jpn., 1994, 27, 727–731 CrossRef CAS .
- B. Ayodele, M. R. Khan, S. S. Lam and C. K. Cheng, Int. J. Hydrogen Energy, 2016, 41, 4603–4615 CrossRef CAS .
- X. Yan, T. Hu, P. Liu, S. Li, B. Zhao, Q. Zhang, W. Jiao, S. Chen, P. Wang, J. Lu, L. Fan, X. Deng and Y.-X. Pan, Appl. Catal., B, 2019, 246, 221–231 CrossRef CAS .
- R. K. Singha, A. Yadav, A. Agrawal, A. Shukla, S. Adak, T. Sasaki and R. Bal, Appl. Catal., B, 2016, 191, 165–178 CrossRef CAS .
- U. Oemar, Y. Kathiraser, L. Mo, X. K. Ho and S. Kawi, Catal. Sci. Technol., 2016, 6, 1173–1186 RSC .
- L. M. Aparicio, J. Catal., 1997, 165, 262–274 CrossRef CAS .
- Z. Zhang, X. E. Verykios, S. M. MacDonald and S. Affrossman, J. Phys. Chem., 1996, 100, 744–754 CrossRef CAS .
- V. A. Tsipouriari and X. E. Verykios, J. Catal., 1999, 187, 85–94 CrossRef CAS .
- A. Slagtern, Y. Schuurman, C. Leclercq, X. Verykios and C. Mirodatos, J. Catal., 1997, 172, 118–126 CrossRef CAS .
- L. Pino, V. Recupero and A. Hernández, ChemEngineering, 2018, 2, 57 CrossRef CAS .
-
M. A. Vannice, Kinetics of catalytic reactions, Springer, New York, 2005, pp. 106–137 Search PubMed .
-
I. Chorkendorff and J. M. Niemantsverdriet, Concept of modern Catalysis and Kinetics, Wiley-VCH
Verlag GmbH & Co. KGaA, 2005, pp. 23–78 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Cristina
Italiano
*,
Cristina
Italiano