
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3d metallaelectrocatalysis for resource economical syntheses
Parthasarathy
Gandeepan†
 ab,
Lars H.
Finger†
a,
Tjark H.
Meyer
a and
Lutz
Ackermann
*acd
ab,
Lars H.
Finger†
a,
Tjark H.
Meyer
a and
Lutz
Ackermann
*acd
aInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de; Web: http://www.ackermann.chemie.uni-goettingen.de/index.html
bDepartment of Chemistry, Indian Institute of Technology Tirupati, Tirupati, Andhra Pradesh 517506, India
cWoehler Research Institute for Sustainable Chemistry (WISCh), Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany
dDepartment of Chemistry, University of Pavia, Viale Taramelli 10, 27100 Pavia, Italy
First published on 27th May 2020
Abstract
Resource economy constitutes one of the key challenges for researchers and practitioners in academia and industries, in terms of rising demand for sustainable and green synthetic methodology. To achieve ideal levels of resource economy in molecular syntheses, novel avenues are required, which include, but are not limited to the use of naturally abundant, renewable feedstocks, solvents, metal catalysts, energy, and redox reagents. In this context, electrosyntheses create the unique possibility to replace stoichiometric amounts of oxidizing or reducing reagents as well as electron transfer events by electric current. Particularly, the merger of Earth-abundant 3d metal catalysis and electrooxidation has recently been recognized as an increasingly viable strategy to forge challenging C–C and C–heteroatom bonds for complex organic molecules in a sustainable fashion under mild reaction conditions. In this review, we highlight the key developments in 3d metallaelectrocatalysis in the context of resource economy in molecular syntheses until February 2020.
 Parthasarathy Gandeepan | Parthasarathy Gandeepan has been Assistant Professor in the Department of Chemistry at the Indian Institute of Technology Tirupati since December 2019. He studied BSc (Chemistry) and MSc (Organic Chemistry) at the University of Madras and obtained his PhD (Chemistry) with Chien-Hong Cheng at the National Tsing Hua University, Taiwan. He did postdoctoral works with Chien-Hong Cheng (NTHU, Taiwan), Lutz Ackermann (University of Göttingen, Germany), and Beatrice Collins (University of Bristol, UK) before starting his independent research career. His research interests include transition metal-catalyzed organic transformations, photoredox catalysis, and sustainable methodology developments for organic synthesis. He is teaching Organic Chemistry courses to graduate and postgraduate students. |
 Lars H. Finger | Lars Finger studied chemistry at the Philipps-University Marburg and spent one semester at the Heriot-Watt University Edinburgh. He received his PhD from the PU Marburg in 2016 under the supervision of Prof. Jörg Sundermeyer, working on organic salts with azolate and chalcogenolate anions for electrochemical applications. In 2017, Lars joined the group of Prof. Lutz Ackermann as a post-doctoral fellow focusing on C–H activation by metallaelectrocatalysis. |
 Tjark H. Meyer | Tjark H. Meyer studied chemistry at the Georg-August-University Göttingen and obtained his MSc in 2016 under the supervision of Prof. L. Ackermann. During his studies he joined the group of Dr Stephen P. Thomas at the University of Edinburgh for a four-month research stay, and later, he performed an industrial placement at Bayer AG, Wuppertal. In 2017, he started his doctoral studies in the group of Prof. L. Ackermann. His current research is focused on synthetic organic electrochemistry, with a major interest on the merger of metal-catalysed C–H activation and electrocatalysis. |
 Lutz Ackermann | Lutz Ackermann studied chemistry at the Christian-Albrechts University Kiel and obtained his PhD in 2001 with Alois Fürstner at the MPI für Kohlenforschung in Mülheim/Ruhr. He was a postdoctoral fellow with Robert G. Bergman (UC Berkeley) before initiating his independent research in 2003 at the Ludwig-Maximilians University München, supported within the Emmy Noether Program of the DFG. In 2007, he became full professor at the Georg-August University Göttingen, where he served as the Dean of Research and Dean of Chemistry as well as the director of the Wöhler Research Institute for Sustainable Chemistry (WISCh). The development of novel concepts for homogeneous catalysis and their applications to sustainable organic synthesis, late-stage peptide diversification, and molecular imaging are among his main current research interests. |
1. Introduction
In the last few decades, significant attention has been paid to the development of sustainable strategies for molecular assembly in terms of both economic and environmental benefits.1 In 1991, Trost proposed the concept of atom economy and highlighted the value of transition metal catalysis towards this goal.2,3 Later, Anastas and Warner introduced the twelve principles of green chemistry in 1998 to provide guidelines for minimizing the environmental footprint of chemical syntheses.4 Further, the principles of step economy5 and redox economy have been put forward.6,7 In contrast, resource economy aims to provide a basis for designing fully sustainable methodologies of molecular syntheses.8 These principles are reflected by the development of the corresponding techniques (Fig. 1a). Considering the transformation of two organic reactants with the corresponding reactive groups, the cross-coupling catalysis was developed. C–H activation allowed the use of even less functionalized starting materials.9–18 Metallaphotocatalysis offers the potential to drive catalytic processes from solar energy.19–21 This is finally accomplished with metallaelectrocatalysis, which in addition has recently gained momentum by outstanding cooperativity effects with base metal catalysts. In this regard, metallaelectrocatalysis has emerged as a potent strategy to achieve selective chemical syntheses (Fig. 1b). In this review, we focus on the recent developments to achieve improved resource economy in molecular syntheses by combining Earth-abundant 3d metal catalysis and electrooxidation towards ideal resource economy until February 2020.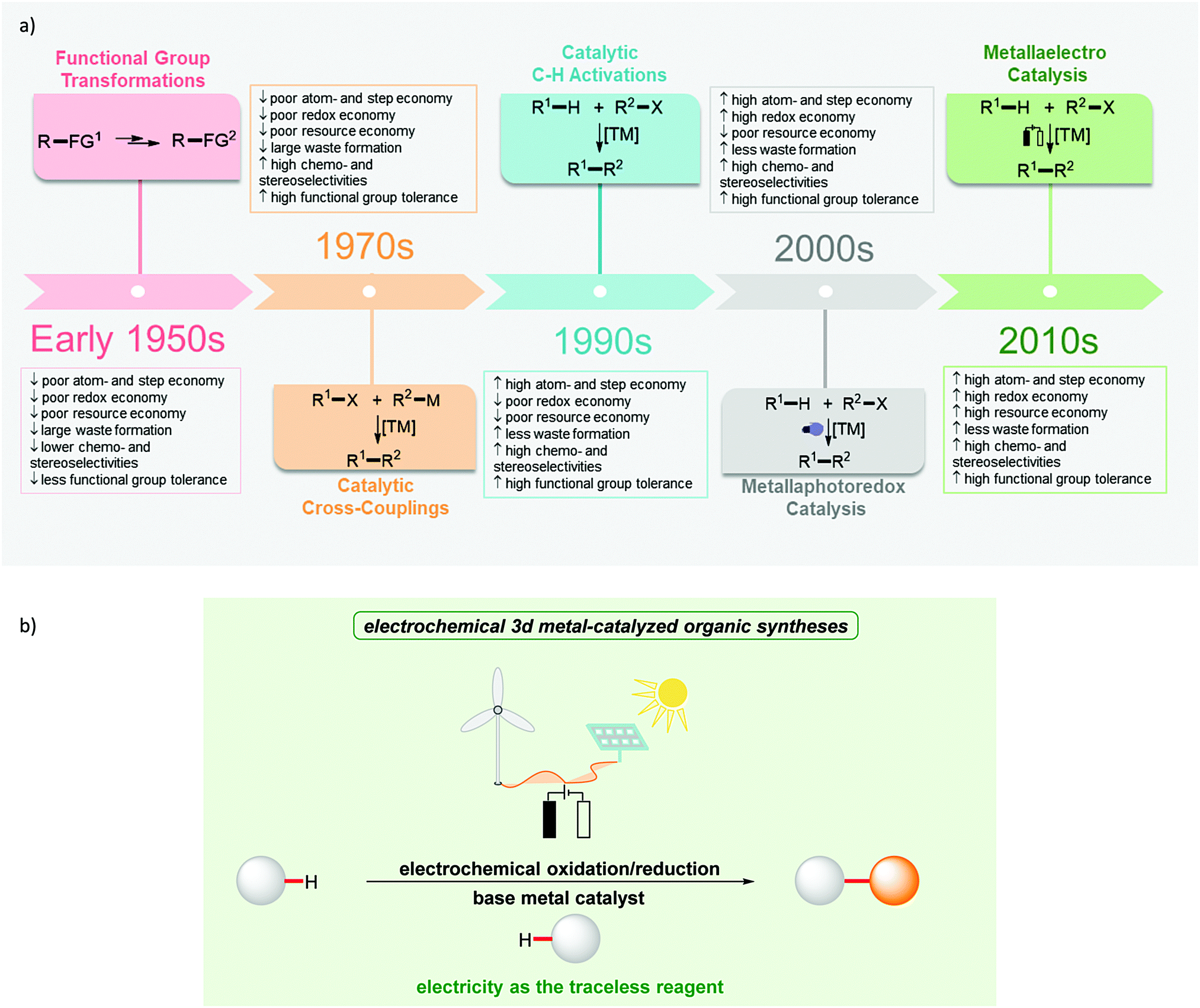 | ||
| Fig. 1 General overview for enhancing the economy of organic syntheses. (a) General paradigm shift in catalytic organic syntheses. (b) Achieving resource economy by electrochemical 3d metal-catalyzed organic transformations. | ||
2. Resource economy in organic syntheses
Organic syntheses based on multi-step functional group transformations should ideally be avoided because of their low atom-and step-economy. In contrast, transition metal-catalyzed C–H activation has been established as a potent tool to achieve high levels of atom- and step-economy by employing non-functionalized starting materials.22–33 Despite significant advances, precious transition metal catalysts, such as palladium, rhodium, ruthenium, and iridium, are predominantly required for achieving high efficiencies. In stark contrast, recent developments in this rapidly evolving arena suggested the use of Earth-abundant 3d transition metal complexes as catalysts in C–H activation reactions.34,35 However, oxidative transition metal-catalyzed transformations usually demand stoichiometric amounts of toxic and expensive metals oxidants. This requirement compromises the overall sustainable nature of the C–H activation strategy by generating often toxic metal wastes, high production costs, and overall poor resource economy.

Considering its natural abundance, the use of molecular oxygen as the terminal oxidant in transition metal-oxidase catalysis continues to be of topical interest.36–38 Despite the improved sustainability and resource economy, the use of oxygen is on the one hand limited by its fixed oxidation potential. On the other hand, and more importantly, major safety hazards in view of flammable reaction media in large scale syntheses need to be addressed, rendering this approach challenging particularly for scale-up.39
As a consequence, in recent years, photoredox catalysis has emerged as a strategy to affect redox processes in transition metal catalysis in a mild and selective manner.40–45 These transformations are mostly based on noble iridium or ruthenium complexes as the photoredox catalyst (PC) to convert light into chemical energy. It is noteworthy that each PC offers only a unique fixed redox potential and therefore the use and synthesis of specifically tailored PCs are essential to realize the desired redox transformation (Fig. 2, top). The synthetic modification of PCs to adjust the redox potential typically demands labor- and cost-intensive multistep transformations. Though the power of photoredox catalysis has evidently been demonstrated for transition metal catalysis,46 the quantum yields of these transformations are typically low when employing visible light, and natural sunlight can normally not be used, because of the narrow absorption range of the PCs. In contrast, high power LEDs or Kessil LED lights are often necessary for satisfactory efficacy.
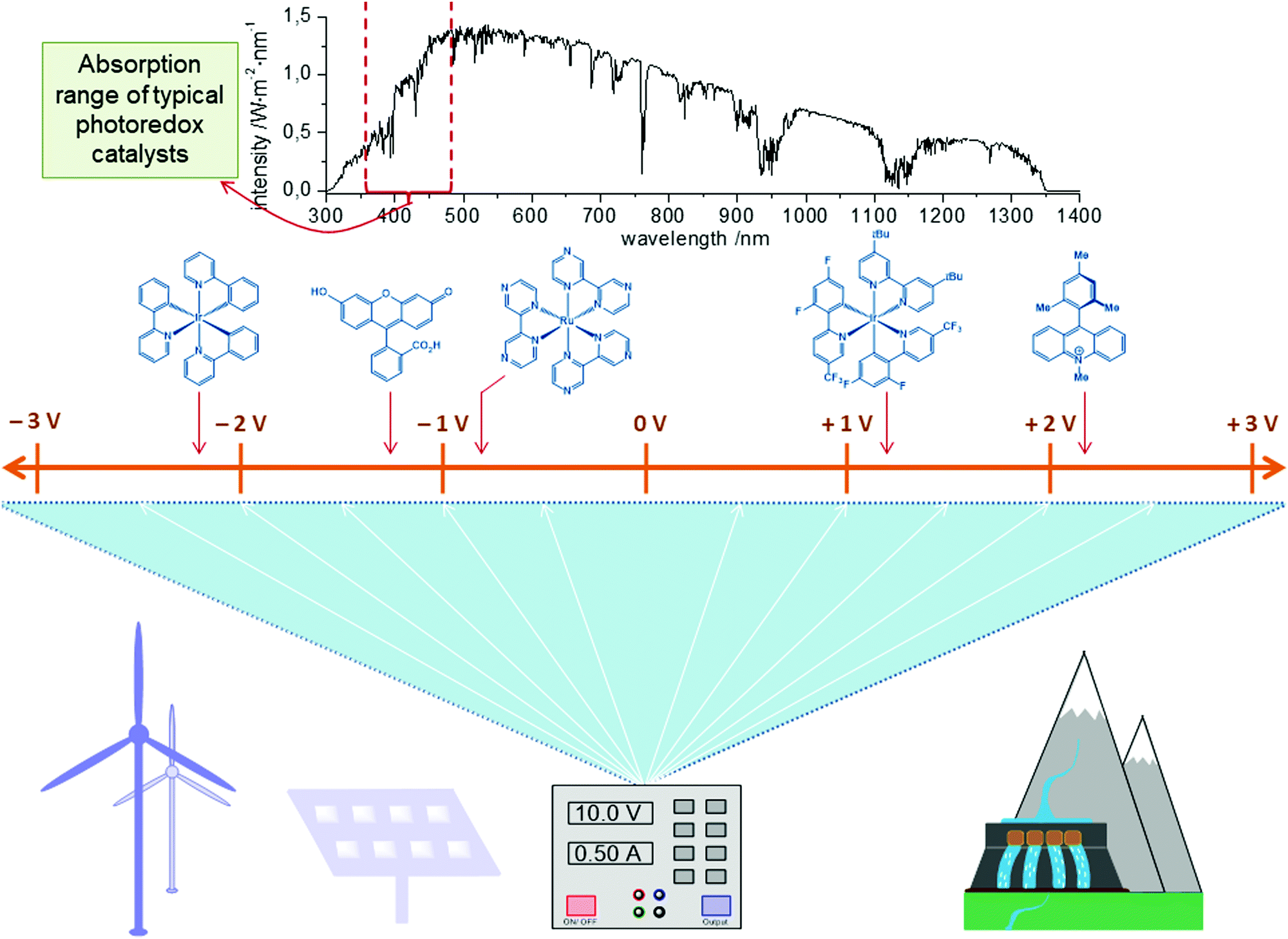 | ||
| Fig. 2 While photo catalysts offer only selected, fixed potentials and can use only a very limited part of the solar spectrum, a potentiostat gives access to the complete potential range with high precision and can be powered exclusively by sustainable energy sources. | ||
In sharp contrast, the direct use of electric current for the development of new redox strategies sets the stage for the efficient utilization of available energy sources, such as solar or wind power. The infinitely variable redox potential is easily set at the flick of a switch (Fig. 2, bottom). In electrocatalysis, electrons serve as an inherently clean redox reagent to drive the redox process and avoid stoichiometric reagent waste. The controllable redox potential generally allows for improved functional group tolerance and high levels of chemo- and regio-selectivity. Realizing the great potential for resource economy, several organic transformations have been developed, which merge traditional oxidative transformations with electrolysis, which has been reviewed elsewhere.47–50
The full potential of dual electron catalysis and transition metal catalysis – coined metallaelectrocatalysis – to enable efficient organic transformations has only recently been recognized.51–53 Until very recently, the use of Earth-abundant 3d metal catalysts in metallaelectrocatalysis for otherwise inert bond activation had proven elusive.54 In addition to their Earth-abundance, 3d metals are cost-effective and generally less toxic in comparison with their noble 4d and 5d homolouges.55 The outstanding capacity for resource economy has made base metal-catalyzed metallaelectrocatalysis an invaluable platform for organic synthesis.8,56
3. Principles, tools, and design of metallaelectrocatalysis
3.1 History
The development of the voltaic pile by Alessandro Volta in Pavia as the first continuous source of electricity57 set the stage for the development of a wealth of further inventions. Importantly it allowed Sir Michael Faraday to quantify electrochemical syntheses by his laws of electrolysis. He also noticed the formation of hydrocarbons during the electrolysis of dilute potassium acetate solution.58 Hermann Kolbe, who was born and educated in Göttingen as a student of Friedrich Wöhler, then identified and isolated ethane, carbon dioxide, hydrogen and dimethyl ether as the products of the electrolysis of a series of carbonic acids.593.2 Tools, design and principles
When an electrode is immersed in a solution of a half-cell, the difference between the solution's and the electrode material's electrochemical potential leads to a charge separation at the electrode–electrolyte interface originating from the systems attempt to reach equilibrium. It is extremely challenging to experimentally determine the value of this electrochemical potential difference. Two distinct half-cells will differ in the amount of charge that accumulates on the electrode surface. If these two half cells are connected, without allowing a flow of electric charge, this difference between the two equilibrium electrode potentials can be measured as an electric potential difference with a voltmeter. The standard hydrogen electrode (SHE) has been chosen as a reference system and its equilibrium electrode potential was defined as 0 V by convention. Therefore, by measuring any cell versus the SHE, the equilibrium electrode potential of the specific half-cell can be determined. The Nernst – eqn (I) relates the potential of a half-cell to the activity of the corresponding species of the redox couple: Instead of the activities, the respective concentrations are commonly used as an approximation. As soon as current flows and an electrochemical reaction occurs, the Nernst-Equation is no longer valid.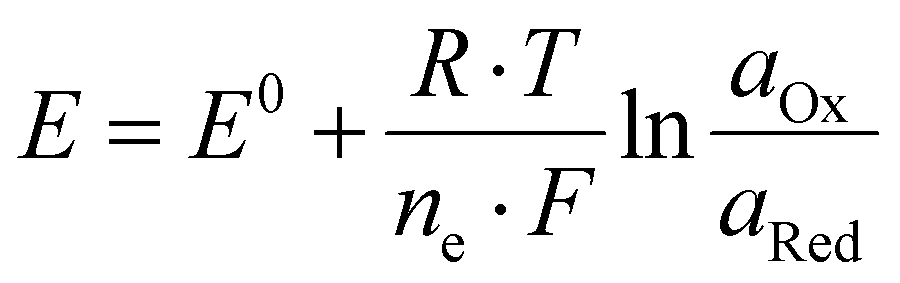 | (I) |
An electrochemical cell consists of at least two electrodes in contact with the electrolyte. Electrons are transported in the electric connections of the electrodes and the circuit is closed by ion movement in the electric field developed between the electrodes in the electrolyte. Electrochemical cells can be divided into two subtypes with respect to their working principle. In a galvanic cell – the voltaic pile consists of several galvanic cells connected in series – electricity is generated until the components of the cell have reached chemical equilibrium. In contrast, in an electrolytic cell a chemical reaction is powered by electricity. In this case, substances are oxidized at a positively charged anode by transferring electrons to the electrode. Simultaneously, at the negatively charged cathode substances are reduced by accepting electrons from the electrode. The two most important characteristics of an electrolytic cell are the potential or voltage (U in Volt) and the current (I in Ampere). Considering a molecular approach, the potential can be viewed as the energy level of the electrons in the electrode. A highly negative value corresponds to a strongly reducing behavior, whereas a high positive value denotes a strong oxidative force. The current relates via Faraday's laws of electrolysis to the number of electrons, which are passed through the solution (eqn (II)).
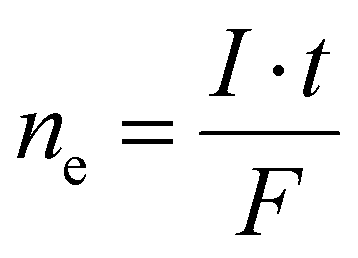 | (II) |
The faradaic yield or often Faraday efficiency (FE) is the percentage value that specifies the ratio of Coulombs consumed in forming the chemical products to the total number of Coulombs passed through the cell. This is expressed in the following formula (eqn (III)):
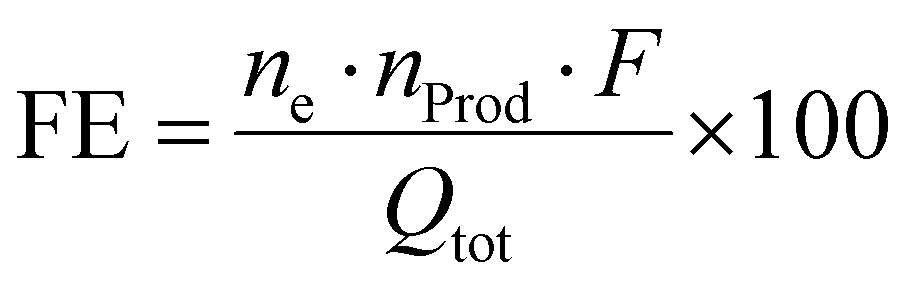 | (III) |
According to Ohm's law, the potential is proportional to the current with the resistance (R) being the constant of proportionality (U = R·I). This implies that only one of these values can be preset in an experiment, while the second one will adapt according to the cell's resistance. The first option is to keep the current constant (CCE = constant current electrolysis). This results in a potential difference between the electrodes, which enables the charge to pass through the cell at this specific rate. This option is advantageous in terms of directly controlling the amount of charge accompanied by a simple setup. However, during an electrochemical synthesis, the potential will typically increase, while the reactants are consumed and the cell resistance rises. This may lead to undesired side reactions and will decrease the current efficiency towards high substrate conversions (Fig. 3a). Alternatively, a fixed potential can be applied (CPE = constant potential electrolysis), which results in an adapting current. This can allow more chemo-selective transformations with high current efficiency so that ideally only the desired transformation takes place. However, during the electrosynthesis the current decreases due to depletion of the reactants and accordingly the reaction slows down (Fig. 3b). Additionally, a CPE requires more sophisticated equipment. A reference electrode as a third electrode in the electrochemical cell is mandatory to reliably control the working electrode's potential. At the reference electrode no electrochemical reaction takes place and no electric current flows. If the potential of one electrode is measured versus the live counter electrode, an unknown overpotential (vide infra) arises. The reference electrode must be chosen according to the system at hand. For aqueous electrolyte mixtures, Hg/Hg2Cl2 (calomel) or Ag/AgCl reference electrodes are well suited. For short reaction times, these can usually be applied in organic reaction media as well. Several organic solvent-based Ag/Ag(I) reference electrodes have also been developed. For a first approximation, it can be sufficient to use an inert metal wire as a pseudo reference electrode. It has to be taken into account, though, that an alteration of the metal surface or the solvent pH and composition may lead to a change of the inherent potential of the reference electrode in the electrolyte. This ultimately leads to a drift of the reference potential and reduces the accuracy with which the potential is kept constant.
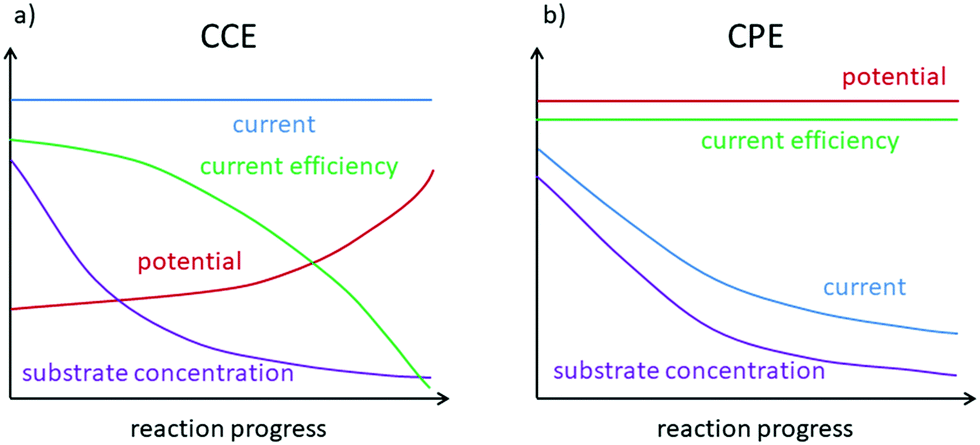 | ||
| Fig. 3 Comparison of theoreatical reaction profiles for CCE and CPE. | ||
The electrolyte in electrolytic cells mostly consists of a conducting agent dissolved in an organic solvent or aqueous media. Overall there are few limitations regarding the choice of solvents for electrosynthesis. However, in addition to the usual aspects of solvent selection for chemical reactions, such as polarity, boiling point, coordination behavior and solubility of reactants and reagents – to name a few – also the stability towards electrolysis and electro-generated reactive intermediates as well as the conductivity are of prime importance. Usually, conducting agents have to be employed to decrease the resistance of the electrolyte. The same restrictions as for the solvent apply. Most importantly, the electrolyte mixture should be electrochemically stable within the potential window that is needed for the desired transformation. Information regarding the stability of common electrolyte mixtures in terms of the applied potential can be found elsewhere.60
Electrolytic cells can be differentiated depending on whether a divided or undivided setup is employed (Fig. 4). In an undivided cell the electrodes are in the same compartment of the reaction vessel and all species in solution can diffuse freely between the electrodes. In contrast, in a divided cell the anode and cathode compartments are separated by a semi-permeable, often ion-selective membrane or a glass frit of very fine porosity. These barriers allow for the diffusion of small ions for the necessary charge transport but hinder the diffusion of larger molecules. Thereby, sensitive species reacting or being formed at the one electrode, are protected from the counter electrode itself and the majority of thus-formed by-products. The undivided cell setup comes in general at a lower price and is easily constructed from almost any reaction vessel in a synthetic lab.
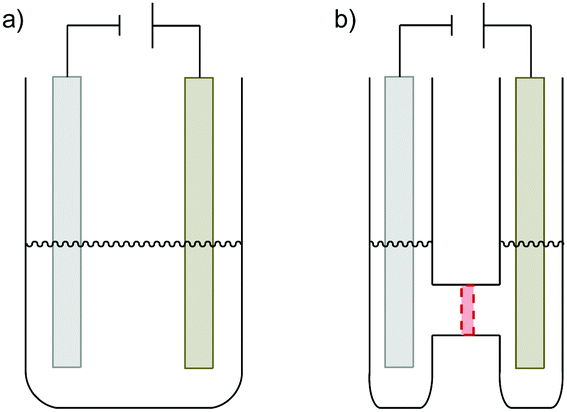 | ||
| Fig. 4 (a) Undivided and (b) divided (H-type) electrochemical cell. | ||
Whereas the reaction between a chemical oxidant (or reductant)61 and a substrate is usually a homogeneous process between two dissolved species, the electrooxidation of a substrate is always occurring at an electrode surface in a heterogeneous surface process. This leads to a series of effects, which result in an overpotential for the respective redox event. The overpotential is the difference between the equilibrium redox potential (the potential difference versus the SHE without current flow) of the species to be oxidized and the actual potential at the electrode, at which the redox event takes place.
The kinetic barrier for the actual electron transfer step from the substrate to the electrode will be different from the barrier observed between two dissolved species. Depending on the electrode material and its interaction with the substrate species, an activation overpotential is observed.
The polarization of the electrode leads to the formation of an electrochemical double layer. At a positively charged anode anionic species will accumulate to achieve microscopic charge compensation. In order to react at the electrode surface, any species has to diffuse through the double layer, potentially rearrange its solvation and after the reaction diffuse back into the bulk solution. In addition, further chemical reaction steps prior to or after the electron transfer may have to be taken into account. All these subsequent steps determine the concentration of the active substrate species at the electrode surface and thereby lead to a concentration overpotential. The activation overpotential and concentration overpotential both increase with the current density at the electrode.
The resistance of the cell components – especially the electrode–electrolyte junctions – and of the electrolyte itself (Ohmic drop or IR drop) have to be taken into account likewise. The resistance can increase drastically, if a precipitate or gas bubbles form at the electrode and thereby decrease the active electrode surface.
However, in certain cases, the overpotential can also be exploited for the tailoring the reactions. For instance, platinum metal is characterized by a very low overpotential for the formation of hydrogen gas by proton reduction. Therefore, using a platinum cathode is effective in promoting hydrogen formation and suppressing other possibly undesired cathodic reductions. The use of platinum as the electrode material, albeit costly, takes a little toll on the resource economy as the electrode is reusable. However, nickel and iron both show low overpotentials for hydrogen formation as well and may be more resource economic alternatives, where possible.
A high overpotential can also be circumvented by using a mediator for indirect electrolysis of the substrate (Fig. 5). In direct electrolysis, the substrate is oxidized at the electrode surface. An indirect electrolysis62 is characterized by the substrate being oxidized by a second dissolved species, which in turn is oxidized at the anodic surface. The redox mediator, which is usually employed in catalytic amounts, acts as an electron shuttle between the electrode and the substrate. This strategy may allow more selective transformations if the overpotential needed for direct electrolysis of the substrate is significantly higher than that for the oxidation of the redox mediator. In terms of resource economy, the use of a redox mediator is in principle unfavorable, as it decreases the atom economy. However, potential benefits like the decreased formation of side products and less energy consumption have to be carefully considered in the individual case.
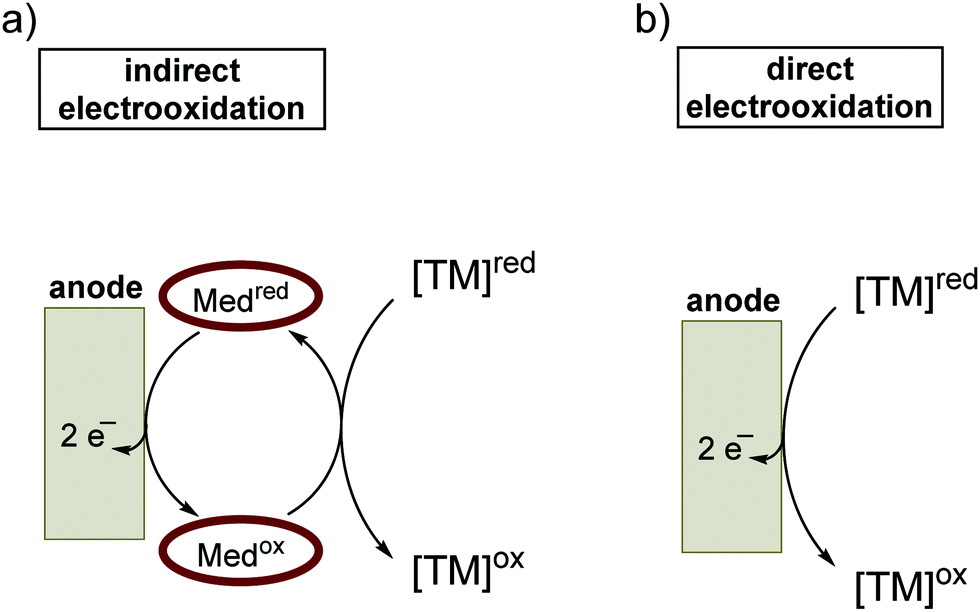 | ||
| Fig. 5 Direct and indirect (mediated) electrolysis. | ||
4. Case studies of resource economical metallaelectrocatalysis
4.1 Case study I: oxidative difunctionalization of alkenes
Transition metal catalysis has the power to improve the control of the reaction chemo-selectivity and thus can expand reaction scope and synthetic utility. In this context, metallaelectrocatalysis provides an innovative platform to realize the oxidative difunctionalization of alkenes in a sustainable fashion.63 The electrochemical approach offers an alternative approach to perform radical alkene functionalization in the absence of hazardous or expensive chemical oxidants.64–73Although a few examples have been known since the 1990's for electrochemical organometallic difunctionalization of olefins, such as epoxidations by manganese salen complexes,74,75 and dihydroxylations using osmium catalysts,76,77 this area of research has gained significant momentum by the exciting work involving manganese-catalyzed diazidation of alkenes 1 as reported by Lin and coworkers in 2017 (Scheme 1).78 Supported by mechanistic studies, a Mn(II)/Mn(III) redox cycle has been proposed for this electrochemical radical alkene difunctionalization (Scheme 1b). The mechanism involves the formation of a [Mn(II)]–N3 coordination compound, which is oxidized to [Mn(III)]–N3 and then transfers ˙N3 to the alkene, regenerating the [Mn(II)] species. In a second analogous sequence the product 2 is generated. The reaction proved compatible with terminal, 1,1- and 1,2-disubstituted, trisubstituted, and tetrasubstituted olefins to provide the desired 1,2-diazides 2 in high yields. The mild electrocatalytic reaction conditions offered high chemoselectivity and various synthetically meaningful functional groups, such as ester, alcohol, ketone, carboxylic acid, and amine, on the alkene moiety were well tolerated (Scheme 1c). The resource economy of the reaction is substantiated by the Earth-abundant manganese catalyst, a high faradaic efficiency (66–87%), high atom economy, and hydrogen and sodium acetate as benign by-products. In a related study, the authors also accomplished the dichlorination of alkenes 1 with MgCl2 as the chlorine source under mild reaction conditions.79 It is worth mentioning that the diazidation of alkenes 1 has subsequently also been realized under transition metal free reaction conditions using aminoxyl radical catalyst, CHAMPO.80
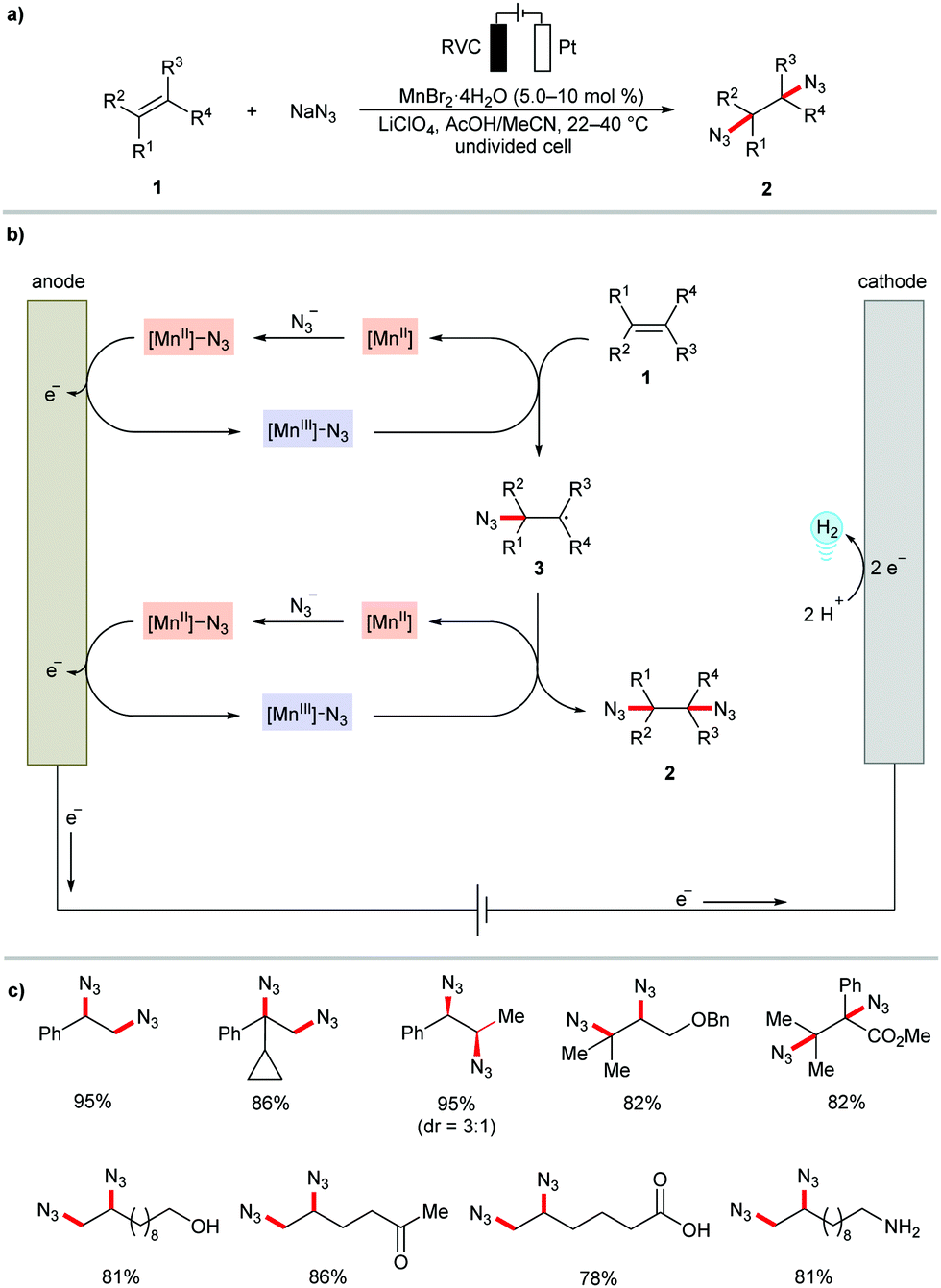 | ||
| Scheme 1 Manganese-catalyzed electrochemical diazidation of alkenes 1. RVC: reticulated vitreous carbon. | ||
The versatile metallaelectrocatalysis was further applied to the heterodifunctionalization of alkenes 1 (Scheme 2).81 Thus, the manganese-catalyzed electrochemical reaction conditions enabled the synthetically valuable chlorotrifluoromethylation of alkenes 1 using commercially available bench-stable MgCl2, and CF3SO2Na (Langlois reagent) as the Cl and CF3 source, respectively. The proposed catalytic cycle involves the anodic generation of the ˙CF3 radical and [Mn(III)]–Cl (Scheme 2b). Subsequently, the ˙CF3 attacks the alkene to generate a transient carbon-centered radical species 5, which is trapped within a radical coupling with [Mn(III)]–Cl to provide the desired chlorotrifluoromethylation product 4. The electrocatalytic conditions conveniently transformed diversely substituted alkenyl substrates 1 into the desired products 4 at ambient reaction temperature (Scheme 2c). Interestingly, the difunctionalization of 1,6-enyne substrates 6 afforded chlorotrifluoromethylated pyrrolidines under only slightly modified reaction conditions.82 Here, the presence of the bidentate ligand 2,2′-bipyridine (bpy) significantly enhanced the stereo control of the alkenyl group in the pyrrolidine products 7 (Scheme 3).
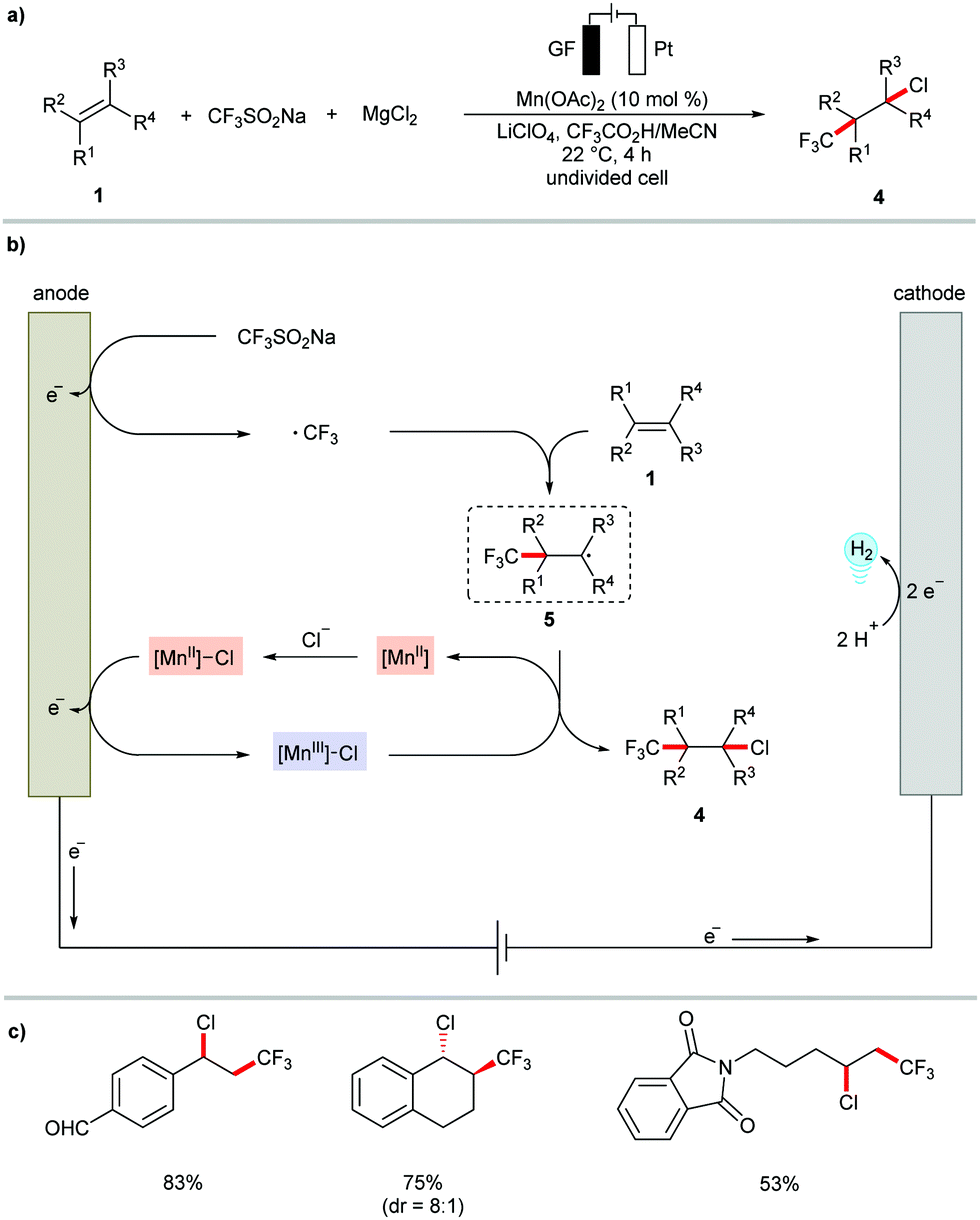 | ||
| Scheme 2 Manganese-catalyzed electrochemical chlorotrifluoromethylation of alkenes 1. GF: Graphite felt. | ||
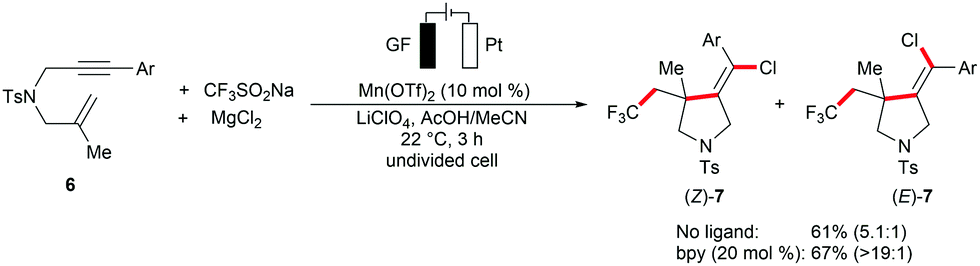 | ||
| Scheme 3 Electrocatalytic synthesis of chlorotrifluoromethylated pyrrolidines 7. | ||
Most recently, the Lin group has further showcased the application of anodically coupled heterodifunctionalization to the chloroalkylation of alkenes 1 (Scheme 4).83 The protocol allowed for the simultaneous construction of vicinal C–C and C–Cl bonds under mild conditions in the presence of catalytic amounts of Mn(OTf)2. Here, malononitriles 8 and cyanoacetates were employed as alkyl substrates and NaCl as the chlorine source. A plausible catalytic cycle was proposed to involve a parallel, manganaelectro-catalyzed generation of alkyl and chlorine radicals followed by selective addition to olefins (Scheme 4b).
 | ||
| Scheme 4 Manganese-catalyzed chloroalkylation of alkenes 1. | ||
4.2 Case study II: cross-coupling reactions
During the past decades, palladium-catalyzed cross-couplings have emerged as promising and widely used reactions to forge C–C and C–heteroatom bonds in drug molecules and functional materials.84–87 Particularly, C–N forming reactions are often required in API syntheses.88–90 In view of the high cost and low natural occurrence of palladium, significant efforts have been devoted to enable these cross-coupling reactions using Earth-abundant nickel catalysts. However, these methods often demand drastic reaction conditions, including high reaction temperatures, strong bases, and air-sensitive nickel(0) pre-catalysts,91–95 which limits their application to late-stage modifications.96In this context, the Baran group recently reported an effective method to construct C–N bonds by cross-coupling of aryl bromides 12 with alkyl amines 13 under electrochemical nickel97 catalysis (Scheme 5).98 The reaction was performed in an undivided cell in the presence of catalytic amounts of air-stable nickel complex NiBr2·glyme and di-tBubpy as the ligand. A variety of (hetero)aryl bromides 12 were efficiently transformed into the corresponding (hetero)arylamines 14 in high yields. Sensitive functional groups, including cyano, ester, hydroxyl, amide and sulfonamide, were thereby well tolerated.
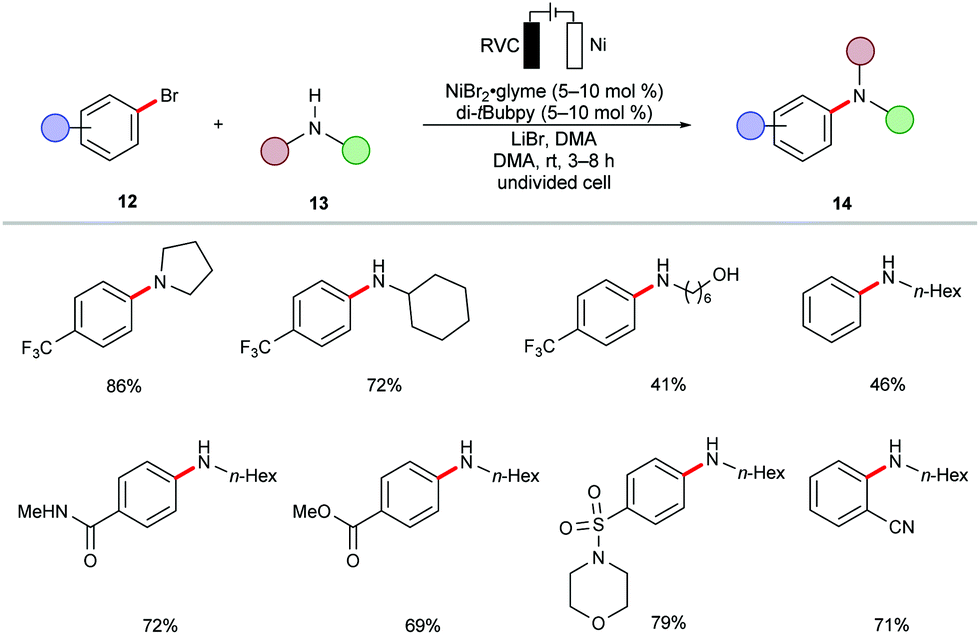 | ||
| Scheme 5 Electrochemical nickel-catalyzed amination of aryl bromides 12. | ||
In subsequent efforts, the same group further expanded the scope of viable amines to include amino acid esters 15 and small oligopeptides under slightly adjusted reaction conditions (Scheme 6a).99 The application of the nickel-catalyzed electrochemical C–H amination protocol was successfully employed as a key step in the total synthesis of Teleocidins B-1–B-4 (Scheme 6b).100 It is worth mentioning that the cross-coupling of aryl bromides with primary alcohols also proved viable under the electrochemical nickel catalysis.99
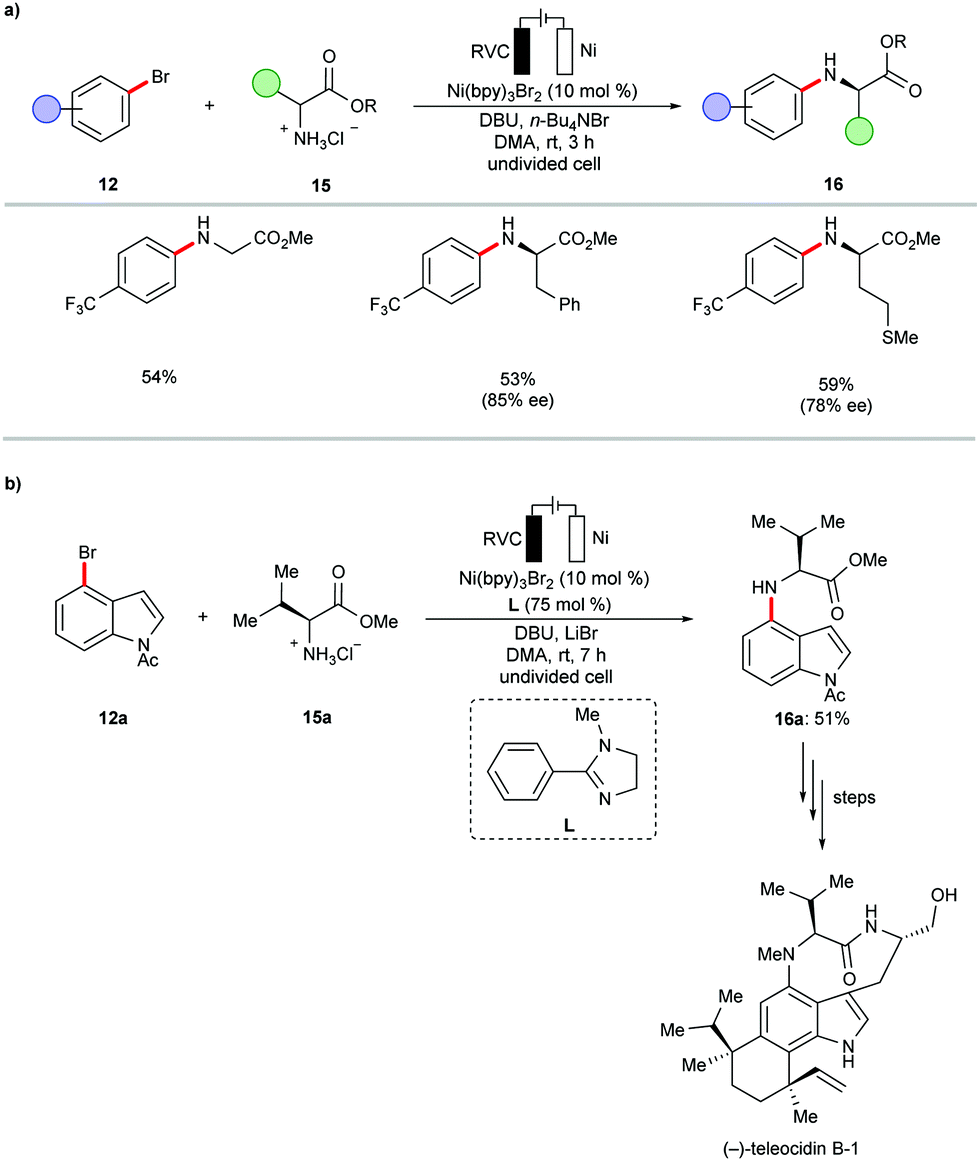 | ||
| Scheme 6 Nickel-catalyzed electrochemical N-arylation of amino acid esters 15. | ||
Based on detailed experimental and computational mechanistic investigations, a plausible catalytic cycle was proposed for the nickel-catalyzed electrochemical amination of aryl halides 12 (Scheme 7).99 It was thus suggested to initiate by cathodic reduction of a dynamically ligated nickel(II) pre-catalyst to generate nickel(I) species 17. Then, the rapid oxidative addition of aryl halide 12 to 17 results in the formation of nickel(III) intermediate 18, which undergoes a second cathodic reduction to form a stable nickel(II) species 19. Coordination of the amine 13 to 19 followed by rate-limiting deprotonation provides intermediate 20. The anodic oxidation of 20 to nickel(III) complex 21, followed by rapid reductive elimination furnishes the desired product 14 and regenerates the catalytically active nickel(I) species 17.
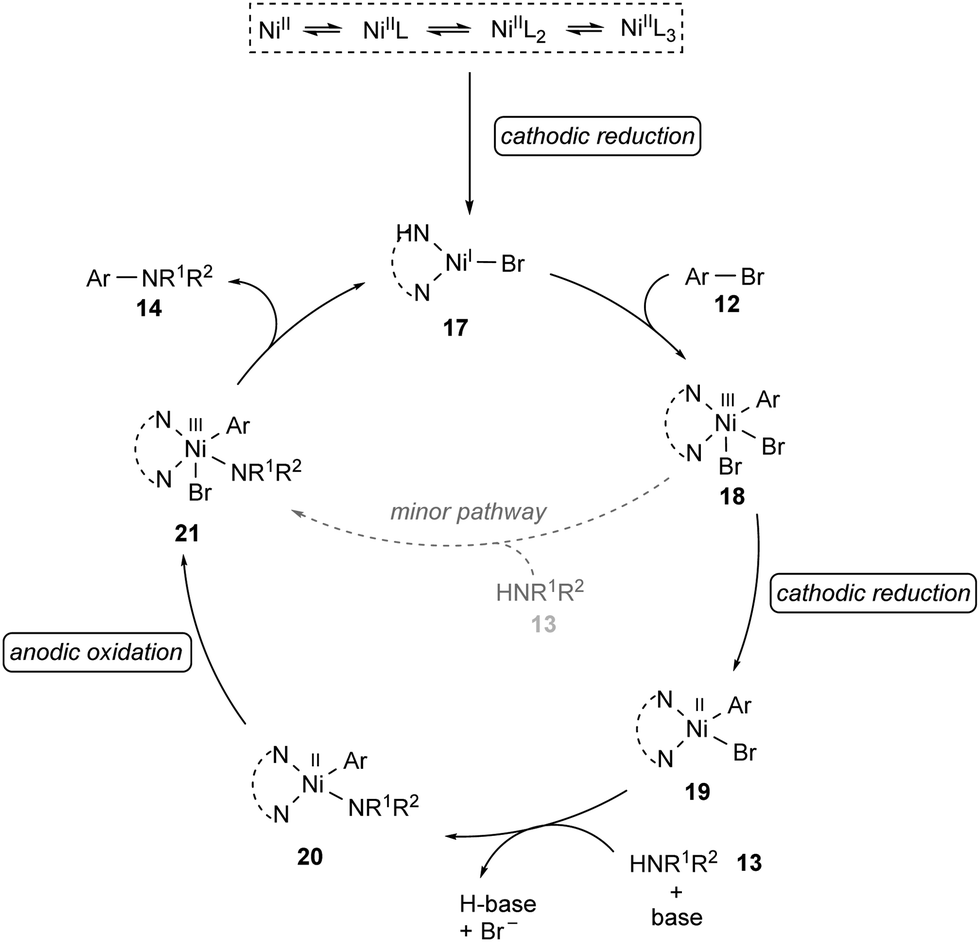 | ||
| Scheme 7 Proposed mechanism for the electrochemical amination. | ||
The versatility of the electrochemical nickel catalysis in cross-coupling was recently showacsed by Leonél and coworkers.101 The authors devised mild electro-assisted nickel catalysis for the synthesis of (hetero)arylphosphonates 23 from (hetero)aryl bromides 12 and dimethyl phosphite 22 (Scheme 8). However, the catalytic cycle was proposed to involve the in situ generation of catalytically active nickel(0) species 24.
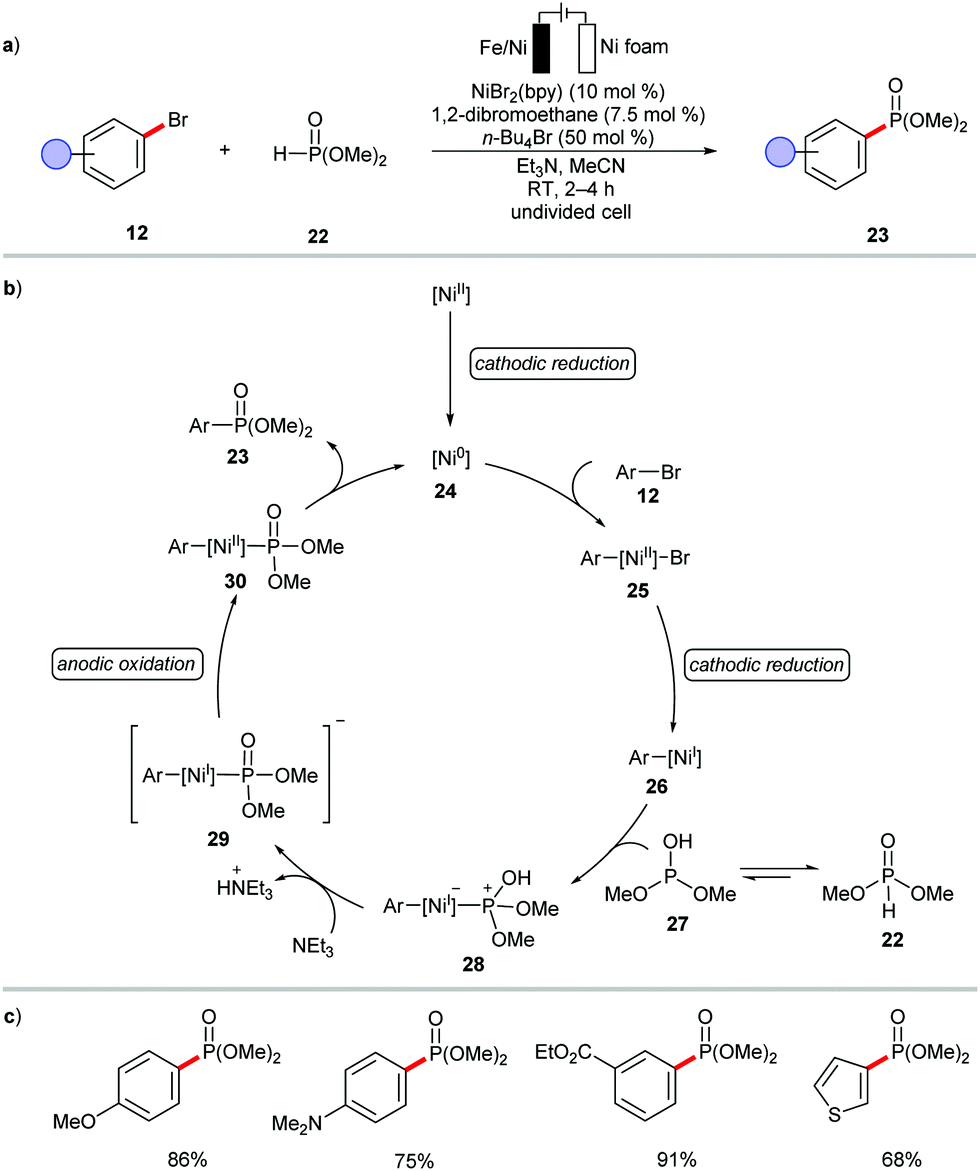 | ||
| Scheme 8 Electrochemical nickel-catalyzed C–P couplings. | ||
The reactivity of nickelaelectrocatalysis is not limited to C–N, C–O, and C–P bond formation. Indeed, the cross-coupling of thiols 32 with aryl iodides 31 was recently disclosed by Wang (Scheme 9).102 The nickel-catalyzed C–S bond formation was realized at an ambient reaction temperature in presence of the air-stable nickel(II) pre-catalyst, NiCl2·glyme. Both aryl- and alkyl thiols 32 were effectively converted under the reaction conditions to offer the desired thioethers 33 in excellent yields. Based on mechanistic findings, the catalytic mode of action was proposed to involve reductive elimination at the nickel(III) intermediate 36 (Scheme 9).102
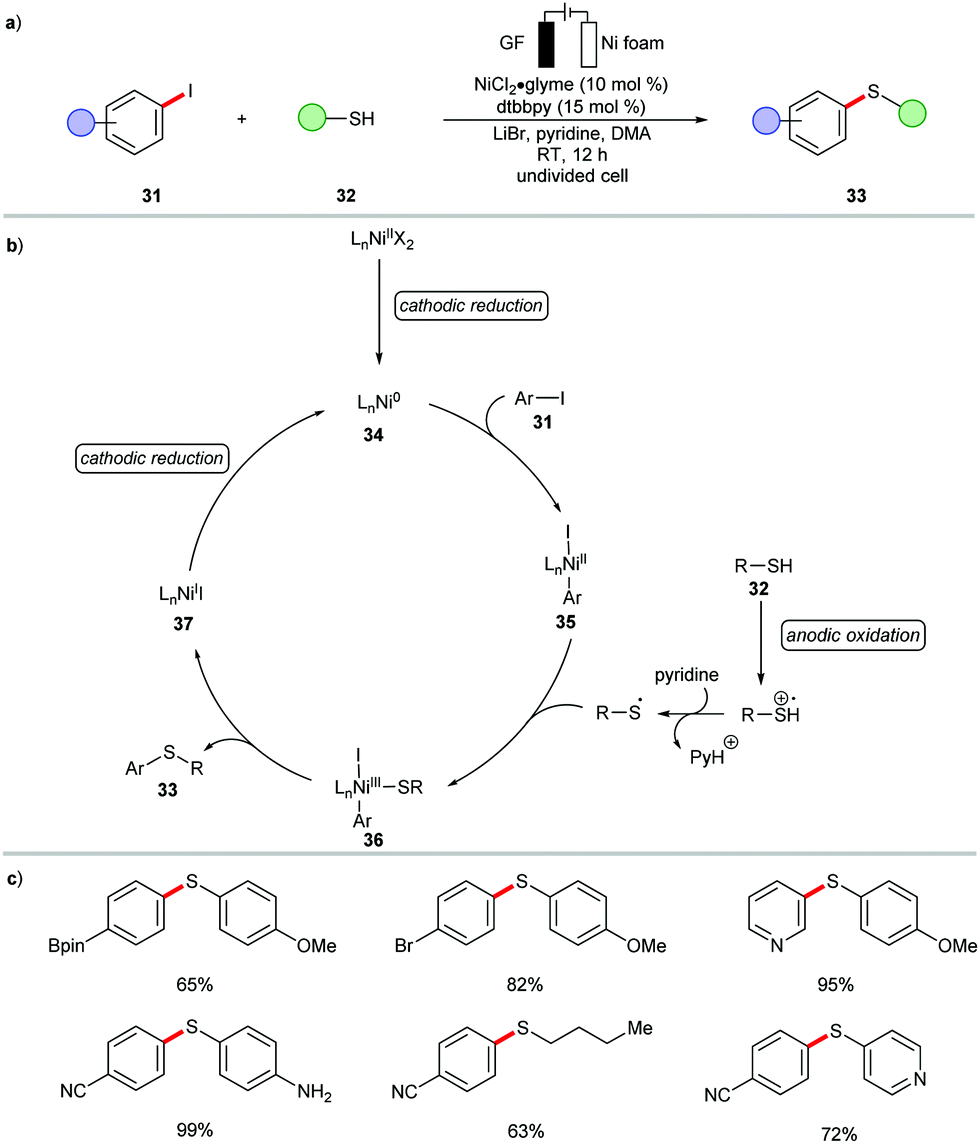 | ||
| Scheme 9 Nickel-catalyzed electrochemical C–S formation. | ||
4.3 Case study III: C–H activation
The merger of C–H activation and electrochemistry set the stage to establish new reactivities with high levels of resource economy. Despite indisputable advances, electrocatalytic organometallic C–H activations were thus far limited to precious palladium catalysts103 and N-coordination as evidenced by Jutand,103 Kakiuchi104–107 and Mei,108–111 among others.52,109,112–115 Very recently, electrochemical organometallic C–H activation reactions employing rhodium(III),116–120 iridium(III),121,122 and less expensive ruthenium(II)123–127 complexes have been accomplished by Ackermann and others. Hence, cost-intensive precious transition metals have until very recently proven mandatory. In contrast, we illustrate the huge potential of merging 3d metal catalysts with electrochemistry to achieve unprecedented reactivities in C–H activation reactions towards full resource economy.56,128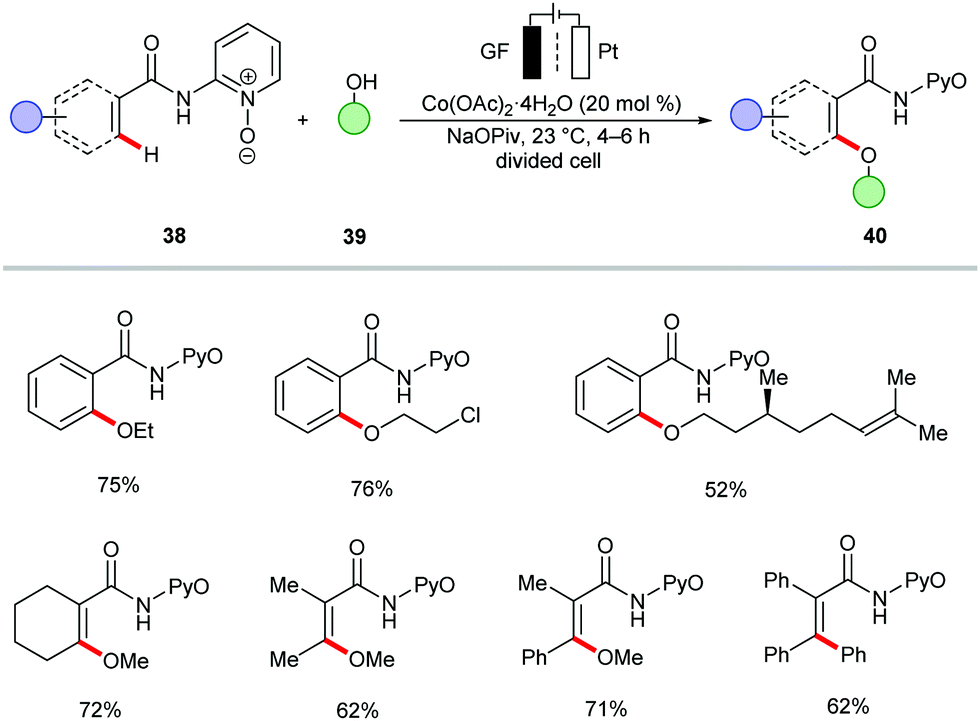 | ||
| Scheme 10 C–H oxygenation via electrochemical cobalt catalysis. | ||
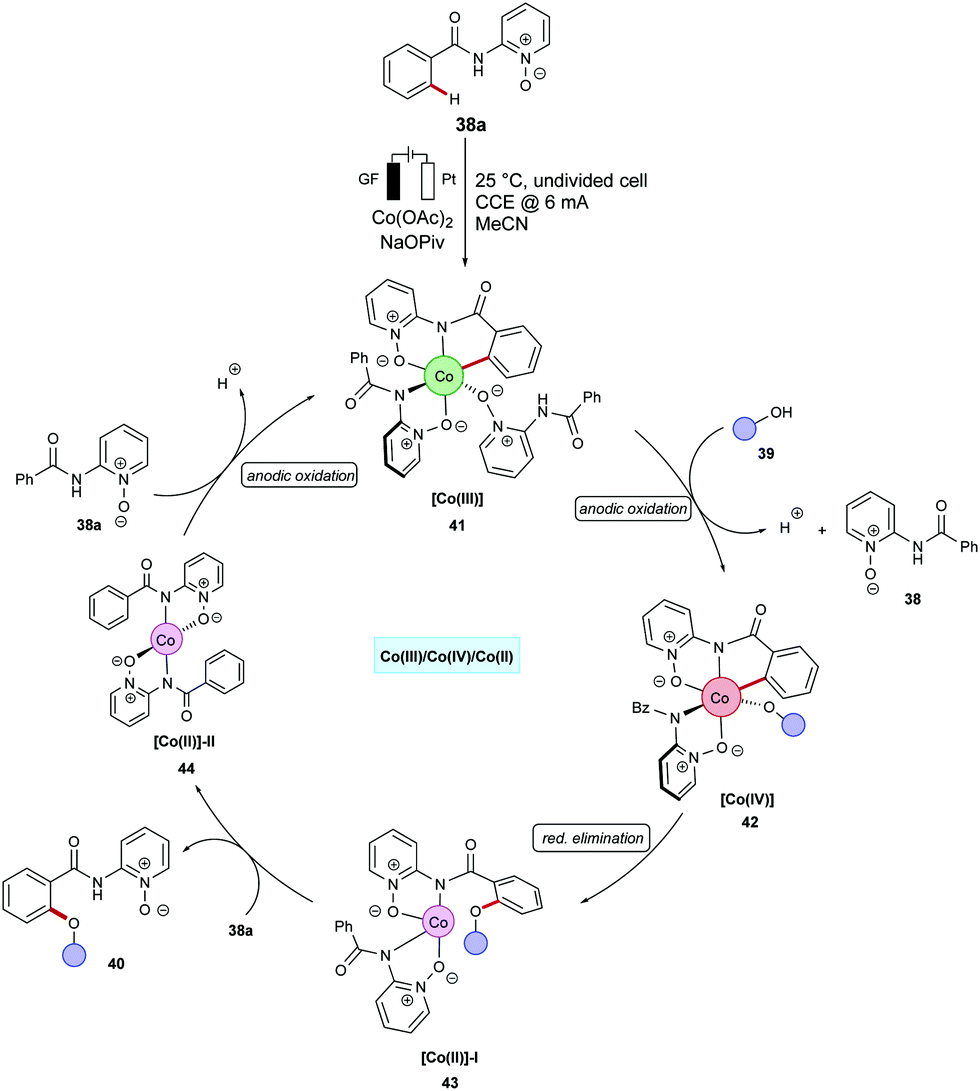 | ||
| Scheme 11 Proposed catalytic cycle for the cobaltaelectro-catalyzed C–H oxygenation. | ||
The cobaltaelectro-catalyzed C–H activation was not limited to C–O bond forming reactions. Thus, Ackermann established an intermolecular C–H amination protocol under mild reaction conditions (Scheme 12).135 A variety of benzamides 38 bearing pyridine N-oxide (PyO) substituents were efficiently ortho-aminated with secondary amines 13 to afford the desired products 45 in good yields, while liberating H2 as the sole by-product. The formation of H2 was unambiguously confirmed by headspace gas chromatography (GC), and could be utilized for future hydrogen evolution reaction strategies (HER). A related C–H amination of amides also proved viable using 8-aminoquinoline (AQ) as the bidentate directing group, as was reported by Lei.136 Here, the cobalt catalyst showed better efficacy in a divided cell using MeCN as reaction medium at 65 °C.
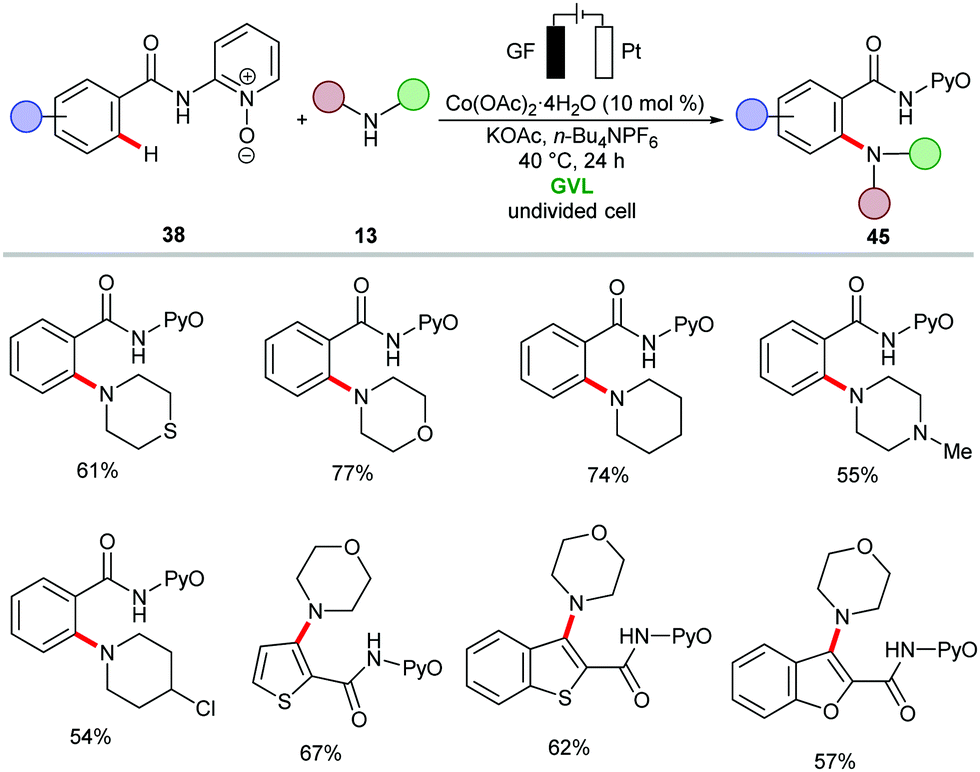 | ||
| Scheme 12 Scope of cobaltaelectro-catalyzed C–H amination. | ||
The versatile electrochemical cobalt catalysis was further-applied to the synthesis of biologically relevant isoquinolones 47 and pyridones 48 from aromatic and vinylic amides 38, respectively (Scheme 13).137 The reaction occurred at ambient temperature in a water-based solvent mixture and displayed ample substrate scope with high yields. The robust cobalt-electrocatalysis manifold for C–H/N–H activation was also achieved in biomass-derived glycerol as reaction medium.138 Remarkably, the resource economy of the transformation was substantiated by the direct use of renewable solar and wind energy.
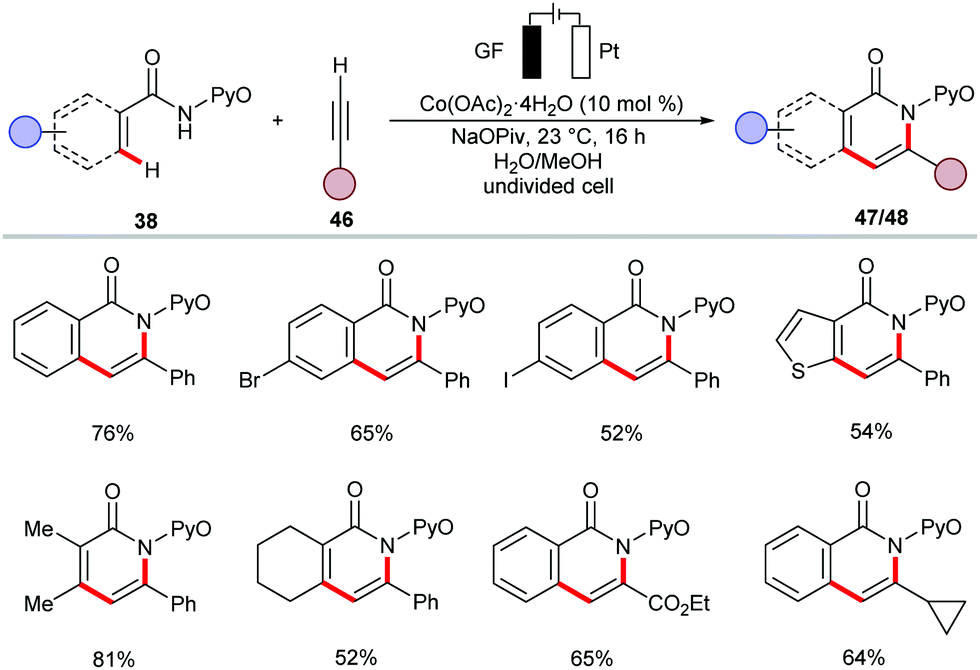 | ||
| Scheme 13 Cobalt-catalyzed electrooxidative annulation reaction by amides 38 and alkenes 46. | ||
Supported by competition experiments, KIE determination, and CV studies, a plausible reaction mechanism was proposed as shown in Scheme 14.137 The anodically generated active cobalt(III) species 49 undergoes C–H cyclometallation to provide key intermediate 50. Then, alkyne insertion into the C–Co bond followed by C–N bond forming reductive elimination delivers the desired product 47 and cobalt(I) species 52. The electrochemical oxidation of 52 regenerates the active cobalt(III) species 49 and completes the catalytic cycle.
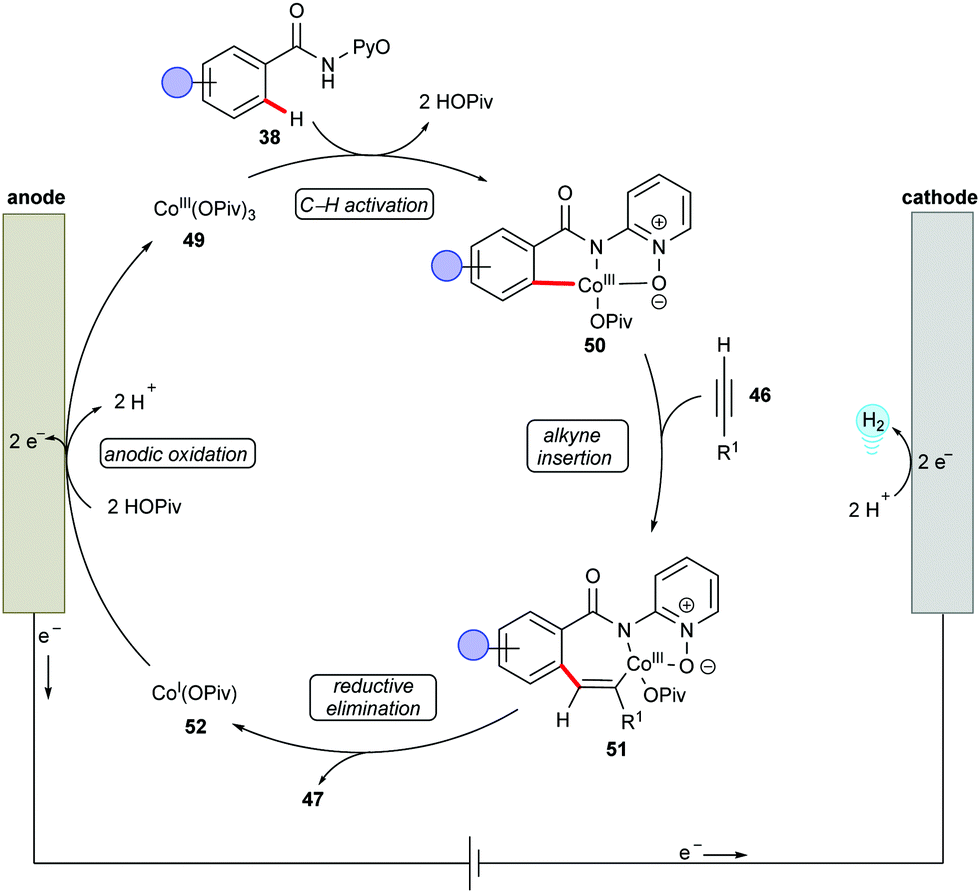 | ||
| Scheme 14 Plausible catalytic cycle for the cobaltaelectro-catalyzed C–H/N–H annulation with terminal alkynes 46. | ||
The cobalt-catalyzed electrochemical C–H/N–H annulation regime was until recently restricted to terminal alkynes 46. In this context, the Ackermann group recently devised a protocol for the cobalt-catalyzed electrochemical C–H/N–H activation of pyridylbenzhydrazides 53 with internal alkynes 54 to access diversely substituted isoquinolones 55 (Scheme 15).139 In the presence of catalytic amounts of Co(OAc)2, a variety of internal alkynes 54 efficiently underwent [4+2] annulation with pyridylbenzhydrazides 53 at 23 °C. The unsymmetrically substituted internal alkynes 54 offered the desired products 55 with high levels of regioselectivity. Further, for the first time, the electrocatalytic removal of the directing group was realized through samarium-catalyzed cathodic reduction (Scheme 16).139
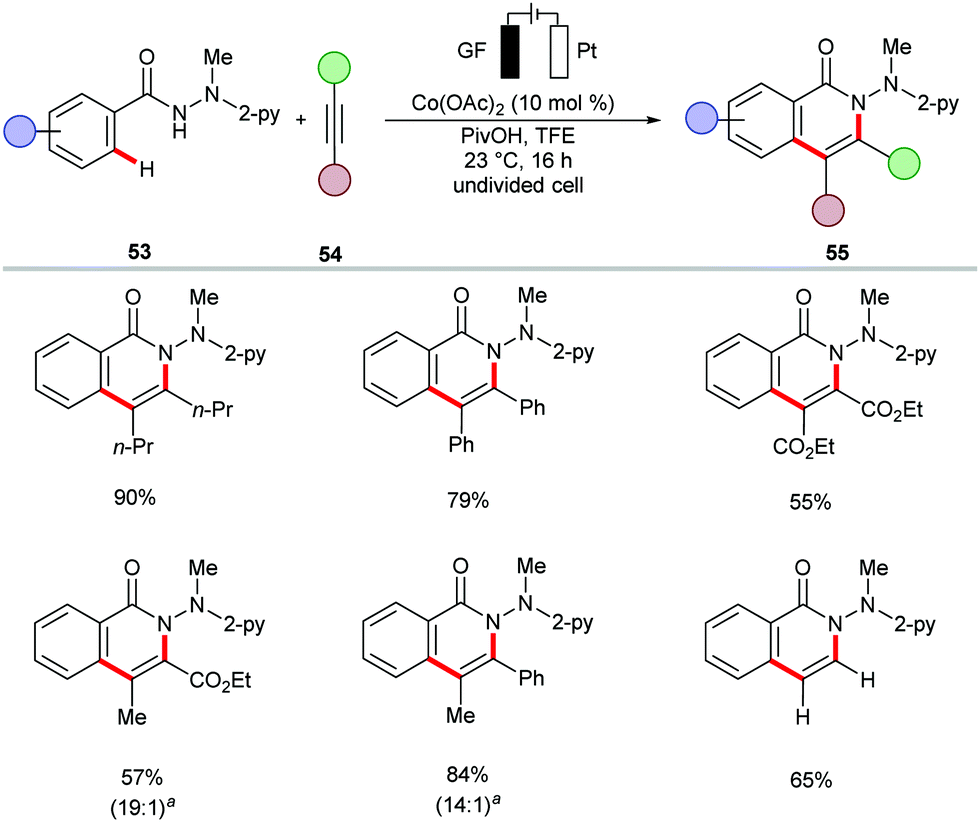 | ||
Scheme 15 Electrochemical C–H/N–H activation with internal alkynes 55. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ratio of regioisomers. Ratio of regioisomers. | ||
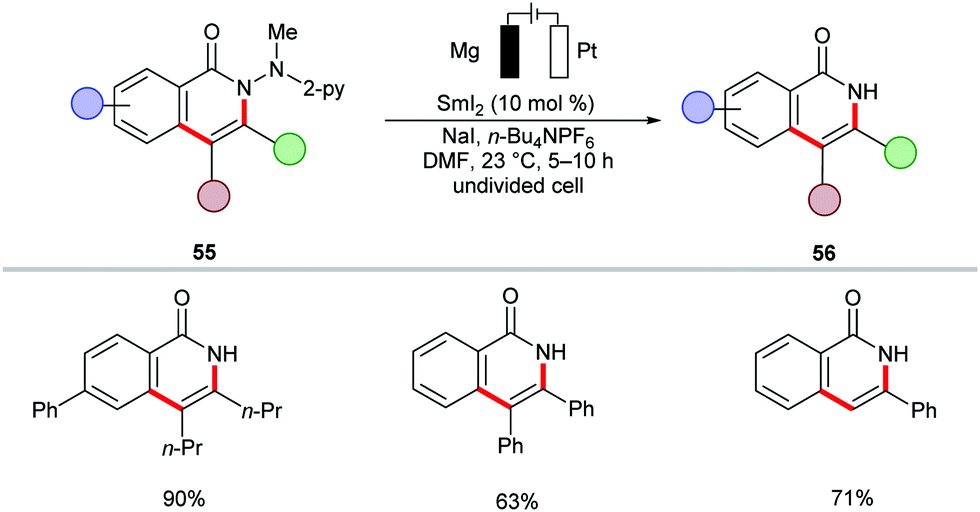 | ||
| Scheme 16 Electroreductive removal of the directing group. | ||
Next, the potency of cobaltaelectro-catalyzed C–H/N–H annulation was probed to include allenes140–149 (Scheme 17).150 The reaction was performed in a user-friendly undivided cell setup at 40 °C. The versatile cobalt catalysis transformed diversely substituted allenes 57 into the desired isoquinolones 58 in high yields. Notably, more challenging internal allenes 57 also proved viable substrates to deliver the corresponding exo-methylene isoquinolones 58. Based on the detailed experimental and computational mechanistic investigations, a plausible catalytic was proposed as shown in Scheme 18. Anodically generated active cobalt(III) species 59 undergoes chelation assisted C–H cobaltation to give intermediate 60. Allene precoordination followed by regioselective migratory insertion results in the seven-membered intermediate 61. The exo-methylene isoquinolones 63 are generated upon reductive elimination. The catalytically active cobalt(III) species 59 is regenerated by anodic oxidation of cobalt(I) species 62.
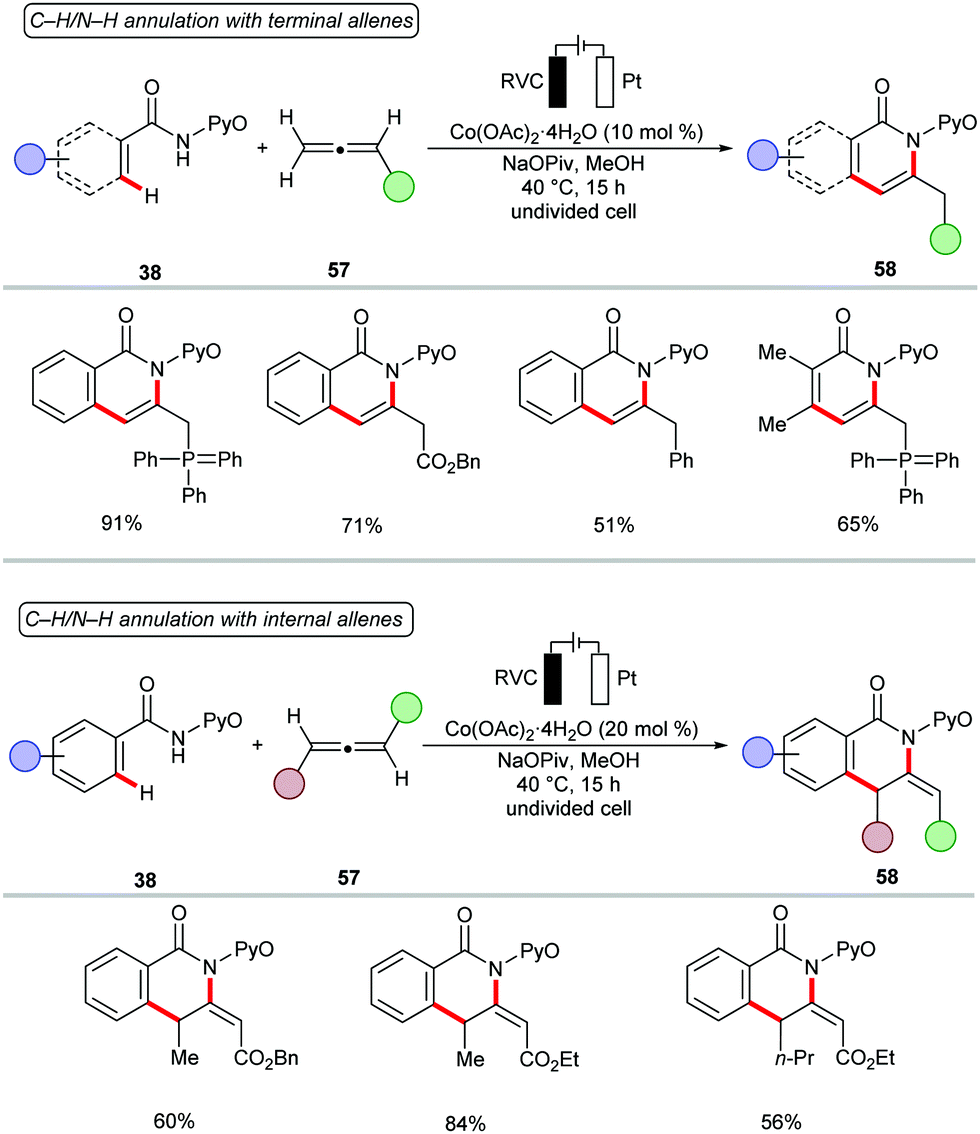 | ||
| Scheme 17 Cobaltaelectro-catalyzed C–H/N–H activation of benz- and acrylamides 38 with allenes 57. | ||
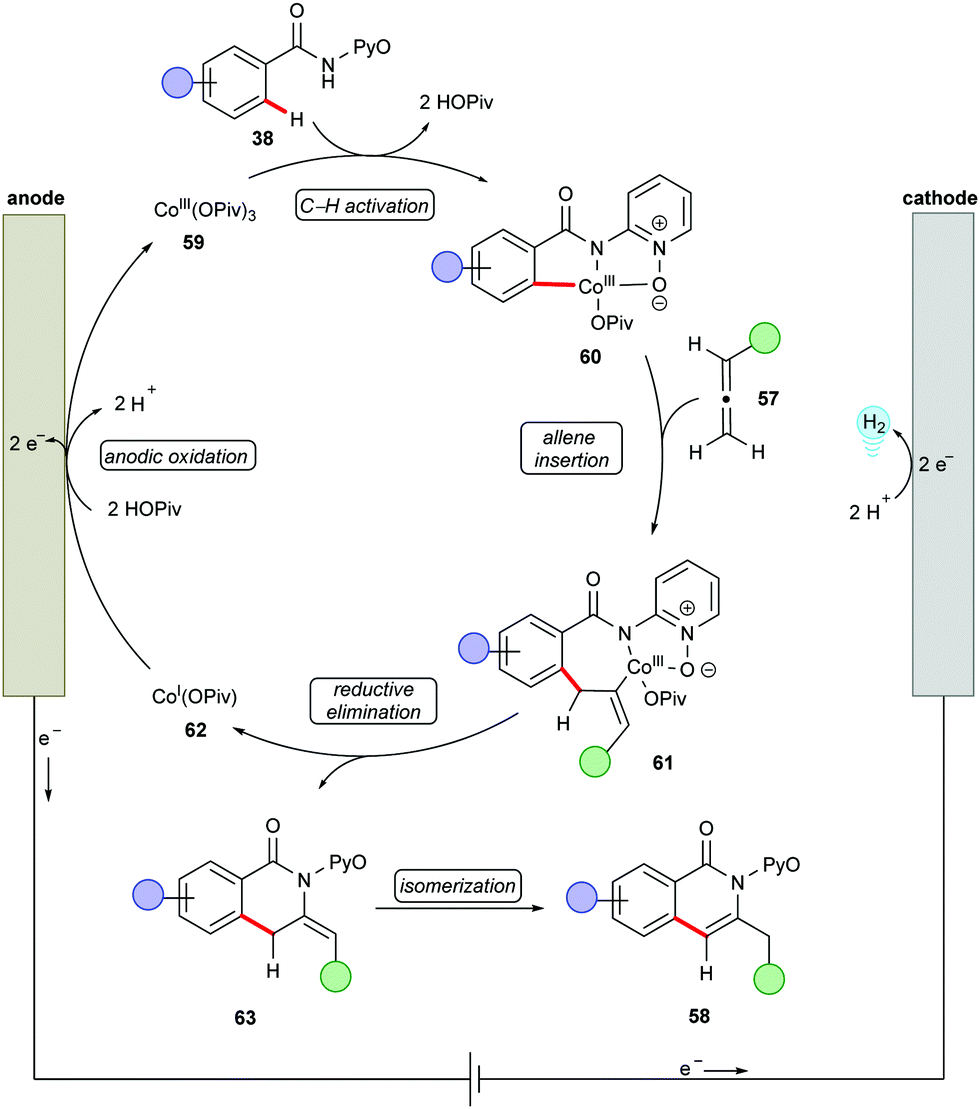 | ||
| Scheme 18 Catalytic cycle for C–H/N–H activation with allenes XX. | ||
The electrochemical cobalt-catalyzed C–H/N–H activation was further extended to a [4+1] annulation of benzhydrazides 53 with isocyanides 64 (Scheme 19).151 Besides, the authors demonstrated the synthesis of phthalimide derivatives 66via annulation of CO (1 atm) by benzhydrazides 53 at 23 °C in a user-friendly undivided cell (Scheme 20).151 This approach was also realized by the group of Lei, albeit with 8-aminoquinoline as the directing group.152
 | ||
| Scheme 19 Cobaltaelectro-catalyzed C–H/N–H annulation with isocyanides 64. | ||
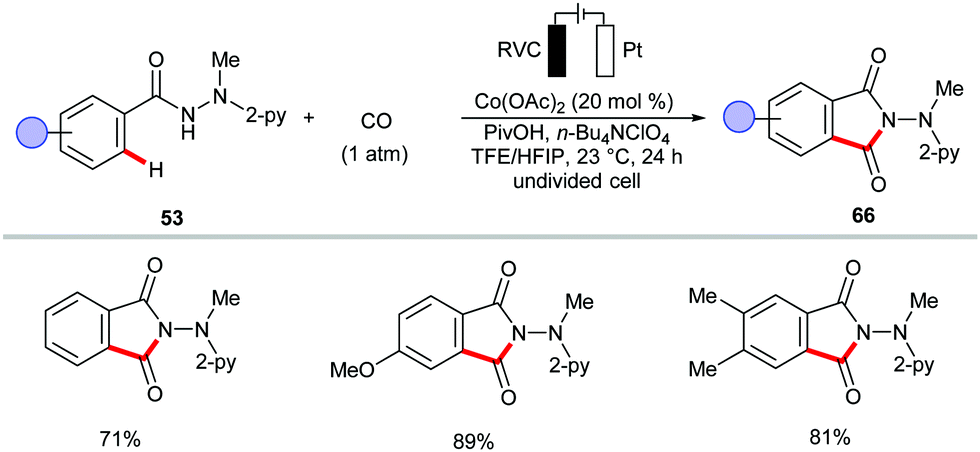 | ||
| Scheme 20 Electrochemical C–H/N–H carbonylation at 23 °C. | ||
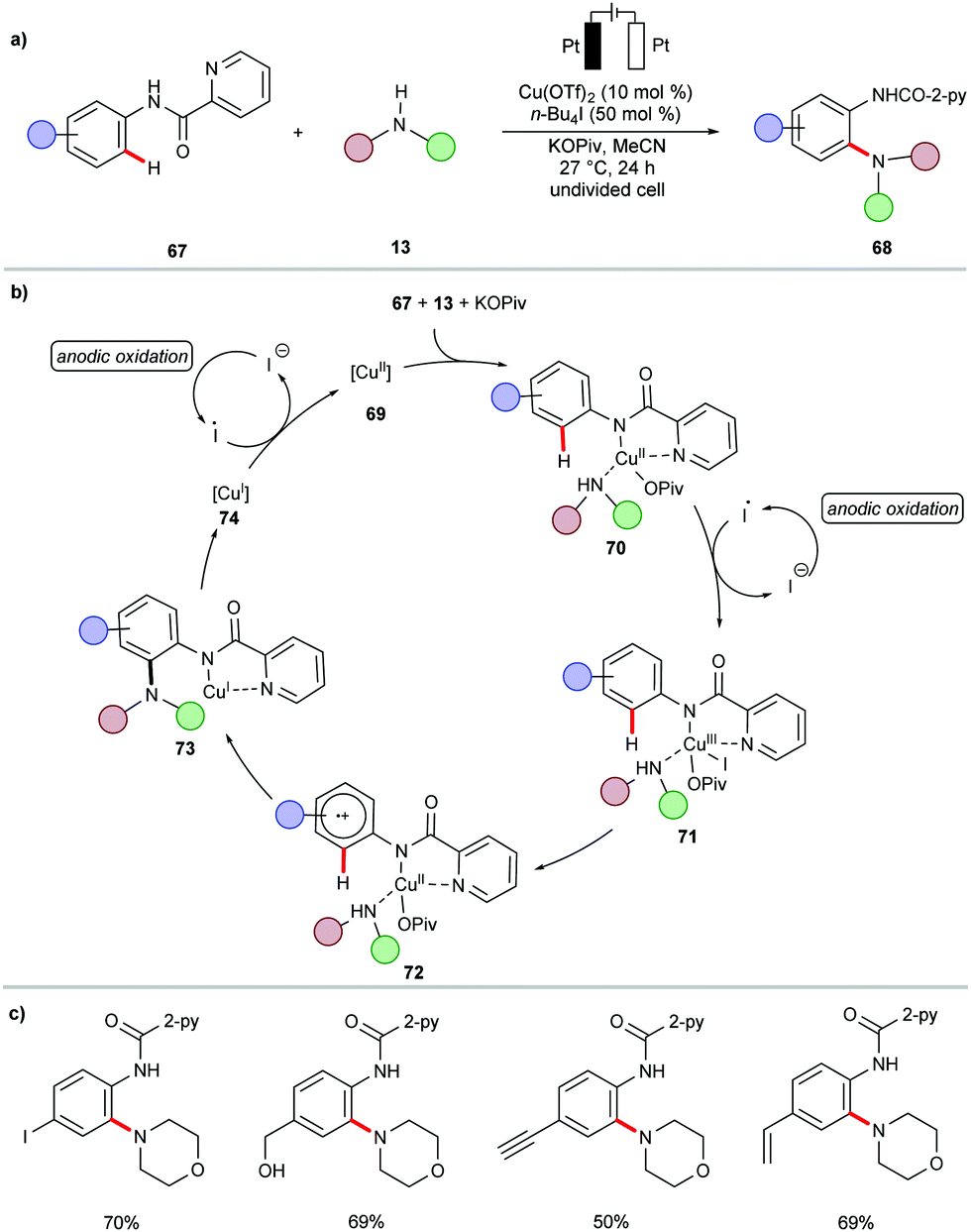 | ||
| Scheme 21 Electrochemical copper-catalyzed C–H amination. | ||
In a subsequent report, Nicholls demonstrated a copper-catalyzed electrochemical C–H amination of benzamides 75 bearing a 8-AQ directing group.156 The synthetic utility of the reaction was well explored for a series of amines containing active pharmaceutical ingredients (APIs) (Scheme 22).
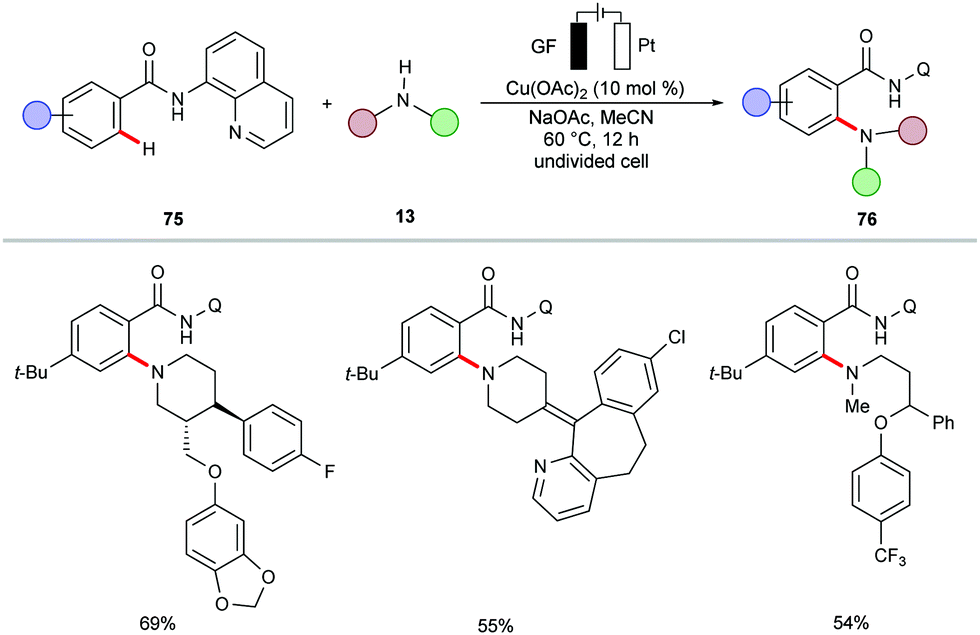 | ||
| Scheme 22 Cupraelectro-catalyzed C–H amination with API molecules. | ||
The cupraelectro catalysis was not limited to C–H aminations. Indeed, the Ackermann group disclosed a first example of cupraelectro-catalyzed alkyne annulation reaction with electricity as the terminal oxidant.157 The transformation provided access to biologically relevant isoindolones 78 from benzamides 75 and terminal alkynes 46via C–H/N–H functionalizations (Scheme 23). The robust cupraelectro-catalysis also proved applicable to alkynyl carboxylic acids 77 to give the desired products 78 in good yields through decarboxylative C–H/C–C cleavage.
 | ||
| Scheme 23 Cupraelectro-catalyzed alkyne 46/77 annulation for isoindolone 78 formation. | ||
Remarkably, the cupraelectro annulation contrasts with related cobaltaelectro annulations (vide infra, Schemes 13 and 15) for isoquinolones 47/55,137,139 delivered the five-membered isoindolones 78. The detailed mechanistic studies including competition experiments, KIE by in operando React-IR supported the C–H alkynylation mechanism via facile C–H scission (Scheme 24). The CV studies revealed an oxidation potential of 0.95 VSCE for the oxidation of copper(II) to copper(III) from the mixture of the copper(II) catalyst and substrate 67 in DMA. The reoxidation of the copper(I) species to copper(II) readily occurred at 0.05 VSCE as observed in CV data.157 The formation of molecular hydrogen as the sole byproduct was confirmed.
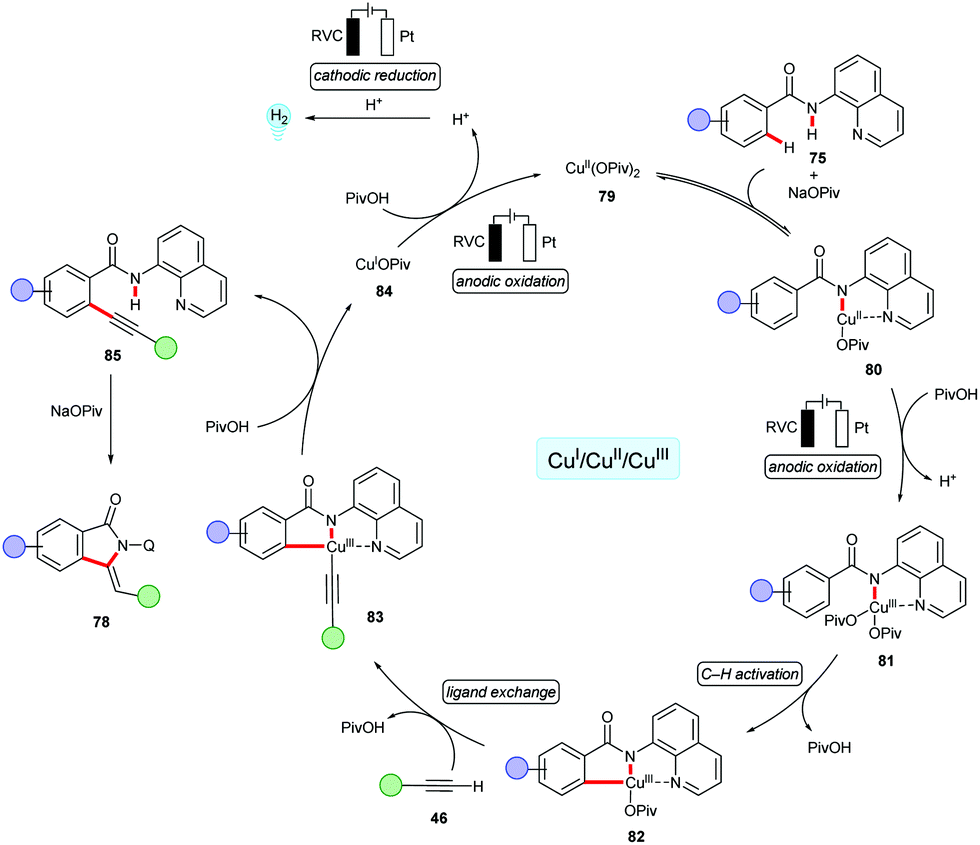 | ||
| Scheme 24 Proposed catalytic cycle for the cupraelectro-catalyzed alkyne 46 annulation. | ||
Very recently, the Ackermann group reported the first example of organometallic C–H activation for C–N formation by nickellaelectrocatalysis.158 Thus, the ortho-selective C–H amination of amides 75 with a variety of secondary amines 13 was achieved using the commercially available nickel salt Ni(DME)Cl2 (Scheme 25). The CV studies performed with a mixture of Ni(DME)Cl2, NaOPiv and substrate 75 in DMA showed a redox potential of 0.26 V vs. Fc+/Fc for the nickel(II)/nickel(III) event. Furthermore, the measurements after preheating at 120 °C for 30 min gave an oxidation wave at Ep = 0.49 V vs. Fc+/0, which is indicative of a nickel(IV) species. Based on these CV data and other kinetic studies, the authors proposed a plausible reaction pathway to involve a Ni(III)/Ni(IV)/Ni(II) manifold (Scheme 25b).158
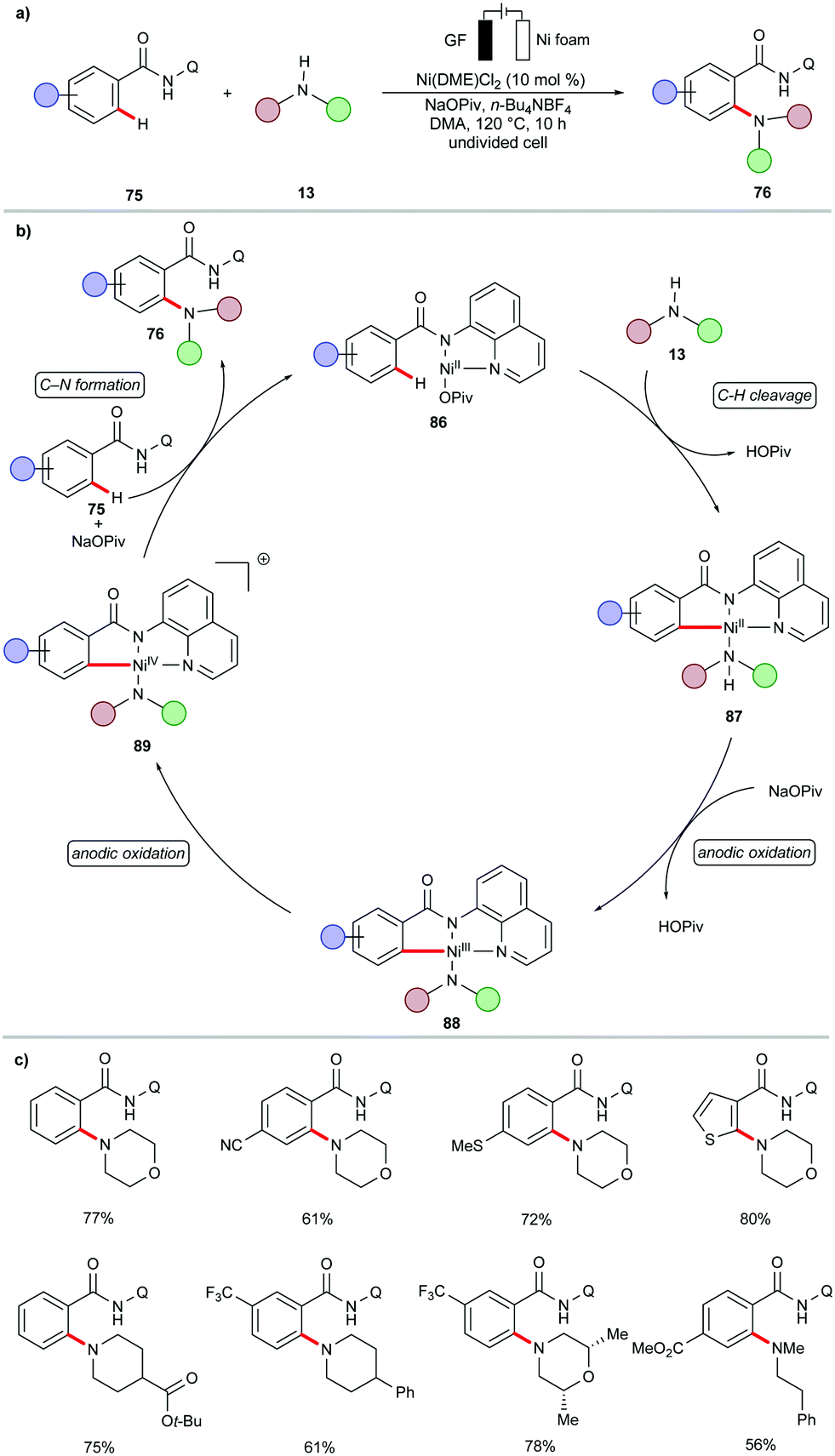 | ||
| Scheme 25 Nickellaelectrocatalytic C–H amination. | ||
C–O forming transformations are of utmost importance for the synthesis of bioactive compounds, pharmaceuticals and functional materials.159–162 In this context, the Ackermann group very recently devised a resource-economical nickellaelectro-catalysis strategy for the oxidative C–H alkoxylations with challenging secondary alcohols 91 with H2 as the only stoichiometric byproduct in lieu of stoichiometric chemical oxidants (Scheme 26).163 The reaction was conveniently achieved using inexpensive and bench-stable nickel salt Ni(DME)Cl2 as the catalyst under economically robust undivided electrochemical cell setup. The versatile nickellaelectro-catalytic reaction conditions compatible with a wide variety of secondary alcohols including alicyclic, cyclic, benzylic and heterocyclic alcohols to give the desired alkoxylation products in excellent yields. Notably, the naturally-occurring alcohols menthol, cholesterol and β-estradiol were found to be compatible without racemization at the stereogenic centers.
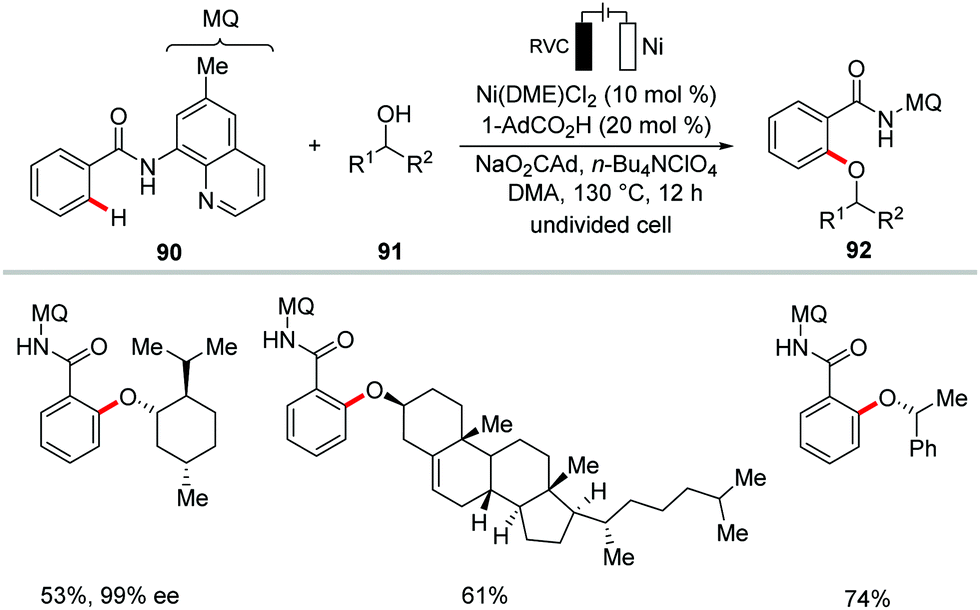 | ||
| Scheme 26 Nickellaelectro-catalyzed secondary alkoxylation. | ||
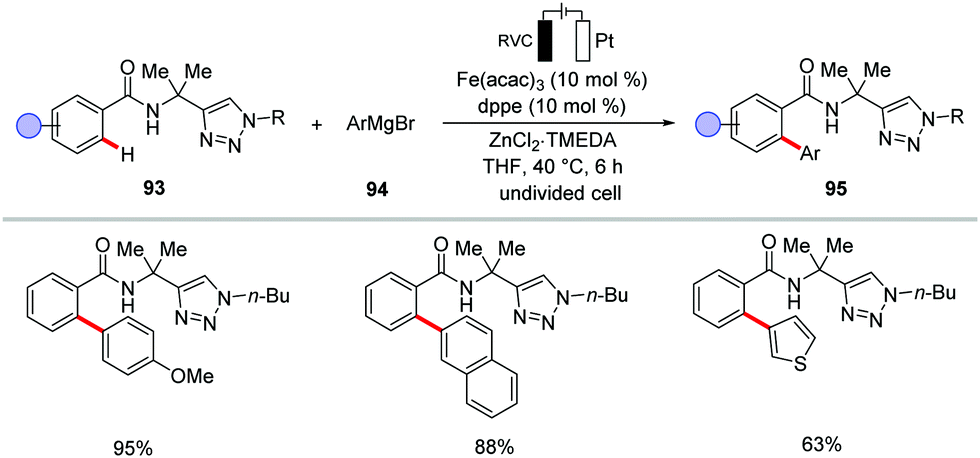 | ||
| Scheme 27 Iron-catalyzed electrochemical C–H arylation. | ||
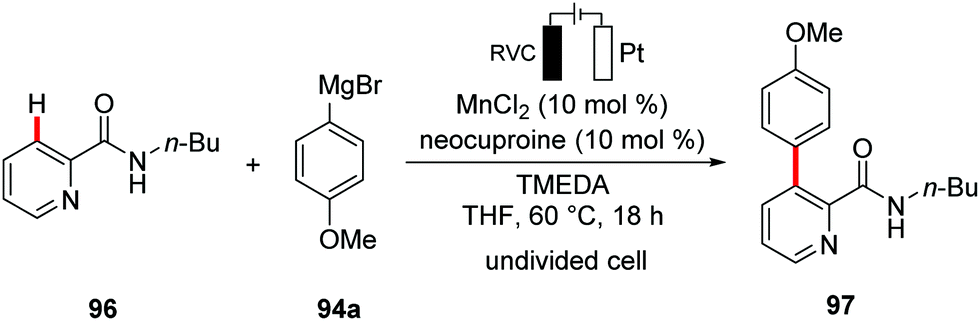 | ||
| Scheme 28 Manganaelectro-catalyzed C–H arylation. | ||
5. Conclusions
Resource economy in molecular syntheses has gained significant momentum in recent years concerning the rising demand for sustainable strategies for molecular syntheses. The merger of electrochemistry with Earth-abundant 3d metal catalysis enabled the construction of challenging C–C and C–heteroatom bonds for complex organic molecules in a sustainable fashion under exceedingly mild reaction conditions. The metallaelectro-catalysis utilizes electro-oxidation/reduction in lieu of reactive chemical redox reagents, and thereby prevents undesired waste formation. Particularly, electricity from renewable energy sources, such as solar and wind power, features unique potential for the ideal step and redox economy. The potential of sustainable electrochemical 3d metal catalysis in molecular synthesis is herein highlighted with three key case studies. Thus, the recent developments of alkene difunctionalizations by manganese catalysts under electrochemical conditions set the stage for the syntheses of synthetically meaningful vicinally difunctionalized products (Case Study I). Electrochemical nickel-catalyzed cross-couplings proceeded under ambient reaction conditions to forge C–N, C–S and C–P bonds (Case Study II). Remarkably, the merger of 3d metal electrochemistry and C–H activation enabled excellent oxidant economy by using electricity as green oxidant and generates molecular hydrogen as the sole by-product (Case Study III). The metallaelectrocatalysis using 3d metal catalysts, such as cobalt, copper, nickel, iron and manganese, proceeded under mild reaction conditions with broad substrate scope. These metallaelectro-catalyzed C–H activations often proceeded in green reaction media, ensuring the high levels of sustainability. In sharp contrast to noble metal electrocatalyses,103,107,110,111,167,168 most of the 3d metallaelectro-catalyzed organic syntheses could be accomplished in an user-friendly, undivided electrochemical cell setup. Likewise, the electrochemical cross-couplings by nickel catalyst involving cathodic reduction of the metal catalyst were conveniently performed in an undivided cell, provided that nickel electrodes were used (Case Study II). Given the unique features of 3d metallaelectrocatalysis in terms of resource economy, further advances are expected in this rapidly emerging research area, including enantioselective transformations, dual metallaelectrocatalysis, and electrocatalytic C–C and C–heteroatom activations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support by the Alexander von Humboldt Foundation (fellowship to P. G.), the DFG (Gottfried-Wilhelm-Leibniz-award to L. A. and SPP1807) is gratefully acknowledged.Notes and references
- S. A. Matlin, G. Mehta, H. Hopf and A. Krief, Nat. Chem., 2016, 8, 393–398 CrossRef CAS PubMed.
- B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505–7511 CrossRef CAS.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63–76 CrossRef.
- W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS PubMed.
- Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921–4942 RSC.
- H. M. L. Davies and K. Liao, Nat. Rev. Chem., 2019, 3, 347–360 CrossRef CAS.
- C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031–1058 CAS.
- G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC.
- S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304–8329 CrossRef CAS PubMed.
- S. Santoro, F. Ferlin, L. Ackermann and L. Vaccaro, Chem. Soc. Rev., 2019, 48, 2767–2782 RSC.
- R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS PubMed.
- H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- M. Oelgemöller, Chem. Rev., 2016, 116, 9664–9682 CrossRef PubMed.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CAS.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- F. Wang, S. Yu and X. Li, Chem. Soc. Rev., 2016, 45, 6462–6477 RSC.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS PubMed.
- P. Gandeepan and C.-H. Cheng, Chem. – Asian J., 2015, 10, 824–838 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- P. Gandeepan and L. Ackermann, in Non-Noble Metal Catalysis: Molecular Approaches and Reactions, ed. R. J. K. Gebbink and M.-E. Moret, Wiley-VCH, Weinheim, 2019, pp. 391–423 Search PubMed.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS PubMed.
- A. Bechtoldt, C. Tirler, K. Raghuvanshi, S. Warratz, C. Kornhaaß and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 264–267 CrossRef CAS PubMed.
- P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1537–1543 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- D. C. Miller, K. T. Tarantino and R. R. Knowles, Top. Curr. Chem., 2016, 374, 30 CrossRef PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- D. C. Fabry and M. Rueping, Acc. Chem. Res., 2016, 49, 1969–1979 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed.
- A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CAS.
- N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- N. Sauermann, T. H. Meyer and L. Ackermann, Chem. – Eur. J., 2018, 24, 16209–16217 CrossRef CAS PubMed.
- A. Volta, Philos. Trans. R. Soc. London, 1800, 90, 403–431 CrossRef.
- M. Faraday, Ann. Phys., 1834, 109, 433–451 CrossRef.
- H. Kolbe, Justus Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwick and D. C. Smith, Org. Chem. Front., 2015, 2, 823–848 RSC.
- In the following text, for the sake of simplicity only the oxidation half-reaction will be used as an example. All general principles are equally valid for the cathodic reduction half-reaction.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS.
- L. Wang, C. Qi, T. Guo and H. Jiang, Org. Lett., 2019, 21, 2223–2226 CrossRef CAS PubMed.
- Y.-X. Dong, Y. Li, C.-C. Gu, S.-S. Jiang, R.-J. Song and J.-H. Li, Org. Lett., 2018, 20, 7594–7597 CrossRef CAS PubMed.
- A. Bunescu, T. M. Ha, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 10555–10558 CrossRef CAS PubMed.
- J. Waser, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 8294–8295 CrossRef CAS PubMed.
- X. Sun, X. Li, S. Song, Y. Zhu, Y.-F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059–6066 CrossRef CAS PubMed.
- R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 8069–8077 CrossRef CAS PubMed.
- K. Shen and Q. Wang, J. Am. Chem. Soc., 2017, 139, 13110–13116 CrossRef CAS PubMed.
- B. B. Snider and J. R. Duvall, Org. Lett., 2004, 6, 1265–1268 CrossRef CAS PubMed.
- B. Zhang and A. Studer, Org. Lett., 2014, 16, 1790–1793 CrossRef CAS PubMed.
- Y.-A. Yuan, D.-F. Lu, Y.-R. Chen and H. Xu, Angew. Chem., Int. Ed., 2016, 55, 534–538 CrossRef CAS PubMed.
- B. H. Nguyen, A. Redden and K. D. Moeller, Green Chem., 2014, 16, 69–72 RSC.
- H. Tanaka, M. Kuroboshi, H. Takeda, H. Kanda and S. Torii, J. Electroanal. Chem., 2001, 507, 75–81 CrossRef CAS.
- S. Torii, P. Liu, N. Bhuvaneswari, C. Amatore and A. Jutand, J. Org. Chem., 1996, 61, 3055–3060 CrossRef CAS PubMed.
- S. Torii, P. Liu and H. Tanaka, Chem. Lett., 1995, 319–320 CrossRef CAS.
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed.
- N. Fu, G. S. Sauer and S. Lin, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef CAS PubMed.
- J. C. Siu, J. B. Parry and S. Lin, J. Am. Chem. Soc., 2019, 141, 2825–2831 CrossRef CAS PubMed.
- K.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef CAS PubMed.
- K.-Y. Ye, Z. Song, G. S. Sauer, J. H. Harenberg, N. Fu and S. Lin, Chem. – Eur. J., 2018, 24, 12274–12279 CrossRef CAS PubMed.
- N. Fu, Y. Shen, A. R. Allen, L. Song, A. Ozaki and S. Lin, ACS Catal., 2018, 9, 746–754 CrossRef PubMed.
- F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, New York, 1998 Search PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2015, 59, 4443–4458 CrossRef PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS PubMed.
- E. C. Hughes, F. Veatch and V. Elersich, Ind. Eng. Chem., 1950, 42, 787–790 CrossRef CAS.
- R. Cramer and D. R. Coulson, J. Org. Chem., 1975, 40, 2267–2273 CrossRef CAS.
- J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 6054–6058 CrossRef CAS.
- S. Ge, R. A. Green and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 1617–1627 CrossRef CAS PubMed.
- N. F. Fine Nathel, J. Kim, L. Hie, X. Jiang and N. K. Garg, ACS Catal., 2014, 4, 3289–3293 CrossRef CAS PubMed.
- M. Marín, R. J. Rama and M. C. Nicasio, Chem. Rec., 2016, 16, 1819–1832 CrossRef PubMed.
- C. Gosmini, J. Y. Nédélec and J. Périchon, Tetrahedron Lett., 2000, 41, 201–203 CrossRef CAS.
- C. Li, Y. Kawamata, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 13088–13093 CrossRef CAS PubMed.
- Y. Kawamata, J. C. Vantourout, D. P. Hickey, P. Bai, L. Chen, Q. Hou, W. Qiao, K. Barman, M. A. Edwards, A. F. Garrido-Castro, J. N. deGruyter, H. Nakamura, K. Knouse, C. Qin, K. J. Clay, D. Bao, C. Li, J. T. Starr, C. Garcia-Irizarry, N. Sach, H. S. White, M. Neurock, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6392–6402 CrossRef CAS PubMed.
- H. Nakamura, K. Yasui, Y. Kanda and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 1494–1497 CrossRef CAS PubMed.
- S. Sengmany, A. Ollivier, E. Le Gall and E. Léonel, Org. Biomol. Chem., 2018, 16, 4495–4500 RSC.
- Y. Wang, L. Deng, X. Wang, Z. Wu, Y. Wang and Y. Pan, ACS Catal., 2019, 9, 1630–1634 CrossRef CAS.
- C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS.
- M. Konishi, K. Tsuchida, K. Sano, T. Kochi and F. Kakiuchi, J. Org. Chem., 2017, 82, 8716–8724 CrossRef CAS PubMed.
- F. Saito, H. Aiso, T. Kochi and F. Kakiuchi, Organometallics, 2014, 33, 6704–6707 CrossRef CAS.
- H. Aiso, T. Kochi, H. Mutsutani, T. Tanabe, S. Nishiyama and F. Kakiuchi, J. Org. Chem., 2012, 77, 7718–7724 CrossRef CAS PubMed.
- F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310–11311 CrossRef CAS PubMed.
- C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC.
- K.-J. Jiao, C.-Q. Zhao, P. Fang and T.-S. Mei, Tetrahedron Lett., 2017, 58, 797–802 CrossRef CAS.
- Y.-Q. Li, Q.-L. Yang, P. Fang, T.-S. Mei and D. Zhang, Org. Lett., 2017, 19, 2905–2908 CrossRef CAS PubMed.
- Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef CAS PubMed.
- F. Kakiuchi and T. Kochi, Isr. J. Chem., 2017, 57, 953–963 CrossRef CAS.
- Y. Budnikova, Y. Dudkina and M. Khrizanforov, Inorganics, 2017, 5, 70 CrossRef.
- J. Chen, S. Lv and S. Tian, ChemSusChem, 2018, 12, 115–132 CrossRef CAS PubMed.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- Y. Qiu, W.-J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef CAS PubMed.
- W. J. Kong, L. H. Finger, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 6342–6346 CrossRef CAS PubMed.
- Y. Qiu, A. Scheremetjew and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 2731–2738 CrossRef CAS PubMed.
- W.-J. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 17198–17206 CrossRef CAS PubMed.
- W.-J. Kong, Z. Shen, L. H. Finger and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 5551–5556 CrossRef CAS PubMed.
- Y. Qiu, M. Stangier, T. H. Meyer, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14179–14183 CrossRef CAS PubMed.
- Q.-L. Yang, Y.-K. Xing, X.-Y. Wang, H.-X. Ma, X.-J. Weng, X. Yang, H.-M. Guo and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 18970–18976 CrossRef CAS PubMed.
- Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef CAS PubMed.
- R. Mei, J. Koeller and L. Ackermann, Chem. Commun., 2018, 54, 12879–12882 RSC.
- F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, ACS Catal., 2018, 8, 3820–3824 CrossRef CAS.
- M.-J. Luo, M. Hu, R.-J. Song, D.-L. He and J.-H. Li, Chem. Commun., 2019, 55, 1124–1127 RSC.
- L. Massignan, X. Tan, T. H. Meyer, R. Kuniyil, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 3184–3189 CrossRef CAS PubMed.
- L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- T. Andou, Y. Saga, H. Komai, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 3213–3216 CrossRef CAS PubMed.
- S. Santoro, A. Marrocchi, D. Lanari, L. Ackermann and L. Vaccaro, Chem. – Eur. J., 2018, 24, 13383–13390 CrossRef CAS PubMed.
- P. Gandeepan, N. Kaplaneris, S. Santoro, L. Vaccaro and L. Ackermann, ACS Sustainable Chem. Eng., 2019, 7, 8023–8040 CrossRef CAS.
- C. Tian, U. Dhawa, J. Struwe and L. Ackermann, Chin. J. Chem., 2019, 37, 552–556 CrossRef CAS.
- T. H. Meyer, J. C. A. de Oliveira, D. Ghorai and L. Ackermann, Angew. Chem., Int. Ed., 2020 DOI:10.1002/ange.202002258.
- N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef CAS PubMed.
- X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195–4199 CrossRef CAS PubMed.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef CAS PubMed.
- T. H. Meyer, G. A. Chesnokov and L. Ackermann, ChemSusChem, 2020, 13, 668–671 CrossRef CAS PubMed.
- R. Mei, N. Sauermann, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 140, 7913–7921 CrossRef CAS.
- J. Mo, T. Müller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 7719–7723 CrossRef CAS.
- S. Nakanowatari, T. Müller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15891–15895 CrossRef CAS.
- S. Nakanowatari, R. Mei, M. Feldt and L. Ackermann, ACS Catal., 2017, 7, 2511–2515 CrossRef CAS.
- R. Boobalan, R. Kuppusamy, R. Santhoshkumar, P. Gandeepan and C.-H. Cheng, ChemCatChem, 2017, 9, 273–277 CrossRef CAS.
- N. Thrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Angew. Chem., Int. Ed., 2016, 55, 12361–12365 CrossRef CAS PubMed.
- P. Gandeepan, P. Rajamalli and C.-H. Cheng, Chem. – Eur. J., 2015, 21, 9198–9203 CrossRef CAS PubMed.
- R. Zeng, J. Ye, C. Fu and S. Ma, Adv. Synth. Catal., 2013, 355, 1963–1970 CrossRef CAS.
- B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef CAS PubMed.
- R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597–9600 CrossRef CAS PubMed.
- Y. J. Zhang, E. Skucas and M. J. Krische, Org. Lett., 2009, 11, 4248–4250 CrossRef CAS PubMed.
- T. H. Meyer, J. C. A. Oliveira, S. C. Sau, N. W. J. Ang and L. Ackermann, ACS Catal., 2018, 8, 9140–9147 CrossRef CAS.
- S. C. Sau, R. Mei, J. Struwe and L. Ackermann, ChemSusChem, 2019, 12, 3023–3027 CrossRef CAS.
- L. Zeng, H. Li, S. Tang, X. Gao, Y. Deng, G. Zhang, C.-W. Pao, J.-L. Chen, J.-F. Lee and A. Lei, ACS Catal., 2018, 8, 5448–5453 CrossRef CAS.
- O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS.
- A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed.
- Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed.
- S. Kathiravan, S. Suriyanarayanan and I. A. Nicholls, Org. Lett., 2019, 21, 1968–1972 CrossRef CAS PubMed.
- C. Tian, U. Dhawa, A. Scheremetjew and L. Ackermann, ACS Catal., 2019, 9, 7690–7696 CrossRef CAS.
- S.-K. Zhang, R. C. Samanta, N. Sauermann and L. Ackermann, Chem. – Eur. J., 2018, 24, 19166–19170 CrossRef CAS PubMed.
- S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC.
- S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- L. Ackermann, Org. Process Res. Dev., 2015, 19, 260–269 CrossRef CAS.
- S.-K. Zhang, J. Struwe, L. Hu and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 3178–3183 CrossRef CAS.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS.
- G. Cera and L. Ackermann, Top. Curr. Chem., 2016, 374, 57 CrossRef.
- C. Zhu, M. Stangier, J. C. A. Oliveira, L. Massignan and L. Ackermann, Chem. – Eur. J., 2019, 25, 16382–16389 CrossRef CAS.
- K. Tsuchida, T. Kochi and F. Kakiuchi, Asian J. Org. Chem., 2013, 2, 935–937 CrossRef CAS.
- K. Mitsudo, T. Kaide, E. Nakamoto, K. Yoshida and H. Tanaka, J. Am. Chem. Soc., 2007, 129, 2246–2247 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |