
Design, synthesis and photoinduced processes in molecular interlocked photosynthetic [60]fullerene systems
Jackson D.
Megiatto
Jr
 *a,
Dirk M.
Guldi
b and
David I.
Schuster
c
*a,
Dirk M.
Guldi
b and
David I.
Schuster
c
aInstitute of Chemistry, University of Campinas (UNICAMP), PO Box 6154, 13083-970, Brazil. E-mail: jdmj@unicamp.br
bDepartment of Chemistry and Pharmacy and Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany
cDepartment of Chemistry, New York University, New York, New York 10003, USA
First published on 6th December 2019
Abstract
In natural photosynthesis, the protein backbone directs and positions primary and secondary electron donor and acceptor moieties in the reaction center to control the yield and kinetics of the sequential electron transfer reactions that transform light energy into chemical potential. Organization of the active cofactors is mainly driven by noncovalent interactions between the protein scaffold and the chromophores. Based on the natural system blueprint, several research efforts have investigated π–π stacking, ionic interactions as well as formation of hydrogen and coordinative bonds as noncovalent organizing principles for the assembly of electron donors and acceptors in artificial reaction centers. Introduction of supramolecular concepts into the organization of electron donor–acceptor in artificial photosynthetic models raises the possibility of applying template-directed synthesis techniques to assemble interlocked systems in which the photo-redox components are mechanically rather than covalently linked. Rotaxanes and catenanes are the leading examples of interlocked molecules, whose recent developments in synthetic chemistry have allowed their efficient preparation. Introduction of mechanical bonds into electron donor–acceptor systems allows the study of the interlocked components’ submolecular motions and conformational changes, which are triggered by external stimuli, on the thermodynamic and kinetic parameters of photoinduced processes in artificial reaction centers. This Tutorial discusses our efforts in the synthesis and photophysical investigation of rotaxanes and catenanes decorated with peripheral electron donors and [60]fullerene as the acceptor. The assembly of our rotaxanes and catenanes is based on the classic 1,10-phenanthroline-copper(I) metal template strategy in conjunction with the virtues of the Cu(I)-catalyzed-1,3-dipolar cycloaddition of azides and alkynes (the CuAAC or “click” reaction) as the protocol for the final macrocyclization or stoppering reactions of the entwined precursors. Time-resolved emission and transient absorption experiments revealed that upon excitation, our multichromophoric rotaxanes and catenanes undergo a cascade of sequential energy and electron transfer reactions that ultimately yield charge separated states with lifetimes as long as 61 microseconds, thereby mimicking the functions of the natural systems. The importance of the Cu(I) ion (used to assemble the interlocked molecules) as an electronic relay in the photoinduced processes is also highlighted. The results of this research demonstrate the importance of the distinct molecular conformations adopted by rotaxanes and catenanes in the electron transfer dynamics and illustrate the versatility of interlocked molecules as scaffolds for the organization of donor–acceptor moieties in the design of artificial photosynthetic reaction centers.
 Jackson D. Megiatto | Jackson Megiatto has been a Professor of Chemistry at the University of Campinas (UNICAMP), Brazil, since 2014. He received his BA from the University of São Paulo (USP) in Brazil in 2001 and his PhD from the Université de Bordeaux-1/University of São Paulo in 2006. He was a postdoctoral fellow at the Université de Bordeaux-1 (2006–2007), and a research associate at New York University (2008–2010) and Arizona State University (2011–2013). His research in Supramolecular Chemistry, Molecular Machines and Artificial Photosynthesis involves the design and self-assembly of interlocked photoactive molecules, polymers and catalysts and their applications in new technologies. |
 Dirk M. Guldi | Dirk M. Guldi completed both his undergraduate studies (1988) and PhD (1990) at the University of Cologne (Germany). Following postdoctoral appointments at the National Institute of Standards and Technology (USA), the Hahn-Meitner Institute Berlin (1992), and Syracuse University, he joined the faculty of the Notre Dame Radiation Laboratory in 1995. He was promoted a year later from assistant to associate professional specialist, and remained affiliated to Notre Dame until 2004. Since 2004, he has been Full Professor in the Department of Chemistry and Pharmacy at the Friedrich-Alexander University in Erlangen. Since 2018, Dirk M. Guldi has been Co-Editor in Chief of Nanoscale and Nanoscale Horizons, RSC journals focused on experimental and theoretical research in all areas of nanotechnology and nanoscience, and he has been named among the world's Highly Cited Researchers by Thomson Reuters. |
 David I. Schuster | David I. Schuster is Professor Emeritus of Chemistry at New York University. He retired in 2005 but continued his research program until 2011. He received his BA from Columbia University in 1956 and his PhD from Caltech in 1961 in the group of John D. Roberts. After leaving Caltech, he was a postdoctoral fellow with Howard Zimmerman at Wisconsin. He joined the chemistry faculty at NYU in 1961 and did research in organic photochemistry and neurochemistry. In the early 1990s, he established the NYU Fullerene Group. The final phase of his research carrier has focused on rotaxanes and catenanes incorporating various electron donor moieties and fullerenes as electron acceptors. He is a Fellow of the American Association for the Advancement of Science and recipient of the Arthur C. Cope Scholar Award from the American Chemical Society. |
Key learning points(1) Importance of supramolecular concepts in the design of biomimetic photosynthetic models.(2) Synthetic techniques to assemble rotaxanes and catenanes bearing electron donor and acceptor moieties. (3) The effects of molecular topology on the rates of electron and energy transfer processes. (4) How the dynamic properties of interlocked photosynthetic systems affect the electron transfer processes. (5) The thermodynamic and kinetic requirements for the design of multi-chromophoric rotaxanes able to undergo sequential energy and electron transfer processes. |
Introduction
In natural photosynthesis, a series of directional energy and electron transfer reactions among several organic and inorganic cofactors imbedded in transmembrane photoredox protein complexes is the fundamental process by which conversion of sunlight energy into chemical potential occurs. More specifically, sunlight excites the natural antenna chromophores to provide electronic excitation, which is funneled to the reaction center with near unity quantum yield. In the reaction center, the electronic excitation drives a cascade of electron transfer reactions among primary and secondary electron donors and acceptors to produce charge separated states extending across the lipid bilayer membrane. The lifetime of the final charge separated state is crucial to the photosynthetic energy conversion process. The kinetic mismatch between the fast photo-induced processes and the relatively slower chemical transformations that ultimately oxidize water molecules and reduce carbon dioxide requires temporal stabilization of the charge separated state. Therefore, recombination of the separated charges through back electron transfer reactions and consequent loss of the stored solar energy as heat must be avoided.1Nature's solution to this problem is to isolate the charges at large distances by moving the photogenerated electron and hole produced in the primary photoexcitation step in opposite directions to secondary donor and acceptor residues. The protein backbone positions the primary and secondary donors and acceptors in a very specific topology to allow efficient orbital overlapping and consequent maximization of electronic coupling. Accordingly, the charge shift reactions among the chromophores are directional, short-range, and thereby fast, and ultimately yield a large spatial separation of the electron and hole in the photosynthetic protein. The large spatial separation leads to weak electronic coupling between the oxidized and reduced secondary donor/acceptor, thereby slowing down the charge recombination processes. Such cascade of vectoral, short-range and fast electron transfer reactions also guarantees the high quantum efficiency of each step when compared to a single long-range electron transfer from the primary donor to the secondary acceptor.2–4
Noncovalent interactions such as hydrogen bonds, π–π stacking and ionic interactions among the redox-photoactive subunits and the protein residues are the main driving force behind the organization process of the natural photosynthetic system. Such protein–cofactor interactions also modulate the potentials of the donors and acceptors to achieve the redox power necessary to drive the further chemical reactions that transform light into biofuel. Therefore, the whole system is fine-tuned both energetically and kinetically by noncovalent interactions.5,6
In the design of artificial reaction centers, early research workers investigated chromophores structurally related to those found in natural systems and replaced the protein scaffold with covalent bonds as the organizing principle. Porphyrins were the natural choice as primary electron donors instead of the synthetically demanding and relatively unstable chlorophyll molecules, while quinone derivatives were commonly used as acceptors.7 However, fullerenes have proven to be superior electron acceptors in photoredox arrays.8–12 Their rigid structure and poor solvation in polar and nonpolar solvents yield small values of the reorganization energy (λ) which, according to Marcus formalism,13 shifts the exergonic back electron transfer reaction into the so-called inverted region of the parabolic relationship between the free energy change of the electron transfer process vs. λ. Therefore, the large free energy change associated with the charge recombination reaction in fullerene-containing systems results in reduced rates. On the other hand, the forward electron transfer processes require small driving forces to reach the apex of the Marcus parabolic profile, thereby affording maximum rate constants for charge separation in donor–acceptor systems incorporating fullerenes.8–12
Despite the progress achieved with the covalent-linked artificial photosynthetic systems,14–19 organization of photoredox arrays capable of absorbing light and generating and transporting charges across large distances through formation of covalent bonds is by its very nature inefficient. The emergence of host–guest20 and supramolecular concepts21 sets the stage for molecular recognition to lead to self-assembly of simple molecular components into large aggregates with well-defined structures. Accordingly, noncovalent interactions have been used as principles for the self-assembly of chromophores in artificial photosynthetic models capable of mimicking the natural photo-induced processes.22,23 The supramolecular approach offers two main advantages over the traditional covalent linkage as the organizing principle: (i) complex arrangements of the photoredox active moieties can be achieved from simple building blocks in a one-pot strategy instead of the stepwise formation of each covalent bond necessary to organize the array; (ii) higher order features such as the effects of molecular topology on the kinetic and thermodynamic aspects of the photoinduced processes can be investigated in these supramolecular photosynthetic arrays.
Within this context, we and others have been interested in the investigation of interlocked photosynthetic models, in which the photoredox active subunits are mechanically rather than covalently linked.24–27 A mechanical bond is an entanglement in space between two or more component parts, such that they cannot be separated without distorting or breaking chemical bonds between atoms.26 Rotaxanes (a dumbbell shaped molecular component in which a molecular ring is trapped) and catenanes (two or more interlocked rings) are prime examples of mechanically linked systems.26 The appealing feature of interlocked molecules as artificial photosynthetic models is the possibility of decorating rotaxanes and catenanes with donor and acceptor moieties and using the inherent circumrotation, shuttling, pirouetting and rocking motions26 of the entangled components to control through external stimuli the relative position and orientation of the chromophores in order to favor forward electron transfer and charge shift reactions, while simultaneously retarding wasteful charge recombination processes.
Efficient formation of mechanical bonds requires the use of template synthesis techniques. The transition metal template synthesis has proven to be a powerful strategy to prepare interlocked molecules. The metal template synthesis of interlocked molecules is based on the ability of metal ions to bind component parts bearing coordination sites through formation of coordinative bonds to create entwined metal complex intermediates. The physical overlapping of the ligands in the entwined complexes orients chemical functionalities in an orthogonal fashion to favor introduction of stoppering groups (to afford rotaxanes) or to effect macrocyclizations (to afford catenanes) through irreversible chemical reactions. Removal of the metal template ion from the systems then reveals the mechanical bond.28–30 In the design of artificial photosynthetic models, the metal template approach also offers a bonus, namely that the entwined metal complexes which serve as building blocks to assemble the interlocked systems can also act as electronic relays.31
In this Tutorial, we discuss the design, synthesis and photophysical investigations of a family of multichromophoric interlocked molecules decorated with electron donor and acceptor moieties. The powerful synthetic strategy developed by us, which combines the virtues of Sauvage's Cu(I)-1,10-phenanthroline template technique28 with those of “click” chemistry protocols,32 has enabled the kinetically controlled syntheses of rotaxanes and catenanes whose size and shape of the ring and thread components as well as the chemical linkages that connect the photoredox active subunits are identical. Therefore, the effects of the distinct molecular topology provided by the two interlocked systems on the electron transfer dynamics could be investigated. To illustrate the versatility of interlocked structures as scaffolds to organize multichromophoric systems, we also report rotaxanes decorated with several distinct chromophores. Excitation of a given subunit by the choice of excitation wavelength as well as investigation of model compounds has enabled the assignment of specific roles to each entity in the multicomponent interlocked structures, while the energy and electron transfer rates have been determined by means of ultrafast spectroscopic techniques. Charge separated states with remarkably long lifetimes have been achieved, thus demonstrating the advantages of using mechanical bonds as the organizing principle in the design of artificial photosynthetic systems.
1. Molecular design and efficient kinetically controlled syntheses of rotaxanes and catenanes decorated with zinc(II)porphyrinate as the electron donor and [60]fullerene as the acceptor
At the outset of our research program, the main synthetic challenge facing us was the combination of the kinetically labile coordinative bonds that hold the component parts in the entwined configuration required for rotaxane and catenane syntheses with the reactivity/solubility issues associated with [60]fullerene (C60). The classical protocols used to functionalize fullerenes required addition of strong bases to drive the reactions.11 The presence of such chemical species in the reaction medium was known to cause dissociation of the entwined metal complex precursors.28,30 Conversely, the reaction conditions for stoppering or for ring closing the entwined complexes to respectively form rotaxanes and catenanes commonly involved in situ generation of nucleophilic species in poor solvents28,30 for the electrophile C60 group.11 Therefore, side reactions with the carbon cage and/or its precipitation during the assembly of interlocked molecules by metal template techniques precluded the efficient preparation of our target C60-based interlocked molecules using standard techniques.To solve those synthetic issues, we proposed33–37 the straightforward synthetic strategy depicted in Fig. 1 to prepare [Cu(phen)2]+-based rotaxanes and catenanes with a peripheral C60 group as the electron acceptor and zinc(II)porphyrinates (ZnP) as electron donors. Our synthetic strategy was composed of three main steps, which were designed to minimize the reactivity/solubility issues of the C60 group and the relative kinetic lability of the [Cu(phen)2]+ complex template. Firstly, the solubility issues related to the C60 synthon were overcome by first attaching the carbon cage to the previously prepared phen-based macrocycle 1 using the classical Bingel-Hirsch reaction11 to afford highly soluble C60-based macrocycle 2. Secondly, the “threading” reaction of phen-string-like fragment 3 through C60-macrocycle 2 was accomplished using the Cu(I) ion as the template species to yield [Cu(phen)2]+–C60 pseudorotaxane precursor 4.28 Finally, pseudorotaxane 4 was submitted to “click”32 stoppering reactions with monoalkynyl ZnP 5 for the preparation of rotaxane 7, or with bis-alkynyl ZnP 6 for the “double click” macrocyclization required for the synthesis of the analogous catenane 8.
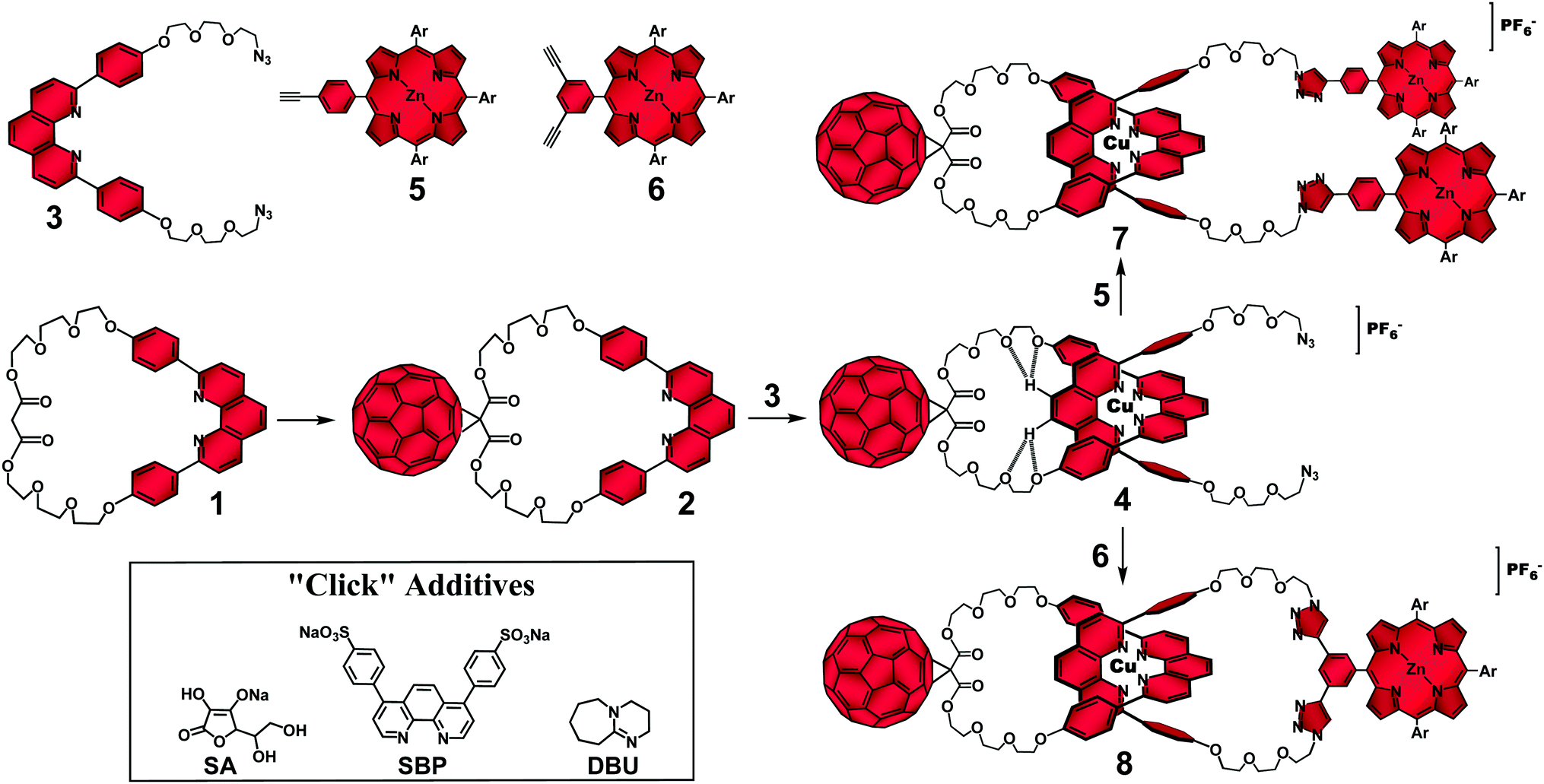 | ||
| Fig. 1 Synthetic strategy for the synthesis of rotaxanes and catenanes using the Cu(I)-template-directed technique in combination with “click” reactions.38 | ||
The hurdle of this strategy resided on kinetic control of the irreversible final “click” reactions. To successfully prepare rotaxane 7 and catenane 8 following our strategy, attachment of the ZnP stoppering groups or the macrocyclization step had to occur without disrupting the [Cu(phen)2]+ coordinative bonds in pseudorotaxane 4. Otherwise, a detrimental unthreading process would take place and the components would react in a random rather than in a templated fashion to yield noninterlocked byproducts. The distribution of interlocked and noninterlocked products is controlled by the relative stability of the corresponding transition states during the “click” reaction. Therefore, our proposed strategy would afford rotaxane 7 and catenane 8 with superior yields only if the kinetics of the final “click” reactions were enhanced. In the case of rotaxane 7, acceleration of the “click” stoppering reactions was achieved by increasing the concentration of the reactants and/or by adding a large excess of ZnP 5 relative to pseudorotaxane 4 in the reaction medium. However, access to catenane 8 required high-dilution and stoichiometric amounts of pseudorotaxane 4 and ZnP 6 to avoid formation of other cyclic and/or oligomeric structures. Equally, the typical homeopathic reactant dilution necessary for the efficient catenane synthesis, when applied to the preparation of rotaxane 7, led to poor yields as the lower “click” reaction rates allowed dissociation of pseudorotaxane 4.
To solve these conflicting reaction conditions and to afford rotaxane 7 and catenane 8 from the common pseudorotaxane precursor 4 under the same “click” conditions, which would render our synthetic strategy more convenient and elegant, we tested39 several reaction conditions to find the best balance between stabilization of pseudorotaxane 4 and acceleration of the final “click” reactions. It turned out that a biphasic reaction medium at room temperature circumvented the incompatible synthetic requirements mentioned above. We demonstrated34,37,40 that by dissolving equimolar amounts of pseudorotaxane 4 and the corresponding ZnP building block in a solvent mixture composed of CH2Cl2/CH3CN (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) at standard concentration levels (∼0.01 M), followed by addition of an innovative homemade “click brew” catalyst composed of copper(I)iodide, sodium ascorbate (SA), sulfonated bathophenanthroline (SBP) and 1,8-diaza[5.4.0]bicycloundec-7-ane (DBU) (Fig. 1), all previously dissolved in a solvent mixture composed of H2O/EtOH (1:1, v/v), afforded rotaxane 7 and analogous catenane 8 in excellent yields, 75% and 57% respectively.34,37,40 We rationalized39 that pseudorotaxane 4 was less susceptible to unthreading in the CH2Cl2 phase as the potentially harmful species present in the “click” brew catalyst remained in the polar phase, while the reactions that connect the pieces together occurred at the interface.
:3, v/v) at standard concentration levels (∼0.01 M), followed by addition of an innovative homemade “click brew” catalyst composed of copper(I)iodide, sodium ascorbate (SA), sulfonated bathophenanthroline (SBP) and 1,8-diaza[5.4.0]bicycloundec-7-ane (DBU) (Fig. 1), all previously dissolved in a solvent mixture composed of H2O/EtOH (1:1, v/v), afforded rotaxane 7 and analogous catenane 8 in excellent yields, 75% and 57% respectively.34,37,40 We rationalized39 that pseudorotaxane 4 was less susceptible to unthreading in the CH2Cl2 phase as the potentially harmful species present in the “click” brew catalyst remained in the polar phase, while the reactions that connect the pieces together occurred at the interface.
2. Structural investigation of the ZnP–[Cu(phen)2]+–C60 rotaxanes and catenanes
A comparison between the 1H NMR spectra of the interlocked molecules39 showed that the signals for the pyrrolic protons of the ZnP moieties (δ = 8–9 ppm) appeared in duplicate in rotaxane 7, while only one set of signals for the same nuclei was observed for catenane 8. The two sets of resonances for the pyrrolic protons informed that the two ZnP groups in rotaxane 7 were not magnetically equivalent, experiencing distinct chemical environments. Careful analyses of 1H NMR spectra of 7 and 8 revealed that one set of pyrrolic signals in rotaxane 7 appeared in the expected region of the spectrum for ZnP moieties and with very similar chemical shifts compared to those observed for catenane 8, while the second set of resonances was shifted upfield. This shielding suggested that one of the ZnP stoppers was most likely engaged in π–π secondary attractive interactions with the C60 and/or [Cu(phen)2]+ subunit, while the other was not.In time-resolved fluorescence experiments, we observed that the ZnP first singlet excited state (1ZnP*) presented a biexponential decay with lifetimes of 61 ps and 400 ps for rotaxane 7,40 while a monoexponential decay with a lifetime of 500 ps was sufficient to fit the data for the same excited state in catenane 8.37 Since the fluorescence lifetime is directly related to the electronic coupling of the chromophores, the biexponential decay observed for the 1ZnP* species in rotaxane 7 confirmed that the ZnP stoppers were at distinctly different distances from the other chromophores. Thus, we concluded that rotaxane 7 was conformationally flexible and folded into a more compact structure in solution with one of the ZnP stopper groups positioned closer to the [Cu(phen)2]+ and C60 subunits, while the other was further away. Computational simulations confirmed the folded structure for rotaxane 7 and also informed that catenane 8 adopted an extended molecular conformation with the chromophores far apart, as shown in Fig. 2.39 The more rigid structure proposed for catenane 8 precluded formation of secondary attractive intramolecular interactions between the chromophores, which was congruent with the single set of resonances observed for the pyrrolic resonances in the 1H NMR spectrum and the monoexponential decay of the 1ZnP* excited state in 8.
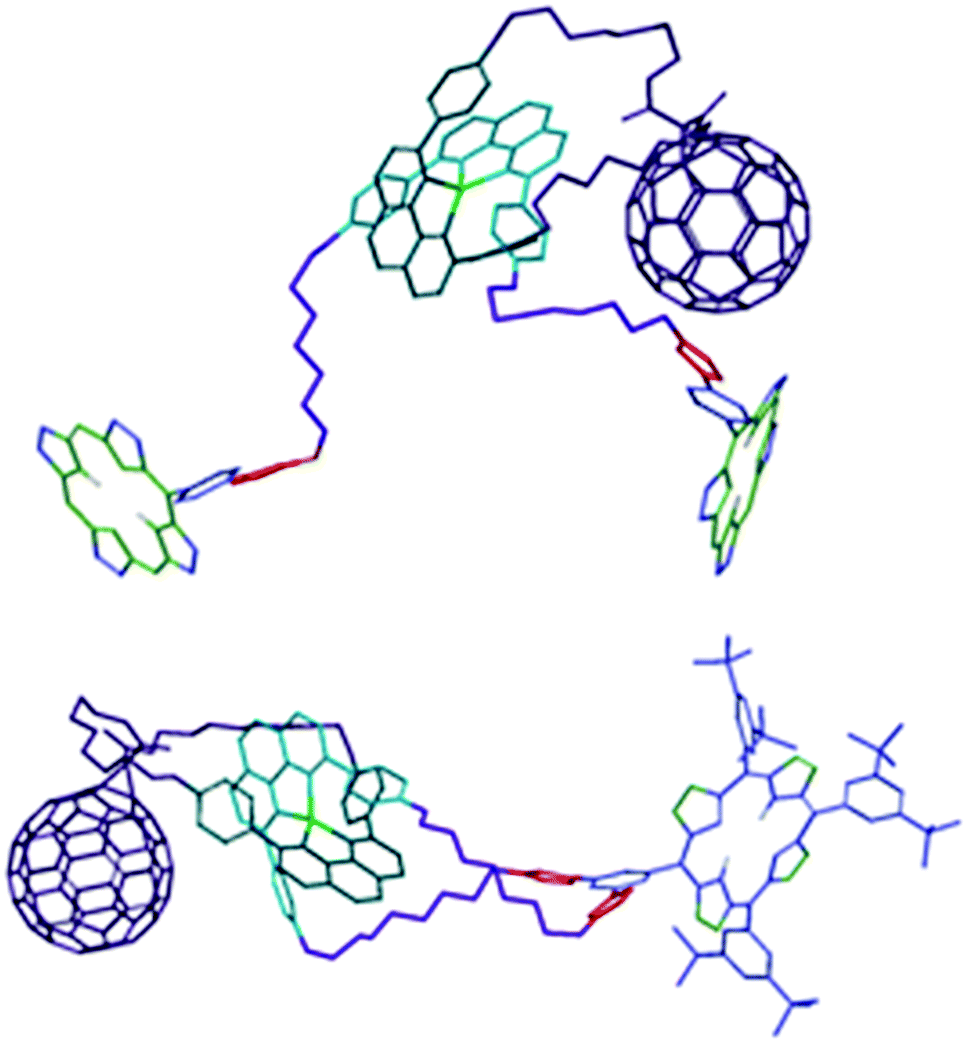 | ||
| Fig. 2 Molecular models of rotaxane 7 (top) and catenane 8 (bottom).41 Adapted with permission from ref. 40. Copyright (2012) American Chemical Society. | ||
3. Investigation of the dynamics of photoinduced processes in ZnP–[Cu(phen)2]+–C60 rotaxanes and catenanes
There are a fair number of photophysical studies on photoactive rotaxanes which have shown that minor structural modifications in the rotaxane structure, such as changes in the size, shape and chemical linkages that connect the chromophores to the rotaxane structure, significantly affect the kinetics of the photoinduced processes.24 Conversely, investigations of photoinduced processes in catenanes are extremely limited,25 probably due to the intrinsic synthetic hurdles imposed by catenane synthesis when compared to the more accessible rotaxanes. No longer constrained by the synthetic difficulties that arise when preparing photoactive C60-based catenanes, a strict comparison of the photophysical properties of rotaxanes and catenanes with identical molecular structural parameters was at reach. Thus, we carried out a detailed and complete electrochemical and photophysical study on rotaxane 7 and catenane 8 as well as structurally related model compounds.37,40,42–44Electrochemical and steady-state absorption and emission spectroscopy informed that there were no significant interactions between the chromophores of rotaxane 740 and catenane 837 in the ground state. From transient absorption37,40 and time-resolved electron paramagnetic resonance42,43 experiments, we found that both interlocked systems presented an identical main sequence of photoinduced processes (Fig. 3) upon exclusive excitation of the ZnP subunits at 420 nm.45 However, the distinct behavior of rotaxane 7 and catenane 8 became evident from the investigation of the dynamics of the main photophysical decay pathways depicted in Fig. 3. Excitation of the ZnP moieties at 420 nm yielded the 1ZnP* state (step 1), which was strongly quenched in both interlocked molecules. The 1ZnP* quenching process was independent of the solvent polarity, thereby signaling a singlet–singlet energy transfer process (EnT) from the 1ZnP* to the [Cu(phen)2]+ subunit (step 2). In principle, EnT should yield the singlet metal-to-ligand charge transfer excited state (1MLCT*) in the complex. However, the 1MLCT* is rapidly converted (in a few hundreds of femtoseconds) into the triplet MLCT state (3MLCT*), which was actually the species detected in our experiments.37,40
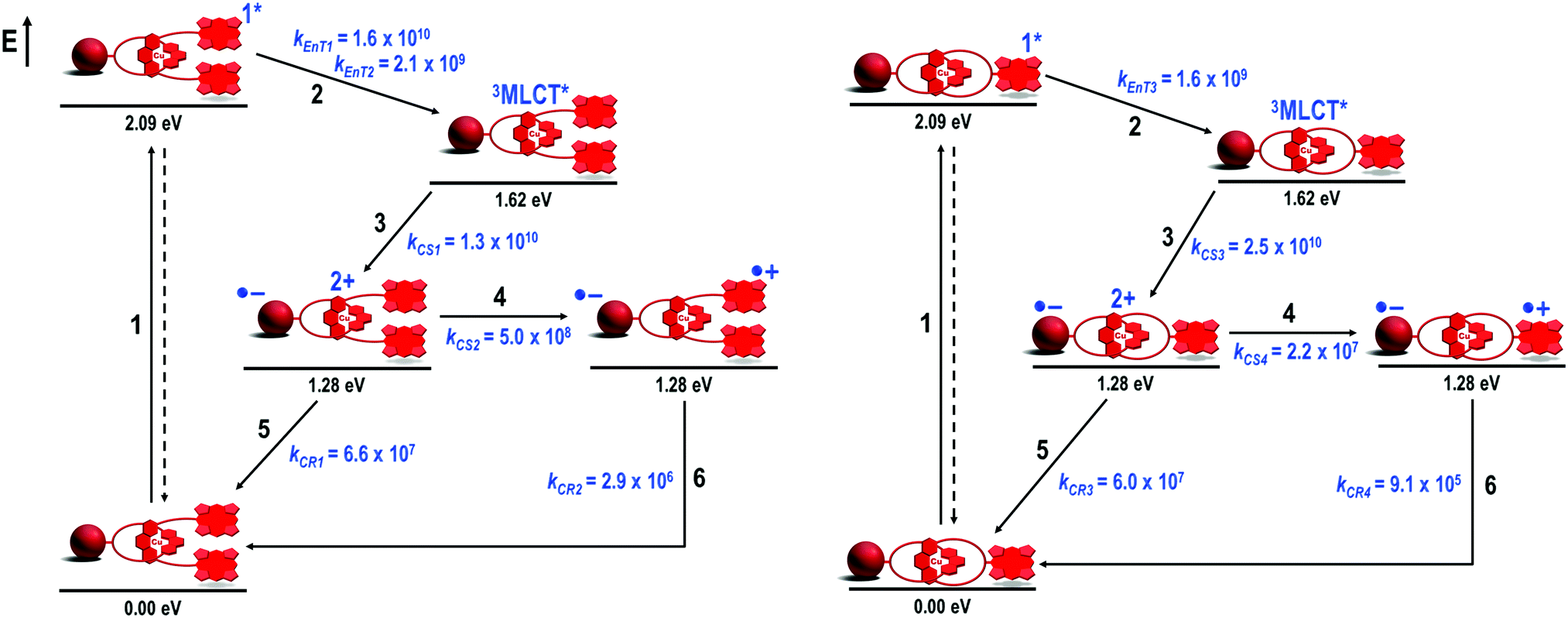 | ||
| Fig. 3 Energy level diagram, proposed decay pathways, and rate constants in s−1 for rotaxane 7 (left) and catenane 8 (right) in benzonitrile following exclusive excitation of the ZnP moiety at 420 nm.46 | ||
From the 1ZnP* fluorescence lifetimes (τ) presented in the previous section, the energy transduction occurred with constant rates of kEnT3 = 1.6 × 109 s−1 (τ = 500 ps) in catenane 8 and kEnT1 = 1.6 × 1010 s−1 (τ = 61 ps) for the close and kEnT2 = 2.1 × 109 s−1 (τ = 400 ps) for the distant ZnP stoppers in rotaxane 7. From the rates,47 we calculated that the energy transfer occurred with 79% quantum yield for catenane 8 and 98% and 83% quantum yields48 for the close and distant ZnP stoppers respectively. The near quantitative yield determined for the EnT event of the close 1ZnP* and the almost identical one obtained for the distant 1ZnP* analog when compared to that afforded for catenane 8 nicely corroborated the structural analyses described in the previous section.
The 3MLCT* manifold was oxidatively quenched by the C60 group via electron transfer (ET) (step 3, Fig. 3) in both structures to afford the intermediate ZnP–[Cu(phen)2]2+–C60˙− charge separated state (CSS). This intermediate CSS could decay to the ground state through charge recombination (CR) (step 5) or undergo a charge shift (CSh) reaction (step 4), which involves donation of an electron from the ZnP moiety to the oxidized [Cu(phen)2]2+ complex to yield the final ZnP˙+–[Cu(phen)2]+–C60˙− CSS. Although the intermediate ZnP–[Cu(phen)2]2+–C60˙− and the final ZnP˙+–[Cu(phen)2]+–C60˙− CSSs are isoenergetic, the final CSS was unambiguously observed in both interlocked molecules by detection of the strong signature transient absorption of ZnP˙+ centered at λmax = 680 nm. The ZnP˙+ lifetime matched the long component decay observed for the biexponential rate law found for the fingerprint absorption of the C60˙− species at λmax = 1040 nm. The C60˙− short component decay was attributed to CR in the ZnP–[Cu(phen)2]2+–C60˙− CSS to give the ground state (step 5), which occurred virtually at the same rate (kCR1 and kCR2 ∼ 6.0 × 107 s−1) in both interlocked molecules. These findings clearly demonstrate that the CSh reaction (step 4) can kinetically compete with CR (step 5) to give the final ZnP˙+–[Cu(phen)2]+–C60˙− CSS in both rotaxane 7 and catenane 8. Fig. 4 shows the differential absorption spectrum afforded from excitation of catenane 8 in benzonitrile, which highlights the transient formation of the ZnP˙+ and C60˙− radical ions.
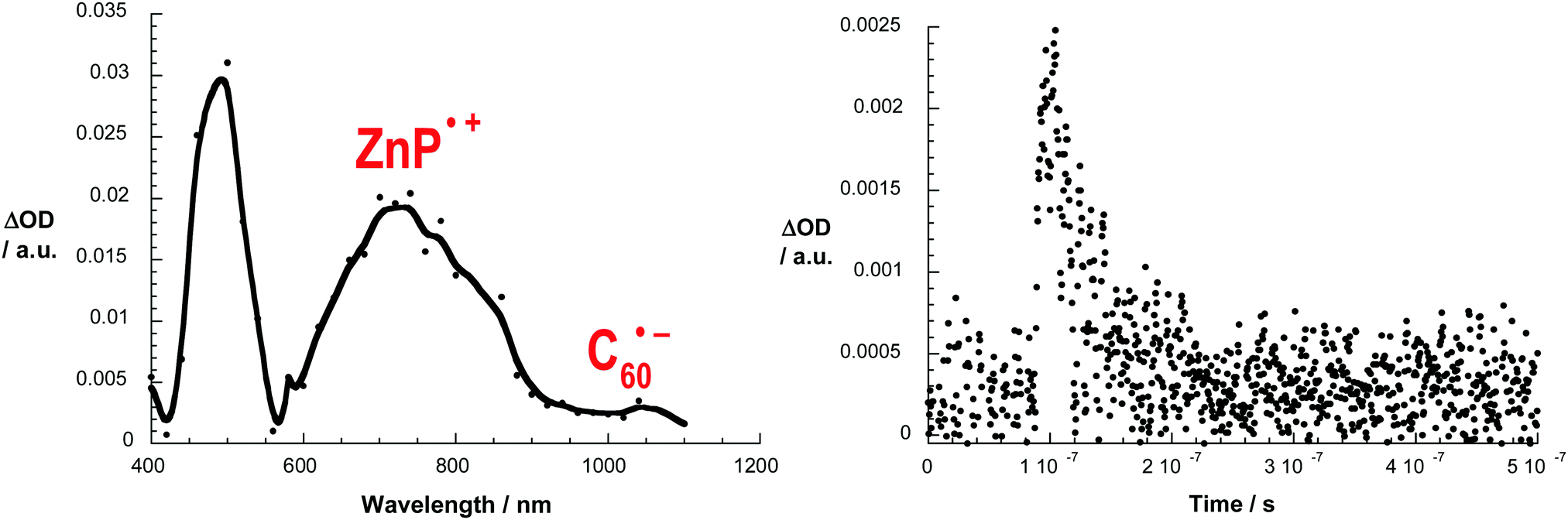 | ||
| Fig. 4 Left: Transient absorption spectra (visible and near-infrared) observed upon nanosecond flash photolysis (355 nm) of ZnP–[Cu(phen)2]+–C60 catenane 8 in benzonitrile with a time delay of 100 ns at room temperature. Right: Time-absorption profiles of the spectrum on the left at 1040 nm, monitoring the charge recombination process. Adapted with permission from ref. 37. Copyright (2010) American Chemical Society. | ||
However, the most dramatic effect of the distinct rotaxane and catenane topology was the rates for the CR reactions of the final ZnP˙+–[Cu(phen)2]+–C60˙− states (step 6, Fig. 3). The lifetime of the final CSS in rotaxane 7 was 0.24 μs (kCR2 = 2.9 × 106 s−1), while it was 1.1 μs (kCR4 = 9.1 × 105 s−1) in catenane 8 under the same conditions. Based on the structural investigation, we attribute the longer lifetime observed for the final ZnP˙+–[Cu(phen)2]+–C60˙− CSS in catenane 8 to its rigid structure, which keeps the active subunits further apart, thus decreasing the electronic coupling of the radical ions when compared to that in the analogous rotaxane 7.
To prove this hypothesis, we carried out the photophysical investigation on rotaxane 7 in the presence of 1,4-diazabicyclo[2.2.2]-octane (DABCO) which transformed rotaxane 7 into the catenane-like structure 9 by coordination to the two ZnP stoppers (Fig. 5).40 The 1:2 DABCO/ZnP ratio was clearly demonstrated by steady state absorption and fluorescence spectroscopy.
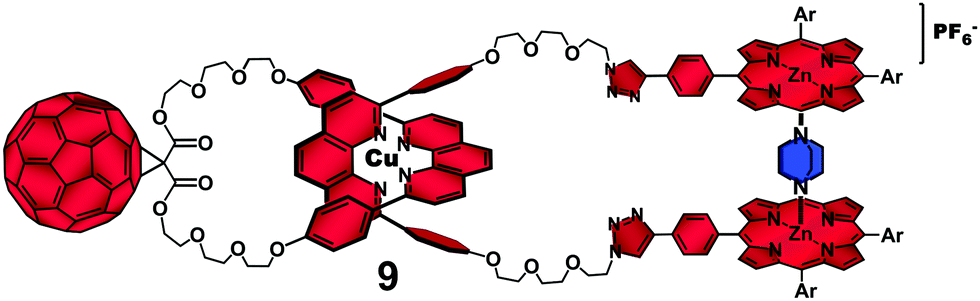 | ||
| Fig. 5 Structure of catenane-like 9 derived from complexation of a DABCO molecule to the ZnP groups of rotaxane 7.38 | ||
Time-resolved transient absorption experiments on 9 indicated a monoexponential decay for the ZnP˙+ transient absorption at λmax = 680 nm with a lifetime of 1.3 μs, which was practically the same as that found for catenane 8 (τ = 1.1 μs). This finding confirmed that formation of coordinative bonds between the ZnP groups and DABCO was sufficiently strong to unfold the interlocked structure by disrupting the π–π secondary interactions between the chromophores originally observed in rotaxane 7.39 Thus, our investigation demonstrated for the first time that the more rigid catenane structure was a more efficient platform than the parent rotaxane to organize ZnP and C60 chromophores in such a way so as to produce long-lived charge separated states.
As described above, the [Cu(phen)2]+ complex plays a central role in the photophysical EnT and ET processes in our rotaxanes and catenanes. Nevertheless, our interest in investigating the relative motions of the interlocked components on the reorientation of the ZnP and C60 groups on the rates of the photoinduced processes required removal of the Cu(I) ion from the phen chelating ligands in our rotaxanes and catenanes. However, removal of the Cu(I) cation from the complex was problematic as the classical demetallation protocol requires the use of nucleophilic cyanide as the active species,28 which is not chemically compatible with the electrophilic C60 subunit.11 Therefore, we developed49 an innovative procedure based on the use of aqueous ammonium hydroxide to remove the Cu(I) template ion from our rotaxanes and catenanes. Inductively coupled plasma mass spectrometry (ICP-MS) revealed that our protocol provided Cu-free interlocked molecules with virtually identical residual copper content as that found for the ones demetallated with cyanide (240 ppm vs. 250 ppm for the cyanide and NH4OH treatments, respectively). However, the kinetics of the demetallation reaction with NH4OH was two-orders of magnitude slower (kNH4OH = 1.6 × 10−3 mol−1 L s−1) than with potassium cyanide (kCN = 0.2 mol−1 L s−1).49
Surprisingly, excitation of the ZnP and/or the C60 groups on the Cu-free interlocked molecules yielded only the corresponding triplet excited states. No evidence was found for ET reactions even in highly polar solvents. 1H NMR structural investigation on rotaxane 10 (Fig. 6, top) revealed that the phen-protons on the macrocycle were strongly shielded, while those on the phen moiety of the thread component were in the usual spectral region. These findings suggested that the phen group on the macrocycle was in the range of the magnetic shielding of the ZnP stoppers. Thus, the ring component seemed not to pirouette about the thread upon demetallation as expected. Instead, the tweezer-like configuration of the ZnP stoppers formed a π–π stacked complex with the phen moiety of the ring in between (Fig. 6, bottom). The π–π intramolecular secondary interactions preclude the expected pirouetting motion promoted by demetallation of rotaxane 10, thus keeping the C60 away from the ZnP donors and shutting down the ET processes. We therefore conclude that the [Cu(phen)2]+ complex is needed to provide the necessary structural organization as well as electronic properties to promote the ET reactions between the ZnP electron donor and the C60 acceptor in our interlocked molecules.40
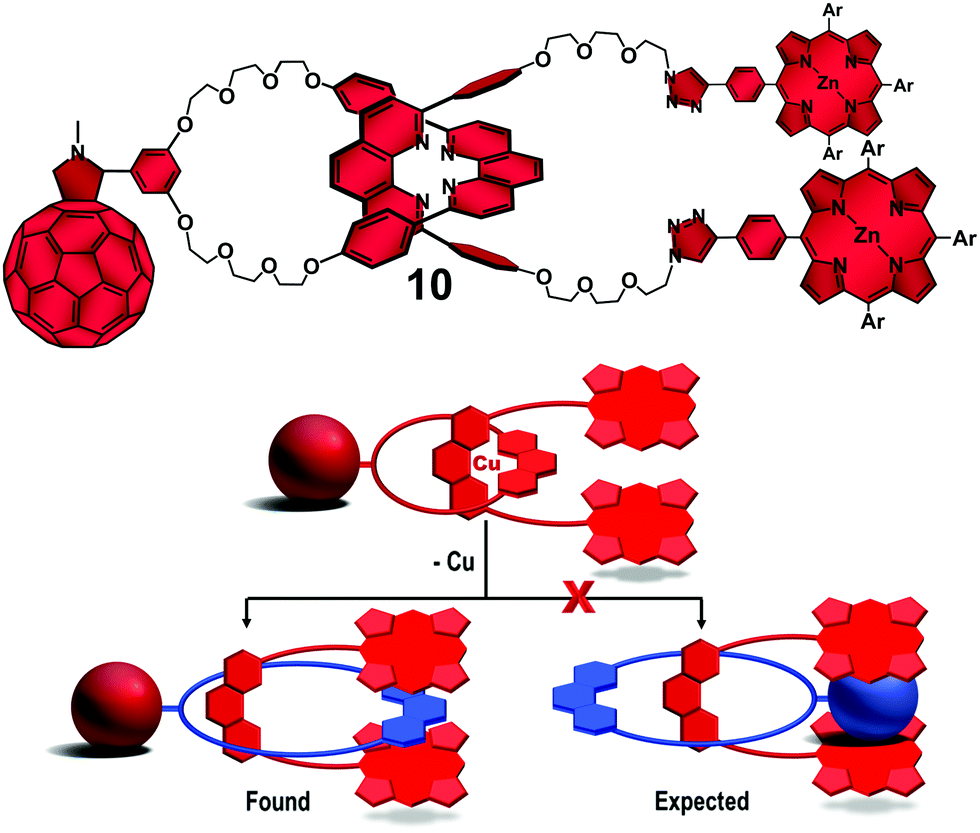 | ||
| Fig. 6 Molecular structure of Cu-free rotaxane 10 (top) and schematic representation of the found and expected rotaxane conformational change upon demetallation (bottom).38 | ||
4. Attempts to improve the yield and lifetime of the photoinduced charge separated states in rotaxanes and catenanes
The lifetime of a specific CSS in artificial photosynthetic models can be improved by three approaches: (1) altering the electronic coupling between the chromophores, (2) modification of the reorganization energy value (λ) of the system and (3) changing the driving force (ΔG0) of the ET reactions.7 In our case, the third approach was the easiest to implement as (1) and (2) would require major structural changes in our molecular design, which in turn could be detrimental to the efficient preparation of the interlocked molecules by our synthetic methodology. Accordingly, we applied the methodology outlined in Fig. 1 to introduce other electron donors into the [Cu(phen)2]+–C60-based interlocked molecules. Ferrocene (Fc) has a low oxidation potential and has proved to be an excellent electron donor for C60-based systems. In a first attempt, we investigated the photophysical properties of (Fc)2–[Cu(phen)2]+–C60 rotaxane 11 (Fig. 7), which was isolated in an impressive 92% yield following our synthetic method.50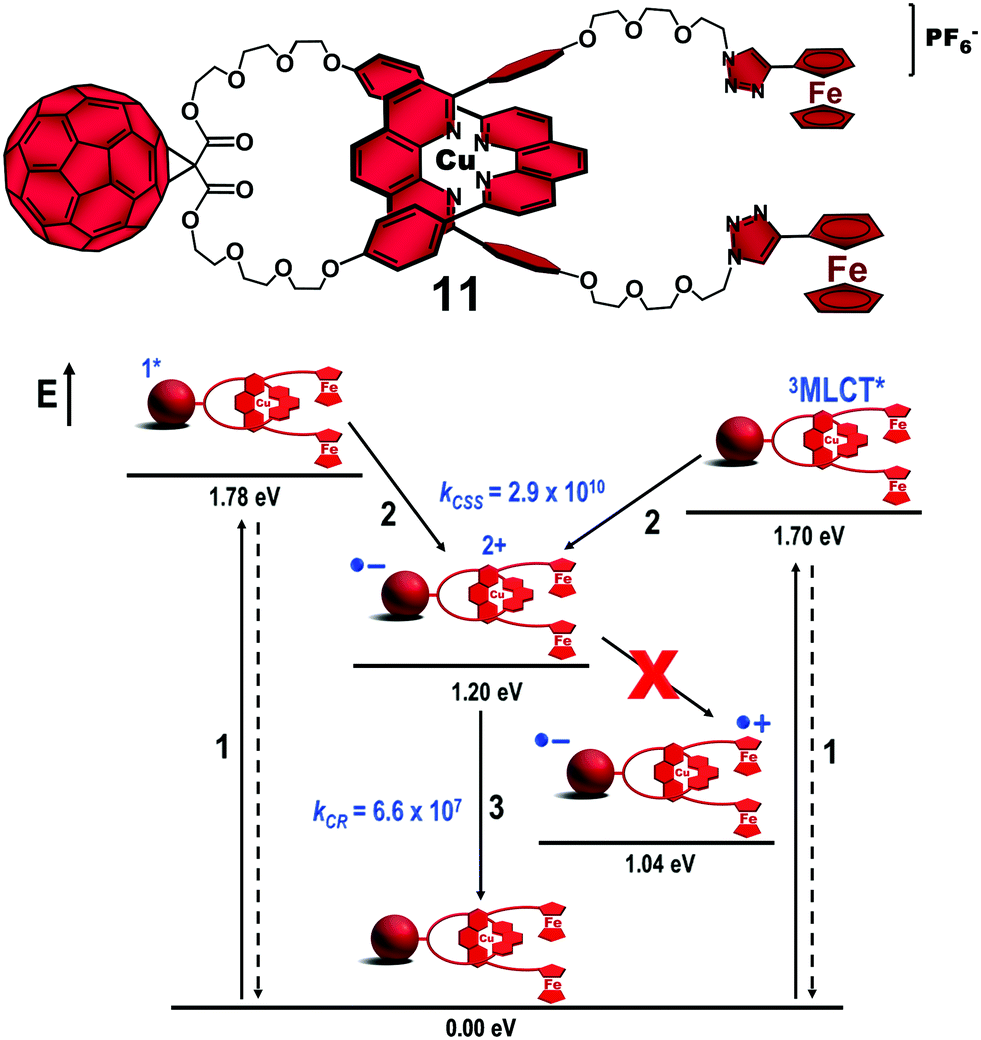 | ||
| Fig. 7 Structure of (Fc)2–[Cu(phen)2]+–C60 rotaxane 11 (top) and its energy level diagram, proposed decay pathways, and rate constants in s−1 following simultaneous excitation of the C60 and [Cu(phen)2]+ subunits in benzonitrile (bottom).46 No unambiguous spectroscopic evidence for the charge shift reaction from the oxidized [Cu(phen)2]2+ central complex to the ferrocene stoppers was found.50 | ||
Excitation of rotaxane 11 at 387 nm simultaneously yielded the 1C60* and the 3MLCT* excited states (step 1, Fig. 7), which rapidly decay (kCSS = 2.9 × 1010 s−1) to afford the intermediate (Fc)2–[Cu(phen)2]2+–C60˙− CSS (step 2). The short component (τ = 15 ns) of the biexponential decay of the C60˙− signature absorption at λmax = 1040 nm was easily assigned to CR in the (Fc)2–[Cu(phen)2]2+–C60˙− CSS (step 3). Based on thermodynamic grounds, the long component (τ = 7.7 μs) could be attributed to CR of the final Fc˙+–[Cu(phen)2]+–C60˙− CSS, whose formation would involve an exergonic CSh reaction (estimated by ΔG0CSh = −0.16 eV) from the oxidized [Cu(phen)2]2+ complex to the Fc subunits. However, the long component was O2-concentration dependent and was quenched to 121 ns in an O2-enriched medium. Due to the weak absorption of the Fc˙+ species at λmax = 680 nm, which was hard to detect in our experiments, we tentatively postulate that the 7.7 μs lifetime corresponds to the decay of the 3C60*, which was formed from the 1C60* excited state by intersystem crossing in competition with the ET reaction that produces the (Fc)2–[Cu(phen)2]2+–C60˙− CSS. The final (Fc)2˙+–[Cu(phen)2]2+–C60˙− CSS was not completely ruled out, but its lifetime, which was 121 ns at best, along with the negligible yield estimated for eventual formation of the final state (at best 0.001%) proved the disadvantageous replacement of the ZnP stoppers by the Fc ones.50
Turning our attention to the catenane architecture, we substituted magnesium(II)porphyrinate (MgP) and the analogous free-base porphyrin (H2P) for the ZnP subunit to afford catenanes 12 and 13 in 55% and 58% yields, respectively (Fig. 8).44 From steady state spectroscopic and electrochemical investigations, we established the energy level diagrams (Fig. 8) for the expected decay pathway of catenanes 12 and 13. As one can see in the energy diagrams, such porphyrin substitution changed the driving force (ΔG0EnT) for the EnT transduction from the porphyrin singlet excited state to the 3MLCT* manifold (step 2) as well as the driving forces for the ET (step 3) and CSh reactions (step 4) that yield the intermediate and final CSSs, respectively.
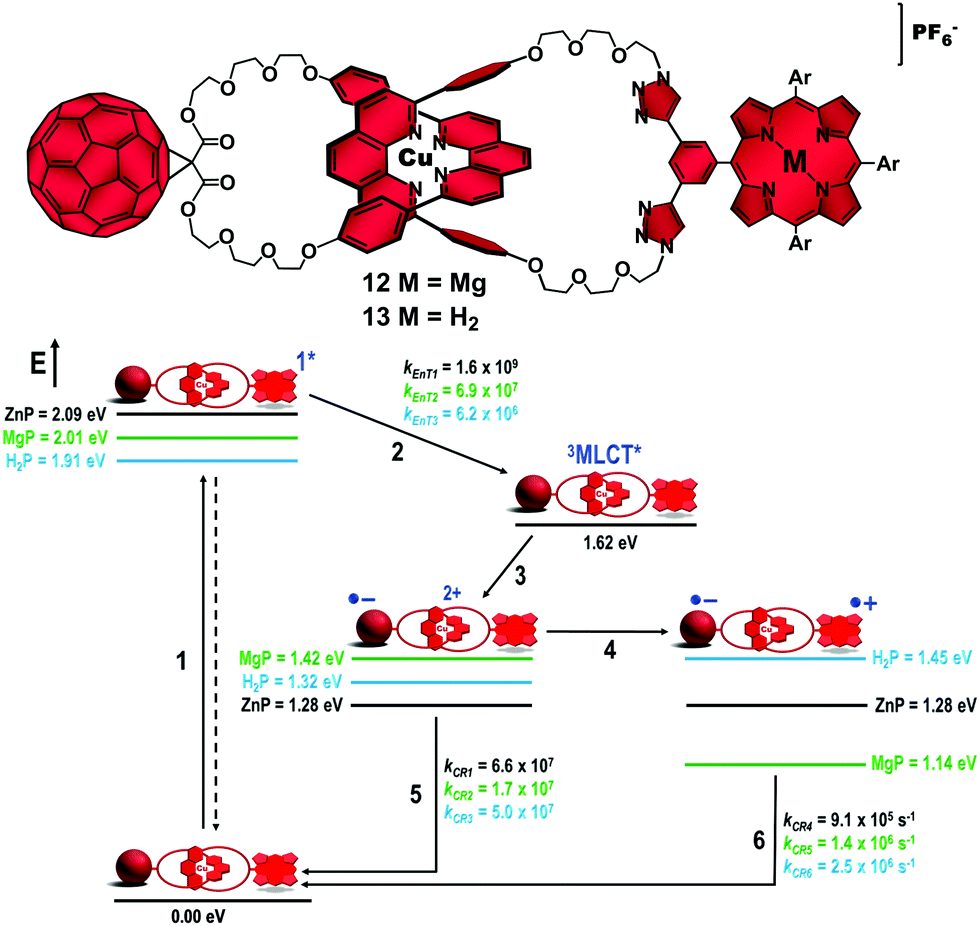 | ||
| Fig. 8 Molecular structures of catenanes 12 and 13 (top). Energy level diagram, proposed decay pathways, and rate constants in s−1 for MgP catenane 12 and H2P catenane 13 following excitation of the porphyrin units in benzonitrile (bottom).38,46 | ||
Energetically, MgP catenane 12 was designed so that the final CSh reaction (step 4, Fig. 8) would be exergonic (ΔG0CSh = −0.28 eV), while it was expected to be endergonic (ΔG0CSh = +0.13 eV) in H2P catenane 13. In ZnP catenane 8, the driving force for step 4 was roughly zero (Fig. 3). Interestingly, the thermodynamic prohibitive CSh reaction in catenane 13 to yield the H2P˙+–[Cu(phen)2]+–C60˙− CSS seemed to take place as the absorption of the H2P˙+ radical cation was observed in our experiments. However, we found that the kinetics of step 5 was 106 times faster than step 4. This pointed to negligible formation of the final H2P˙+–[Cu(phen)2]+–C60˙− CSS in catenane 13. As expected, substitution of the H2P moiety for the ZnP analog was detrimental for the yield and lifetime of the final CSS. On the other hand, the downhill CSh reaction in MgP catenane 11 occurred one order of magnitude faster (kCSh = 6.6 × 108 s−1) than in the ZnP analog (kCSh = 2.2 × 107 s−1).47 However, the efficiency of step 4 in ZnP catenane 8 was estimated to be 46%, while it was only 26% in MgP catenane 12.48
This poorer performance of the MgP catenane 12 to produce the final CSS was surprising and our rationale is as follows. Introduction of the MgP moiety in our catenanes destabilizes the MgP–[Cu(phen)2]2+–C60˙− intermediate CSS (1.42 eV) by 0.14 eV compared to the ZnP–[Cu(phen)2]2+–C60˙− counterpart (1.28 eV). As the 3MLCT* state is at 1.62 eV for both catenanes, the driving force for step 3 is smaller in MgP catenane 12 (ΔG0CSS = −0.20 eV) than in ZnP catenane 8 (ΔG0CSS = −0.34 eV). Using Marcus formalism, both ET reactions are in the normal region as we estimate a reorganization energy (λ) of 0.57 eV for the intermediate CSSs in our catenanes. Therefore, reduction of the driving force for this ET reaction results in a slow rate and low yield for step 3 in MgP catenane 12 compared to ZnP catenane 8.
On the other hand, CRs (step 5) for the intermediate CSSs are in the inverted region (λ = 0.57 eV) for both MgP catenane 12 (ΔG0CR = −1.42 eV) and ZnP catenane 8 (ΔG0CR = −1.28 eV). Consequently, the larger driving force for step 5 results in a longer lifetime for the intermediate CSS in MgP catenane 12 (τ = 60 ns) compared to ZnP catenane 8 (τ = 15 ns). We expected that this low CR rate in conjunction with the faster CSh reaction would result in more efficient formation of the final CSS in MgP catenane 12 than in ZnP catenane 8. Thus, the anticipated decrease in the efficiency of step 3 that would be compensated for by an increase in the efficiency of step 4 was not observed.
1H NMR structural analyses indicated no conformational changes upon substitution of MgP for ZnP, with MgP catenane 12 appearing to adopt the same extended conformation with the chromophores far apart. Therefore, the electronic coupling between the chromophores is similar if not identical in both catenanes. These findings forced us to conclude that the initial EnT process from the porphyrinates to the [Cu(phen)2]+ complex to afford the 3MLCT* state was the efficiency determining step (efficiency order 8 > 12 > 13). The 1ZnP* excited state lies at 2.09 eV, while the parent 1MgP* state is at 2.01 eV. The 3MLCT* was estimated to be at 1.62 eV for both catenanes. From the emission experiments, the EnT transduction occurred with rate constants of kEnT = 6.9 × 107 s−1 and kEnT = 1.6 × 109 s−1 for catenanes 12 and 8, respectively. The quantum yield for the EnT reaction was 42% for MgP catenane 12, which was significantly lower than the 79% yield determined for the ZnP catenane 8. Therefore, the large driving force for step 2 in ZnP catenane 8 (ΔG0CR = −0.49 eV) compared to that in MgP catenane 12 (ΔG0CR = −0.41 eV) results in a higher efficiency for EnT in the former system.
Taken together, the results for the new family of catenanes demonstrated that decreasing the energy level of the final CSS relative to the level of the intermediate CSS to increase the driving force of the CSh reaction led to faster kinetics. However, the energetics and dynamics of our catenanes pointed to the importance of step 2 in the overall yield of the final CSS. Thus, ZnP was a better electron donor compared to MgP and H2P for the efficient formation of the long-lived final CSS in our catenanes. Our investigation proves that decoration of [Cu(phen)2]+ catenane structures with ZnP and C60 groups efficiently yields the final ZnP˙+–[Cu(phen)2]+–C60˙− CSS, which preserves about 1.28 eV of the 2.09 eV originally stored in the 1ZnP* excited state, and with lifetimes in the microsecond time domain.
5. Exploring rotaxane structures as platforms to position four distinct chromophores in space
As previously described, the photoinduced processes between the ZnP, [Cu(phen)2]+ complex and C60 that ultimately generate the long distance and long-living ZnP˙+–[Cu(phen)2]+–C60˙− CSS in our interlocked molecules are sequential. In such systems, yields and lifetimes of the CSSs are dictated by the kinetic competition between the corresponding forward ET reaction that moves the positive and negative charges apart and the wasteful CR processes that promote CR to the ground state. In our case, the lifetime of the intermediate ZnP–[Cu(phen)2]2+–C60˙−CSS is critical for the efficient formation of the ZnP˙+–[Cu(phen)2]+–C60˙− CSS.Based on the classical work by Gust, Moore and Moore,7 we reasoned that introduction of an extra electron donor for the ZnP subunits in our interlocked molecules would lead to the same photophysical events that produced the intermediate ZnP–[Cu(phen)2]2+–C60˙−CSS. However, a judicious choice of the electron donors might result in additional CSh reactions that could compete with the CR reaction of the intermediate CSS more efficiently. The first parallel reaction would be direct CSh from the oxidized complex to the new electron donor, which would work in parallel with the previously described charge shift to the ZnP subunit. The second one would be the CSh reaction from ZnP˙+ to the new electron donor. If the thermodynamic requirements were satisfied, both parallel CSh reactions could converge to generate the more stable and final CSS, characterized by the oxidized new electron donor and the reduced fullerene. Thus, longer-living CSSs could be achieved with better yields at the expense of losing some of the original energy stored in the porphyrin singlet excited states.
To explore this tactic, the rotaxane architecture was ideal as the structural platform as two distinct electron donors could function as stoppers. We designed a variety of rotaxanes,51 assembled as before, i.e. through the [Cu(phen)2]+ complex and with the C60 acceptor on the ring component, but bearing several combinations of zinc(II)phthalocyanates, ferrocenes (Fc) and zinc(II)porphyrinates (ZnP) as stoppers. All stopper combinations yielded the proposed final CSSs with remarkable lifetimes. For illustration, only Fc–ZnP–[Cu(phen)2]+–C60 rotaxane 18 (Fig. 9) will be discussed here. Fc was chosen as the extra electron donor due to its low oxidation potential,50 which allowed for the convergent decay of both intermediate Fc–ZnP–[Cu(phen)2]2+–C60˙− (1.31 eV) and Fc–ZnP˙+–[Cu(phen)2]+–C60˙− (1.31 eV) CSSs to give the same final and most stable Fc˙+–ZnP–[Cu(phen)2]+–C60˙−CSS at 1.13 eV (Fig. 10).
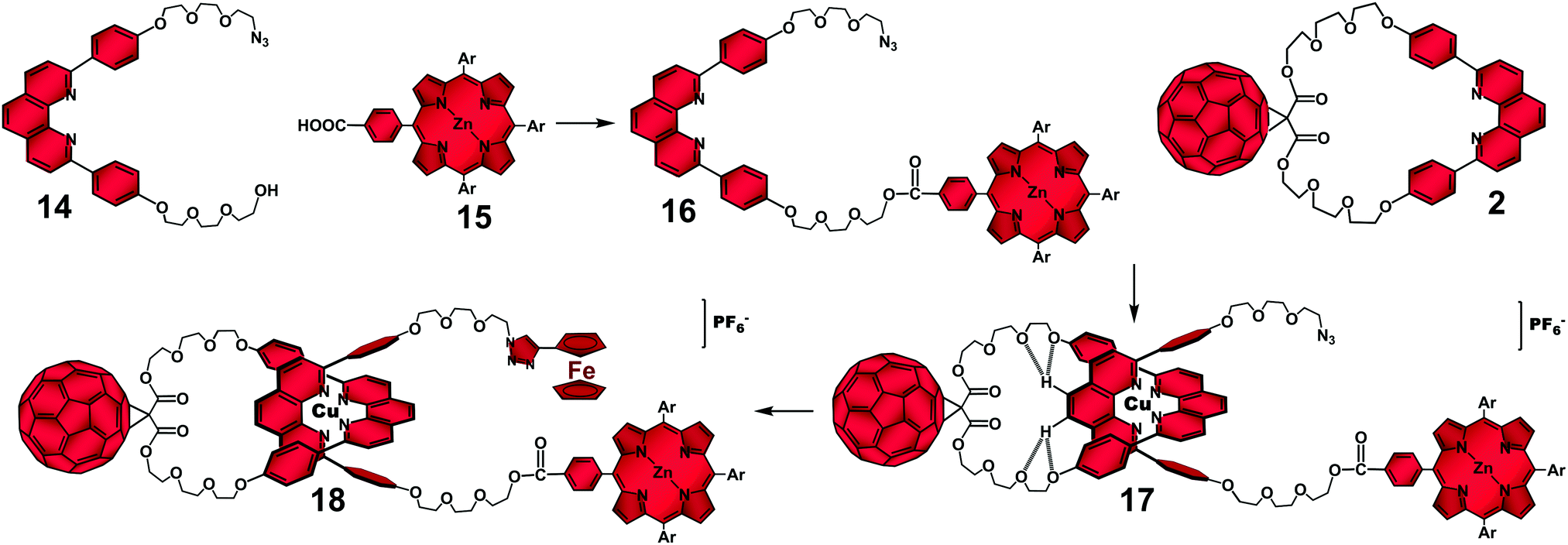 | ||
| Fig. 9 Sequential stoppering–threading–stoppering synthetic strategy for the synthesis of rotaxane 18.38 | ||
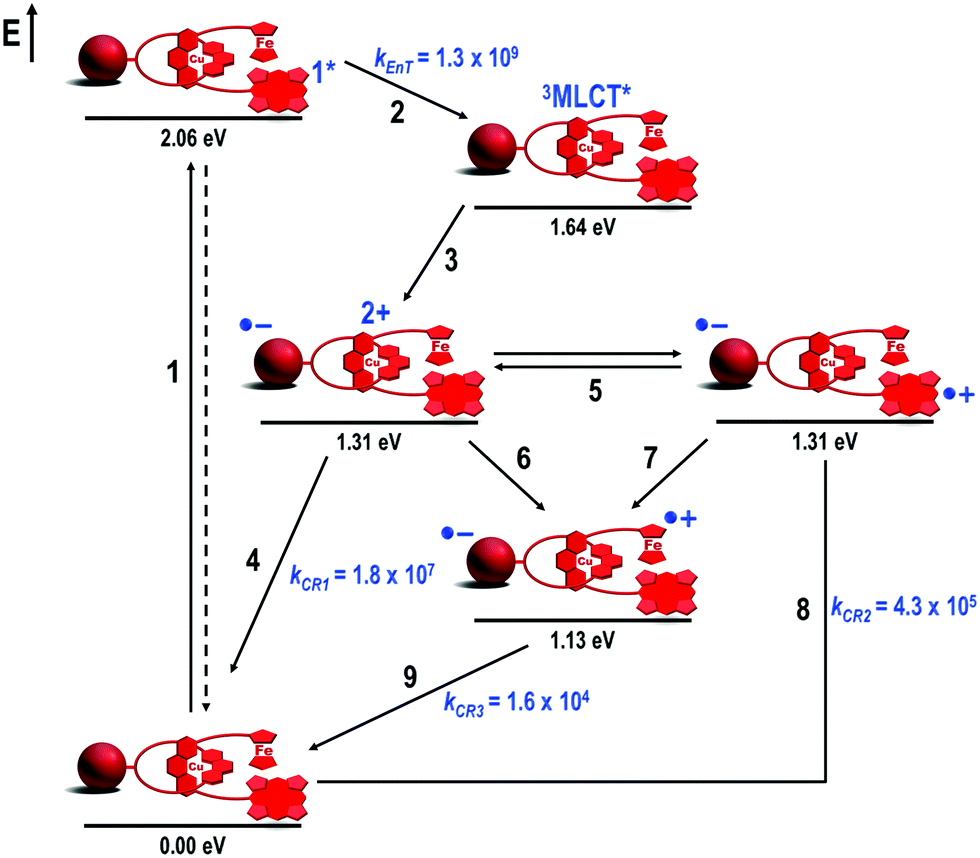 | ||
| Fig. 10 Energy level diagram, proposed decay pathways, and rate constants in s−1 following exclusive excitation of the ZnP subunit at 425 nm in Fc–ZnP–[Cu(phen)2]+–C60 rotaxane 18 in THF.46 | ||
Rotaxane 18 was efficiently synthesized as depicted in Fig. 9 using a slight variation of our original “click” strategy (Fig. 1). In this case, a sequential stoppering–threading–stoppering approach was used, which involved (i) performing an esterification reaction between the hydroxyl functionality in 14 and monocarboxyphenyl Zn(II)porphyrinate derivative 15 to produce monostoppered phen-thread 16; (ii) threading of 16 through macrocycle 2 by means of the Cu(I) templation to afford monostoppered pseudorotaxane 17, and (iii) a second “click” stoppering reaction between the azide functionality on pseudorotaxane 17 and ethynylferrocene. The excellent 90% isolated yield obtained for multichromophoric rotaxane 18 highlighted the versatility of our stoppering–threading–stoppering stepwise approach when incorporating multiple chromophores on the rotaxane.
Exclusive excitation of the ZnP stopper at 425 nm yielded 1ZnP* (step 1, Fig. 10), which partially decayed by EnT (kEnT = 1.3 × 109 s−1) to the 3MLCT* centered on the [Cu(phen)2]+ complex (step 2) along with intersystem crossing to the 3ZnP* manifold (not shown in Fig. 10).51 Formation of the 3MLCT* state triggered ET to produce the first Fc–ZnP–[Cu(phen)2]2+–C60˙− CSS (step 3). The decay of the C60˙− fingerprint transient absorption (λmax = 1040 nm) required three components to fit the data. The short component with a lifetime of 55 ns corresponded to the CR of the first Fc–ZnP–[Cu(phen)2]2+–C60˙− CSS (kCR1 = 1.8 × 107 s−1, step 4). The intermediate component had a lifetime of 2.3 μs, which matched the lifetime determined for the ZnP˙+ ion at λmax = 680 nm, and was therefore assigned to CR of the Fc–ZnP˙+–[Cu(phen)2]+–C60˙− CSS (kCR2 = 4.3 × 105 s−1, step 8). The longest and O2-concentration independent component with a lifetime of 61 μs was attributed to CR of the target Fc˙+–ZnP–[Cu(phen)2]+–C60˙− CSS (kCR3 = 1.6 × 104 s−1, step 9).47
From these data, the combination of Fc and ZnP stoppers increased the lifetimes of the Fc–ZnP–[Cu(phen)2]2+–C60˙− (τ = 55 ns) and Fc–ZnP˙+–[Cu(phen)2]+–C60˙− (τ = 2.3 μs) CSSs when compared to the analogous CSSs in the parent (ZnP)2–[Cu(phen)2]+–C60 rotaxane 7 (τ = 15 ns and 0.24 μs, respectively). We anticipated that both downhill (ΔG0CSh = −0.18 eV) CSh processes from the [Cu(phen)2]2+ (step 6) and ZnP˙+ (step 7) subunits to the Fc moiety would rapidly occur to quench the lifetimes of both CSSs in rotaxane 18. Our explanation for this longer lifetime of Fc–ZnP–[Cu(phen)2]2+–C60˙− and Fc–ZnP˙+–[Cu(phen)2]+–C60˙− CSSs in rotaxane 18 compared to rotaxane 7 was based on the inherent flexibility of the rotaxane backbone, which can fold into more compact structures in solution, driven by intramolecular attractive π–π secondary interactions between the chromophores as previously observed in the structural investigation of rotaxane 7.
In rotaxane 18, the folding process most likely brings the ZnP subunit closer to the [Cu(phen)2]+ complex than the Fc moiety, as the former (with a larger π-system) would establish stronger π–π secondary interactions with the [Cu(phen)2]+ complex than Fc (smaller π-system). Such close contact between the ZnP and [Cu(phen)2]+ subunits allowed those two chromophores to rapidly exchange the positive charge (step 5), which would stabilize both the ZnP˙+ and the oxidized [Cu(phen)2]2+ complex, hence explaining the longer lifetimes observed for the Fc–ZnP–[Cu(phen)2]2+–C60˙− and Fc–ZnP˙+–[Cu(phen)2]+–C60˙− CSSs. Such exchange of positive charges has no thermodynamic penalty as the two CSSs appear to be isoenergetic. The CShs from the [Cu(phen)2]2+ and ZnP˙+ groups to the Fc stopper (steps 6 and 7, respectively) both occur as thermodynamically predicted. Furthermore, both charge transfer processes take place with comparable rates as their driving forces are similar (ΔG0CSh = −0.18 eV), while the proposed folded rotaxane structure keeps the ZnP and [Cu(phen)2]+ subunits approximately equidistant from the Fc stopper.
Confirmation of this plausible interpretation would come from structural analyses. However, the 1H NMR spectrum of rotaxane 18 was complex with multiple signals in different solvents and temperatures. This complexity arose from the low symmetry of the interlocked molecule in conjunction with the proposed conformational flexibility. Despite the lack of structural evidence for the folded conformation of rotaxane 18, we conclude that formation of the target Fc˙+–ZnP–[Cu(phen)2]+–C60˙− CSS occurs from two parallel CSh reactions (steps 6 and 7, Fig. 10), which is consistent with the thermodynamic and kinetic parameters found. This state conserves 1.13 eV and has the longest lifetime (τ = 61 μs at room temperature) ever reported for interlocked artificial photosynthetic model systems.
Conclusions
From the development of a mild chemical protocol compatible with porphyrins, [Cu(phen)2]+ complexes and C60, we were able to efficiently prepare previously inaccessible photoactive rotaxanes and catenanes that function as artificial photosynthetic model systems. We expect that these advances in the template-directed syntheses of interlocked molecules will aid and abet the design and preparation of even more complex photosynthetic model systems. Future investigations should bring further insights into electron transfer reactions in covalently unlinked but nonetheless well-associated donor–acceptor systems with well-defined structures. Our family of mechanically linked photosynthetic model systems absorb light over a wide range of the visible spectrum to trigger rapid energy transfer processes along with sequential electron transfer reactions to afford highly energetic, but still long-lived charge separated states. These features indeed mimic the early principal aspects of the natural photosynthetic energy conversion process. We also demonstrated that the inherent submolecular motions and conformational flexibility provided by the mechanical bond can be used as tools to reposition and reorient the chromophores on the interlocked molecules. As the field of mechanically linked molecules has matured to give birth to elegant molecular machines, in which the interlocked components can move relative to each other through the influence of external stimuli, we reason that such external control of the motions of interlocked components provides interesting possibilities for the design of new interactive donor–acceptor photosynthetic model systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JDMJ is grateful to Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for financial support (grant# 2013/22160-0) and to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for research fellowship (grant# 307635/2018-0). The work on this project at New York University was supported by the US National Science Foundation (NSF) under Grant # CHE-0647334.Notes and references
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Science, Oxford, 2002 Search PubMed.
- G. R. Fleming, J. L. Martin and J. Breton, Nature, 1988, 333, 190–192 CrossRef CAS.
- G. Feher, J. P. Allen, M. Y. Okamura and D. C. Rees, Nature, 1989, 339, 111–116 CrossRef CAS.
- J. Deisenhofer and H. Michel, Angew. Chem., Int. Ed. Engl., 1989, 28, 829–847 CrossRef.
- L. Kalman, R. LoBrutto, J. P. Allen and J. C. Williams, Nature, 1999, 402, 696–699 CrossRef.
- P. L. Dutton and C. C. Mosser, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10247–10250 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198–205 CrossRef CAS.
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695–703 CrossRef CAS PubMed.
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC.
- G. F. Moore, J. D. Megiatto, Jr., M. Hambourger, M. Gervaldo, G. Kodis, T. A. Moore, D. Gust and A. L. Moore, Photochem. Photobiol. Sci., 2012, 11, 1018–1025 RSC.
- A. Hirsch and M. Brettreich, Fullerenes, Wiley-VCH, Weinheim, 2005 Search PubMed.
- D. I. Schuster and J. D. Megiatto, Jr., Nat. Chem., 2009, 1, 182–183 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- J. D. Megiatto, Jr., D. D. Méndez-Hernández, M. Tejeda-Ferrari, A.-L. Teillout, M. J. Llansola-Portolés, G. Kodis, O. G. Poluektov, T. Rajh, V. Mujica, T. L. Groy, D. Gust, T. A. Moore and A. L. Moore, Nat. Chem., 2014, 6, 423–428 CrossRef PubMed.
- A. Antoniuk-Pablant, Y. Terazono, B. J. Brennan, B. D. Sherman, J. D. Megiatto, Jr., G. W. Brudvig, A. L. Moore, T. A. Moore and D. Gust, J. Mater. Chem. A, 2016, 4, 1–10 RSC.
- J. D. Megiatto, Jr., A. Antoniuk-Pablant, B. D. Sherman, G. Kodis, M. Gervaldo, T. A. Moore, A. L. Moore and D. Gust, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15578–15583 CrossRef PubMed.
- J. Ravensbergen, A. Antoniuk-Pablant, B. D. Sherman, G. Kodis, J. D. Megiatto, Jr., D. D. Méndez-Hernández, R. N. Frese, R. Van Grondelle, T. A. Moore, A. L. Moore, D. Gust and J. T. M. Kennis, J. Phys. Chem. B, 2015, 119, 12156–12163 CrossRef CAS PubMed.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, Jr., B. D. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- J. D. Megiatto, Jr., D. Patterson, B. D. Sherman, T. A. Moore, D. Gust and A. L. Moore, Chem. Commun., 2012, 48, 4558–4560 RSC.
- D. J. Cram and J. M. Cram, Acc. Chem. Res., 1978, 11, 8–14 CrossRef CAS.
- J. M. Lehn, Supramolecular Chemistry, Wiley-VCH, Weinheim, 1995 Search PubMed.
- V. Balzani and F. Scandola, Supramolecular Photochemistry, Horwood, Chichester, 1991 Search PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- J. D. Megiatto, Jr. and D. I. Schuster, in Fullerenes: Principles and Applications, ed. F. L. de la Puente and J.-F. Nierengarten, Royal Society of Chemistry, Cambridge, 2012, ch. 10, vol. 1 Search PubMed.
- J. D. Megiatto, Jr. and D. I. Schuster, in Handbook of Carbon Nanomaterials, ed. F. D'Souza and K. M. Kadish, World Scientific Press, London, 2011, ch. 7, vol. 1 Search PubMed.
- C. J. Burns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Inc., Hoboken, 2017 Search PubMed.
- Stoddart defines a mechanical bond as an entanglement in space between two or more component parts, such that they cannot be separated without breaking or distorting chemical bonds between atoms. Rotaxanes and catenanes bear at least one mechanical bond as separation of the ring components in a catenane structure requires rupture of a chemical bond between atoms, while the component parts of a rotaxane can be separated by either rupture of chemical bonds or by simple distortion of the chemical bonds between atoms (for example, expansion of the ring or compression of the stoppers without breaking chemical bonds). Hence, rotaxanes and catenanes are described as mechanomolecules. However, molecular knots also possess entanglements, but not mechanical bonds as they lack component parts. Therefore, molecular knots are classified as mechanically interlocked molecules (MIMs) and mechanomolecules (rotaxanes and catenanes) are a subset of MIMs that possess mechanical bonds. Accordingly, we herein often use the terms mechanically-linked or interlocked molecules to refer to rotaxanes and catenanes.
- J.-P. Sauvage, Acc. Chem. Res., 1990, 23, 319–327 CrossRef CAS.
- A. F. P. Alcantara, L. A. Fontana, V. H. Rigolin, Y. S. F. Andrade, M. A. Ribeiro, W. P. Barros, C. Ornelas and J. D. Megiatto, Angew. Chem., Int. Ed., 2018, 57, 8979–8983 CrossRef CAS PubMed.
- J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- L. Flamigni, J.-P. Collin and J.-P. Sauvage, Acc. Chem. Res., 2008, 41, 857–871 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J. D. Megiatto, Jr. and D. I. Schuster, J. Am. Chem. Soc., 2008, 130, 12872–12873 CrossRef PubMed.
- J. D. Megiatto, Jr., R. Spencer and D. I. Schuster, Org. Lett., 2009, 11, 4152–4155 CrossRef PubMed.
- J. D. Megiatto, Jr. and D. I. Schuster, Chem. – Eur. J., 2009, 15, 5444–5448 CrossRef PubMed.
- J. D. Megiatto, Jr. and D. I. Schuster, New J. Chem., 2010, 34, 276–286 RSC.
- J. D. Megiatto, Jr., D. I. Schuster, S. Abwandner, G. de Miguel and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 3847–3861 CrossRef PubMed.
- Ar = 3,5-di-tert-butylphenyl groups located at the meso positions of the porphyrins.
- J. D. Megiatto, Jr., R. Spencer and D. I. Schuster, J. Mater. Chem., 2011, 21, 1544–1550 RSC.
- J. D. Megiatto, Jr., D. I. Schuster, G. de Miguel, S. Wolfrum and D. M. Guldi, Chem. Mater., 2012, 24, 2472–2485 CrossRef PubMed.
- Molecular modeling was afforded from Spartan’06 software using the parametric semi-empirical method 3 (PM3 minimization). For clarity, the hydrogen atoms have been removed from the structures along with the 3,5-di-tert-butylphenyl groups at the meso positions of the porphyrin subunits in rotaxane 7.
- M. Jakob, A. Berg, H. Levanon, D. I. Schuster and J. D. Megiatto, Jr., J. Phys. Chem. A, 2011, 115, 5044–5052 CrossRef CAS PubMed.
- M. Jakob, A. Berg, H. Levanon, D. I. Schuster and J. D. Megiatto, Jr., J. Phys. Chem. C, 2011, 115, 24555–24563 CrossRef CAS.
- S. V. Kirner, D. M. Guldi, J. D. Megiatto, Jr. and D. I. Schuster, Nanoscale, 2014, 7, 1145–1160 RSC.
- We also investigated our rotaxanes and catenanes upon excitation of the [Cu(phen)2]+ and C60 moieties (at 387 and 355 nm) to gather further information about the generation and fate of the proposed intermediate states shown in Fig. 3. However, the high absorption and emission coeficients of ZnP rendered this chromophore the best probe to investigate the photophysical decay pathways of our interlocked molecules. For simplicity, only excitation at 420 nm is discussed in this Tutorial.
- The energy levels (in eV) were derived from steady-state absorption, fluorescence, and electrochemical measurements and are not in scale. The rate constants (k) in s−1 were obtained from transient absorption and fluorescence measurements. kEnT = rate constant for energy transfer, kCS = rate constant for charge separation and kCR = rate constant for charge recombination.
- We used the relation
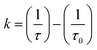 , in which τ is the lifetime of a quenched excited state and τ0 is the unquenched lifetime of the same state in a model compound, to calculate the rate constants (k) for the relevant intermediates in the photophysical decay of our rotaxanes and catenanes. A zinc(II)porphyrinate reference compound had a singlet lifetime of τ0 = 2.4 ns under exactly the same conditions.
, in which τ is the lifetime of a quenched excited state and τ0 is the unquenched lifetime of the same state in a model compound, to calculate the rate constants (k) for the relevant intermediates in the photophysical decay of our rotaxanes and catenanes. A zinc(II)porphyrinate reference compound had a singlet lifetime of τ0 = 2.4 ns under exactly the same conditions. - Quantum yield (ϕ) defined as ϕ = k × τ, in which τ is the lifetime of the state and k is the rate constant associated to its formation. In our efficiency calculations, we did not consider intersystem crossing (ISC), although the 3ZnP* manifold was marginally detected in our O2-experiments for rotaxane 7 and catenane 8.
- J. D. Megiatto, Jr. and D. I. Schuster, Org. Lett., 2011, 13, 1808–1811 CrossRef PubMed.
- J. D. Megiatto, Jr., K. Li, D. I. Schuster, A. Palkar, M. A. Herranz, L. Echegoyen, S. Abwandner, G. de Miguel and D. M. Guldi, J. Phys. Chem. B, 2010, 114, 14408–14419 CrossRef PubMed.
- S. V. Kirner, C. Henkel, D. M. Guldi, J. D. Megiatto, Jr. and D. I. Schuster, Chem. Sci., 2015, 6, 1–12 RSC.
| This journal is © The Royal Society of Chemistry 2020 |