
PtNixCoy concave nanocubes: synthesis and application in photocatalytic hydrogen generation†
Xinjia
Jia
a,
Shuang
Liu
a,
Li
Huang
a,
Perumal
Devaraji
a,
Laxman
Walekar
a,
Wei
Chen
 ab,
Xiying
Li
ab,
Shanhu
Liu
b and
Liqun
Mao
*a
ab,
Xiying
Li
ab,
Shanhu
Liu
b and
Liqun
Mao
*a
aHenan Engineering Research Center of Resource & Energy Recovery from Waste, Henan University, Kaifeng 475004, PR China. E-mail: mlq@henu.edu.cn; Tel: +86 13513781969
bInstitute of Functional Polymer Composites, College of Chemistry and Chemical Engineering, Henan University, Kaifeng 475004, PR China
First published on 18th November 2019
Abstract
PtNixCoy concave nanocubes (CNCs) are synthesized using a hydrothermal method and decorated on the surface of CdS via a fast photo-deposition method (denoted as PtNixCoy/CdS). The photocatalytic hydrogen evolution activity is investigated under visible light irradiation, and Pt/CdS exhibits a good hydrogen generation rate of about 37.80 mmol h−1 g−1 (QE = 44.64%, λ = 420 nm). Moreover, the performance of the PtNixCoy CNCs is enhanced with the addition of Ni and Co, and the hydrogen generation rate of PtNiCo/CdS is as high as 50.60 mmol h−1 g−1 (QE = 56.82%, λ = 420 nm), which is 1.3-fold higher than that of Pt/CdS and also significantly higher than those of PtNi/CdS (42.71 mmol h−1 g−1) and PtCo0.5/CdS (40.33 mmol h−1 g−1). The high photocatalytic performance could be explained by the high surface index and the highly unsaturated atom coordination of PtNiCo CNCs, and the synergistic effects of the tri-metals, which enhance the capturing ability of photo-generated electrons, and finally result in high charge separation and high charge transport between the alloy CNCs and CdS at the interface.
1 Introduction
Nowadays, the use of semiconductors as photocatalysts to split water for hydrogen generation using solar energy is regarded as one of the ideal ways to solve energy poverty and environmental pollution issues.1–5 However, the bottleneck of the rapid electron–hole recombination kinetics of photocatalysts resulting in low energy conversion efficiency is impeding industrial applications, so the key for breaking through this bottleneck is to develop high-efficiency catalysts in the visible light region.6,7 As an excellent photocatalyst candidate, CdS has received great attention in photocatalytic hydrogen generation reactions due to its low band gap (about 2.4 eV) and suitable band edge position for proton reduction.8,9 Nevertheless, CdS alone is inactive for photocatalytic hydrogen generation via water-splitting using solar energy, and is always coupled with noble metals to improve its performance.10–12 It has been well demonstrated that among all metals, Pt nanoparticles (NPs) are well-recognized as the most effective co-catalyst in multifarious photocatalytic reactions, but their wide-scale application is greatly limited because of the high price and scarcity of Pt resources. Therefore, minimizing the amount of Pt used while increasing the catalytic activity and stability has always been a focus of researchers.13–16There is no doubt that the catalytic properties of Pt nanocrystals are dependent on their size, shape and composition, which has been proven in previous reports.17,18 In recent years, the fabrication of Pt concave nanocubes (Pt CNCs) has become of great interest. Concave nanocubes usually have high-index facets with a high density of atomic steps, ledges, and kinks, so it is easy for them to interact with the reactant molecules and become a catalytically active center, thus exhibiting excellent catalytic performance.19–22 Up to now, many synthetic approaches have been successfully exploited for preparing concave nanocubes, such as anisotropic etching methodology,23 one-pot hydrothermal methods,20,24 seed-mediated growth25 and galvanic replacement reactions.26 As everyone knows, the high-index facets disappear during the growth of crystals thanks to their high surface energy. In a facile one-pot hydrothermal synthesis process, high-index facets could be preserved by a preferential adsorption surface control agent in solution.27 More recently, Zheng and co-workers reported the preparation of Pt CNCs with high index {411} and {100} facets, which exhibit a highly superior electrocatalytic activity compared to commercial Pt black or Pt/C in the electro-oxidation of formic acid and ethanol.19 Xia et al. also used Pt CNCs enclosed by {720} facets as an electrocatalyst for the oxygen reduction reaction (ORR), and compared the obtained results with that of Pt nanocubes, cuboctahedrons and commercial Pt/C catalysts as well. It should be noted that Pt CNCs exhibit substantially high specific activity, which is three times higher than that of commercial Pt/C, showing promise to serve as the next-generation ORR electrocatalysts in proton exchange membrane (PEM) fuel cells.22 Also, it has been reported that Pt-based alloy CNCs (Pt-M CNCs) can be synthesized with other low cost non-noble metals (M) (such as M = Ni, Co, Cu, Fe, etc.), which not only reduces the cost of the catalyst, but also enhances the catalytic performance.28–33 Sun and co-workers reported that a Pt–Ni CNC alloy with high-index facets was synthesized via a wet-chemical method, and the experimental results revealed that the Pt–Ni CNC alloy has substantially higher oxygen reduction activity and stability relative to those of Pt–Ni nanocubes and Pt black.28 Fang and co-worker also prepared Pt–Co CNCs with an open structure and demonstrated that the active atomic sites stand on high-index facets and show an enhanced catalytic activity as compared to the activity of Pt–Co nanocubes and Pt nanocubes of the same size with a low-index surface for styrene hydrogenation reaction.29
Although there are several reports on Pt-based alloys functioning as a co-catalyst of a semiconductor for water-splitting,12–14,16 the use of Pt-based alloy CNCs as a co-catalyst has not yet been reported. Herein, in our work, PtNixCoy CNCs are successfully synthesized via a simple one-pot hydrothermal method and deposited on the surface of CdS nanorods by photo-induced deposition. With (NH4)2SO3 acting as the sacrificial agent of photo-induced holes, the hydrogen generation rate of Pt/CdS is 37.80 mmol h−1 g−1 (QE = 44.64%, λ = 420 nm). When Ni or Co is introduced to form binary alloy CNCs, the hydrogen generation rates of PtNi/CdS and PtCo0.5/CdS are 42.71 mmol h−1 g−1 and 40.33 mmol h−1 g−1, respectively. Surprisingly, when Ni and Co are simultaneously introduced to form ternary alloy CNCs, the hydrogen generation rate of PtNiCo/CdS is as high as 50.60 mmol h−1 g−1 (QE = 56.82%, λ = 420 nm), which is 1.3 times higher than that of Pt/CdS and also higher than those of PtNi/CdS and PtCo0.5/CdS. In addition, a comparative experiment shows that the hydrogen generation rate of PtNixCoy CNC-decorated CdS is significantly higher than that of sphere-like PtNixCoy NP-modified CdS. Therefore, it's clearly seen that the concave structure of PtNixCoy benefits the hydrogen production rate of CdS.
2 Experimental section
2.1 Preparation of photocatalysts
All the chemicals were analytical grade and used as such without further purification. H2PtCl6, NiCl2 and CoCl2 were used as Pt, Ni and Co precursors, respectively. Glycine and polyvinyl pyrrolidone (PVP) were used as reducing and capping agents, respectively.Pt CNCs were synthesized by a hydrothermal method. In a typical synthesis procedure, 1 mL of H2PtCl6 aqueous solution (0.1 mol L−1) was added into 20 mL of glycine solution (6.6 × 10−3 mol L−1), and then the mixture was sonicated for 20 min. Afterwards, 65 mg of glycine and 400 mg of PVP aqueous solution were added to the above mixture and sonicated for 10 more min. The resulting yellow homogeneous solution was further diluted to 25 mL using DI (deionized water), and then it was transferred to a 50 mL Teflon-lined stainless-steel autoclave and subjected to heat treatment in the range of 433–513 K for 5–15 h. The as-resultant black product was centrifuged and washed with acetone and absolute alcohol several times, and dried at 333 K for 12 h in a vacuum furnace. PtNi CNCs, PtCo CNCs and PtNiCo CNCs were prepared with the same hydrothermal method as that of Pt CNCs at 473 K for 10 h. The sphere-like Pt NPs and PtNiCo NPs were prepared with the same hydrothermal method as that of Pt CNCs at 473 K for 10 h except that no PVP was added.
CdS nanorods were synthesized through a solvothermal method at 433 K for 48 h.34,35 The as-resultant CdS nanorods are of single-crystal nanorod structure, with an estimated diameter and length of 40 nm and dozens of microns, respectively.
2.2 Characterization
Powder X-ray diffraction (XRD) (Philips, XPert Pro) with Cu Kα radiation was used to determine the crystalline structure of the as-prepared Pt CNCs. The diffraction scanning range was from 5 to 90 (2θ°) with a scan rate of 0.06° s−1. A high-resolution transmission electron microscope (HRTEM) (JEM-2100, Jeol Ltd., Japan) was used to analyze the grain size and surface microstructure of the Pt NPs. The surface element composition was measured with X-ray photoelectron spectroscopy (XPS, AlKα source). The Brunauer–Emmett–Teller (BET Micromeritics NOVA1000, America) specific surface area values were obtained from nitrogen adsorption data at 77 K. A UV-2600 UV/vis spectrophotometer (Shimadzu, Japan) was utilized to record UV/vis diffuse reflection spectra (DRS) in order to determine the absorbance via the Kubelka–Munk method. An FLS 980 (Edinburgh analytical instruments, UK) was used to recorded the PL spectra and lifetime of the prepared materials with a 450 W xenon lamp and a μF900H high-energy microsecond flash lamp as a source.2.3 Evaluation of photo-catalytic activity
The photocatalytic properties were measured by using a photoreactor with a water-jacketed stainless vessel with a diameter and height of 14.0 cm × 10.0 cm. In the above setup, a quartz window (diameter 7.0 cm, exposed area 38.0 cm2) was used to pass the light. A 300 W xenon lamp (Beijing Perfect light Company; Beijing, China) and visible light filter (λ > 420 nm) were used as the light source. The photolyte reaction temperature was maintained at 283 ± 1 K with the help of circulating isothermal water. For the photocatalytic water splitting reaction, 0.1 g of as-prepared CdS photocatalyst was taken and mixed well with PtNixCoy CNCs; after that, the slurry was transferred into 1.0 M (NH4)2SO3 aqueous solution, and finally deionized water was added to keep the reaction solution at 50 mL. The final solution was treated with N2 flow (>99.999%) for 30 min with constant stirring to remove the dissolved oxygen in the solution. Under visible light illumination, the surface of the CdS photocatalyst was decorated by PtNixCoy CNCs through a photo-induced electron deposition method.36,37 The measurement and calculation of the quantum efficiency are available in the ESI.†2.4 Electrochemical measurements
Linear sweep voltammetry (LSV) and transient photocurrent measurements were conducted in a standard three-electrode system using 300 W xenon lamp light irradiation. A saturated calomel electrode and platinum wire were used as a reference and counter electrode, respectively. A glassy-carbon electrode (diameter: 3 mm, area: 0.07 cm2) with the catalyst was used as the working electrode, and 0.5 M aqueous Na2SO4 solution was used as an electrolyte for all of the electrochemical measurements. The measured potentials are all converted with reference to a reversible hydrogen electrode (RHE) for convenience of comparison.3 Results and discussion
3.1 Photocatalytic activity for the hydrogen evolution reaction
Firstly, the effect of synthesis temperature and time of the Pt CNCs on the hydrogen production rate is presented in Fig. S1a–d.† The experimental testing results suggested that the synthesis conditions might have an effect on the structure of the Pt CNCs, which results in a difference in catalytic performance, and also this observation indicates the optimal conditions of Pt CNC synthesis (473 K, 10 h). The structural characterization of the Pt CNCs subsequently confirmed this deduction. The hydrogen production rate curves of Pt/CdS photocatalysts with different loading amounts of Pt CNCs are shown in Fig. S1e and f;† the optimal loading of Pt CNCs is 2% and the hydrogen production rate of Pt/CdS is 37.80 mmol h−1 g−1, (QE = 44.64%, λ = 420 nm). Secondly, Fig. S2a–d† shows the hydrogen generation activity of PtNix/CdS and PtCoy/CdS. At a mole ratio of Pt/Ni = 1/1 and Pt/Co = 1/0.5, the hydrogen generation rates of PtNi/CdS and PtCo0.5/CdS reached a maximum of about 42.71 mmol h−1 g−1 and 40.33 mmol h−1 g−1, respectively, which was higher than that of the Pt/CdS photocatalyst. Thirdly, to further enhance the hydrogen generation rate of the CdS photocatalyst, PtNixCoy ternary alloy CNC co-catalysts were synthesized and loaded on the surface of CdS. Fig. 1a–d exhibits the hydrogen evolution rates of PtNixCo/CdS and PtNiCoy/CdS photocatalysts. With the increase of Ni, the hydrogen evolution rate of PtNixCo/CdS increased initially, followed by a decrease. Similarly, the hydrogen evolution rate of PtNiCoy/CdS also shows a trend of increasing first and then decreasing with the increase of Co. When the mole ratio of Pt–Ni–Co is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, the hydrogen generation rate of PtNiCo/CdS is as high as 50.60 mmol h−1 g−1 (QE = 56.82%, λ ≥ 420 nm), which is 1.2 times that of PtNi/CdS, 1.3 times that of PtCo0.5/CdS, and 1.3 times that of Pt/CdS. The high photocatalytic activity for PtNiCo/CdS is attributed to the electronic interaction of Pt, Ni and Co, which accelerates the separation of photo-generated carriers to minimize their recombination. Hence, addition of Ni and Co to form ternary alloy CNCs not only further enhances the hydrogen evolution rate of CdS, but also further decreases the cost of the catalyst. Finally, a comparison of the hydrogen production rate of PtNixCoy modified CdS with different structures is shown in Fig. S3.† The experimental results imply that the hydrogen production rate of PtNixCoy CNC-decorated CdS is significantly higher than that of sphere-like PtNixCoy NP-modified CdS. The N2 adsorption–desorption isotherms (Fig. S4†) and the BET surface area (Table S1†) indicate that PtNixCoy CNC-decorated CdS has more active sites than sphere-like PtNixCoy NP-modified CdS, and facilitates good photocatalytic performance.
:1:1, the hydrogen generation rate of PtNiCo/CdS is as high as 50.60 mmol h−1 g−1 (QE = 56.82%, λ ≥ 420 nm), which is 1.2 times that of PtNi/CdS, 1.3 times that of PtCo0.5/CdS, and 1.3 times that of Pt/CdS. The high photocatalytic activity for PtNiCo/CdS is attributed to the electronic interaction of Pt, Ni and Co, which accelerates the separation of photo-generated carriers to minimize their recombination. Hence, addition of Ni and Co to form ternary alloy CNCs not only further enhances the hydrogen evolution rate of CdS, but also further decreases the cost of the catalyst. Finally, a comparison of the hydrogen production rate of PtNixCoy modified CdS with different structures is shown in Fig. S3.† The experimental results imply that the hydrogen production rate of PtNixCoy CNC-decorated CdS is significantly higher than that of sphere-like PtNixCoy NP-modified CdS. The N2 adsorption–desorption isotherms (Fig. S4†) and the BET surface area (Table S1†) indicate that PtNixCoy CNC-decorated CdS has more active sites than sphere-like PtNixCoy NP-modified CdS, and facilitates good photocatalytic performance.
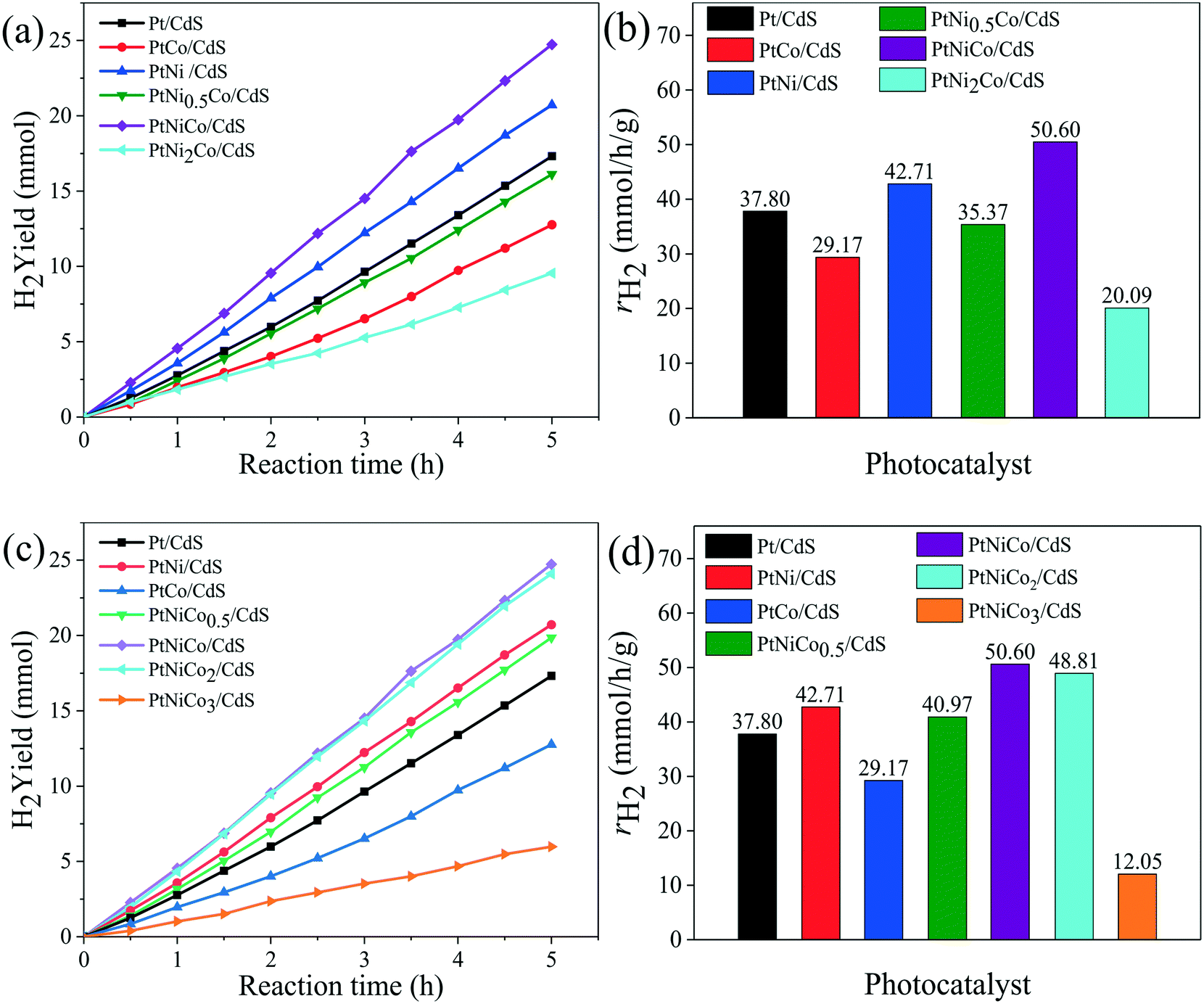 | ||
| Fig. 1 The hydrogen production rate of (a) PtNixCo/CdS (mole ratio of x varied from 0 to 2 with mole ratio of Pt/Co = 1) and (c) PtNiCoy/CdS (mole ratio of y varied from 0 to 3 with mole ratio of Pt/Ni = 1) photocatalysts. The hydrogen production rate of (b) PtNixCo/CdS (x = 0, 0.5, 1, 2) and (d) PtNiCoy/CdS (y = 0, 0.5, 1, 2, 3) when its activity becomes stable. The reaction conditions: 0.1 g CdS photocatalyst (loading amount of PtNixCoy CNCs is 2 wt%), 50 mL of 1 M (NH4)2SO3 solution, and a 300 W xenon lamp (λ ≥ 420 nm). | ||
To further analyze the stability of the photocatalyst, four repeated 20 h cycling experiments of the PtNiCo/CdS photocatalyst were performed under visible light irradiation. At the end of each cycle the photocatalyst was filtered, dried and reused for the next cycle. As shown in Fig. 2, after each cycle, the hydrogen production activity of the PtNiCo/CdS photocatalyst slightly decreased because of the loss of photocatalyst during washing or photocorrosion of CdS. After four cycles, the hydrogen production rate of the PtNiCo/CdS photocatalyst decreased to 40.03 mmol h−1 g−1, which is 81% of the original rate (49.55 mmol h−1 g−1). Therefore, this result (without obvious decline) indicates that our PtNiCo/CdS material can act as a stable and effective photocatalyst for photocatalytic hydrogen production.
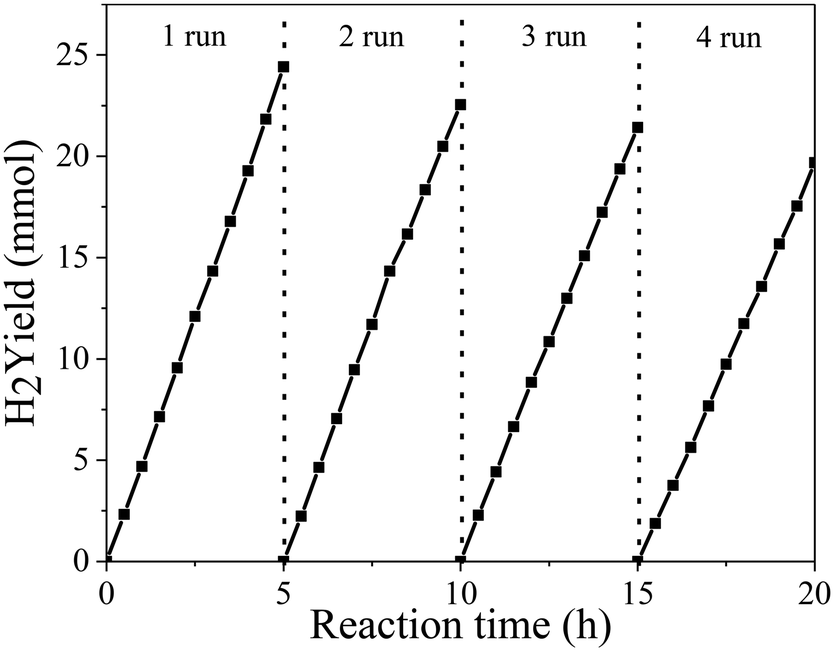 | ||
| Fig. 2 Recycling activity test of photocatalytic hydrogen production of the PtNiCo/CdS photocatalyst under visible light irradiation. | ||
3.2 Electrochemical measurement
To reveal the nature of the PtNixCoy CNCs in increasing the hydrogen production activity of CdS, the hydrogen evolution reaction of CdS and PtNixCoy/CdS was performed by a LSV method (shown in Fig. 3a). It is very clear that the hydrogen evolution current density of PtNixCoy/CdS is significantly enhanced as compared to that of bare CdS. The increasing order of photocurrent density is as follows: CdS < Pt/CdS < PtNi/CdS < PtNiCo/CdS, which is consistent with the hydrogen evolution rate curves (Fig. 1), implying that PtNixCoy CNCs as co-catalysts can effectively improve the hydrogen evolution activity of CdS.38 Furthermore, Fig. 3b shows the Tafel plots from LSV data, and the overpotential value is shown in Table S2.† The PtNixCoy/CdS photocatalysts display much lower overpotential than bare CdS. Specifically, the low overpotential after PtNixCoy CNC loading demonstrated that it has a significant role in decreasing the hydrogen evolution overpotential of CdS. More clearly, the overpotential of the PtNiCo/CdS (0.63 V) photocatalyst is also lower than that of the PtNi/CdS (0.65 V) and Pt/CdS (0.66 V) photocatalysts, which indicated that compared to the PtNi/CdS and Pt/CdS photocatalysts, the hydrogen generation reaction is more likely to happen on the PtNiCo/CdS photocatalyst.39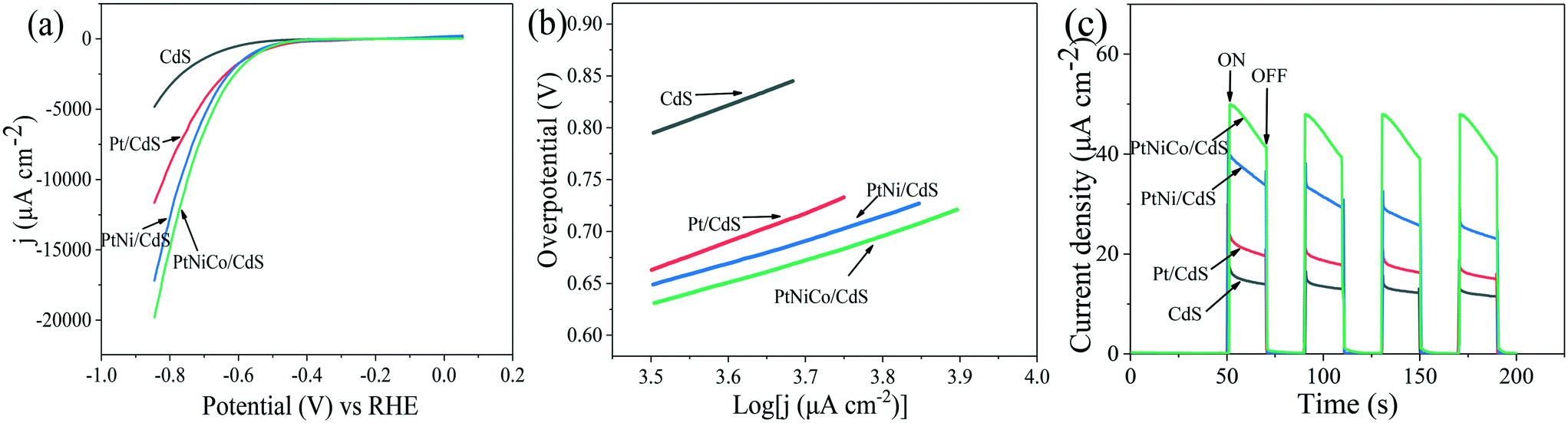 | ||
| Fig. 3 (a) LSV spectra, (b) Tafel plot and (c) I–t curves of the CdS, Pt/CdS, PtNi/CdS and PtNiCo/CdS photocatalysts using 0.5 M Na2SO4 aqueous solution as an electrolyte at pH = 7. | ||
The transient photocurrent responses of the CdS and PtNixCoy/CdS photocatalysts were determined to demonstrate the enhanced photo-induced electron–hole separation. Fig. 3c displays that the photocurrent of PtNixCoy/CdS is distinctly higher than that of bare CdS, implying that the separation efficiency of photo-generated electron–hole pairs is improved after the PtNixCoy CNCs were deposited on the CdS nanorods. Moreover, the PtNiCo/CdS exhibits the strongest photocurrent compared to other photocatalysts, indicating the highest separation and transfer efficiency of photo-induced electrons and holes, further proving that PtNiCo/CdS has the best photocatalytic performance. This is mainly attributed to the fact that the concave nanocube structure of the PtNiCo CNCs and synergistic ternary metal result in an enhanced ability to capture electrons.40,41
3.3 Structure characterization
Fig. 4a–f show the TEM images of Pt CNCs under different synthesis temperatures and times. It can be seen that the average diameter of the Pt nanocubes is 11–13 nm at a low temperature (Fig. 4a, 433 K). As the synthesis temperature increases, Pt nanocubes (the red arrow in Fig. 4b) and Pt octopods (the blue arrow in Fig. 4b) were generated in the reaction solution, and the average apex-to-apex diameters of the Pt CNCs are 18–19 nm for 453 K (Fig. 4b), 12–14 nm for 473 K (Fig. 4c), and 14–16 nm for 493 K (Fig. 4d), respectively. Moreover, keeping the synthesis temperature at 473 K and increasing the synthesis time from 5 h to 15 h, both the morphology and the particle size didn't exhibit obvious changes (Fig. 4c, e, and f). These test results indicate that the synthesis temperature is a key factor in shape-controlled manufacture. The preparation of a concave structure is thermodynamically forbidden because of the low Gibbs free energy in the concave region. However, this forbidden nature can be overcome by using a proper capping agent or performing synthesis in a kinetically controlled manner. Xia et al. found that PVP can selectively passivate {100} facets and reduce their growth rate, which results in the formation of nanocubes or five-fold twinned nanorods.42 Zheng et al. synthesized Pt nanocrystals with a concave structure on the {411} plane. A unique octopod morphology was obtained by introducing methylamine as a capping agent.19 They concluded that the preferential {100} growth orientation of the Pt nanocrystals was responsible for the unidirectional overgrowth and thus helps to form concave structures. In this case, the typical product synthesized at 433 K is a cube in shape. As the synthesis temperature increased to 453 K, the products were mainly in a concave nanocube shape. The synthesis temperature (high) is responsible for the highest rate of conversion from precursor (H2PtCl6) to atoms. In the growth processes, the binding of PVP and amine groups on the side faces of CdS occurs, and then it forms a CNC-like structure.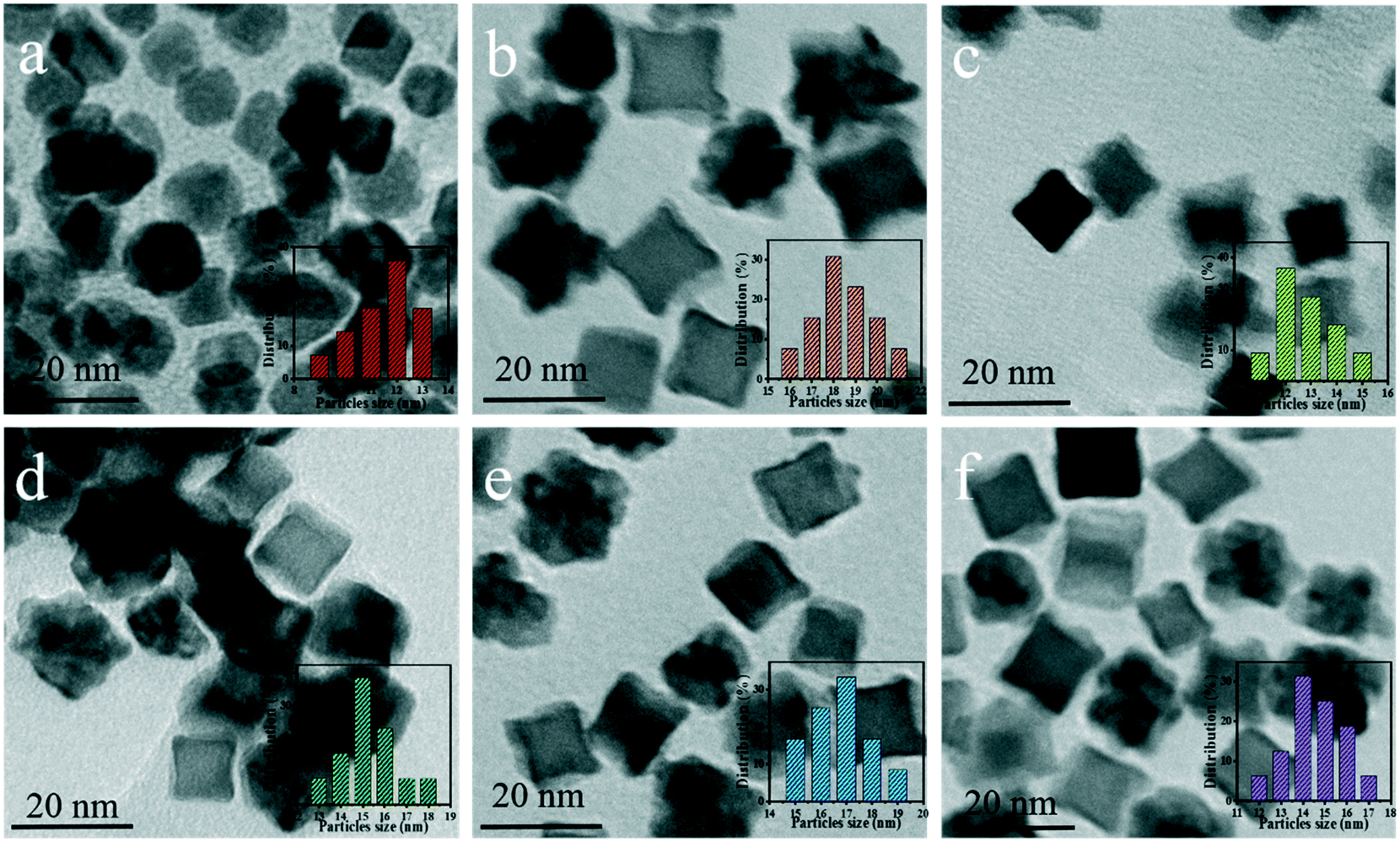 | ||
| Fig. 4 TEM images of the as-prepared Pt CNCs under different reaction temperatures and times: (a) 10 h at 433 K, (b) 10 h at 453 K, (c) 10 h at 473 K, (d) 10 h at 493 K, (e) 5 h at 473 K, and (f) 15 h at 473 K. | ||
Typical high-resolution images (HRTEM) of Pt CNCs and PtNiCo CNCs synthesized at 473 K for 10 h are shown in Fig. 5. The Pt nanoparticles display an obvious concave structure with a diameter of 17 nm (vertex-to-vertex). The surface in Fig. 5a is the projective plane of Pt CNCs and the labeled crystalline interplanar spacings of 0.19 nm and 0.22 nm are attributed to Pt (200) and Pt (111), respectively. Similarly, PtNiCo nanoparticles also exhibit a regular concave structure, but slightly reduced in size (shown in Fig. 5b). The angles between the vertical facets were measured and found to be 8° and 12°, indicating the presence of some highly orientated plane.
 | ||
| Fig. 5 HRTEM images of (a) Pt CNCs and (b) PtNiCo CNCs synthesized at 473 K for 10 h. | ||
Fig. 6a–c display transmission electron microscopy (TEM) images of the as-prepared PtNixCoy CNCs. As shown in Fig. 6a, the product consisted of Pt CNCs with high yields and an average apex-to-apex diameter of 14 nm. It can be seen from Fig. 6b and c that the concave structure yields of PtNi and PtNiCo decreased slightly, but the size is also around 14 nm. The reason for the decline in the yield of concave structures may be due to the incorporation of Ni and Co atoms, which affects the growth of Pt nanocrystals, resulting in the lattice shrinkage of Pt, such that some nanocrystals do not form concave structures. TEM images of sphere-like Pt NPs and PtNiCo NPs are shown in Fig. S5,† exhibiting slight agglomeration, and the size is about 0.5–1 μm. Combined with the hydrogen production activity of PtNixCoy/CdS, we think that PtNixCoy CNCs exhibit a concave cube structure with a high surface index and highly unsaturated atom coordination, and can provide more active sites on the surface of CdS, which will facilitate more H+ to be reduced to H2.
 | ||
| Fig. 6 TEM images of (a) Pt CNCs, (b) PtNi CNCs and (c) PtNiCo CNCs prepared at 473 K for 10 h, and TEM images of (d) Pt/CdS, (e) PtNi/CdS and (f) PtNiCo/CdS after photocatalytic reaction. | ||
Fig. 6d–f show TEM images of Pt/CdS, PtNi/CdS and PtNiCo/CdS after photocatalytic reaction. It can be clearly seen that CdS synthesized via a solvothermal method displays a rod-like structure with 25–40 nm diameter and 200–900 nm length. In addition, the PtNixCoy CNCs showed a concave nanocube structure, which was consistent with the HRTEM analysis (Fig. 5) and the PtNixCoy CNCs were deposited on the surface of the rod-like CdS (Fig. 6d–f).
Fig. 7a and b show TEM and HRTEM images of PtNiCo/CdS after photocatalytic reaction, where the PtNiCo CNCs were highly dispersed on the surface of the CdS nanorods (Fig. 7a). The spacing of the lattice fringes is found to be 0.22 nm and 0.32 nm (Fig. 7b), corresponding to the (111) crystal face of PtNiCo and (002) crystal face of CdS, respectively. More importantly, the interface between the PtNiCo CNCs and CdS can be clearly seen in the HRTEM image (Fig. 7b), which suggested that the CdS nanorods were electronically interconnected with the PtNiCo CNCs and this is a crucial factor for fast carrier transport and minimizing their recombination by trapping of excited electrons. Additionally, to confirm the presence of elements and their distribution on the surface of the CdS nanorods, the elemental mapping was analyzed and the mapping images of the selected area in Fig. 7c are shown in Fig. 7d–i. It can be found that Cd, S, Pt, Ni, and Co are all present in the PtNiCo/CdS photocatalyst and also the uniform distribution of PtNiCo on the surface of the CdS was revealed. The valence state of Pt, Ni, and Co elements will be further discussed in the XPS analysis.
 | ||
| Fig. 7 (a) TEM and (b) HRTEM images and (c–i) EDS mappings of PtNiCo/CdS. | ||
The structure of the PtNixCoy CNCs was further characterized by X-ray diffraction (XRD). As shown in Fig. 8a, the Pt CNCs have five diffraction peaks at 2 theta of 39.88°, 46.25°, 67.43°, 81.62° and 85.83°, which correspond to the (111), (200), (220), (311) and (222) plane of the Pt face-centered cubic structure (JCPDS No. 87-0647), respectively. When compared with Pt CNCs, the diffraction peak positions of the PtNi CNCs and PtNiCo CNCs were slightly shifted to a higher angle, and no characteristic diffraction peaks of Ni and Co were observed. This observation confirms the formation of PtNi and PtNiCo alloys. The shifting of the diffraction peaks was mainly due to the introduction of Ni and Co (which have smaller atomic radii) decreasing the interatomic distances between adjacent Pt sites, leading to lattice contraction. Thus, the diffraction peak positions of the PtNi CNCs and PtNiCo CNCs shift toward a higher angle.43,44 In addition, the diffraction peaks of the PtNi CNCs and PtNiCo CNCs were weaker in intensity and broader than that of the Pt CNCs, which might be stipulated by the lattice contraction as a result of substitution of smaller Ni with Co atoms for larger Pt atoms, and thus the crystallinity of the alloy decreases.
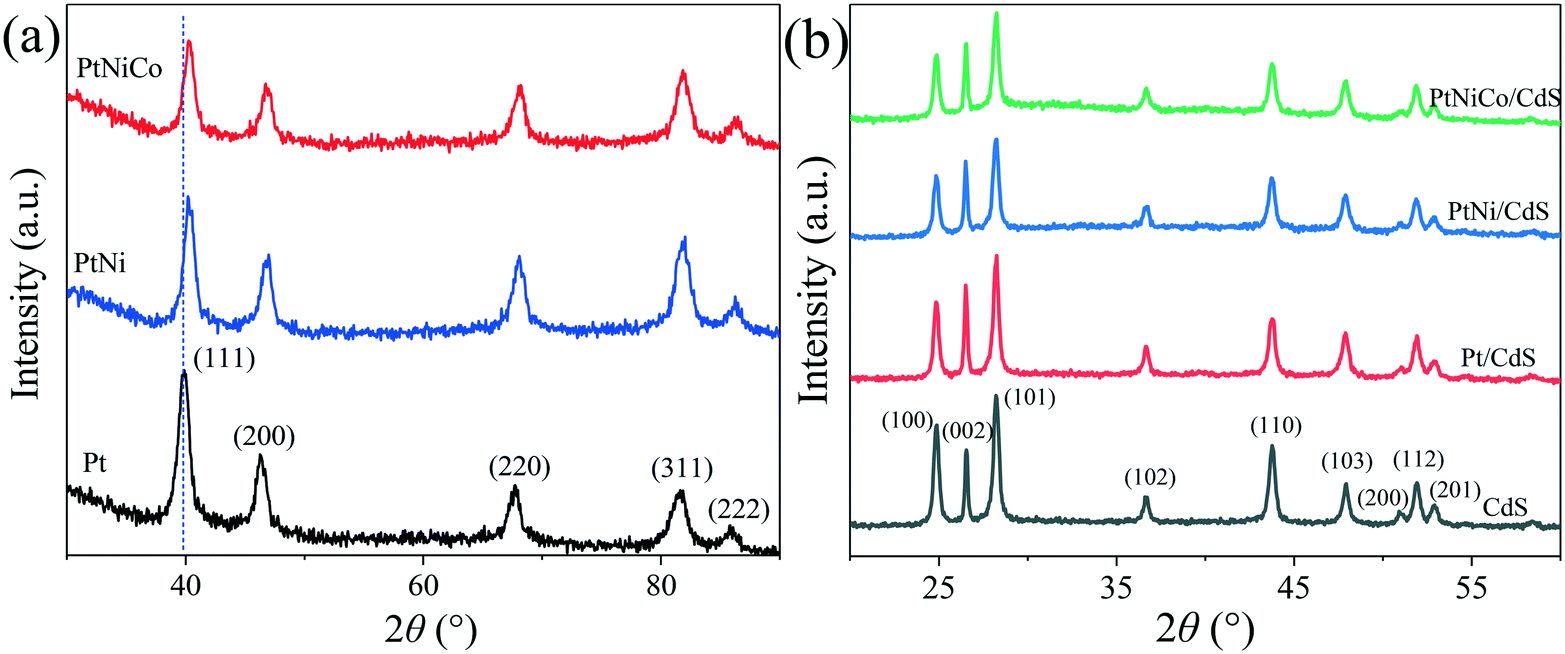 | ||
| Fig. 8 (a) XRD spectra of Pt CNCs, PtNi CNCs and PtNiCo CNCs synthesized at 473 K for 10 h, and (b) XRD spectra of CdS, Pt/CdS, PtNi/CdS and PtNiCo/CdS photocatalysts after photocatalytic reaction. | ||
Fig. 8b displays the XRD spectra of pure CdS and PtNixCoy/CdS after photocatalytic reaction. It can be found that the pattern for CdS has a number of peaks that can be well indexed to hexagonal CdS phase (JCPDS No. 77-2306). The characteristic peaks of CdS are sharp and strong, implying their highly crystalline nature. For the PtNixCoy/CdS samples, no characteristic diffraction peaks of PtNixCoy CNCs were observed because of their lower loading content. On the other hand, it also indicated the good dispersion of PtNixCoy CNCs on the CdS surface, which is consistent with TEM analyses. Compared with the XRD spectra of the CdS, Pt/CdS, PtNi/CdS and PtNiCo/CdS photocatalysts before photocatalytic reaction (Fig. S6†), the crystallinity and structure of the CdS and PtNixCoy/CdS were kept well after photocatalytic reaction. Moreover, compared with pure CdS, the peak intensity of each crystal plane of PtNixCoy/CdS changed to varying degrees, as listed in Table S3.† The peak intensity of PtNixCoy/CdS is significantly decreased for the (100), (101) and (110) crystal planes. We can infer that PtNixCoy CNCs are selectively loaded on the CdS nanorods and preferentially deposited on the (100), (101) and (110) crystal planes of CdS under visible light irradiation.
The chemical states and surface chemical composition of PtNiCo/CdS were investigated using X-ray photoelectron spectroscopy (XPS). In Fig. 9a, the binding energies of 411.8 eV and 405.1 eV are assigned to the Cd 3d3/2 and Cd 3d5/2, respectively. As shown in Fig. 9b, two peaks appeared at 162.6 eV and 161.5 eV, which can be attributed to S 2p1/2 and S 2p3/2 for S2− in the CdS, respectively. Additionally, the Pt 4f (Fig. 9c) spectrum has been fitted into two peaks at 73.8 eV and 70.5 eV, which belong to the Pt 4f5/2 and Pt 4f7/2 of zero valence state Pt, respectively. The Ni 2p spectrum in Fig. 9d shows that the binding energies of 873.7 eV and 855.5 eV correspond to the Ni 2p1/2 and Ni 2p3/2 of Ni(0), respectively. It can be clearly seen from the Co 2p spectrum in Fig. 9e that the two peaks located at 796.7 eV and 781.3 eV are associated with Co 2p1/2 and Co 2p3/2 of Co(0), respectively. The XPS spectra of Pt CNCs, PtNi CNCs and PtNiCo CNCs are plotted in Fig. S7,† indicating that Pt, Ni and Co elements all exist in zero valence state. The presence of Cd, S, Pt, Ni and Co elements in the PtNiCo/CdS photocatalyst suggests that PtNiCo CNCs have been successfully loaded on the surface of CdS. Besides, these results also confirm that a composite photocatalyst is formed between PtNiCo and CdS rather than a simple physical mixture.
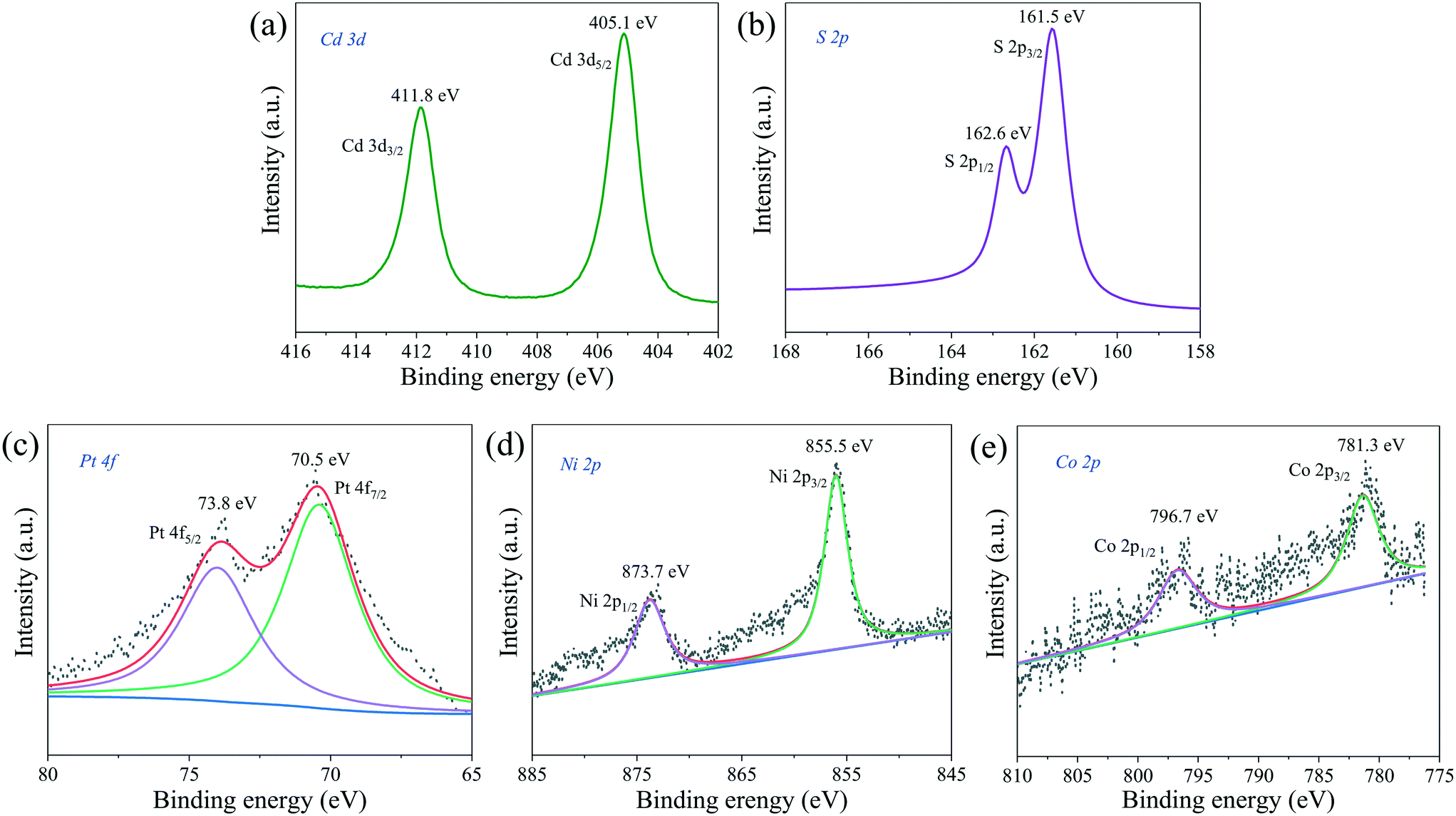 | ||
| Fig. 9 XPS spectra of (a) Cd 3d, (b) S 2p, (c) Pt 4f, (d) Ni 2p and (e) Co 2p of the PtNiCo/CdS photocatalyst. | ||
3.4 Spectral properties
UV-vis diffuse reflectance spectra (DRS) of the CdS, Pt/CdS, PtNi/CdS, and PtNiCo/CdS photocatalysts are shown in Fig. 10a. The absorption edges of the CdS, Pt/CdS, PtNi/CdS, and PtNiCo/CdS photocatalysts are all at about 520 nm (optical band gap 2.4 eV), indicating that the introduction of PtNixCoy CNCs caused no significant structural changes of the CdS and they were deposited only on its surface. However, compared with pure CdS, the PtNixCoy/CdS photocatalyst exhibits an obvious absorption enhancement phenomenon in the range of λ ≥ 520 nm, which is mainly caused by the light absorption of black metallic PtNixCoy CNCs on the surface of CdS.3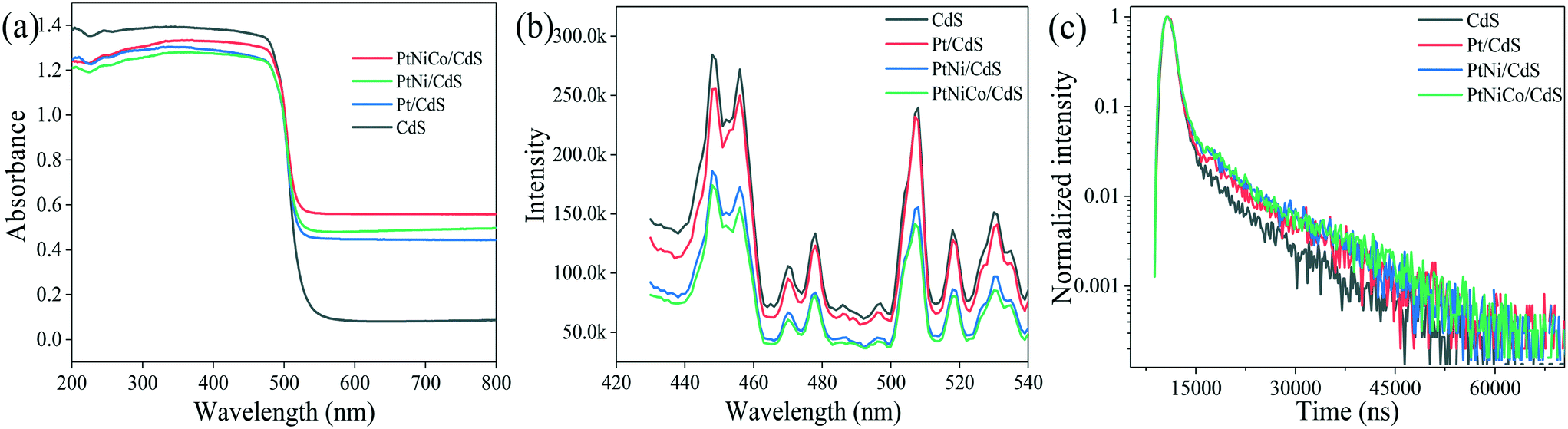 | ||
| Fig. 10 (a) UV-vis DRS, (b) PL spectra and (c) lifetimes of the CdS, Pt/CdS, PtNi/CdS and PtNiCo/CdS photocatalysts after photocatalytic reaction. | ||
Fig. 10b shows the photoluminescence (PL) spectra of CdS, Pt/CdS, PtNi/CdS and PtNiCo/CdS photocatalysts excited at 407 nm. The PL behaviors of the CdS nanostructured materials have been studied intensively. As shown in Fig. 10b, two PL emission peaks of CdS nanostructures are observed, corresponding to the band-edge and surface-defect structure of CdS. The PL peaks arising through band-edge emission are positioned usually in the wavelength range of 440–460 nm,45–47 while peaks at 500–540 nm are caused by surface states such as sulphur vacancies or sulphur dangling bonds.48–50 CdS exhibits strong PL emission features as compared with all other materials, and this higher emission property indicates high recombination of photo-generated charge carriers, which lowers the photocatalytic activity. On the other hand, introduction of Pt (in Pt/CdS), and PtNi (in PtNi/CdS) to the CdS surface decreases the emission features. However, PtNiCo introduction in PtNiCo/CdS drastically suppresses the emission and low intensity features are observed; this indicates that the photo-generated electrons and holes are well separated, which decreases their recombination. In the presence of surface decorated PtNiCo CNCs for the PtNiCo/CdS photocatalyst, the photo-generated charge carriers are synergistically trapped by the metallic alloy at the interface, which suppresses their recombination, resulting in high photocatalytic hydrogen generation performance.
For a more detailed study, we measured the lifetime of electrons through time resolved PL decay. The lifetime curves of CdS, Pt/CdS, PtNi/CdS and PtNiCo/CdS samples were monitored with an excitation wavelength of 407 nm (Fig. 10c). According to the double exponential fitting formula, the lifetime value (τ1, τ2) and the pre-exponential factor (α1, α2) could be obtained. Besides, the average lifetime value (τave) could be calculated using the formula [τave = (α1τ12 + α2τ22)/(α1τ1 + α2τ2)]. The obtained lifetimes of CdS, Pt/CdS, PtNi/CdS and PtNiCo/CdS are 5592.79, 7004.48, 7354.31 and 7612.59 ns (shown in Table S4†), respectively. Among all photocatalysts, PtNiCo/CdS shows significantly higher lifetime (7612.59 ns), which combined with the TEM, XPS, PL, and photocurrent measurements well supports the high photocatalytic hydrogen generation activity of PtNiCo/CdS. The high lifetime of electrons for PtNiCo/CdS was associated with enhanced charge separation and transformation of the photo-induced carriers,45,51,52 which resulted in the higher photocatalytic hydrogen generation rate.
3.5 Photocatalytic mechanism
Based on the photocatalytic experimental results and from various spectroscopic analyses, a possible mechanism for PtNixCoy CNC loading onto CdS increasing the hydrogen production activity was proposed and is shown in Fig. 11. CdS can be excited to yield photo-generated electrons and holes under visible light irradiation, the electrons in the CB of CdS can easily migrate to the surface of the PtNixCoy CNCs, and a large amount of H+ is reduced to H2.53–55 At the same time, the holes in the VB of CdS would be consumed with the assistance of the sacrificial agent (NH4)2SO3. In addition, owing to the low overpotential of hydrogen production after PtNixCoy CNC loading, the electrons on the PtNixCoy CNC surface can effectively reduce H+ to produce H2.56 In other words, PtNixCoy CNCs can effectively promote the separation of photo-generated electrons and holes, minimizing their recombination, and thus enhance the photocatalytic performance. These results are also consistent with the transient photocurrent response and photoluminescence.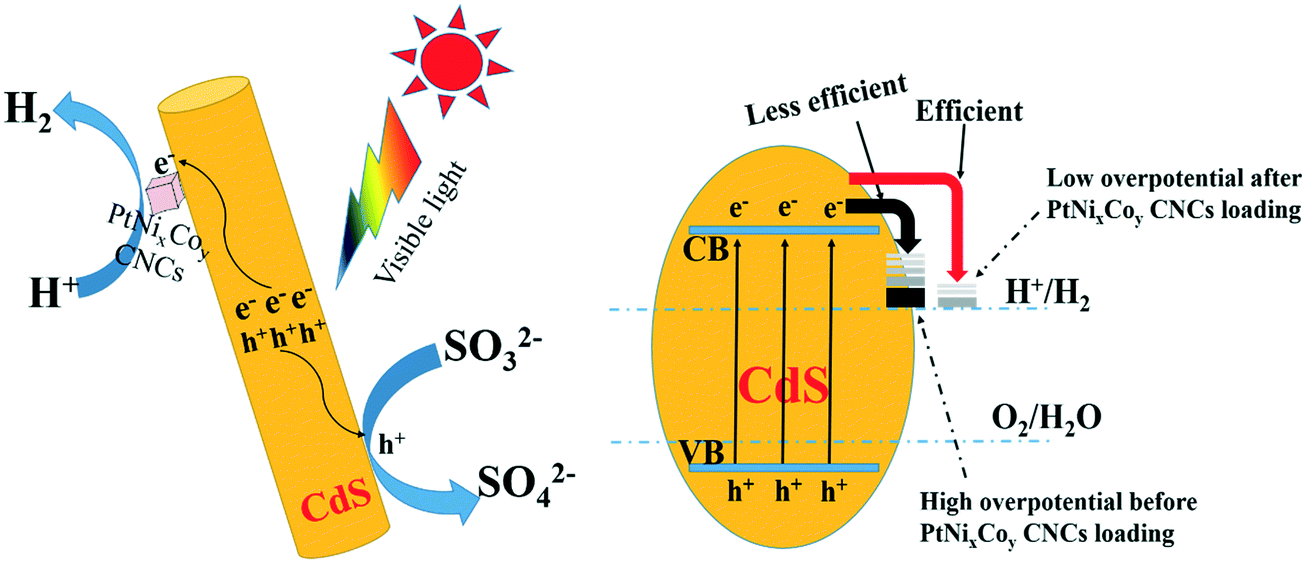 | ||
| Fig. 11 Hydrogen generation mechanism of Pt-based CNCs deposited on CdS. | ||
4 Conclusions
Pt CNCs were successfully synthesized by a hydrothermal method using H2PtCl6 as a precursor, and photo-deposited on the surface of CdS nanorods for photocatalytic decomposition of water to hydrogen. When the Pt CNCs were synthesized at 473 K for 10 h, the hydrogen generation rate of Pt/CdS (Pt dosage: 2 wt%) was 37.80 mmol h−1 g−1 (QE = 44.64%, λ = 420 nm). When Ni and Co were simultaneously introduced to form ternary alloy CNCs, the hydrogen generation rate of PtNiCo/CdS is as high as 50.60 mmol h−1 g−1 (QE = 56.82%, λ = 420 nm), which is 1.3-fold higher than that of Pt/CdS and also significantly higher than those of PtNi/CdS (42.71 mmol h−1 g−1) and PtCo0.5/CdS (40.33 mmol h−1 g−1). Moreover, LSV and Tafel tests showed that PtNixCoy/CdS not only enhances the hydrogen evolution current density, but also reduces the hydrogen evolution overpotential compared to bare CdS. The structural characterizations displayed that the PtNixCoy CNCs achieved at 473 K for 10 h exhibit a clear cubic concave structure and the average crystallite size is 14 nm, and they are preferentially deposited on the (100), (101) and (110) crystal planes of the CdS nanorod surface. Steady-state fluorescence exhibited that the fluorescence intensity of the PtNixCoy/CdS photocatalyst is weaker than that of CdS, and the average fluorescence lifetime of PtNixCoy/CdS is significantly higher than that of CdS. The as-prepared PtNixCoy CNCs have a high-index crystal plane, and the atomic coordination is highly unsaturated. Therefore, PtNixCoy CNCs have a high-energy surface and have a stronger electron trapping capacity. After being loaded on the surface of CdS, they capture the conduction band electrons of CdS, effectively suppress the recombination of photo-generated electrons and holes, and promote the photocatalytic decomposition of hydrogen into aquatic products. Hence, PtNixCoy CNCs have good application prospects in the field of photocatalytic hydrogen generation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the International Science Foundation of China (51602091) and the International Cooperation Project of Department of Science and Technology of Henan Province (162102410011).References
- J. Yang, P. Jiang, M. Yue, D. Yang, R. Cong, W. Gao and T. Yang, J. Catal., 2017, 345, 236–244 CrossRef CAS.
- L. Wang, N. Xu, X. Pan, Y. He, X. Wang and W. Su, Catal. Sci. Technol., 2018, 8, 1599–1605 RSC.
- H. Wang, W. Chen, J. Zhang, C. Huang and L. Mao, Int. J. Hydrogen Energy, 2015, 40, 340–345 CrossRef CAS.
- S. Cao, C. Wang, X. Lv, Y. Chen and W. Fu, Appl. Catal., B, 2015, 162, 381–391 CrossRef CAS.
- S. Ma, Y. Deng, J. Xie, K. He, W. Liu, X. Chen and X. Li, Appl. Catal., B, 2018, 227, 218–228 CrossRef CAS.
- L. Zhang and Y. Zhu, Catal. Sci. Technol., 2012, 2, 694–706 RSC.
- R. Wei, Z. Huang, G. Gu, Z. Wang, L. Zeng, Y. Chen and Z. Liu, Appl. Catal., B, 2018, 231, 101–107 CrossRef CAS.
- X. Zhang, N. Li, J. Wu, Y. Zheng and X. Tao, Appl. Catal., B, 2018, 229, 227–236 CrossRef CAS.
- N. Li, J. Wu, Y. Lu, Z. Zhao, H. Zhang, X. Li, Y. Zheng and X. Tao, Appl. Catal., B, 2018, 238, 27–37 CrossRef CAS.
- K. Lee, R. Hahn, M. Altomare, E. Selli and P. Schmuki, Adv. Mater., 2013, 25, 6133–6137 CrossRef CAS.
- W. Chen, Y. Wang and W. Shanguan, Int. J. Hydrogen Energy, 2019, 44, 4123–4132 CrossRef CAS.
- D. Liang, F. Cheng and Q. Xiang, Catal. Sci. Technol., 2016, 6, 6207–6216 RSC.
- M. Luo, W. Yao, C. Huang, Q. Wu and Q. Xu, J. Mater. Chem. A, 2015, 3, 13884–13891 RSC.
- I. Vamvasakis, B. Liu and G. S. Armatas, Adv. Funct. Mater., 2016, 26, 8062–8071 CrossRef CAS.
- P. Navajas, M. Serra and H. Garcia, Catal. Sci. Technol., 2013, 3, 2252–2258 RSC.
- Y. Wang, Y. Wang and R. Xu, J. Phys. Chem. C, 2013, 117, 783–790 CrossRef CAS.
- X. Tan, S. Prabhudev, A. Kohandehghan, D. Karpuzov, G. Botton and D. Mitlin, ACS Catal., 2015, 5, 1513–1524 CrossRef CAS.
- P. Wang, Q. Shao, X. Cui, X. Zhu and X. Huang, Adv. Funct. Mater., 2018, 28, 1705918–1705928 CrossRef.
- X. Huang, Z. Zhao, J. Fan, Y. Tan and N. Zheng, J. Am. Chem. Soc., 2011, 133, 4718–4721 CrossRef CAS.
- Z. Zhang, J. Hui, Z. Liu, X. Zhang, J. Zhuang and X. Wang, Langmuir, 2012, 28, 14845–14848 CrossRef CAS.
- H. Zhang, M. Jin and Y. Xia, Angew. Chem., 2010, 132, 268–274 Search PubMed.
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 2773–2777 CrossRef CAS.
- M. J. Mulvihill, X. Ling, J. Henzie and P. Yang, J. Am. Chem. Soc., 2011, 133, 4718–4721 CrossRef.
- J. Zhang, M. R. Langille, M. L. Personick, K. Zhang, S. Li and C. A. Mirkin, J. Am. Chem. Soc., 2010, 132, 14012–14014 CrossRef CAS.
- X. Xia, J. Zeng, B. Mcdearmon, Y. Zheng, Q. Li and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 12542–12546 CrossRef CAS.
- H. Zhang, M. Jin, J. Wang, W. Li, P. H. C. Camargo, M. J. Kim, D. Yang, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2011, 133, 6078–6089 CrossRef CAS.
- L. Zhang, D. Chen, Z. Jiang, J. Zhang, S. Xie, Q. Kuang, Z. Xie and L. Zheng, Nano Res., 2012, 5, 181–189 CrossRef CAS.
- X. Xu, X. Zhang, H. Sun, Y. Yang, X. Dai, J. Gao, X. Li, P. Zhang, H. Wang, N. Yu and S. Sun, Angew. Chem., Int. Ed., 2014, 53, 12522–12527 CAS.
- C. Wang, C. Lin, L. Zhang, Z. Quan, K. Sun, B. Zhao, F. Wang, N. Porter, Y. Wang and J. Fang, Chem. – Eur. J., 2014, 20, 1753–1759 CrossRef CAS PubMed.
- Y. Qin, X. Zhang, X. Dai, H. Sun, Y. Yang, X. Li, Q. Shi, D. Gao, H. Wang, N. Yu and S. Sun, Small, 2016, 12, 524–533 CrossRef CAS.
- A. Yin, X. Min, W. Zhu, W. Liu, Y. Zhang and C. Yan, Chem. – Eur. J., 2012, 18, 777–782 CrossRef CAS.
- Y. Qi, T. Bian, S. L. Choi, Y. Jiang, C. Jin, M. Fu, H. Zhang and D. Yang, Chem. Commun., 2014, 50, 560–562 RSC.
- J. Wu, J. Zhu, M. Zhou, Y. Hou and S. Gao, CrystEngComm, 2012, 14, 7572–7575 RSC.
- T. Chu, H. Wang and L. Mao, J. Mater. Eng. Perform., 2014, 23, 3413–3417 CrossRef CAS.
- H. Wang, W. Chen, J. Zhang, C. Huang and L. Mao, Int. J. Hydrogen Energy, 2015, 40, 340–345 CrossRef CAS.
- L. Mao, H. Liu, S. Liu, Q. Ba, H. Wang, L. Gao, X. Li, C. Huang and W. Chen, Mater. Res. Bull., 2017, 93, 9–15 CrossRef CAS.
- X. Li, H. Liu, S. Liu, J. Zhang, W. Chen, C. Huang and L. Mao, Int. J. Hydrogen Energy, 2016, 41, 23015–23021 CrossRef CAS.
- W. Sheng, Y. Song, M. Dou, J. Ji and F. Wang, Appl. Surf. Sci., 2018, 436, 613–623 CrossRef CAS.
- J. Li, F. Li, S. Guo, J. Zhang and J. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 8151–8160 CrossRef CAS.
- Q. Ba, X. Jia, L. Huang, X. Li, W. Chen and L. Mao, Int. J. Hydrogen Energy, 2019, 44, 5872–5880 CrossRef CAS.
- Z. Fang, L. Zhang, T. Yang, L. Su, K. Chou and X. Hou, Phys. E, 2017, 93, 116–123 CrossRef CAS.
- B. Wiley, Y. Sun, B. Mayers and Y. Xia, Chem. – Eur. J., 2005, 11, 454–463 CrossRef CAS.
- R. Sriphathoorat, K. Wang, S. Luo, M. Tang, H. Du, W. Du and P. Shen, J. Mater. Chem. A, 2016, 4, 18015–18021 RSC.
- D. Liang, J. Gao, J. Wang, P. Chen, Y. Wei and Z. Hou, Catal. Commun., 2011, 12, 1059–1062 CrossRef CAS.
- Q. Pan, K. Huang, S. Ni, Q. Wang, F. Yang and D. He, Mater. Lett., 2007, 61, 4773–4776 CrossRef CAS.
- W. S. Chae, J. H. Ko, I. W. Hwang and Y. R. Kim, Chem. Phys. Lett., 2002, 365, 49–56 CrossRef CAS.
- T. Orii, S. I. Kaito, K. Matsuishi, S. Onari and T. Arai, J. Phys.: Condens. Matter, 2002, 14, 9743–9752 CrossRef CAS.
- P. V. Kameat, N. M. Dimitrijevic and R. W. Fessenden, J. Phys. Chem., 1987, 91, 396–401 CrossRef.
- G. Xu, B. Liu, S. Xu, C. H. Chew, S. J. Chua and L. M. Gana, J. Phys. Chem. Solids, 2000, 61, 829–836 CrossRef CAS.
- J. Zhang, L. Sun, C. Liao and C. Yan, Solid State Commun., 2002, 124, 45–48 CrossRef CAS.
- T. Cai, M. Yue, X. Wang, Q. Deng, Z. Peng and W. Zhou, Chin. J. Catal., 2007, 28, 10–16 CrossRef CAS.
- J. Tang, Z. Zou and J. Ye, ChemInform, 2004, 35, 14265–14269 Search PubMed.
- H. Yao, J. Zhang, K. Dai, Q. Li, J. Lv, G. Zhu and C. Liang, Appl. Catal., B, 2019, 241, 528–538 CrossRef.
- T. Hu, K. Dai, J. Zhang, G. Zhu and C. Liang, Appl. Surf. Sci., 2019, 481, 1385–1393 CrossRef CAS.
- Z. Li, Y. Yang, K. Dai, J. Zhang and L. Lu, Appl. Surf. Sci., 2019, 469, 505–513 CrossRef CAS.
- W. Chen, Y. Wang, M. Liu, L. Gao, L. Mao, Z. Fan and W. Shangguan, Appl. Surf. Sci., 2018, 444, 485–490 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01958h |
| This journal is © The Royal Society of Chemistry 2020 |