
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A PMMA-based heterogeneous photocatalyst for visible light-promoted [4 + 2] cycloaddition†
Niklas
Huber
a,
Run
Li
a,
Calum T. J.
Ferguson
a,
Dominik W.
Gehrig
a,
Charusheela
Ramanan
 a,
Paul W. M.
Blom
a,
Katharina
Landfester
a and
Kai A. I.
Zhang
*ab
a,
Paul W. M.
Blom
a,
Katharina
Landfester
a and
Kai A. I.
Zhang
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: kai.zhang@mpip-mainz.mpg.de
bDepartment of Materials Science, Fudan University, Shanghai 200433, P. R. China. E-mail: kai_zhang@fudan.edu.cn
First published on 21st February 2020
Abstract
Macromolecular organic photocatalysts consisting of a completely conjugated network have broad and promising applications in visible light-promoted photoredox catalysis. Precise reproduction and control of exact conjugation length remains an important challenge for fully conjugated and macromolecular photocatalysts. Here, we introduce a new photocatalytic material based on classical PMMA copolymerised with defined electron donor and acceptor units with precisely controllable redox potential and conjugation length, creating a promising class of metal-free, stable and low-cost heterogeneous photocatalysts. Furthermore, swelling of the PMMA copolymer matrix in organic solvents led to enhanced substrate diffusion and thereby increased catalytic efficiency. High efficiency and selectivity was achieved for photocatalytic [4 + 2] cycloaddition reactions in air with low effective photocatalyst loading. The photocatalytic efficiency of the PMMA photocatalyst was comparable with the state-of-the-art metal or non-metal catalysts while facilitating easy recyclability.
Introduction
Metal-free heterogeneous photocatalysts empowering chemical synthesis under sustainable and environmentally benign reaction conditions have attracted much attention.1–6 They have shown wide-scale use in catalysing visible light-promoted photoredox reactions in many applications including energy for water splitting,7–11 organic redox reactions12–15 and environmental photocatalysis,16–19 or in biological applications for photodynamic therapy20–23 and enzyme regeneration.24–26 Among fully conjugated systems being used, exact control of conjugation length, and thereby electronic and optical properties within the polymer network, is not easily achievable. Moreover, inflexible conjugated networks hinder swelling in solvents, and as a result prevent substrate diffusion. A key challenge is therefore to construct a cheap and flexible polymer-based photocatalyst with reproducible precisely defined optical and electronic properties.The classical polymer chemistry toolbox provides access to tuneable polymeric materials with variable solubility, biological compatibility, mechanical properties, and responses to external triggers. However, the role of classical polymers as a platform for photocatalytic systems has barely been explored and studies are limited to attaching photoactive metal complexes27,28 or organic dyes29 to linear polymers. Recently, our group published temperature-responsive photocatalytic nanogels based on poly-N-isopropylacrylamide, demonstrating the versatility of combining photoactive units with well-known polymeric systems.30 In general, the incorporation of photocatalytic properties into classical polymer networks via copolymerisation of photoactive monomers could be an effective strategy towards a novel class of efficient, cheap and metal-free photocatalysts.31–33 Moreover, this combination may yield photocatalytic materials with considerable advantages in terms of stability and long-term usage for visible light-promoted redox catalysis.
In this work, we designed a cross-linked copolymer based on poly(methyl methacrylate) (PMMA) with integrated photocatalytic units consisting of 4,7-diphenylbenzothiadiazole (cPMMA-BTPh2). The resulting polymer is an efficient metal-free and redox-active heterogeneous photocatalyst for visible light-promoted chemical transformations. This material must exhibit optical and redox properties similar to the molecular photocatalytic analogue unit, whilst demonstrating advantageous chemical robustness and recyclability attributed to polymer materials. [4 + 2] cycloaddition reactions (the Diels–Alder reaction) could be efficiently catalysed by the PMMA photocatalytic material with low catalyst loading, in particular, 0.06 mol% of the effective photocatalytic unit with respect to the substrate. The copolymer photocatalyst exhibited comparable photocatalytic efficiency and selectivity to the state-of-the-art molecular and transition metal-containing photocatalysts. Specifically, the photocatalytic copolymer material exhibited comparable efficiency when compared to its small molecule analogue. No photobleaching effect of the polymer photocatalyst was observed, as confirmed by recycling experiments. Furthermore, a detailed mechanistic study employing advanced photophysical methods, such as time-resolved photoluminescence and transient absorption spectroscopy, was conducted. Further to the detailed studies on the benchmark reaction, the scope of the reactivity of the copolymer photocatalyst was expanded to a broader range of substrates, proving its excellent efficiencies and functional group tolerance (Scheme 1).
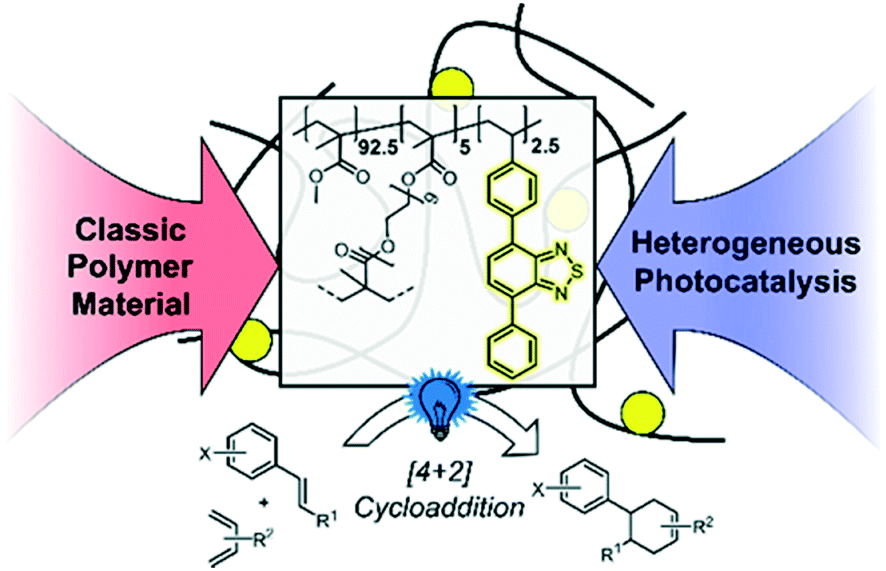 | ||
| Scheme 1 Illustration of the design concept of the PMMA-based photocatalyst copolymer and its application in the photocatalytic [4 + 2] cycloaddition. | ||
Results and discussion
The photocatalytic copolymer (cPMMA-BTPh2) in this study was synthesized via free-radical polymerisation of methyl methacrylate (MMA), polyethylene glycol dimethacrylate (PEGDMA) and 4-phenyl-7-(4-vinylphenyl)benzothiadiazole (BTPh2) in low concentrations. Details on synthetic procedures and characterization are given in the Experimental section and ESI.†In order to confirm the incorporation of the photoactive BTPh2 unit into the cross-linked PMMA, solid-state nuclear magnetic resonance (ssNMR), UV/vis, photoluminescence (PL) and cyclic voltammetry (CV) analyses were performed. The ssNMR spectra of cPMMA-BTPh2 and a photoinactive cPMMA analogue show clear signals of the photoactive unit (Fig. S1 and S2†). The UV/vis absorption spectrum of cPMMA-BTPh2 showed an absorption range up to 475 nm with a maximum at 386 nm, while the emission area between 425 nm and 650 nm with a maximum at 512 nm was determined (Fig. 1a). The result is consistent with the optical properties of the corresponding small molecule BTPh2 and previous reports.34,35 The slight red shift of cPMMA-BTPh2 compared to BTPh2 can be attributed to hyperconjugation effects of the photoactive unit in cPMMA-BTPh2, which is one-sidedly attached to the polymer backbone. The optical properties of cPMMA-BTPh2 in terms of emission and absorption behaviors are similar to those of fac-[Ir(ppy)3], a well-established transition metal photocatalyst.36
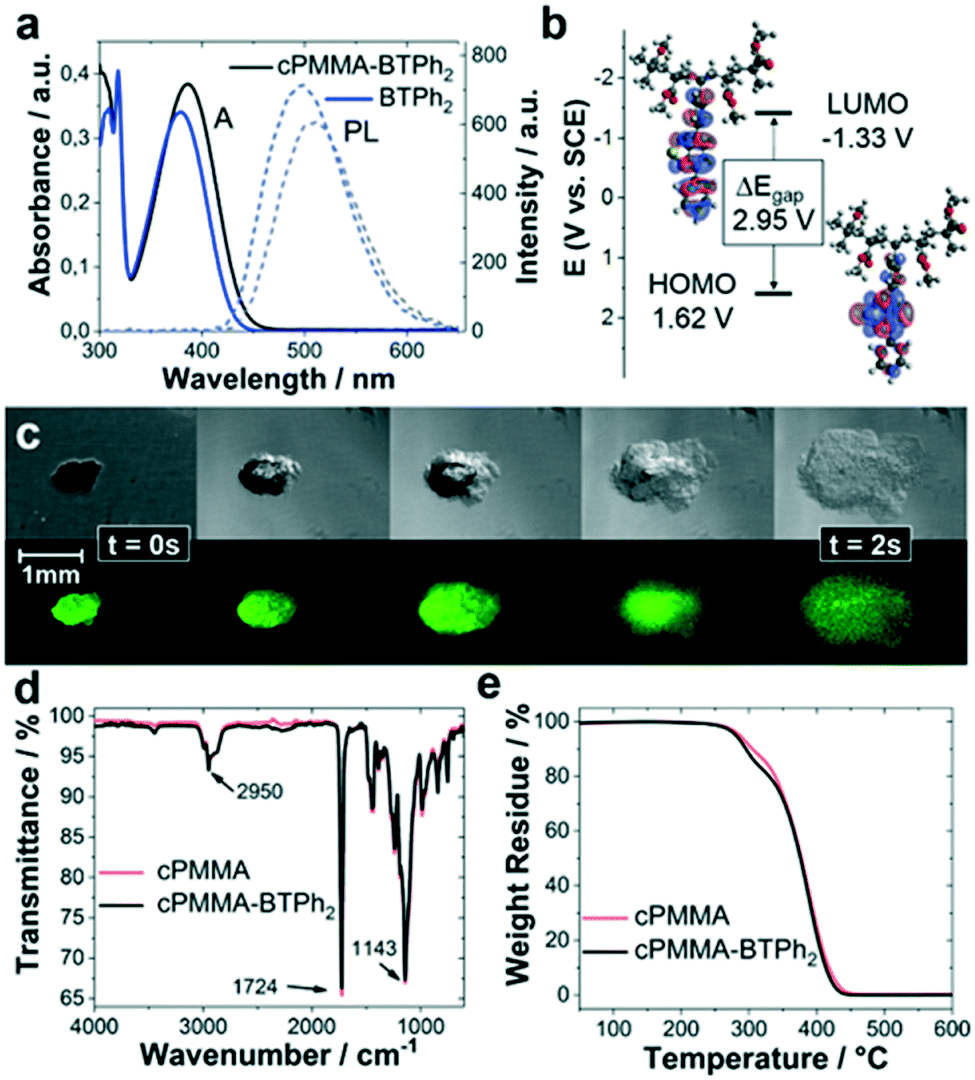 | ||
| Fig. 1 (a) Comparison of the photoluminescence and UV/vis spectra of BTPh2 units and the polymer cPMMA-BTPh2 in the solid state and (b) DFT/B3LYP 6-31G(d) calculated electronic structures for the HOMO and LUMO of a simplified cPMMA-BTPh2. Potentials resulting from the CV measurements on cPMMA-BTPh2. (c) Optical microscopy images of cPMMA-BTPh2, swelling after exposure to CH3NO2. (d) FTIR and (e) TGA spectra of cPMMA-BTPh2 and the blank cPMMA sample. | ||
Cyclic voltammetry (CV) measurements revealed that the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of cPMMA-BTPh2 are at +1.62 eV and −1.33 eV vs. SCE (Fig. 1b and S3†), which matches with the potentials of the analogous small molecular organic photocatalyst BTPh2. Theoretical calculations for cPMMA-BTPh2 using the density functional theory (DFT) at B3LYP/6-31G(d) level (Table S1†) revealed that the electron densities of HOMO and LUMO are mainly located on the diphenylbenzothiadiazole unit. This indicates that the electronic structure is determined with a side-chain consisting of the D–A-type unit BTPh2. The small deviations between the UV/vis, PL and CV spectra of BTPh2 and cPMMA-BTPh2 were reproducible and we attributed them to the hyperconjugation effects in cPMMA-BTPh2.
The swelling behavior of cPMMA-BTPh2 was investigated by live-imaging the photocatalytic material after exposure to CH3NO2 with a bright-field/fluorescence microscope. The photocatalytic gel was observed to swell to about 4 times its original size within 2 s. The ability of the gel to swell is controlled by the cross-linking density and the cross-linker length. The long ethylene glycol chains in the cross-linker PEGDMA allow the gel to expand, enabling easy diffusion of substrates and products in and out of the photocatalytic material. Further swelling experiments showed the strong dependency of the swelling behaviour on the solvent choice (Fig. S4†).
Fourier transform infrared (FTIR) spectroscopy and thermogravimetric analysis (TGA) characterization were performed with the cPMMA-BTPh2 photocatalyst and compared to a non-photoactive cPMMA analogue. The FTIR spectrum (Fig. 1d) showed typical signals, for example, at 2992 and 2950 cm−1, which are characteristic of the C–H bond stretching vibrations of the –CH3 and –CH2– groups, respectively. The signal at 1724 cm−1 indicates the presence of an acrylate carboxyl group. The broad signals at 1239 and 1143 cm−1 can be assigned to the C–O–C stretching vibrations. The two bands at 1387 cm−1 and 754 cm−1 can be attributed to the α-methyl group vibrations. The band at 1441 cm−1 can be attributed to the bending vibrations of the C–H bonds of the –CH3 group. Interestingly, the influence of the photoactive unit on the FTIR spectrum is minimal (Fig. S5†), showing that the chemical constitution and connectivity of the catalyst is mainly determined by the PMMA network. Thermogravimetric analysis (TGA) showed that the photocatalyst remained intact up to 295 °C under nitrogen, comparable to the blank cPMMA sample. FTIR and TGA measurements prove the robustness of the cPMMA-BTPh2 photocatalyst, which shows that the properties of the cross-linked PMMA network are directly transferred to the polymeric cPMMA-BTPh2 system.
A rough estimation of the material cost of the cPMMA-BTPh2 photocatalyst gave a value of approximately 7€ per g based on the current commercial prices of the chemicals used in the synthesis (see Fig. S6†). The low photoactive unit loading in the catalyst, as well as using PMMA as a commodity polymer, gives the system a cost advantage.
To demonstrate the photocatalytic activity of the cPMMA-BTPh2 system, the stereochemically highly challenging [4 + 2] reaction was selected. The screening and control experiments of the model reaction between trans-anethole and isoprene are listed in Table 1. Significantly, the [4 + 2] cycloaddition of trans-anethole and isoprene was achieved with high selectivity (>99%) in a quantitative manner within 4 h at room temperature in air (entry 1). An apparent quantum yield of 2.27% could be observed, with 0.6 μmol mL−1 photoactive unit present in the model reaction, which is 0.06 mol% with respect to the substrate. A first order kinetic reaction was observed (Fig. S7†). A control experiment conducted under light, then in the dark, and then under light again yielded only a trace amount of the product in the dark period. This indicated that the generated radical cation of trans-anethole was only active during light irradiation (Fig. S8†), suggesting temporal and spatial limitations of the radical-based reaction.37
|
|
|||
|---|---|---|---|
| Entry | Reaction condition variations | Time | Conv.b (%) |
| a Reaction conditions: [trans-anethole] = 0.1 M, [isoprene] = 1 M, 1.5 mL nitromethane, [cPMMA-BTPh2] = 4 mg mL−1 (effective concentration of the photocatalytic unit = 0.6 mM), blue LED lamp (460 nm, 0.16 W cm−2), room temperature, air. b Conversion determined by GC-MS. c [CuCl2] = 1.5 M. d [KI] = 1.5 M. e [BTPh2] = 0.6 mM. | |||
| 1 | Standard conditions | 4 h | >99 |
| 2 | CH3CN as solvent | 4 h | 91 |
| 3 | DCM as solvent | 4 h | 54 |
| 4 | No cPMMA-BTPh2 | 4 h | <1 |
| 5 | No light | 4 h | <1 |
| 6 | Under nitrogen | 4 h | <1 |
| 7c | With CuCl2/under nitrogen | 4 h | 71 |
| 8d | With KI | 4 h | 4 |
| 9e | BTPh2 as the catalyst | 2 h | >99 |
Nitromethane outperformed both acetonitrile and dichloromethane for the model reaction (entries 2 and 3). The solvent dependency could be ascribed to the polarity-dependent stabilization of the excited state. Additionally, the solvent-dependent swelling and accessibility of the photocatalytic units could play a major role. The reaction did not proceed without light irradiation or the use of cPMMA-BTPh2 (entries 4 and 5). In the absence of oxygen, no product formation could be observed after 4 h (entry 6). When using CuCl2 as an electron scavenger under a nitrogen atmosphere, an increased reaction conversion (71%) was obtained, indicating that the copper salt can take the role of oxygen as the catalyst regenerator (entry 7). Addition of KI as a photogenerated hole scavenger only led to traces of the desired product (entry 8). The small molecular photocatalyst BTPh2 (applied with a similar loading) shows full conversion of the [4 + 2] adduct within 2 h of reaction time, demonstrating the efficiency of homogeneous catalysis. The PMMA photocatalyst in this study works efficiently on a similar timescale to its small molecular analogue, which hints at the good accessibility of photocatalytically active moieties and diffusion of reactants throughout the network.
In general, the benchmark reaction demonstrates the high efficiency of cPMMA-BTPh2 in catalysing organic reactions and the successful application of the concept. The cPMMA-BTPh2 photocatalyst could be repeatedly used for several cycles without losing its photocatalytic efficiency (Fig. S9†), outperforming the molecular analogue BTPh2 (Fig. S10†). No photobleaching was observed in the UV/vis absorption spectra of cPMMA-BTPh2 after the reaction (Fig. S11†). Solubility tests with nitromethane, chloroform and THF underlined the heterogeneity of the photocatalyst (Fig. S12†). These findings demonstrate the robustness, high stability and reusability of the photocatalytic material designed.
We propose a reaction mechanism based on our observations and previous reports.38–40 Three reaction pathways are possible, as illustrated in Fig. 2. Under light irradiation, the substrate trans-anethole was oxidised by the photogenerated hole of cPMMA-BTPh2 to its cationic radical intermediate, which reacts with isoprene to form a cyclic intermediate. The cyclic intermediate could be reduced by cPMMA-BTPh2 formed via reductive quenching, regenerating the photocatalyst back to its ground state and forming the final product to complete the catalytic cycle (pathway i). In addition, the final product may also be obtained via either oxidation of another neutral trans-anethole by the cyclic intermediate (pathway iii), or oxidation of O2˙−,5 generated by electron transfer from cPMMA-BTPh2 to oxygen (pathway ii). The formation of the electron-activated superoxide radical (O2˙−) was confirmed by electron paramagnetic resonance (EPR) experiments. In a control experiment using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a superoxide radical trapping agent, typical EPR patterns for DMPO–O2˙− adducts were obtained (Fig. 3a).
 | ||
| Fig. 2 Proposed reaction mechanism of the photocatalytic [4 + 2] cycloaddition using cPMMA-BTPh2 as the photocatalyst. | ||
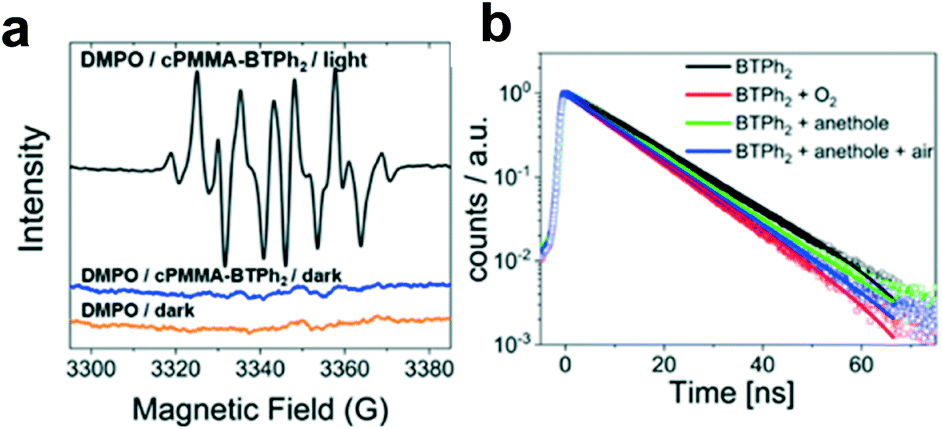 | ||
| Fig. 3 (a) EPR spectra of DMPO–O2˙− adducts with cPMMA-BTPh2 as the photocatalyst in darkness and under blue light irradiation (λ = 460 nm, 1.2 W cm−2). (b) Time-resolved photoluminescence spectra of BTPh2 under N2 (black), under O2 (red), and with trans-anethole under N2 (green) and in air (blue) with 20 mg mL−1 of BTPh2 and 2 mM trans-anethole in acetonitrile. | ||
Time-resolved photoluminescence (TRPL) was used to gain a deeper insight and elucidate the electron transfer between BTPh2 as the photocatalytic unit and substrates. As shown in Fig. 3b, the fluorescence lifetime of BTPh2 in CH3CN after purging with N2 was determined to be 12.9 ns, which was comparable to that of the well-applied organic photocatalysts, such as Acr+–Mes (15 ns)41 and eosin Y (6 ns).42 In the presence of O2, the excited state of BTPh2 could be quenched to a decreased fluorescence lifetime of 10.7 ns. A similar phenomenon was observed when using trans-anethole as a quencher, leading to a reduced fluorescence lifetime of 11.5 ns. This indicated a hole filling process by electron transfer between BTPh2 and trans-anethole.5 By simulating the conditions of the model reaction with trans-anethole in air, a similar decay lifetime of BTPh2 (11.2 ns) was observed, further confirming that the [4 + 2] cycloaddition is hole initiated. Transient absorption (TA) spectra were recorded and are consistent with the TRPL data shown (Fig. S13–S18†).
The versatility of the photocatalytic material was investigated by employing different dienes and dienophiles in the [4 + 2] cycloaddition reaction (Fig. 4). High reaction yields from 76 to 99% were obtained, confirming the general applicability of cPMMA-BTPh2 for organic photoredox reactions. The conversions were found to be selective towards the [4 + 2] cycloaddition. The formation of (2 + 2) cycloaddition products could not be observed.
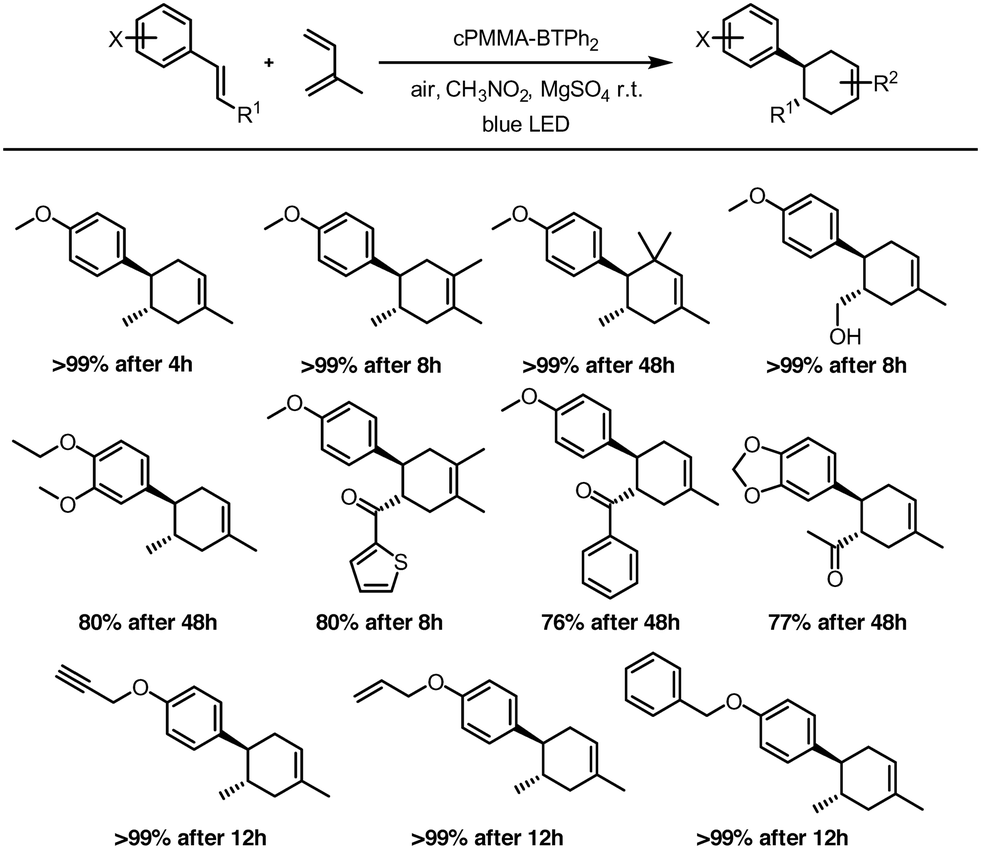 | ||
| Fig. 4 Scope of the photocatalytic Diels–Alder reaction using cPMMA-BTPh2 as the photocatalyst. The reactions were irradiated with a blue LED lamp (460 nm, 0.16 W cm−2). | ||
Conclusions
In summary, we have introduced photocatalytic units into classical polymeric materials as a new class of metal-free, stable and low-cost photocatalysts. Photoactive copolymer organogels based on PMMA show similar optical and redox properties compared to the molecular photocatalytic analogue unit, whilst retaining advantageous chemical robustness and recyclability attributed to their parent polymeric material. With a very low effective photocatalyst loading, 0.06 mol% of the effective photocatalytic unit with respect to the substrate, [4 + 2] cycloaddition reactions could be catalysed with high conversion and selectivity. The PMMA photocatalyst demonstrated excellent stability and recoverability. A deeper mechanistic insight study revealed the electron transfer between the photocatalyst and substrates, involving the electron and hole migration during the catalytic processes. In general, we believe that the concept of introducing photocatalytic units into classical polymeric systems provides a platform for a broad range of new versatile photocatalytic materials. The approach elaborated in this study may open the door to future large-scale applications and photocatalytic materials with improved recyclability and stability.Experimental
Materials and physical methods
All chemicals and solvents were purchased from commercial sources and used as received unless otherwise noted. For the photocatalytic Diels–Alder reaction, all dienes and reaction solvents, i.e. nitromethane, were purified by elution through neutral aluminium oxide (50–200 um) and anhydrous CaCl2 (w/w, 95/5). Flash column chromatography was conducted over silica 60 (0.063–0.2 mm). Reaction yields refer to the pure compounds after being purified via column chromatography. Solid-state diffuse reflectance UV-vis absorption and fluorescence spectra were recorded on a PerkinElmer Lambda 100 spectrophotometer and J&M TIDAS spectrofluorometer at ambient temperature, respectively. EPR (electron paramagnetic resonance) was conducted on a Magnettech MiniScope MS200 spectrometer at room temperature. DMPO (0.1 M) and the catalyst (1 mg mL−1) in acetonitrile were used for the spin trap experiment. Irradiation was conducted with an integrated blue light source. Cyclic voltammetry measurements were carried out on a Metrohm Autolab PGSTAT204 potentiostat/galvanostat with a three-electrode-cell system, glassy carbon electrode as the working electrode, Hg/HgCl2 electrode as the reference electrode, platinum wire as the counter electrode, and Bu4NPF6 (0.1 M in acetonitrile) as the supporting electrolyte, with a scan rate of 100 mV s−1 in the range of −2 eV to 2 eV. GC-MS measurements were performed on a Shimadzu GC-2010 plus gas chromatograph and QP2010 ultra mass spectrometer with a fused silica column (Phenomenex, Zebron-5ms nonpolar) and flame ionization detector. 1H and 13C NMR spectra of all the compounds were measured using a Bruker Avance 300 MHz. Solid State 13C CP MAS NMR measurements were carried out using a Bruker Avance II solid state NMR spectrometer operating at 300 MHz Larmor frequency equipped with a standard 4 mm magic angle spinning (MAS) double resonance probe head. FT-IR measurements were conducted with a Bruker Tensor II FTIR spectrometer. Bright field and fluorescence images were acquired on a Leica DMi8 inverted light microscope. The morphology was recorded with a scanning electron microscope (SEM) (LEO Gemini 1530, Germany) with an in-lens SE detector. Thermogravimetric analysis (TGA) was conducted in a nitrogen atmosphere with temperature increasing from room temperature to 1000 °C at a rate of 10 K min−1. All DFT calculations were carried out using the Gaussian 09 package.43 The structures were optimized at the B3LYP level of theory,44 with the basis set of 6-31G*.45,46 TD-DFT results were obtained from excited state calculations at the same level of theory. Time-resolved photoluminescence (TR-PL) spectra were obtained with a C4742 Hamamatsu streak camera system in slow sweep mode. Excitation pulses at 400 nm were provided by frequency doubling the output of a commercial femtosecond amplifier laser system (Coherent LIBRA-HE).General procedures and synthesis
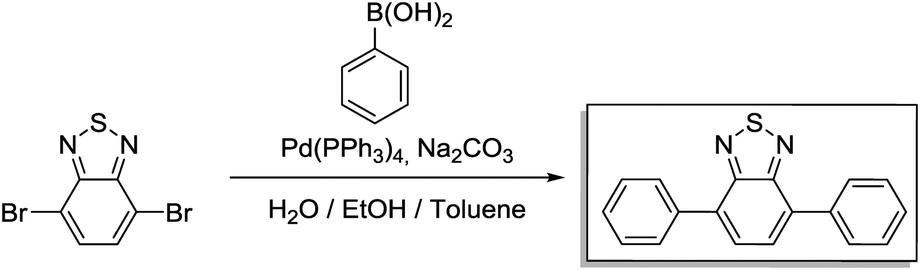 A 250 mL three-necked flask was loaded with 4,7-dibromo-2,1,3-benzothiadiazole (1.47 g 5 mmol), phenylboronic acid (2.68 g, 22 mmol) and toluene (25 mL). Then, a solution of potassium carbonate (2.76 g, 20 mmol) in H2O (10 mL) was added into the flask. After degassing for 30 min, Pd(PPh3)4 (0.047 g, 0.04 mmol) was added, and the reaction mixture was heated to 90 °C and reacted overnight under a N2 atmosphere. After cooling to room temperature, the mixture was poured into water and extracted with dichloromethane. The organic layer was washed with water and dried over anhydrous MgSO4. After concentrating with a rotary evaporator, the crude product was purified via column chromatography on silica with dichloromethane as an eluent. For further purification, the crude product was recrystallized with methanol to afford BTPh2 as yellow needles. Yield: 1.17 g (82%).
A 250 mL three-necked flask was loaded with 4,7-dibromo-2,1,3-benzothiadiazole (1.47 g 5 mmol), phenylboronic acid (2.68 g, 22 mmol) and toluene (25 mL). Then, a solution of potassium carbonate (2.76 g, 20 mmol) in H2O (10 mL) was added into the flask. After degassing for 30 min, Pd(PPh3)4 (0.047 g, 0.04 mmol) was added, and the reaction mixture was heated to 90 °C and reacted overnight under a N2 atmosphere. After cooling to room temperature, the mixture was poured into water and extracted with dichloromethane. The organic layer was washed with water and dried over anhydrous MgSO4. After concentrating with a rotary evaporator, the crude product was purified via column chromatography on silica with dichloromethane as an eluent. For further purification, the crude product was recrystallized with methanol to afford BTPh2 as yellow needles. Yield: 1.17 g (82%).
1H NMR (300 MHz, CDCl3) δ 8.00 (m, 4H), 7.81 (s, 2H), 7.58 (m, 4H), 7.49 (t, 2H). 13C NMR (75 MHz, CDCl3) δ 128.13, 128.38, 128.63, 129.25, 133.39, 137.44, 154.11.
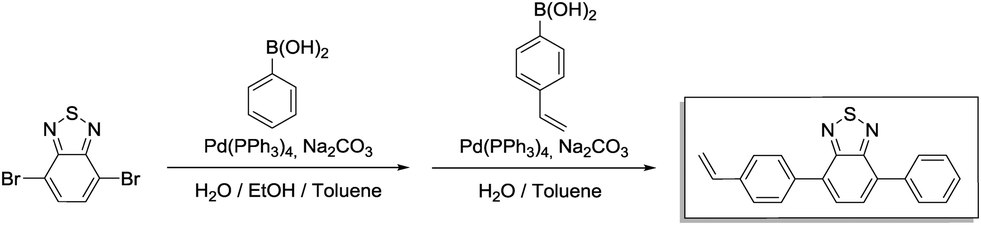 Step 1 (4-bromo-7-phenylbenzo[c][1,2,5]thiadiazole): phenylboronic acid (509 mg, 4.18 mmol, 1.00 eq.), 4,7-dibromobenzo[c][1,2,5]thiadiazole (1.84 g, 6.26 mmol, 1.50 eq.), 17 mL of toluene, 7 mL of 2.0 M aqueous Na2CO3 solution and 7 mL of ethanol were placed in a 100 mL Schlenk flask. After degassing by Ar bubbling for 20 min, Pd(PPh3)4 (144 mg, 125 μmol, 0.03 eq.) was added in an Ar counterstream. The solution was vigorously stirred at 90 °C for 48 h. After cooling to room temperature, the resulting mixture was extracted with dichloromethane (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/dichloromethane 3
Step 1 (4-bromo-7-phenylbenzo[c][1,2,5]thiadiazole): phenylboronic acid (509 mg, 4.18 mmol, 1.00 eq.), 4,7-dibromobenzo[c][1,2,5]thiadiazole (1.84 g, 6.26 mmol, 1.50 eq.), 17 mL of toluene, 7 mL of 2.0 M aqueous Na2CO3 solution and 7 mL of ethanol were placed in a 100 mL Schlenk flask. After degassing by Ar bubbling for 20 min, Pd(PPh3)4 (144 mg, 125 μmol, 0.03 eq.) was added in an Ar counterstream. The solution was vigorously stirred at 90 °C for 48 h. After cooling to room temperature, the resulting mixture was extracted with dichloromethane (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/dichloromethane 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The light green solid (1.75 g) was used as is in the next step.
:1). The light green solid (1.75 g) was used as is in the next step.
Step 2 (4-phenyl-7-(4-vinylphenyl)benzo[c][1,2,5]thiadiazole): the crude product from step 1 (1.75 g), (4-vinylphenyl)boronic acid (1.15 g, 7.83 mmol, 1.3 eq.), 16 mL of toluene and 12 mL of 2.0 M aqueous Na2CO3 were combined in a 100 mL Schlenk tube. After degassing by Ar bubbling for 20 min, Pd(PPh3)4 (139 mg, 120 μmol, 0.02 eq.) was added in an Ar counterstream. The solution was vigorously stirred at 90 °C for 48 h. After cooling to room temperature, water was added (40 mL) and the resulting mixture was extracted with dichloromethane (4 × 20 mL). The combined organic phases were washed with 1 M NaOH (50 mL) and brine (50 mL). After drying over anhydrous MgSO4 and filtration, rotary evaporation yielded the crude product. The residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 20:1 → 0:1). After recrystallization in H2O/MeCN (1:9), the product was obtained as bright yellow needles (950 mg, 2.99 mmol, 72% over 2 steps).
1H NMR (300 MHz, CDCl3) δ 7.97 (d, 4H), 7.79 (s, 2H), 7.58 (m, 4H), 7.48 (q, 1H), 6.82 (q, 1H), 5.86 (d, 1H), 5.34 (d, 1H). 13C NMR (75 MHz, CDCl3) δ 154.27, 154.21, 137.78, 137.56, 136.91, 136.53, 133.50, 133.05, 129.52, 129.38, 128.77, 128.52, 128.27, 127.99, 126.61, 114.67.
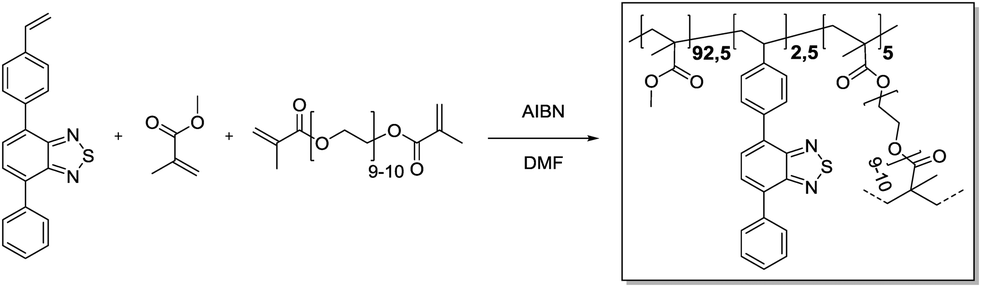 4-Phenyl-7-(4-vinylphenyl)benzo[c][1,2,5]thiadiazole (25.5 mg, 81.1 μmol, 0.025 eq.), methyl methacrylate (315 μL, 3.00 mmol, 0.93 eq.) and polyethylene glycol dimethacrylate (93.0 mg, 55.3 μmol, 0.05 eq.) were added in a 20 mL vial and dissolved in DMF (3.6 mL). Azobisisobutyronitrile (4.26 mg, 25.9 μmol, 0.80 eq.) was added. Then, the vial was capped and the solution was purged with Ar under harsh stirring for 10 min. After heating to 70 °C overnight, gel formation could be observed. The gel was transferred to an extraction thimble, purified by Soxhlet extraction with DCM/MeOH (1:1) for 12 h and dried under a nitrogen stream. After subsequent high vacuum drying, the polymer was obtained as a bright yellow solid (300 mg).
4-Phenyl-7-(4-vinylphenyl)benzo[c][1,2,5]thiadiazole (25.5 mg, 81.1 μmol, 0.025 eq.), methyl methacrylate (315 μL, 3.00 mmol, 0.93 eq.) and polyethylene glycol dimethacrylate (93.0 mg, 55.3 μmol, 0.05 eq.) were added in a 20 mL vial and dissolved in DMF (3.6 mL). Azobisisobutyronitrile (4.26 mg, 25.9 μmol, 0.80 eq.) was added. Then, the vial was capped and the solution was purged with Ar under harsh stirring for 10 min. After heating to 70 °C overnight, gel formation could be observed. The gel was transferred to an extraction thimble, purified by Soxhlet extraction with DCM/MeOH (1:1) for 12 h and dried under a nitrogen stream. After subsequent high vacuum drying, the polymer was obtained as a bright yellow solid (300 mg).
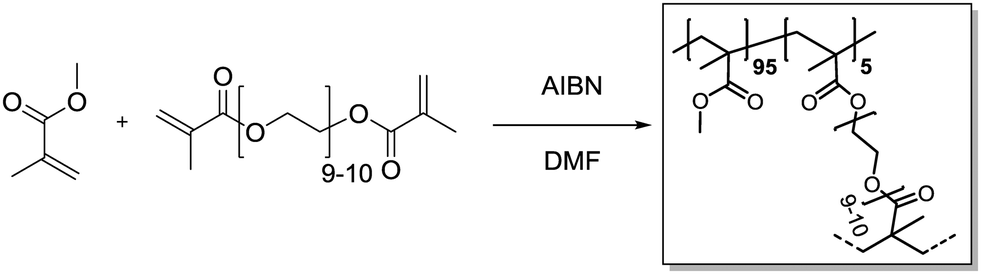 Methyl methacrylate (315 μL, 3.00 mmol, 0.95 eq.) and polyethylene glycol dimethacrylate (101 mg, 60.0 μmol, 0.05 eq.) were added in a 20 mL vial and dissolved in DMF (3.6 mL). Azobisisobutyronitrile (4.26 mg, 25.9 μmol, 0.80 eq.) was added. Then, the vial was capped and the solution was purged with Ar under harsh stirring for 10 min. After heating to 70 °C overnight, gel formation could be observed. The gel was transferred to an extraction thimble, purified by Soxhlet extraction with DCM/MeOH (1:1) for 12 h and dried under a nitrogen stream. After subsequent high vacuum drying, the polymer was obtained as a bright yellow solid (321 mg).
Methyl methacrylate (315 μL, 3.00 mmol, 0.95 eq.) and polyethylene glycol dimethacrylate (101 mg, 60.0 μmol, 0.05 eq.) were added in a 20 mL vial and dissolved in DMF (3.6 mL). Azobisisobutyronitrile (4.26 mg, 25.9 μmol, 0.80 eq.) was added. Then, the vial was capped and the solution was purged with Ar under harsh stirring for 10 min. After heating to 70 °C overnight, gel formation could be observed. The gel was transferred to an extraction thimble, purified by Soxhlet extraction with DCM/MeOH (1:1) for 12 h and dried under a nitrogen stream. After subsequent high vacuum drying, the polymer was obtained as a bright yellow solid (321 mg).
a) (E)-1-(Prop-1-en-1-yl)-4-(prop-2-yn-1-yloxy)benzene.
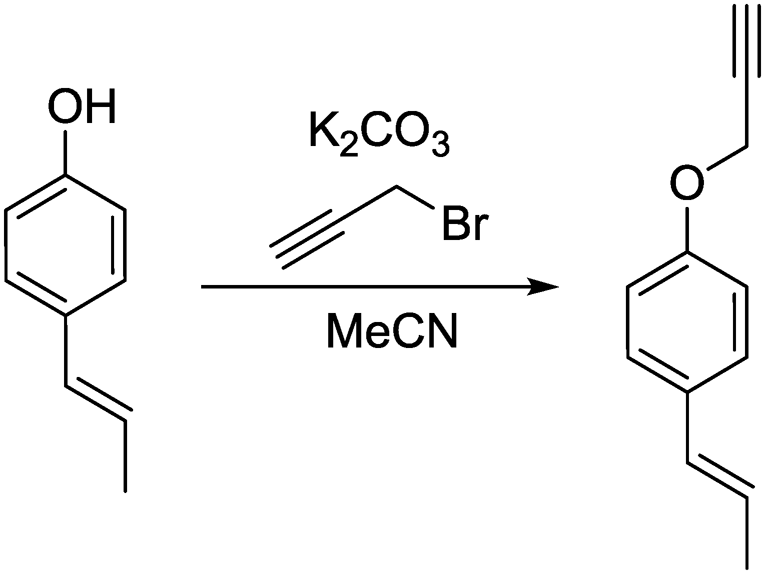 (E)-1-(4-Hydroxyphenyl)propene (200 mg, 1.49 mmol, 1.00 eq.), potassium carbonate (206 mg, 1.49 mmol, 1.00 eq.) and acetonitrile (5 mL) were added to a 20 mL vial. Propargyl bromide (213 mg, 1.79 mmol, 1.20 eq.) was added slowly and the mixture was stirred overnight. The next day, the sample was extracted with ethyl acetate (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 1:0 → petroleum ether/ethyl acetate 3:1). Small amounts of the Z isomer could be detected in the NMR spectra, probably originating from the impurities in the starting material.
(E)-1-(4-Hydroxyphenyl)propene (200 mg, 1.49 mmol, 1.00 eq.), potassium carbonate (206 mg, 1.49 mmol, 1.00 eq.) and acetonitrile (5 mL) were added to a 20 mL vial. Propargyl bromide (213 mg, 1.79 mmol, 1.20 eq.) was added slowly and the mixture was stirred overnight. The next day, the sample was extracted with ethyl acetate (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 1:0 → petroleum ether/ethyl acetate 3:1). Small amounts of the Z isomer could be detected in the NMR spectra, probably originating from the impurities in the starting material.
The resulting oil (234 mg, 91%) was used as a substrate in the photocatalytic reaction.
1H NMR (300 MHz, CDCl3) δ 7.28 (d, 2H), 6.92 (d, 2H), 6.36 (d, 1H), 6.12 (m, 1H), 4.69 (s, 2H), 2.53 (s, 1H), 1.87 (d, 3H). 13C NMR (75 MHz, CDCl3) δ 156.63, 131.84, 130.33, 126.98, 124.14, 115.06, 78.75, 75.58, 55.97, 18.55.
b) (E)-1-(Benzyloxy)-4-(prop-1-en-1-yl)benzene.
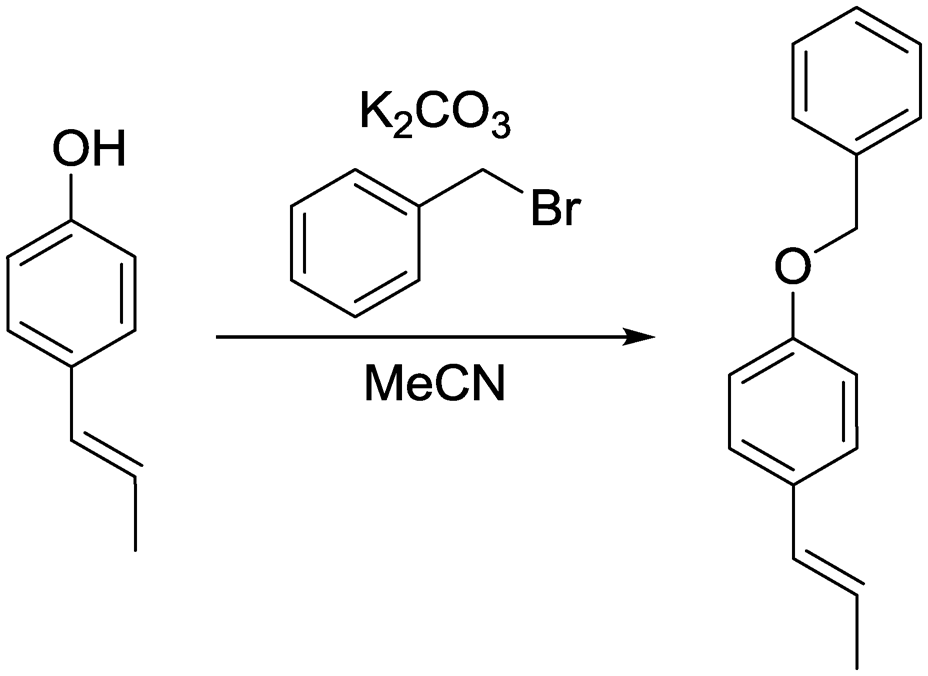 (E)-1-(4-Hydroxyphenyl)propene (201 mg, 1.50 mmol, 1.00 eq.), potassium carbonate (311 mg, 2.25 mmol, 1.50 eq.) and acetonitrile (4 mL) were added to a 20 mL vial. Benzyl bromide (308 mg, 1.80 mmol, 1.20 eq.) was added and the mixture was heated to 80 °C overnight. The next day, the sample was extracted with ethyl acetate (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 1:0 → petroleum ether/ethyl acetate 7:1). The resulting oil (320 mg, 95%) was used as a substrate in the photocatalytic reaction.
(E)-1-(4-Hydroxyphenyl)propene (201 mg, 1.50 mmol, 1.00 eq.), potassium carbonate (311 mg, 2.25 mmol, 1.50 eq.) and acetonitrile (4 mL) were added to a 20 mL vial. Benzyl bromide (308 mg, 1.80 mmol, 1.20 eq.) was added and the mixture was heated to 80 °C overnight. The next day, the sample was extracted with ethyl acetate (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 1:0 → petroleum ether/ethyl acetate 7:1). The resulting oil (320 mg, 95%) was used as a substrate in the photocatalytic reaction.
1H NMR (300 MHz, CDCl3) δ 7.32 (m, 5H), 7.20 (d, 2H), 6.85 (d, 2H), 6.28 (d, 1H), 6.02 (m, 1H), 4.99 (s, 2H), 1.80 (d, 3H). 13C NMR (75 MHz, CDCl3) δ 157.92, 137.20, 131.21, 130.45, 128.70, 128.06, 127.59, 127.02, 123.76, 115.01, 70.16, 18.56.
c) (E)-1-(Allyloxy)-4-(prop-1-en-1-yl)benzene.
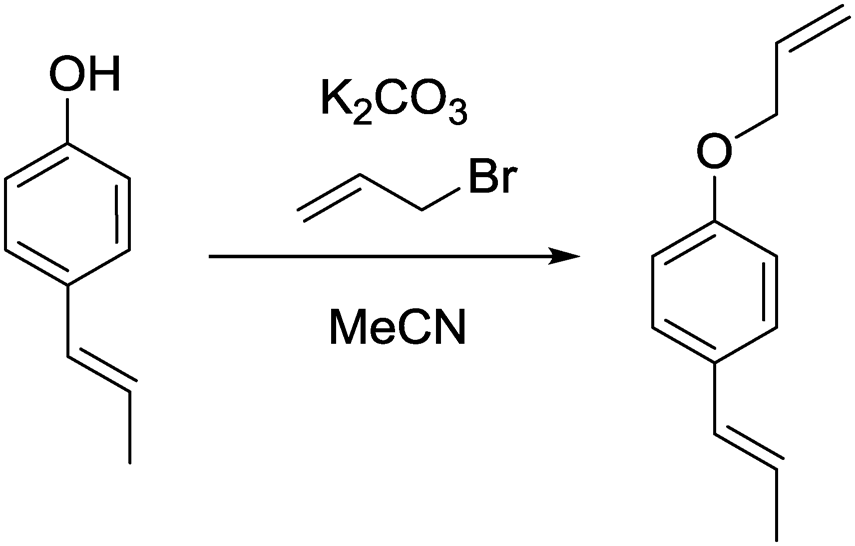 (E)-1-(4-Hydroxyphenyl)propene (200 mg, 1.49 mmol, 1.00 eq.), potassium carbonate (206 mg, 1.49 mmol, 1.00 eq.) and acetonitrile (5 mL) were added to a 20 mL vial. Allyl bromide (216 mg, 1.79 mmol, 1.20 eq.) was added and the mixture stirred overnight. The next day, the sample was extracted with ethyl acetate (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 1:0 → petroleum ether/ethyl acetate 7:1). The resulting oil (244 mg, 94%) was used as a substrate in the photocatalytic reaction.
(E)-1-(4-Hydroxyphenyl)propene (200 mg, 1.49 mmol, 1.00 eq.), potassium carbonate (206 mg, 1.49 mmol, 1.00 eq.) and acetonitrile (5 mL) were added to a 20 mL vial. Allyl bromide (216 mg, 1.79 mmol, 1.20 eq.) was added and the mixture stirred overnight. The next day, the sample was extracted with ethyl acetate (4 × 20 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. After filtration and rotary evaporation of solvents, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 1:0 → petroleum ether/ethyl acetate 7:1). The resulting oil (244 mg, 94%) was used as a substrate in the photocatalytic reaction.
1H NMR (300 MHz, CDCl3) δ 7.28 (d, 2H), 6.88 (d, 2H), 6.37 (d, 1H), 6.10 (m, 2H), 5.45 (d, 2H), 5.31 (d, 2H), 4.56 (d, 2H), 1.89 (d, 3H). 13C NMR (75 MHz, CDCl3) δ 157.71, 133.47, 131.09, 130.45, 126.97, 123.67, 117.72, 114.87, 68.95, 18.54.
General procedure of photocatalysis
A 20 mL vial with a magnetic stir bar was loaded with cPMMA-BTPh2, alkene (1.0 eq.), diene (10 eq.) and nitromethane. Then, the vial was capped and placed under irradiation with a blue LED lamp (0.061 W cm−2). The reaction mixture was stirred at room temperature and the conversion determined by GC-MS. Afterwards, the mixture was transferred into a separatory funnel containing DCM and H2O (v/v, 1/1). The organic layers were separated and extracted thrice with DCM. The combined organic layers were washed with brine, dried over anhydrous MgSO4 and concentrated by rotary evaporation. The crude product was purified via column chromatography on silica to afford the pure compound.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Max Planck Society for the financial support. N. H. acknowledges the support from the Fonds der Chemischen Industrie (FCI) for a Kekulé fellowship as well as the Gutenberg-Akademie of JGU Mainz for a junior membership. K. A. I. Z. thanks the Deutsche Forschungsgemeinschaft (DFG) for funding. This work is part of the research conducted by the MaxSynBio consortium that is jointly funded by the Federal Ministry of Education and Research of Germany (BMBF) and the Max Planck Society (MPG). Open Access funding provided by the Max Planck Society.Notes and references
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. H. Xu, S. B. Jin, H. Xu, A. Nagai and D. L. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. J. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- L. Wang, R. Fernandez-Teran, L. Zhang, D. L. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- A. Hayat, N. Shaishta, S. K. B. Mane, A. Hayat, J. Khan, A. U. Rehman and T. H. Li, J. Colloid Interface Sci., 2020, 560, 743–754 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- C. L. Su, R. Tandiana, B. B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- Y. Chen, J. S. Zhang, M. W. Zhang and X. C. Wang, Chem. Sci., 2013, 4, 3244–3248 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Mater., 2015, 27, 1921–1924 CrossRef CAS.
- S. Ghasimi, S. Prescher, Z. J. Wang, K. Landfester, J. Yuan and K. A. I. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14549–14553 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- C. Lu, P. Zhang, S. Jiang, X. Wu, S. Song, M. Zhu, Z. Lou, Z. Li, F. Liu, Y. Liu, Y. Wang and Z. Le, Appl. Catal., A, 2017, 200, 378–385 CrossRef CAS.
- A. Hayat, F. Raziq, M. Khan, I. Ullah, M. U. Rahman, W. U. Khan, J. Khan and A. Ahmad, J. Photochem. Photobiol., A, 2019, 379, 88–98 CrossRef CAS.
- J. Geng, C. Sun, J. Liu, L.-D. Liao, Y. Yuan, N. Thakor, J. Wang and B. Liu, Small, 2015, 11, 1603–1610 CrossRef CAS PubMed.
- L. Xu, L. Cheng, C. Wang, R. Peng and Z. Liu, Polym. Chem., 2014, 5, 1573–1580 RSC.
- K. Yang, H. Xu, L. Cheng, C. Sun, J. Wang and Z. Liu, Adv. Mater., 2012, 24, 5586–5592 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486–1493 RSC.
- J. Liu, R. Cazelles, Z. P. Chen, H. Zhou, A. Galarneau and M. Antonietti, Phys. Chem. Chem. Phys., 2014, 16, 14699–14705 RSC.
- K. T. Oppelt, J. Gasiorowski, D. A. M. Egbe, J. P. Kollender, M. Himmelsbach, A. W. Hassel, N. S. Sariciftci and G. Knör, J. Am. Chem. Soc., 2014, 136, 12721–12729 CrossRef CAS PubMed.
- H. Shimakoshi, M. Nishi, A. Tanaka, K. Chikama and Y. Hisaeda, Chem. Commun., 2011, 47, 6548–6550 RSC.
- D. Rackl, P. Kreitmeier and O. Reiser, Green Chem., 2016, 18, 214–219 RSC.
- C. Boussiron, M. Le Bechec, L. Petrizza, J. Sabalot, S. Lacombe and M. Save, Macromol. Rapid Commun., 2019, 40, 1800329 CrossRef PubMed.
- C. T. J. Ferguson, N. Huber, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10567–10571 CrossRef CAS PubMed.
- W. Y. Lu, C. M. Sun, Q. Lu, N. Li, D. Z. Wu, Y. Y. Yao and W. X. Chen, Sci. China: Chem., 2012, 55, 1108–1114 CrossRef.
- W. J. Yoo and S. Kobayashi, Green Chem., 2014, 16, 2438–2442 RSC.
- J. M. Tobin, T. J. D. McCabe, A. W. Prentice, S. Holzer, G. O. Lloyd, M. J. Paterson, V. Arrighi, P. A. G. Cormack and F. Vilela, ACS Catal., 2017, 7, 4602–4612 CrossRef CAS.
- F. S. Mancilha, B. A. DaSilveira Neto, A. S. Lopes, P. F. Moreira, F. H. Quina, R. S. Gonçalves and J. Dupont, Eur. J. Org. Chem., 2006, 2006, 4924–4933 CrossRef.
- D. Aldakov, M. A. Palacios and P. Anzenbacher, Chem. Mater., 2005, 17, 5238–5241 CrossRef CAS.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS PubMed.
- S. S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed.
- Y. B. Zhao and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 9336–9340 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715–722 RSC.
- A. U. Meyer, T. Slanina, C. J. Yao and B. Konig, ACS Catal., 2016, 6, 369–375 CrossRef CAS.
- M. Frisch, G. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, revision D. 01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data such as fluorescence, EPR, UV-vis, additional transient absorption spectra, and 1H, 13C NMR spectra are shown. See DOI: 10.1039/d0cy00016g |
| This journal is © The Royal Society of Chemistry 2020 |