
The luminescent and photophysical properties of covalent organic frameworks
W. Karl
Haug†
 ,
Erica M.
Moscarello†
,
Eric R.
Wolfson†
and
Psaras L.
McGrier
*
,
Erica M.
Moscarello†
,
Eric R.
Wolfson†
and
Psaras L.
McGrier
*
Department of Chemistry & Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: mcgrier.1@osu.edu
First published on 20th January 2020
Abstract
Covalent organic frameworks (COFs) are an emerging class of crystalline porous polymers that have attracted significant attention due to their tunable properties and structural robustness. As a result, COFs with luminescent properties are of great interest for fields such as chemical sensing, solid-state light emitters, photocatalysis, and optoelectronics. However, the bottom-up synthesis of luminescent COF systems remains a challenge in the field due to an abundance of competing non-radiative pathways, including phenomena such as aggregate caused quenching (ACQ). To overcome these obstacles, there has been a burgeoning investigation into the luminescent and photophysical properties of COFs. This review will highlight methods used to fabricate luminescent COFs and discuss the factors that are critical for their production. A collection of known luminescent COF systems will be featured. In addition, the ability to utilize the photophysical properties of COFs for applications related to photocatalysis, solid-state light emitters, and chemical sensing will be addressed. An outlook will address the current progress and remaining challenges facing the field to ultimately expand the scope of their applications.
 W. Karl Haug | W. Karl Haug is a PhD candidate advised by Psaras L. McGrier at The Ohio State University. He received his bachelor's degree from University of Denver in 2016 where he worked on photo-induced cyclizations of ortho-oxaxylylenes, in the lab of Andrei Kutateladze. His current research interests are focused on the utilization of metal-doped covalent organic frameworks as a platform for heterogeneous catalysis. |
 Erica M. Moscarello | Erica Moscarello received her BS from Rowan University in 2017 where she worked under the supervision of Professor Gustavo Moura-Letts. She is currently in the third year of her PhD at The Ohio State University under the supervision of Professor Psaras McGrier. As a doctoral student, her work focuses on investigating the thermal properties of benzobisoxazole-linked covalent organic frameworks. |
 Eric R. Wolfson | Eric Wolfson received his bachelor's degree in Chemistry in 2015 from University of Maryland Baltimore County, where he studied the cure kinetics of fast-setting epoxy resins under Dr Lisa Kelly. Eric also worked on the synthesis of CdSe and PbSe nanowires under Dr Narsingh Singh. He has conducted his PhD research under the guidance of Psaras McGrier since 2016 at The Ohio State University. As a doctoral student, his work focuses on functional porous organic polymers for use as cutting-edge energy storage materials. |
 Psaras L. McGrier | Psaras is an Assistant professor in the Department of Chemistry and Biochemistry at The Ohio State University. He received his BS in Chemistry from the University of South Carolina-Aiken in 2004 and his PhD in Organic/Polymer chemistry from the Georgia Institute of Technology (Georgia Tech) in 2010 under the direction of Professor Uwe Bunz. After finishing his PhD, he moved to Northwestern University (2010–2013) to join the group of Professor Sir Fraser Stoddart as a postdoctoral scholar on a fellowship from Georgia Tech Facilitating Academic Careers in Engineering and Science (FACES) committee and the National Science Foundation (NSF). His current research interests include the synthesis and utilization of 2D and 3D porous polymers for practical applications. He is a recipient of the 2018 ACS Division of Polymeric Materials: Science and Engineering (PMSE) Young Investigator Award. |
1. Introduction
Light is essential for life on earth. The investigation of light and light-emitting processes has long been of interest to society, resulting in significant advances in modern technology such as light-emitting diodes (LEDs),1,2 photography,3 and forensics.4 Photoluminescence (PL) is a phenomenon wherein a photon is absorbed by a chromophore through a photoexcitation event, followed by photon emission as a consequence of electron relaxation. However, there are a number of different pathways through which a photoexcited state can return to its ground state. These relaxation pathways may be as fast as 10−12 s,5 or as slow as hours,6 depending on the photophysical properties of the chromophore. Radiation from a transition between states of the same multiplicity (i.e., S1 → S0 + hν) is referred to as fluorescence. In some instances, the excited electron can undergo a spin–flip. This intersystem-crossing (ISC) event can produce a longer-lived photon emission process, in a phenomenon known as phosphorescence. This longer lifetime of emission, or afterglow, can be attributed to the ‘forbidden’ nature of the transition from the meta-stable triplet state to the singlet ground state (i.e., T1 → S0 + hν). The continued investigation of photophysical properties is paramount to the development of more advanced technologies, and therefore society as a whole.The examination and practical use of luminescent polymers has seen a recent boom due to the development of advanced materials such as hyper-crosslinked polymers (HCPs),7,8 polymers of intrinsic microporosity (PIMs),9 porous organic polymers,10–13 π-conjugated polymers,14 and metal organic frameworks (MOFs).15,16 In particular, the advancement of luminescent porous materials has been largely due to (a) the ability to incorporate fluorescent probes directly into the polymer skeleton, (b) the tunability of optoelectronic properties through careful selection of monomers, and (c) a growing interest in heterogeneous sensors which can be used repeatedly without loss of activity or contamination of the sample. As a direct result of this research, significant advances in fields such as chemical sensing,17–19 organic light emitting polymers,20–22 and phototherapy23–26 have been made. However, due to the often-amorphous nature of these materials, fluorescent polymers can be challenged by poor solvent permeability leading to decreased sensitivities. In addition, in the case of MOFs, high densities and poor hydrolytic stabilities are major issues challenging their use in commercial applications. There is an ongoing need for the further development of novel, stable, and highly performing luminescent materials that can be processed into a variety of forms while still retaining their desired photophysical properties and activity.
Covalent organic frameworks (COFs)27–29 are an advanced class of crystalline porous polymers that enable the integration of various molecular building blocks into highly ordered periodic arrays. The modular nature of COFs has made them of immense interest in a variety of applications including energy storage,30,31 catalysis,32,33 gas storage,34 and carbon capture.35 A large portion of COF research has been focused on developing methods to incorporate novel linkages and building blocks, allowing for COFs with different functional architectures.36 A large area of research has since been focused on working towards utilizing these innovative materials and their unique properties in different applications. One such area of research focuses on exploiting the photophysical properties of COFs. COFs that exhibit tunable photophysical properties are particularly desirable for applications in chemical sensing,37 photocatalysis,38 and organic light emitting diodes (OLEDs).39 The interactions between the monomer units and adjacent layers of COFs can greatly influence their photophysical properties, giving them a distinct advantage over amorphous materials with the same components. Therefore, it is important to understand the underlying principles that govern the luminescent properties of these materials.
The following review will discuss COFs in the context of their luminescent and photophysical properties. Specifically, this review will include a summary of methods used to fabricate luminescent COFs, a collection of known luminescent COF systems, and a discussion about the factors that are critical for producing luminescent COFs. In addition, the use of COFs for applications related to chemical sensing, photocatalysis, as well as light emitters and photodetectors will also be addressed.
2. COF synthesis and design
Initially, COFs were primarily designed utilizing dynamic covalent chemistry in conjunction with favorable non-covalent interactions to form ordered and permanently porous materials. In 2005, Yaghi and co-workers utilized the high reversibility of boroxine and boronate ester condensation reactions to develop the first COFs.27 However, these linkages exhibited weak hydrolytic stability. The challenge to synthesize more robust frameworks using different connectivities soon led to the development of more chemically stable linkages such as imines,40 oxazoles,35,41 and olefins42 (Chart 1).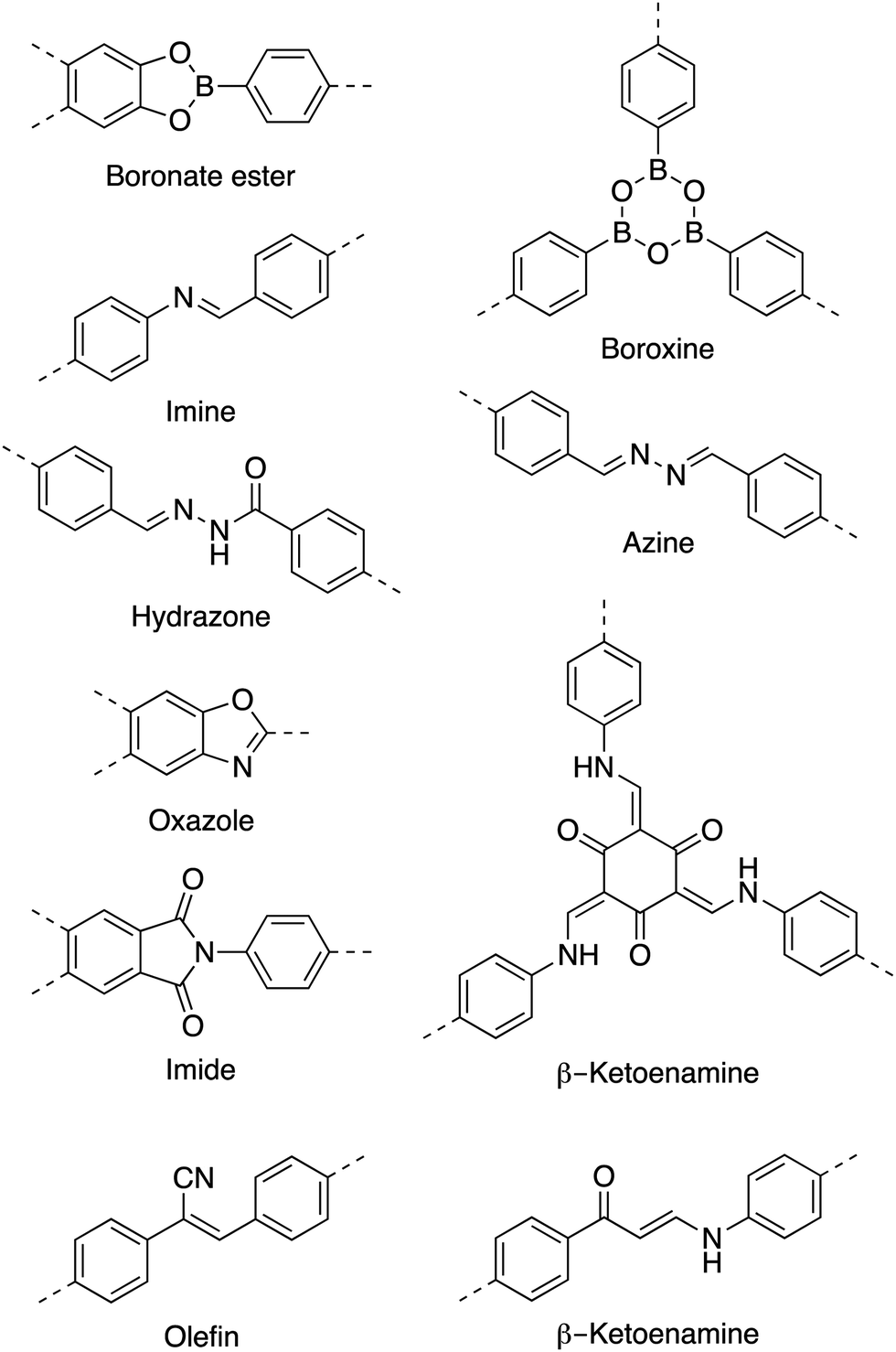 | ||
| Chart 1 Common linkages used in COFs. | ||
As a result of using only organic building blocks and covalent linkages, COFs are also often characterized as having low densities and high thermal stabilities.29 By selecting π-electron rich monomers with specific symmetries, a variety of 2D and 3D43,44 architectures have been achieved (Fig. 1).45 This strategy not only allows one to have precise control over the topology of the material, but also allows for the ability to pre-tailor bulk properties and performance for specific applications through the careful choice of suitable monomers (Chart 2). In addition, the ability to incorporate functional moieties that remain inert during COF synthesis allows one to further post-functionalize the molecular skeleton. As a consequence, these design strategies have enabled the creation of various functional COFs with a high degree of tunability.
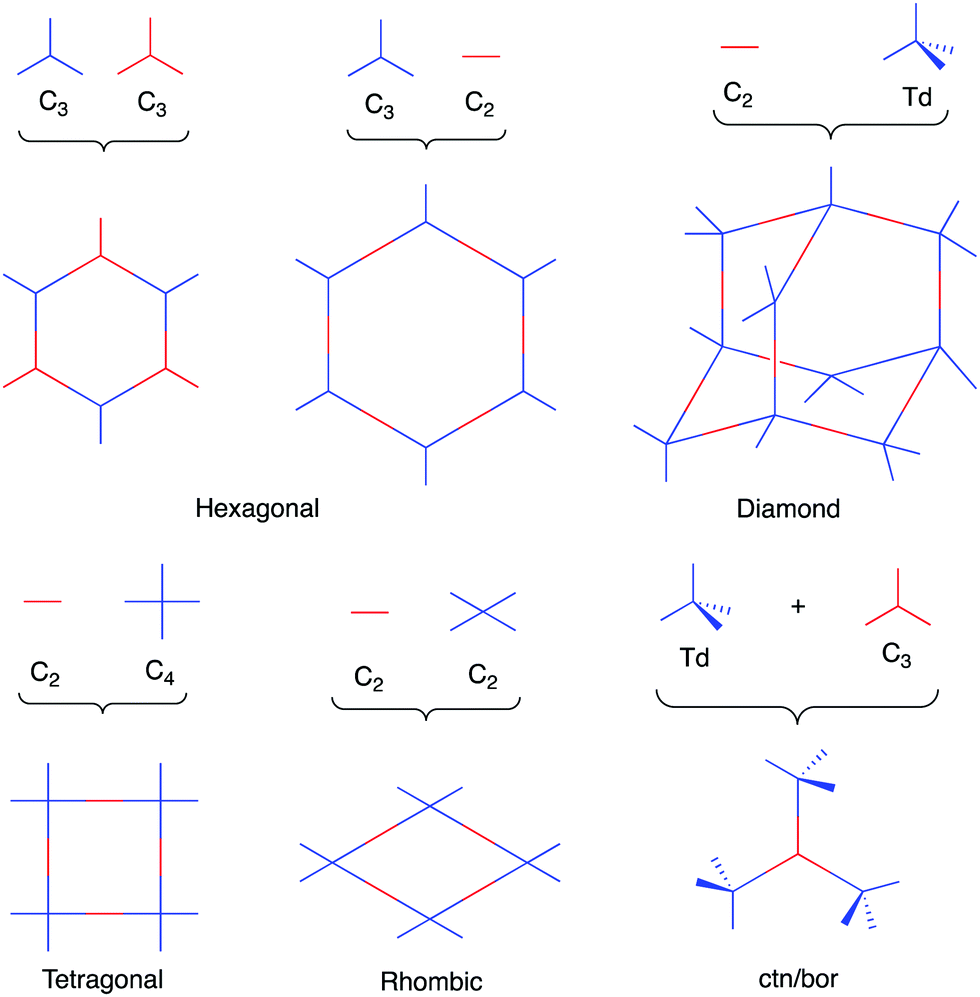 | ||
| Fig. 1 Sample of topologies accessible through COF synthesis. | ||
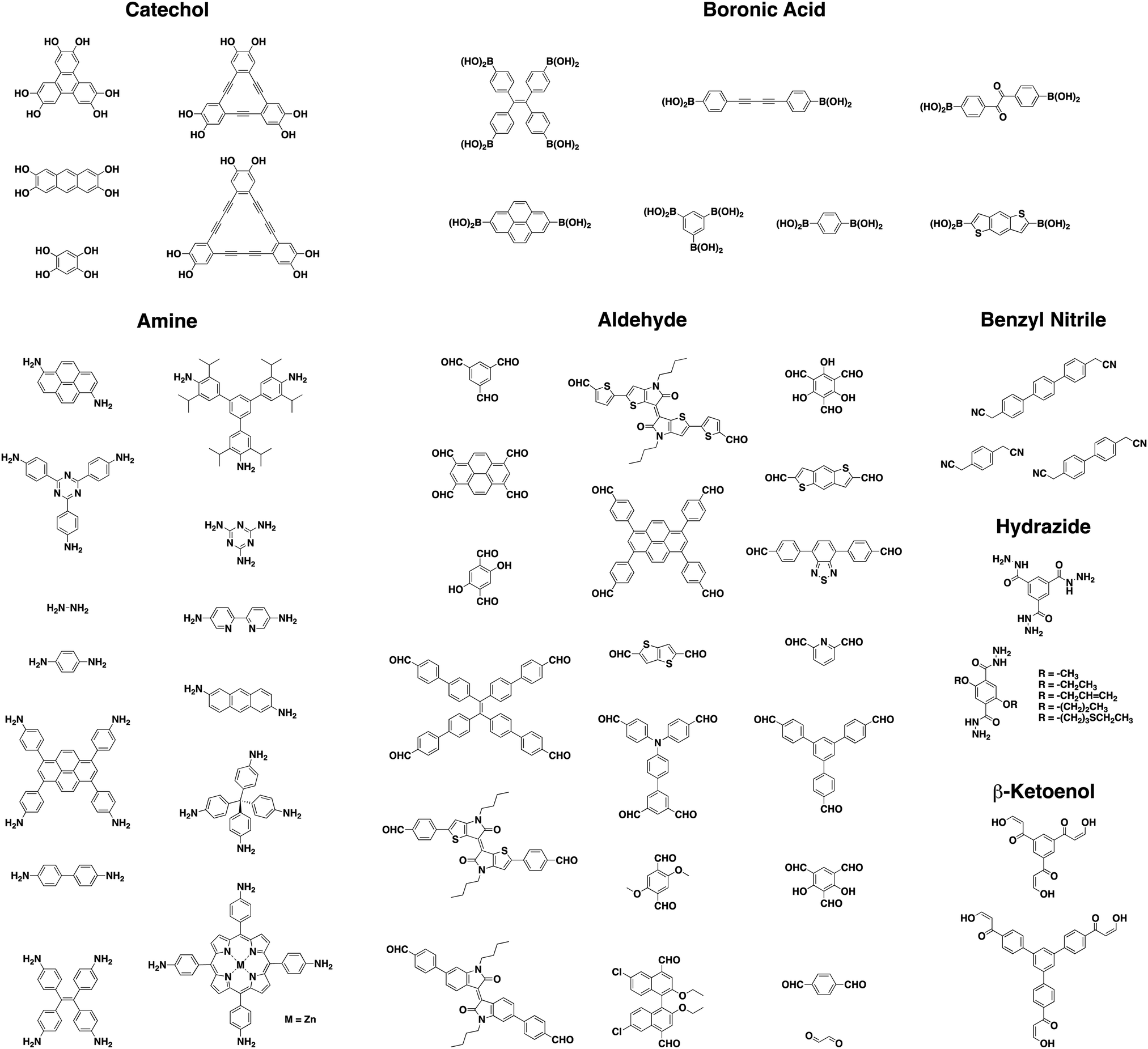 | ||
| Chart 2 Examples of monomers used in luminescent COFs referenced in this review. | ||
Initially, solvothermal methods were utilized in the synthesis of COFs. The use of high temperatures in a closed system promotes reversibility and eventually drives product formation towards the thermodynamically favored product. Due to the poor solubility of COF crystallites in solution, the solvothermal method typically limits applications to those where a bulk powder is sufficient. As a result, a key challenge is improving the scalability and processability of COF materials. Interest in expanding the scope of COF-based applications has led to methodologies such as mechanochemical, microwave, ionothermal, and substrate-templated synthesis.45 As a result, COFs have now been processed into a variety of forms including nanoparticles, hollow microspheres/rods, and membranes.46,47 A number of these forms such as thin films and exfoliated sheets have yielded materials suitable for photoluminescent applications.32
While new and more robust linkages led to substantial progress in the development of new COF systems, the polymerization and crystallization processes remained poorly controlled during their syntheses. COFs are predominantly formed as polycrystalline aggregates with small crystalline domains, and a major hurdle in the synthesis of more crystalline materials was a poor fundamental understanding of nucleation and crystallite growth processes. Therefore, it was of great interest to develop and investigate synthetic methods that yield single-crystalline COFs. As a result, single-crystalline COFs with 3D48 and 2D49 topologies have been successfully developed via the use of modulators and directed growth, respectively. In addition, mechanistic investigation of COF 550 formation led to new insights on the impact of reversible bond formation. Dichtel and co-workers eventually utilized this new information to form single-crystalline COFs via a seeded-growth method.51
To control the orientation of growth, extensive research has been dedicated to growing COFs as oriented thin films.52–56 This often leads to a more uniform material with less defects allowing for the study of anisotropic properties. Additionally, this processing method results in materials viable for a larger range of applications where bulk powder is not sufficient. Dichtel and co-workers reported the growth of a crystalline covalent organic film on a substrate-supported single layer graphene in 2011.57 Bein and co-workers have also made significant contributions towards advancing the field both in the fabrication of thin films as well as their utilization in applications.58–60 It was initially believed that favorable π-interactions between the substrate surface and COF monomers was necessary for successful COF thin film growth. Bein and co-workers showed that this was not necessarily true by growing thin films of a benzodithiophene COF (BDT-COF) on multiple polycrystalline inorganic substrates including indium-doped tin oxide substrates, gold surfaces, and fused silica substrates.61
3. Origins of luminescence in COFs
3.1. Extended conjugation
It is well understood that π-systems undergo electronic excitations more easily than their saturated counterparts.62 This is due to the lower energy difference (ΔE) for a π–π* transition than for a σ–σ* transition, thus leading to a higher λmax. This effect is enhanced for conjugated systems where the π–π* transitions are redshifted to longer wavelengths (lower ΔE).Yoshida and co-workers were able to establish a relationship between the length of π-conjugation and fluorescence emission efficiency of a system by comparing fluorescent quantum yields (Φf), and other photophysical properties (λem, λabs), to the π-structure of common fluorophores.63 Their results demonstrated a nearly linear relationship between Φf (for 0.1 > Φf > 0.9) and the magnitude of π-conjugation length (Aπ) in the S1 state of the investigated hydrocarbon fluorophores. Additionally, it was revealed that a positive linear relationship between Aπ and λem exists for linear oliogophenylenes. This discovery highlighted how extending the conjugation of chromophores can significantly affect their luminescent properties.
1D polymers have taken advantage of extended conjugation for applications including photovoltaics, sensors, field-effect transistors, photodetectors, and organic light-emitting diodes.64 The ability to further propagate π-conjugation in two spatial directions has the potential to greatly improve upon the abilities and functions of these materials. 2D COFs present an ideal platform to achieve this goal, as their planar sheets have the potential to extend π-conjugation in multiple directions, while their modular nature allows for the precise placement of functional units within the framework.
In an effective demonstration of the influence that extended conjugation has on 2D COFs, Jiang and co-workers incorporated conjugated anthracene units into Ph-An-COF,65 yielding a fluorescent system (λem = 429 nm, Φf = 5.4%). Due to the AA-stacking of the COF layers, the photoactive anthracene units were aligned in a face-on-face π-stacking pattern with an intermolecular (interlayer) distance of 3.4 Å. The proximity (>4 Å) and alignment of the π-stacked anthracene units allowed for their photo-induced dimerization (Fig. 2). Upon irradiation under Ar (xenon lamp, 360 nm light), the newly formed Ph-AnCD-COF displayed quenching of the material's fluorescence. The significant change in the photophysical properties of the COF can be attributed to the loss of planarity and extended π-conjugation within the 2D layers upon the formation of the anthracene dimers. It was further demonstrated that the fluorescence could be regained when the anthracene units were returned to their planar state via the thermally-reversible cycloaddition.
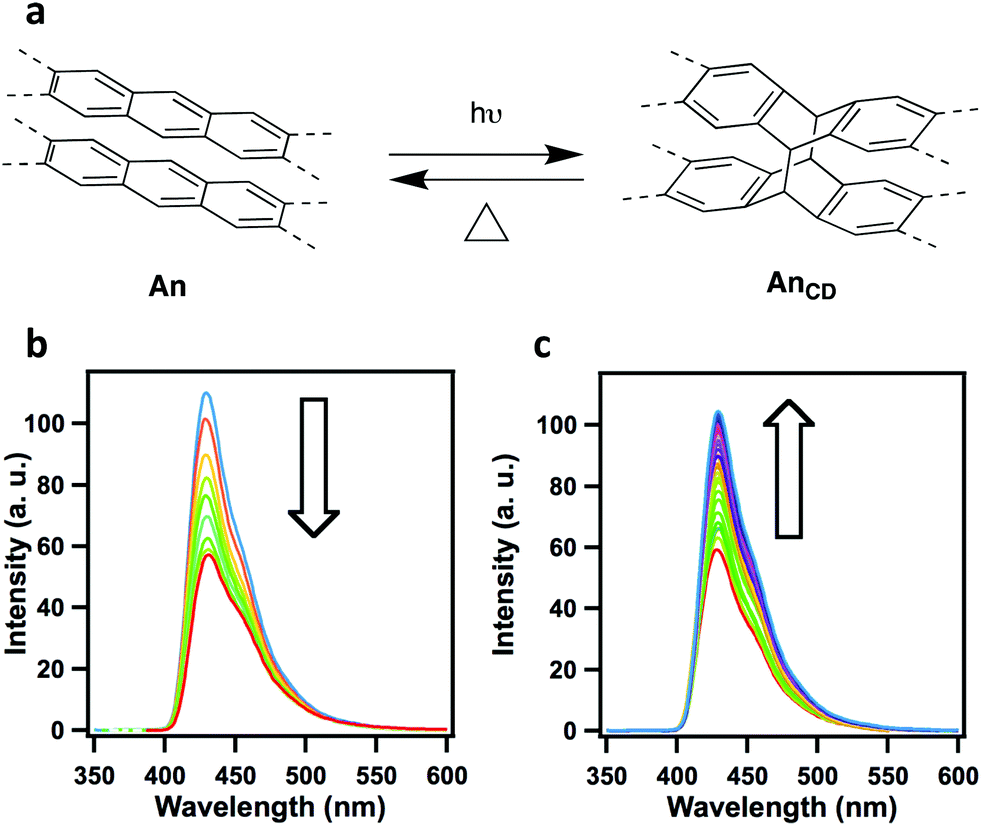 | ||
| Fig. 2 (a) Photo-induced and thermally reversible dimerization of anthracene units. Changes in the fluorescence spectra of (b) Ph-An-COF upon photoirradiation, and (c) recovery of Ph-An-COF from the heating of Ph-AnCD-COF, 100 °C in the dark. Adapted with permission from Angew. Chem., Int. Ed., 2015, 54, 8704–8707. | ||
In an attempt to increase the extent of π-conjugation throughout the layers in 2D-COFs, Perepichka and co-workers developed a novel linkage by taking advantage of the reversibility of Michael addition–elimination reactions.66 Several chemically stable and ordered frameworks were achieved through the condensation of various β-ketoenols and aromatic amines. The resulting COFs were linked through β-ketoenamine functionalities. This linkage strategy demonstrated improved chemical stability as compared to their imine-linked counterparts, while simultaneously providing extended conjugation in the system. The effects of the π-conjugation could be observed as an orange, solid-state luminescence (λmax = 547–560 nm) which was redshifted from that of the reference compounds (λmax = 480–510 nm).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds. However, these linkages often lead to relatively weak electron delocalization due to the high polarization of the CN bonds.66 Switching from Schiff base to Knoevenagel condensations allows for the synthesis of olefin (CC) linked COFs.67–71 These olefin linkages not only allow for significant enhancements in chemical stability, but also increase the effectiveness of π-conjugation between monomer units in the fully sp2-carbon-linked 2D systems.
N bonds. However, these linkages often lead to relatively weak electron delocalization due to the high polarization of the CN bonds.66 Switching from Schiff base to Knoevenagel condensations allows for the synthesis of olefin (CC) linked COFs.67–71 These olefin linkages not only allow for significant enhancements in chemical stability, but also increase the effectiveness of π-conjugation between monomer units in the fully sp2-carbon-linked 2D systems.
When Li and co-workers employed a Knoevenagel condensation between 2,4,6-tris(4-formylphenyl)-1,3,5-triazine (TFPT) and 2,2′-(1,4-phenylene)diacetonitrile (PDAN), a fully conjugated 2D sp2-carbon linked COF (TP-COF) was obtained.70 TP-COF was isolated as a yellow crystalline solid that exhibited bright solid-state fluorescence (λem = 543 nm). The observed solid-state fluorescence for TP-COF showed a noticeable redshift (37 nm) when compared to the synthesized model compound (λem = 506 nm) (Fig. 3). This shift towards a longer wavelength was attributed to the successful synthesis of an extended π-conjugated framework.
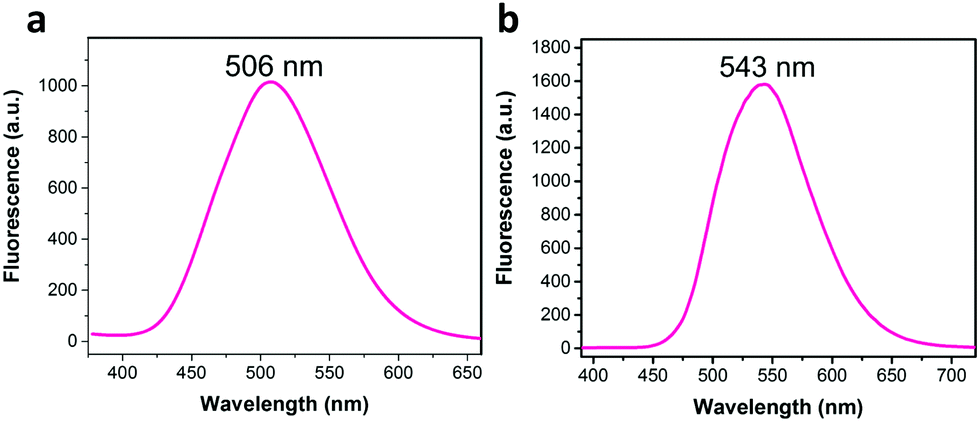 | ||
| Fig. 3 The 37 nm redshift in solid-state fluorescence spectra of (a) the model compound, and (b) TP-COF. Adapted with permission from Angew. Chem., Int. Ed., 2019, 58, 5376–5381. | ||
Jiang and co-workers were able to synthesize a series of sp2 carbon-conjugated 2D-COFs (sp2c-COFs) through the condensation between a tetraphenylpyrene aldehyde unit and linear C2-symmetric phenyl diacetonitrile linkers (Fig. 4).71 Each sp2c-COF was fluorescent and exhibited linker-dependent emission colors. The extent of the π-conjugation derived from the incorporation of olefin linkages was investigated by comparing the electronic absorption bands of sp2c-COF, the synthesized model compound, and an imine-linked pyrene COF with a similar skeleton to sp2c-COF (Fig. 4b). As expected, when compared to the synthesized model compound, sp2c-COF exhibited a large (53 nm) redshift. When the imine-linked version of sp2c-COF was investigated, it only showed a redshift of 21 nm compared to the model compound. This larger bathochromic shift in the sp2c-COF demonstrated that the CC linkage is much more effective than the CN linkage at creating an extended π-conjugated framework. Although both sp2c-COF-2 and sp2c-COF-3 exhibited redshifts when compared to the model compound, they were blueshifted from sp2c-COF by 17 and 36 nm, respectively. These hypochromic shifts were associated with lessened efficiency of π-conjugation throughout the framework, caused by the twisted nature of the biphenyl and terphenyl moieties disrupting the π-conjugation. The twisted structures of sp2c-COF-2 and sp2c-COF-3 could also be observed through their increased interlayer separation distances (3.74 and 3.79 Å compared to 3.58 Å for sp2c-COF). In addition to tuning the λabs, the switching of linkers also led to a change in the structure's band gap. As the efficiency of π-conjugation decreased, the band gap increased.
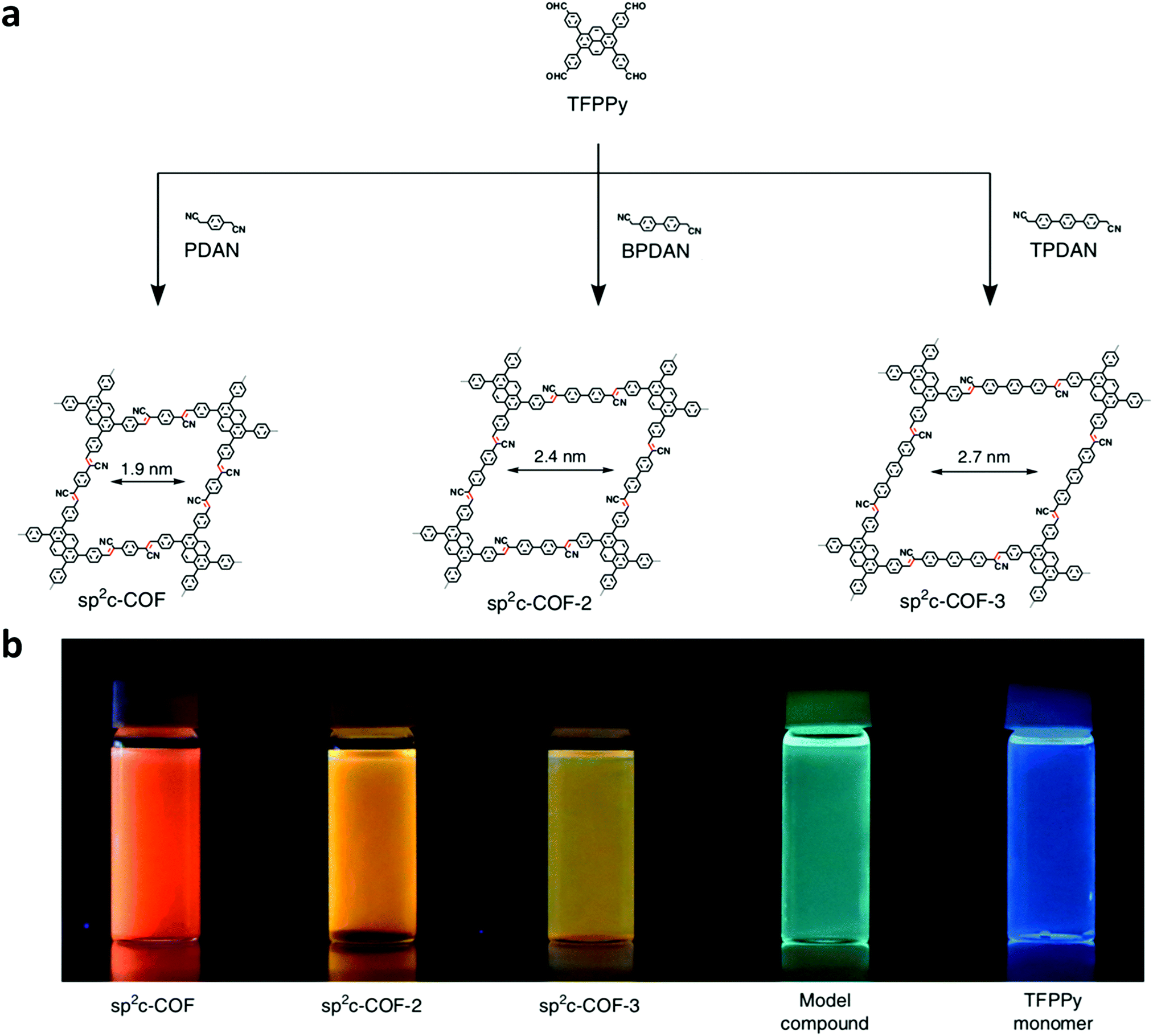 | ||
| Fig. 4 (a) Design and structures of sp2c-COFs from the condensation of TFPPy with PDAN, BPDAN, and TPDAN. (b) Images of sp2c-COF, sp2c-COF-2, sp2c-COF-3, model compound, and TFPPy monomer dispersed in water under a UV lamp. Adapted with permission from Nat. Commun., 2018, 9, 4143. | ||
3.2. Excited-state intramolecular proton transfer
Excited-state intramolecular proton transfer (ESIPT) is a photochemical process that can be utilized to construct luminescent materials. This photoinduced process has been exploited and investigated for numerous applications72–74 since it was first reported by Weller in 1955.75 ESIPT is a phototautomerization that occurs when a chromophore contains an intramolecular hydrogen bond donor (–OH, –NH2) and a hydrogen bond acceptor (–CO, –CN–) in close proximity. Upon photoexcitation, many systems exhibit dual emission bands that encompass the entire visible spectrum due to the rapid four-step enol (E–E*)–keto (K–K*) phototautomerization process (Fig. 5). This four-step photochemical process has proven to be beneficial for sensory-based applications.
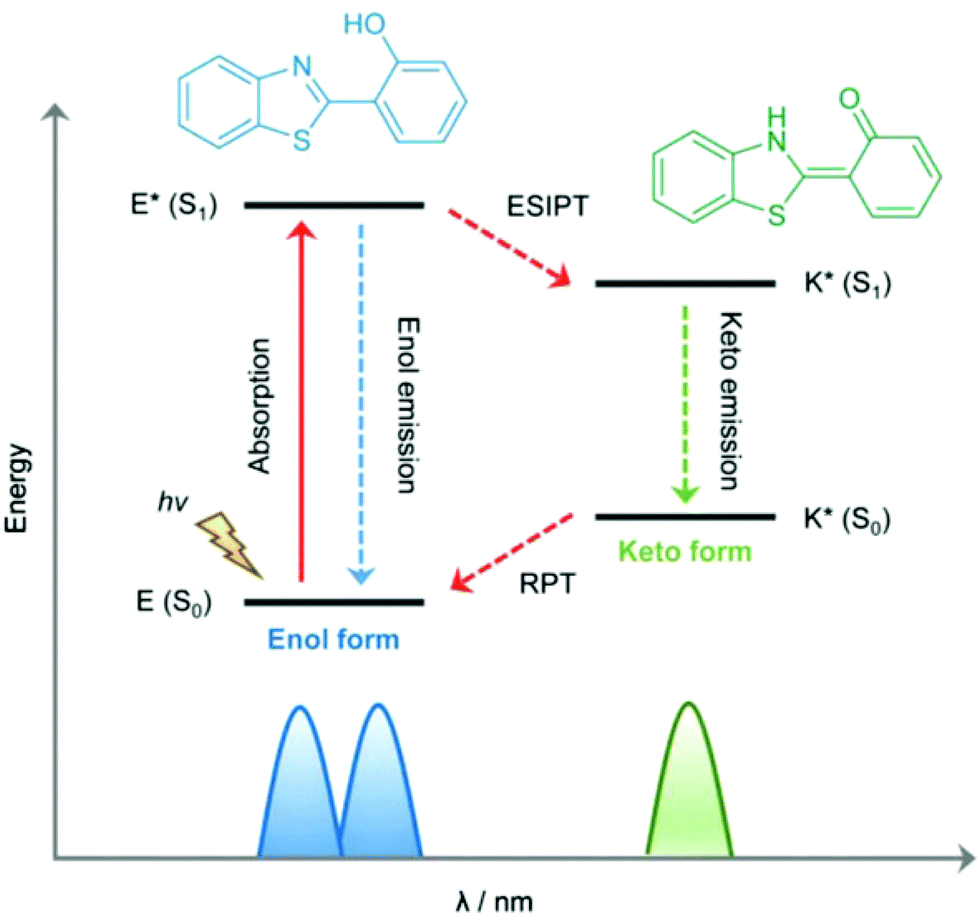 | ||
| Fig. 5 Mechanistic representation of excited state intramolecular proton transfer. Reproduced from Chem. Soc. Rev. 2018, 47, 8842–8880, with permission from the Royal Society of Chemistry. | ||
In 2017, Yan and co-workers developed TzDa, a highly crystalline COF with dual emissions to detect trace water in organic solvents.76 4,4′,4′′-(1,3,5-Triazine-2,4,6-triyl)trianiline (Tz) and 2,5-dihydroxyterephthalaldehyde (Da) were chosen as monomers, to incorporate both an intramolecular charge transfer (ICT) and an ESIPT moiety, respectively, into the polymeric system. The investigation of the effect of water in these two processes enabled the success of the ratiometric fluorescent sensor. The increase of water content was observed to lead to a higher fluorescence intensity of the longer wavelength emission and a reduction in intensity of the shorter wavelength emission. It was hypothesized that this result arose from the interference of water on the COF's intramolecular processes. The docking of a water molecule on the Tz moiety prevented the ICT process from the triazine to the phenyl group from taking place. Similarly, the interaction of water molecules with the –OH group on the Da moiety led to the interruption of intramolecular hydrogen bonding (Fig. 6). The synergetic effect resulted in the intensity changes of both emission peaks and ultimately the detection of trace water in organic solvents.
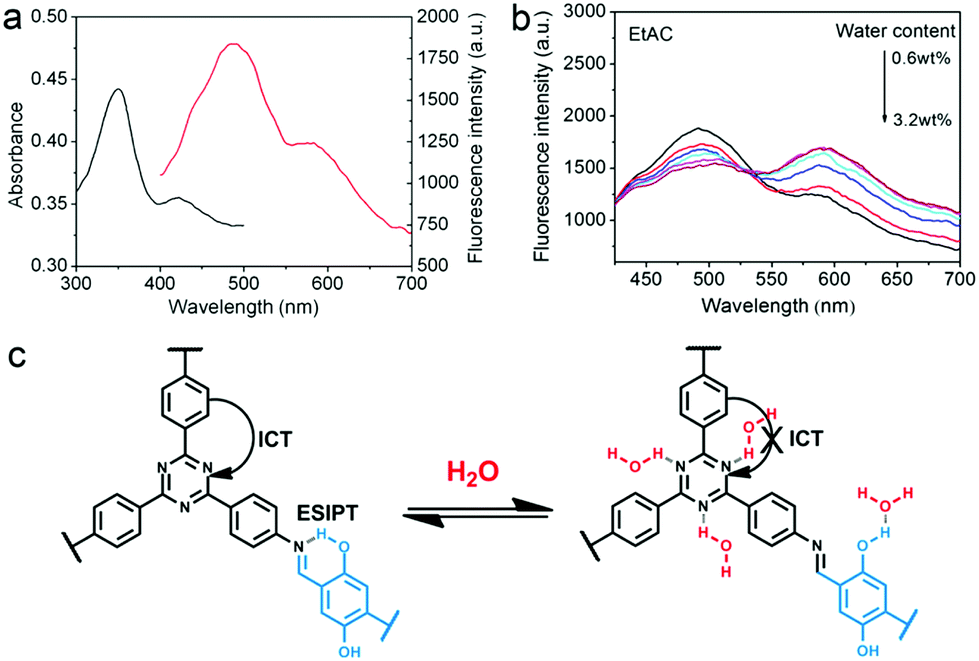 | ||
| Fig. 6 (a) Absorption (black) and fluorescence (red) spectra of TzDa in EtOAc. (b) Ratiometric fluorescence sensing of TzDa in EtOAc with varying water content. (c) Interruption of intramolecular hydrogen bonding in TzDa upon introduction of water. Reprinted with permission from ACS Appl. Mater. Interfaces, 2017, 9, 24999–25005. Copyright 2017 American Chemical Society. | ||
Vaidhyanathan and co-workers developed an anthracene-resorcinol-based COF that exhibited white-light emission.77 IISERP-COF7 was synthesized as a black powder that emitted white light when dispersed in N-methylpyrrolidone (NMP). Upon excitation at 340 nm, the emission spectrum exhibited three bands corresponding to blue, green, and red. It was hypothesized that the green and red light were both originating from the resorcinol trialdehyde unit. This unit exhibits yellow light under UV, which lies between red and green on the color map. Keto–enol tautomerization was expected between the resorcinol trialdehyde and anthracene unit due to the geometry between the two moieties connected via an imine bond. The color of the IISERP-COF7 could be manipulated through dispersion in different solvents. By utilizing O- or N-donating solvents, the intensity of the blue-green or yellow-orange band, respectively, could be increased. O-Donating solvents stabilized the enol form via hydrogen bonding, effectively increasing the intensity of the blue-green band. N-Donating solvents stabilized the keto form by acting as a base to assist in the transfer of the enol proton to the nitrogen, resulting in the increase in intensity of the yellow-orange band. NMP contains both an oxygen and basic nitrogen and was therefore found to stabilize both forms, resulting in near-white-light emission (Fig. 7). UV-vis spectra were taken of the IISERP-COF7 dispersed in aprotic toluene and protic ethanol. No new bands appeared due to any potential intermolecular proton transfer between solvent molecules and the COF, indicating the presence of an ESIPT process.
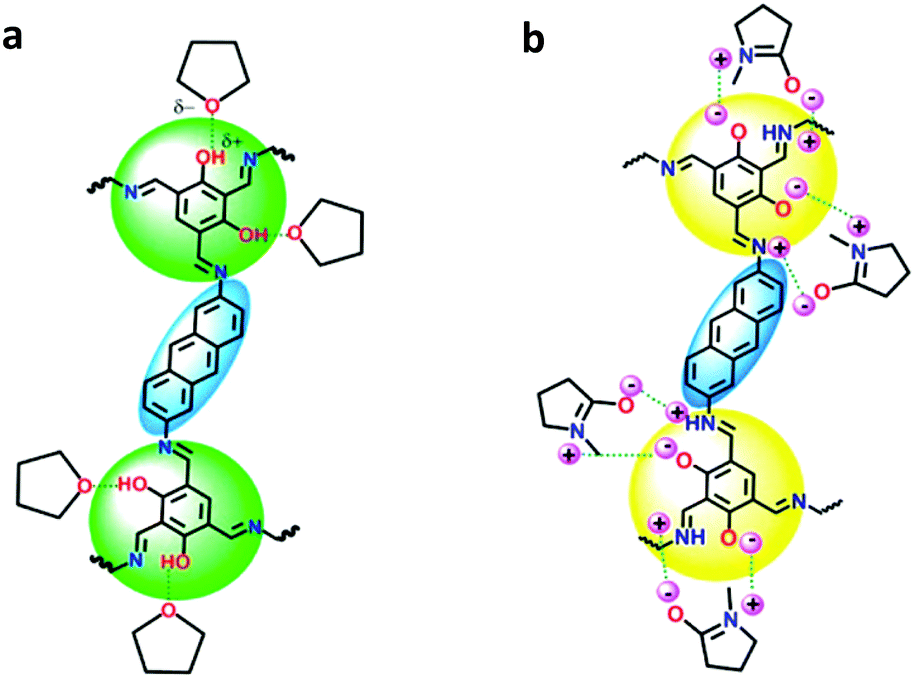 | ||
| Fig. 7 (a) Stabilization of the enol-form due to hydrogen bonding by THF. (b) Stabilization of the intermediate keto–enol form by the polarized NMP. Adapted with permission from J. Am. Chem. Soc., 2018, 140, 13367. Copyright 2018 American Chemical Society. | ||
3.3. Aggregation induced emission
In a highly fluorescent system, radiative decay outcompetes the many non-radiative pathways available. However, for some systems, steric and/or electronic interactions inhibit radiative decay while solvated. In some of these cases, it is possible to sequester the faster, non-radiative pathways via aggregation. This aggregation allows the radiative decay processes to compete effectively, and thus improves the emission quantum yield. This aggregate-induced emission (AIE) process can be attributed to various factors such as reduced intramolecular rotations (RIR), reduced intramolecular vibrations (RIV), and reduced intramolecular motions (RIM).78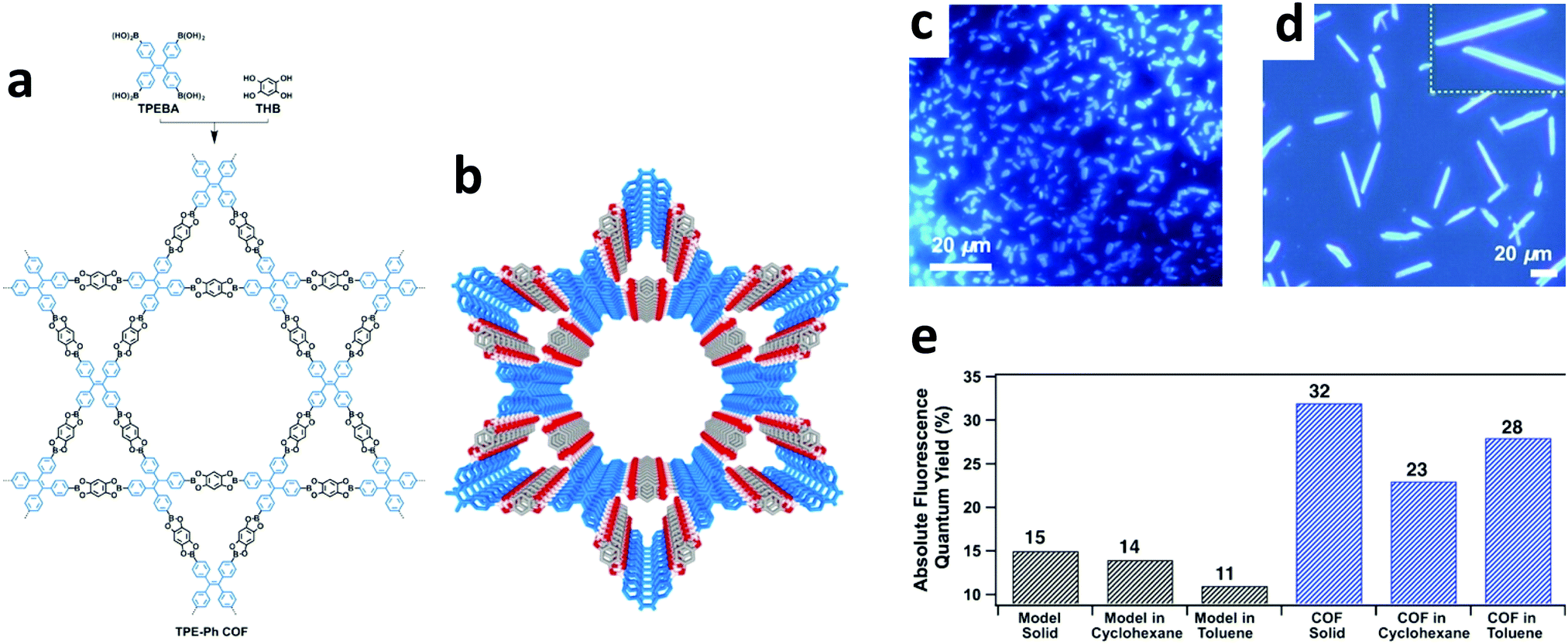 | ||
| Fig. 8 (a) Synthesis of TPE-Ph COF from the condensation of TPEBA and THB. (b) Crystal structure of TPE-Ph COF with AA stacking mode. Fluorescence microscopy images of TPE-Ph COF samples synthesized in (c) 3 days (d) 30 days (the yellow insert depicts an enlarged view of the belts). (e) Comparative fluorescence quantum yields of TPE-Ph COF and the model compound dispersed in cyclohexane and toluene, as well as in the solid state. Adapted with permission from J. Am. Chem. Soc., 2016, 138, 5797–5800. Copyright 2016 American Chemical Society. | ||
In 2018, Wang and co-workers synthesized a 3D imine-linked COF, 3D-TPE-COF, which adopted a 7-fold interpenetrated unit-cell.39 The COF's photoluminescence quantum yield (ΦPL = 20%) was markedly higher than its model compound (ΦPL = 6.6%), which was ascribed to both the rotational and vibrational relaxations being restricted when packed in the COF structure.
Zhao and co-workers investigated the AIE characteristics of TPE with regard to extended structures.80 When aniline was condensed with an aldehyde-functionalized TPE unit (TFBE) to form the small molecule TMBE, the fluorescence enhancement was greatly reduced compared to free TFBE. Specifically, when comparing their fluorescence intensities in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 H2O/THF mixture versus in pure THF, TFPE's enhancement ratio was ∼164; however, TMBE demonstrated only a 4.4-fold increase (Fig. 9b). This was attributed to TMBE's inability to planarize due to steric effects with its neighboring phenyl groups. By conformationally locking the COF system both laterally and vertically, it was thought that the AIE masking could be enhanced (Fig. 9c). Thus, azine-linked NUS-30, imine-linked NUS-32, and mixed imine/azine-linked NUS-31 were synthesized through condensation of an aldehyde-functionalized TPE with either hydrazine, 1,4-diaminobenzene, or a 1:1 ratio of each, respectively, which were then processed into nanosheets. All three polymeric systems showed almost complete AIE suppression with fluorescence enhancements of only 1.2-, 1.1-, and 1.5-fold for NUS-30, NUS-31, and NUS-32, respectively. Additionally, when suspended in 90:10 water/MeCN solutions, a bathochromic shift was observed for all three nanosheet systems, which was attributed to the π–π restacking of the nanosheets. This strongly corroborates with the findings of Jiang and co-workers79 that the arms of TPE can be effectively locked in the COF structure. In this case, however, the restricted planarization of TPE locked it in a conformation that prohibits enhanced fluorescence.
:10 H2O/THF mixture versus in pure THF, TFPE's enhancement ratio was ∼164; however, TMBE demonstrated only a 4.4-fold increase (Fig. 9b). This was attributed to TMBE's inability to planarize due to steric effects with its neighboring phenyl groups. By conformationally locking the COF system both laterally and vertically, it was thought that the AIE masking could be enhanced (Fig. 9c). Thus, azine-linked NUS-30, imine-linked NUS-32, and mixed imine/azine-linked NUS-31 were synthesized through condensation of an aldehyde-functionalized TPE with either hydrazine, 1,4-diaminobenzene, or a 1:1 ratio of each, respectively, which were then processed into nanosheets. All three polymeric systems showed almost complete AIE suppression with fluorescence enhancements of only 1.2-, 1.1-, and 1.5-fold for NUS-30, NUS-31, and NUS-32, respectively. Additionally, when suspended in 90:10 water/MeCN solutions, a bathochromic shift was observed for all three nanosheet systems, which was attributed to the π–π restacking of the nanosheets. This strongly corroborates with the findings of Jiang and co-workers79 that the arms of TPE can be effectively locked in the COF structure. In this case, however, the restricted planarization of TPE locked it in a conformation that prohibits enhanced fluorescence.
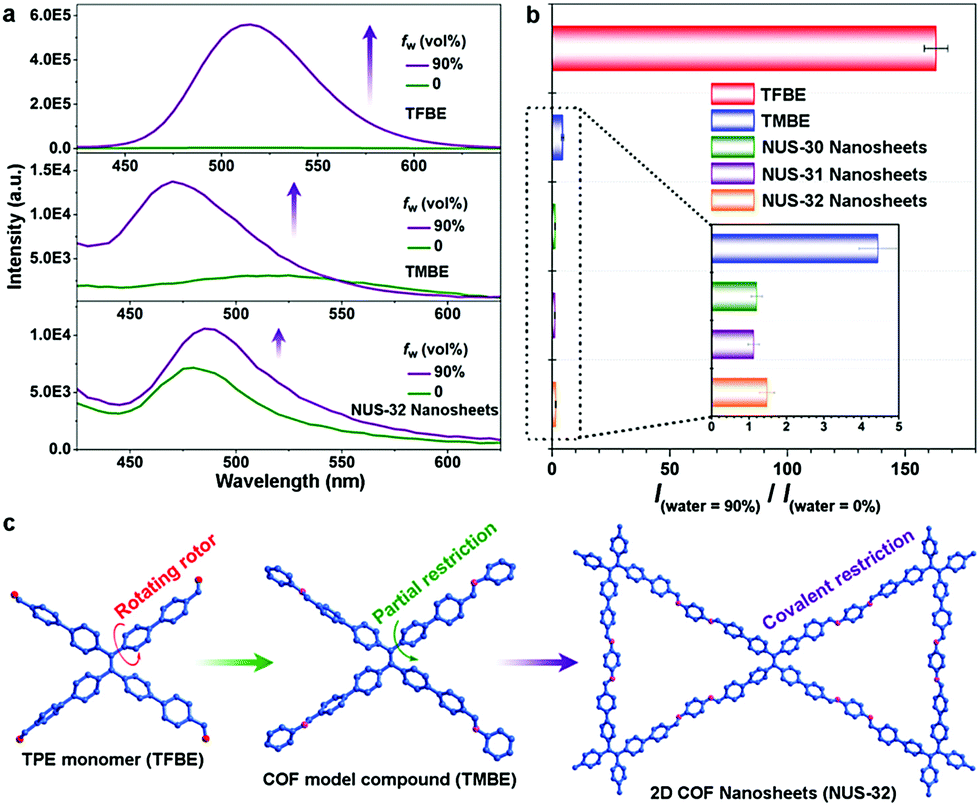 | ||
| Fig. 9 (a) Fluorescence spectra (λex = 365 nm) of TFBE, TMBE, and NUS-32 in dry solvent or 90% water fractions. (b) Relative fluorescence intensities of TFBE, TMBE, and NUS 30–32 in solvent/water mixtures compared to dry solvent [I(water = 90%)/I(water = 0%)]. (c) Representation of the increasing restriction of AIE molecular rotor from monomer (TFBE) to COF model compound (TMBE) to 2D COF nanosheets (NUS-32). Reprinted with permission from Chem. Mater., 2019, 31, 146–160. Copyright 2019 American Chemical Society. | ||
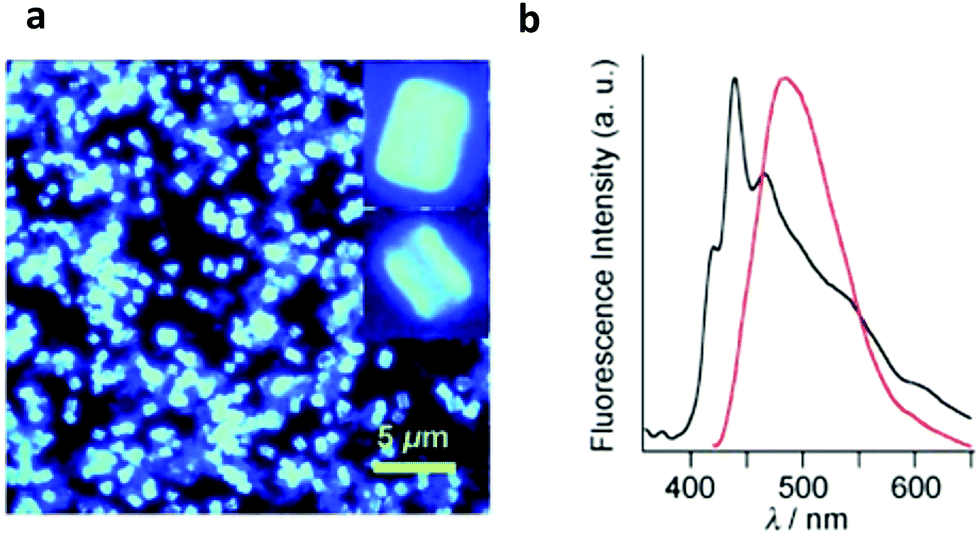 | ||
| Fig. 10 (a) Fluorescence microscopic image of PPy-COF (the inset depicts an enlarged image of individual cubes). (b) Normalized fluorescence spectra (λex = 414 nm, 25 °C) of PDBA (black curve) and PPy-COF (red curve). Adapted with permission from Angew. Chem., Int. Ed., 2009, 48, 5439–5442. | ||
In 2016, a variety of imine-linked, pyrene-containing COFs were synthesized by condensing 1,3,6,8-tetrakis(4-aminophenyl)pyrene with a variety of linear dialdehydes.83 The synchronization effect of the twisted phenyl substituents led to a highly crystalline COF whose intra- and interlayer electronics could be tuned based on the dialdehyde linkers employed. Despite exhibiting poor photoluminescence quantum yields (ΦPL < 1%), it was revealed that the COF systems were capable of delocalizing excitations along the pyrene stacks and that interlayer charge-transfer excitations via the imine bonds were taking place.
In 2016, three dehydrobenzoannulene (DBA)-containing COF systems were synthesized by McGrier and co-workers via co-condensation between PDBA and either DBA[12], DBA[18], or a 1:1 ratio of each to give Py-DBA-COF 1, Py-DBA-COF 2, or Py-MV-DBA-COF, respectively (Fig. 11).84 All three systems showed strong fluorescence due to AIE from the DBA monomers. The fluorescence emission for Py-DBA-COF 2 (λem = 483 nm) was notably higher in energy than that of Py-DBA-COF 1 (λem = 530 nm). This was attributed to the symmetry-forbidden nature of excitations for the DBA[12] vertex in Py-DBA-COF 1. Interestingly, the mixed vertex variant, Py-MV-DBA-COF, also exhibited a redshifted (λem = 528 nm) emission spectrum, indicating that DBA[12] primarily dominated its excited-state properties (Fig. 12).
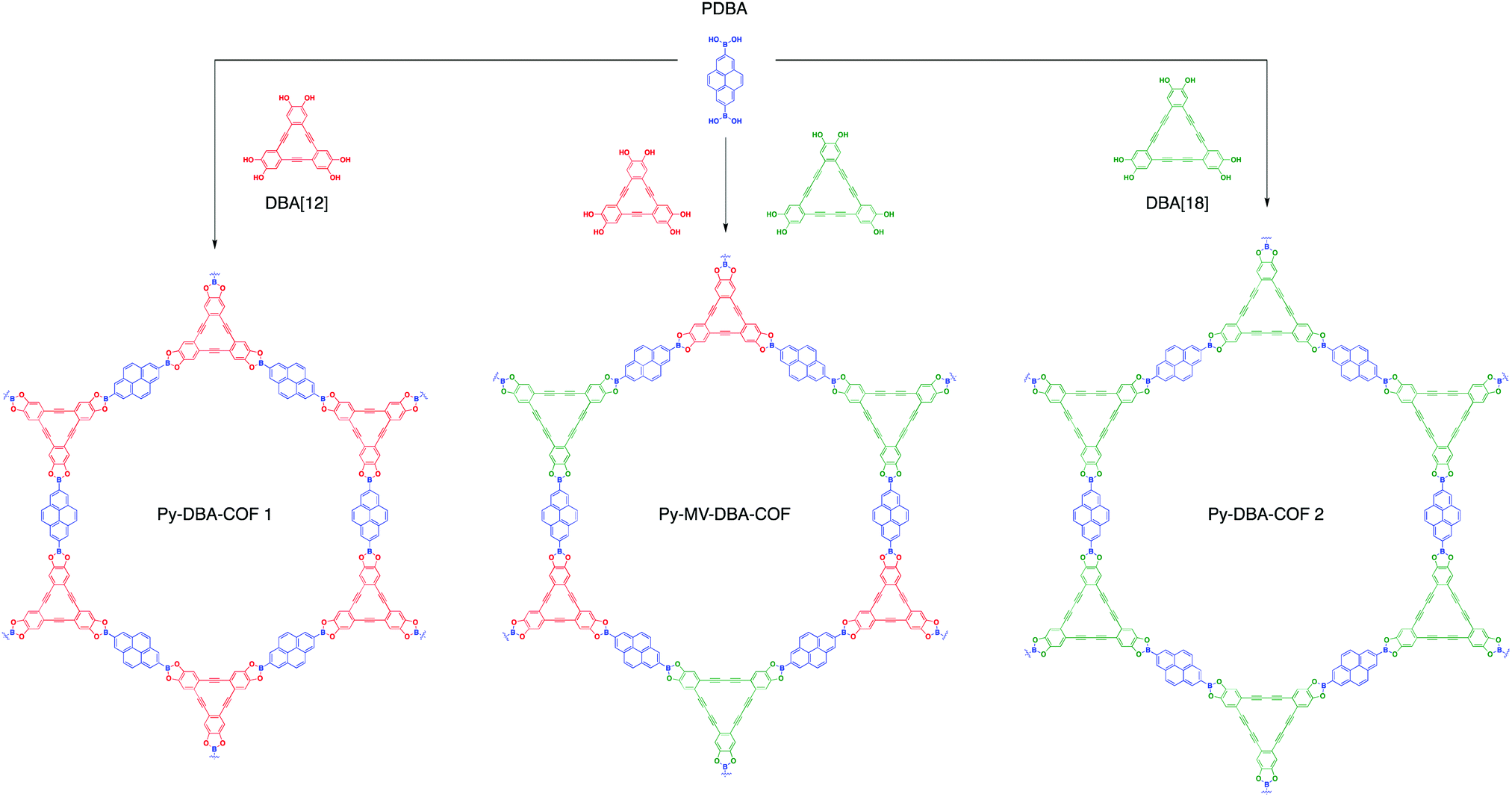 | ||
| Fig. 11 Synthesis of Py-DBA-COF 1, Py-MV-DBA-COF, and Py-DBA-COF 2. | ||
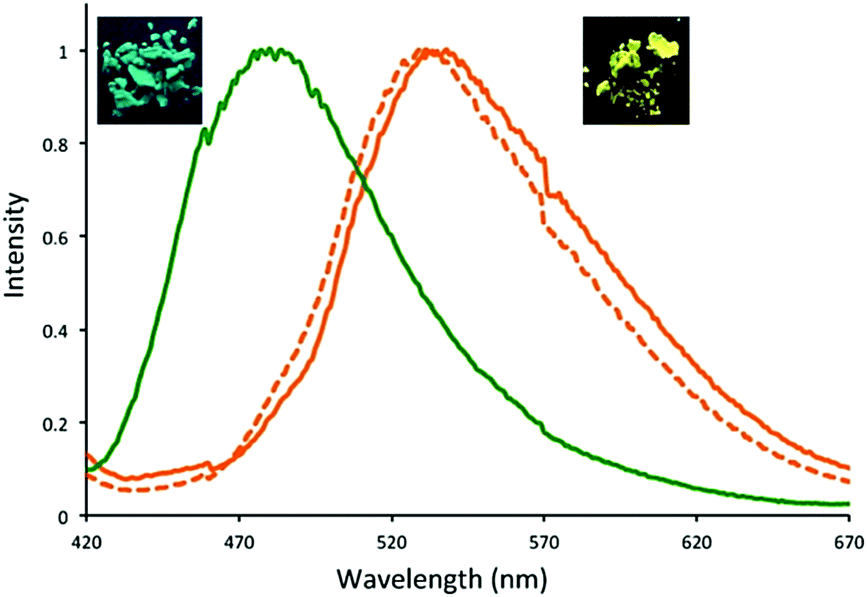 | ||
| Fig. 12 Normalized emission spectra of Py-DBA-COF 1 (orange, λex = 350 nm), Py-DBA-COF 2 (green, λex = 365 nm), and Py-MV-DBA-COF (dashed orange, λex = 350 nm). Photographs of the fluorescent solids were taken using a hand-held UV-lamp at 365 nm. Reprinted with permission from J. Am. Chem. Soc. 2016, 138, 10120–10123. Copyright 2016 American Chemical Society. | ||
In 2018, Lotsch and co-workers85 synthesized three azine-linked COFs by condensing hydrazine with 1,3,6,8-tetrakis(4-ethynylbenzaldehyde)-pyrene (TEBPY), 1,3,6,8-tetrakis(6-ethynylnicotinaldehyde)-pyrene (TENPY), and 1,3,6,8-tetrakis(2-ethynylpyrimidin-5-carb-aldehyde)pyrene (TEPPY) to form A-TEBPY-COF, A-TENPY-COF, and A-TEPPY-COF, respectively (Fig. 13). According to DFT calculations, the addition of alkynes between the porphyrin and phenyl units allowed for improved planarization of the overall monomers. As a result, all three COF systems exhibited a reduced interlayer stacking distance (3.43 Å) compared to previously reported, less planar systems (3.9–4.33 Å).86,87 The increased planarity also led to the formation of both in-plane extended conjugation and out-of-plane excimers, which allowed the system to emit a broad low energy emission band (λem = 670–700 nm).
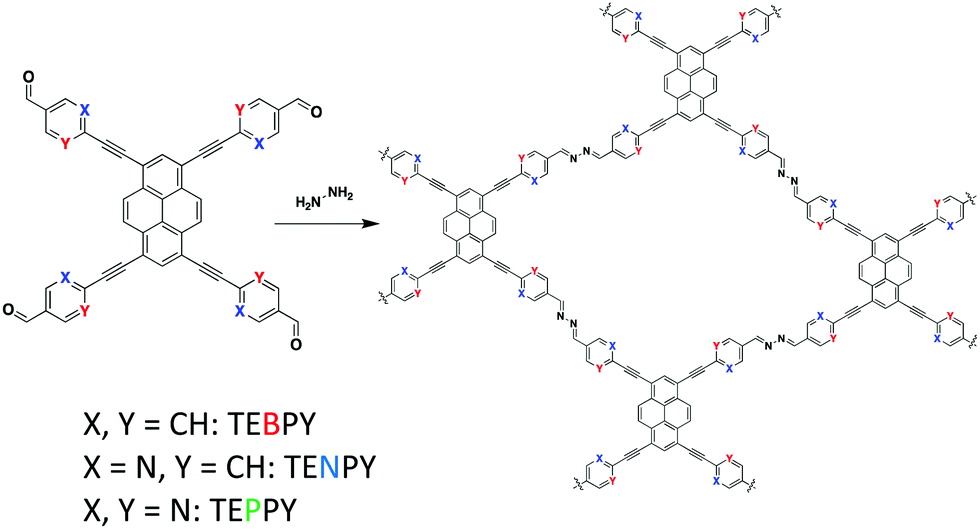 | ||
| Fig. 13 The acetic acid catalyzed condensation of hydrazine and pyrene-based aldehyde linkers to form various azine-linked COFs. | ||
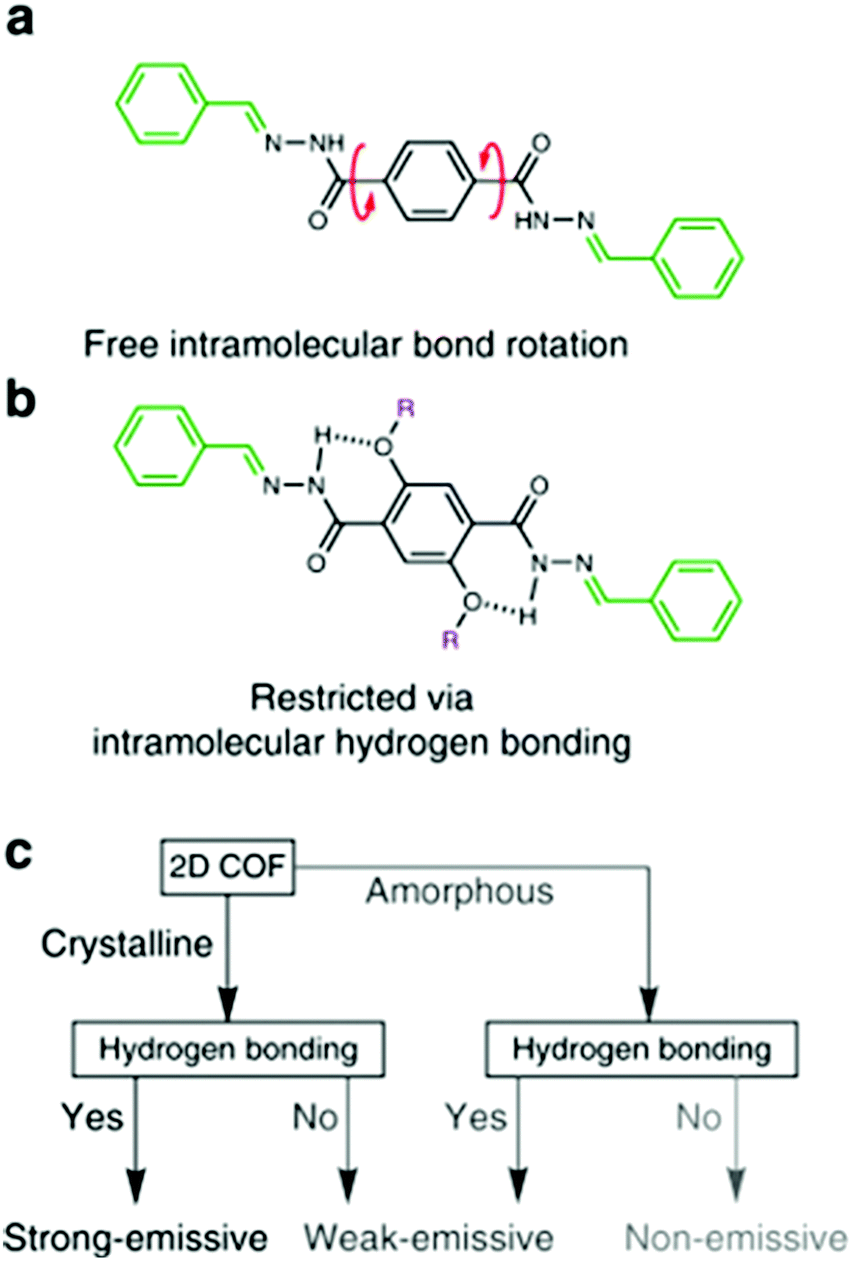 | ||
| Fig. 14 (a) Free intramolecular rotation in the amorphous solid state makes it non-emissive. (b) Intramolecular hydrogen bonding restricts rotation, causing enhanced emission in the UV to violet region. (c) Flow chart depicting the strategy to achieve strong solid state emission in 2D COFs. Adapted with permission from Nat. Commun., 2018, 9, 233. | ||
4. Exploiting the photophysical properties of COFs for practical applications
4.1. Chemical sensing
One application that luminescent COFs are particularly suited for is chemical sensing. The porous nature of COFs allows potential analytes to diffuse through the pore channels. This property not only allows for molecular recognition, but also the potential removal of analytes. Furthermore, the modular nature of COFs allows for the design of polymeric materials that are useful for chemical sensing. Explosives and metal cations have both been popular analytes in the field of COF-based chemical sensing. Several groups have also investigated the detection of other analytes such as ammonia, chiral molecules, and hydroxyl radicals.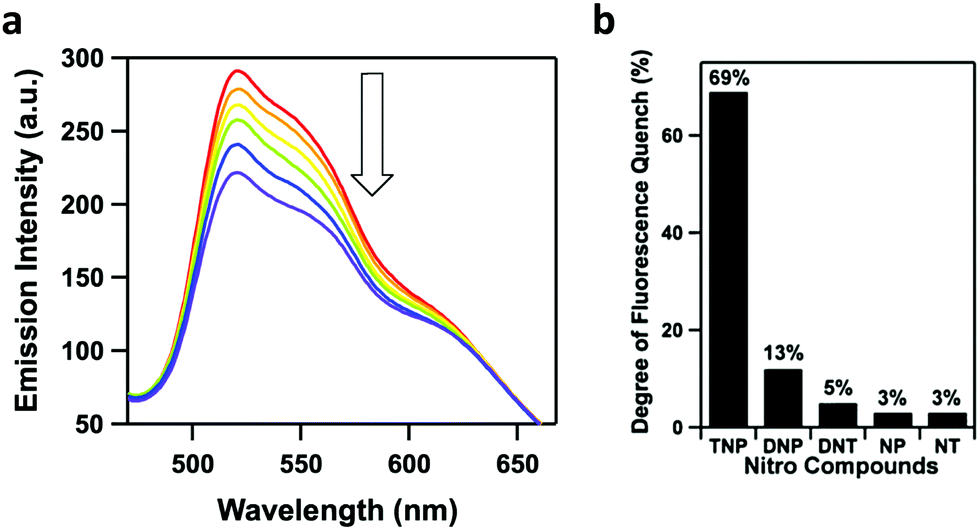 | ||
| Fig. 15 (a) Fluorescence quenching of Py-azine COF in MeCN (0.01 mg mL−1) upon exposure to TNP vapors for various times (0–70 min). (b) Degree of fluorescence quenching after the addition of various nitro compounds (70 ppm). Adapted with permission from J. Am. Chem. Soc. 2013, 135, 17310–17313. Copyright 2013 American Chemical Society. | ||
In 2018, Zhao and co-workers developed two imine-linked, heteroporous, fluorescent COFs that exhibited fluorescence quenching in the presence of small quantities of TNP.90 The two COFs were designed by the condensation of a C2v symmetric building block with a linear linker yielding COF-BABD-BD and COF-BABD-BZ. Powder X-ray diffraction (PXRD) and pore size distribution analysis was employed to confirm the AB stacking pattern of the 2D COF layers. It was hypothesized that the slip-stacked arrangement may have reduced any potential aggregate caused quenching (ACQ). During the sensing experiments, a nitroaromatic compound (TNP, DNP, DNT, 1,3-dinitrobenzene, p-nitrotoluene, or nitrobenzene) was slowly introduced into a suspension of the COF in acetonitrile. TNP induced significant fluorescence quenching of each COF while other nitroaromatic compounds had no significant effect on the fluorescence of the COFs (Fig. 16a–c) indicating that they may be used as effective chemical sensors for the presence of TNP with good selectivity and sensitivity. The acetonitrile suspensions changed color upon the introduction of TNP from golden yellow to dark brown indicating the material could be used as visual chemosensors with the naked eye (Fig. 16d). Stern–Volmer quenching constants (KSV) were calculated to quantify the sensitivity of the COFs towards different nitroaromatic compounds resulting in KSV values of 5.7 × 105 M−1 and 4.5 × 105 M−1 for COF-BABD-BD and COF-BABD-BZ, respectively, for TNP. These constants are ten to one hundred times larger than for the other tested nitroaromatic compounds and support the observation that the COFs are highly selective towards TNP sensing.
 | ||
| Fig. 16 Fluorescence spectra (λex = 370 nm) of (a) COF-BABD-DB and (b) COF-BABD-BZ dispersed in MeCN with the addition of varying amounts of TNP. (c) Fluorescence quenching (%) of COF-BABD-BZ with different nitroaromatics (12.5 μM). (d) Photographs of COF-BABD-DB dispersed in MeCN with the addition of various nitroaromatics (4.0 μM). Adapted from Chem. Commun., 2018, 54, 2308–2311, with permission from the Royal Society of Chemistry. | ||
A photoluminescent TPE-based imine-linked COF was reported by the Loh and co-workers in 2018.91 This COF was also utilized to sense TNP. 1,3,6,8-Tetrakis(4-aminophenyl)-pyrene and 1,1,2,2-tetrakis(4-formylphenyl)ethane were chosen as the building blocks due to their fluorescent and AIE properties yielding Py-TPE-COF. Transmission electron microscopy (TEM) images showed that Py-TPE-COF crystallized as sphere-like nanoparticles with domain sizes of 300–500 nm. The nanoparticles displayed solvatochromism when dispersed in THF, acetone, acetonitrile, or DMF, emitting either a bright blue or blue-green luminescence with PL bands centered at 462, 469, 474, and 493 nm, respectively (Fig. 17). The fluorescence was attributed to the use of the nonplanar TPE groups that should reduce π–π interactions, prevent ACQ, and thus enhance PL. Furthermore, as spherical COF nanoparticles, a curvature-induced strain was introduced, that may have acted to prevent any rotations in the imine linkages. Fluorescence quenching experiments with TNP were performed in acetone. An addition of 1 ppm of TNP solution caused a sharp decrease in fluorescence of the COF nanoparticles. Several other nitroaromatic compounds were tested to investigate the selectivity of the fluorescence quenching towards TNP including DNP, DNT, NT, and NP. Only small decreases in PL were measured indicating Py-TPE-COF could be used as a chemical sensor selective for TNP detection. It was suggested that the fluorescence quenching may result from the hydroxy group on TNP hydrogen-bonding with a nitrogen on the pore walls of the COF. This non-emissive complex could then trap the excitation energy, resulting in static fluorescence quenching.
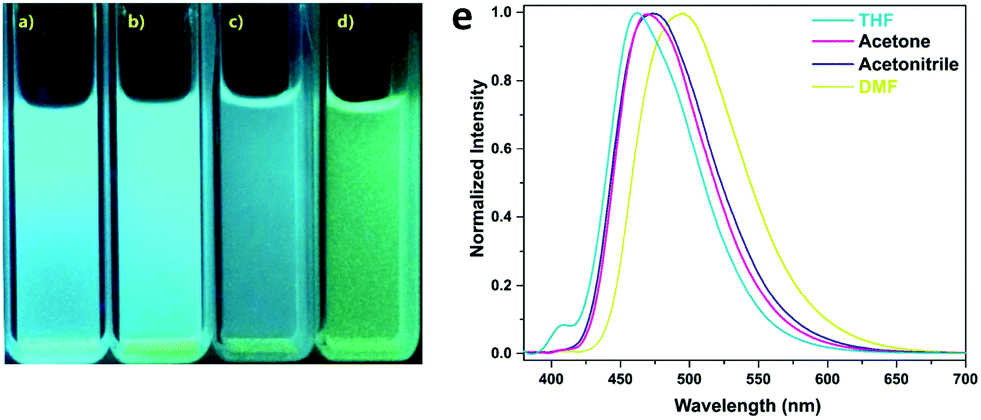 | ||
| Fig. 17 Photographs of Py-TPE COF dispersed in (a) THF, (b) acetone, (c) acetonitrile, and (d) DMF, while illuminated by UV-light (365 nm). (e) Normalized fluorescence spectra (λex = 365 nm) of Py-TPE-COF dispersed in different solvents. Adapted from Chem. Commun., 2018, 54, 2349–2352, with permission from the Royal Society of Chemistry. | ||
Wang and co-workers used a luminescent 3D COF (3D-Py-COF) for explosives detection (Fig. 18).92 Tetra(p-aminophenyl)methane and 1,3,6,8-tetrakis(4-formylphenyl) pyrene were chosen as the tetrahedral (3D-Td) and rectangular (2D-C2) building blocks, respectively. Their condensation yielded an imine-linked 3D-COF with a pts topology that displayed a yellow-green luminescence. TNP was employed to investigate the chemical sensing properties of the 3D COF. TNP was gradually added to a suspension of 3D-Py-COF in DMF. The COF exhibited 75% fluorescence quenching after the addition of 20 ppm of TNP with a quenching constant (KSV) of 3.1 × 104 M−1. The fluorescence quenching is most likely due to an electron transfer process. The energy levels of the HOMOs and LUMOs of 3D-Py-COF and TNP were investigated using the DFTB-D method. Results indicated that the electron transfer from the HOMO of 3D-Py-COF to the LUMO of TNP is facilitated upon photoexcitation.
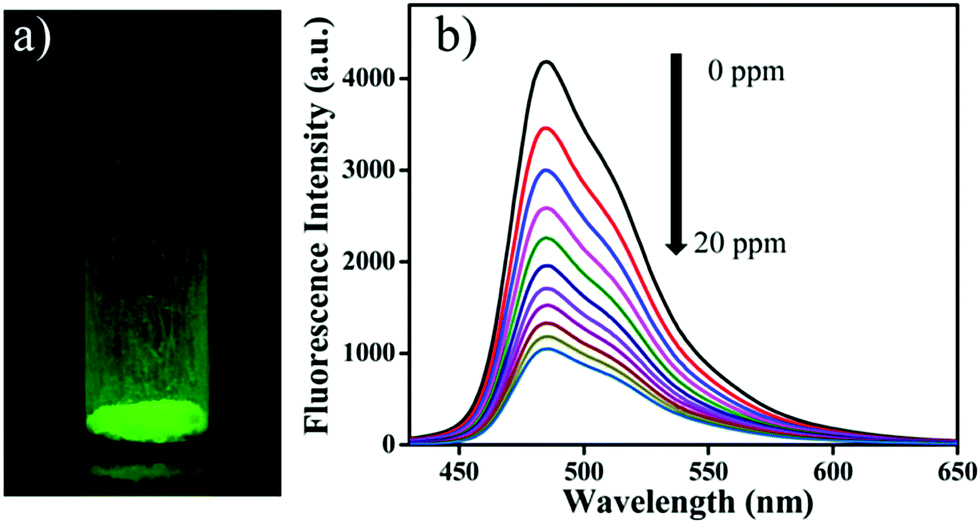 | ||
| Fig. 18 (a) Photograph of 3D-Py-COF powder under UV light irradiation. (b) Fluorescence quenching of 3D-Py-COF in DMF upon addition of TNP (0–20 ppm). Reprinted with permission from J. Am. Chem. Soc., 2016, 138, 3302–3305. Copyright 2016 American Chemical Society. | ||
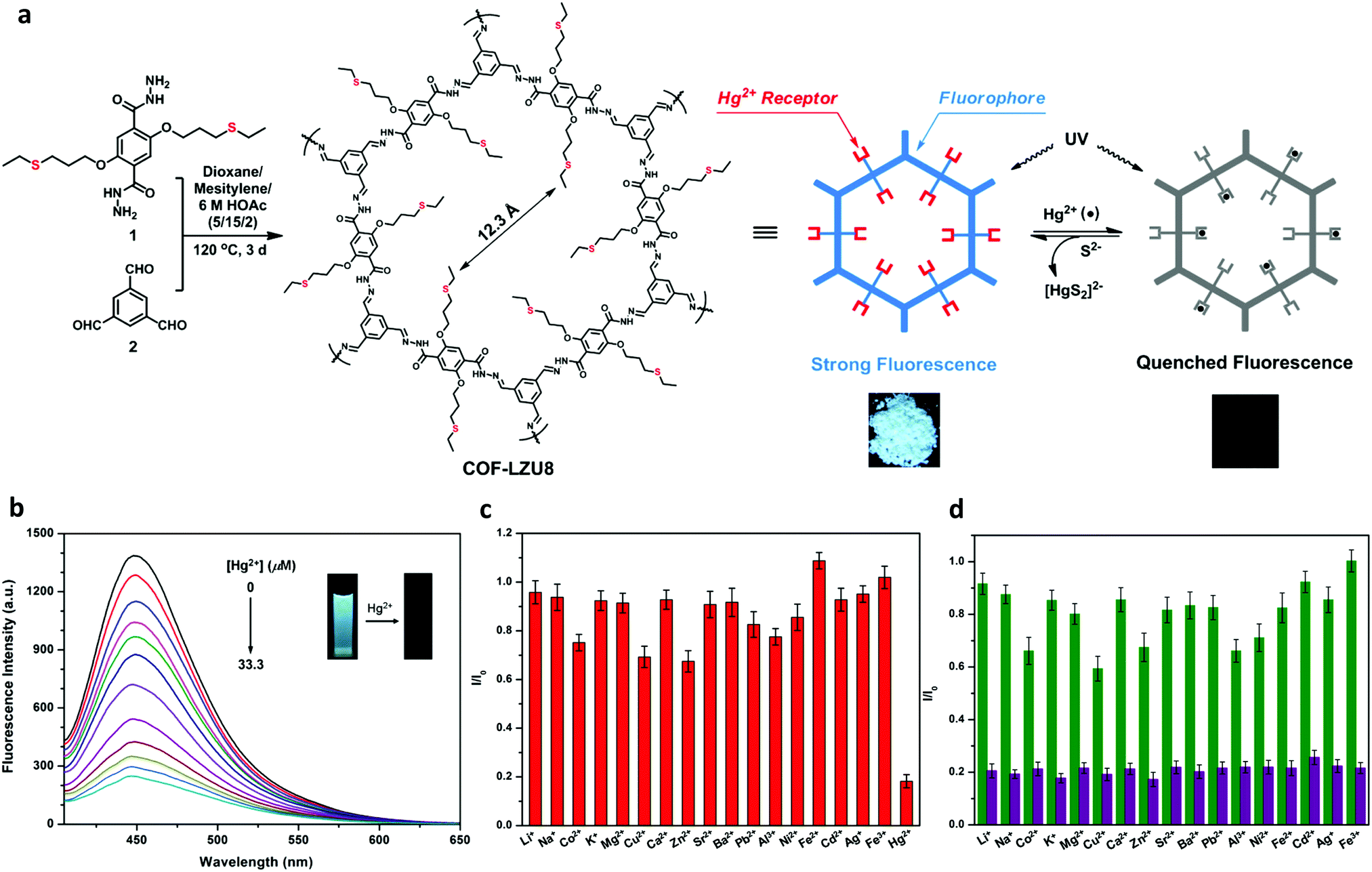 | ||
| Fig. 19 (a) Synthesis of COF-LZU8 and the proposed mechanism for fluorescence quenching by Hg2+ (photographs taken of COF-LZU8 under UV lamp, 365 nm). (b) Fluorescence quenching of COF-LZU8 dispersed in MeCN upon the addition of Hg2+ (λex = 390 nm). (c) Fluorescence of COF-LZU8 with different metal ions in MeCN (λex = 390 nm). (d) Competition experiment of the emission intensity of COF-LZU8 in the presence of competitive ions (2.0 equiv., green bars) and the resulting intensity upon the addition of Hg2+ (1.0 equiv., purple bars). Adapted with permission from J. Am. Chem. Soc., 2016, 138, 3031–3037. Copyright 2016 American Chemical Society. | ||
Liu and co-workers constructed a hydrogen bond assisted azine-linked COF (COF-JLU3) for the sensing of Cu2+ cations.37 1,3,5-tris(3′-tert-butyl-4′-hydroxy-5′-formylphenyl)benzene and hydrazine hydrate were used as the building blocks. The inclusion of –OH groups on the trialdehyde monomer allowed for an intramolecular hydrogen-bonding interaction with a nitrogen from the nearby azine linkage, which imparted enhanced crystallinity and chemical stability (Fig. 20a). The tert-butyl groups were shown to impact the π–π interactions between the 2D layers, effectively enhancing the luminescence of the material. The redshift in the absorption band of COF-JLU3 in comparison to the monomers indicated the role of extended conjugation in the material's fluorescent properties. These structural characteristics were possibly responsible for the sensing of Cu2+. For the sensing experiments, the COF was dispersed in THF before the introduction of M(NO3)x with a variety of metal ions (Li+, Na+, K+, Mg2+, Ca2+, Ba2+, Zn2+, Cd2+, Ni2+, Pb2+, Co2+, Cu2+, Ag+, Fe3+, Al3+) at 2 × 10−1 mol L−1. It was observed that alkaline, alkaline-earth, and transition metal ions with filled d shells had little effect on the luminescence intensity of the COF. Metal ions with different electronic configurations such as Fe3+, Cu2+, and Co2+ exhibited a larger influence on luminescence of the COF. Cu2+ caused the largest change. After the addition of two equivalents of the Cu2+ ion, COF-JLU3 retained only 18.3% of its original luminescence intensity. XPS measurements indicated Cu2+ ions were successfully immobilized in the COF pore which contained multiple binding sites such as the hydroxyl groups and nitrogen atoms on the pore wall. The binding of the metal leads to the fluorescence quenching of the system via a photoinduced electron transfer (PET) process. Interference studies were employed with competing cations and different counterions were also investigated. Ultimately, it was determined that the addition of other cations and the nature of the anion had no significant effect on the luminescence of COF-JLU3. Recyclability studies showed that fluorescence could be regained after the removal of Cu2+ ions and quench again upon reintroduction (Fig. 20b).
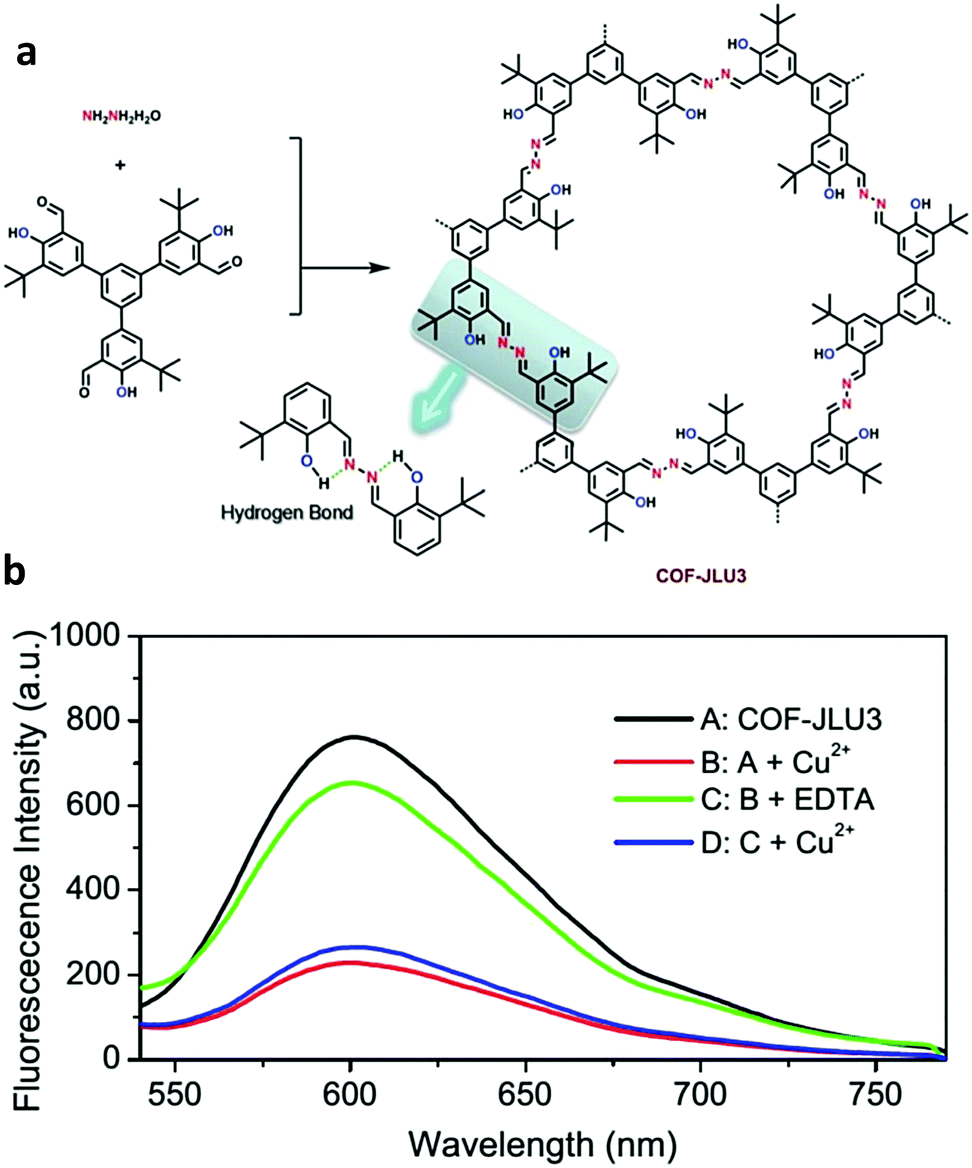 | ||
| Fig. 20 (a) Synthesis of COF-JLU3 depicting the intramolecular hydrogen bonding. (b) Fluorescence spectra of COF-JLU3 demonstrating its effective reversible quenching by Cu2+ ions. Adapted from Chem. Commun., 2016, 52, 6613, with permission from the Royal Society of Chemistry. | ||
A series of four 2D imine-linked COFs were investigated for their Fe3+ sensing abilities by Yan and co-workers.94 1,3,5-Tri-(4-aminophenyl) benzene (TaPb), terephthaldehyde (TpTa), 1,3,5-tri-(4-aminophenyl) triazine (Tz) and 2,5-dihydroxyterephthalaldehyde (Da) were employed as the building blocks to obtain the four COFs (TaTa, DhaTab, TRITER-1 and TzDa). Multiple metal cations were utilized to probe the possibility of metal ion detection (Na+, Mg2+, K+, Ca2+, Cu2+, Zn2+, Pb2+, Ag+, Al3+, Fe3+, and Cd2+). Of these only Fe3+ showed a significant effect on the fluorescence quenching of each COF. The KSV values for COFs TaTa, DhaTab, TRITER-1 and TzDa were determined to be 1.4 × 104, 1.0 × 105, 5.6 × 103, and 3.3 × 103 M−1, respectively. It was suggested that this fluorescence quenching was caused by absorption competition quenching. In contrast to the other metal cations tested, Fe3+ had a UV-vis absorption from 250–400 nm. This absorption overlapped with those of the COFs. The order of increasing overlap was DhaTab > TaTa > TRITER-1 > TzDa, which matched the pattern of the quenching constants indicating this could very likely have been the source of fluorescence quenching.
Zhang and co-workers sought to develop a COF that could detect Fe3+ by exploiting the fact that Fe3+ will chelate to the O,N,O′ moiety of salicylaldehyde benzoyl hydrazone (SBH) to form a strong coordination complex (Fig. 21).95 O,N,O′-Chelating units were introduced into a COF structure by employing benzene-1,3,5-tricarbohydrazide (Bth) with 2,5-dihydroxyterephthalaldehyde (Dha) or 2,5-dimethoxyterephthalaldehyde as building units yielding COFs Bth-Dha and Bth-Dma, respectively. Bth-Dma displayed a strong fluorescence, attributed to restricted bond rotation, therefore, this COF was utilized in Fe3+ detection experiments. The metal sensing experiments were carried out by introduction of chloride salts at 100 μM to Bth-Dma suspensions. The addition of Na+, K+, Mg2+, Ba2+, Mn2+, Ca2+, Gd3+, or Al3+ to the COF suspension led to almost no change in luminescence intensity while the addition of Zn2+, Ni2+, Co2+, La3+, Nd3+, Fe2+, or Cu2+ led to a marginal decrease in luminescence. However, the introduction of Fe3+ led to a significant quenching of luminescence. A KSV of 2.3 × 104 M−1 was obtained from Stern–Volmer plots with a detection limit of 0.17 μM. Changes in pH and anion had no effect on the fluorescence quenching indicating Bth-Dma is selective towards Fe3+. The fluorescence quenching was thought to arise from energy or electron transfer that occurs after the O,N,O′-Fe3+ coordination with excited states involving d-orbital electrons. This binding event was supported by XPS measurements after soaking Bth-Dma in FeCl3 for two days.
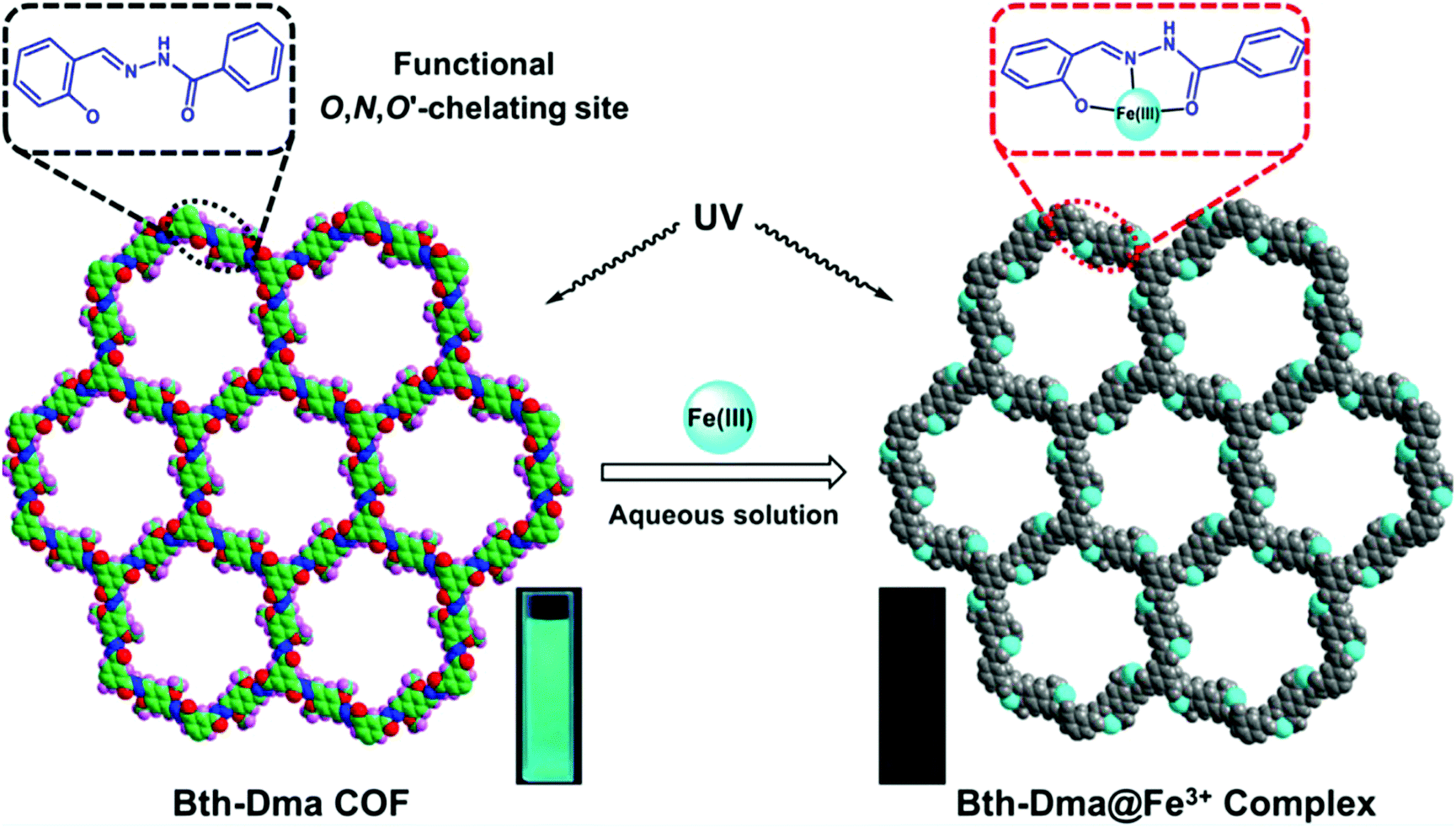 | ||
| Fig. 21 Structure of Bth-Dma COF with and without the addition of Fe3+. Inset photographs show the fluorescence quenching upon the formation of Bth-Dma@Fe3+ complex. Reprinted with permission from ACS Appl. Mater. Interfaces, 2019, 11, 12830–12837. Copyright 2019 American Chemical Society. | ||
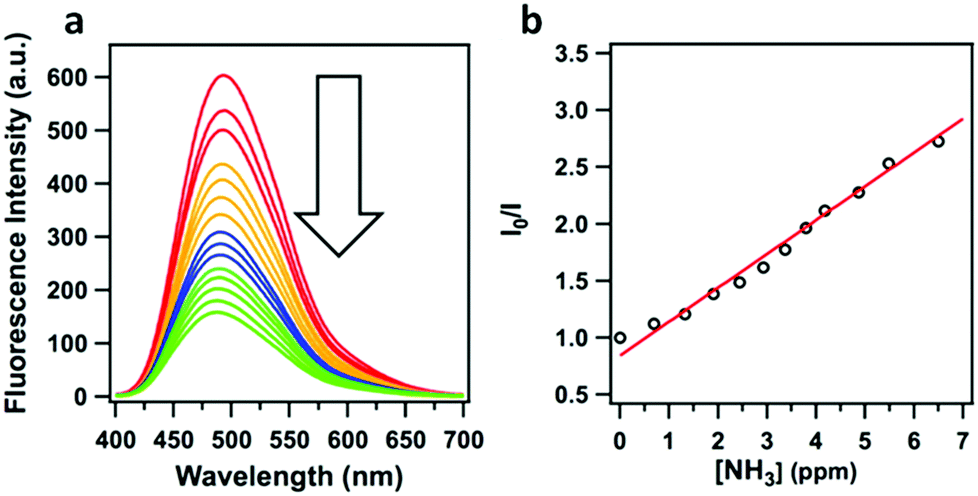 | ||
| Fig. 22 (a) Fluorescence quenching of TPE-Ph COF upon the addition of ammonia. (b) Stern–Volmer plot of the ammonia driven fluorescence quenching of TPE-Ph COF. Adapted with permission from J. Am. Chem. Soc., 2016, 138, 5797–5800. Copyright 2016 American Chemical Society. | ||
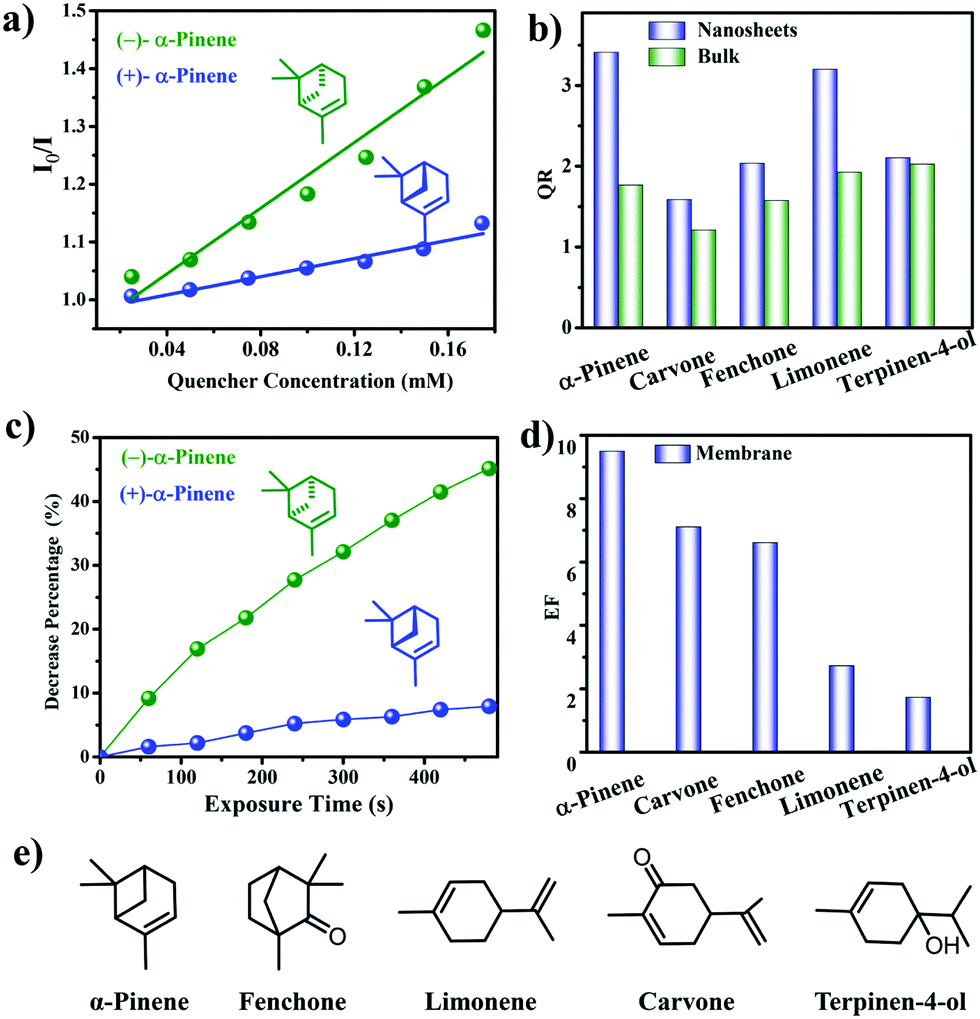 | ||
| Fig. 23 (a) Stern–Volmer plots of 7-NS in MeCN after titration of α-pinene. (b) Enantioselective quenching ratios for various terpenes. (c) Decreased fluorescence (%) of 7@PVDF upon introduction of α-pinene. (d) Enantioselective quenching ratios for various terpene vapors. (e) Structures of terpenes used. Reprinted with permission from J. Am. Chem. Soc., 2019, 141, 7081–7089. Copyright 2019 American Chemical Society. | ||
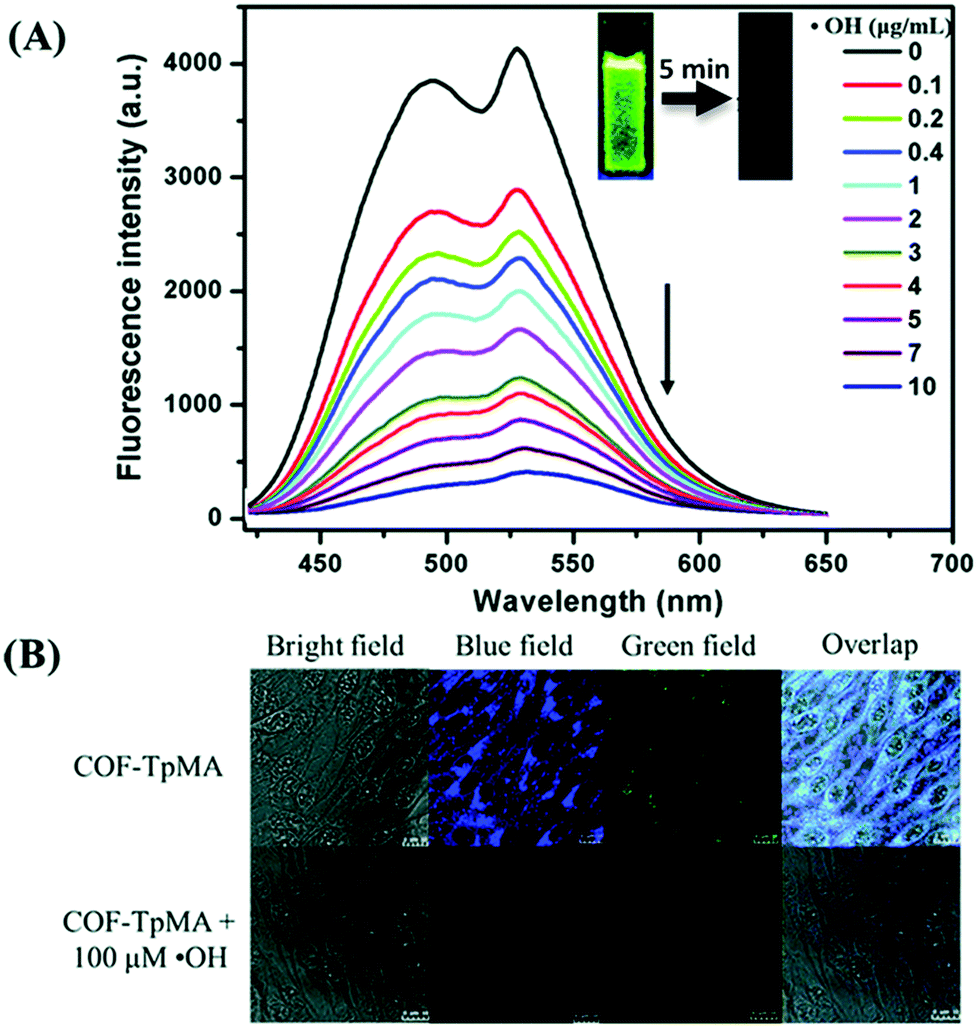 | ||
| Fig. 24 (A) Fluorescence quenching of COF-TpMA upon the addition of ˙OH. (B) Confocal laser scanning microscopy images of BHK cells treated with COF-TpMA (top) and pretreated with COF-TpMA and then treated with ˙OH (bottom). Scale bar = 10 μm. Reproduced from Chem. Commun., 2019, 55, 167–170, with permission from the Royal Society of Chemistry. | ||
4.2. Photocatalysis
The utilization of COFs as photocatalysts has seen significant interest. The insolubility and stability of COF frameworks allows for the facile use as heterogeneous catalysts, while the modular nature of their design allows for the precise integration of functional building units. Due to the large number of works in this area, this section will highlight recent progress in a few of the investigated transformations.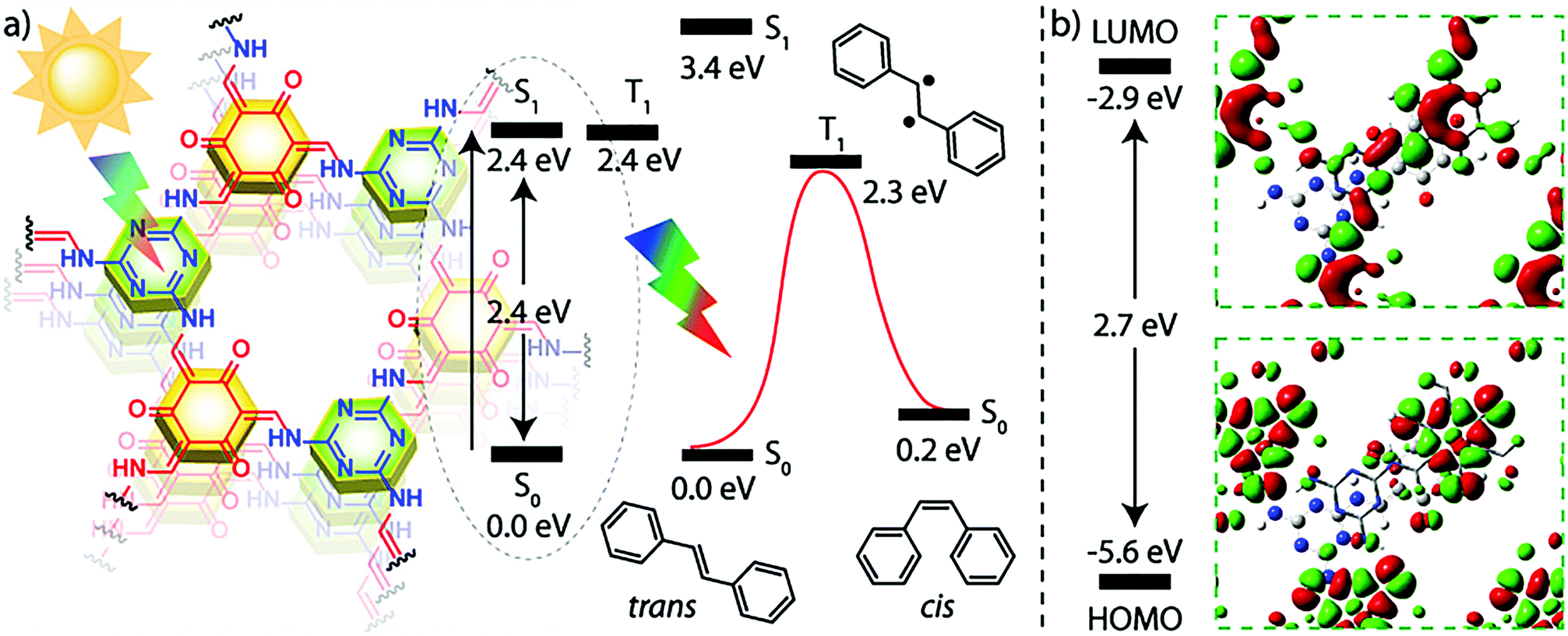 | ||
| Fig. 25 (a) Representation of the proposed mechanism for the TpTt COF photocatalyzed E–Z isomerization of stilbene. (b) Graphic representation of the HOMO and LUMO orbitals of TpTt and their energy levels (the orbital energy levels are not to scale). Reprinted with permission from J. Am. Chem. Soc., 2019, 141, 6152–6156. Copyright 2019 American Chemical Society. | ||
In 2018, Bein and co-workers reported the utilization of a COF as a photocathode for light-driven water electrolysis.59 The synthesized BDT–ETTA COF combined the geometric, crystalline, and interlayer stacking influences of 1,1′,2,2′-tetra-p-aminophenylethylene (ETTA)100 with benzo[1,2-b:4,5-b′]dithiophene-2,6-dicaroxaldehyde (BDT), a donor-type dithiophene. When grown as thin films, the combination of nearly planar p-type functional monomers with polarizable imine linkages resulted in a conjugated BDT–ETTA film capable of acting as a photocathode to facilitate photoelectrochemical water splitting without the use of a sacrificial agent or a co-catalyst. The BDT–ETTA films showed strong absorbance of visible light, with a direct optical band gap (2.47 eV), HOMO (−5.51 eV), and LUMO (−3.34 eV) that are favorable for photoelectrochemical water splitting. The photoexcited electrons on the COF film's surface were capable of spontaneously transferring to the electrolyte, due to the LUMO being higher in energy than the H2O/H2 redox pair, resulting in water splitting. As intended, the photoactivity of the system came from the BDT building units, while the high orientation of the COF films not only improved stability, but also amplified the photoresponsiveness of the system.
Lotsch and co-workers utilized a series of triphenylarylaldehydes, with varying concentrations of nitrogens (Nx) in the central aryl ring, to tune a 2D-COF for photocatalytic water reduction (Fig. 26).101 The hydrogen evolution activity increased with the nitrogen content of the monomers, reaching a maximum of 1703 μmol h−1 g−1 for N3-COF (Fig. 26c). This trend was attributed to a combination of several factors including increasing planarity to facilitate exciton migration within the COF plane, the electronic character of the central aryl ring, the ability of the sacrificial electron donor to efficiently remove the hole from the photoexcited COF, and the relative stabilities of the radical formed in the catalytic process. Despite the complexity of the effects that the structural changes had on the photocatlytic activity of the COFs, this work demonstrates the significant impact that structural and electronic properties have on the photocatalytic properties of the resulting COFs.
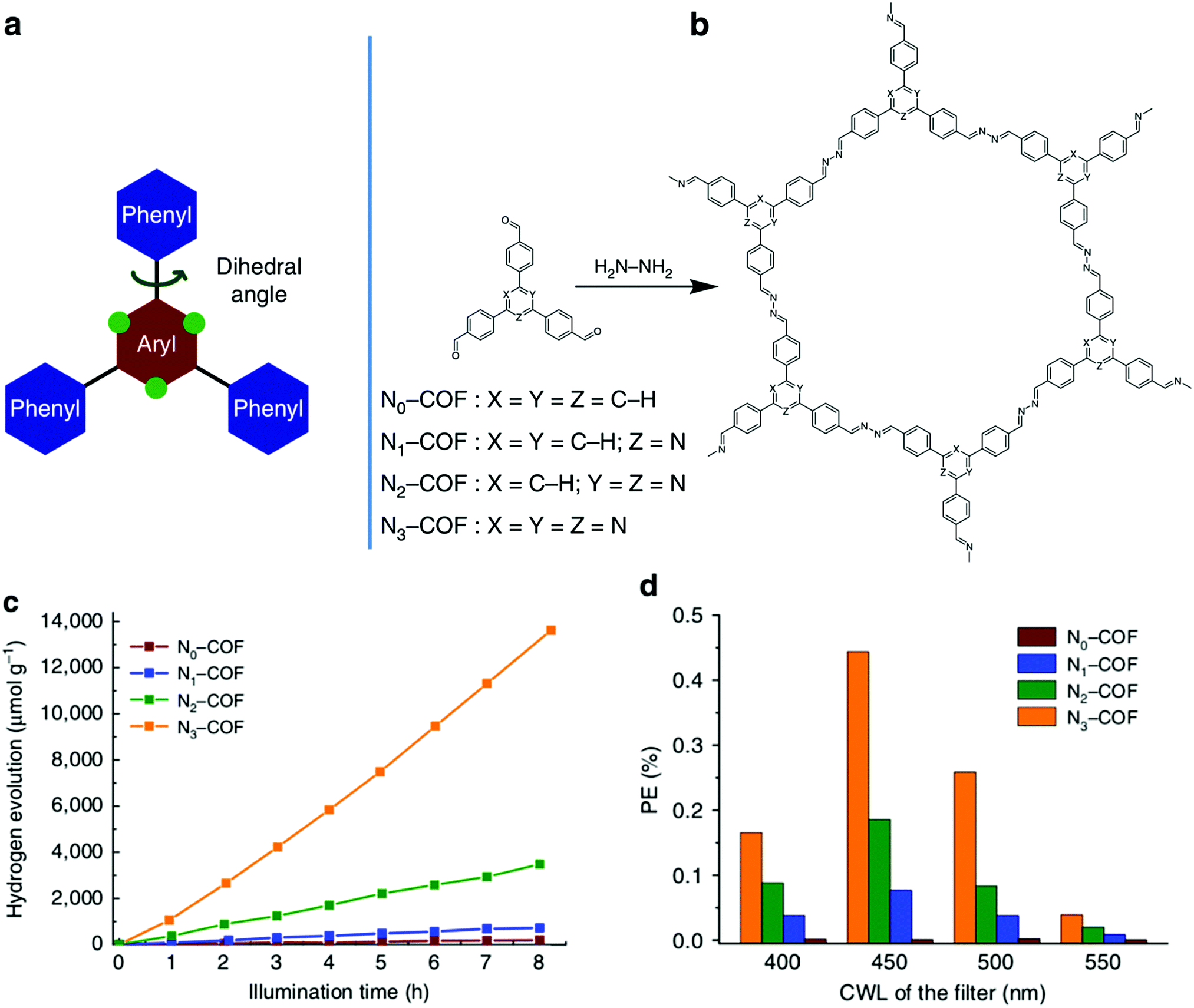 | ||
| Fig. 26 (a) Depiction of the tunable nature of the triphenylarene platform. Swapping ‘C–H’ groups to nitrogen atoms at the green dots effects the planarity of the aryl monomer. (b) Synthesis of Nx-COFs from hydrazine and Nx-Ald monomers. (c) Hydrogen evolution by Nx-COF photocatalysts over 8 h, using triethanolamine as the sacrificial electron donor. (d) The photonic efficiency (PE) of Nx-COFs using different band-pass filters with central wavelengths (CWLs) at 400, 450, 500, and 550 nm. Adapted with permission from Nat. Commun., 2015, 6, 8508. | ||
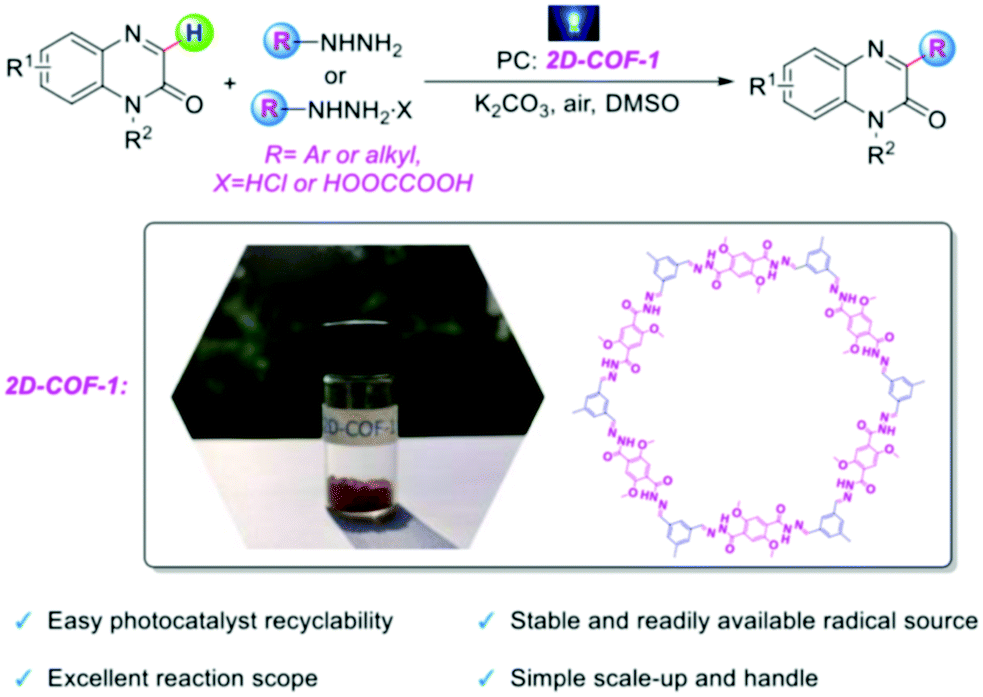 | ||
| Fig. 27 The use of 2D-COF 1 as a photocatalyst for the direct C3 arylation and alkylation of quinoxalin-2(1H)-ones. Adapted with permission from Chem. – Eur. J., 2020, 26, 369–373. | ||
The alteration of electron donor–acceptor pairs in polymeric structures is known to strengthen charge carrier transport mobility. Thomas and co-workers104 utilized this principle in the design of two thiophene-based 2D-COFs (TTT-DTDA and TTT-BTDA). Both COFs proved to be effective radical initiators for visible light driven free radical polymerization (Fig. 28). Irradiation of the COF samples led to charge separation as seen by electron paramagnetic resonance (EPR). When combined with triethylamine (TEA) as a co-initiator, the system promoted the radical polymerization of methyl methacrylate (MMA) to form poly-methyl methacrylate (PMMA) with conversions as high as 63% after 12 h for TTT-DTDA, compared to ∼54% for TTT-BTDA under the same conditions. The heightened yield for TTT-DTDA could be attributed to better charge transfer separation as in situ EPR analysis showed a slower rate for undesired electron–hole pair recombination events, while also showing a more pronounced radical formation in the photocatalyst.
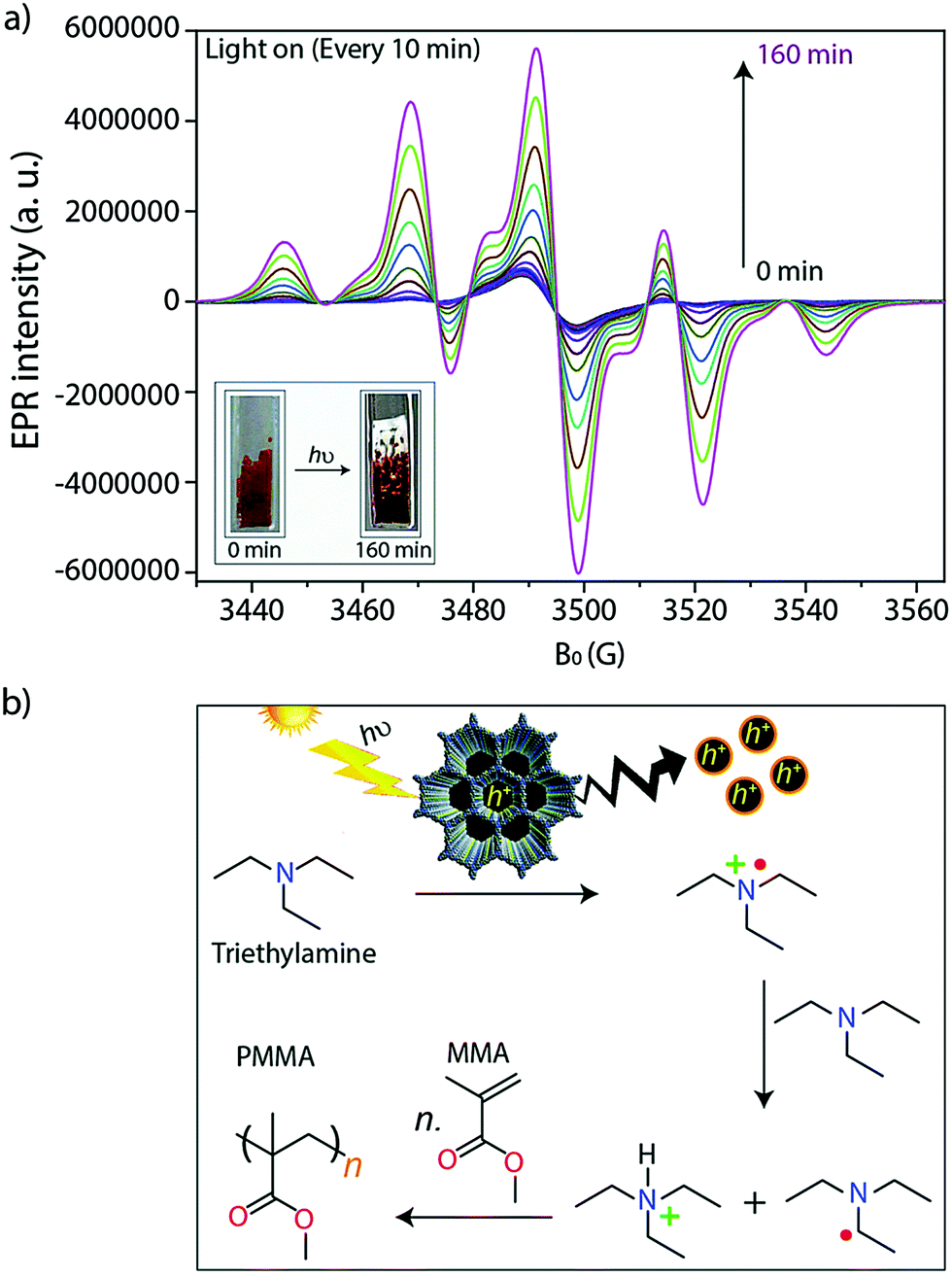 | ||
| Fig. 28 (a) In situ EPR spectra during the photocatalyzed free radical polymerization of MMA to PMMA. (b) Proposed mechanism for the TTT-DTDA (or TTT-BDTA) catalyzed conversion of MMA to PMMA. Reproduced from Chem. Sci., 2019, 10, 8316–8322, with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 29 Proposed mechanistic representation of the COF-JLU22 catalyzed photoreductive dehalogenation. Adapted with permission from Appl. Catal., B, 2019, 245, 334–342. | ||
 | ||
| Fig. 30 (a) Visible light irradiated photocatalytic degradation of phenol. (b) Linear simulation −ln(C/C0) = kt of the kinetic curves. (c) Recyclability of TpMA. (d) Scavenger effects on the photo-degradation of phenol. Reprinted with permission from J. Hazard. Mater., 2019, 369, 494–502. | ||
Jiang and co-workers were able to substantially enhance the catalytic activity of a porphyrin-based COF through the incorporation of graphitic carbon nitride (g-C3N4) to form CuPor-Ph-COF/g-C3N4 hybrid composites.107 The heterojunction-containing composites significantly outperformed both the pure g-C3N4 and CuPor-Ph-COF for the photo-degradation of rhodamine B (RhB), a common organic pollutant, showing removals of 86, 36, and 23%, after 90 min of visible-light irradiation, respectively. This increased activity was attributed to an increased visible light absorption and faster charge carrier transfer within the hybrid material. XPS and UV-vis spectra showed that CuPor-Ph-COF had a valence band (VB) and CB that were 0.79 eV and 0.61 eV lower in energy than g-C3N4, respectively. These results suggested that the photoexcited electrons on the CuPor-Ph-COF migrate to the g-C3N4 through the interface. Simultaneously, the holes generated on the g-C3N4 transfer to the CuPor-Ph-COF, resulting in efficient charge separation and increased photocatalytic activity (Fig. 31).
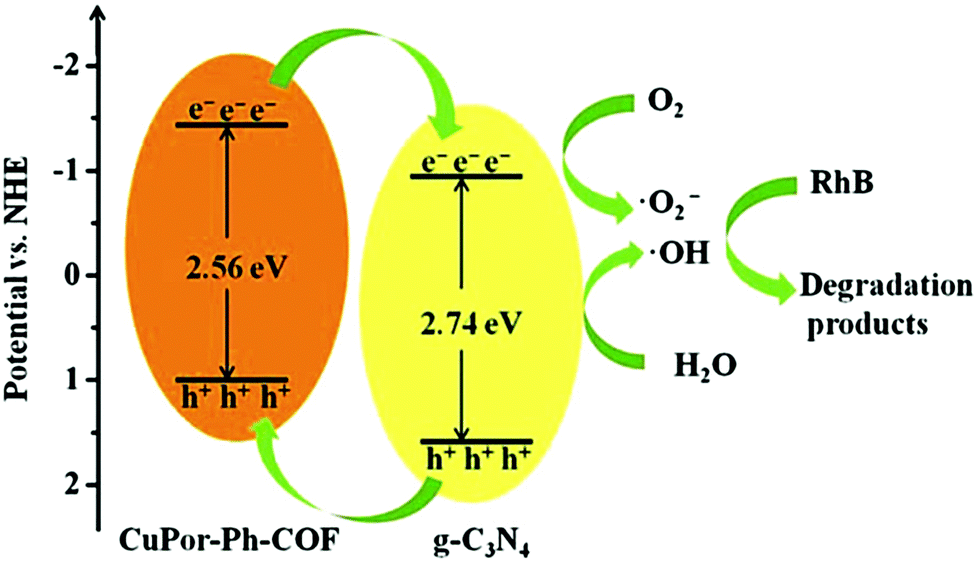 | ||
| Fig. 31 Proposed mechanism for the photocatalytic degradation of rhodamine B by CuPor-Ph-COF/g-C3N4 hybrid composite. Reproduced from Dalton Trans., 2019, 48, 14989–14995, with permission from the Royal Society of Chemistry. | ||
4.3. White-light emitters
In addition to chemical sensors and photocatalysts, COFs are of significant interest for solid-state luminescent devices. This is particularly the case for white-light emitters, due to the ability to control the photophysical properties with molecular precision. Loh and co-workers demonstrated this level of tunability by synthesizing hydrogen-bonding COF systems using a variety of linkers (Fig. 32).89 Due to the increased conjugation, the systems with longer linkers showed a redshifted emission. In addition, dual emissions were observed for some systems depending on the extent of inter- and intralayer hydrogen bonding. The highest absolute photoluminescence quantum yield observed for the 9 reported systems was 16.3%. It was found that a dual emission (ΦPL = 7.3%) could also be induced by incorporating allyl groups into a multi-component system and that, by doing so, CIE coordinates of (0.30, 0.38) were obtained. These coordinates closely approach those of pure white light (0.33, 0.33). | ||
| Fig. 32 (a) Synthetic scheme for obtaining the series of multicomponent COFs for the fine-tuning of PL emission. (b) CIE diagram of various COFs. (c) Absolute PL quantum yields for various COFs; inset depicts fluorescence microscopic images of COFs under visible (top) and UV (bottom) light (scale bar = 50 μm). Adapted with permission from Nat. Commun., 2018, 9, 2335. | ||
Another method for obtaining white-light emission is by simultaneously emitting blue, red, and green light from one source. Van der Voort and co-workers utilized this via co-condensation of 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) and 2,6-diformylpyridine (DFP) to form TTA-DFP-COF as a blue-emitting, imine-linked COF.108 The material was subsequently grafted with Eu3+ and Tb3+ ions chelated with β-diketonate ligands (Fig. 33).108 Due to the particular chelation environment around the lanthanide ions, excitation of 10%Eu90%Tb@TTA-DFP-COF (Φf = 3.1%) and 1%Eu99%Tb@TTA-DFP-COF (Φf = 2.8%) gave calculated CIE coordinates of (0.3636, 0.3355) and (0.3442, 0.3801), respectively.
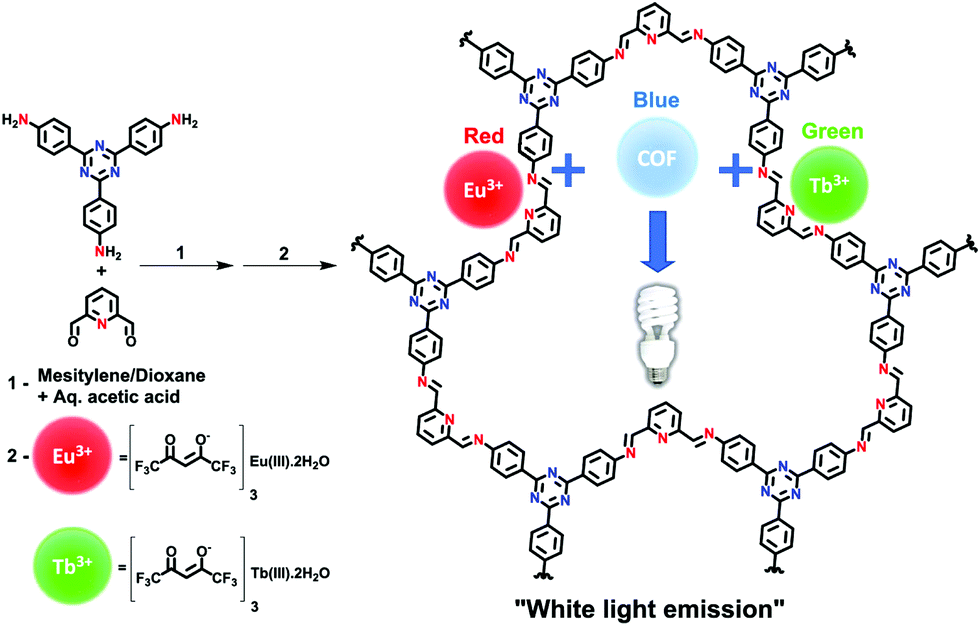 | ||
| Fig. 33 Synthetic scheme for the formation of TTA-DFP COF and the subsequent grafting of lanthanides. Reprinted with permission from ACS Appl. Mater. Interfaces, 2019, 11, 27343–27352. Copyright 2019 American Chemical Society. | ||
Solvatochromism can be utilized to induce white-light emission. Vaidhyanathan and co-workers achieved this by incorporating 2,4-dihydroxybenzene-1,3,5-tricarbaldehyde and anthracene-2,6-diamine to form IISERP-COF7 (Fig. 34a).77 When dispersed in various common solvents, IISERP-COF7 displayed a wide range of emission wavelengths. It was determined through UV-vis experiments that the anthracene units provided a blue fluorescence. The hydroxyl functionalities yielded a dual emission as a result of enol and β-ketoenamine tautomers. The tautomerization equilibrium was strongly affected by the base or hydrogen-bonding capability of the solvent, as discussed in a previous section, which influenced the overall emission energy. When dispersed in NMP, a white-light emission was observed due to NMP's ability to satisfy a 50:50 ratio of tautomers. In addition, NMP improved the fluorescence lifetime via transition state stabilization. Flexible thin films could be fabricated (COF@PMMA) using 0.32 wt% COF, providing a single-source white-light emitter with CIE coordinates of (0.35, 0.36) which could be adjusted to (0.29, 0.33) simply by reducing the amount of COF present in the film to 0.30% (Fig. 34b and c).
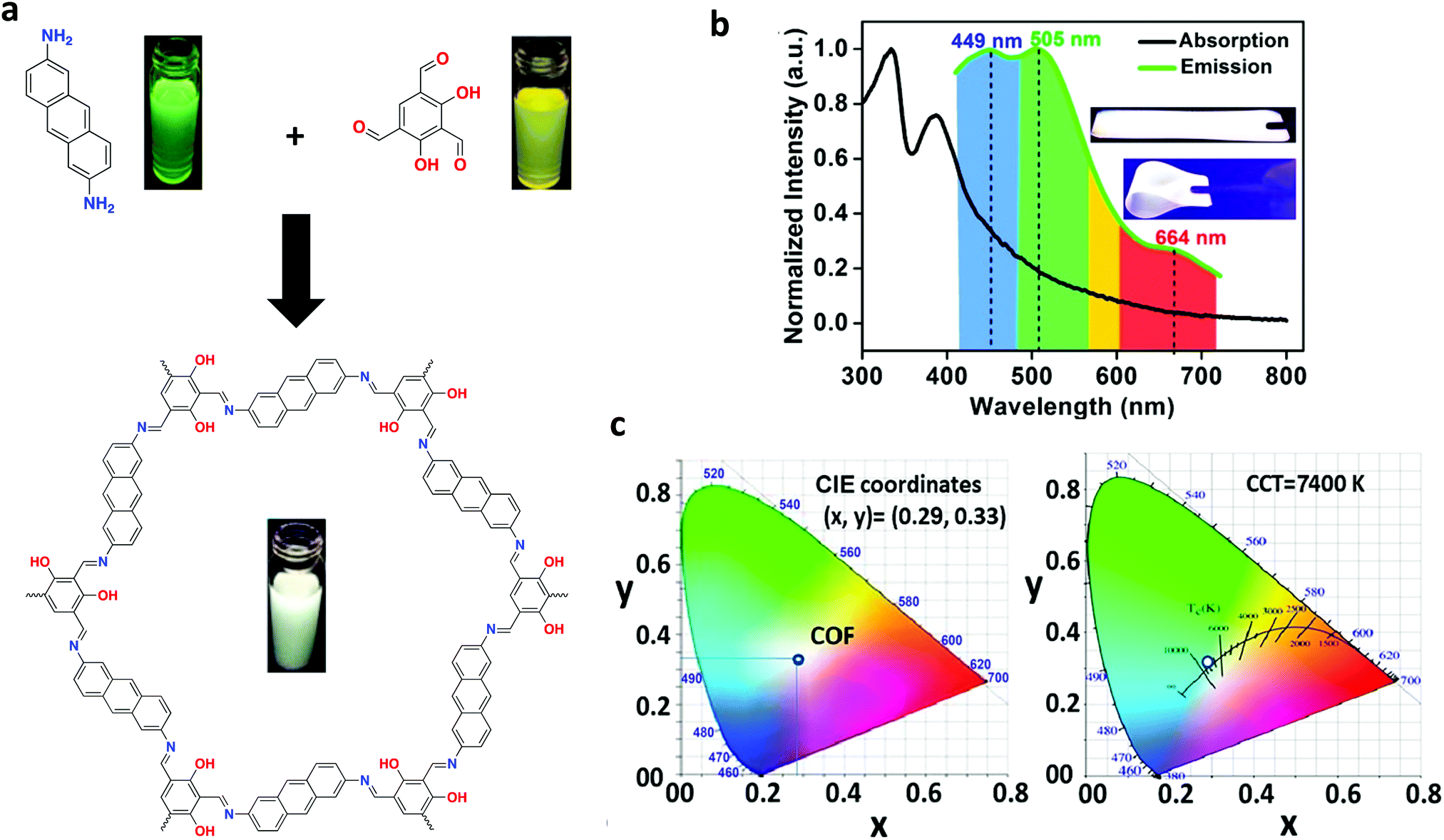 | ||
| Fig. 34 (a) Synthesis of IISERP-COF7, including photographs of monomers and COF dispersed in NMP under a UV lamp. (b) Solid state absorption and emission spectra of COF@PMMA (λex = 420 nm), with inset photographs of COF@PMMA film under UV irradiation. (c) CIE coordinate and CCT diagram of COF film with a thickness of 2 mm. Adapted with permission from J. Am. Chem. Soc., 2018, 140, 13367–13374. Copyright 2018 American Chemical Society. | ||
Recently, Weng and co-workers successfully fabricated a COF-coated LED for white-light emission.39 By co-condensing TPE with the tetrahedral-shaped tetrakis-(4-aminophenyl)methane (TAPM), 3D-TPE-COF was constructed and exhibited a two-fold interpenetrated structure with a bright yellow emission (ΦPL = 20%) was constructed. 3D-TPE-COF was then mixed with an epoxy resin to act as a coating for a blue LED (Fig. 35), which yielded calculated CIE coordinates of (0.30, 0.35). Interestingly, the stability of the system persisted over 1200 h of continuous emission at ambient conditions.
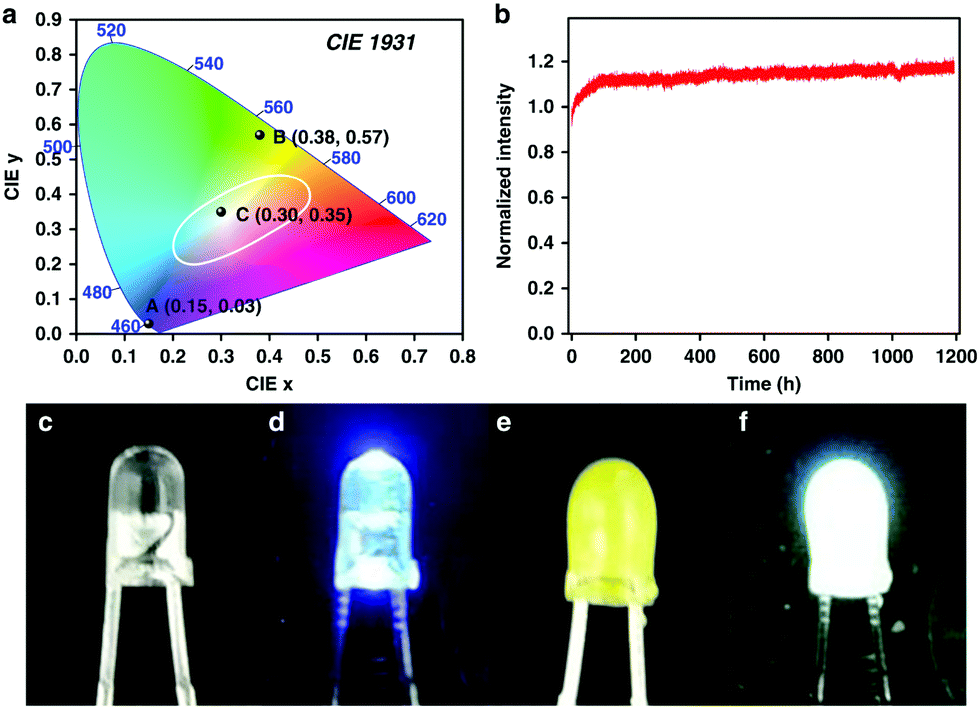 | ||
| Fig. 35 (a) CIE diagram of blue LED (A), 3D-TPE-COF (B), and COF-coated LED (C). (b) Normalized emission intensity for the COF-coated LED over 1200 h, at 2 mA. Photographs of reference blue LED when off (c) and on (d), COF-coated LED when off (e) and on (f). Reprinted with permission from Nat. Commun., 2018, 9, 5234. | ||
4.4. Photodetectors
In 2017, Bein and co-workers109 sought to develop a COF that possessed absorption capabilities in the near-infrared (NIR) spectral regions to employ in the fabrication of a COF-based NIR photodetector. This was accomplished by combining electron-rich and -deficient moieties in the same building block to hopefully allow charge-transfer transitions to take place at energies below the π–π* transition. A material absorbing in the NIR spectral region should enhance its light harvesting capabilities in photovoltaic devices. Isoindigo and thieneoisoindigo-based building blocks were utilized to construct a series of 2D imine-linked COFs. Ultimately, 5,5′-bis(2-formylthiophen5-yl)-N,N′-dibutyl-thienoisoindigo (tTII) was paired with 1,3,6,8-tetrakis(4-aminophenyl)pyrene (Py) yielding Py-tTII COF, which was utilized in thin film growth. The UV-vis spectra showed strong absorption bands in the blue and red-NIR regions. Py-tTII COF was grown on MoOx-modified indium-tin oxide transparent electrodes with [6,6]-phenyl C71 butyric acid methyl ester (PC71BM) to form an interdigitated heterojunction as part of a COF-based NIR photodetector. The device, COF:PC71BM, exhibited an absorbance from 300 to 1100 nm. This work revealed that it is possible to switch the spectral response from blue- and red-sensitive to green- and NIR-sensitive by changing the bias voltage made possible by the growth of the thin COF film.4.5. Thermometers
Van Der Voort and co-workers demonstrated the use of COFs as luminescent ratiometric thermometers via a lanthanide grafting process. 1,3,5-Triformylphloroglucinol (Tp) and 2,2′-bipyridine-5,5′-diamine (Bpy) were first combined to form TpBpy, which produced a broad emission band between 400–550 nm at room temperature.110 The material was soaked in solutions containing excess amounts of lanthanide ions complexed with acetylacetone, which was chosen as a ligand due to its high-lying triplet level, thus making it energetically compatible with the lanthanide ions used. The resulting lanthanide-grafted COFs, TpBpy-Ln_acac (Ln = Eu3+, Tb3+, Eu3+/Tb3+, Dy3+), were able to function as solid-state thermometers in the 10–360 K range for Eu/Tb systems, as well as the 280–440 K temperature ranges for Dy systems (Fig. 36). Interestingly, in Eu/Tb systems, there was a noted lack of thermal quenching. This phenomenon was attributed to the absence of ion-to-ligand/host energy transfer. It was also determined that no Tb3+-to-Eu3+ energy transfer was occurring within the material, which was attributed to large spatial separation between ions within TpBpy. For a summary of COFs discussed in the application section see Table 1.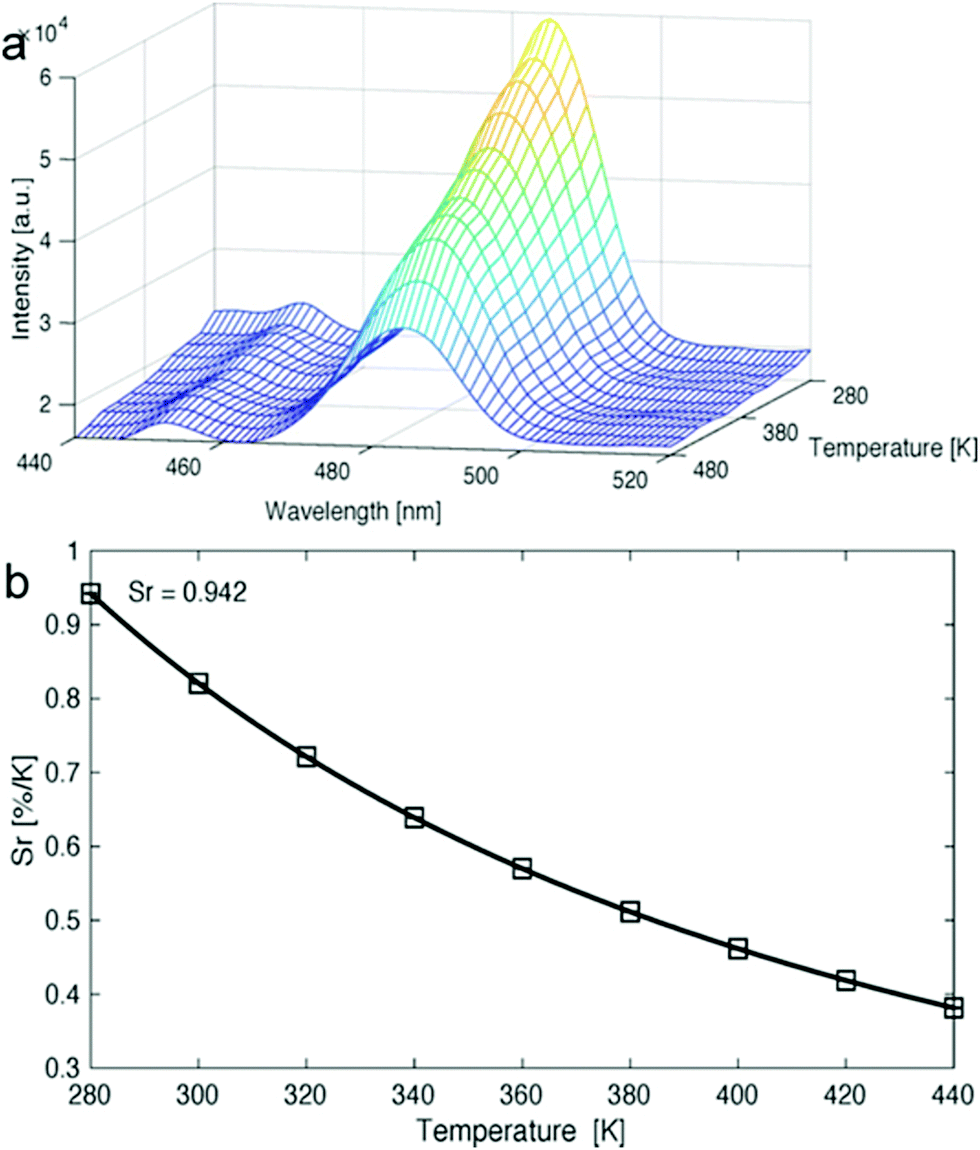 | ||
| Fig. 36 (a) Emission map of Dy(acac)3-grafted TpBpy COF material recorded at 280–480 K. (b) Represenation of the relative sensitivity values for the same compound at varying temperatures. Reprinted with permission from Angew. Chem., Int. Ed., 2020, 59, 2–11. | ||
| COF | Processed form | Application | Ref. |
|---|---|---|---|
| Py-azine COF | Powder | Explosives detection | 86 |
| COF-BABD-BD, COF-BABD-BZ | Powder | Explosives detection | 90 |
| Py-TPE-COF | Nanospheres | Explosives detection | 91 |
| 3D-Py-COF | Powder | Explosives detection | 92 |
| COF-LZU8 | Powder | Hg2+ sensing | 93 |
| COF-JLU3 | Powder | Cu2+ sensing | 37 |
| TaTa, DhaTab, TRITER-1, TzDa | Powder | Fe3+ sensing | 94 |
| Bth-Dha, Bth-Dma | Powder | Fe3+ sensing | 95 |
| TPE-Ph COF | Powder | Ammonia sensing | 79 |
| CCOF 7, CCOF 8 | Nanosheets, membrane | Chirality sensing | 96 |
| COF-TpMA | Powder | Hydroxyl radical sensing, photo-degradation | 97 and 106 |
| TpTt | Powder | E–Z isomerization | 38 |
| TFPT-COF | Powder | H2 evolution | 98 |
| BDT–ETTA COF | Thin film | H2 evolution | 59 |
| N3-COF | Powder | H2 evolution | 101 |
| 2D-COF-1 | Powder | C–C bond forming | 103 |
| TTT-DTDA, TTT-BTDA | Powder | Radical polymerization | 104 |
| COF-JLU22 | Powder | Dehalogenation | 105 |
| CuPor-Ph-COF | Hybrid composite | Photo-degradation | 107 |
| Tf-DHzDPr, Tf-DHzDM, TFPB-DHzDPr, Tf-DHzDM, TFPB-DHZDM, TFPB-DHzDS, TFPB-THz, TFPB-DHzDAll, DFDM-THz | Powder | White-light emission | 89 |
| TTA-DFP-COF | Powder | White-light emission | 108 |
| IISERP-COF7 | Powder, thin film | White-light emission | 77 |
| 3D-TPE-COF | Powder (in epoxy) | White-light emission | 39 |
| Py-tTII COF | Thin film | Photodetection | 109 |
| TpBpy | Powder | Thermometer | 110 |
5. Conclusions
In summary, the modular nature of COFs permits the integration of various π-conjugated monomers to construct 2D and 3D polymeric materials with unique photophysical properties. However, the conditions necessary for obtaining good quantum yields are highly linkage-dependent, and it is now understood that fluorescent imine-linked COFs are particularly challenging due to the rotational and vibrational relaxation pathways that lead to non-radiative decay. By reinforcing the planarity of the imine bond, incorporating highly fluorescent subunits like anthracene, or utilizing AIE phenomena with monomers such as TPE, the radiative decay pathways can be reprogrammed to overcome the non-radiative decay processes. Extended conjugation through CC linkages is also successful in producing fluorescent systems, and further extension with longer linkers can be a useful method to tune the photophysical properties of the overall system. Bulk COF powders dispersed in various solvents are also capable of exhibiting solvatochromism.
With the development of novel methodologies came a diverse set of processing alternatives that allowed for applications of not only powdered materials, but also thin films, nanosheets, and nanofibers. When processed into nanoparticles, COF-based chemical sensors for explosives can achieve sensitivities down to the ppb level. Other sensors are now available for metal ions, organic contaminants, and even hydroxyl radicals. By immobilizing chiral moieties onto the framework and then exfoliating into thin sheets, highly selective enantiomeric sensors can be fabricated into membranes for the detection of odor vapors.
Due to the photoactivity of the bulk system, COFs are also of great interest for photocatalysis. The band gaps of some systems are optimal for use in hydrogen evolution reactions and provide continuous hydrogen evolution, even in the case of one system's auto-exfoliation in its aqueous solution. COFs are also useful for photocatalyzing C–C bond forming reactions by aiding in processes such as radical initiation and SET, leading to improved radical polymerizations and drug derivatizations, respectively. In addition to molecule derivatizations, the degradation of toxic compounds has now been demonstrated with moderate to good conversions in relatively short timescales.
Multiple COF-based systems have been reported to achieve white-light emissions in solution and in the solid-state. Simultaneous emission of red, green, and blue light can be achieved via lanthanide doping or solvent variation to approach near-white-light emissions. These systems can also exhibit robust structural properties when coated on blue LEDs and illuminated for >1000 h, highlighting their potential as solid-state emitters for commercial applications.
Up to this point, a wide variety of luminescent COF systems have been introduced. However, because of the inconsistent and sometimes unpredictable nature of these materials, investigation into their fundamental properties remains a topic of interest. At present, it remains difficult to predict whether a COF architecture that includes a chromophore will yield a luminescent system, or if quenching phenomena will render it non-emissive. In addition, it has been observed that COF emissions can be completely quenched as a result of changing the covalent linkage. Therefore, there is a strong need for further investigation into quenching mechanisms and covalent linkage-dependent fluorescence in order to accurately predict the success of future system designs.
In addition to these challenges, at present, there are a limited number of building blocks available for luminescent COF synthesis, considering the vast number of well-studied chromophores. It is important to continue introducing new building blocks not only to modulate the architectural properties of the COF skeleton, but also to tune the luminescent behavior of the system. Similarly, expanding the scope of covalent linkages has shown great success in enabling the use of more complex functionalized building blocks in COF synthesis. With the new insights that have come from recent mechanistic investigations, it may be possible to synthesize even more complex frameworks with inherent functionality. Taken together, these advancements will allow for the intentional design of a vast range of photoactive systems, and therefore diversify the scope of their applications.
In summary, significant progress has been made towards understanding the translation from small-molecule luminescent behavior to 2D and 3D COF structures. As a direct result, emerging systems have demonstrated great potential in a wide variety of fields. However, close multidisciplinary collaboration between fields including biomedicine, chemical engineering, and materials science should be encouraged for successful implementation of luminescent COF systems for practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Science Foundation (CHE-1856442).References
- H. Zhu, C. C. Lin, W. Luo, S. Shu, Z. Liu, Y. Liu, J. Kong, E. Ma, Y. Cao, R.-S. Liu and X. Chen, Nat. Commun., 2014, 5, 4312 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- C. Yang, D. Qi, J. Liang, X. Wang, F. Cao, Y. He, X. Ouyang, B. Zhu, W. Wen, T. Jia, J. Tian, L. Gao, Z. Sun, S. Zhang and L. V. Wang, Laser Phys. Lett., 2018, 15, 116202 CrossRef CAS.
- P. F. Jones, Forensic Sci., American Chemical Society, 1975, ch. 19, vol. 13, pp. 183–196 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry – An Introduction, University Science Books, USA, 2009 Search PubMed.
- Z. Pan, Y.-Y. Lu and F. Liu, Nat. Mater., 2011, 11, 58 CrossRef PubMed.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- M. Dressler, J. Chromatogr. A, 1979, 165, 167–206 CrossRef CAS.
- P. Jagadesan, T. Whittemore, T. Beirl, C. Turro and P. L. McGrier, Chem. – Eur. J., 2017, 23, 917–925 CrossRef CAS PubMed.
- Y. Li, Y. He, F. Guo, S. Zhang, Y. Liu, W. P. Lustig, S. Bi, L. J. Williams, J. Hu and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 27394–27401 CrossRef CAS PubMed.
- D. Chen, C. Liu, J. Tang, L. Luo and G. Yu, Polym. Chem., 2019, 10, 1168–1181 RSC.
- K. Watanabe and K. Akagi, Sci. Technol. Adv. Mater., 2014, 15, 044203 CrossRef PubMed.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- A. Deshmukh, S. Bandyopadhyay, A. James and A. Patra, J. Mater. Chem. C, 2016, 4, 4427–4433 RSC.
- L. Chen, D. W. McBranch, H.-L. Wang, R. Helgeson, F. Wudl and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12287 CrossRef CAS PubMed.
- S. Bi, Y. Li, S. Zhang, J. Hu, L. Wang and H. Liu, J. Mater. Chem. C, 2018, 6, 3961–3967 RSC.
- P. Pallavi, S. Bandyopadhyay, J. Louis, A. Deshmukh and A. Patra, Chem. Commun., 2017, 53, 1257–1260 RSC.
- C. Gu, Y. Chen, Z. Zhang, S. Xue, S. Sun, K. Zhang, C. Zhong, H. Zhang, Y. Pan, Y. Lv, Y. Yang, F. Li, S. Zhang, F. Huang and Y. Ma, Adv. Mater., 2013, 25, 3443–3448 CrossRef CAS PubMed.
- T. Mondal, S. Mondal, S. Bose, D. Sengupta, U. K. Ghorai and S. K. Saha, J. Mater. Chem. C, 2018, 6, 614–621 RSC.
- R. Chen, W.-C. Chen, L. Yan, S. Tian, B. Liu, X. Chen, C.-S. Lee and W. Zhang, J. Mater. Chem. B, 2019, 7, 4763–4770 RSC.
- G. Lan, K. Ni and W. Lin, Coord. Chem. Rev., 2019, 379, 65–81 CrossRef CAS PubMed.
- K. Lu, C. He and W. Lin, J. Am. Chem. Soc., 2014, 136, 16712–16715 CrossRef CAS PubMed.
- Z. Li, X. Feng, S. Gao, Y. Jin, W. Zhao, H. Liu, X. Yang, S. Hu, K. Cheng and J. Zhang, ACS Appl. Bio Mater., 2019, 2, 613–618 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- C. R. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruña and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed.
- F. Xu, S. Yang, X. Chen, Q. Liu, H. Li, H. Wang, B. Wei and D. Jiang, Chem. Sci., 2019, 10, 6001–6006 RSC.
- Z.-J. Mu, X. Ding, Z.-Y. Chen and B.-H. Han, ACS Appl. Mater. Interfaces, 2018, 10, 41350–41358 CrossRef CAS PubMed.
- X. Wu, X. Han, J. Zhang, H. Jiang, B. Hou, Y. Liu and Y. Cui, Organometallics, 2019, 38, 3474–3479 CrossRef CAS.
- L. Zhu and Y.-B. Zhang, Molecules, 2017, 22, 1149–1178 CrossRef PubMed.
- D. A. Pyles, J. W. Crowe, L. A. Baldwin and P. L. McGrier, ACS Macro Lett., 2016, 5, 1055–1058 CrossRef CAS.
- C. R. DeBlase and W. R. Dichtel, Macromolecules, 2016, 49, 5297–5305 CrossRef CAS.
- Z. Li, Y. Zhang, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 6613–6616 RSC.
- M. Bhadra, S. Kandambeth, M. K. Sahoo, M. Addicoat, E. Balaraman and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 6152–6156 CrossRef CAS PubMed.
- H. Ding, J. Li, G. Xie, G. Lin, R. Chen, Z. Peng, C. Yang, B. Wang, J. Sun and C. Wang, Nat. Commun., 2018, 9, 5234 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- P. J. Waller, Y. S. AlFaraj, C. S. Diercks, N. N. Jarenwattananon and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 9099–9103 CrossRef CAS PubMed.
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS PubMed.
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 15134–15137 CrossRef CAS PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- D. Rodríguez-San-Miguel and F. Zamora, Chem. Soc. Rev., 2019, 48, 4375–4386 RSC.
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48 CrossRef CAS PubMed.
- B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2014, 136, 8783–8789 CrossRef CAS PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52 CrossRef CAS PubMed.
- E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416–19421 CrossRef CAS PubMed.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodríguez-Calero, R. P. Bisbey, H. D. Abruña and W. R. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS PubMed.
- R. P. Bisbey, C. R. DeBlase, B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2016, 138, 11433–11436 CrossRef CAS PubMed.
- J. I. Feldblyum, C. H. McCreery, S. C. Andrews, T. Kurosawa, E. J. G. Santos, V. Duong, L. Fang, A. L. Ayzner and Z. Bao, Chem. Commun., 2015, 51, 13894–13897 RSC.
- K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228 CrossRef CAS PubMed.
- J. M. Rotter, S. Weinberger, J. Kampmann, T. Sick, M. Shalom, T. Bein and D. D. Medina, Chem. Mater., 2019, 31, 10008–10016 CrossRef CAS.
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. Evans, M. Döblinger, S. Herbert, K. Peters, D. Böhm, P. Knochel, D. D. Medina, D. Fattakhova-Rohlfing and T. Bein, J. Am. Chem. Soc., 2018, 140, 2085–2092 CrossRef CAS PubMed.
- D. D. Medina, M. L. Petrus, A. N. Jumabekov, J. T. Margraf, S. Weinberger, J. M. Rotter, T. Clark and T. Bein, ACS Nano, 2017, 11, 2706–2713 CrossRef CAS PubMed.
- D. D. Medina, V. Werner, F. Auras, R. Tautz, M. Dogru, J. Schuster, S. Linke, M. Döblinger, J. Feldmann, P. Knochel and T. Bein, ACS Nano, 2014, 8, 4042–4052 CrossRef CAS PubMed.
- C. E. Wayne and R. P. Wayne, Photochemistry, Oxford University Press, 1996 Search PubMed.
- Y. Yamaguchi, Y. Matsubara, T. Ochi, T. Wakamiya and Z.-I. Yoshida, J. Am. Chem. Soc., 2008, 130, 13867–13869 CrossRef CAS PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- N. Huang, X. Ding, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 8704–8707 CrossRef CAS PubMed.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, Polym. Chem., 2016, 7, 4176–4181 RSC.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 849–853 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- Y. Zhao, H. Liu, C. Wu, Z. Zhang, Q. Pan, F. Hu, R. Wang, P. Li, X. Huang and Z. Li, Angew. Chem., Int. Ed., 2019, 58, 5376–5381 CrossRef CAS PubMed.
- E. Jin, J. Li, K. Geng, Q. Jiang, H. Xu, Q. Xu and D. Jiang, Nat. Commun., 2018, 9, 4143 CrossRef PubMed.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- A. Weller, Naturwissenschaften, 1955, 42, 175–176 CrossRef CAS.
- H.-L. Qian, C. Dai, C.-X. Yang and X.-P. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 24999–25005 CrossRef CAS PubMed.
- S. Haldar, D. Chakraborty, B. Roy, G. Banappanavar, K. Rinku, D. Mullangi, P. Hazra, D. Kabra and R. Vaidhyanathan, J. Am. Chem. Soc., 2018, 140, 13367–13374 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed.
- J. Dong, X. Li, S. B. Peh, Y. D. Yuan, Y. Wang, D. Ji, S. Peng, G. Liu, S. Ying, D. Yuan, J. Jiang, S. Ramakrishna and D. Zhao, Chem. Mater., 2019, 31, 146–160 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed.
- F. Auras, L. Ascherl, A. H. Hakimioun, J. T. Margraf, F. C. Hanusch, S. Reuter, D. Bessinger, M. Döblinger, C. Hettstedt, K. Karaghiosoff, S. Herbert, P. Knochel, T. Clark and T. Bein, J. Am. Chem. Soc., 2016, 138, 16703–16710 CrossRef CAS PubMed.
- J. W. Crowe, L. A. Baldwin and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 10120–10123 CrossRef CAS PubMed.
- L. Stegbauer, S. Zech, G. Savasci, T. Banerjee, F. Podjaski, K. Schwinghammer, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2018, 8, 1703278 CrossRef.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- X. Chen, N. Huang, J. Gao, H. Xu, F. Xu and D. Jiang, Chem. Commun., 2014, 50, 6161–6163 RSC.
- A. C. Pratt, Chem. Soc. Rev., 1977, 6, 63–81 RSC.
- X. Li, Q. Gao, J. Wang, Y. Chen, Z.-H. Chen, H.-S. Xu, W. Tang, K. Leng, G.-H. Ning, J. Wu, Q.-H. Xu, S. Y. Quek, Y. Lu and K. P. Loh, Nat. Commun., 2018, 9, 2335 CrossRef PubMed.
- M.-W. Zhu, S.-Q. Xu, X.-Z. Wang, Y. Chen, L. Dai and X. Zhao, Chem. Commun., 2018, 54, 2308–2311 RSC.
- Q. Gao, X. Li, G.-H. Ning, K. Leng, B. Tian, C. Liu, W. Tang, H.-S. Xu and K. P. Loh, Chem. Commun., 2018, 54, 2349–2352 RSC.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed.
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- L.-L. Wang, C.-X. Yang and X.-P. Yan, Sci. China: Chem., 2018, 61, 1470–1474 CrossRef CAS.
- G. Chen, H.-H. Lan, S.-L. Cai, B. Sun, X.-L. Li, Z.-H. He, S.-R. Zheng, J. Fan, Y. Liu and W.-G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12830–12837 CrossRef CAS PubMed.
- X. Wu, X. Han, Q. Xu, Y. Liu, C. Yuan, S. Yang, Y. Liu, J. Jiang and Y. Cui, J. Am. Chem. Soc., 2019, 141, 7081–7089 CrossRef CAS PubMed.
- W. Liu, Y. Cao, W. Wang, D. Gong, T. Cao, J. Qian, K. Iqbal, W. Qin and H. Guo, Chem. Commun., 2019, 55, 167–170 RSC.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- R. van de Krol, Y. Liang and J. Schoonman, J. Mater. Chem., 2008, 18, 2311–2320 RSC.
- T.-Y. Zhou, S.-Q. Xu, Q. Wen, Z.-F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS PubMed.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- R. Ballini, S. Gabrielli and A. Palmieri, Synlett, 2009, 965–967 CrossRef CAS.
- M. Tian, S. Liu, X. Bu, J. Yu and X. Yang, Chem. – Eur. J., 2020, 26, 369–373 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M.-Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC.
- Z. Li, Y. Zhi, P. Shao, H. Xia, G. Li, X. Feng, X. Chen, Z. Shi and X. Liu, Appl. Catal., B, 2019, 245, 334–342 CrossRef CAS.
- H. Lv, X. Zhao, H. Niu, S. He, Z. Tang, F. Wu and J. P. Giesy, J. Hazard. Mater., 2019, 369, 494–502 CrossRef CAS PubMed.
- Y. Hou, C.-X. Cui, E. Zhang, J.-C. Wang, Y. Li, Y. Zhang, Y. Zhang, Q. Wang and J. Jiang, Dalton Trans., 2019, 48, 14989–14995 RSC.
- C. Krishnaraj, A. M. Kaczmarek, H. S. Jena, K. Leus, N. Chaoui, J. Schmidt, R. Van Deun and P. Van Der Voort, ACS Appl. Mater. Interfaces, 2019, 11, 27343–27352 CrossRef CAS PubMed.
- D. Bessinger, L. Ascherl, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 12035–12042 CrossRef CAS PubMed.
- A. M. Kaczmrek, Y. Liu, M. K. Kaczmarek, A. H. Liu, L. D. Carlos and P. Van Der Voort, Angew. Chem., Int. Ed., 2020, 59, 2–11 CrossRef.
Footnote |
| † W. K. H., E. M. M., and E. R. W. contributed equally to this review. |
| This journal is © The Royal Society of Chemistry 2020 |