
MOF-derived nanostructured catalysts for low-temperature ammonia synthesis†
Ignacio
Luz
 *,
Sameer
Parvathikar
,
Michael
Carpenter
,
Timothy
Bellamy
,
Kelly
Amato
,
John
Carpenter
and
Marty
Lail
*
*,
Sameer
Parvathikar
,
Michael
Carpenter
,
Timothy
Bellamy
,
Kelly
Amato
,
John
Carpenter
and
Marty
Lail
*
RTI International, 3040 E. Cornwallis Rd, Research Triangle Park, NC 27709, USA. E-mail: iluzminguez@rti.org; mlail@rti.org
First published on 14th November 2019
Abstract
Nanostructured catalysts for low-temperature ammonia synthesis have been developed via thermal treatment under nitrogen of Ru-containing MOFs. The resulting catalysts reveal high concentration of small Ru nanocrystals (<4 nm) well-dispersed on a graphitic carbon shell demonstrating stable ammonia formation rates at low temperature. Additional acetylene pre-treatment leads to more dispersed active catalysts containing stable sub-nanometric Ru clusters.
Introduction
Ammonia is vital as an active nitrogen source for important chemicals including fertilizers, polymers, dyes and explosives, and has also been proposed as a futuristic hydrogen storage material.1 Despite the technological developments within the past century, ammonia production by the conventional Haber–Bosch process over promoted magnetite (Fe3O4) catalysts remains a high-energy and capital-intensive industry due to the high operating temperatures (400–500 °C) and pressures (150–300 bar) required, which results in high energy consumption.2 As an alternative process, high ammonia yields of approximately 40–50% have been achieved under lower pressure and temperature conditions (370–400 °C and 50–100 atm) by using Ru-based catalysts supported on graphitic carbon and promoted by combination of alkali or alkaline-earth metal oxides (such as Na, Cs and Ba), which lower the energy barrier for N2 dissociation by electron injection (Kellogg advanced ammonia process).3,4 Approaches for reducing while stabilizing the Ru crystalline domains as well as for providing better control over the interactions between the Ru active surface and promoters are still challenging, which are key factors for the further development of active and stable commercial catalysts for low-temperature ammonia synthesis.In heterogeneous catalysts, dispersing the catalytically active metallic phase by reducing its crystalline domain down to a few nanometers, or even further to sub-nanometric clusters,5 typically leads to an enhancement of the catalytic performance.6 Nevertheless, in the absence of strong metal–support interactions to immobilize them, these nanometric metallic species tend to sinter in reactive environments, thus reducing their active surface area and decreasing the catalytic performance over time. Several attempts to provide them with more stability via strong bonding between the support and metal,7 or even by encapsulation within support cavities,8 have not been sufficient under harsh reaction conditions. Fortunately, a couple of recent approaches revealed excellent stabilization of these metal species, such as trapping them during the crystallization of a small pore CHA zeolite (8-member ring window), or covering them with a carbon shell by thermal decomposition of metal–organic precursors,9 including metal–organic frameworks (MOFs).10
MOFs have been recently used as versatile precursors for the preparation of advanced heterogeneous catalysts and electrocatalysts.11,12 The versatility of MOFs as precursors is mainly due to their unique and highly tunable features, such as well-defined metal sites spaced by organic struts displayed along a crystalline structure with permanent porosity, which can play two simultaneous roles acting as a template and precursor upon applying the suitable conditions at high temperature, such as controlled pyrolysis under nitrogen, calcination under air, or reduction under hydrogen.13 Upon this transformation, MOFs can lead to well-defined and nanostructured catalytically active species, which are well-dispersed within a microporous carbonaceous matrix.14–16
In this work, active and stable nanostructured catalysts for low-temperature ammonia synthesis have been developed via thermal treatment under nitrogen of Ru-containing MOFs. The resulting catalysts reveal small Ru nanocrystals (<4 nm) well-dispersed on a graphitic carbon shell demonstrating stable ammonia formation rates. Furthermore, the additional pre-treatment with acetylene leads to more active catalysts containing stable sub-nanometric Ru clusters.
Experimental
MOF synthesis
(Ru)HKUST-1 containing 100% of benzene tricarboxylic acid (RuMOF), and (Ru)HKUST-1 containing 50% of benzene tricarboxylic acid and 50% of pyridine-3,5-dicarboxylic acid (Ru(N)MOF) were prepared according to the literature.17 In a typical synthesis, 1 g of the Ru precursor, 600 mg of the appropriate mixture of organic ligands 1,3,5-benzenetricarboxylic acid and pyridine-3,5-dicarboxylic acid, 5 mL of acetic acid and 25 mL of H2O were loaded into a 100 mL autoclave and heated at 160 °C for 24 hours. The resulting powder was thoroughly washed with H2O in a filtration funnel and purified in a Soxhlet apparatus with MeOH. All the samples were evacuated at 150 °C under vacuum.Ru precursor [Ru2(CH3COO)4Cl] was prepared according to the literature18 by mixing 10 g of RuCl3·xH2O (40 wt% Ru) and 12 g of LiCl (anhydrous), previously evacuated at 80 °C overnight in a vacuum oven, with 70 mL of acetic anhydride and 350 mL of glacial acetic acid. The mixture was stirred and refluxed for 2–4 days until the solution turned reddish. After cooling down, the Ru precursor was collected by filtration and was thoroughly washed with acetone.
Characterization
Catalyst testing
The catalytic performance for ammonia synthesis was tested using a Micromeritics PID Effi microreactor. The microreactor is a stand-alone, fully automated system with inlet gas flows metered through mass flow controllers. Feed gases consisted of nitrogen and hydrogen, with argon used as an internal standard. The reactor consisted of a 9 mm stainless-steel tube in a furnace. The reactor and the furnace are contained in a hot box, with pressure controlled downstream by a back-pressure regulator. Pressure, temperature and gas flow rates were all controlled by the microreactor's process control software. The catalyst was diluted with silicon carbide (SiC) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio and was placed between two beds of SiC in the reactor, such that the reactor thermocouple was in the middle of the catalyst bed. All the catalysts were reduced in 75% H2/25% N2 at 370 °C overnight, prior to catalytic activity tests at 95 bar. Downstream of the reactor system, the composition of the process gases was analysed by gas chromatography (for quantifying nitrogen, hydrogen and argon) and using an MKS multiGas FTIR continuous gas analyzer (for quantifying ammonia).
:2 ratio and was placed between two beds of SiC in the reactor, such that the reactor thermocouple was in the middle of the catalyst bed. All the catalysts were reduced in 75% H2/25% N2 at 370 °C overnight, prior to catalytic activity tests at 95 bar. Downstream of the reactor system, the composition of the process gases was analysed by gas chromatography (for quantifying nitrogen, hydrogen and argon) and using an MKS multiGas FTIR continuous gas analyzer (for quantifying ammonia).
Results and discussion
MOF-derived Ru nanostructured catalysts
A series of nanostructured catalysts were developed for ammonia synthesis via controlled pyrolysis of microporous (Ru)HKUST-1 under nitrogen at increasing temperature up to 900 °C. The resulting catalysts exhibit high concentration of Ru nanocrystals (up to 80 wt%) homogeneously dispersed and stabilized along a porous carbon scaffold. The transformational mechanism occurring during the pyrolysis of the MOF precursor was elucidated via combination of characterization techniques, such as X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis coupled to mass spectroscopy (TGA-MS), scanning transmission electron microscopy (STEM), Raman spectroscopy, N2 sorption isotherm measurements and CO pulse chemisorption. This understanding enables the optimization of the physicochemical properties of the resulting nanostructured Ru catalysts in terms of the nanocrystal size, surface area, active surface and level of graphitization, which have a crucial impact on their reaction rates and long-term stability.Monitoring the thermal transformation of the MOF precursor by TGA-MS (Fig. 1a) shows the release of CO2 molecules between 200 and 450 °C, consistent with decarboxylation of benzene– and pyridine–tricarboxylic acid linkers. This may lead to the aggregation of Ru dimers into clusters while decarboxylated phenyl rings are converted into an amorphous carbonaceous matrix, as previously proposed for carboxylate-containing MOF-5.19 XPS analysis of the resulting metal clusters confirms the complete reduction to metallic Ru nanocrystals of the MOF precursor (Fig. 1b). The direct reduction of Ru2+/3+ into Ru0 under these conditions is characteristic of elements having positive reduction potential, such as Cu, Co or Fe, in the presence of carbonaceous species acting as reducing agents.16
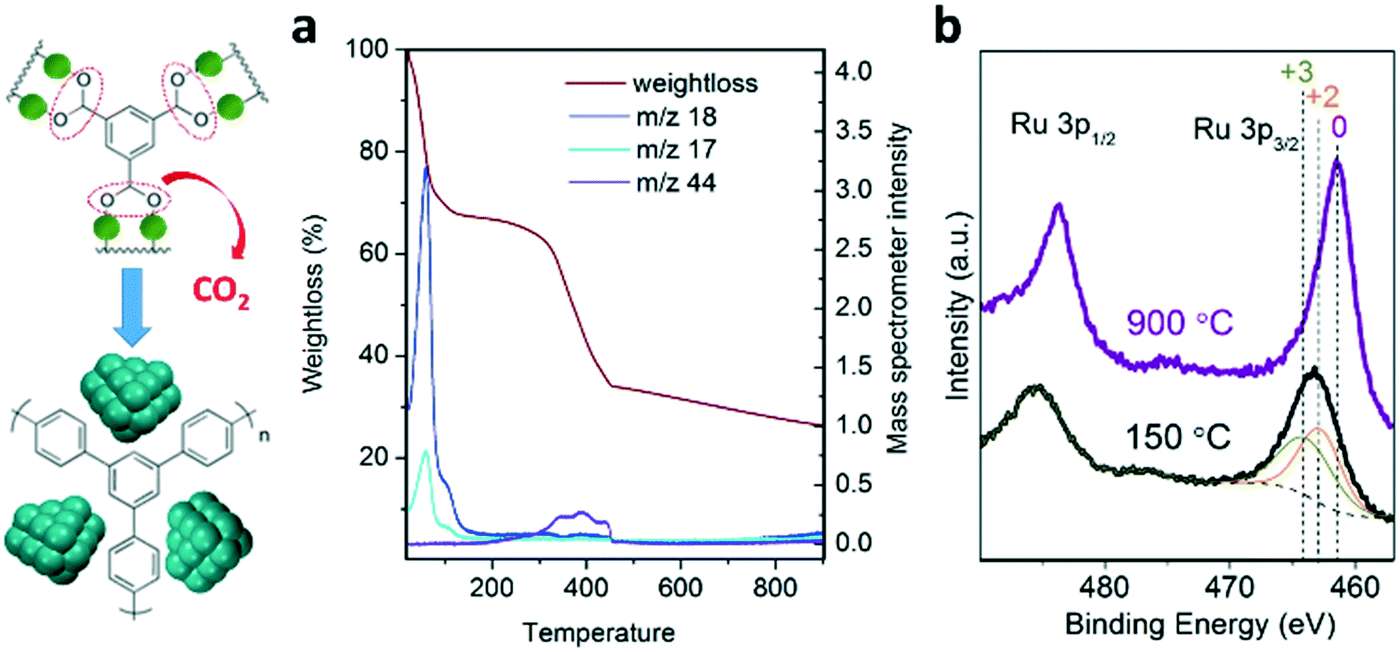 | ||
| Fig. 1 a) TGA-MS analysis of the transformation treatment of the MOF precursor into Ru nanostructured catalysts. b) XPS analysis of the Ru3p signals for the MOF precursor (150 °C) and nanostructured catalysts (900 °C). | ||
As shown in Fig. 2a, in situ XRD monitoring of the MOF transformation into Ru nanostructured catalysts as a function of temperature reveals an advanced attenuation of the characteristic diffraction peaks corresponding to the MOF crystalline structure between 400 and 450 °C. An amorphization of the MOF crystalline phase may occur in this temperature range followed by the appearance of broad diffraction peaks at 2θ = 35–50° corresponding to the metallic Ru crystalline phase. The broadness of the signals suggests the initial formation of sub-nanometric clusters upon full decarboxylation which evolve into larger Ru nanocrystals with temperature, as the diffraction peaks converge into sharper peaks, such as the one centered at 2θ = 44° attributed to the Ru crystalline plane (101). At 900 °C, the dwelling time promotes the aggregation of the small nanocrystals into larger ones as indicated by the progressive sharpening of the diffraction peaks (see Fig. 2b). This Ru nanocrystal sintering is attributed to a rearrangement of the carbonaceous matrix, as discussed in more detail below (see Fig. 3). The absence of additional XRD peaks (such as RuO2) also confirms the full reduction of cationic Ru2+/3+ into metallic Ru0 during the pyrolytic transformation, thus supporting the results obtained by XPS. STEM analysis of the material obtained at 900 °C with no holding time (0 h) reveals a high concentration of well-dispersed small Ru nanocrystals (85% of Ru, see Fig. S1†), suggesting the existence of a thin carbonaceous matrix separating the nanocrystals. A close look at the nanostructure by STEM suggests the existence of a multilayered graphitic carbon scaffold stabilizing the Ru nanocrystals, as identified by the typical spacing for a multilayered structure (0.4 nm) as well as the presence of graphitic carbon confirmed by Raman spectroscopy (Fig. 3d) and elemental mapping analysis (see Fig. 3d and S1†). This specific nanostructure has been recently reported for other MOF-derived materials, such as CoMOF-74.20
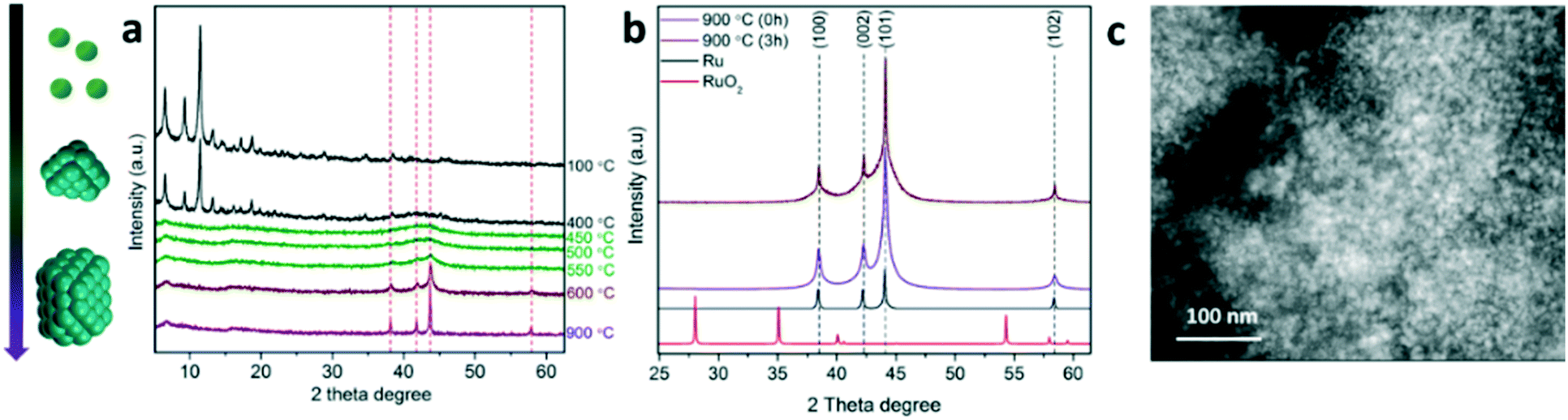 | ||
| Fig. 2 a) In situ XRD monitoring of the transformation of the MOF precursor into nanostructured catalysts. b) XRD pattern of catalysts obtained at 900 °C at varying holding times (0 and 3 h) compared to Ru and RuO2 crystalline patterns. c) STEM-HAADF image of nanostructured catalysts. | ||
 | ||
| Fig. 3 (a) CO chemisorption at 35 °C, (b) TEM analysis, (c) N2 sorption isotherms at 77 K (BET surface area and carbon elemental analysis), and (d) Raman spectra of MOF-derived catalysts obtained by pyrolysis at 900 °C and different holding times (0, 3 and 12 h). | ||
In order to provide more insights into the effect of holding the pyrolysis temperature on the nanostructure of the Ru nanocrystals as well as the nature of the carbonaceous matrix acting as a support, the samples treated at 900 °C for different holding times (0, 3 and 12 h) were characterized by a combination of techniques, such as TEM, N2 sorption, CO chemisorption, Raman spectroscopy and elemental analysis. Longer holding times during pyrolysis (12 h) lead to the partial sintering of Ru nanocrystals accompanied by apparent changes in the composite morphology (see Fig. 3b and S2†). According to the results obtained by CO pulse chemisorption, holding times higher than 3 hours result in the reduction of the active surface (Fig. 3a) due to the aforementioned Ru sintering. The catalyst surface area and pore size distribution also decrease from 210 m2 g−1 for 0 h, to 183 or 91 m2 g−1 for 3 or 12 h, respectively (Fig. 3c and S3†). This suggests a rearrangement of the carbon matrix into a less porous (but more stable) morphology which involves a proportional loss of carbon (each sample contains 16.5, 14.3 and 10.5 wt% C, respectively). In the same way, full release of hydrogen also indicates the advanced fusion of benzylic rings into 2D-graphitic structures even at low holding times (0 h), as determined by elementary analysis.
As shown in Fig. 3d, the Raman spectra of the MOF-derived catalysts exhibit two peaks located at 1326 and 1562 cm−1, which are attributed to the vibration bands of carbon in disordered graphite (D band) and the E2g mode of graphite (G band).21 The G to D band intensity ratio (IG/ID) is normally used to assess the crystalline structure of graphitic carbon. This result reveals that the crystallinity and the concentration (intensity of the peaks) of graphitic carbon in the catalyst improve by holding for a longer time (12 h) at 900 °C, as the IG/ID ratio increases from 0.7 for no holding time (0 h) to 1.5 for 12 h. This supports the hypothesis that the loss of the catalyst surface area as a function of holding time may be explained by a rearrangement of carbon into more ordered graphitic carbon. Better definition and higher intensity of the Raman signals G and D at longer holding times are consistent with the removal of non-stable amorphous carbonaceous species, reducing the background signal and the carbon contained therein.
Mechanism of formation of MOF-derived nanostructured catalysts
As illustrated in Scheme 1, the hypothetical mechanism for the thermal transformation of (Ru)HKUST-1 into Ru nanostructured catalysts under inert conditions involves first the progressive cleavage of the carboxylate group and release of CO2 between 200 and 400 °C,12 which may lead to the formation of metallic Ru sub-nanometric clusters embedded within a thin amorphous carbonaceous matrix resulting from the polymerization of the benzylic rings. Subsequently, the Ru sub-nanometric clusters may start sintering to form larger nanocrystals between 400 and 600 °C, which are still well-dispersed on the microporous amorphous carbon matrix. Finally, the amorphous carbonaceous matrix may be progressively rearranged into multilayer graphitic carbon at 900 °C, which leads to a material exhibiting a lower carbon content and lower surface area, but higher order, after long holding times (12 h).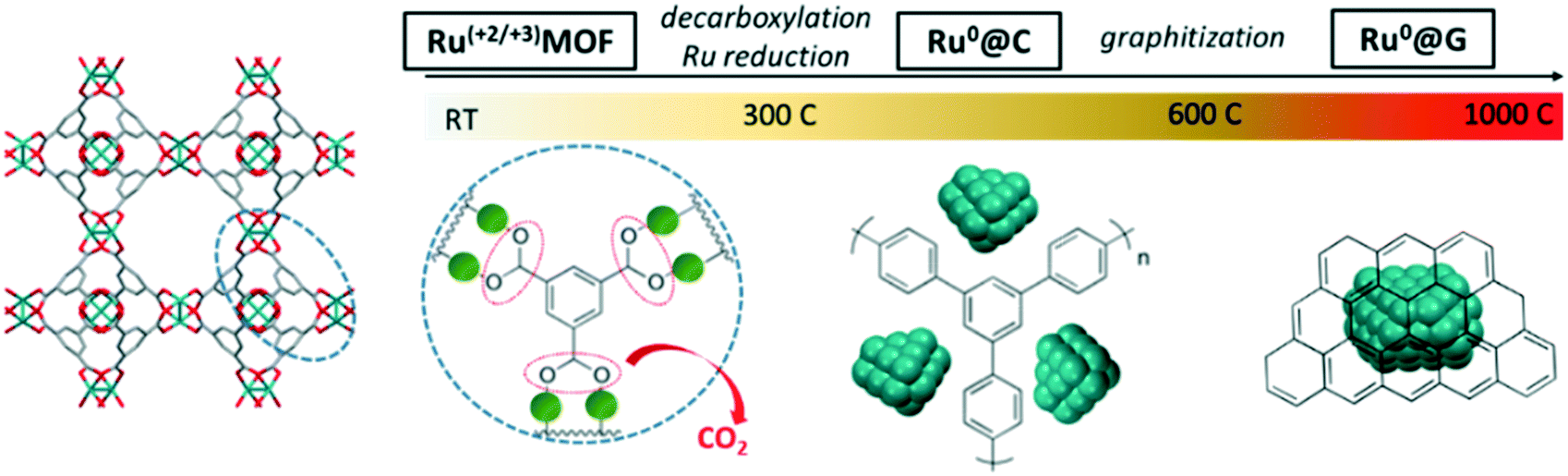 | ||
| Scheme 1 Transformational mechanism of the (Ru)HKUST-1 precursor into nanostructured catalysts. | ||
Stability of MOF-derived Ru nanostructured catalysts for ammonia synthesis
The thermal and chemical stability of the MOF-derived catalysts have been evaluated under ammonia synthesis conditions for 4 hours (H2 at 400 °C) by in situ and ex situ characterization techniques, such as TEM, Raman spectroscopy and XRD. TEM analysis of the material before and after exposure to reaction conditions reveals a morphological change in terms of the apparent Ru nanocrystal size. After H2 activation, the Ru nanocrystals for the sample prepared with 0 h holding time seem to be smaller (3–4 nm) than the as-synthetized materials (6–7 nm). Furthermore, Raman spectroscopy shows better definition and higher intensity of the signals after H2 activation (Fig. 4b) while thermo-XRD reveals no change in the size of the crystalline domain (Fig. 4c). The combination of these results suggests that only amorphous carbonaceous species are released during H2 activation, as the Raman relative intensities and the Ru nanocrystal sizes are maintained during the treatment. Evidence for amorphous carbon loss is obtained by mass spectroscopy detection of a small amount of CH4 during the activation stage of CO pulsed-chemisorption experiments, which leads to the enhancement of the available Ru sites upon H2 activation (see Fig. S4†). This finding supports the hypothesis that the Ru active species catalyse the methanation of the non-stable amorphous carbon under reaction conditions, which leads to a highly stable catalyst composed of 3–4 nm Ru nanocrystals stabilized by highly pure graphitic carbon. | ||
| Fig. 4 Ru nanostructured catalyst stability under ammonia synthesis conditions characterized by TEM (a), Raman spectroscopy (b) and thermo-XRD (c). | ||
Catalytic performance of MOF-derived Ru nanostructured catalysts for ammonia synthesis
The MOF-derived catalysts were promoted with Ba and Cs salts following literature reports for other carbon-supported Ru catalysts.22 The Ru:Ba:Cs promotion ratio of 1:0.05:0.2 was found optimal for achieving the highest ammonia production rate at 350 °C, a gas hourly space velocity (GHSV) of 15000 h−1 and 95 barg (Table S1†). The reaction was also carried out with the optimal promotion at higher GHSVs (30000 and 60000 h−1) to eliminate equilibrium constraints, as shown in Fig. S5 and S7.† The MOF-derived nanostructured catalysts show ammonia production rates per gram of catalyst of ca. 5 gNH3 gcat−1 h−1 (see Table S3†), similar to some of the best Ru catalysts that have been used for commercial ammonia production, such as Ba–Cs–Ru/C.4,23 These production rates per gram of catalyst are attributed to the high concentration of well-dispersed Ru nanocrystals, which is crucial to reduce the reactor dimensions compared to the catalysts exhibiting low Ru loadings.
In order to probe the potential of the graphitic carbon environment to improve the catalytic performance of the RuMOF-derived catalysts, we also evaluated two alternative approaches to reduce and preserve the metal nanocrystal size of the MOF-derived materials. In the first approach, the RuMOF precursor is doped with nitrogen via incorporation of defective ligands, and in the second approach, the RuMOF precursor is pre-treated with C2H4 at 500 °C prior to the pyrolytic treatment at 900 °C with N2.
Alternative MOF precursors: N-containing (Ru)HKUST-1
According to the literature,14 the presence of N-containing ligands in MOF precursors provides improved stabilization of sub-nanometric metal clusters upon pyrolysis. Here, the pyrolysis of defect-engineered (Ru)HKUST-1 (Ru(N)MOF) with a combination of ligands (3,5-pyridinedicarboxylate and 1,3,5-pyridinetricarboxylate17) has been evaluated as an alternative route to obtain smaller Ru nanocrystals, and therefore, more active MOF-derived catalysts. As shown in Fig. 5, TEM analysis reveals the presence of sub-nanometric Ru clusters for the Ru(N)MOF precursor pyrolyzed at 500 °C compared to the N-free MOF precursor exhibiting nanocrystals of 2–3 nm. In addition, the surface area of the material containing N ligands (533 m2 g−1) is more than 2 times greater than in the case of the N-free MOF pyrolyzed at 500 °C (250 m2 g−1). Therefore, this confirms the role of N-doped carbonaceous species as excellent stabilizers of sub-nanometric Ru clusters during the early stage of the thermal transformation (between 400 and 600 °C).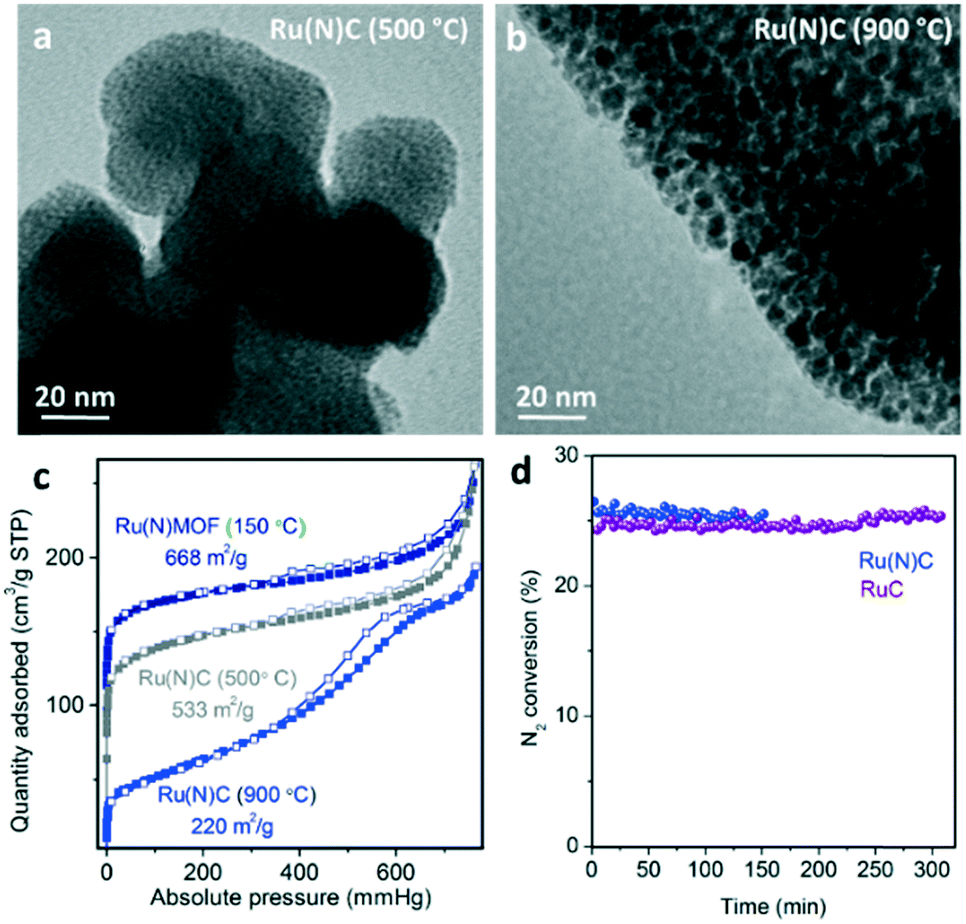 | ||
| Fig. 5 TEM images of nanostructured catalysts prepared from alternative Ru(N)MOF precursors at a) 500 °C and b) 900 °C. c) Effect of the temperature of the treatment on the N2 sorption isotherms at 77 K. d) N2 conversion for the Ru(N)MOF compared with the RuMOF at 350 °C and 95 barg. | ||
Unfortunately, pyrolysis of the Ru(N)MOF precursor at 900 °C, which is required for carbon graphitization, leads to less porous materials (200 m2 g−1) exhibiting larger Ru nanocrystals (2–3 nm). This may be caused by the carbon rearrangement during graphitization at 900 °C, as seen for the N-free RuMOF precursor. Promotion with barium and cesium leads to catalysts with similar activity levels to the N-free RuMOF catalysts as demonstrated in Fig. 5d. Additionally, the Ru(N)MOF-derived catalysts showed higher deactivation (3% per h) compared to the RuMOF catalysts (0.3% per h). Unfortunately, the stability test performed over the Ru(N)C material prepared at 500 °C (without additional treatment at 900 °C) under ammonia synthesis conditions shows full degradation of the non-graphitic matrix and sintering of the sub-nanometric Ru domains. Nevertheless, this highly dispersed Ru catalyst may be stable for other heterogeneously catalyzed reactions operating at lower temperatures.
Acetylene pretreatment
Acetylene deposition has been well studied for the preparation of carbon nanotubes or carbon-encapsulated metal particles.24 Recently, the use of acetylene deposition on the pores of MOFs resulted in an efficient approach for the precisely-controlled manufacture of ultra-small metal nanoparticles within porous carbon shells (core/shell metal/C).20 As shown in Fig. 6, the pre-treatment of the RuMOF precursor with acetylene at 500 °C resulted in the formation of carbonaceous species filling the MOF micropores, which stabilize the sub-nanometric Ru clusters upon pyrolysis at 900 °C under N2 in contrast to non-pre-treated samples (Fig. 6b).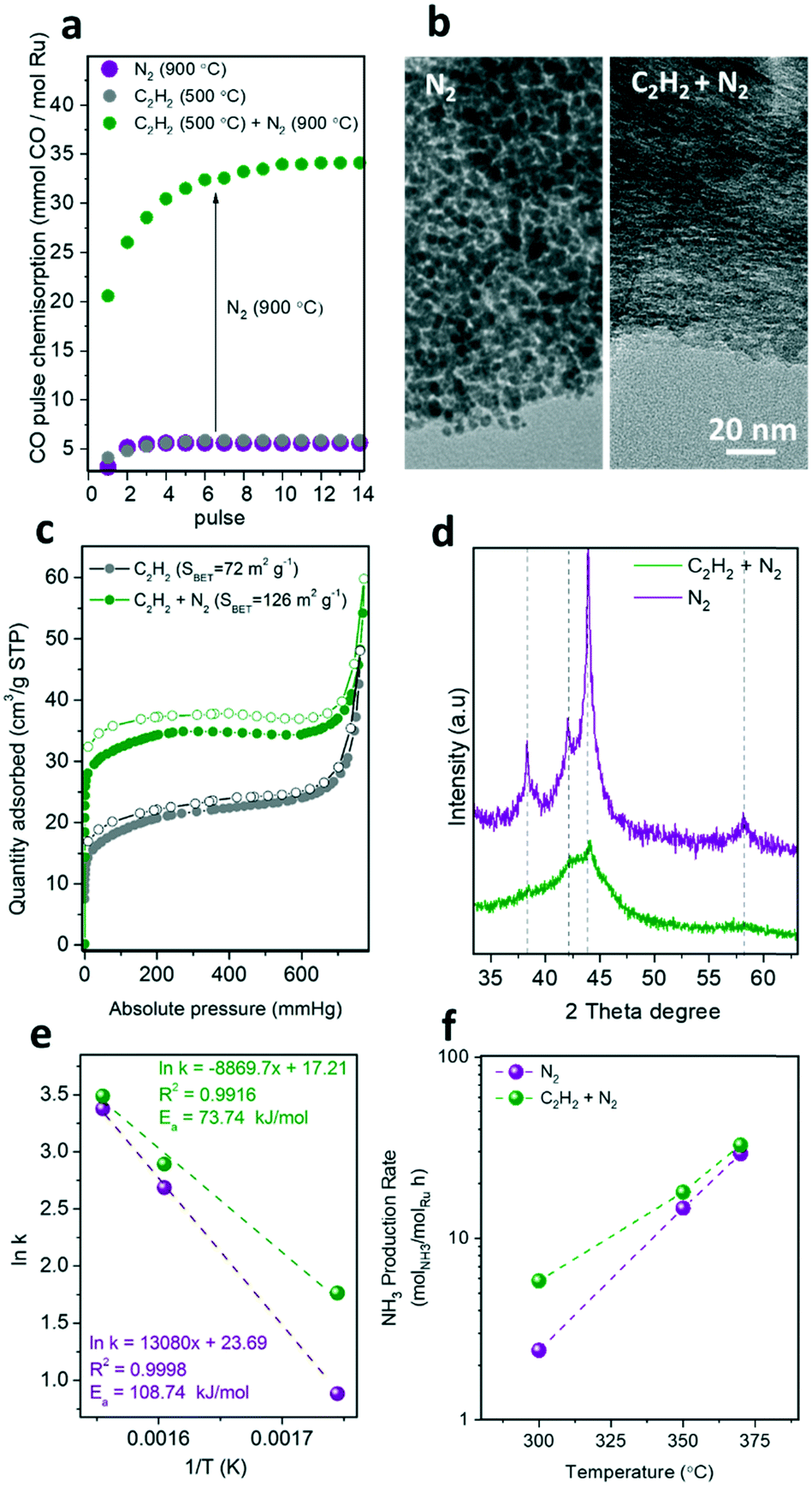 | ||
| Fig. 6 (a) CO chemisorption at 35 °C, (b) TEM analysis, (c) N2 sorption isotherms at 77 K, (d) XRD pattern, e) activation energy, and f) ammonia production rates for MOF-derived catalysts treated with C2H2 at 500 °C (grey), treated with C2H2 at 500 °C and also with N2 at 900 °C (green), and treated with only N2 at 900 °C (purple). | ||
The CO chemisorption study reveals that the pyrolytic treatment at 900 °C is crucial to uncover the available active surface of Ru (Fig. 6a), which also leads to an increment of the surface area (Fig. 6c). This can be attributed to the release of amorphous carbon species that were blocking the Ru surface. The stabilization of the sub-nanometric Ru clusters via acetylene pre-treatment was confirmed by TEM and XRD. The TEM images in Fig. 6b highlight the effect of the additional acetylene pre-treatment on reducing the Ru particle size, as suggested by the absence of larger Ru nanocrystals (>1 nm). This is supported by the highly broad XRD diffraction peaks collected for acetylene-treated samples (Fig. 6d). As shown in Fig. 6e and f and Table S2,† this increment in the active surface area of the C2H2-treated catalysts resulted in a 2-fold enhancement of the catalytic activity. For instance, a TOF of 50 × 10−3 s−1vs. 20 × 10−3 s−1 at 430 °C, and 1.6 × 10−3 s−1vs. 0.7 × 10−3 s−1 at 300 °C were measured for RuC (C2H2 + N2) vs. RuC (N2), respectively.
This result shows that the enhancement of the ruthenium active surface (Fig. 6a) by reducing the particle size does not result in a proportional enhancement of the catalytic activity. This suggests that the accessibility of the gas reagents to the active sites may be reduced by mass transport limitations within the bulky catalyst particles. This limitation can be overcome by further pre-dispersing MOF nanocrystals on a stable support prior to carbonization, as recently demonstrated by our group for the transformation of single MOF nanocrystals into single nanostructured catalysts.13,25 Moreover, the activity of the MOF-derived catalysts may still be limited by the N2 dissociation step occurring over the promoter surface rather than the hydrogenation step over Ru active sites, as recently demonstrated by the use of barium-doped calcium amides (Ba-Ca(NH2)2) as a dual support/promoter (Table S3†).26
Conclusions
Active and stable nanostructured Ru catalysts for low-temperature ammonia synthesis have been developed via pyrolytic treatment of Ru containing MOFs. The optimization of the thermal treatment conditions resulted in small Ru nanocrystals (<4 nm) well-dispersed on a graphitic carbon matrix demonstrating good activity and stability for ammonia synthesis. An additional pretreatment with acetylene allows stabilization of smaller and more active sub-nanometric Ru clusters upon an additional pyrolytic treatment at 900 °C in N2 required to release the available catalytically active surface. This two-step strategy provides an efficient and general route to develop highly stable and active MOF-derived heterogeneous catalysts, serving as a versatile platform to improve the interactions between the Ru active surface and alkali/alkaline-earth promoter via atomic design of appropriate multi-metallic MOF precursors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support from the U.S Department of Energy's Advanced Research Project Agency Energy (ARPA-E) Renewable Energy to Fuels Through Utilization of Energy-Dense Liquids (REFUEL) program (DE-AR-0000811). The authors would also like to acknowledge Mr. Ching-Chang Chung, Mr. Rohan Dhall and Mr. Fred Stevie from the Analytical Instrumentation Facility, College of Engineering, North Carolina State University (Raleigh, NC).References
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS; A. Klerke, C. H. Christensen, J. K. Norskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- N. Saadatjou, A. Jafari and S. Sahebdelfar, Chem. Eng. Commun., 2015, 202, 420–448 CrossRef CAS.
- Z. Kowalczyk, S. Jodzis and J. Sentek, Appl. Catal., A, 1996, 138, 83–91 CrossRef CAS; R. Schlogl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- H. Bielawa, O. Hinrichsen, A. Birkner and M. Muhler, Angew. Chem., Int. Ed., 2001, 40, 1061–1063 CrossRef CAS PubMed.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Dominguez, A. Leyva-Perez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- S. T. Hunt, M. Milina, A. C. Alba-Rubio, C. H. Hendon, J. A. Dumesic and Y. Román-Leshkov, Science, 2016, 352, 974–978 CrossRef CAS PubMed; H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef PubMed.
- C. P. Canlas, J. Lu, N. A. Ray, N. A. Grosso-Giordano, S. Lee, J. W. Elam, R. E. Winans, R. P. Van Duyne, P. C. Stair and J. M. Notestein, Nat. Chem., 2012, 4, 1030 CrossRef CAS PubMed; J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef PubMed; G.-H. Wang, J. Hilgert, F. H. Richter, F. Wang, H.-J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schüth, Nat. Mater., 2014, 13, 293 CrossRef PubMed.
- L. Liu, P. Concepción and A. Corma, J. Catal., 2016, 340, 1–9 CrossRef CAS; K. J. Lee, Y. J. Sa, H. Y. Jeong, C. W. Bielawski, S. H. Joo and H. R. Moon, Chem. Commun., 2015, 51, 6773–6776 RSC.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS; S. Dang, Q.-L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef; B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef PubMed.
- J. L. Shui, C. Chen, L. Grabstanowicz, D. Zhao and D. J. Liu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10629–10634 CrossRef CAS; X. W. Liu, T. J. Sun, J. L. Hu and S. D. Wang, J. Mater. Chem. A, 2016, 4, 3584–3616 RSC; K. Shen, X. D. Chen, J. Y. Chen and Y. W. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef; X. Sun, A. I. O. Suarez, M. Meijerink, T. van Deelen, S. Ould-Chikh, J. Zečević, K. P. de Jong, F. Kapteijn and J. Gascon, Nat. Commun., 2017, 8, 1680 CrossRef; K. J. Lee, J. H. Lee, S. Jeoung and H. R. Moon, Acc. Chem. Res., 2017, 50, 2684–2692 CrossRef; A. Dokania, A. Ramirez, A. Bavykina and J. Gascon, ACS Energy Lett., 2019, 4, 167–176 CrossRef; T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. E. Warringa, A. I. Dugulan, A. Chojecki, A. C. J. Koeken, M. Ruitenbeek, G. Meima, H.-U. Islam, G. Sankar, M. Makkee, F. Kapteijn and J. Gascon, ACS Catal., 2016, 6, 3236–3247 CrossRef.
- I. Agirrezabal-Telleria, I. Luz, M. A. Ortuño, M. Oregui-Bengoechea, I. Gandarias, N. López, M. A. Lail and M. Soukri, Nat. Commun., 2019, 10, 2076 CrossRef.
- I. Luz, M. Soukri and M. Lail, Chem. Commun., 2018, 54, 8462–8465 RSC.
- S. Ji, Y. Chen, Q. Fu, Y. Chen, J. Dong, W. Chen, Z. Li, Y. Wang, L. Gu, W. He, C. Chen, Q. Peng, Y. Huang, X. Duan, D. Wang, C. Draxl and Y. Li, J. Am. Chem. Soc., 2017, 139, 9795–9798 CrossRef CAS; X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng, W. Yan, X. Zheng, Z. Li, X. Wang, J. Yang, D. He, Y. Wang, Z. Deng, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef.
- J. Tang, R. R. Salunkhe, H. Zhang, V. Malgras, T. Ahamad, S. M. Alshehri, N. Kobayashi, S. Tominaka, Y. Ide, J. H. Kim and Y. Yamauchi, Sci. Rep., 2016, 6, 30295 CrossRef CAS; R. Lippi, S. C. Howard, H. Barron, C. D. Easton, I. C. Madsen, L. J. Waddington, C. Vogt, M. R. Hill, C. J. Sumby, C. J. Doonan and D. F. Kennedy, J. Mater. Chem. A, 2017, 5, 12990–12997 RSC; W. Cao, W. Luo, H. Ge, Y. Su, A. Wang and T. Zhang, Green Chem., 2017, 19, 2201–2211 RSC.
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC.
- O. Kozachuk, I. Luz, F. X. Llabrés i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS.
- M. Mukaida, T. Nomura and T. Ishimori, Bull. Chem. Soc. Jpn., 1967, 40, 2462 CrossRef CAS.
- L. Zhang and Y. H. Hu, J. Phys. Chem. C, 2010, 114, 2566–2572 CrossRef CAS.
- C. Zhang, X. Guo, Q. Yuan, R. Zhang, Q. Chang, K. Li, B. Xiao, S. Liu, C. Ma, X. Liu, Y. Xu, X. Wen, Y. Yang and Y. Li, ACS Catal., 2018, 7120–7130 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- Z. Kowalczyk, M. Krukowski, W. Raróg-Pilecka, D. Szmigiel and J. Zielinski, Appl. Catal., A, 2003, 248, 67–73 CrossRef CAS.
- C. Liang, Z. Wei, Q. Xin and C. Li, Appl. Catal., A, 2001, 208, 193–201 CrossRef CAS.
- A. K. M. Fazle Kibria, Y. H. Mo, K. S. Nahm and M. J. Kim, Carbon, 2002, 40, 1241–1247 CrossRef CAS; T.-C. Liu and Y.-Y. Li, Carbon, 2006, 44, 2045–2050 CrossRef.
- I. Luz, M. Soukri and M. Lail, Chem. Mater., 2017, 29, 9628–9638 CrossRef CAS; F. G. Cirujano, I. Luz, M. Soukri, C. Van Goethem, I. F. J. Vankelecom, M. Lail and D. E. De Vos, Angew. Chem., Int. Ed., 2017, 56, 13302–13306 CrossRef.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Angew. Chem., Int. Ed., 2018, 57, 2648–2652 CrossRef CAS; P. Peng, Y. L. Cheng, R. Hatzenbeller, M. Addy, N. Zhou, C. Schiappacasse, D. J. Chen, Y. N. Zhang, E. Anderson, Y. H. Liu, P. Chen and R. Ruan, Int. J. Hydrogen Energy, 2017, 42, 19056–19066 CrossRef; M. Hara, M. Kitano and H. Hosono, ACS Catal., 2017, 7, 2313–2324 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01303b |
| This journal is © The Royal Society of Chemistry 2020 |