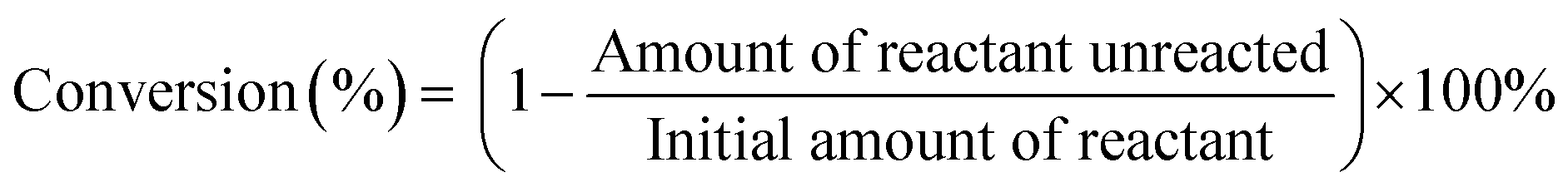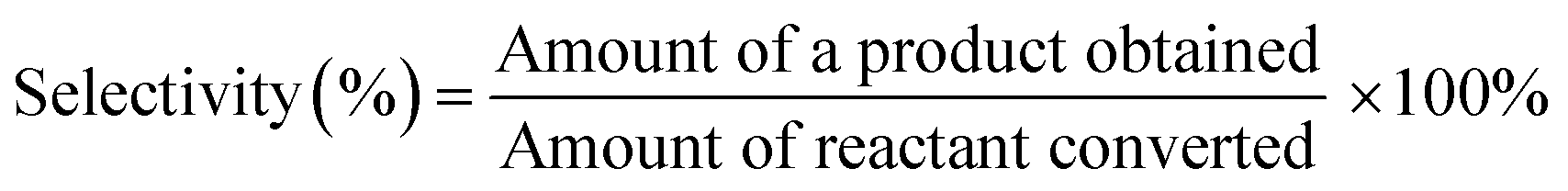Hydrodeoxygenation of ethyl stearate over Re-promoted Ru/TiO2 catalysts: rate enhancement and selectivity control by the addition of Re†
Received
21st September 2019
, Accepted 22nd November 2019
First published on 23rd November 2019
Abstract
The hydrodeoxygenation of a model compound, ethyl stearate, of biomass-derived resources into long chain alkanes was investigated with Re-modified Ru/TiO2 catalysts. A monometallic Ru(1)/TiO2 catalyst, in which Ru was loaded in 1 wt%, gave a selectivity of >95% to the long chain alkanes of n-octadecane and n-heptadecane at almost 100% conversion at 220 °C, while another monometallic Re(10)/TiO2 catalyst was less active in spite of its higher metal loading (10 wt%). It is interesting that the addition of less active Re species to Ru(1)/TiO2 not only enhanced the rate of hydrodeoxygenation and increased the ratio of n-octadecane/n-heptadecane, while the high total selectivity towards alkanes remained unchanged. The alkane ratio depended on the content of Re added, increasing significantly from 0.5 for Ru(1)/TiO2 to 3.2 for Ru(1)Re(10)/TiO2. The impacts of Re addition on the catalytic performance of Ru/TiO2 were discussed on the basis of physicochemical properties characterized for Re-modified and unmodified Ru/TiO2 catalysts. It is suggested that the improved hydrodeoxygenation activity of Re-modified catalysts depends on the presence of Lewis acid sites, and the weak acid sites are important for determining catalytic selectivity. In addition, reaction pathways in the hydrodeoxygenation of ethyl stearate to n-octadecane and n-heptadecane were examined by control reactions with possible intermediates such as 1-octadecanol and stearic acid. The Re-modified Ru/TiO2 catalysts were shown to be recyclable during repeated runs when the same mole ratio of catalyst to substrate was used.
Introduction
The utilization of biomass-based resources, which can replace fossil feedstock, is highly desirable owing to supply reliance, limited availability of fossil feedstock and global warming concerns.1,2 Among various biomass resources such as cellulose, lignin, fats, oils, and carbohydrates, vegetable oils are considered as one of the promising resources. These are mainly composed of triglycerides and free fatty acids with serious drawbacks such as high viscosity, low oxidation stability, poor cold flow properties and incompatibility with fossil fuel, so it is difficult to use them directly as fuel.3,4 Therefore, several approaches have been used for upgrading triglycerides, including transesterification, pyrolysis, hydro-treatment, hydrocracking, hydrodeoxygenation and so on.5 One of the often used methods is transesterification because of its relatively easy and simple operation, but its product suffers, when used as the transportation fuel, from engine problems due to incomplete removal of oxygen. For pyrolysis and hydrocracking, it is difficult to regulate the product distribution and undesired carbon deposition occurs under harsh conditions (350–600 °C, 10–20 MPa). Hydrodeoxygenation has been attracting more attention as one of the effective approaches for catalytic upgrading of biomass-derived resources to produce renewable, diesel-range hydrocarbons, which are often acquired from petroleum chemistry. It is demonstrated that conventional hydro-treating catalysts used in petroleum refineries such as NiMo and CoMo sulfide catalysts are effective in producing long-chain alkanes in the range of C12 to C18 from triglyceride at 260–450 °C in the presence of 4–15 MPa H2. It is unavoidable, however, that the alkane products would be polluted through sulfur leaching of the catalysts during the upgrading process. Moreover, most sulfide-based catalysts are moisture-sensitive and they are rapidly deactivated through sulfur leaching in the existence of trace amounts of water.6–8 Thus, some sulfur-free catalyst systems have been designed to catalyze the hydrodeoxygenation of vegetable fats, oils, fatty acid methyl esters and their model compounds for the production of desired alkanes.9 Supported transition metal catalysts containing Fe,10,11 Co,12,13 metal phosphides14,15 and metal carbides16–18 have been used in the hydrodeoxygenation of fatty acids and esters. For all these non-noble metal catalyst systems, however, harsh reaction conditions, in particular high reaction temperature (>260 °C), are required for satisfactory performance and cause some carbon loss for decarboxylation and/or decarbonylation of fatty acids and esters. In the last few years, supported Ni,19,20 Pt,21–23 Rh,24 Pd,25–27 and Ru28–31 based catalysts were reported to have high activity and selectivity for the conversion of bio-oils to alkanes. Among Ru catalysts supported on SiO2–Al2O3, SiO2, HZSM-5, ZSM-5, Mg(OH)2, La(OH)3 and La2O3, Ru/La(OH)3 showed remarkable performance, 95.9% selectivity to n-heptadecane at 100% conversion of stearic acid.32 Ru/NCNT33 and Ru/HZSM-529 were also reported in the hydrodeoxygenation of stearic acid and esters to diesel-range alkanes. Although monometallic catalysts are active, the use of metal–acid bifunctional catalysts and bimetallic catalysts was reported to enhance the deoxygenation of fatty acids and esters remarkably due to synergy between the two active components. For hydro-treating Jatropha oil, for instance, Pt/H-ZSM-5 modified with Re was much more active with a 67 wt% yield of long-chain alkanes than monometallic Pt/H-ZSM-5 at a high Jatropha oil/catalyst weight ratio (up to 10).34 A ReOx-modified Ir/SiO2 catalyst showed high-efficiency performance for the hydrodeoxygenation of various vegetable oils and waste cooking oil to alkanes.35 Although those multi-component catalysts may give good results, the loading of noble metals is >3 wt%. It is still required to reduce the loading of precious metals. Previously, our study reported that 1 wt% Ru/TiO2 catalyzed the hydrodeoxygenation of ethyl stearate to n-heptadecane although it needed 17 h to obtain 95% conversion.30 It is interesting, however, that the rate of hydrodeoxygenation was enhanced when the physical mixture of γ-Al2O3 and Ru/TiO2 was used, in which a conversion of 98.6% was obtained in a shorter reaction time of 4 h. These also worked for decarbonylation and/or decarboxylation routes than hydrodeoxygenation, unfortunately resulting in one carbon loss than the feedstock. RuRe/AC (activated carbon-supported Ru catalyst promoted with ReOx)36 and Re–Ru@SiO2 (Re promoted Ru catalyst encapsulated in porous silica nanoparticles)37 were reported to be effective catalysts for the hydrodeoxygenation of lignin-derived phenols and hydrogenolysis of glycerol, respectively. Hensen et al. reported that acidity induced by the presence of Re facilitated dehydration (C–O bond cleavage) over C–C bond cleavage.38 Liu et al. also assumed that partially reduced ReOx exhibited Lewis acidity and interacted with the oxygen atom of the carbonyl group of fatty acids and esters.35 This interaction weakens the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and promotes acyl C–O bond cleavage.
O bond and promotes acyl C–O bond cleavage.
Those results allow us to expect that Re-modified Ru/TiO2 can be an effective catalyst for the hydrodeoxygenation of fatty acid esters to alkanes with high carbon efficiency. In the present work, various Ru/TiO2 catalysts, modified and unmodified with Re, were applied for the hydrodeoxygenation of model compound ethyl stearate, CH3(CH2)16COOCH2CH3, at a temperature of 220 °C. It is interesting to note that although Re itself is less active, the addition of Re to Ru/TiO2 enhances the rate of ethyl stearate transformation and increases the ratio of n-C18H38/n-C17H36 that are produced in a total selectivity of >95%. The physicochemical properties of Re-modified and unmodified Ru/TiO2 catalysts were characterized by several methods and the features of the surface of supported metal particles effective for the target reaction were discussed. In addition, control reactions were also made with possible reaction intermediates, stearic acid (CH3(CH2)16COOH) and 1-octanedecanol (CH3(CH2)17OH), to examine possible reaction pathways over Re-modified Ru catalysts, involving hydrogenolysis, hydrogenation, decarbonylation, and dehydration steps.
Results and discussion
Hydrodeoxygenation performance
Under the reaction conditions used, n-octadecane, n-heptadecane and 1-octadecanol were observed to form with very minor products n-hexadecane and n-pentadecane and no other products were detected. The product selectivity for ethyl stearate hydrodeoxygenation over the Ru/TiO2 catalyst was found to change significantly with the addition of Re species. Table 1 presents the results with various catalysts (entries 2–7) collected at high conversion levels close to 100% except for monometallic Re(10)/TiO2 (entry 8). All these catalysts tested were reduced at 300 °C. A conversion of 98% and a total selectivity to n-octadecane and n-heptadecane of 93% were obtained with Ru(1)/TiO2, with the ratio of the long chain alkanes of n-C18H38/n-C17H36 being 0.5 (entry 1). The addition of Re in the amounts of 0.5–10 wt% increased the long chain alkane ratio (entries 2–6), and the ratio of n-C18H38/n-C17H36 reached 3.2 for Ru(1)Re(10)/TiO2. A further increase in the Re content to 15 wt% did not affect the ethyl stearate conversion but decreased the alkane ratio to 2.8 (entry 7). In contrast to Re-modified and unmodified Ru(1)/TiO2 catalysts, Re(10)/TiO2 containing a larger amount of metal species was even less active and gave a larger amount of 1-octadecanol compared to the long chain alkanes at a total conversion of 51% in 8 h (entry 8). The reaction rate was compared at a conversion lower than 30% in Table S1.† The reaction rate was 53.6 mol min−1 gcat−1 for Ru(1)/TiO2 and 25.8 mol min−1 gcat−1 for Re(10)/TiO2, and it gradually increased with increasing Re content from 1 to 10 wt% and reached 413 mol min−1 gcat−1 for Ru(1)Re(10)/TiO2, which was 7 times higher than that of Ru(1)/TiO2. That is, the addition of Re to Ru(1)/TiO2 not only enhanced the rate of hydrodeoxygenation but also increased the long chain alkane ratio of n-C18H38/n-C17H36.
Table 1 Results of hydrodeoxygenation of ethyl stearate on various Re-modified Ru(1)/TiO2 catalysts at high conversion levels
| Entry |
Catalyst |
Time (h) |
Conversion (%) |
Selectivitya (%) |
Carbon balanceb (%) |
C18/C17c (−) |
| C18H37OH |
n-C17H36 |
n-C18H38 |
| Reaction conditions: 1 mmol ethyl stearate, 60 mg catalyst (reduced at 300 °C), 10 mL n-hexane, 3 MPa H2, 220 °C. Small amounts of other alkane products n-C15H32 and n-C16H34 were detected. “n.d.” indicates “not detected”. The total amount of C18H37OH, n-C17H36, n-C18H38, n-C15H32 and n-C16H34 against the amount of ethyl stearate consumed. A deviation from 100% carbon balance may be due to the undesired consumption of the substrate ethyl stearate via transesterification with product 1-octadecanol producing a larger molecule that cannot be detected by the present GC analysis.39 n-C18H38/n-C17H36. |
| 1 |
Ru(1) |
6 |
98.4 |
2.3 |
63.8 |
29.5 |
98.9 |
0.5 |
| 2 |
Ru(1)Re(0.5) |
4 |
99.9 |
n.d. |
50.9 |
47.3 |
99.1 |
0.9 |
| 3 |
Ru(1)Re(1) |
4 |
99.5 |
0.9 |
33.4 |
51.3 |
88.2 |
1.5 |
| 4 |
Ru(1)Re(2) |
2 |
96.5 |
3.6 |
30.4 |
57.5 |
94.7 |
1.9 |
| 5 |
Ru(1)Re(5) |
2 |
92.2 |
3.8 |
21.8 |
60.9 |
88.8 |
2.8 |
| 6 |
Ru(1)Re(10) |
2 |
99.9 |
n.d. |
22.2 |
69.7 |
94.3 |
3.2 |
| 7 |
Ru(1)Re(15) |
2 |
99.9 |
n.d. |
24.8 |
68.2 |
97.7 |
2.8 |
| 8 |
Re(10) |
8 |
50.5 |
27.8 |
0.5 |
4.0 |
32.3 |
— |
The influence of the reduction temperature on the catalytic performance was examined using Ru(1)Re(10)/TiO2 as the most effective catalyst (Table S2†). It was indicated that the reduction temperature (200, 300, and 400 °C) little affected the rate of ethyl stearate hydrodeoxygenation but did alter the long chain alkane ratio of n-C18H38/n-C17H36, which was 2.7, 3.2 and 2.3 at conversion levels of >96% for the catalysts reduced at 200, 300, and 400 °C, respectively. The catalyst reduced at 300 °C was used in subsequent experiments.
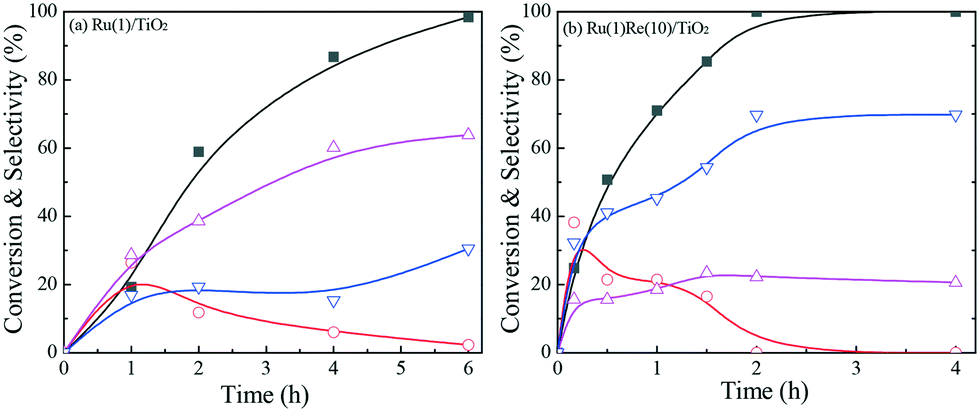
Variation in ethyl stearate conversion and product selectivity with reaction time was examined with two selected catalysts, the most active Ru(1)Re(10)/TiO2 and unmodified reference Ru(1)/TiO2, as shown in Fig. 1. For either catalytic reaction, n-heptadecane (n-C17H36), n-octadecane (n-C18H38) and 1-octadecanol were found to form with even smaller amounts of by-products; the amounts of the long chain alkanes (n-C17H36 and n-C18H38) increased with time while that of the 1-octadecanol was maximized at a certain time and then decreased. This indicates that 1-octadecanol is a possible intermediate in the hydrodeoxygenation of ethyl stearate into these alkanes. The ratio of the two alkanes (n-C18H38/n-C17H36) does not change with time (conversion) so much. As mentioned above, Ru(1)/Re(10)/TiO2 has a higher n-C18H38/n-C17H36 ratio of 3.2, by a factor of >6, compared to 0.5 Ru(1)/TiO2 (Table 1). Besides, the activity of Ru(1)Re(10)/TiO2 was compared with the commercial catalysts Ru/C and Ru/Al2O3. n-C18H38 was produced as the main product over the former one; while the main product changed to n-C17H36 over the latter, and the activity of Ru/C was much lower compared with those of Ru(1)Re(10)/TiO2 and Ru/Al2O3 (Table S3†).
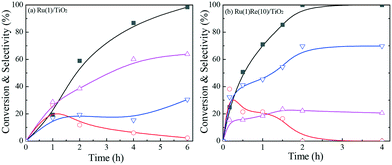 |
| | Fig. 1 Variation in ethyl stearate conversion and product selectivity with reaction time over (a) Ru(1)/TiO2 and (b) Ru(1)Re(10)/TiO2. Reaction conditions: 1 mmol ethyl stearate, 60 mg catalyst, 10 mL n-hexane, 3 MPa H2, 220 °C. Conversion (■), n-C17H36 ( ), n-C18H38 ( ), n-C18H38 ( ) and 1-octadecanol ( ) and 1-octadecanol ( ). The data are fitted to B-type spline curves using the Origin software. ). The data are fitted to B-type spline curves using the Origin software. | |
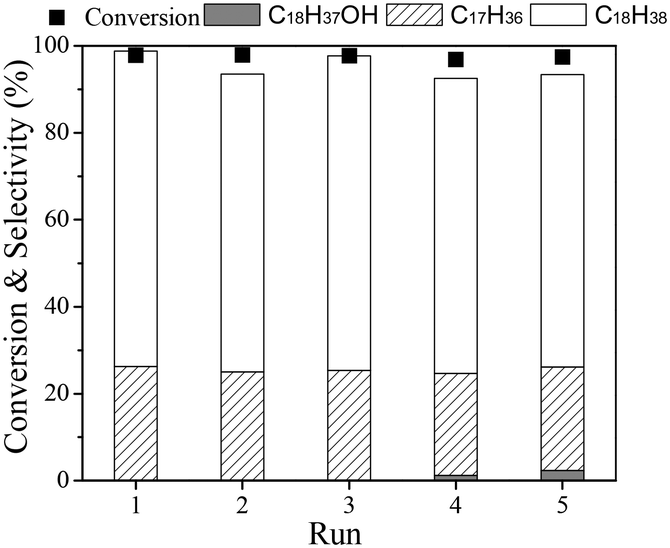
Finally, the stability of the Ru(1)Re(10)/TiO2 catalyst was examined. For the recycling tests, the catalyst was separated from the reaction mixture after a run, washed with n-hexane several times, dried in air, reduced at 300 °C, and then reused for the next run. Recycling was carried out with the recovered catalyst under the same reaction conditions with the same mole ratio of catalyst to substrate, and the results are shown in Fig. 2. It is observed that the catalyst was stable during the repeated runs; the total conversion of ethyl stearate was >98% and the selectivity to n-octadecane was around 67% for the five runs. The characterization results (XRD and TGA in Fig. S1†) of the used catalyst after being recycled five times showed no obvious difference between the fresh and used catalysts, and the amount of carbon deposit was lower, about 5.5%.
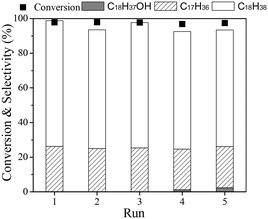 |
| | Fig. 2 Recycling test of Ru(1)Re(10)/TiO2 catalyst in hydrodeoxygenation of ethyl stearate. Reaction conditions: mole ratio of catalyst to substrate (0.006![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), T 220 °C, n-hexane 10 mL, H2 3 MPa, time 2 h. :1), T 220 °C, n-hexane 10 mL, H2 3 MPa, time 2 h. | |
Catalyst characterization
As mentioned above, the addition of less active Re species to Ru/TiO2 could enhance the catalytic performance in the hydrodeoxygenation of ethyl stearate into long chain alkanes, n-C18H38 and n-C17H36, which were produced in a total selectivity of >98.2% at almost 100% conversion (Table 1, entry 2). In addition, the alkane ratio of n-C18H38/n-C17H36 was controlled by the content of Re added, which increased from 0.5 for Ru(1)/TiO2 to 3.2 for Ru(1)Re(10)/TiO2. To discuss these impacts of Re addition to Ru/TiO2, the physicochemical properties of Re-modified and unmodified Ru(1)/TiO2 catalysts were characterized by several methods.
The structural features were examined for the support and monometallic and bimetallic catalysts TiO2, Ru(1)/TiO2, Re(10)/TiO2 and Ru(1)Re(10)/TiO2 reduced at 300 °C, as listed in Table 2. The surface area and pore volume are similar among these catalysts but the average pore diameter is decreased by the loading of a larger amount of Re (entries 2 and 3) compared to Ru(1)/TiO2 (entry 1). The TEM images show that the sizes of metal particles are similar (about 1 nm) for the monometallic catalysts and the bimetallic ones (1.2 nm) (Fig. S2†). The XRD data show only diffraction peaks due to the TiO2 support but no diffraction peaks of Ru and Re species, indicating that these are finely dispersed on the support (Fig. S3†). The three catalysts showed the same diffraction peak positions and intensities; the diffraction peaks at 25.2°, 37.8°, 48.0° and 62.7° were assigned to the anatase phase and those at 27.4°, 54.3° and 69.0° to the rutile phase. Ru(1)/TiO2 can adsorb CO in a volume as expected from its metal particle size determined by TEM (entry 1) while a very small volume of CO is adsorbed on Re(10)/TiO2 than expected (entry 2). However, the bimetallic catalyst can adsorb CO in a larger volume compared to Ru(1)/TiO2 and Re(10)/TiO2, implying the presence of some interactions between Ru and Re species. The same catalysts were further subjected to H2-TPD. As shown in Fig. S4,† either Ru(1)/TiO2 or Re(10)/TiO2 showed a H2 desorption peak around 198 °C. However, Ru(1)Re(10)/TiO2 had a peak at 73 °C and a shoulder one at 183 °C similar to the monometallic catalysts. The amount of H2 desorbing from Ru(1)/TiO2 was smaller than that from Re(10)/TiO2. The addition of Re to Ru(1)/TiO2 increased the amount of H2 desorbed in a low temperature range up to 150 °C but the H2 desorption profile at higher temperatures was similar to that of Re(10)/TiO2. Some Re species added to Ru(1)/TiO2 should produce H2 adsorption sites from which H2 desorbs easily at low temperatures, in addition to the adsorption sites similar to those of monometallic Re(10)/TiO2. Hence, the properties of the surface of supported metal particles, characterized by CO and H2 adsorption, of the Re-modified Ru/TiO2 catalyst are significantly different from those of the corresponding monometallic Ru(1)/TiO2 and Re(10)/TiO2 catalysts, which should be responsible for its enhanced activity in the hydrodeoxygenation of ethyl stearate.
Table 2 Physicochemical properties of the Ru(1)/TiO2, Re(10)/TiO2, Ru(1)Re(10)/TiO2 and TiO2
| Entry |
Catalyst |
Amount of CO chemisorbeda (mmol gcat−1) |
Metal particle sizeb (nm) |
Surface areac (m2 g−1) |
Pore volumec (cm3 g−1) |
Average pore diameterc (nm) |
|
Figure in parentheses indicates the degree of metal dispersion determined from CO chemisorption. The CO/metal atom was assumed to be 1:1.40
Determined by TEM. Figure in parentheses indicates the degree of metal dispersion estimated by (0.9/size in nm) × 100.41
Determined by nitrogen adsorption.
|
| 1 |
Ru(1) |
0.067 (68) |
1.0 (90) |
55.5 |
0.34 |
4.3 |
| 2 |
Re(10) |
0.021 (4.8) |
0.9 (100) |
51.8 |
0.31 |
3.1 |
| 3 |
Ru(1)Re(10) |
0.116 (18) |
1.2 (75) |
49.9 |
0.39 |
3.1 |
| 4 |
TiO2 |
— |
— |
56.2 |
0.35 |
3.8 |
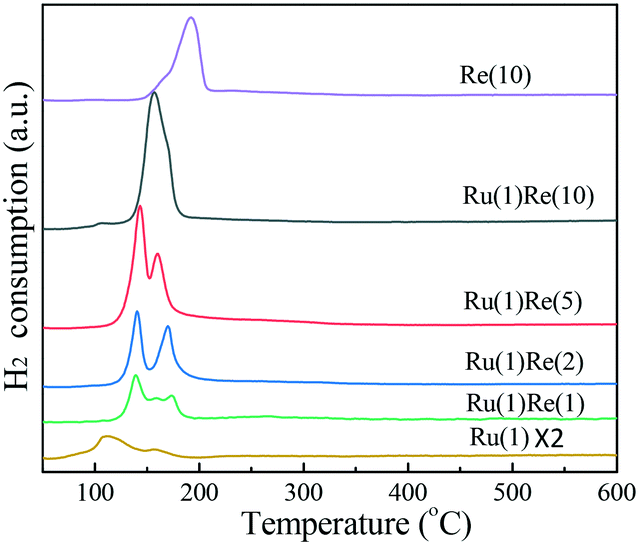
Then, the reduction behaviour of supported Ru catalysts was examined by H2-TPR. Fig. 3 shows that Ru(1)/TiO2 has a major reduction peak at 110 °C and a shoulder peak at around 157 °C and these peaks are ascribed to the reduction of RuOx. Meanwhile, Re(10)/TiO2 shows a broad peak at a slightly higher temperature of 192 °C. Ru(1)Re(2)/TiO2 exhibits two peaks at around 140 °C and 170 °C. The amount of H2 consumed for the former is larger than that for Ru(1)/TiO2, and so this peak may be attributed to the reduction of RuOx and partial reduction of ReOx species in intimate contact with Ru. The latter peak at around 170 °C was assigned to the reduction of isolated ReOx species. Ru(1)Re(5)/TiO2 also shows two peaks at 143 °C and 160 °C, similar to Ru(1)Re(2)/TiO2. When the Re content was increased to 10 wt%, however, the H2 consumption took place as indicated by one peak, centred at 156 °C, which was due to the reduction of RuOx and ReOx. In comparison with Re(10)/TiO2, the reduction peak of ReOx species in the Re-modified Ru(1)/TiO2 samples shifted from 192 °C to 137–174 °C, indicating that the presence of Ru facilitated the reduction of ReOx moieties. It is likely that the reduction of Re species is promoted by more easily reducible Ru species, and the two metal species produce the surface features of metal particles different from those of the monometallic catalysts and give the high performance of the bimetallic catalyst. It was reported that the existence of Rh lowered the reduction temperature of Re precursors.42 Henson et al. reported that the Pt component in Pt–Re/TiO2 favoured the reduction of ReOx oxide species.43 Tomishige and co-workers also reported that the addition of Ru, Pt, Pd, and Rh to Re/SiO2 could lower the reduction temperature of ReOx.44
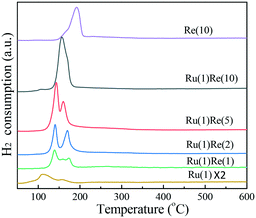 |
| | Fig. 3 H2-TPR results of different TiO2-supported catalysts. | |
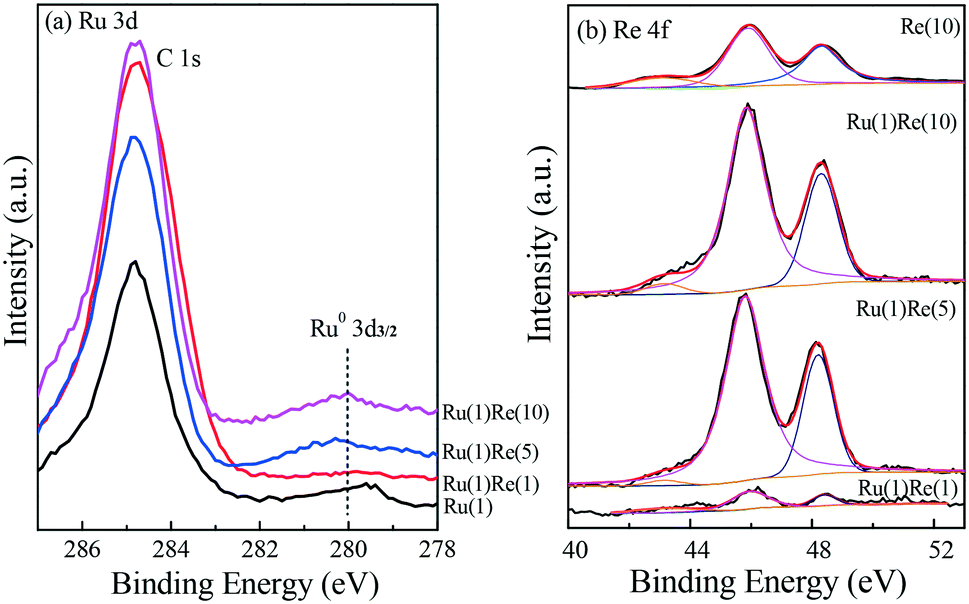
The catalysts were characterized by XPS to further examine the chemical state of their component species. Fig. 4(a) shows the Ru 3d5/2 results, in which the strength was weak due the small metal loading (1 wt%) and the nature of the metal itself. A small peak was observed to exist at a binding energy (BE) of 279.5 eV and so Ru was almost reduced to metallic zero-valent Ru0.45–47 The peaks of Re 4f observed can be attributed to Re7+ (48.3 eV), Re6+ (45.9 eV) and Re4+ (43.1 eV), indicating that Re is not completely reduced to Re0 by the reduction at 300 °C, being mainly in the form of ReOx.48,49 The presence of such ReOx species will provide acid sites on the surface of the Re-modified Ru/TiO2 catalyst, as described below.
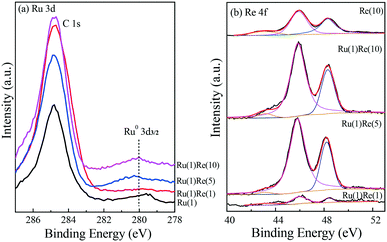 |
| | Fig. 4 XPS results of (a) Ru 3d and (b) Re 4f for reduced TiO2-supported metal catalysts. | |
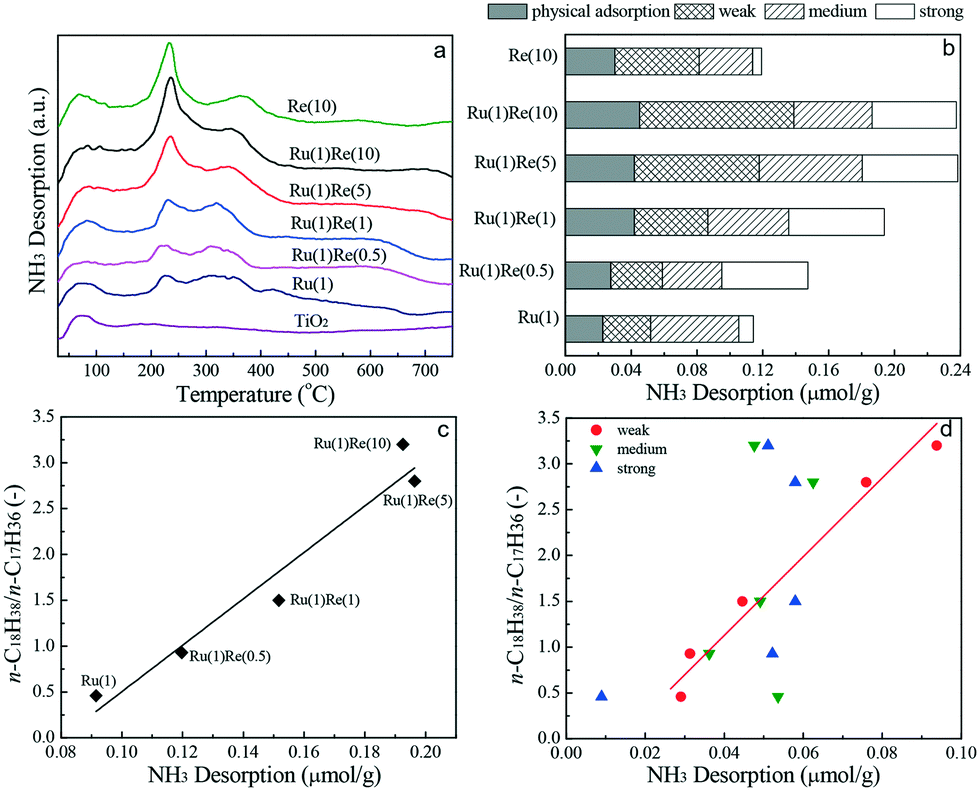
Finally, NH3-TPD analysis is carried out to evaluate the strength and distribution of acid sites existing on the catalysts. The NH3-TPD profiles collected are shown in Fig. 5(a). The NH3 desorption profile for TiO2 shows only one peak with a maximum at 80 °C, which is mainly the physical adsorption of NH3. For the monometallic and Re-modified Ru(1)/TiO2 catalysts, NH3 desorption occurred in a wide temperature range and the desorption profiles were separated (deconvoluted) into four desorption regions characterized by the corresponding peak desorption temperatures at 80, 250, 330 and 500 °C (Fig. S5†). These peaks were physical adsorption sites (80 °C), and weak (250 °C), medium (330 °C) and strong (500 °C) acid sites. The NH3-TPD profiles showed similar adsorption peaks for all the catalysts but the addition of Re increased the strength of acid sites and the total amount of acid sites. Fig. 5b shows the distribution of different acid sites estimated based on the TPD peak areas. It was found that the n-C18H38/n-C17H36 ratio increased linearly with the total amount of acid sites (Fig. 5c) and it was correlated well with the amount of weak acid sites. The importance of Lewis acid sites is consistent with the literature.38,49–54
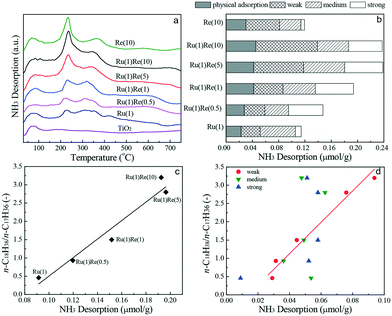 |
| | Fig. 5 (a) NH3-TPD profiles of TiO2 and TiO2-supported catalysts, (b) relative amounts of acid sites different in strength characterized by desorption temperatures, (c) relationship between the ratio of n-C18H38/n-C17H36 and the amount of total acid sites, and (d) relationship between the ratio of n-C18H38/n-C17H36 and the amount of weak, medium, strong acid sites. The line in (c) is the correlation of the ratio of n-C18H38/n-C17H36 with the amount of total acid sites. The line in (d) is the correlation of the ratio of n-C18H38/n-C17H36 with the amount of weak acid sites. | |
Reaction pathways: control reactions of possible intermediates
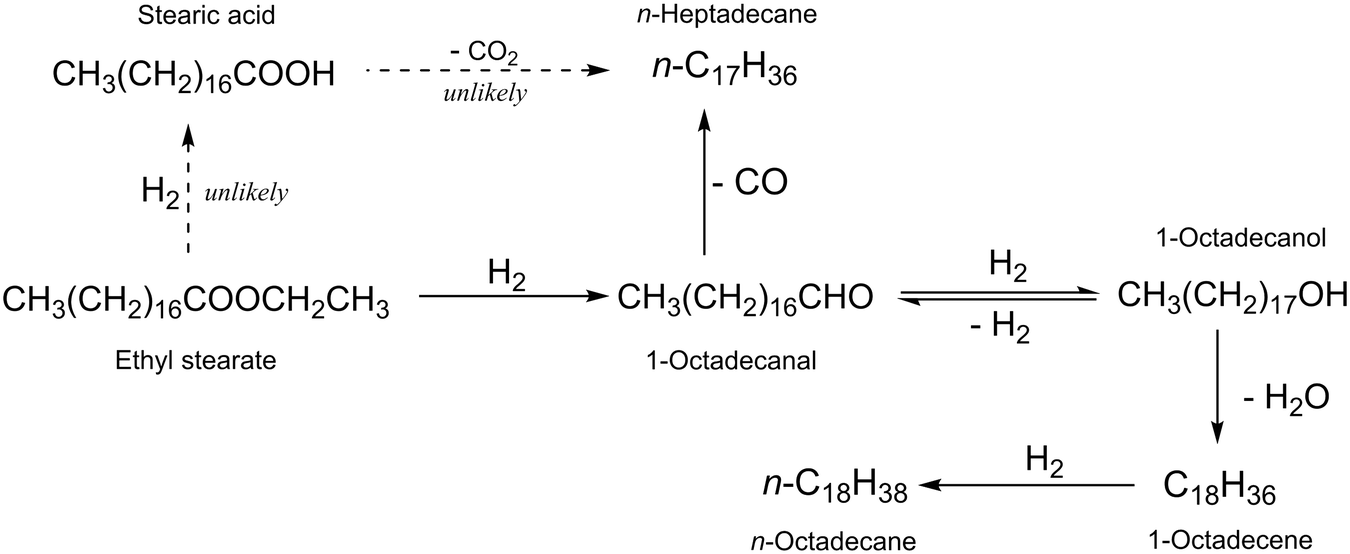
The hydrodeoxygenation of ethyl stearate into n-octadecane and n-heptadecane occurred through several reaction pathways. To clarify these pathways, possible intermediates were tested as starting substrates under the same reaction conditions as used for the target reaction. As shown in Fig. 1, the amount of 1-octadecanol increased, was maximized and then decreased with reaction time. Table 3 shows the results with several TiO2-supported catalysts for the reaction with 1-octadecanol as a starting reactant, and the results collected at high conversion levels indicated that the long chain alkane ratio of n-C18H38/n-C17H36 with Ru(1)/TiO2 was 0.4 at a conversion of 73% (entry 1) and increased with the increasing amount of Re added (entries 2–6). The alkane ratios observed in the reactions with 1-octadecanol and ethyl stearate were comparable to the same catalyst (Tables 1 and 3). That is, 1-octadecanol should be an important intermediate for the formation of the two long chain alkanes, n-C18H38 and n-C17H36. Possible reaction pathways are explained in Scheme 1. The former is obtained through dehydration of 1-octadecanol followed by hydrogenation while the latter is formed through dehydrogenation of 1-octadecanol and subsequent decarbonylation of 1-octadecanal. Under the reaction conditions used, 1-octadecanal was unstable and reactive, which was then not detected by GC analysis. The rate of 1-octadecanol conversion was higher than that of ethyl stearate under the same conditions (Tables 3 and S4†) for all the catalysts examined. It is assumed that the rates of hydrogenation and decarbonylation of 1-octadecanal to 1-octadecanol and n-heptadecane, respectively, are faster than the rate of hydrogenation of ethyl stearate into 1-octadecanal. There is a possibility that the ethyl stearate hydrogenation occurs and stearic acid is an intermediate to be produced. However, stearic acid was not detected in the reactions with ethyl stearate; in addition, when stearic acid was used as a starting reactant, the long chain alkane ratio was <2.0 at a high conversion of >99% over Ru(1)Re(10)/TiO2 (Table S5†), which is much lower than the value (3.2) obtained in the reaction of ethyl stearate with the same catalyst (Table 1). Thus, the reaction pathways via stearic acid are unlikely.
Table 3 Hydrodeoxygenation of a possible intermediate 1-octadecanol (C18H37OH) on various catalysts
| Entry |
Catalyst |
Time (h) |
Conversion (%) |
Selectivitya (%) |
C18/C17b |
|
n-C17H36 |
n-C18H38 |
| Reaction conditions:1 mmol 1-octadecanol, 60 mg catalyst (reduced at 300 °C), 10 mL n-hexane, 3 MPa H2, 220 °C. There are other very minor products (n-C15H32 and n-C16H34) detected. n-C18H38/n-C17H36. |
| 1 |
Ru(1) |
2 |
72.8 |
64.4 |
25.2 |
0.4 |
| 2 |
Ru(1)Re(0.5) |
1 |
99.8 |
29.6 |
52.9 |
1.8 |
| 3 |
Ru(1)Re(1) |
1 |
99.9 |
26.7 |
57.5 |
2.2 |
| 4 |
Ru(1)Re(2) |
1 |
99.3 |
28.4 |
63.2 |
2.2 |
| 5 |
Ru(1)Re(5) |
1 |
99.9 |
23.4 |
60.5 |
2.6 |
| 6 |
Ru(1)Re(10) |
1 |
99.9 |
12.9 |
77.2 |
6.0 |
| 7 |
Re(10) |
2 |
12.0 |
6.2 |
67.4 |
10.9 |
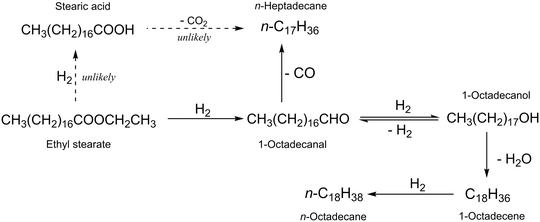 |
| | Scheme 1 Possible reaction pathways in the hydrodeoxygenation of ethyl stearate to long chain alkanes. n-Heptadecane is produced via decarbonylation of 1-octadecanal while n-octadecane from 1-octadecanal via hydrogenation, dehydration, and hydrogenation. The other route via hydrogenation of ethyl stearate to stearic acid is unlikely. | |
The possible reaction pathways are proposed above (Scheme 1). The roles of the component species of the Re-modified Ru/TiO2 catalysts active in the hydrodeoxygenation of ethyl stearate will then be considered, such as metallic Ru species modified by Re additives and acid sites created by ReOx species. One of the reactants, H2, is adsorbed on the monometallic Ru(1)/TiO2 and Re(10)/TiO2 catalysts and the Re-modified Ru/TiO2 one based on the H2-TPD results (Fig. S4†). The H2 desorption temperature is similar between the two monometallic catalysts, in which a larger amount of H2 is adsorbed on Re(10)/TiO2 due to its higher metal loading compared to Ru(1)/TiO2. For Ru(1)Re(10)/TiO2, in contrast, H2 was observed to desorb at low temperatures of 30–100 °C in a larger amount, in addition to the H2 desorption at the same higher temperatures as for the two monometallic catalysts. This indicates that more reactive (less stable) hydrogen species exist on the surface of the Re-modified Ru(1)Re(10)/TiO2 catalyst, being one of the factors determining its higher rate of ethyl stearate hydrodeoxygenation. The metallic zero-valent Ru species exposed on the surface of the supported metal particles in the Ru(1)/TiO2 and Ru(1)Re(10)/TiO2 catalysts are the main active sites for the hydrogenation reactions involved in hydrodeoxygenation (Scheme 1). The other reactant, ethyl stearate, may be adsorbed on the surface of the metal particles and the TiO2 support in the Re-modified and -unmodified Ru/TiO2 catalysts. For the target reactions, the surface of TiO2 close to the metal particles should be important. A possible factor for the lowest activity of Re(10)/TiO2 is the inability of the adsorption of ethyl stearate, which is difficult to be adsorbed on its surface as inferred from the very small amount of CO chemisorbed (Table 2); the catalyst interacts little with the substrate including the CO group attributable to its molecular adsorption with the long aliphatic chain. K. Shimizu and co-workers reported that the amount of Lewis acid sites increased by the addition of MoOx to Pt/TiO2 and the cooperation of Pt with the Lewis acid sites produced the higher activity of Pt–MoOx/TiO2.55 It was also reported that the Lewis acidity increased after Sn addition to Pt/SAPO-11 due to the presence of partially reduced SnOx and these Lewis acid sites could improve the hydrodeoxygenation rate over PtSn/SAPO-11.56 Moreover, the addition of WOx to Pt/Al2O3 could enhance the catalytic activity and C18 selectivity, due to the modification of the structural (electronic and particle size) and acidic properties of the catalyst.57 For the monometallic catalyst, the amount of acid sites is lower as compared with that of the Ru(1)Re(10)/TiO2 catalyst, which could adsorb the substrate with its CO group. It is believed that the ratio of n-C18H38/n-C17H36 depends on the acid sites (Fig. 5) which will affect the decarbonylation of 1-octadecanal and dehydration of 1-octadecanol. The formation of these two long chain alkanes is promoted by these species and so their total selectivity increases with an increase of these species. When the amount of metallic Re species is large, the rate of hydrogenation of 1-octadecanal to 1-octadecanol relatively becomes more significant and so the decarbonylation of 1-octadecanal to n-C17H36 was relatively lowered, resulting in the increase in the formation of n-C18H38 and enhancing the n-C18H38/n-C17H36 ratio. Therefore, the addition of Re species to Ru/TiO2 enhances the degree of metal (Ru) dispersion (positive for the hydrogenation) and produces more acid sites (positive for the dehydration), which results in the enhancement in the overall rate of reaction and the long chain alkane selectivity. The increase in the relative amount of hydrogenation sites and the acid sites enhances the n-C18H38/n-C17H36 ratio.
Conclusions
The hydrodeoxygenation of ethyl stearate was investigated with Re-modified Ru/TiO2 catalysts in n-hexane at 220 °C. A monometallic Ru(1)/TiO2 catalyst showed almost 100% conversion in 6 h, in which the total selectivity to the long chain alkanes n-octadecane and n-heptadecane was >95% and the n-C18H38/n-C17H36 ratio was 0.5. The addition of less active Re species to Ru(1)/TiO2 enhanced not only the rate of hydrodeoxygenation but also the n-C18H38/n-C17H36 ratio while its high total selectivity remained unchanged. The alkane ratio was significantly increased from 0.5 to 3.2 with an increasing Re content from 0 to 10 wt% in weight. It is worth noting that the present transformation exhibits higher atom efficiency due to lower carbon loss. The amount of weak acid sites is the main factor affecting the change of catalyst selectivity. For the present hydrodeoxygenation of ethyl stearate, it is likely that the substrate is hydrogenated through 1-octadecanal to 1-octadecanol but little to stearic acid; the transformation between 1-octadecanal and 1-octadecanol is reversible; n-octadecane is formed via the dehydration of 1-octadecanol and the subsequent hydrogenation of 1-octadecene while n-heptadecane is formed via the decarbonylation of 1-octadecanal.
Experimental
Preparation of the catalysts
All chemicals used in this work were of analytical grade and used without further purification. Commercial TiO2 (P25, Acros) was used as a support and RuCl3·3H2O and NH4ReO4 (Sigma-Aldrich) as metal precursors. A Ru/TiO2 sample with Ru 1 wt% loading (= metal/(metal + support)) was prepared by an impregnation method. Briefly, RuCl3·3H2O (26.2 mg) was dissolved in water and the solution was added drop by drop onto the TiO2 support (1.0 g) at room temperature. Afterwards, the sample obtained was dried overnight at 80 °C and calcined in air at 400 °C for 2 h. This sample will be labeled as Ru(1)/TiO2. Bimetallic catalysts including Ru and Re species were prepared by a co-impregnation method. TiO2 was impregnated with RuCl3·3H2O and NH4ReO4 together and the sample was dried in an oven at 80 °C overnight and calcined at 400 °C for 2 h in air. The loading of Ru was fixed to 1.0% in weight and that of Re was changed, being 0.5%, 1%, 2%, 5%, 10% and 15% in weight. In the following, the sample will be abbreviated as, for example, Ru(1)Re(10)/TiO2, where 1 and 10 stand for the mass fraction of Ru and Re, respectively. Besides, the Re(1)/TiO2 sample was also prepared from NH4ReO4 by the impregnation method following the procedures described above. The sample was dried in an oven at 80 °C overnight and calcined at 400 °C for 2 h. Prior to use, in the hydrodeoxygenation runs, all samples were reduced under a pure H2 flow (20 mL min−1) at 300 °C for 2 h.
Characterization of the catalysts
The loading of Ru and Re was measured by inductively coupled plasma optical emission spectrometry (ICP-OES) and the actual contents observed were very similar to the nominal values within ±95%. Powder X-ray diffraction (XRD) was performed to measure the crystal phases of the catalysts using a Bruker D8 Advance X-ray diffractometer with a Cu Kα source at 40 kV and 40 mA in a 2θ range from 10° to 90°. Transmission electron microscopy (TEM) was performed on a JEOL JEM-2010 instrument at an accelerating voltage of 188 kV. Hydrogen temperature programmed reduction (H2-TPR) was carried out in a flow of 10 vol% H2/Ar (30 mL min−1) on a Micromeritics AutoChem II 2920 equipped with a thermal conductivity detector (TCD). Both hydrogen temperature programmed desorption (H2-TPD) and ammonia temperature programmed desorption (NH3-TPD) experiments were performed on a Tianjin XQ TP-5080 chemisorption instrument with a TCD. As for H2-TPD, 100 mg catalyst was loaded into a quartz reactor and then reduced with a H2 flow (30 mL min−1) at 300 °C for 1 h. After reduction, the catalyst was cooled to room temperature in a H2 flow (30 mL min−1) and the reactor was flushed with Ar for 1 h to reach a stable background. Then, H2-TPD was carried out with Ar at a flow rate of 30 mL min−1 and at a temperature ramp rate of 10 °C min−1. As for NH3-TPD, 100 mg pre-reduced catalyst was loaded into a quartz reactor and treated with a He flow (30 mL min−1) at 120 °C for 30 min to remove surface moisture. Then, the reactor was cooled to 30 °C and the catalyst was exposed to a pure NH3 flow (20 mL min−1) for 30 min. After the NH3 adsorption, the reactor was flushed with He for 1 h to reach a stable background and then, NH3-TPD was carried out with He at a flow rate of 30 mL min−1 and a temperature ramp rate of 10 °C min−1. Nitrogen adsorption–desorption experiments were performed on a Micromeritics ASAP 2020 surface area and porosity analyzer. X-ray photoelectron spectroscopy (XPS) measurements were conducted using a VG Microtech 3000 Multilab equipped with an Al Kα X-ray exciting source (1486.6 eV). The C1s peak at 248.8 eV (accuracy within ±0.3 eV) was used as the charge-up correction reference. The thermogravimetric analysis (TGA) curve was recorded using a Perkin-Elmer Thermal Analysis at a heating rate of 10 °C min−1 in an air flow, from room temperature to 800 °C.
Activity test of the catalysts
The TiO2-supported Ru, Re, and RuRe catalysts prepared were tested for the hydrodeoxygenation of ethyl stearate in a 100 mL stainless steel autoclave reactor. Prior to the reaction, the catalyst sample was reduced with a H2 flow (30 mL min−1) at 300 °C for 2 h. Measured amounts of ethyl stearate (1 mmol), solvent (n-hexane 10 mL) and catalyst (60 mg) were added into the reactor and it was sealed and flushed with 2 MPa H2 at least three times to remove the air. Then, the reactor was pressurized with 3 MPa H2 and put into an oil bath at the desired temperature for 15 min. The reaction was started by magnetic stirring at 1200 rpm, under which the reaction was in a kinetic regime. After the reaction, the reactor was cooled using an ice-water bath and the gas mixture was then vented to ambient pressure. The liquid products were analyzed using a gas chromatograph (Shimadzu GC-14C) equipped with a capillary column (Restek Stabilwax 30 m × 0.53 mm × 1 μm) and a flame ionization detector, in which eicosane was used as the internal standard. The products were identified by gas chromatography/mass spectrometry (GC/MS, Agilent 5890). The conversion was determined as the amount of reactant converted against the initial amount of reactant. The product selectivity was determined by the amount (mole) of product obtained against the amount (mole) of reactant converted.| |  | (1) |
| |  | (2) |
The catalyst reusability was also examined. The catalyst was collected by centrifugation after the first run, washed with n-hexane several times, dried in air overnight, and then reduced at 300 °C for 2 h before the next run. A slight loss in the catalyst mass (<10%) was observed during catalyst separation and collection in each run. As the conversion was linearly increased with the amount of catalyst used (Table S6†), for the recycling test, the same mole ratio of catalyst to substrate (0.006:1) was used for each run.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Key Research and Development Program of China (2016YFA0602900), the National Natural Science Foundation of China (NSFC 21603212, 21672204), and the Chinese Academy of Sciences President's International Fellowship Initiative (2018VCA0012).
Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- B. P. Pattanaik and R. D. Misra, Renewable Sustainable Energy Rev., 2017, 73, 545–557 CrossRef CAS.
- M. Ameen, M. T. Azizan, S. Yusup, A. Ramli and M. Yasir, Renewable Sustainable Energy Rev., 2017, 80, 1072–1088 CrossRef CAS.
- S. Chen, G. L. Zhou and C. X. Miao, Renewable Sustainable Energy Rev., 2019, 101, 568–589 CrossRef CAS.
- A. Vonortas and N. Papayannakos, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 3–23 CAS.
- R. Tiwari, B. S. Rana, R. Kumar, D. Verma, R. Kumar, R. K. Joshi, M. O. Garg and A. K. Sinha, Catal. Commun., 2011, 12, 559–562 CrossRef CAS.
- A. Centeno, E. Laurent and B. Delmon, J. Catal., 1995, 154, 288–298 CrossRef CAS.
- X. Li, X. Y. Luo, Y. B. Jin, J. Y. Li, H. D. Zhang, A. P. Zhang and J. Xie, Renewable Sustainable Energy Rev., 2018, 82, 3762–3797 CrossRef CAS.
- J. Li, J. J. Zhang, S. Wang, G. Y. Xu, H. Wang and D. G. Vlachos, ACS Catal., 2019, 9, 1564–1577 CrossRef CAS.
- K. Kandel, J. W. Anderegg, N. C. Nelson, U. Chaudhary and I. Slowing, J. Catal., 2014, 314, 142–148 CrossRef CAS.
- G. J. Wu, N. Zhang, W. L. Dai, N. J. Guan and L. D. Li, ChemSusChem, 2018, 11, 2179–2188 CrossRef CAS PubMed.
- W. D. Jia, G. Y. Xu, X. H. Liu, F. Zhou, H. X. Ma, Y. Zhang and Y. Fu, Energy Fuels, 2018, 32, 8438–8446 CrossRef CAS.
- Y. Yang, J. X. Chen and H. Shi, Energy Fuels, 2013, 27, 3400–3409 CrossRef CAS.
- Y. X. Yang, C. Ochoa-Hernandez, V. A. D. O'Shea, J. M. Coronado and D. P. Serrano, ACS Catal., 2012, 2, 592–598 CrossRef CAS.
- L. S. Macedo, R. R. Oliveira, T. van Haasterecht, V. T. da Silva and H. Bitter, Appl. Catal., B, 2019, 241, 81–88 CrossRef.
- D. R. Stellwagen and J. H. Bitter, Green Chem., 2015, 17, 582–593 RSC.
- S. K. Kim, D. Yoon, S.-C. Lee and J. Kim, ACS Catal., 2015, 5, 3292–3303 CrossRef CAS.
- C. M. Pichler, D. Gu, H. Joshi and F. Schuth, J. Catal., 2018, 365, 367–375 CrossRef CAS.
- P. Kumar, S. R. Yenumala, S. K. Maity and D. Shee, Appl. Catal., A, 2014, 471, 28–38 CrossRef CAS.
- J. M. Crawford and M. A. Carreon, Ind. Eng. Chem. Res., 2018, 57, 15991–15997 CrossRef CAS.
- L. Zhou and A. Lawal, Appl. Catal., A, 2017, 532, 40–49 CrossRef CAS.
- Y. X. Liu, X. J. Yang, H. Y. Liu, Y. H. Ye and Z. J. Wei, Appl. Catal., B, 2017, 218, 679–689 CrossRef CAS.
- Y. W. Bie, J. Lehtonen and J. Kanervo, Appl. Catal., A, 2016, 526, 183–190 CrossRef CAS.
- N. A. Grosso-Giordano, T. R. Eaton, Z. Y. Bo, S. Yacob, C. C. Yang and J. M. Notestein, Appl. Catal., B, 2016, 192, 93–100 CrossRef CAS.
- E. Meller, U. Green, Z. Aizenshtat and Y. Sasson, Fuel, 2014, 133, 89–95 CrossRef CAS.
- D. P. Serrano, J. M. Escola, L. Briones and M. Arroyo, Microporous Mesoporous Mater., 2019, 280, 88–96 CrossRef CAS.
- S. Mondal, R. Singuru, S. C. Shit, T. Hayashi, S. Irle, Y. Hijikata, J. Mondal and A. Bhaumik, ACS Sustainable Chem. Eng., 2018, 6, 1610–1619 CrossRef CAS.
- J. Z. Chen and Q. Y. Xu, Catal. Sci. Technol., 2016, 6, 7239–7251 RSC.
- L. M. He, C. Y. Wu, H. Y. Cheng, Y. C. Yu and F. Y. Zhao, Catal. Sci. Technol., 2012, 2, 1328–1331 RSC.
- G. Y. Xu, Y. Zhang, Y. Fu and Q. X. Guo, ACS Catal., 2017, 7, 1158–1169 CrossRef CAS.
- J. H. Guo, G. Y. Xu, F. Shen, Y. Fu, Y. Zhang and Q. X. Guo, Green Chem., 2015, 17, 2888–2895 RSC.
- J. Li, S. Wang, H. Y. Liu, H. J. Zhou and Y. Fu, ChemistrySelect, 2017, 2, 33–41 CrossRef CAS.
- K. Murata, Y. Y. Liu, M. Inaba and I. Takahara, Energy Fuels, 2010, 24, 2404–2409 CrossRef CAS.
- S. B. Liu, T. Simonetti, W. Q. Zheng and B. Saha, ChemSusChem, 2018, 11, 1446–1454 CrossRef CAS PubMed.
- B. K. Jung, J. Lee, J. M. Ha, H. Lee, D. J. Suh, C. H. Jun and J. Jae, Catal. Today, 2018, 303, 191–199 CrossRef.
- K. T. Li and R. H. Yen, Nanomaterials, 2018, 8, 14 Search PubMed.
- A. Ciftci, D. Ligthart and E. J. M. Hensen, Green Chem., 2014, 16, 853–863 RSC.
- J.-h. Guo, G.-y. Xu, F. Shen, Y. Fu, Y. Zhang and Q.-x. Guo, Green Chem., 2015, 17, 2888–2895 RSC.
- E. Gonzalez, C. Marchant, C. Sepulveda, R. Garcia, I. T. Ghampson, N. Escalona and J. L. Garcia-Fierro, Appl. Catal., B, 2018, 224, 368–375 CrossRef CAS.
-
G. D.-M. M. Boudart, Kinetics of Heterogeneous Catalytic Reactions, Princeton Univ Press, Princeton, N.J., 1984, pp. 25–26 Search PubMed.
- M. Chia, Y. J. Pagan-Torres, D. Hibbitts, Q. H. Tan, H. N. Pham, A. K. Datye, M. Neurock, R. J. Davis and J. A. Dumesic, J. Am. Chem. Soc., 2011, 133, 12675–12689 CrossRef CAS PubMed.
- J. Pritchard, A. Ciftci, M. Verhoeven, E. J. M. Hensen and E. A. Pidko, Catal. Today, 2017, 279, 10–18 CrossRef CAS.
- Y. Takeda, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2012, 2, 2221–2223 RSC.
- H. Ojagh, D. Creaser, S. Tamm, P. Arora, S. Nystrom, E. L. Grennfelt and L. Olsson, Ind. Eng. Chem. Res., 2017, 56, 5547–5557 CrossRef CAS.
- S. A. Singh and G. Madras, Appl. Catal., A, 2016, 518, 102–114 CrossRef CAS.
- C. Elmasides, D. I. Kondarides, S. G. Neophytides and X. E. Verykios, J. Catal., 2001, 198, 195–207 CrossRef CAS.
- H. Iida and A. Igarashi, Appl. Catal., A, 2006, 303, 192–198 CrossRef CAS.
- S. B. Liu, W. Q. Zheng and B. Saha, ChemSusChem, 2018, 11, 1446–1454 CrossRef CAS PubMed.
- Q. R. Tian, K. Qiao, F. Zhou, K. Q. Chen, T. F. Wang, J. Fu, X. Y. Lu and P. K. Ouyang, Energy Fuels, 2016, 30, 7291–7297 CrossRef CAS.
- S. B. Liu, S. Dutta, W. Q. Zheng, N. S. Gould, Z. W. Cheng, B. J. Xu, B. Saha and D. G. Vlachos, ChemSusChem, 2017, 10, 3225–3234 CrossRef CAS PubMed.
- K. Kon, W. Onodera, S. Takakusagi and K.-i. Shimizu, Catal. Sci. Technol., 2014, 4, 3705–3712 RSC.
- Y. Qin, L. L. He, J. Z. Duan, P. Chen, H. Lou, X. M. Zheng and H. P. Hong, ChemCatChem, 2014, 6, 2698–2705 CrossRef CAS.
- M. H. Lu, F. W. Lu, J. Zhu, M. S. Li, J. J. Zhu and Y. H. Shan, React. Kinet., Mech. Catal., 2015, 115, 251–262 CrossRef CAS.
- K. Kon, T. Toyao, W. Onodera, S. Siddiki and K. Shimizu, ChemCatChem, 2017, 9, 2822–2827 CrossRef CAS.
- N. Chen, Y. X. Ren and E. W. Qian, J. Catal., 2016, 334, 79–88 CrossRef CAS.
- S. Janampelli and S. Darbha, Catal. Today, 2018, 309, 219–226 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01909j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ac,
Ke
Liu
ac,
Zhuangqing
Wang
abc,
Qiang
Liu
ac,
Haiyang
Cheng
*ac,
Ke
Liu
ac,
Zhuangqing
Wang
abc,
Qiang
Liu
ac,
Haiyang
Cheng
 ), n-C18H38 (
), n-C18H38 ( ) and 1-octadecanol (
) and 1-octadecanol ( ). The data are fitted to B-type spline curves using the Origin software.
). The data are fitted to B-type spline curves using the Origin software.