
Molecular and nanoengineering approaches towards activatable cancer immunotherapy
 *a
*a
Abstract
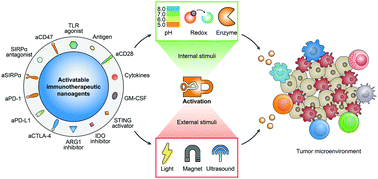
Cancer immunotherapy is an emerging treatment strategy that modulates the immune system to fight against cancer. Although several immunotherapeutic agents have been utilized in the clinic for cancer treatment, low patient response rates and potential immune-related adverse events remain two major challenges. With the merits of delivery controllability and modular flexibility, nanomedicines provide opportunities to facilitate immunotherapies for clinical translation in a safe and effective manner. In this review, we discuss the convergence of nanomedicine with immunotherapy with a focus on molecular and nanoengineering approaches towards activatable cancer immunotherapy. These activatable nanoagents exert immunotherapeutic action only in response to internal or external stimuli. This allows them to locally reprogram the tumor microenvironment and activate antitumor immunity while reducing the incidence of immune-related adverse events. The category of activatable immunotherapeutic nanoagents are discussed along with an overview of their clinical translation prospects and challenges.
 Please wait while we load your content...
Please wait while we load your content...

