
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed oxidative dehydrogenative carbonylation reactions using carbon monoxide and mechanistic overviews
Can
Zhu†
 *a,
Jie
Liu†
a,
Man-Bo
Li†
ab and
Jan-E.
Bäckvall
*a
*a,
Jie
Liu†
a,
Man-Bo
Li†
ab and
Jan-E.
Bäckvall
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: jeb@organ.su.se; zhucaner@gmail.com
bInstitute of Physical Science and Information Technology, Anhui University, Hefei 230601, China
First published on 18th December 2019
Abstract
Carbon monoxide, which is an abundant and inexpensive carbonyl source, has been widely applied to synthesize carbonyl-containing compounds, for example ketones, esters, and amides. These types of compounds are ubiquitous in natural products, pharmaceuticals, as well as in functional materials. This review focuses on the palladium-catalyzed dehydrogenative C–H/X–H (X = C, N, O) carbonylation transformations under oxidative conditions. The related C–H bonds here include C(sp)–H, C(sp2)–H, and C(sp3)–H bonds. From a step- and atom-economy perspective, transition metal-catalyzed oxidative dehydrogenative C–H/X–H carbonylation reactions with CO constitute one of the most efficient strategies for the construction of versatile carbonyl groups, without the requirement of pre-functionalized substrates.
 Can Zhu | Can Zhu received his PhD degree under the supervision of Prof. Shengming Ma from Shanghai Institute of Organic Chemistry, China in 2014. After that, he started his postdoctoral studies in the group of Prof. Jan-E. Bäckvall at Stockholm University. In Sep. 2016, he was awarded Alexander von Humboldt Foundation Postdoctoral Fellowship to join Prof. Frank Glorius's research group, Westfälische Wilhelms-Universität Münster, working on non-noble metal catalysis. Since July 2018, he moved back to Stockholm University as a researcher. Currently, his interests focus on the development of green oxidation methodology via a biomimetic approach. |
 Jie Liu | Jie Liu was born in 1990 in Hunan, China. He studied at Wuhan University, China, where he obtained his BS and MSc degree in 2012 and 2014 under the supervision of Prof. Aiwen Lei. Then he joined Prof. Matthias Beller's group at the Leibniz Institute for Catalysis, Germany and received his PhD degree in 2017. He is currently a postdoctoral researcher in Prof. Jan-E. Bäckvall's research group at Stockholm University, Sweden. He is interested in the development of homogenous catalysts for oxidation chemistry. |
 Man-Bo Li | Man-Bo Li was born in 1986. He finished his bachelor studies and received his PhD degree at the University of Science and Technology of China in 2013 under the supervision of Prof. Shi-Kai Tian. After three years of research work on metal nanoclusters at the Chinese Academy of Sciences and a short stay at KAUST on heterogeneous catalysis, he joined Prof. Jan-E. Bäckvall's research group at Stockholm University to develop efficient heterogeneous Pd-catalyzed oxidations. In November of 2019, he finished his postdoctoral research in Sweden and was appointed professor at Anhui University, China. |
 Jan-E. Bäckvall | Jan-Erling Bäckvall was born in Malung, Sweden, in 1947. He received his PhD from the Royal Institute of Technology, Stockholm, in 1975 with Prof. B. Åkermark. After postdoctoral work (1975–1976) with Prof. K. B. Sharpless at Massachusetts Institute of Technology, he joined the faculty at Royal Institute of Technology. He was appointed Professor of Organic Chemistry at Uppsala University in 1986. In 1997, he moved to Stockholm University where he is currently Professor of Organic Chemistry. He is a member of the Royal Swedish Academy of Sciences, Finnish Academy of Science and Letters, and Academia Europaea. His current research interests include transition-metal-catalyzed organic transformations, biomimetic oxidations, and enzyme catalysis. |
Key learning points1. Palladium-catalyzed carbonylative coupling reactions using CO2. Oxidative dehydrogenative coupling reactions 3. Four different means for the palladation of C–H bonds 4. Pathways of carbonylation step using CO with weak/strong nucleophiles 5. Oxidation of Pd(0) to Pd(II) by various oxidants |
1. Introduction
The carbonyl group, a basic yet crucial functional group in organic chemistry, widely exists in natural products, pharmaceutical compounds, and functional materials. A carbonyl group can be easily transformed into many other functional groups, such as alcohols, imines, amines, amides, and olefins.1Classic transition metal-catalyzed cross-coupling reactions provide efficient pathways for construction of complex molecules from pre-functionalized electrophiles and nucleophiles (Scheme 1a).2 In these cross-coupling reactions, palladium catalysts are highly efficient.3 Since the identification of carbon monoxide (CO) by William Cruikshank in 1800, exploration of the utility of this cheap and abundant C1 feedstock in organic chemistry has been at the core of organic synthesis.4 The bond-dissociation energy of CO is stronger than that of N2 (1072 kJ mol−1vs. 942 kJ mol−1) and represents the strongest chemical bond known, which makes CO relatively inert. It can be activated by various transition metals and here the identity of the metal is extremely important for imparting reactivity. In recent years, CO has become an attractive renewable building block in carbonylation reactions, in particular in those catalyzed by palladium. The reason why palladium is frequently used is that it can also participate in many other fundamental organometallic reactions, such as oxidative addition, reductive elimination, migratory insertion, β-elimination, and nucleophilic attack on coordinated ligands as well as in oxidation reactions. In the presence of a transition metal catalyst, reactions of electrophiles, nucleophiles, and carbon monoxide result in carbonylative coupling reactions (Scheme 1b).5,6 This type of carbonylation reaction has been demonstrated as a direct strategy for the synthesis of various carbonyl-containing compounds and their derivatives. However, these reactions require pre-functionalized substrates, for example, aromatic halides or triflates as electrophiles, and boronic acids or zinc reagents as nucleophiles.
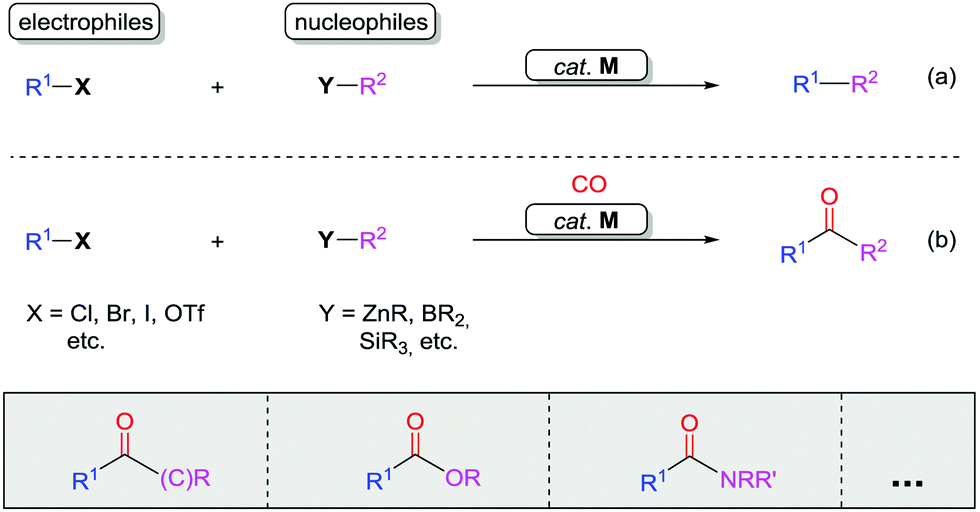 | ||
| Scheme 1 (a) Transition metal-catalyzed coupling reactions. (b) Transition metal-catalyzed carbonylative coupling reactions. M = transition metal. | ||
Oxidative dehydrogenative coupling reactions constitute a direct approach for the construction of C–X (X = C, N, or O) bonds by only using C–H/X–H bonds (Scheme 2).7 The related C–H bonds here include C(sp)–H, C(sp2)–H, and C(sp3)–H bonds. These dehydrogenative coupling reactions do not require the preparation of pre-functionalized substrates, which can shorten synthetic routes. Oxidative dehydrogenative coupling reactions are therefore considered as one of the most efficient synthetic methods in the field of chemical synthesis.
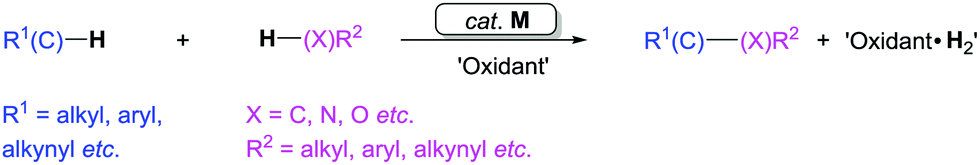 | ||
| Scheme 2 Transition metal-catalyzed oxidative dehydrogenative cross-coupling reactions. | ||
Oxidative dehydrogenative carbonylation reactions using carbon monoxide combine the concept of dehydrogenative coupling and carbon monoxide-based carbonylation. These reactions lead to significant increase of efficiency in the synthesis of carbonyl-containing compounds (Scheme 3). In the past decade, palladium catalysis has played a major role in oxidative dehydrogenative carbonylation.6,8,9 This review focuses on the research progress achieved in recent years within the area of palladium-catalyzed oxidative dehydrogenative carbonylation.
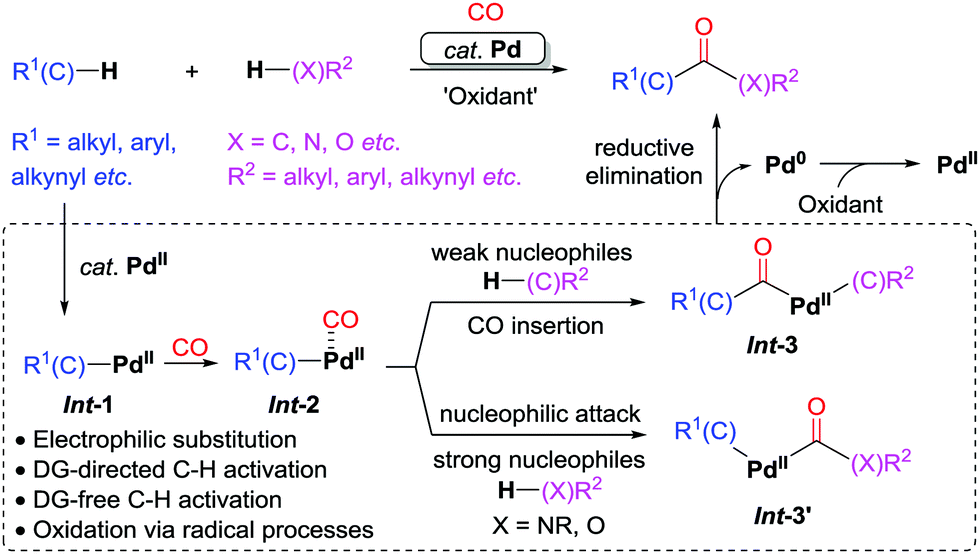 | ||
| Scheme 3 Palladium-catalyzed oxidative dehydrogenative carbonylation reactions. DG = directing group. | ||
As the initial step in Scheme 3, C–H palladation would produce the active species Int-1via mechanistically different pathways depending on the interaction of the substrates and the palladium catalyst, including electrophilic substitution, DG (DG = directing group)-directed C–H activation, DG-free C–H activation, or oxidation via radical processes.10 Coordination of CO to palladium leads to Int-2, which would undergo a subsequent CO insertion followed by reaction with a weak nucleophile to give Int-3. Reductive elimination from Int-3 would furnish the product. When N- or O-containing strong nucleophiles are employed, nucleophilic attack on the coordinated CO may occur to give Int-3′, which would undergo reductive elimination forming the carbonyl products. The released Pd(0) is reoxidized to Pd(II) with a suitable oxidant, which closes the catalytic cycle. We believe that this strategy will have a positive impact on the development of organic synthesis.
2. Palladium-catalyzed oxidative dehydrogenative carbonylation reactions
2.1 C(sp)–H/(X)–H oxidative dehydrogenative carbonylation
In 1980, Tsuji et al. reported the first example on a palladium-catalyzed oxidative dehydrogenative carbonylation reaction of terminal alkynes with alcohols in the presence of CO (Scheme 4).11a Stoichiometric amounts of CuCl2 were employed to reoxidize the palladium catalyst. Terminal acetylenes were successfully converted to acetylenecarboxylates in high yields under an atmospheric pressure of carbon monoxide at room temperature. This oxidative coupling approach, using readily available alkynes and alcohols, provided a direct and efficient access to acetylenecarboxylates, which are usually prepared from the oxidation of the corresponding propargylic alcohols, or the corresponding olefinic acids.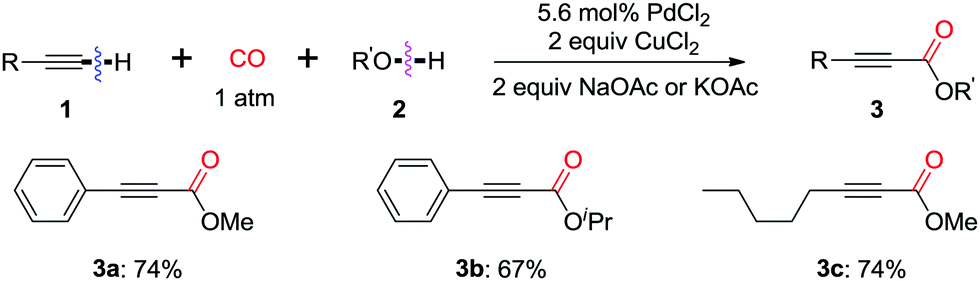 | ||
| Scheme 4 Palladium-catalyzed oxidative carbonylation of terminal acetylenes. | ||
In 1999, Ishii and co-workers modified the catalytic system, by using molybdovanadophosphate (NPMoV) as the oxidation catalyst together with oxygen as the terminal oxidant (Scheme 5).11b It was observed that the reaction gave the acetylenecarboxylate product in MeOH, while a double insertion of CO was observed in dioxane to afford the corresponding maleic anhydride 4via cyclopalladium(II) intermediate Int-4.
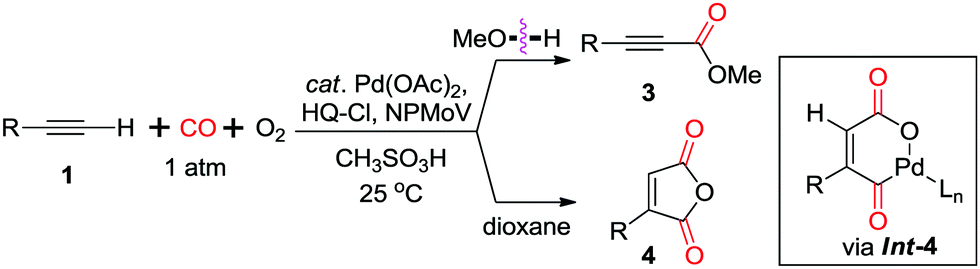 | ||
| Scheme 5 Palladium-catalyzed aerobic oxidative carbonylation of terminal acetylenes. HQ-Cl = chlorohydroquinone. | ||
In 2013, the group of Bhanage found that a heterogeneous catalyst, palladium-on-carbon (Pd/C) was efficient for promoting the oxidative carbonylation using CO as the C1 source (Scheme 6).11c Interestingly, the reaction in dioxane gave acetylenecarboxylate product 3, while the reaction under solvent-free reaction conditions afforded cis-diester product 5. Moreover, the heterogeneous catalyst, Pd/C showed remarkable recyclability, being used in up to six consecutive cycles without significant loss of activity or selectivity.
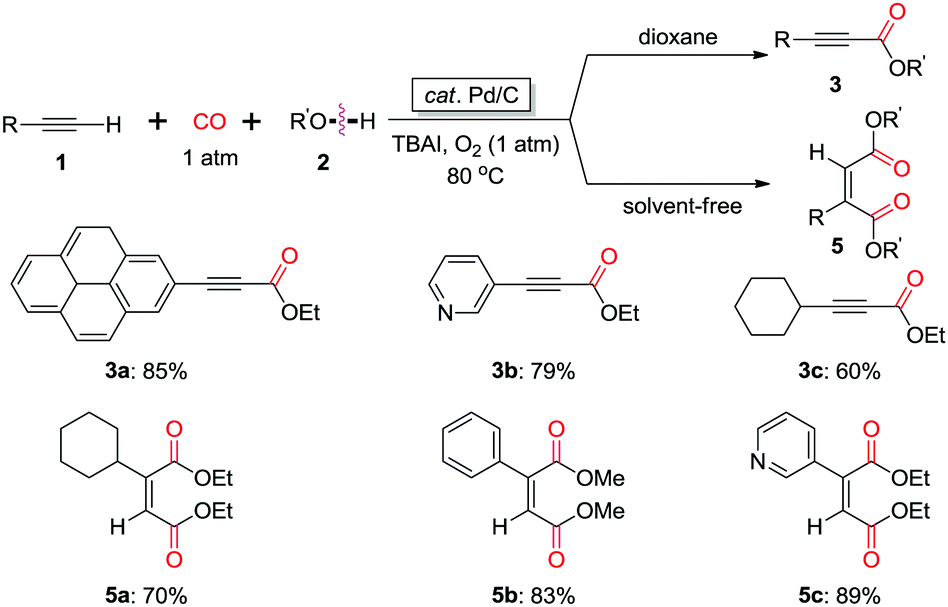 | ||
| Scheme 6 Synthesis of α,β-alkynyl esters and unsymmetrical maleate esters catalyzed by heterogeneous catalyst Pd/C. | ||
2.2 C(sp2)–H/(X)–H oxidative dehydrogenative carbonylation
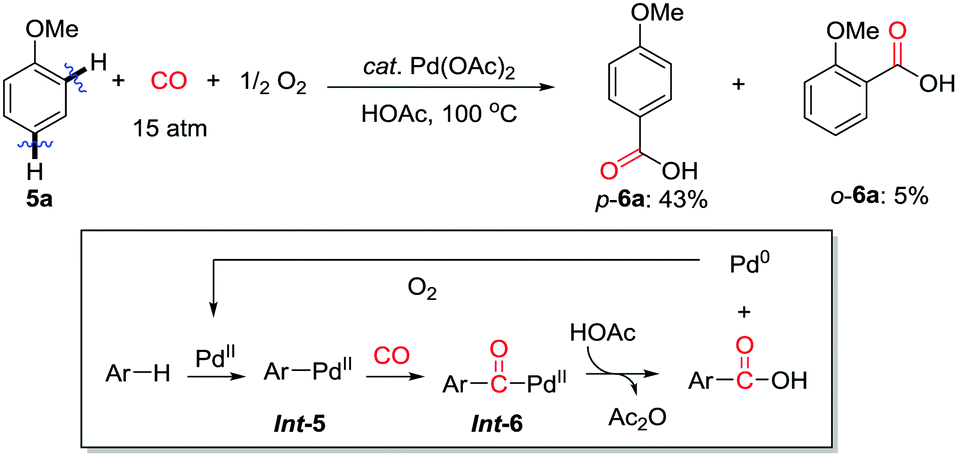 | ||
| Scheme 7 Carboxylation of anisole derivatives with CO and O2 catalyzed by Pd(OAc)2. | ||
In 2008 the first example of regioselective Pd(II)-catalyzed carbonylation of unactivated aryl and vinyl C(sp2)–H bonds was reported by the Yu group (Scheme 8).13 A carboxylic acid group was used as the directing group for the C(sp2)–H activation forming a C–Pd bond, therefore dicarboxylic acid derivatives were produced from benzoic and phenylacetic acids. In an analogous manner cis-1,2-dicarboxylic acids were successfully obtained when an α,β-unsaturated carboxylic acid was employed. Treatment of benzoic acid with sodium acetate produces sodium carboxylate, in which the carboxylate group functions as the directing group for the Pd-catalyzed C–H carbonylation. Subsequent intramolecular cyclization of the carboxylate directing group leads to the formation of phthalic anhydride derivatives, which are in situ hydrolyzed to give dicarboxylic acid derivatives.
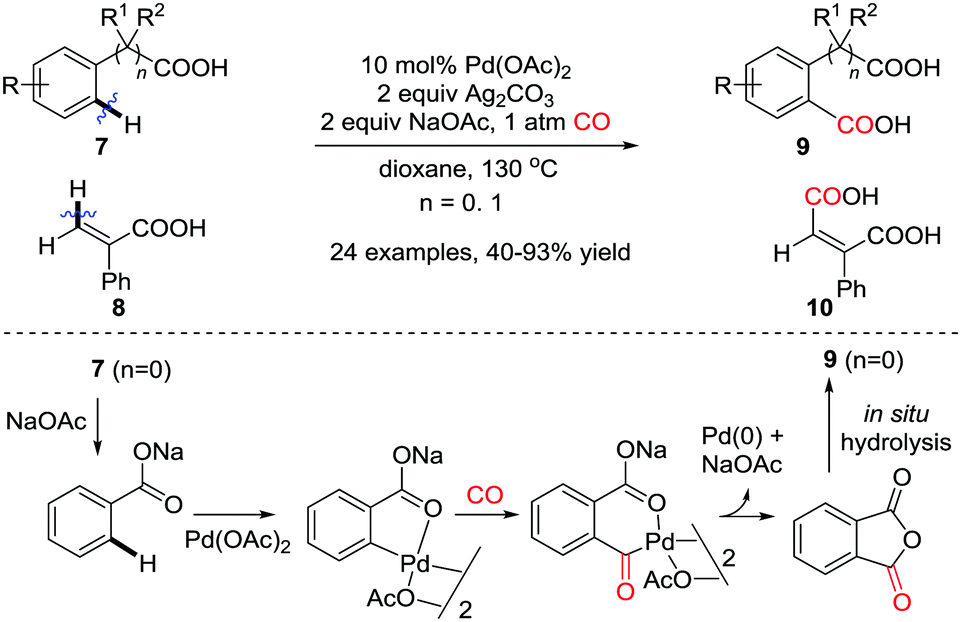 | ||
| Scheme 8 Pd(II)-Catalyzed carboxylation of aryl and vinyl C–H bonds. | ||
Shortly thereafter, in 2009, the Booker-Milburn group developed a room-temperature palladium-catalyzed C–H activation/carbonylation/alcohol-quenching of anilines for the synthesis of o-aminobenzoic acid esters (Scheme 9a).14 Ureas were efficient directing groups for the ortho-C–H activation. The reaction leads to imidates 12 in DCM, while methyl anthranilates 13 are selectively produced in a mixed solvent of THF/MeOH via methanolysis of imidates 12. Later, an amine was also employed as the directing group to promote the C–H activation/carbonylation/alcohol-quenching for the synthesis of o-aminobenzoates (Scheme 9b).15
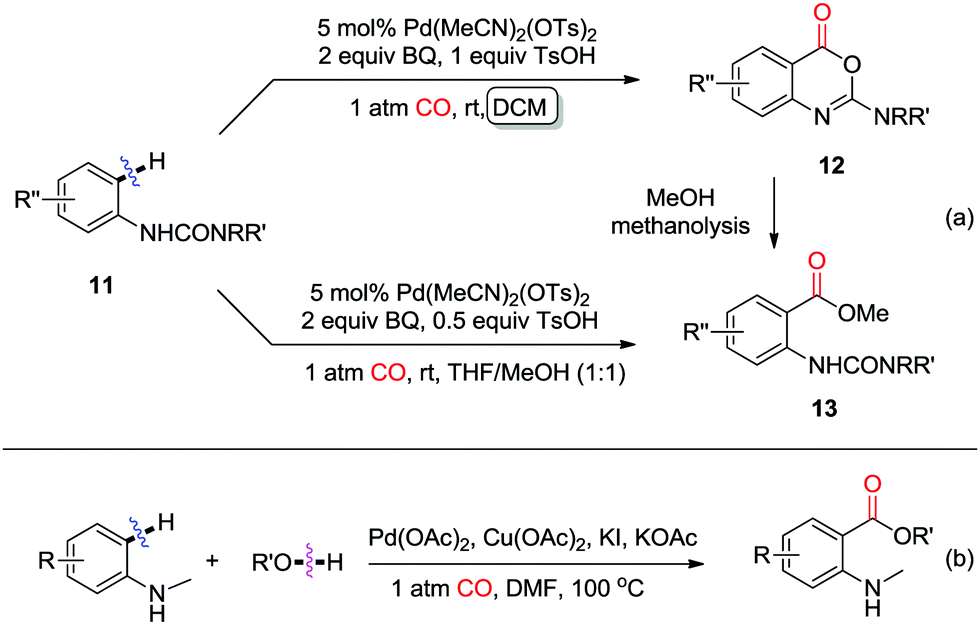 | ||
| Scheme 9 Palladium-catalyzed C–H activation/carbonylation of aniline derivatives. | ||
In 2010, the Shi group extensively developed a Pd(II)-catalyzed directed ortho-carbonylation of N,N-dimethylbenzylamines 14 to give 15 using CO (Scheme 10).16 It was demonstrated that LiCl is crucial for enhancing the product yield. It is worth mentioning that the directing group can be efficiently removed under catalytic hydrogenation conditions, as shown by the formation of 16 in 90% yield from 15a.
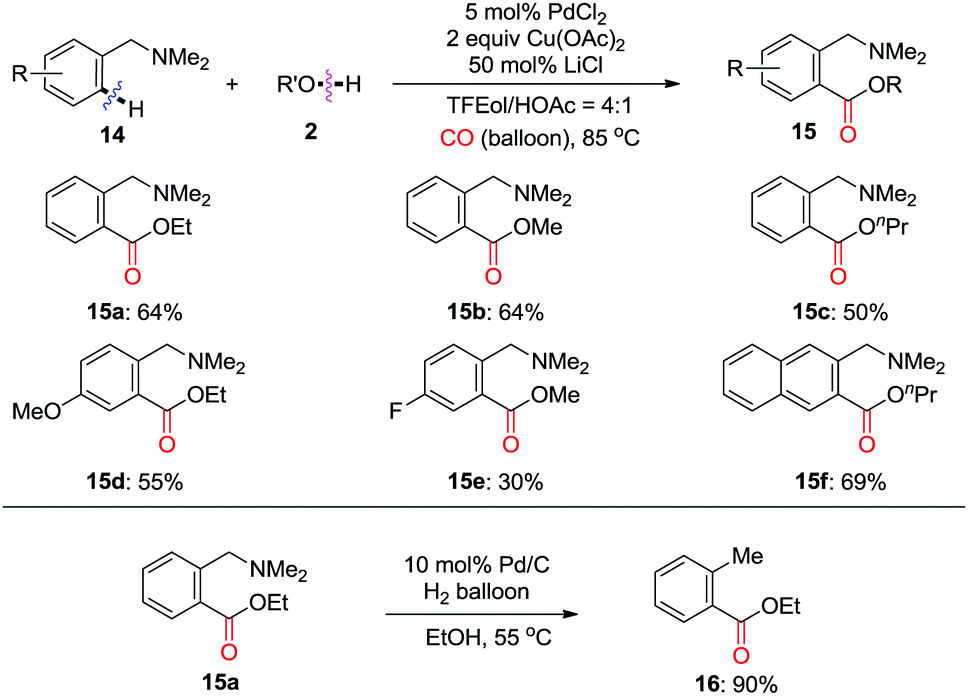 | ||
| Scheme 10 Pd(II)-Catalyzed directed ortho-carbonylation of N,N-dimethylbenzylamines using CO. | ||
Other nitrogen-containing groups were also reported to be useful directing groups for ortho-C–H activation/carbonylation. Shi and co-workers observed that, when the DG is a 2-pyridyl group, CuBr2 is the best oxidant for the esterification (Scheme 11).17 However, AgTFA performs much better than CuBr2 for phenol substrates.18 Therefore, with these methods, a variety of benzoic acid esters were obtained with high efficiency, avoiding the use of pre-functionalized substrates, such as aryl halides. Moreover, it is interesting to note that, in both cases, atmospheric pressure of CO/O2 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was employed. A lower yield was obtained in the absence of O2, indicating that oxygen participates as a co-oxidant in the reactions.
:1) was employed. A lower yield was obtained in the absence of O2, indicating that oxygen participates as a co-oxidant in the reactions.
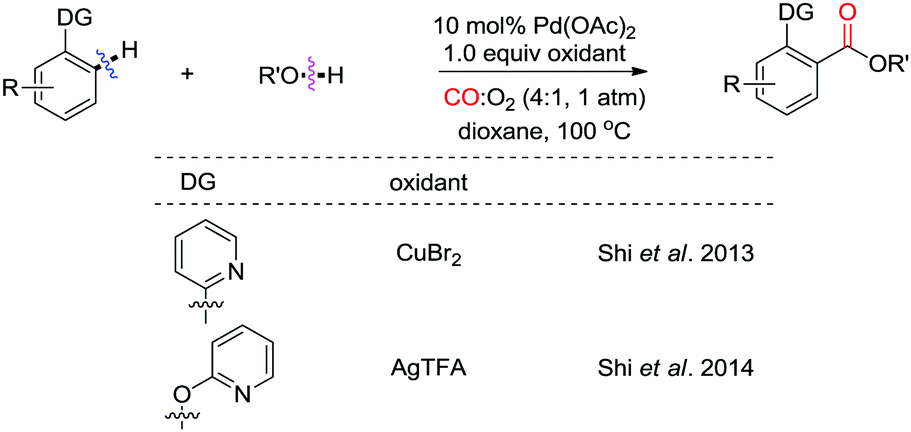 | ||
| Scheme 11 Palladium(II)-catalyzed direct alkoxycarbonylation of arenes with CO and alcohols. | ||
Weak coordination of a hydroxyl group to the palladium catalyst was demonstrated to trigger the ortho-carbonylation in the presence of CO (Scheme 12). Intramolecular nucleophilic attack on Int-2 to give Int-3′ (see Scheme 3) and subsequent reductive elimination would generate the corresponding lactones as the products. In 2011, the Yu group reported a Pd(II)-catalyzed ortho-C–H carbonylation reaction with phenethyl alcohol derivatives for the synthesis of benzoic acid esters (Scheme 12).19 A mono-N-protected amino acid was employed as an efficient ligand to ensure the C–H carbonylation transformation. Subsequently, Shi et al. successfully applied an ortho-C–H carbonylation strategy for the synthesis of dibenzopyranones from 2-phenylphenol derivatives (Scheme 12).20 Air was used as the terminal oxidant together with Cu(OAc)2 as catalytic oxidant/electron transfer mediator. At the same time, the group of Chuang independently developed a similar approach with stoichiometric amounts of AgOAc as the oxidant (Scheme 12).21
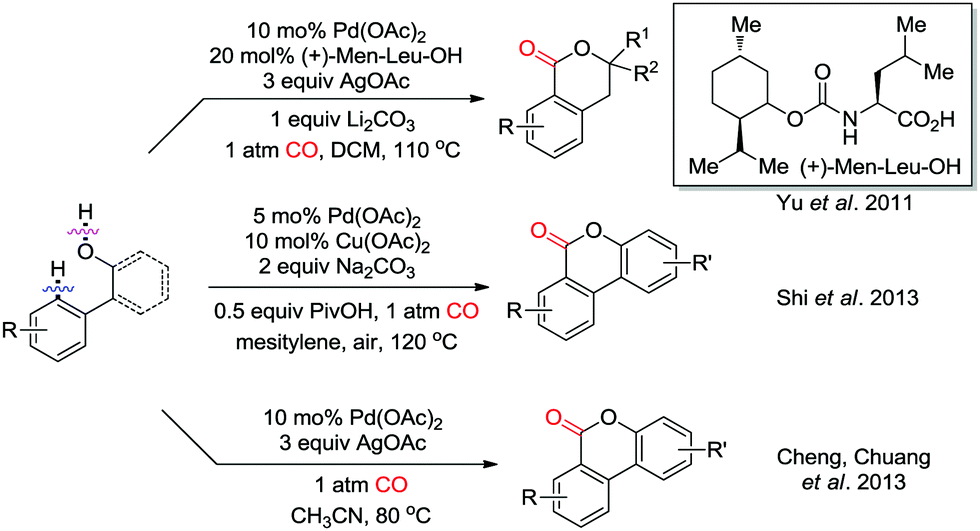 | ||
| Scheme 12 Hydroxyl group-directed Pd(II)-catalyzed ortho-C–H carbonylation reactions. | ||
In 2011, the Lei group developed an efficient protocol for aerobic oxidative C–H carbonylation of heteroarenes for the synthesis of various heterocyclic esters in the presence of CO (Scheme 13).22 An electrophilic palladation mechanism was proposed to explain the regioselective carbonylation reaction. Air was used as the terminal oxidant, with catalytic amounts of Cu(II) as electron transfer mediator. The carbonylation approach can be extended to other heteroarene systems, including thiophene, benzo[b]thiophene, and pyrrolo[2,3-b]pyridine.
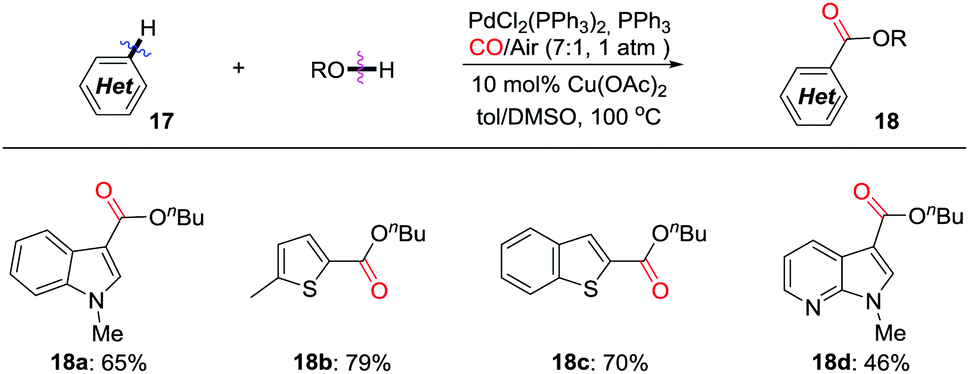 | ||
| Scheme 13 Palladium-catalyzed regioselective aerobic oxidative C–H/O–H carbonylation of heteroarenes. | ||
Intramolecular alkenyl C(sp2)–H/(O)–H cyclocarbonylation was demonstrated as a convenient approach for the synthesis of α,β-unsaturated lactones by the group of Alper in 2012 (Scheme 14).23 2-Vinylphenols as substrates afforded the corresponding coumarins as products with a variety of functional groups tolerated. Instead of the initial C(sp2)–H palladation pathway, the reaction of Pd(II) with 2-vinylphenol generates a palladium phenoxide species Int-7via ligand exchange. Subsequent CO insertion and intramolecular olefin insertions would give Int-9, which undergoes β-H-elimination to furnish coumarin as the final product. The released Pd(0) is reoxidized to Pd(II) by BQ (BQ = benzoquinone) or air, which closes the catalytic cycle.
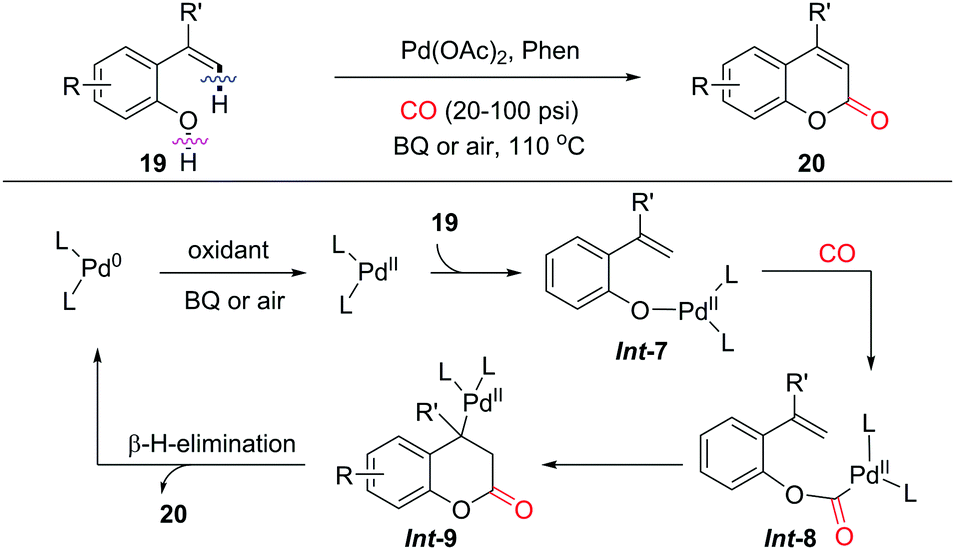 | ||
| Scheme 14 Synthesis of coumarins via Pd-catalyzed oxidative cyclocarbonylation of 2-vinylphenols. | ||
In 2014, the groups of Lei and Kočovský independently reported the intermolecular alkenyl C(sp2)–H/(O)–H cyclocarbonylation for the synthesis of acrylate derivatives (Scheme 15).24,25 In both cases, Cu(OAc)2 and molecular oxygen were used as the oxidation system. The reaction of alcohol with Pd(II) in the presence of CO generates an (alkoxycarbonyl)palladium species, which undergoes olefin insertion and β-H-elimination to give acrylate derivatives.
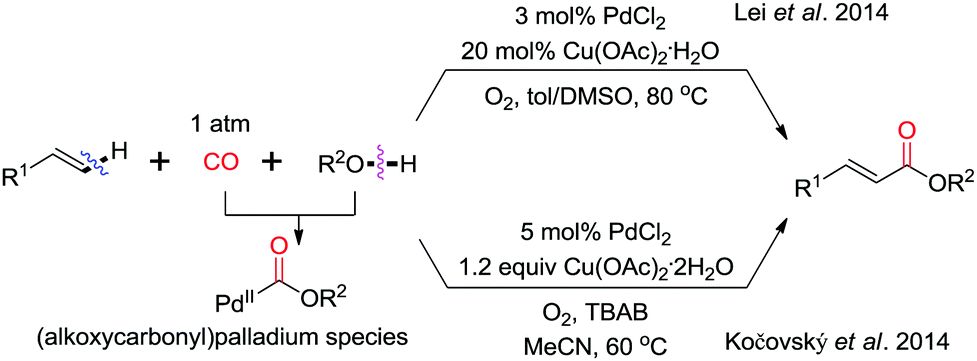 | ||
| Scheme 15 Synthesis of acrylate derivatives via Pd-catalyzed oxidative intermolecular vinyl C(sp2)–H/(O)–H cyclocarbonylation. | ||
:1 (Scheme 16b). In 2011, the group of Garcia and Granell developed an unprecedented NH2-directed Pd(II)-catalytic carbonylation of quaternary aromatic α-amino esters to yield 6-membered benzolactams (Scheme 16c).26b The reaction shows a strong bias to 6-membered lactams over 5-membered ones, as shown by the selective formation of 6-membered lactam 21e in 87% yield from 14e.
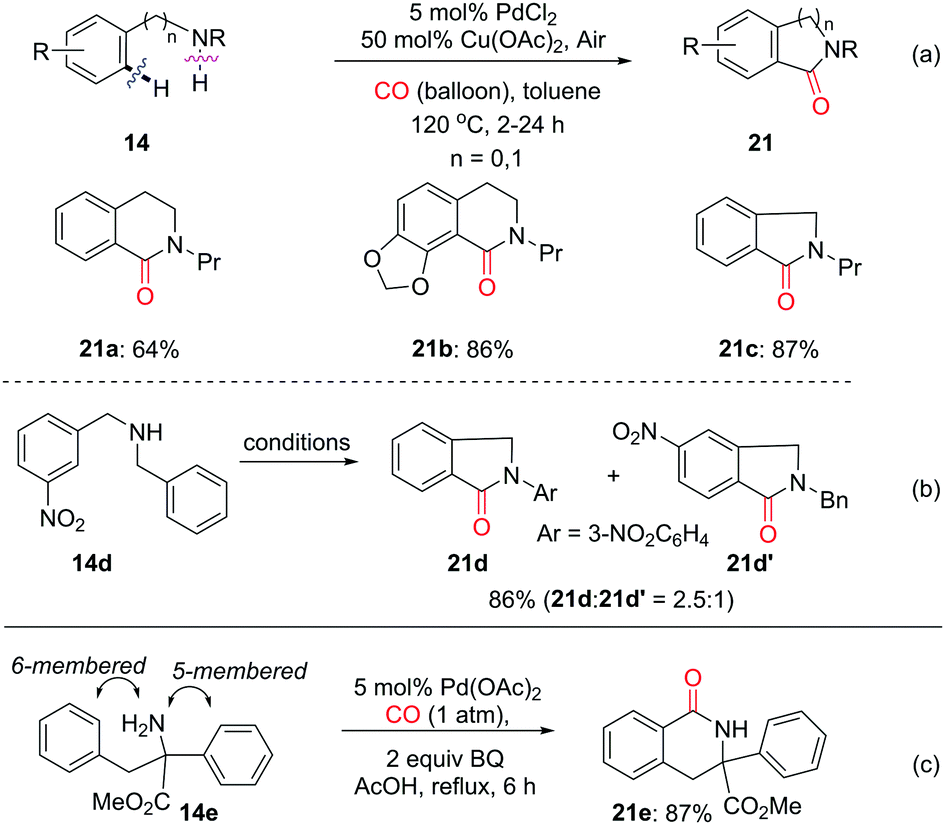 | ||
| Scheme 16 Palladium-catalyzed C–H carbonylation with CO for the synthesis of benzolactams. | ||
In 2012, the Guan group reported on the palladium-catalyzed regioselective carbonylation of N-alkyl anilines for the synthesis of isatoic anhydrides in the presence of CO (Scheme 17a).26c Various isatoic anhydrides were produced from N-alkyl anilines under mild conditions. The key intermediate, Int-10 was isolated from the reaction of N-methyl aniline with stoichiometric amounts of Pd(OAc)2 under CO atmosphere (1 atm). The transformation of cyclic organopalladium intermediate Int-10 to product 23a was achieved via double CO insertion with Cu(OAc)2 as the oxidant. Pd-catalyzed oxidative dehydrogenative double carbonylation was described by the Lei group (Scheme 17b).26d Various isatins 24 were prepared by using the same substrate 22.
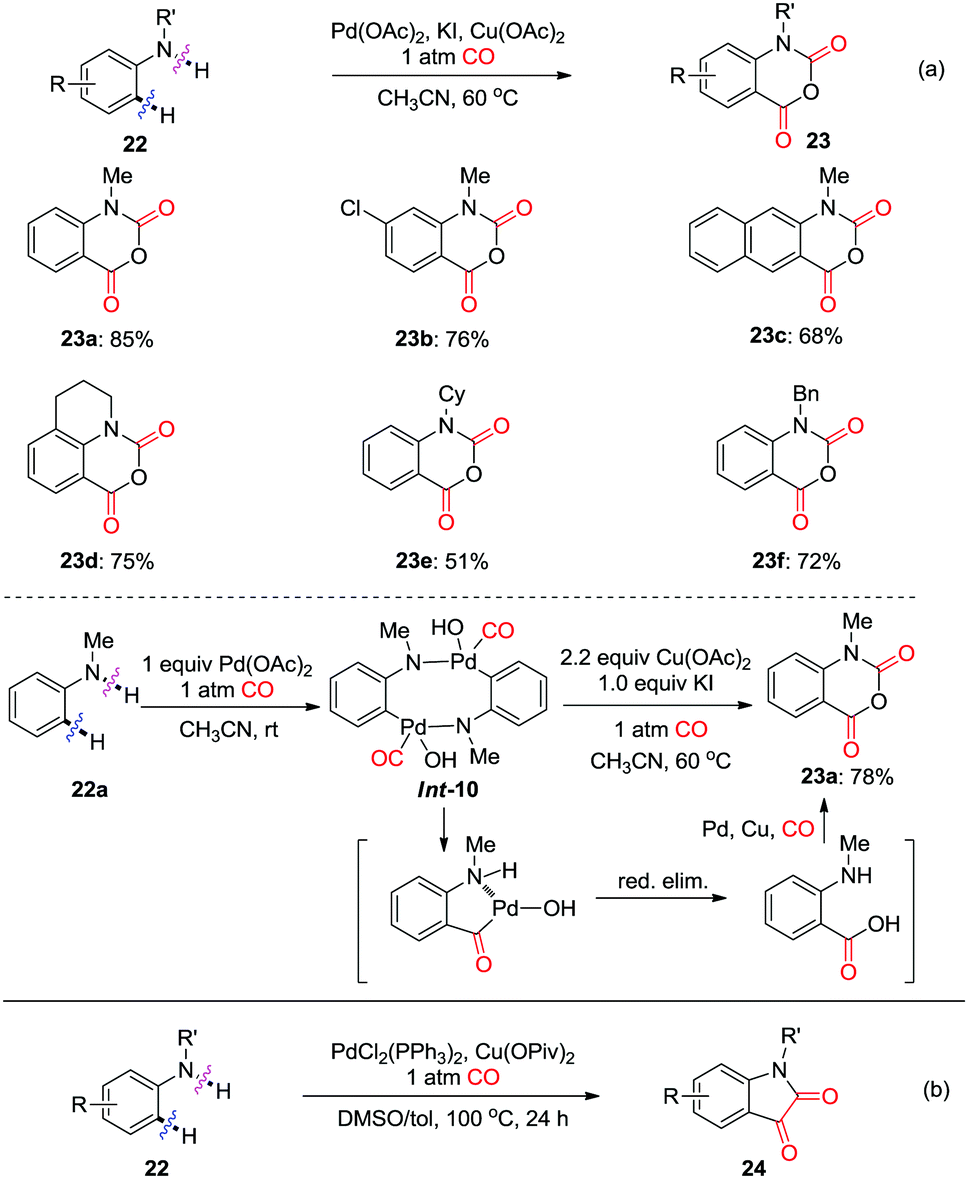 | ||
| Scheme 17 Palladium-catalyzed regioselective carbonylation of N-alkyl anilines for the synthesis of isatoic anhydrides (a) and isatins (b) in the presence of CO. | ||
In 2013, the Lei group reported an aerobic palladium/copper-catalyzed oxidative C–H alkenylation/N-dealkylative carbonylation of tertiary anilines 25, which afforded 3-methyleneindolin-2-ones derivatives 26 (Scheme 18).26e Intermolecular Heck-type reaction of dimethylaniline 25 with the terminal olefin gives olefination intermediate 27, which undergoes C–N bond cleavage in the presence of copper and O2 to afford Int-11. Subsequent transmetalation, carbonylation and intramolecular Heck reaction furnish the final product 26viaInt-12.
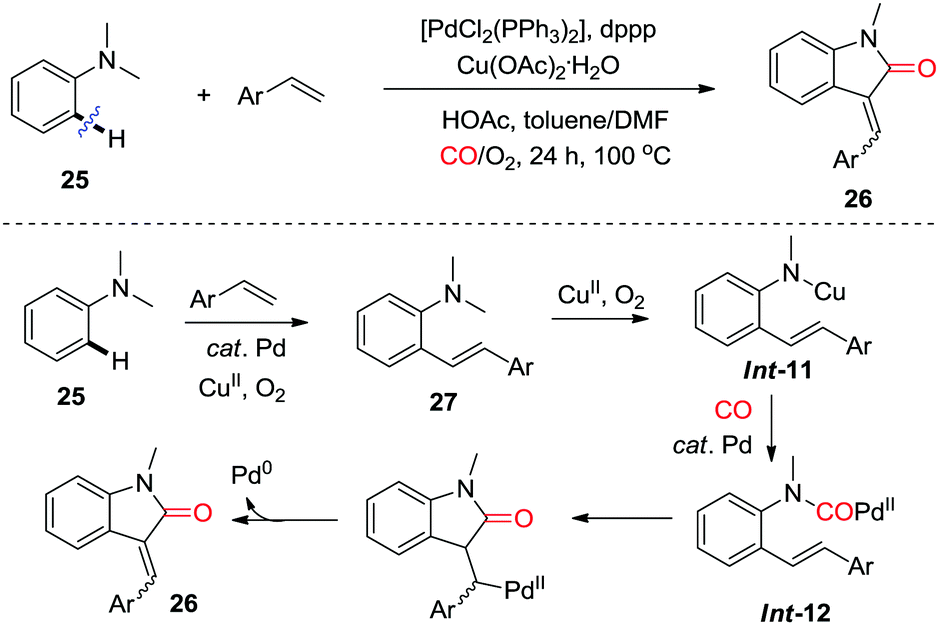 | ||
| Scheme 18 Palladium/copper-catalyzed oxidative C–H alkenylation/N-dealkylative carbonylation of tertiary anilines. | ||
Later, Zhu et al. developed an efficient palladium-catalyzed C(sp2)–H pyridocarbonylation of N-aryl-2-aminopyridines (Scheme 19).27 A 2-pyridyl group was used as the directing group to trigger C(sp2)–H activation forming a C–Pd bond, followed by insertion of CO. Intramolecular nucleophilic attack and subsequent reductive elimination give fused tricycles 28 selectively. K2S2O8 was found to be the best oxidant for the overall transformations.
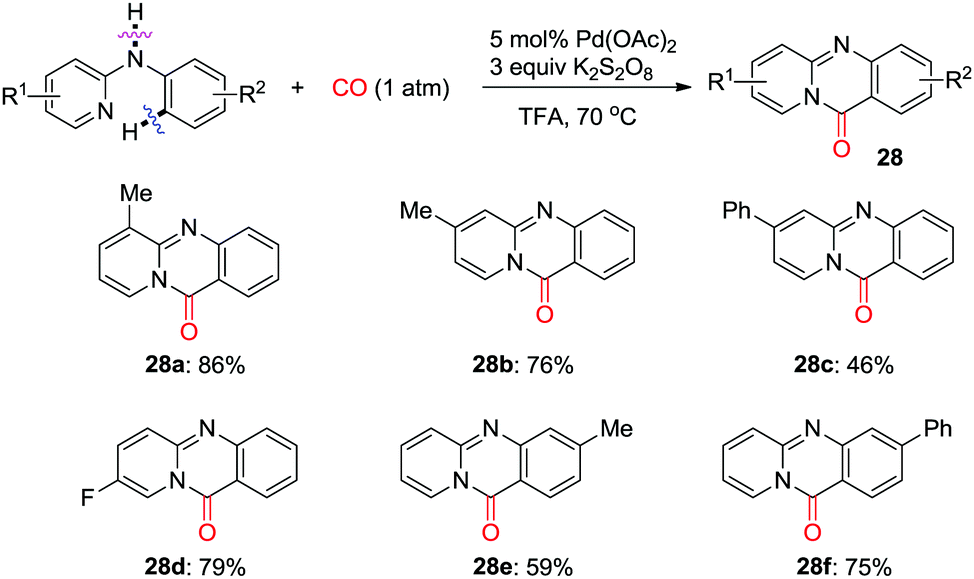 | ||
| Scheme 19 Palladium-catalyzed C(sp2)–H pyridocarbonylation of N-aryl-2-aminopyridines. | ||
At the same time, Jiang and co-workers reported a palladium-catalyzed oxidative carbonylation for the synthesis of polycyclic aromatic compounds, 6H-isochromeno[4,3-c]-quinolin-6-ones (Scheme 20).28 The reaction proceeds via Pd-catalyzed oxidative carbonylation, and intramolecular reductive elimination with high atom-efficiency. However, when a 1,8-naphthyridine derivative was used as the substrate, none of the corresponding product (30d) could be detected under the optimized reaction conditions.
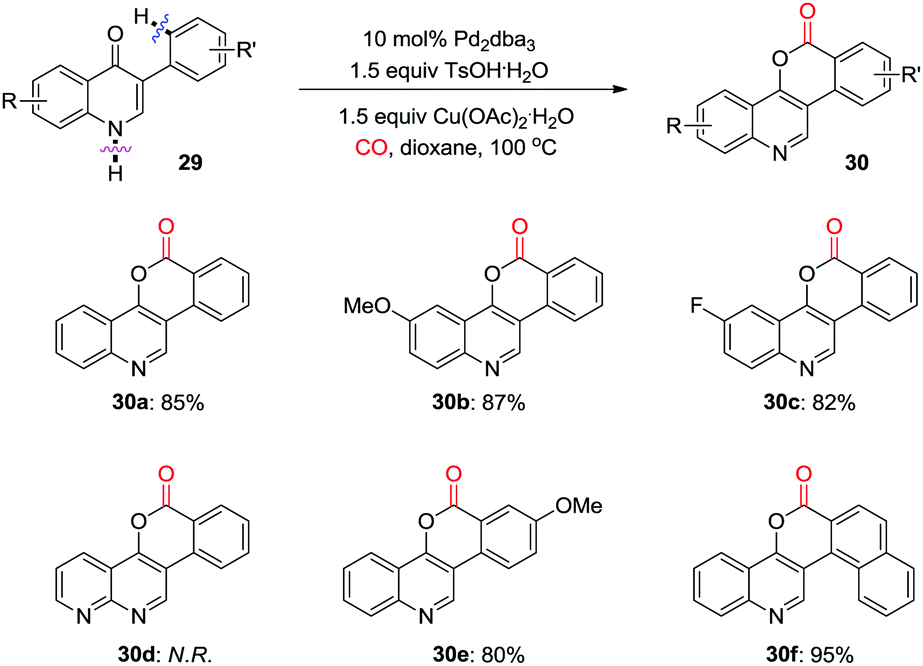 | ||
| Scheme 20 Palladium-catalyzed oxidative carbonylation for the synthesis of polycyclic aromatic cycles. | ||
In 2016, the group of Zhang reported a practical intermolecular aminocarbonylation of anilines for the construction of o-aminobenzamides (Scheme 21).29 The employment of C–H activation for aminocarbonylation with CO as C1 source is an elegant solution for the synthesis of amides. In this work, both aromatic and aliphatic primary amines were utilized as the coupling partners. However, secondary amines, as well as bulky primary amines, were not tolerated under the reaction conditions.
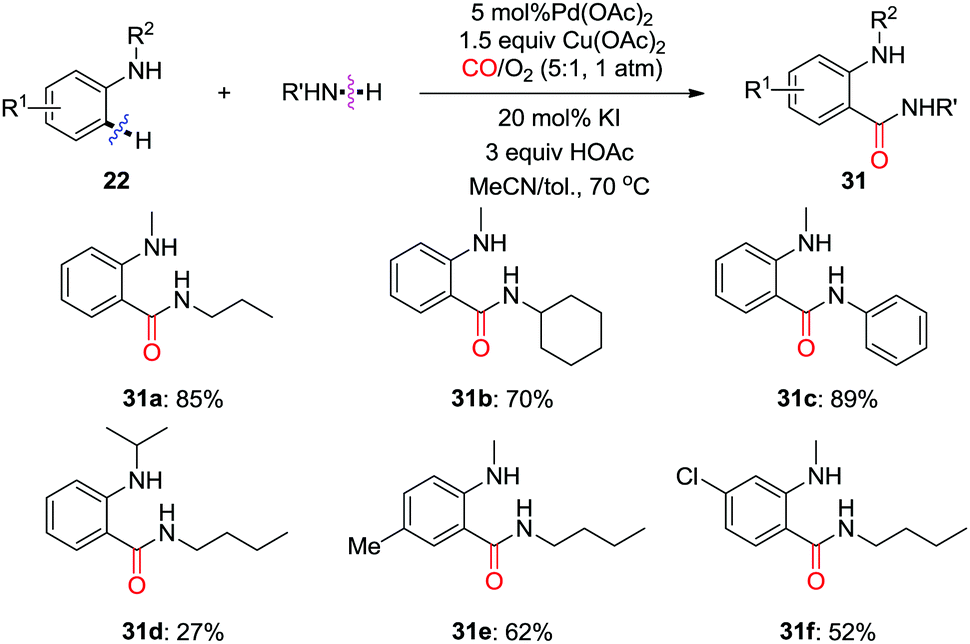 | ||
| Scheme 21 Palladium-catalyzed C(sp2)–H carbonylation of anilines with CO and primary amines for the synthesis of o-aminobenzamides. | ||
2.3 C(sp3)–H/(X)–H oxidative dehydrogenative carbonylation
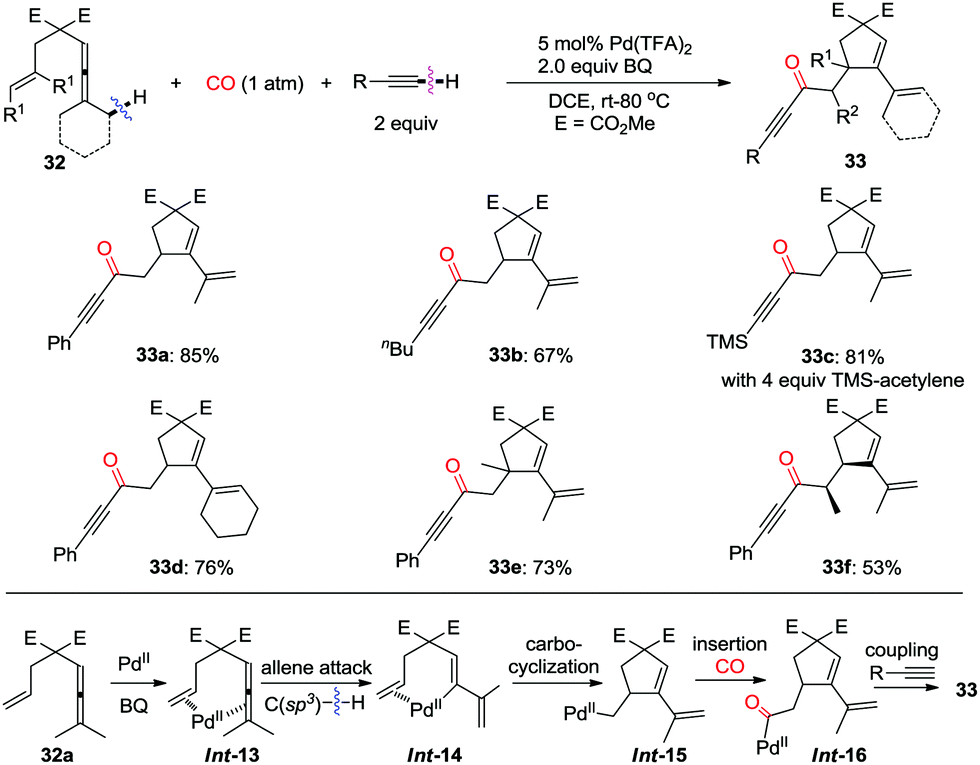 | ||
| Scheme 22 Oxidative coupling reaction of enallenes with terminal alkynes via C(sp3)–H cleavage in the presence of carbon monoxide. | ||
In 2015, a one-pot palladium-catalyzed oxidative cascade reaction of enallenes via carbonylation–carbocyclization–carbonylation–alkynylation was reported by the same group (Scheme 23).31 The insertion cascade is highly efficient and gives products 35 with exclusive chemoselectivity, which proceeds via sequential CO–olefin–CO insertion, involving overall formation of four C–C bonds.
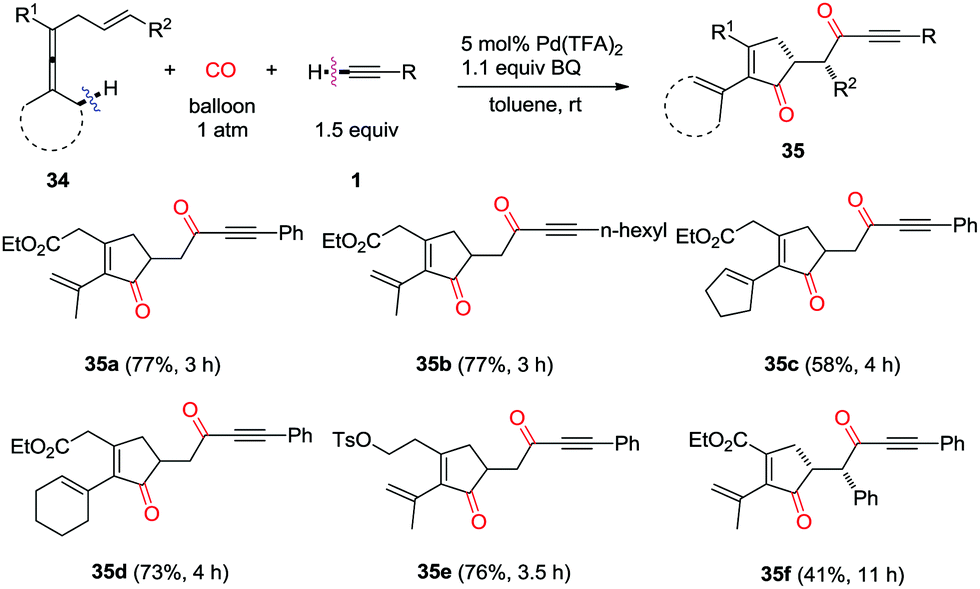 | ||
| Scheme 23 Insertion cascade forming cyclopentenones. | ||
The pending olefin was demonstrated to be indispensable for triggering the initial allene attack.32 This olefin acts as the directing/assisting group in Int-17 to give Int-18via C(sp3)–H bond cleavage, which is the rate-limiting step during the overall transformations as confirmed by kinetic isotope effect studies (Scheme 24). The insertion cascade proceeds via sequential CO-olefin–CO insertion to generate Int-21 from Int-18. Final products 35 were produced from the oxidative coupling reaction of Int-21 with terminal alkynes. Four C–C bonds were formed in the overall cascade reaction.
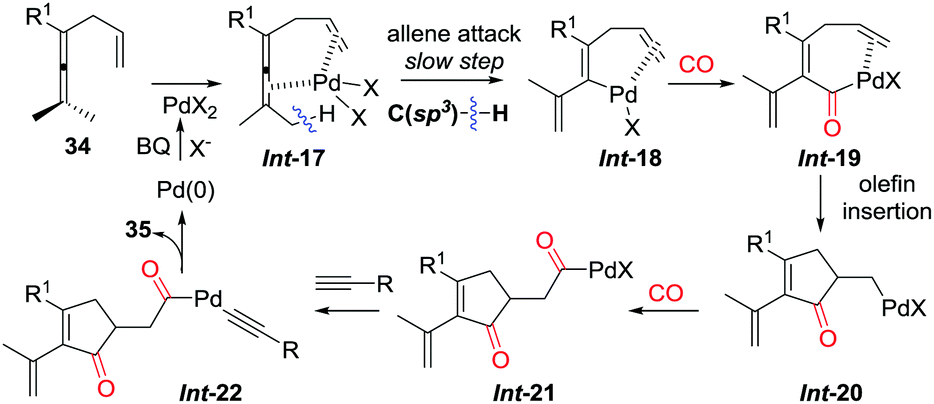 | ||
| Scheme 24 Proposed mechanism for the insertion cascade reactions. | ||
Moreover, the Bäckvall group found that, under the catalysis of PdII and VAPOL phosphoric acid (CPA), the asymmetric version of the dehydrogenative carbonylation–carbocyclization reaction was realized to afford ketones bearing α-chirality from enallenes (Scheme 25).33 Vaulted biaryl-type chiral phosphoric acids served as useful co-catalysts for this asymmetric transformation, therefore ketone products were generated in good yields with up to 95.5:4.5 e.r. In the catalytic cycle, enantioselective migratory insertion of the olefin into the C–Pd bond produces the carbocyclic intermediate Int-20, introducing the chirality at the α-position of the ketone. However, a racemization pathway in Int-20 probably lowered the e.r. of the ketone products 35via reversible β-hydride elimination-hydropalladation.
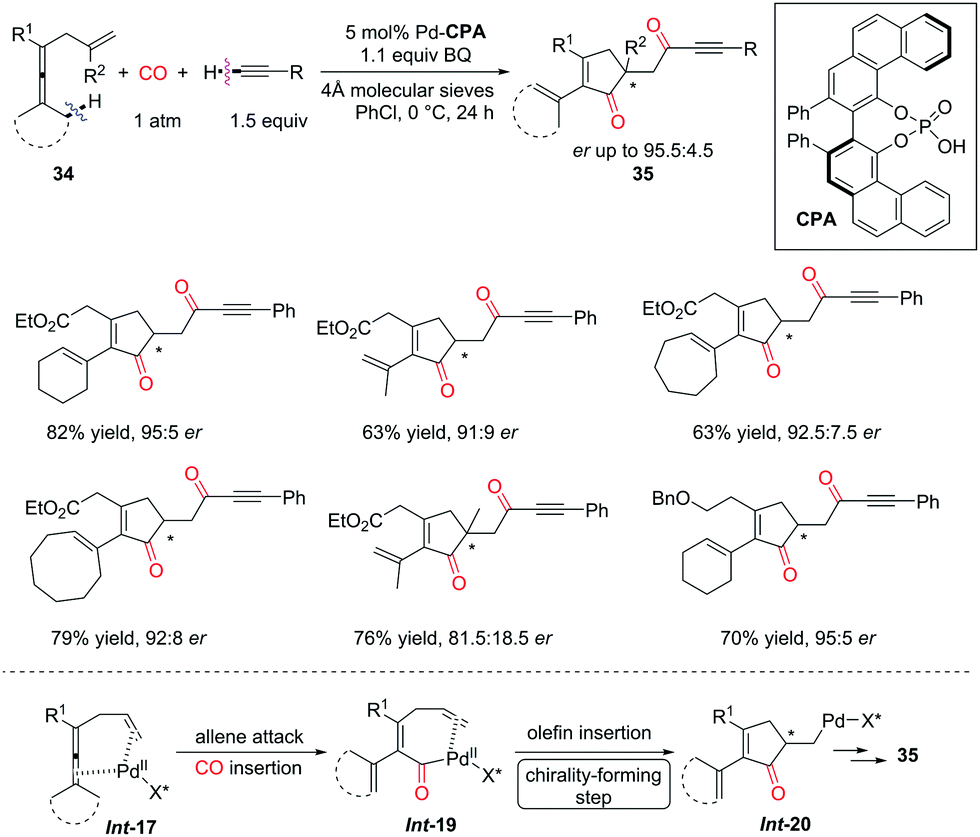 | ||
| Scheme 25 Asymmetric version of the dehydrogenative carbonylation-carbocyclization reaction. | ||
Spirocarbocyclic scaffolds bearing a quaternary carbon center, have attracted an increasing interest recently, because they occur as structural elements in a range of natural products, pharmaceutical ingredients, and chiral ligands. By introducing an additional distal olefin unit, spiro[3.4]octenes 37 and spiro[4.4]nonene 38 derivatives were obtained chemoselectively by controlling the reaction conditions (Scheme 26).34 Lower temperature in DCE favored double insertion of CO into C–Pd bond to provide compounds 38, while spirocyclobutene derivatives 37 were generated as single diastereoisomers at a higher temperature in MeCN.
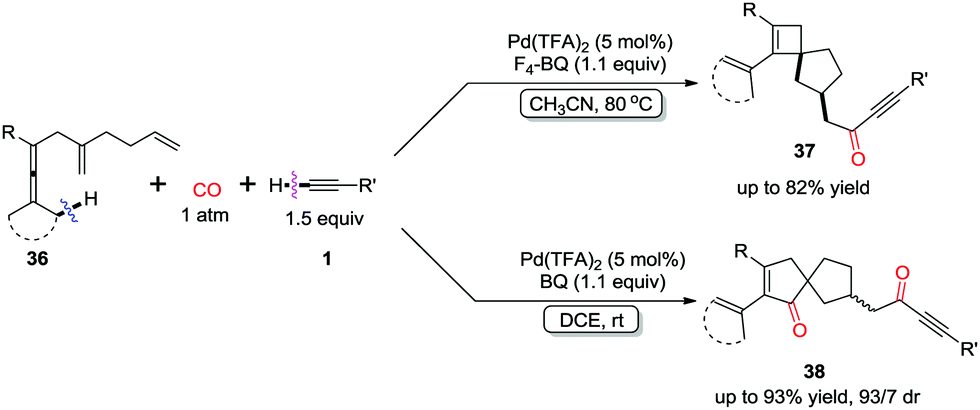 | ||
| Scheme 26 Selective formation of spirocarbocyclic scaffolds. | ||
The coordination of dienallene to PdII generates the chelate Int-23, in which the pending olefin is an indispensable element for the subsequent allene attack to afford vinylpalladium intermediate Int-24 (Scheme 27).32,34 Direct insertion of the pending olefin into the C–Pd bond gives Int-25, which proceeds via sequential olefin–CO insertion to produce carbonyl-palladium intermediate Int-26. Spirocyclobutene derivatives 37 is subsequently obtained from the coupling reaction of Int-26 with a terminal alkyne. However, by using DCE as the solvent at lower temperature, sequential insertions of CO–olefin–olefin–CO give spiro[4.4]nonene derivatives 38 in high selectivity.
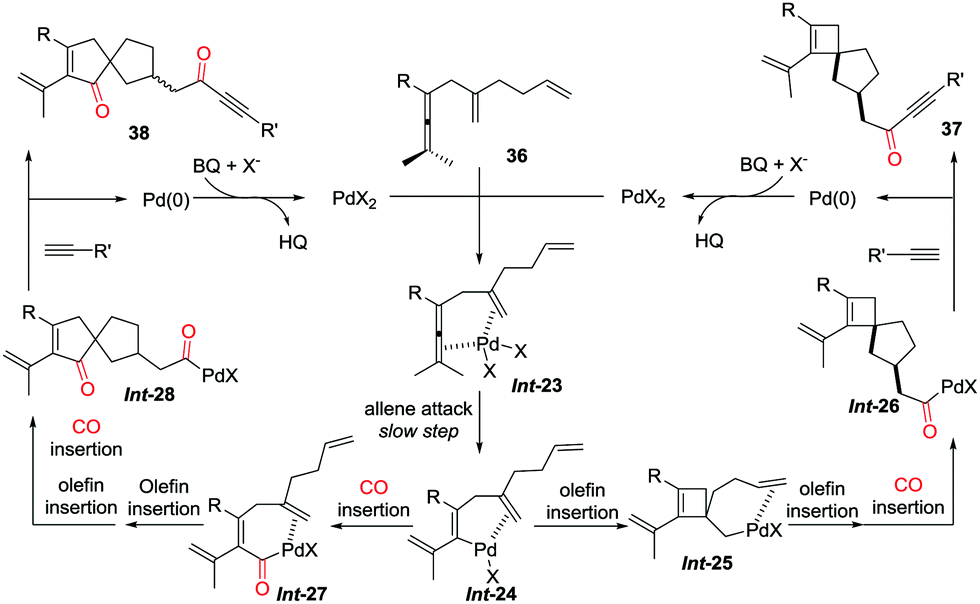 | ||
| Scheme 27 Proposed mechanism for the selective formation of spirocarbocyclic scaffolds. | ||
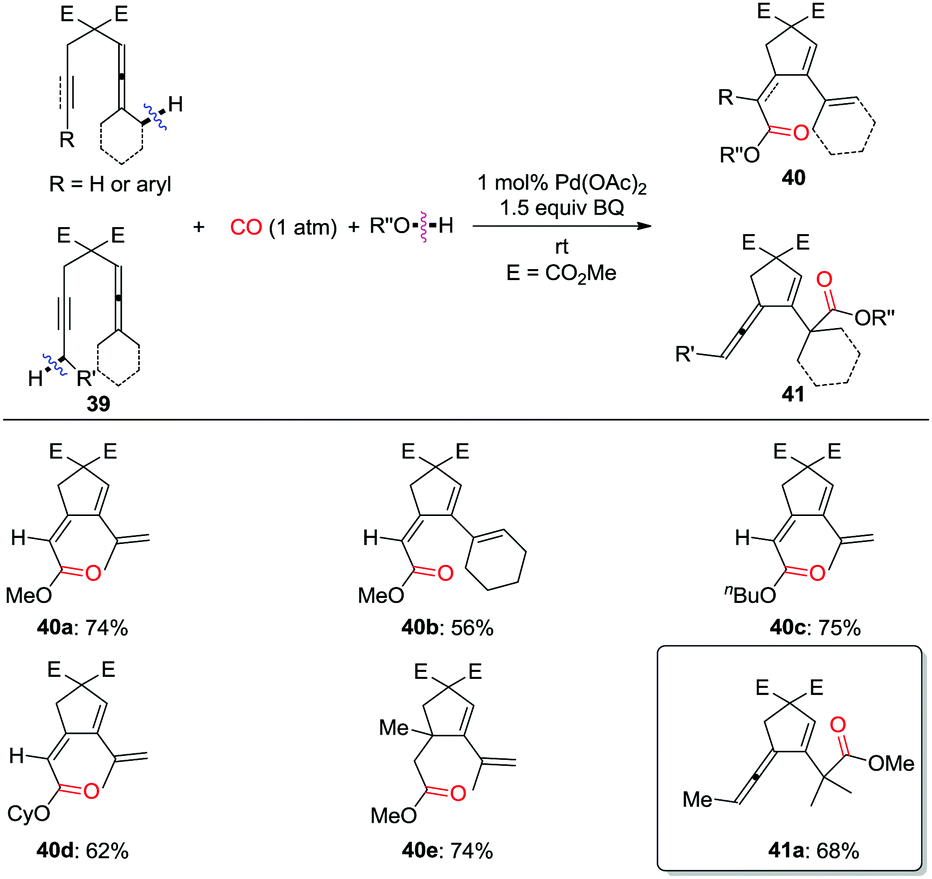 | ||
| Scheme 28 Oxidative palladium-catalyzed carbocyclization–carbonylation of allenynes and enallenes. | ||
The cascade insertion can be extended to synthesize spirolactones. By using an internal hydroxyl group as an intramolecular quencher, cascade carbonylative spirolactonization of enallenols was realized to afford spirolactones 43.36 After the sequential insertion of CO–olefin–CO into the C–Pd bond in Int-29 (Scheme 29), the internal hydroxyl group quenching of the carbonyl-palladium intermediate Int-32 leads to the selective formation of spirolactones 43, bearing an all-carbon quaternary center.
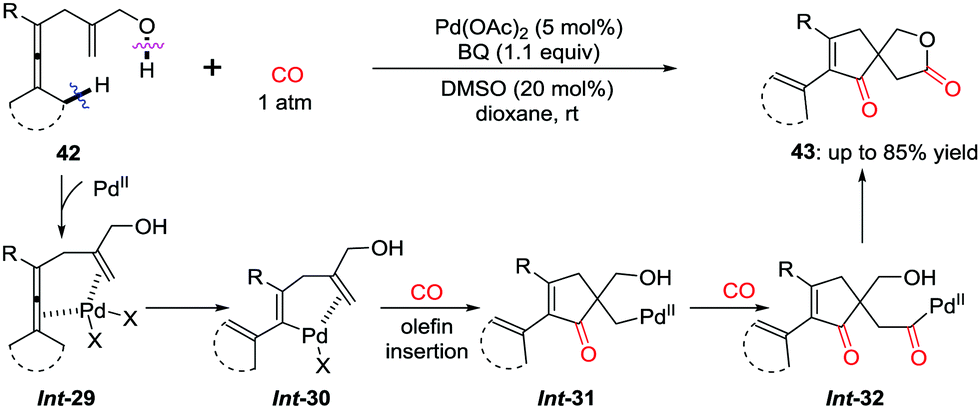 | ||
| Scheme 29 Oxidative cascade carbonylative spirolactonization of enallenols. | ||
Recently, a pending olefin was successfully employed as the assisting group for the carbocyclization–alkoxycarbonylation of dienallenes or bisallenes, and in this manner six- or seven-membered carbocycles were generated selectively and efficiently (Scheme 30).37,38 The olefin-ligand exchange from the pending olefin in Int-33 to the remote olefin/allene forming Int-34 or Int-34′ was found to be the key step for the overall transformations.
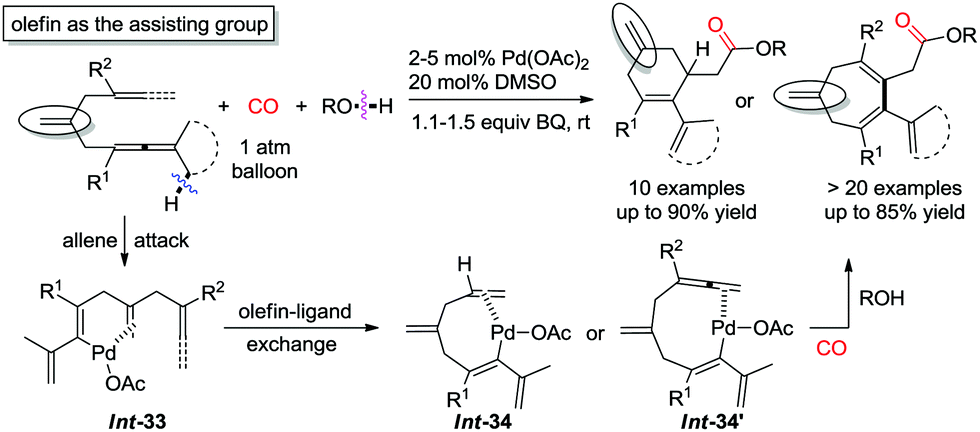 | ||
| Scheme 30 Oxidative carbocyclization–alkoxycarbonylation of dienallenes or bisallenes. | ||
More recently, this group developed a palladium-catalyzed oxidative carbonylation reaction of enallenols for the chemodivergent synthesis of γ-lactone and γ-lactam derivatives (Scheme 31).39 Interestingly, the catalyst-controlled chemodivergent carbonylation forming products 45 and 46, respectively, was achieved by switching between homogeneous and heterogeneous catalysts.
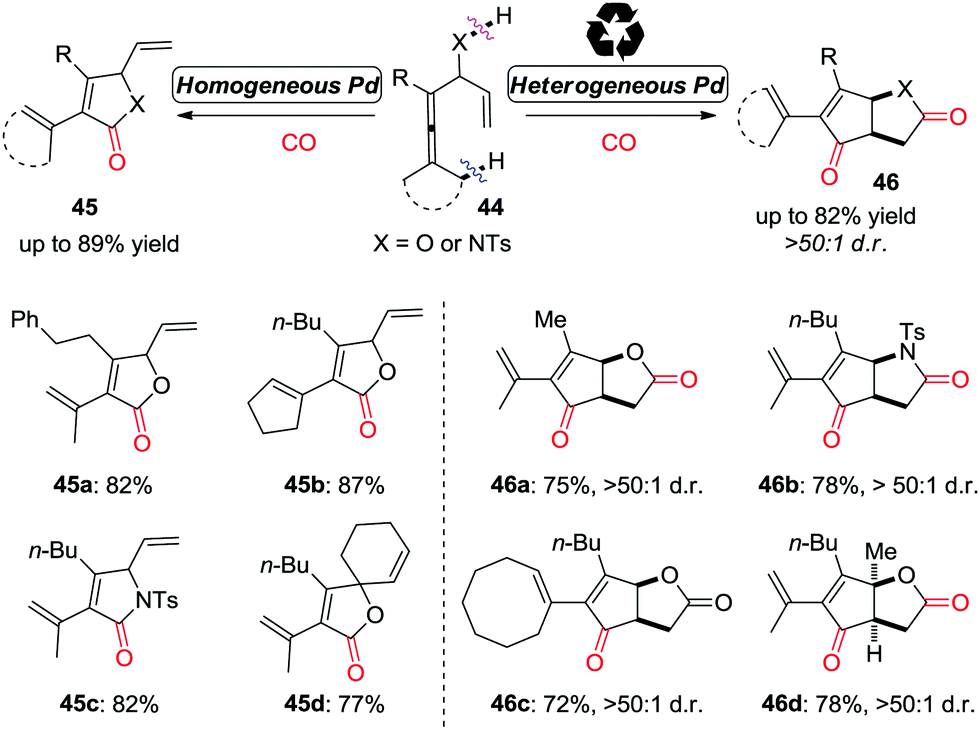 | ||
| Scheme 31 Oxidative tandem reaction of enallenols for the chemodivergent and diastereoselective synthesis of γ-lactone and γ-lactam derivatives. | ||
Allylic oxidation followed by subsequent nucleophilic attack presents a highly efficient approach for the synthesis of functionalized olefins. In 2011, the group of Jiang found that, in the presence of CO, allylic oxidation for the formation of linear β,γ-unsaturated ester 48 was realized using alcohol as the quencher (Scheme 32).40 Mechanistic studies on the kinetic isotope effects indicated that the initial cleavage of the allylic C(sp3)–H bond is involved in the rate-determining step. This allylic C(sp3)–H oxidation/carbonylation approach provides a new route for accessing more synthetically useful β-enoic acid esters with high regioselectivity.
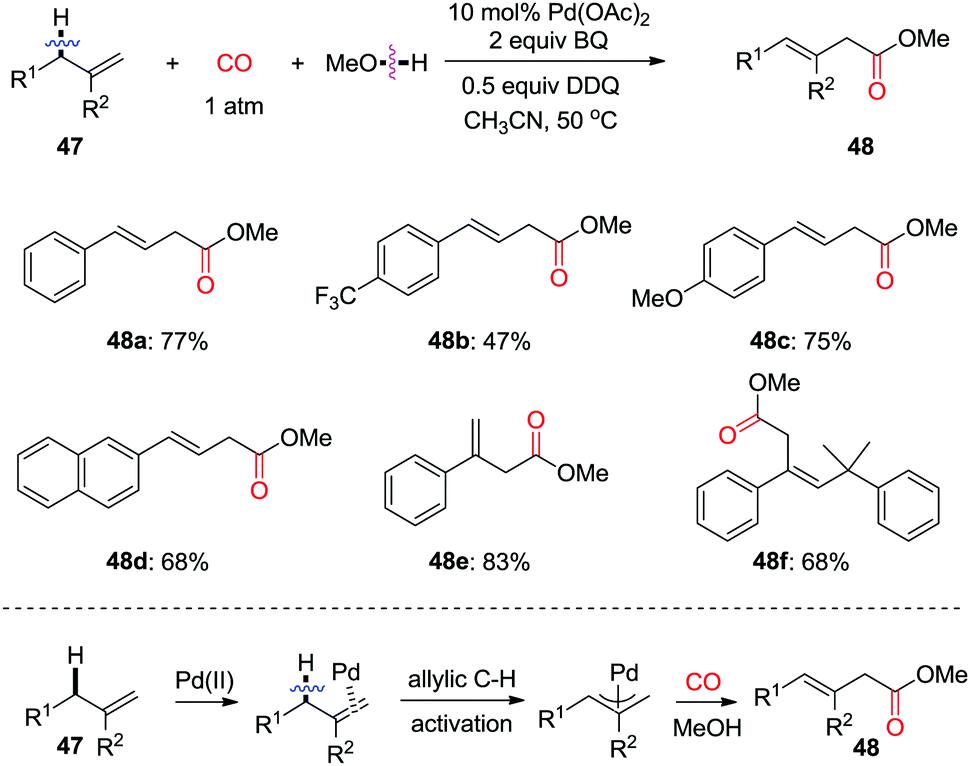 | ||
| Scheme 32 Regioselective palladium-catalysed oxidative allylic C–H carbonylation of alkenes. | ||
In 2012, Huang et al. reported a palladium-catalyzed oxidative carbonylation of benzylic C(sp3)–H bonds to benzylic esters in the presence of a high pressure of CO (10 atm) (Scheme 33).41a In this study, they developed a new strategy for generating reactive benzylpalladium species from toluenes via nondirected C(sp3)–H activation using TBP (di-tert-butyl peroxide) as the oxidant. High TONs (turnover numbers) were observed for almost all of the substrates. This approach produces ethyl esters via transmetalation/reductive elimination as the major products. However, the corresponding tert-butyl ester was also detected as a minor product due to the reductive elimination from Int-38.
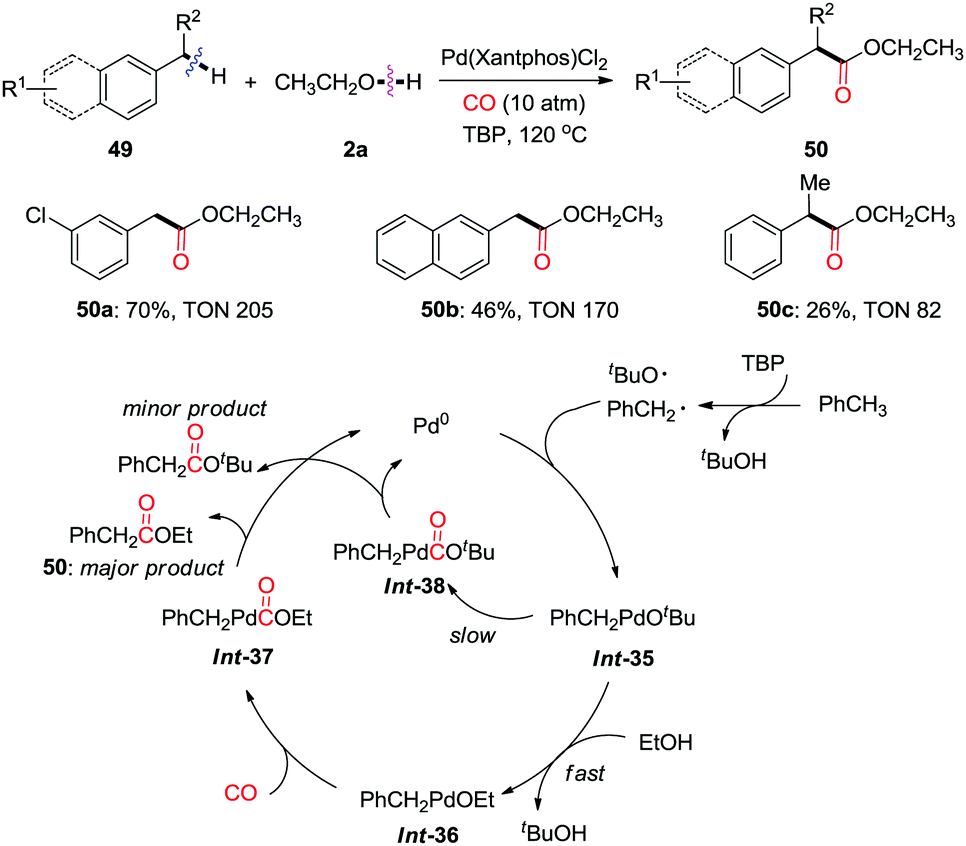 | ||
| Scheme 33 Palladium-catalyzed oxidative carbonylation of benzylic C–H bonds to benzylic esters in the presence of CO. | ||
Very recently, Yu and colleagues reported the design of a hemilabile directing group, which exploited the chelation of a readily removable benzyl ether moiety to direct γ- or δ-C(sp3)–H carbonylation of alcohols (Scheme 34).41b Under an atmospheric pressure of carbon monoxide, the remote methyl C–H bonds, as well as the methylene C–H bonds in cyclopropane and cyclobutane substrates were successfully converted to ester functionalities in the products under the catalysis of Pd(II). δ-C(sp3)–H carbonylation of alcohols through remote palladation was also realized as shown by the formation of 52f in 33% yield.
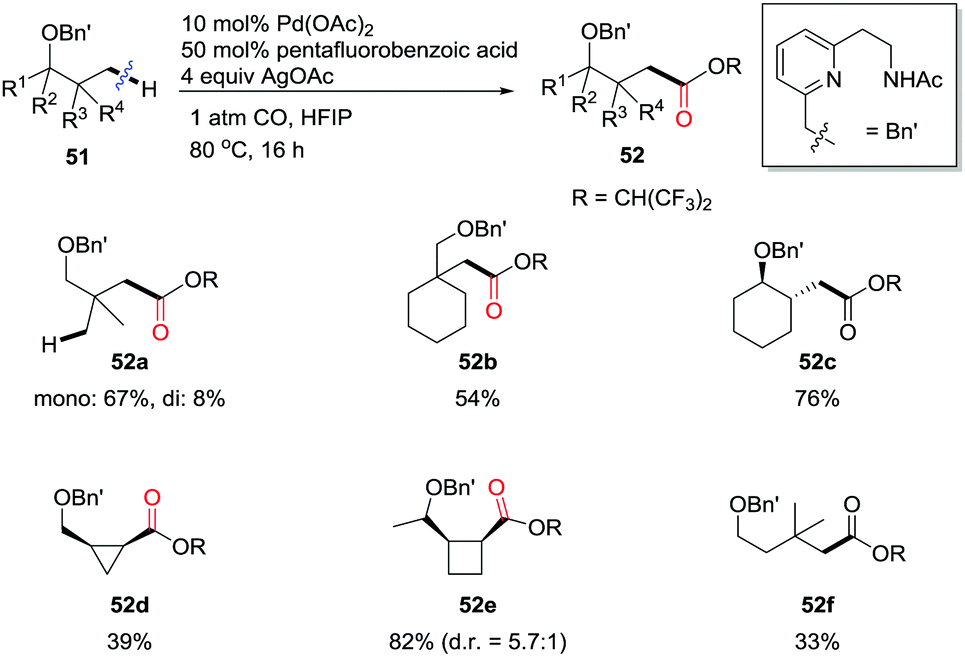 | ||
| Scheme 34 Palladium-catalyzed C(sp3)–H carbonylation of alcohols. | ||
The directing group coordinated to Pd(II) triggers the cyclopalladation of the C–H bond to give Int-39, which is accelerated by participation of the NHAc motif (Scheme 35). The dissociation of the hemilabile ether from the palladium center allows the binding of carbon monoxide to furnish Int-40. Subsequent migratory insertion and oxidative coupling with the solvent give the final product 52viaInt-41. The authors concluded that both the acceleration caused by the internal ligand and the lability of the ether to dissociate during the catalytic cycle were crucial for the reaction to occur.
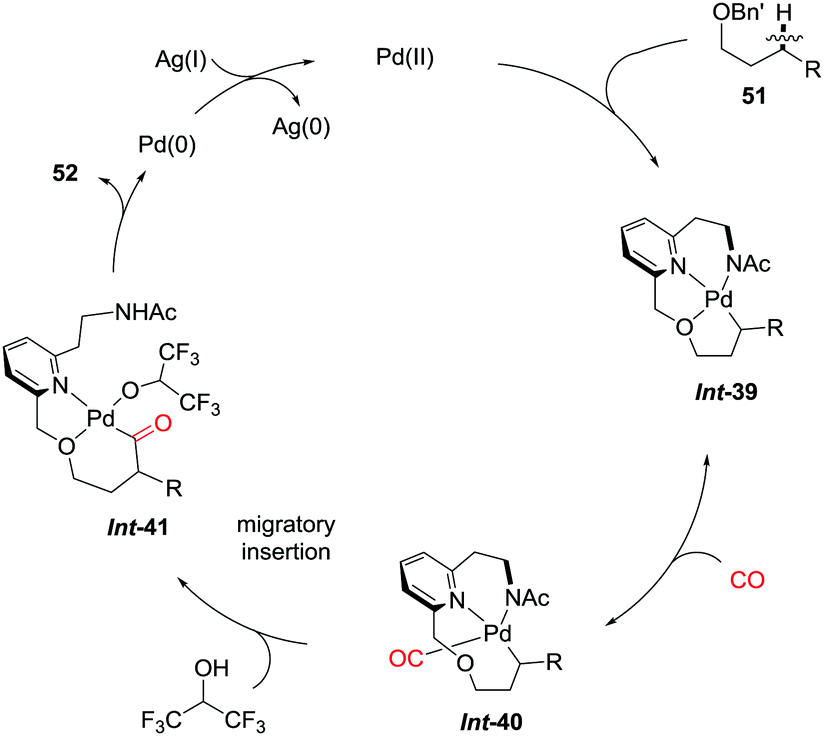 | ||
| Scheme 35 Proposed catalytic cycle for the palladium-catalyzed C(sp3)–H carbonylation of alcohols. | ||
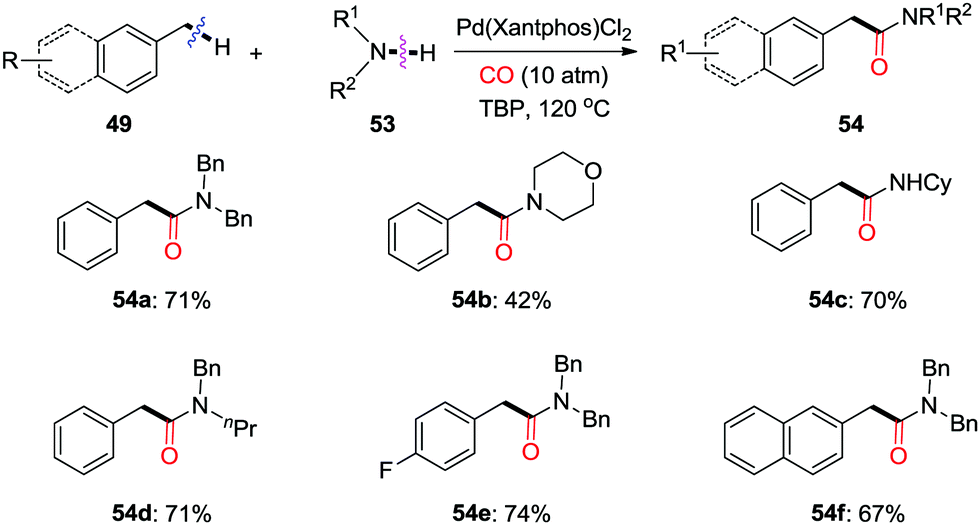 | ||
| Scheme 36 Palladium-catalyzed oxidative aminocarbonylation of benzylic C–H bonds to amides in the presence of CO. | ||
As early as 2010, the Yu group found that, when an amide was used as the directing group, C(sp3)–H carbonylation catalyzed by Pd(II) was achieved under atmospheric pressure of CO (Scheme 37).43 Following amide-directed C(sp3)–H cleavage and insertion of CO into the formed C–Pd bond, intramolecular C–N reductive elimination provides the corresponding succinimides. These products could be readily converted to acyclic 1,4-dicarbonyl compounds under basic conditions. This method can be also extended to cyclopropane substrates.
 | ||
| Scheme 37 Pd(II)-Catalyzed carbonylation of C(sp3)–H bonds to 1,4-dicarbonyl compounds. | ||
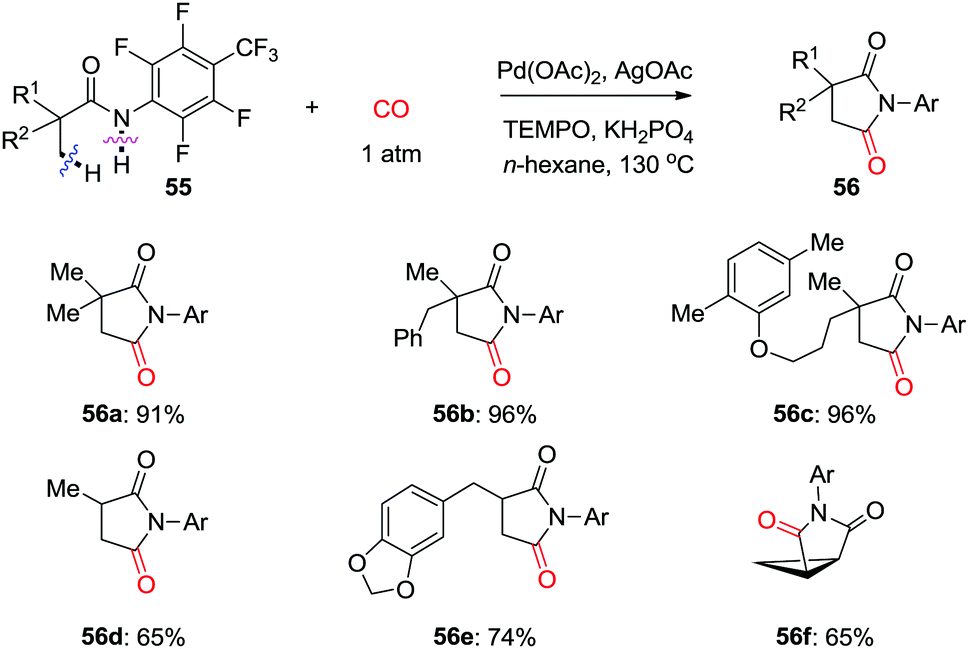
In 2015, the groups of Wang and Zhao independently reported Pd-catalyzed C(sp3)–H carbonylation of aliphatic amine substrates for the synthesis of γ-lactams and γ-amino acids (Scheme 38a and b).44,45 When the 2-pyridyl group was used as the directing group, TEMPO was found to be an efficient oxidant for the carbonylation transformations (Scheme 38a). However, in case of substrates having oxalyl amide as bidentate directing group, AgOAc performed better (Scheme 38b). Therefore, γ-lactams were obtained from γ-carbonylation reactions, and γ-amino acids were formed after subsequent hydrolysis in the presence of HCl. Gaunt et al. used N-alkyl amines as the substrates and developed diastereoselective C–H carbonylative annulation of aliphatic amines (Scheme 38c).46,47 Here, Cu(OAc)2 was employed as the co-catalyst with air as the terminal oxidant.
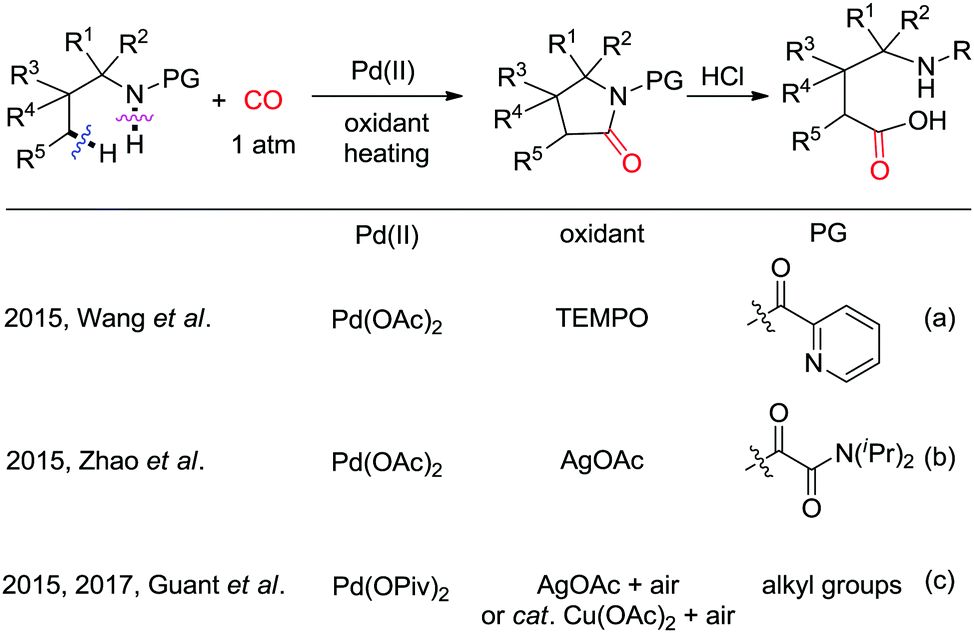 | ||
| Scheme 38 Amine-directed Pd(II)-catalyzed carbonylation of C(sp3)–H bonds to γ-lactams. | ||
In 2014, the Gaunt group developed a palladium-catalyzed C(sp3)–H activation of hindered aliphatic amines for the synthesis of strained nitrogen heterocycles 58 (Scheme 39).48 Mixing 2,2,6,6-tetramethylpiperidine (TMP) with stoichiometric Pd(OAc)2 gave rise to a four-membered ring palladacycle (Int-42), which was directly characterized by X-ray diffraction. When this complex was treated with carbon monoxide, fused β-lactam 58a was obtained in high yield. A number of β-lactams were prepared using the catalytic method. This methodology should find further application as this structural motif is considered important in the design of pharmaceutical agents.
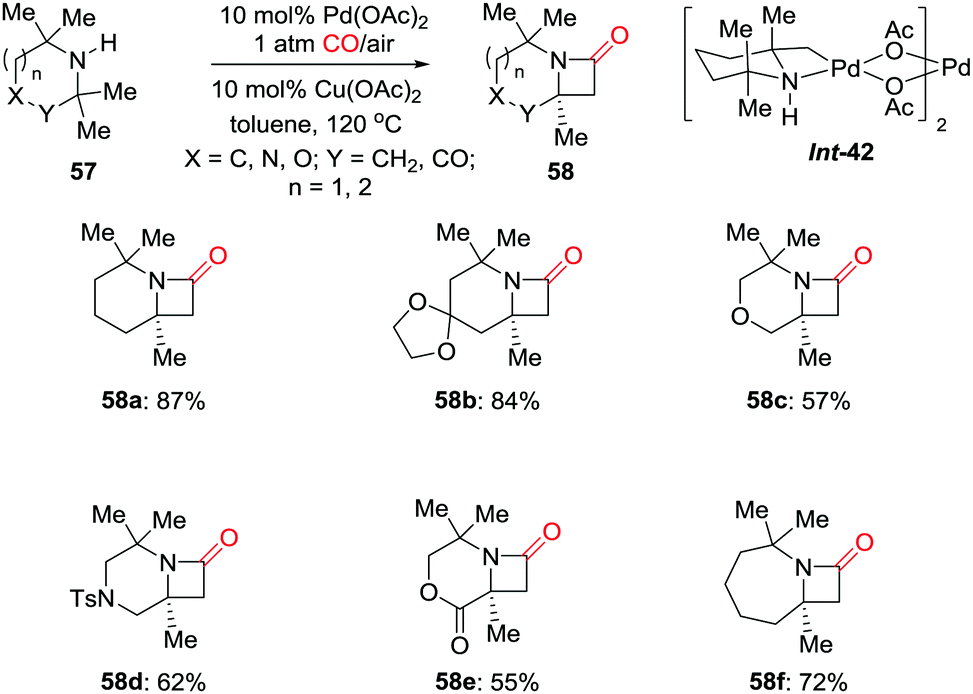 | ||
| Scheme 39 Palladium-catalyzed C–H activation of aliphatic amines for the synthesis of strained nitrogen heterocycles. | ||
Subsequently, the same group presented more general reaction conditions for the synthesis of diverse β-lactams from readily available aliphatic amines (Scheme 40).49 In contrast to acetate as the anion in the palladium complex, a sterically hindered carboxylate ligand orchestrated an amine attack on the proximal carbonyl in a palladium anhydride Int-43 more efficiently, therefore transformed aliphatic amines into β-lactams 59via C–H activation and subsequent reductive elimination (path b). Under optimal reaction conditions, a wide range of secondary amines were successfully employed as substrates, and the corresponding β-lactams were produced in good to high yield.
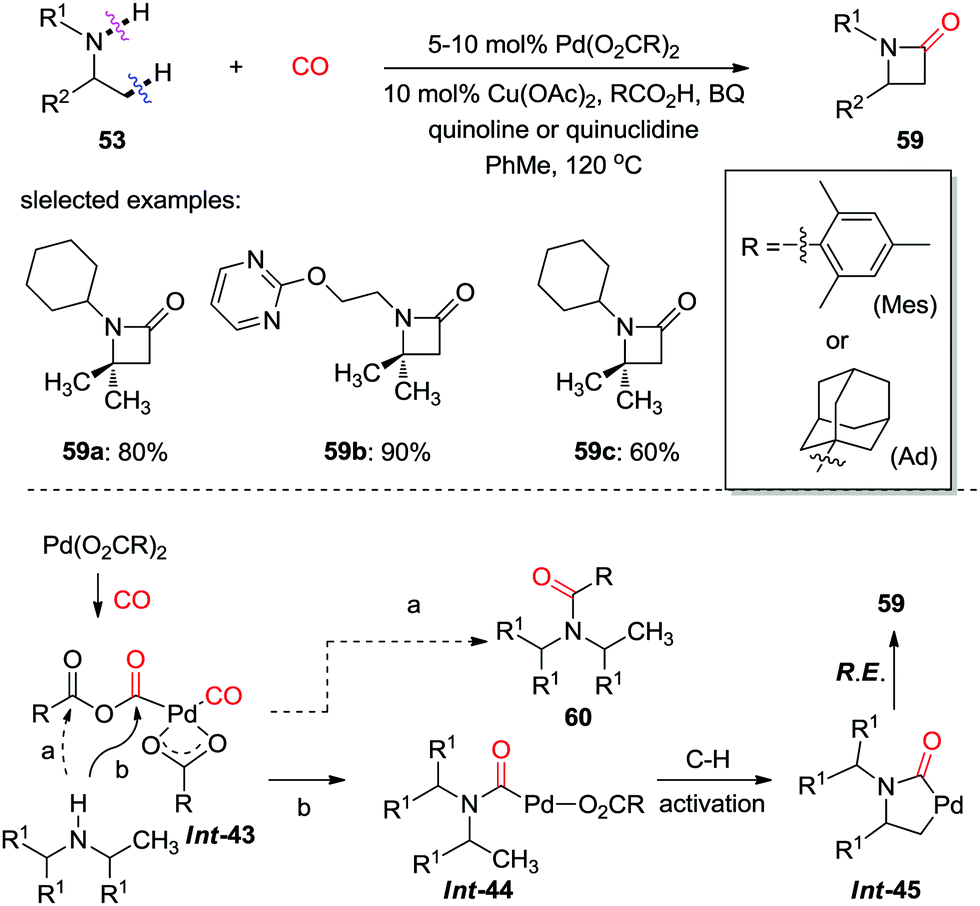 | ||
| Scheme 40 A general catalytic β-C–H carbonylation of aliphatic amines to β-lactams. | ||
3. Conclusions and perspectives
In conclusion, the main achievements of palladium-catalyzed oxidative dehydrogenative carbonylations have been summarized and discussed. This strategy has proven to be a powerful tool for the construction of carbonyl-containing compounds, including ketones, esters, and amides. However, there are still many disadvantages of this chemistry, and these drawbacks have to be overcome. First of all, although many groups have reported aerobic oxidations with air or molecular oxygen as the terminal oxidant, there are many limitations associated with these processes. In most cases, stoichiometric amounts of a nonbenign oxidant are still used to reoxidize Pd(0) to Pd(II). Therefore, environmentally benign oxidants are highly desirable in this field. For example, using biomimetic oxidations with the assistance of electron-transfer mediators (ETMs) will allow the use of benign terminal oxidant such as O2 or H2O2.50 Secondly, similar to Pd-catalyzed C–H functionalization reactions, the majority of the reported carbonylation examples use high catalyst loadings (5–10 mol%), therefore systems with higher catalytic activity has to be developed. Moreover, due to the presence of π backbonding, CO is a strong field ligand to transition metals. The occupation of orbitals of palladium by carbon monoxide prevents the coordination of various nucleophiles to palladium, which results in the limited number of C–H carbonylation reactions compared to other C–H functionalization reactions. Above all, the oxidative dehydrogenative carbonylation reactions have been demonstrated as a powerful tool for the synthesis of carbonyl compounds, and it will remain as a hot topic in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swedish Research Council (2016-03897), the Olle Engkvist Foundation, the Berzelii Center EXSELENT, and the Knut and Alice Wallenberg Foundation (KAW 2016.0072) is gratefully acknowledged. We thank Dr Ivana Némethová for fruitful discussions.Notes and references
- J. D. Rawn and R. J. Ouellette, Organic Chemistry, Academic Press, Elsevier, 2nd edn, 2018 Search PubMed.
- New Trends in Cross-Coupling – Theory and Applications, ed. T. J. Colacot, RSC Publishing, Cambridge, 2014 Search PubMed.
- C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- Y. Li, Y. Hu and X.-F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC.
- X.-F. Wu, H. Neumann and M. Beller, ChemSusChem, 2013, 6, 229–241 CrossRef CAS PubMed.
- J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed.
- (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (c) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC.
- Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS PubMed.
- B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 1863–1881 CrossRef CAS.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- (a) J. Tsuji, M. Takahashi and T. Takahashi, Tetrahedron Lett., 1980, 21, 849–850 CrossRef CAS; (b) Y. Sakurai, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 1999, 40, 1701–1704 CrossRef CAS; (c) S. T. Gadge and B. M. Bhanage, Synlett, 2013, 981–986 CAS.
- Y. Fujiwara, T. Kawauchi and H. Taniguchi, J. Chem. Soc., Chem. Commun., 1980, 220 RSC.
- R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082–14083 CrossRef CAS PubMed.
- C. E. Houlden, M. Hutchby, C. D. Bailey, J. G. Ford, S. N. G. Tyler, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830–1833 CrossRef CAS PubMed.
- M. Chen, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, J. Org. Chem., 2015, 80, 1258–1263 CrossRef CAS PubMed.
- H. Li, G.-X. Cai and Z.-J. Shi, Dalton Trans., 2010, 39, 10442–10446 RSC.
- B. Liu and B.-F. Shi, Synlett, 2013, 2274–2278 CAS.
- B. Liu, H.-Z. Jiang and B.-F. Shi, Org. Biomol. Chem., 2014, 12, 2538–2542 RSC.
- Y. Lu, D. Leow, X. Wang, K. M. Engle and J.-Q. Yu, Chem. Sci., 2011, 2, 967–971 RSC.
- S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598–10601 CrossRef CAS PubMed.
- T.-H. Lee, J. Jayakumar, C.-H. Cheng and S.-C. Chuang, Chem. Commun., 2013, 49, 11797–11799 RSC.
- H. Zhang, D. Liu, C. Chen, C. Liu and A. Lei, Chem. – Eur. J., 2011, 17, 9581–9585 CrossRef CAS PubMed.
- J. Ferguson, F. Zeng and H. Alper, Org. Lett., 2012, 14, 5602–5605 CrossRef CAS PubMed.
- L. Wang, Y. Wang, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 5657–5661 CrossRef CAS PubMed.
- A. V. Malkov, N. Derrien, M. Barłóg and P. Kočovský, Chem. – Eur. J., 2014, 20, 4542–4547 CrossRef CAS PubMed.
- (a) K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342–14343 CrossRef CAS PubMed; (b) B. López, A. Rodriguez, D. Santos, J. Albert, X. Ariza, J. Garcia and J. Granell, Chem. Commun., 2011, 47, 1054–1056 RSC; (c) Z.-H. Guan, M. Chen and Z.-H. Ren, J. Am. Chem. Soc., 2012, 134, 17490–17493 CrossRef CAS PubMed; (d) W. Li, Z. Duan, X. Zhang, H. Zhang, M. Wang, R. Jiang, H. Zeng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 1893–1896 CrossRef CAS PubMed; (e) R. Shi, L. Lu, H. Zhang, B. Chen, Y. Sha, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 10582–10585 CrossRef CAS PubMed.
- D. Liang, Y. He and Q. Zhu, Org. Lett., 2014, 16, 2748–2751 CrossRef CAS PubMed.
- F. Ji, X. Li, W. Wu and H. Jiang, J. Org. Chem., 2014, 79, 11246–11253 CrossRef CAS PubMed.
- X. Zhang, S. Dong, X. Niu, Z. Li, X. Fan and G. Zhang, Org. Lett., 2016, 18, 4634–4637 CrossRef CAS PubMed.
- C. M. R. Volla and J.-E. Bäckvall, Org. Lett., 2014, 16, 4174–4177 CrossRef CAS PubMed.
- C. Zhu, B. Yang and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 11868–11871 CrossRef CAS PubMed.
- C. Zhu, B. Yang, T. Jiang and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2015, 54, 9066–9069 CrossRef CAS PubMed.
- B. Yang, Y. Qiu, T. Jiang, W. D. Wulff, X. Yin, C. Zhu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2017, 56, 4535–4539 CrossRef CAS PubMed.
- Y. Qiu, B. Yang, C. Zhu and J.-E. Bäckvall, J. Am. Chem. Soc., 2016, 138, 13846–13849 CrossRef CAS PubMed.
- C. M. R. Volla, J. Mazuela and J.-E. Bäckvall, Chem. – Eur. J., 2014, 20, 7608–7612 CrossRef CAS PubMed.
- Y. Qiu, B. Yang, T. Jiang, C. Zhu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2017, 56, 3221–3225 CrossRef CAS PubMed.
- Y. Qiu, B. Yang, C. Zhu and J.-E. Bäckvall, Chem. Sci., 2017, 8, 616–620 RSC.
- C. Zhu, B. Yang, Y. Qiu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2016, 55, 14405–14408 CrossRef CAS PubMed.
- M.-B. Li, A. K. Inge, D. Posevins, K. P. J. Gustafson, Y. Qiu and J.-E. Bäckvall, J. Am. Chem. Soc., 2018, 140, 14604–14608 CrossRef CAS PubMed.
- H. Chen, C. Cai, X. Liu, X. Li and H. Jiang, Chem. Commun., 2011, 47, 12224–12226 RSC.
- (a) P. Xie, Y. Xie, B. Qian, H. Zhou, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 9902–9905 CrossRef CAS PubMed; (b) K. Tanaka, W. R. Ewing and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 15494–15497 CrossRef CAS PubMed.
- P. Xie, C. Xia and H. Huang, Org. Lett., 2013, 15, 3370–3373 CrossRef CAS PubMed.
- E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378–17380 CrossRef CAS PubMed.
- P.-L. Wang, Y. Li, Y. Wu, C. Li, Q. Lan and X.-S. Wang, J. Am. Chem. Soc., 2018, 140, 14604–14608 CrossRef PubMed.
- C. Wang, L. Zhang, C. Chen, J. Han, Y. Yao and Y. Zhao, Chem. Sci., 2015, 6, 4610–4614 RSC.
- J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009–1016 CrossRef CAS PubMed.
- Z. M. Png, J. R. Cabrera-Pardo, J. P. Cadahía and M. J. Gaunt, Chem. Sci., 2018, 9, 7628–7633 RSC.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed.
- (a) D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851–857 CrossRef CAS PubMed; (b) K. F. Hogg, A. Trowbridge, A. Alvarez-Pérez and M. J. Gaunt, Chem. Sci., 2017, 8, 8198–8203 RSC.
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |