
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Förster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents
Luling
Wu
 ab,
Chusen
Huang
*ab,
Ben P.
Emery
b,
Adam C.
Sedgwick
c,
Steven D.
Bull
b,
Xiao-Peng
He
d,
He
Tian
*d,
Juyoung
Yoon
*e,
Jonathan L.
Sessler
*c and
Tony D.
James
*ab
ab,
Chusen
Huang
*ab,
Ben P.
Emery
b,
Adam C.
Sedgwick
c,
Steven D.
Bull
b,
Xiao-Peng
He
d,
He
Tian
*d,
Juyoung
Yoon
*e,
Jonathan L.
Sessler
*c and
Tony D.
James
*ab
aThe Education Ministry Key Laboratory of Resource Chemistry, Shanghai Key Laboratory of Rare Earth Functional Materials, and Shanghai Municipal Education Committee Key Laboratory of Molecular Imaging Probes and Sensors, Department of Chemistry, Shanghai Normal University, 100 Guilin Road, Shanghai 200234, China. E-mail: huangcs@shnu.edu.cn
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: t.d.james@bath.ac.uk
cDepartment of Chemistry, The University of Texas at Austin, 105 E 24th Street A5300, Austin, 78712-1224, USA. E-mail: sessler@cm.utexas.edu
dKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Rd., Shanghai 200237, China. E-mail: tianhe@ecust.edu.cn
eDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea. E-mail: jyoon@ewha.ac.kr
First published on 22nd July 2020
Abstract
In this tutorial review, we will explore recent advances in the construction and application of Förster resonance energy transfer (FRET)-based small-molecule fluorescent probes. The advantages of FRET-based fluorescent probes include: a large Stokes shift, ratiometric sensing and dual/multi-analyte responsive systems. We discuss the underlying energy donor–acceptor dye combinations and emphasise their applications for the detection or imaging of cations, anions, small neutral molecules, biomacromolecules, cellular microenvionments and dual/multi-analyte responsive systems.
 From left to right: Luling Wu, Tony D. James, Steven D. Bull and Adam C. Sedgwick | Luling Wu received his MSc in 2017 from Shanghai Normal University. In 2017, he was awarded scholarships by the China Scholarship Council (CSC) and The University of Bath to carry out research towards a PhD at The University of Bath. Tony D. James is a Professor at The University of Bath and Fellow of the Royal Society of Chemistry. He was awarded the Daiwa-Adrian Prize (2013), Inaugural CASE Prize (2015), MSMLG Czarnik Award (2018) and currently holds a prestigious Royal Society Wolfson Research Merit Award (2017–2022). Steven D. Bull is a Professor of Organic Chemistry at The University of Bath. He was awarded a Royal Society Industrial Fellowship (2002–2006), the Daiwa-Adrian Prize (2013) and University of Bath prize for outstanding PhD supervision (2013). Adam C. Sedgwick is a postdoctoral research fellow working under the supervision of Prof. Jonathan L. Sessler at The University of Texas in Austin. His research interests are in the realms of stimuli-responsive materials, molecular imaging agents, and theranostic agents. |
 Chusen Huang | Chusen Huang is an Associate Professor at Shanghai Normal University (SHNU). In 2017, he was awarded a Humboldt Research Fellowship to carry out his Postdoctoral Research with Prof. Werner Nau at Jacobs University Bremen (2017–2019). His research focuses on chemical biotechnology and synthesis of new supramolecular ring structures. |
 Ben P. Emery | Ben P. Emery completed his undergraduate master's degree in chemistry for Drug Discovery at The University of Bath in 2017. In 2017 he began his PhD in synthetic organic chemistry under the supervision of Prof. Steven D. Bull. |
 Xiao-Peng He | Xiao-Peng He is professor at Feringa Nobel Prize Scientists Research Center, School of Chemistry and Molecular Engineering, ECUST. He obtained his BSc (2006) and PhD (2011) from ECUST. He completed a co-tutored doctoral program at ENS Cachan (France) (2008 to 2009) and postdoctoral research with Kaixian Chen (SIMM, CAS) from 2011 to 2013 at ECUST. |
 He Tian | He Tian was born in China in 1962. In 1999, he was appointed Cheung Kong Distinguished Professor by the Education Ministry of China. In 2011, he was selected as a member of the Chinese Academy of Science. He has been listed by Thomson Reuters as a Highly Cited Researcher in Chemistry (2014) and Materials Science (2014–2019). |
 Juyoung Yoon | Juyoung Yoon received his PhD (1994) from The Ohio State University. After completing post-doctoral research at UCLA and at Scripps Research Institute, he joined the faculty at Silla University in 1998. In 2002, he moved to Ewha Womans University, where he is currently a Distinguished Professor of the Department of Chemistry and Nano Science. Department of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea. |
 Jonathan L. Sessler | Jonathan L. Sessler received a BSc degree in Chemistry in 1977 from the University of California, Berkeley. He obtained his PhD from Stanford University in 1982. After postdoctoral stays in Strasbourg and Kyoto, he accepted a position as an Assistant Professor of Chemistry at the University of Texas at Austin, where he is currently a Doherty-Welch Chair. |
Key learning points1. The major classes of fluorophores that can act as donors or acceptors in a FRET combination.2. A focus on biological applications, including sensing and imaging of cellular environments. However, the understanding we hope to impart could be applied to the development of any new FRET-based sensing system. 3. The construction of FRET-based systems where the acceptors are quenchers. Such combinations are typically found in enzyme-based sensor systems. 4. The design and construction of dual/multiple-responsive FRET-based systems. |
Introduction
Fluorescent sensors have been widely applied in diverse fields, such as biology, physiology, medicine, and pharmacology. They have also been the subject of intense research interest on the part of many chemists and biologists. Fluorescent sensor-based detection methods possess many advantageous features, such as simplicity, low cost, high sensitivity, facile adaptation to automated analysis, an ability to support spatially resolved imaging, and provide for diverse signal output modes. More broadly, fluorescent sensors offer a unique route to detecting biologically and/or environmentally important analytes and can help to uncover physiological and pathological functions of these analytes.1 Within the cadre of fluorescent sensors, those based on Förster resonance energy transfer (FRET) involving changes in the electronic interactions between a donor and an acceptor, have seen wide applicability.2,3 Particularly, FRET-based small-molecule probes used as chemosensors and imaging agents are often the chosen detection method for biological applications due to their fast uptake by cells, the non-destructive visualisation techniques whose use they permit, and the potential for in situ detection with little interference to the structure–function of key biomacromolecules, and their readily tuned structural features.2,3The term FRET is named after Theodor Förster, who proposed an equation to quantify the electronic excitation transfer efficiency from an energy donor to acceptor in 1948.4 Förster was a theoretical physicist by training but devised his theory while working at the Max Planck Institute for Physical Chemistry in Göttingen. FRET is a process of non-radiative energy transfer through long-range dipole–dipole interactions between the donor–acceptor pair. Upon photoexcitation, the electronic excitation energy of the donor in its excited state can be transferred to the acceptor in its ground state. When the donor and acceptor are both fluorophores, FRET is often referred to “fluorescence resonance energy transfer” (Scheme 1).
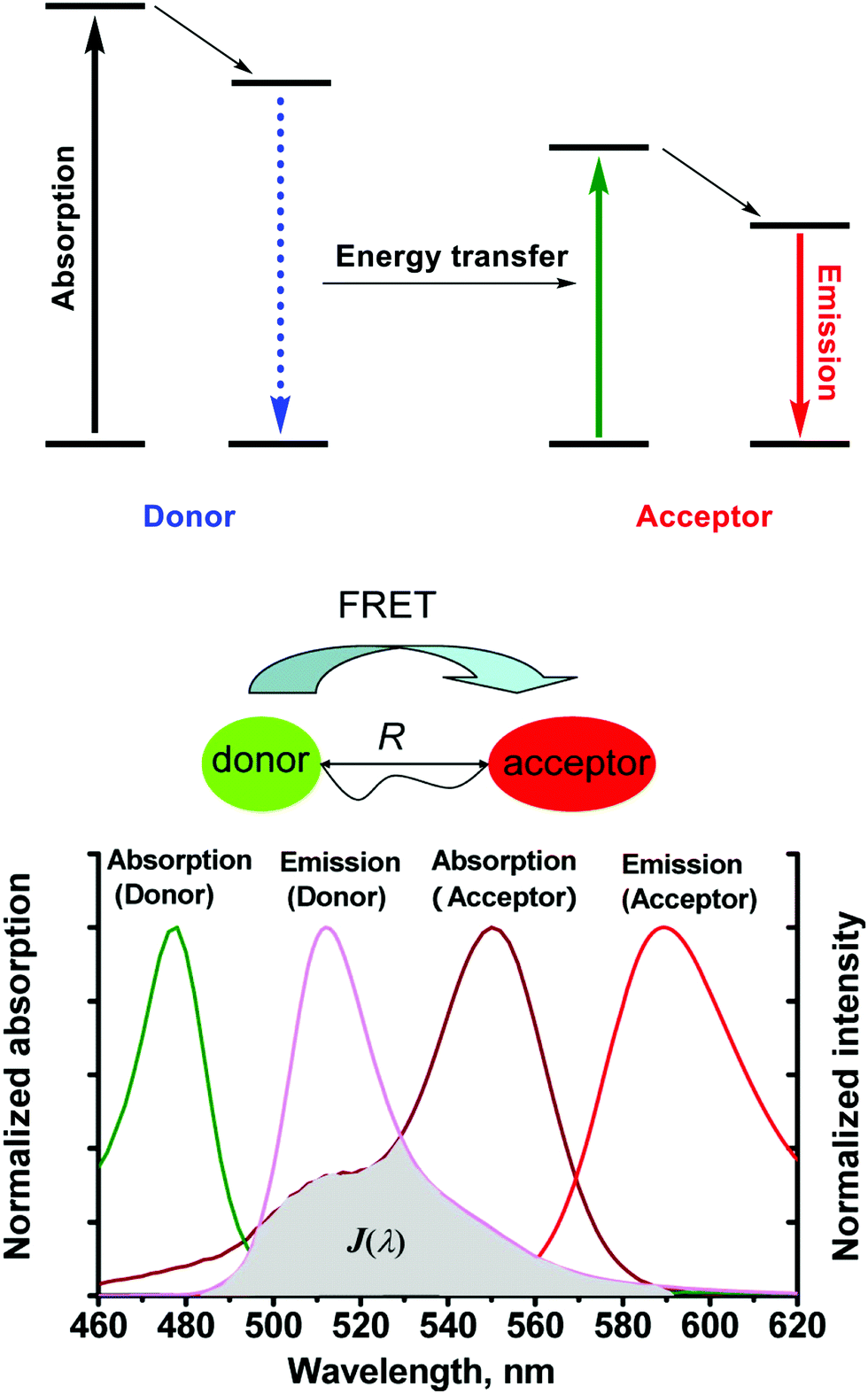 | ||
| Scheme 1 The mechanism of Förster resonance energy transfer (FRET). R is the distance between the energy donor and acceptor, J(λ) represents the degree of spectral overlap between the donor emission and the acceptor absorption. Reproduced with permission from ref. 3 (Acc. Chem. Res., 2013, 46, 1462–1473). Copyright (2013) American Chemical Society. | ||
As a general rule, molecules being considered for use in FRET applications should have the following prerequisites: (1) The donor–acceptor pair should be positioned in proximity of each other (typically 10–100 Å), a requirement that helps rationalise the distance dependency of FRET and its efficiency. (2) The emission spectra of the donor should overlap with the absorption spectra of the acceptor. (3) The donor emission moment, the acceptor absorption moment, and their separation vector must be in a favorable mutual orientation.
Because they rely on a distance-dependent energy transfer process, FRET-based fluorescent probes typically display an inherent sensitivity to the environment and probe structure that has made them attractive for use in a wide range of biological studies. For instance, they have enabled explorations of various biomacromolecules and biological phenomena where function and events can lead to changes in molecular proximity or, more precisely, donor–acceptor overlap.
The general strategy of designing FRET-based small-molecule fluorescent probes is anchoring the donor–acceptor pair in close proximity to one another in one single system or molecule. A reactive unit that can be used as a specific recognition group is then introduced synthetically to construct the final FRET system. When the FRET system or molecules are exposed to specific target analytes, the FRET process is initiated or prevented. As a result, a change in fluorescence signal can be observed, allowing for confirmation of the presence (or absence) of the target analytes.
To date, four common approaches have been used for the construction of FRET probes (Scheme 2). One approach involves alteration of the chemical structure of an acceptor from the non-fluorescent form to fluorescent form (Scheme 2a). Initially, there is relatively little overlap between the absorption band of the non-fluorescent acceptor and the emission band of the donor. After interaction between the non-fluorescent form of the acceptor and the target analyte, the absorption band of the acceptor is shifted such that it increases overlap with the emission band of the donor, facilitating FRET.
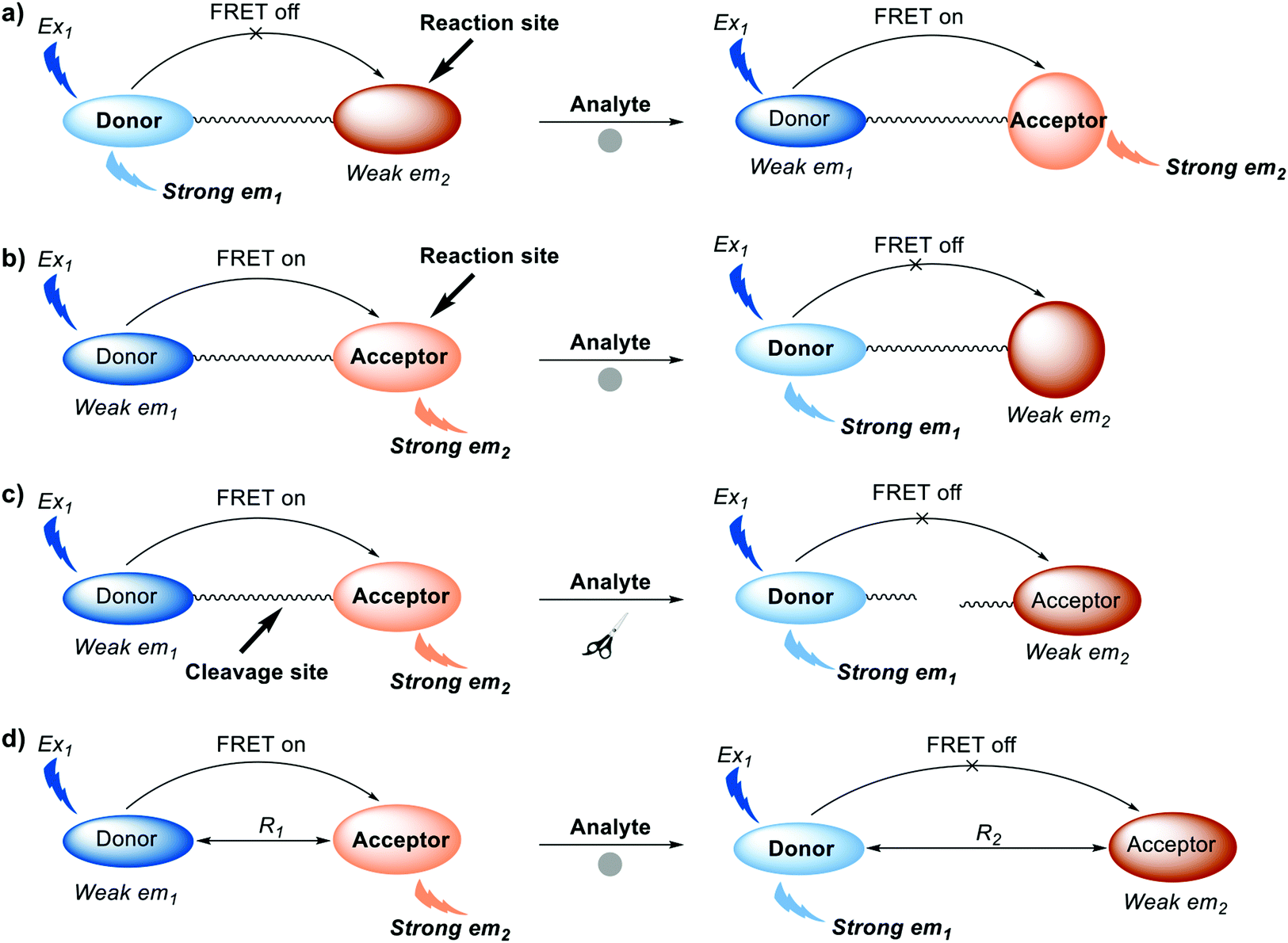 | ||
| Scheme 2 Diagrammatic representation of the four common strategies used to generate FRET-based small-molecule fluorescent probes. (a) Turn-on FRET systems: alteration of the chemical structure of an acceptor from the non-fluorescent form. (b) Turn-off FRET systems: alteration of the chemical structure of an acceptor from the fluorescent form to the non-fluorescent form. (c) Turn-off FRET systems: chemical or enzymatic cleavage of the covalent linker between donor and acceptor. (d) Turn-off FRET systems: intermolecular dissociation of FRET donor and acceptor. Where Ex is excitation wavelength and em is maximum emission wavelength. R1 and R2 represent two different distances between energy donor and energy acceptor. Note: donor and acceptor are fluorophores. | ||
This review will summarise recent progress in the development of small-molecule FRET-based chemosensors and imaging agents. Since to many end users, these systems operate as a “black box”, we will discuss how they work, what they can do, and where they can be applied. The goal is to help readers gain insight into the importance of this vibrant area and understand both the opportunities it affords, its basis for action, and merits and demerits relative to the sensing field as a whole. Specifically, this review will describe the design principles required for the construction of FRET-based small-molecule probes and provide a basic tutorial on how they work. A number of donor–acceptor combinations used to prepare FRET scaffolds will be detailed and their utility linked to recent leading literature references since 2008. Potential biological applications of FRET-based small-molecule probes will also be highlighted. Then, this review will include personal perspectives from the authors and insights into how and where FRET-based fluorescent sensors are important. Finally, key challenges that need to be overcome for the future development of FRET-based systems will be outlined.
FRET-based sensors for cations
Metal cations play numerous roles in regulating various biological process and many are recognised environmental pollutants. Many fluorescent sensors have been developed for sensing cations in recent decades. Compared to other commonly used design strategies for the detection of cations, such as photoinduced electron transfer (PeT) and intramolecular charge transfer (ICT), FRET-based fluorescent sensors exhibit properties ideal for ratiometric sensing. They also benefit from facile structural design. In this section some representative FRET-based fluorescent sensors are showcased to illustrate the thought processes used in the construction of FRET-based sensors. The coverage is not exhaustive; however, it is hoped that these examples will help the reader access the primary literature with a greater level of understanding.Mercury ions (Hg2+) can cause serious damage to the central nervous and endocrine systems. Therefore, the first example we discuss is the FRET-based fluorescent probe 1 for the detection of Hg2+ (Fig. 1).5 Probe 1 was constructed by covalently linking BODIPY and rhodamine as a fluorescent donor–acceptor system to generate a FRET pair. Upon interaction with Hg2+, the thiosemicarbazide moiety present in 1 irreversibly cyclised to form a 1,3,4-oxadiazole. This caused the spirolactam of rhodamine to ring open, allowing FRET to occur. As prepared, 1 exhibited a maximum emission at 514 nm upon excitation at 488 nm, which was ascribed to the emission profile of the fluorescent donor BODIPY. Such an observation was consistent with the design expectation for 1, namely that prior to interaction with Hg2+, no FRET occurred. On the other hand, addition of Hg2+ caused a maximum absorption at 560 nm to appear and a concurrent reduction in the BODIPY-based fluorescence emission at 514 nm and an increase in the emission intensity at 589 nm. Such observations were consistent with the “turning on” of a FRET process, presumably resulting from Hg2+-promoted formation of the oxadiazole and induced ring opening of the spirolactam group of the rhodamine subunit. The FRET efficiency of the product was calculated to be 99%. Additionally, 1 could be shown to detect Hg2+ down to ppb (parts per billion) in mixed aqueous media (C2H5OH/H2O (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2); pH 7.0). Probe 1 was further used for detection of Hg2+ in MCF-7 cancer cells via ratiometric fluorescent imaging, wherein the ratio of the two emission maxima, rather than the overall change in intensity, was monitored.
:2); pH 7.0). Probe 1 was further used for detection of Hg2+ in MCF-7 cancer cells via ratiometric fluorescent imaging, wherein the ratio of the two emission maxima, rather than the overall change in intensity, was monitored.
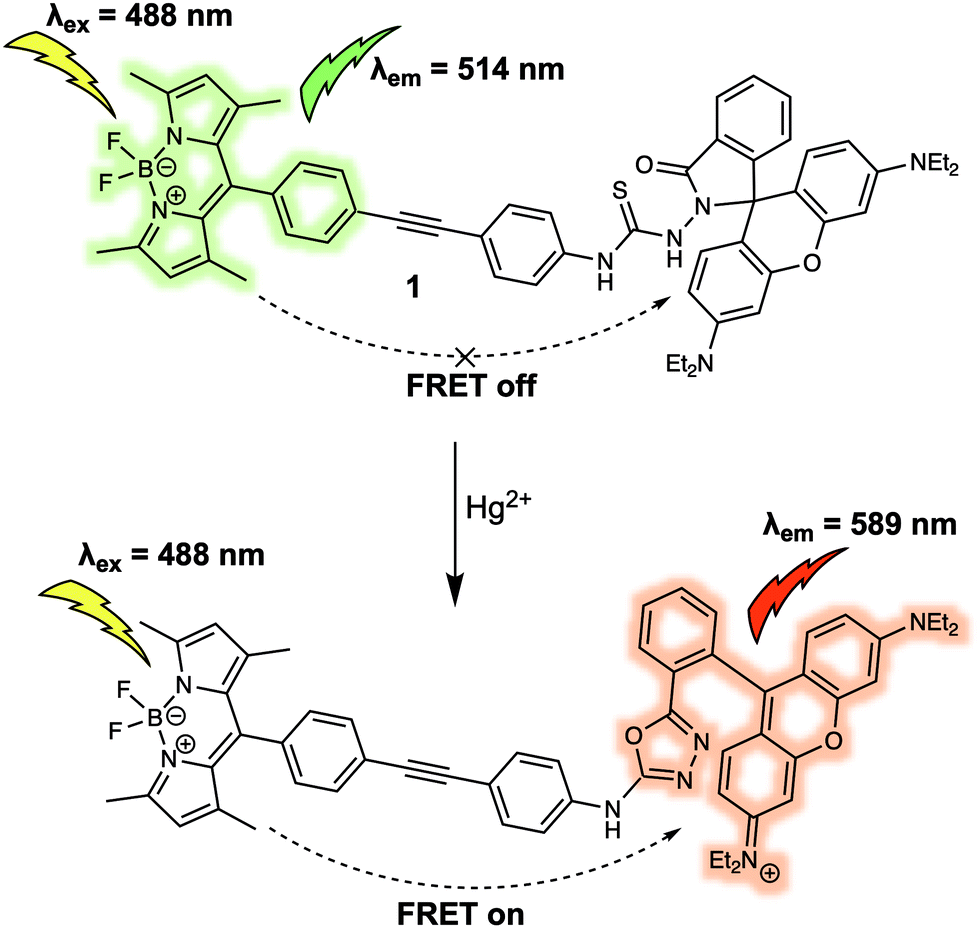 | ||
| Fig. 1 BODIPY/rhodamine (energy donor/acceptor)-based FRET fluorescent probe 1 used for Hg2+ sensing. | ||
Poisonous cadmium ions (Cd2+) may enter the body through the consumption of contaminated food, water, or inhalation of cigarette smoke. The exposure to Cd2+ can cause renal dysfunction, mutations, calcium metabolism disorders, as well as Itai-itai disease. In 2015, Goswami et al. developed probe 2 bearing the quinoline–benzothiazole based donor and rhodamine-based acceptor, and demonstrated its use in Cd2+ sensing (Fig. 2).6 Upon excitation at 360 nm, 2 displayed a strong emission at 470 nm that was ascribed to the emission of the quinoline–benzothiazole fluorophore. However, no emission peak at around 585 nm was present, which led the authors to suggest that the rhodamine moiety existed predominantly in its spirolactam form. Upon addition of Cd2+, a new absorption peak at 565 nm developed. This was taken as an indication that ring opening of the spirolactam of the rhodamine moiety occurred, which in turn allowed for FRET from the quinoline–benzothiazole to the rhodamine moiety, shifting the emission to 585 nm. The emission intensity ratio (I585/I470) displayed a linear relationship with increasing Cd2+ concentration (from 0 to 9.5 μM). The proposed binding mode was further supported by a single-crystal X-ray analysis of the 2-Cd2+ complex, which revealed a six-coordinate Cd2+ centre. The design strategy of 2 resulted in high selectivity towards Cd2+ even in the presence of other potential interferants, such as Zn2+, Cr3+, and Pb2+. Probe 2 was used to detect Cd2+ in live RAW264.7 cells through ratiometric fluorescence cell imaging. It was also developed into a portable qualitative test kit using TLC plates soaked with probe 2, which allowed for the rapid sensing of Cd2+.
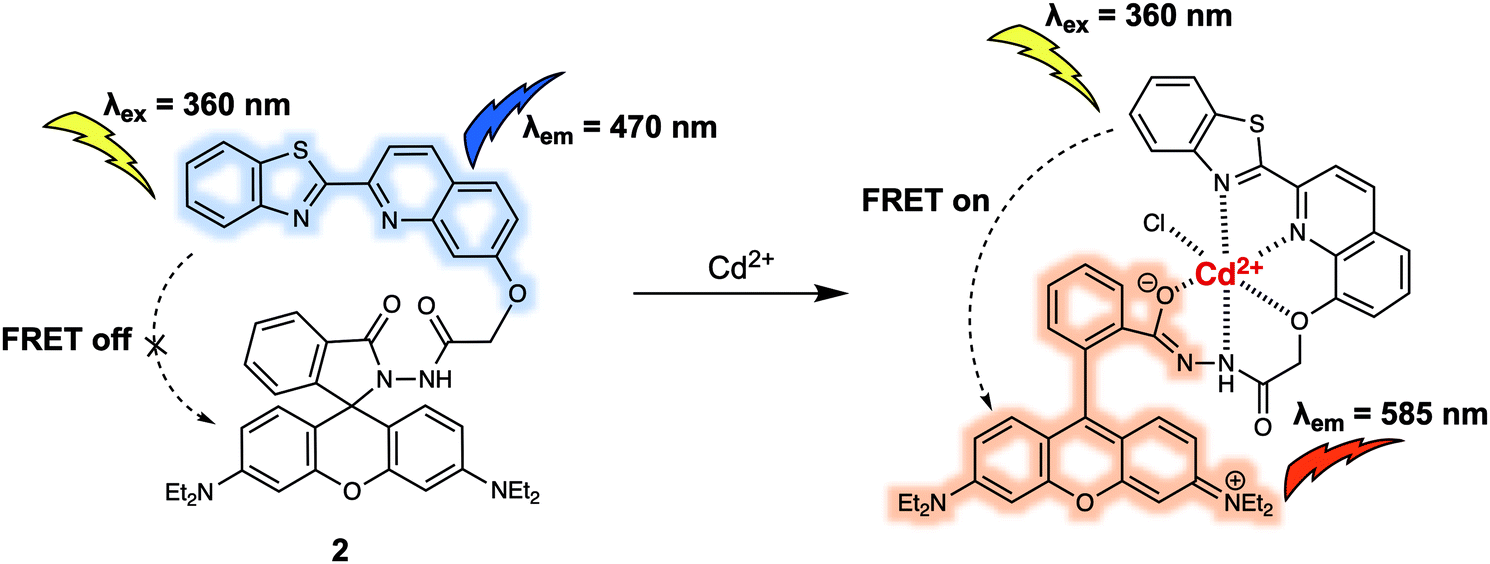 | ||
| Fig. 2 Quinoline–benzothiazole/rhodamine (energy donor/acceptor)-based FRET probe 2 used for the ratiometric detection of Cd2+. | ||
Both 1 and 2 are typical FRET systems that rely on a rhodamine dye as the energy acceptor. In these systems, ring opening of the spirolactam portion of the rhodamine dye is controlled by the coordination of heavy metal cations to produce a turn-on FRET corresponding to the emission of rhodamine, something made possible because of the spectral overlap between the absorption of the fluorescence acceptor and the emission of the donor. The turn-off FRET design strategy has been widely used in FRET-based systems for the detection of various cations in biological systems. For instance, Chang et al. reported a new FRET system (probe 3) for the detection of elevated Fe2+ levels in living cells.7 Despite its importance in numerous regulatory systems involved in cellular iron homeostasis, the real time monitoring of Fe(II)/Fe2+ concentrations in live biological systems remains a largely unsolved problem in part because of the potent fluorescence quenching ability of Fe2+. Many current approaches employ fluorescence on–off systems, many of which fail to provide critical spatiotemporal information. Probe 3 was designed to overcome these limitations. It was constructed by attaching a 5-aminomethyl fluorescein (5-AMF) donor and a cyanine 3 (Cy3) acceptor to an endoperoxide core that is known to be an Fe2+ responsive trigger (Fig. 3). As prepared, 3 displayed maximum emissions at 515 and 556 nm, corresponding to the 5-AMF donor and Cy3 acceptor, respectively, when the probe was excited with 488 nm radiation. In this case, the default state of the probe is FRET on before addition of an analyte. Addition of Fe2+ resulted in an increased emission at 515 nm, corresponding to the emission of the 5-AMF donor, and a loss at emission at 556 nm was observed. This result could be rationalised by the Fe2+-promoted cleavage of the endoperoxide core, resulting in the donor/acceptor pair being broken apart and the FRET process quenched. Probe 3 exhibited selectivity for Fe2+ over other metal cations such as Fe3+, Cu2+ and Zn2+. It was then tested in cells. Firstly, exogenously induced changes in labile Fe2+ levels in HEK 293T cells were tracked using probe 3 by exploiting its ratiometric fluorescence imaging features. Then, endogenous changes in the labile Fe2+ concentrations in different cell types were tracked using 3, leading the authors to suggest its potentially generalisable utility for exploring the role of labile Fe2+ in biological systems.
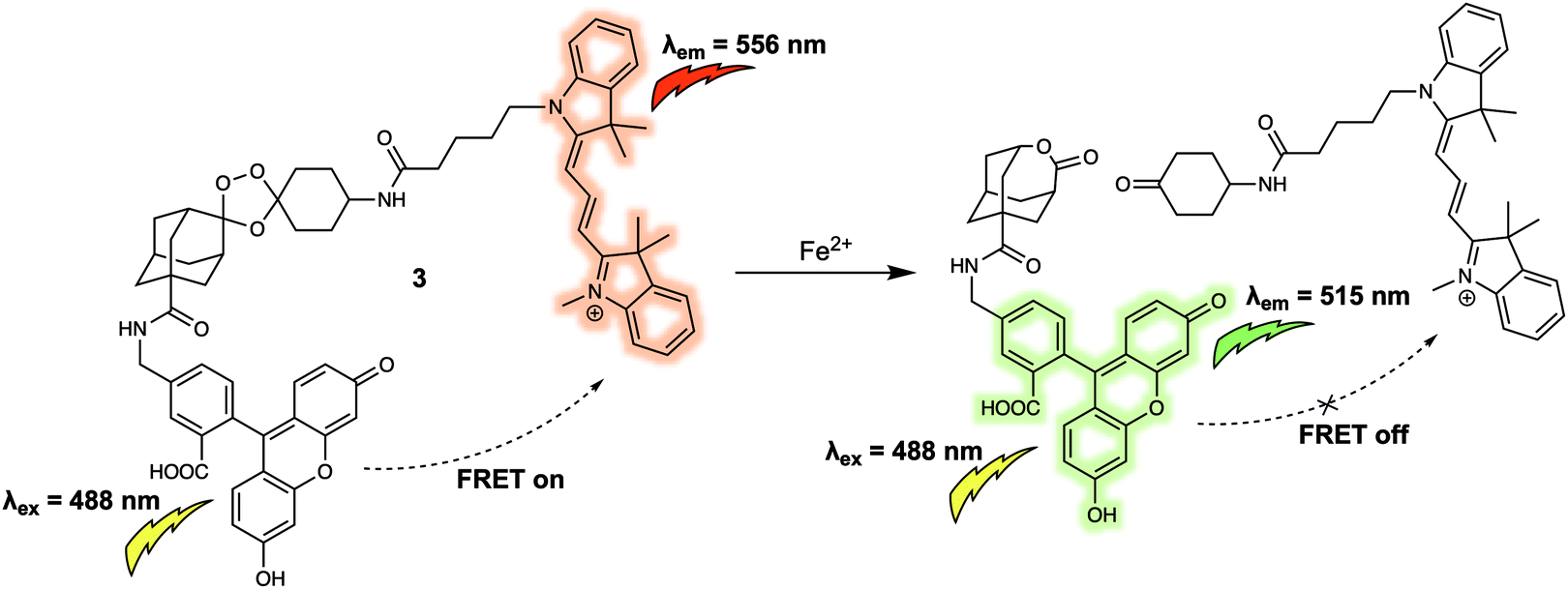 | ||
| Fig. 3 5-Aminomethyl fluorescein/cyanine 3 (energy donor/acceptor)-based FRET fluorescent probe 3 that permits the ratiometric detection of Fe2+. | ||
Copper ions (Cu+) exist in labile pools, weakly bound to low-molecular weight ligands, which can control numerous transition metal signalling processes including neural communication and olfaction, lipolysis, rest–activity cycles, and kinase pathways crucial in tumour development. A FRET platform (probe 4) for the detection of labile Cu+ was recently reported.8 In probe 4, fluorescein and rhodamine moieties are used as the energy donor and acceptor respectively. These subunits were anchored to a tris[(2-pyridyl)methyl]amine (TPA) group that was designed to serve a dual roles as the linker and as the selective recognition site for Cu+ (Fig. 4). As prepared, probe 4 displayed emissions at 526 and 576 nm corresponding to the fluorescein and rhodamine when excited at 458 nm, respectively. The presence of emission at 576 nm was taken as evidence of energy transfer from the excited fluorescein moiety to the rhodamine subunit present in 4. Upon addition of Cu+, a strong emission at 526 nm was observed whose increase in intensity over time was accompanied by a reduction in the fluorescence intensity at 576 nm. On this basis it was proposed that the FRET process was inhibited in the presence of Cu+ as the result of fragmentation of the fluorescein/rhodamine FRET pair. Support for this proposed mechanism of FRET quenching came from LC-MS analyses that confirmed the appearance of separate fluorescein methyl ester and rhodamine-TPA fragments in solution after treatment with Cu+ under aerobic conditions, because, the Cu+ promotes oxidative cleavage of the benzylic ether C–O bond of the TPA group within the probe. The fluorescence intensity at 526 vs. 576 nm (I526/I576) displayed a linear dose–response towards Cu+ (from 0.01 to 1 μM). Consistent with the excellent performance of 4 in detecting Cu+ in aqueous buffered solution, labile cellular Cu+ could be tracked via ratiometric fluorescence imaging. Specifically, dynamic changes in the labile Cu+ concentrations under the stimulus of reagents could be monitored directly, an ability that could prove useful in elucidating the potential relationships between labile Cu+ homeostasis and endogenous central redox mediators in the cell. It is known, for instance, that glutathione (GSH) can increase and/or reduce labile Cu+ pools in a GSH-linked manner subject to total GSH levels, in addition to GSH/GSSG redox ratios. In addition, 4 may have a role to play as a chemical visualisation tool useful in exploring the connections within labile Cu+ pools, GSH metabolism, and oncogenic transformation.
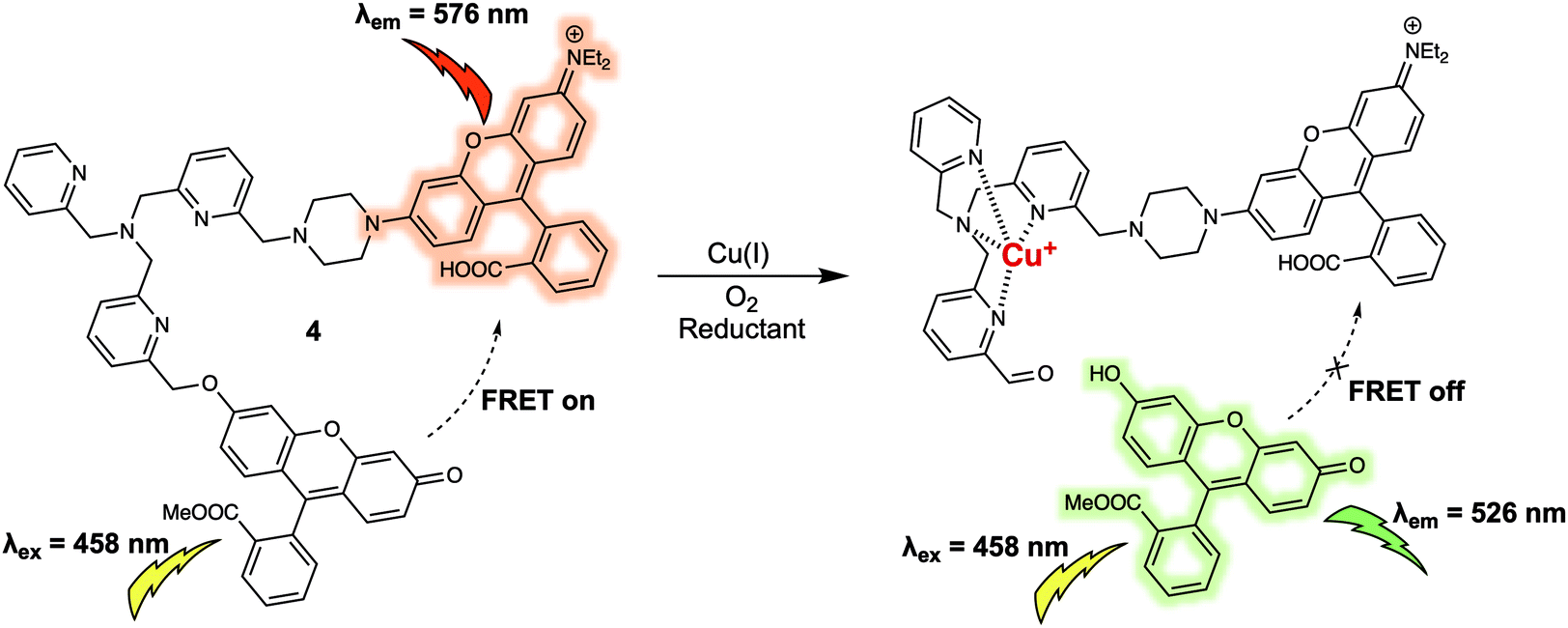 | ||
| Fig. 4 Fluorescein and rhodamine (energy donor/acceptor)-based FRET fluorescent probe 4 used for the ratiometric detection of Cu+. | ||
FRET-based sensors for anions
Similar to cations, anions are crucial for a wide variety of biological processes. For instance, hypochlorite (ClO−) has high reactivity towards numerous biomolecules including amino acids, nucleotides, carbohydrates, and lipids etc. which, if unregulated, can contribute to a variety of diseases. Additionally, as a reactive oxygen species (ROS), ClO− works as a powerful microbicidal agent towards invading microbes. Therefore, the development of suitable chemical tools for monitoring dynamic changes in biologically relevant concentrations of ClO− in living systems is likely to aid the exploration of ClO− related biological processes. In 2014, Zhao and colleagues designed a new FRET sensor (probe 5) for the ratiometric detection of ClO−.9 Probe 5 was constructed using a coumarin/rhodamine dyad as the FRET report pair, and a diacylhydrazine group coupled to the rhodamine moiety as the specific recognition site for ClO− (Fig. 5). In the absence of analyte, 5 displayed an emission at 470 nm characteristic of coumarin when excited at 410 nm. This fluorescence behaviour was ascribed to the predominant existence of the leuco-spirolactam form of the rhodamine moiety of the probe. Addition of ClO− to 5 was found to induce ring opening of rhodamine, and concurrent selective cleavage of the diacylhydrazine to afford the carboxylic acid. As a result of the increased overlap between the absorption band of the ring-opened rhodamine moiety and emission of the coumarin part, a strong fluorescence emission centred at 580 nm was seen upon excitation at 410 nm. This was ascribed to a FRET process. Analysis of the fluorescence intensity ratio (I580/I470), facilitated the quantitative detection of ClO− with a linear range from 170 to 230 μM in Na2HPO4 (0.1 M, pH = 8):DMF (6:4, v/v) solution. To further improve the water solubility and achieve mitochondrial localisation in live cells, the phenyl group in 5 was replaced synthetically by a quaternized pyridine moiety.10 This gave 6, which exhibited a fluorescent response towards ClO− in buffered aqueous solution that was similar to that of 5. Additionally, 6 was found to localise in the mitochondria of RAW264.7 cells. Changes in the endogenous production of ClO− stimulated by lipopolysaccharide (LPS) in RAW264.7 cells could be tracked via ratiometric fluorescence imaging using 6; moreover, the fluorescence signals produced in this way proved consistent with mitochondrial targeting.
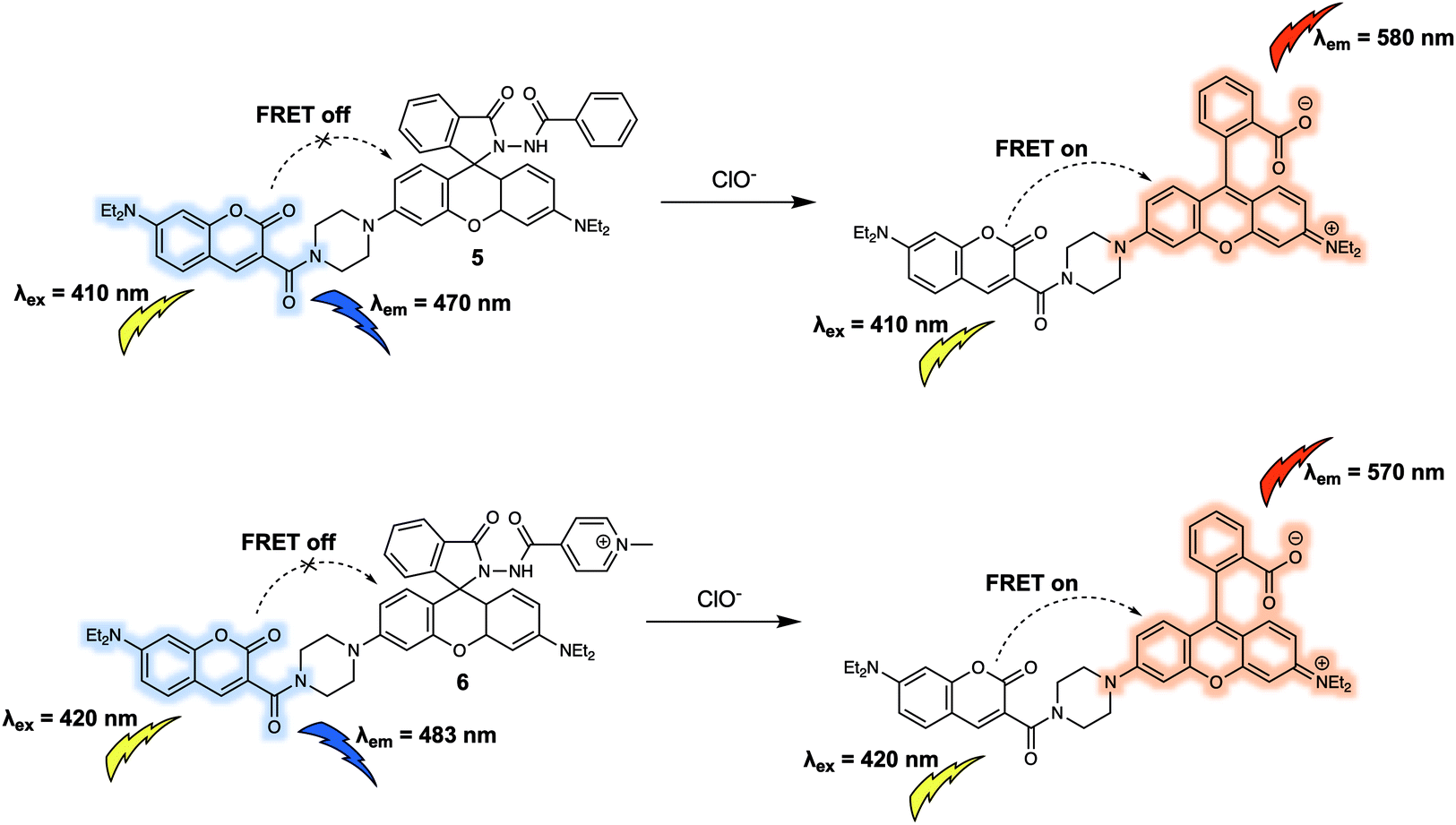 | ||
| Fig. 5 Coumarin/rhodamine (energy donor/acceptor)-based FRET fluorescent probes 5 and 6 developed for the ratiometric detection of ClO−. | ||
Peroxynitrite (ONOO−) is an important reactive nitrogen species (RNS) that plays critical roles in living systems.11 Excessive accumulation of ONOO− can cause adverse effects to organisms due to its high reactivity towards various biomolecules. For instance, peroxynitrite has been linked to many pathological conditions including chronic inflammation, Alzheimer's disease, cancer, and autoimmune diseases. In 2016, Qian et al. designed a new FRET platform (probe 7) for the selective detection of ONOO− (Fig. 6).12 To create 7, a Cy3/Cy5 dyad, designed to serve as the fluorescence energy donor and acceptor pair, respectively, were linked via an acetyl-piperazyl-hexanoyl linker. The proximity of the Cy3 and Cy5 cores in the parent structure facilitated FRET and enabled the probe to emit a strong fluorescence emission at 660 nm upon excitation at 530 nm. Upon addition of ONOO−, the Cy5 moiety was selectively oxidised to its oxindole derivative, quenching the FRET process. This chemical transformation is shown in terms of the characteristic fluorescence emission of Cy3 at 560 nm increasing in intensity, while the fluorescence intensity at 660 nm is seen to undergo a commensurate decrease. The proposed cleavage-based mechanism of action was supported by mass spectrometric analyses, which confirmed the formation of the oxidised daughter product 8. A linear relationship between the fluorescence intensity ratio (I560/I660) and concentration of ONOO− (0–700 nM) was observed in 0.1 M phosphate buffer (pH = 7.4, 0.2% DMF, v/v) solution. Probe 7 was then used as a ratiometric sensor for both exogenous and endogenous ONOO− in RAW264.7 macrophages via fluorescence imaging. The intracellular fluorescence signal from 7 was found to be largely localised in the mitochondria, allowing the authors to suggest that it could be used as a chemical tool for exploring mitochondrial ONOO− concentrations in living systems.
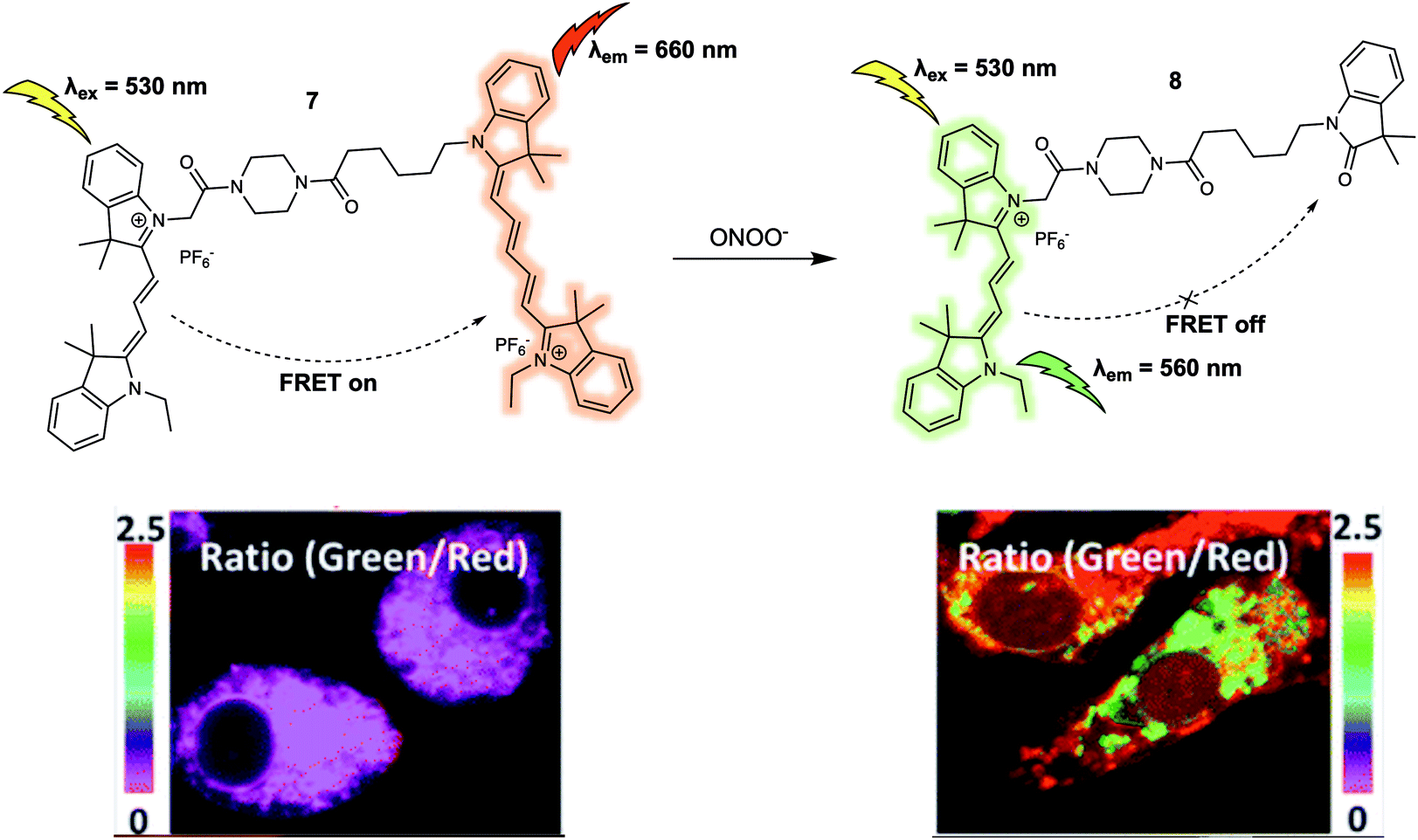 | ||
| Fig. 6 Cy3/Cy5 (energy donor/acceptor)-based FRET fluorescent probe 7 developed for the ratiometric detection of ONOO−. Also shown is the structure of its daughter product, 8. Reproduced with permission from ref. 12 (J. Am. Chem. Soc., 2016, 138, 10778–10781). Copyright (2016) American Chemical Society. | ||
Another example of a FRET-based probe for the ratiometric detection of ONOO− is probe 10 (Fig. 7) designed by Yuan et al.13 They took a screening approach and explored the sensitivity and selectivity of 19 commercially available or rationally synthesized dyes as putative sensors for ONOO−. Based on their results, these authors suggested that the monomeric dye 9 was selectively oxidized by ONOO− but was unreactive towards other ROS, such as hypochlorous acid (HOCl) and hydrogen peroxide (H2O2). In addition, 9 exhibited relatively long wavelength fluorescence emission, enabling it to act as a good candidate as an energy acceptor in the construction of a FRET-based sensor system. After introducing a coumarin group as the energy donor, these researchers constructed the FRET probe 10. As prepared, 10 displayed a strong emission at 651 nm and a weak emission at 473 nm upon excitation at 420 nm, a finding considered consistent with a FRET process. The addition of ONOO− resulted in a decrease of the emission at 651 nm and concomitant increase in emission at 473 nm, characteristic of the free coumarin dye. The response of 10 towards ONOO− in PBS buffer (25 mM, cosolvent 30% ethanol) solution could be observed within 20 seconds. The detection limit was determined to be 11.3 nM on the basis of reading out the ONOO−-dependent ratiometric fluorescence signal (I473/I651), which was taken as evidence that 10 would display high sensitivity in applications involving the quantitative detection of ONOO−. Probe 10 was then used for the ratiometric imaging of endogenous ONOO− concentrations in live HepG-2 and RAW264.7 cells. The dynamic changes in the endogenous ONOO− levels stimulated by LPS and IFN-γ (interferon-gamma) were successfully tracked using 10via ratiometric fluorescence imaging. The fluorescence signal proved to be strongly localised in the mitochondria, a finding attributed to the positive charge of the probe. Additionally, 10 could be used as a two-photon fluorescent probe. Upon excitation at 800 nm, fluorescence emission consistent with the response seen under single photon excitation at 405 nm was observed. These favourable attributes allowed 10 to be used for the detection of endogenous ONOO− in living hepatic tissue and for the observation of elevated peroxynitrite levels in the artificially inflamed leg tissues of live mice.
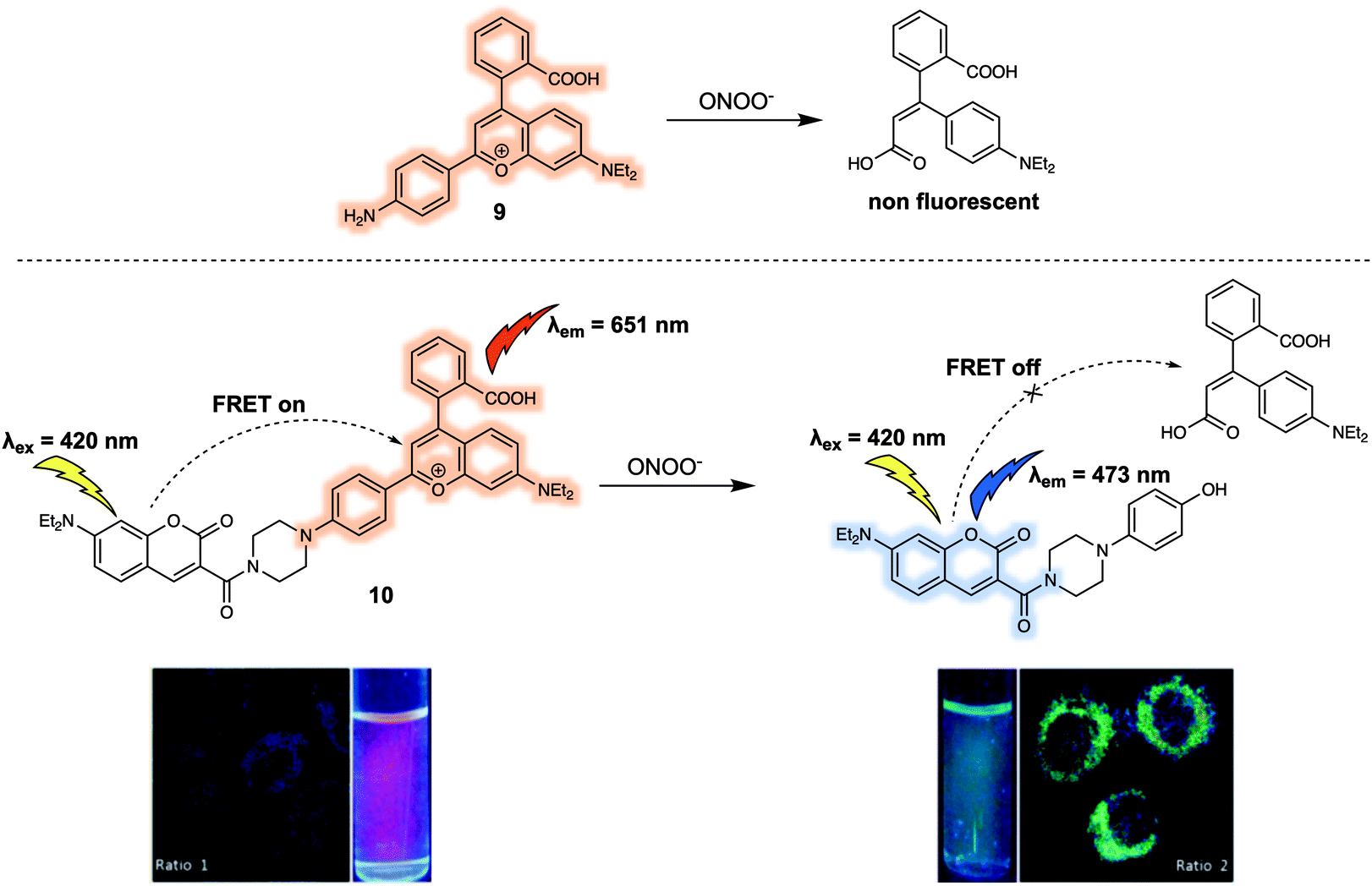 | ||
| Fig. 7 FRET fluorescent probe 10 developed for the ratiometric detection of ONOO−. Also shown is the structure of 9 upon which it was based. Reproduced with permission from ref. 13 (J. Am. Chem. Soc., 2017, 139, 285–292). Copyright (2016) American Chemical Society. | ||
The sulfite (SO32−) and bisulfite (HSO3−) anions constitute another important type class of negatively charged analytes that play recognised roles in the environment and in living systems. For instance, aqueous concentrations of HSO3−/SO32− are directly related to the concentration of sulfur dioxide (SO2), which has been reported to be involved in febrile seizures associated with brain injury. In 2016, Zhao et al. reported the FRET probe 11 (Fig. 8) designed to permit the ratiometric detection of HSO3−/SO32− in cancer cells.14 In this probe, dansyl and hemicyanine fluorophores were used as the energy donor and acceptor, respectively. When excited at 410 nm, probe 11 gave rise to an emission centred at 582 nm leading the authors to propose that it operated with a FRET efficiency of 95%. Treatment of 11 with NaHSO3 (source of HSO3−/SO32−) resulted in a decrease of emission intensity at 582 nm, accompanied by an increase in emission intensity centred at 530 nm. The sensing mechanism behind the efficacy of 11 was ascribed to nucleophilic addition of HSO3−/SO32− to the conjugated alkene of the hemicyanine acceptor. This addition interrupts the π-conjugated system, leading to a reduced overlap between the absorption of the acceptor and emission of the donor, thus interfering with the FRET process. Having established that 11 could selectively detect HSO3−/SO32−via the ratiometric fluorescence changes in solution, the probe was then used to successfully track exogenous HSO3−/SO32− in live HeLa cells. Probe 11 was also shown to be capable of differentiating endogenous bisulfite in human liver cancer cells (HepG2 cells) and healthy human liver cells (L-02 cells). The distribution of the fluorescence signal was consistent with the probe being localised in the mitochondria. On this basis the authors concluded that 11 could potentially be used to investigate mitochondrial HSO3−/SO32− concentrations in living systems.
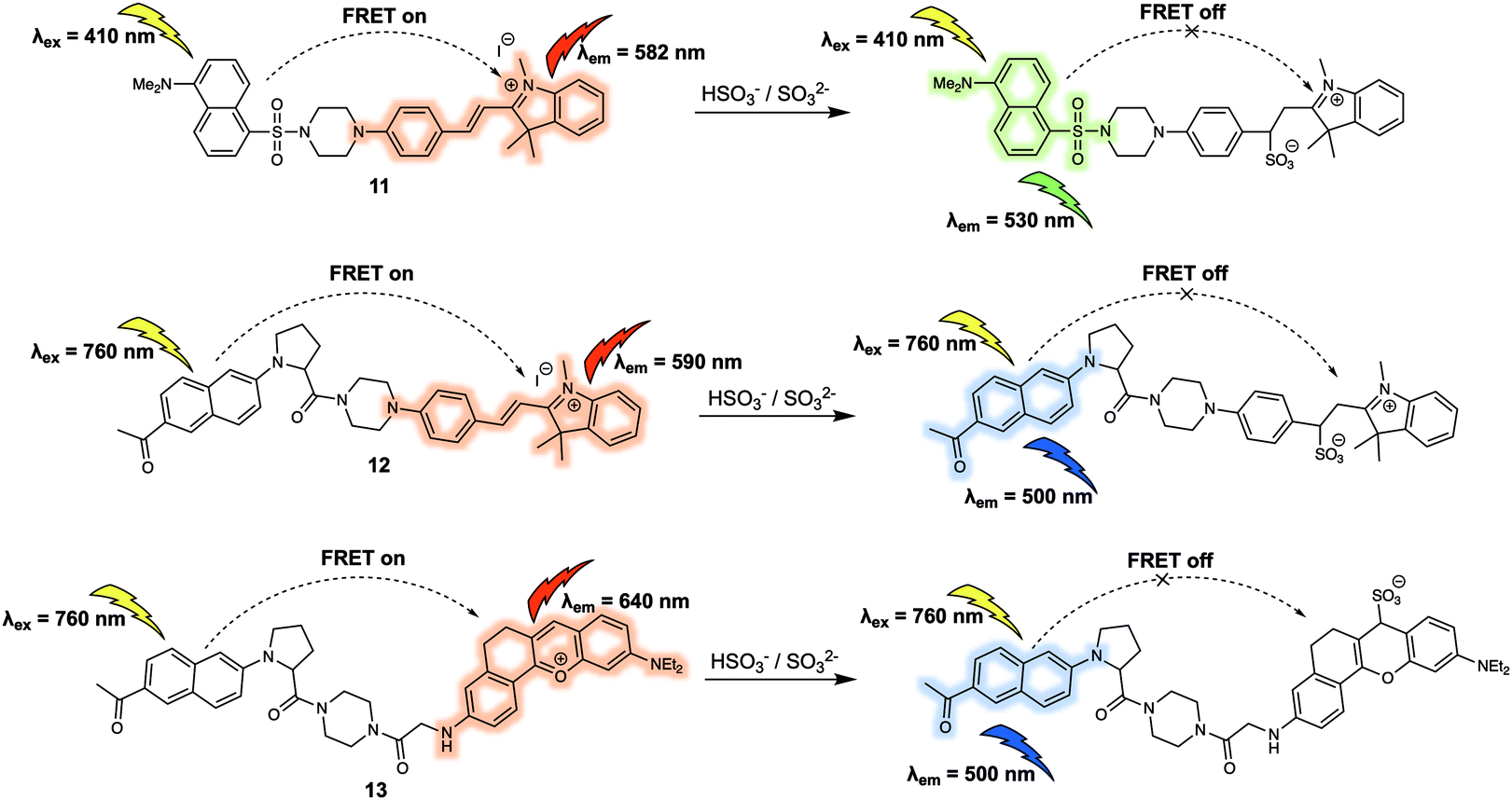 | ||
| Fig. 8 FRET-based probes (11, 12 and 13) developed for the ratiometric detection of HSO3−/SO32−. | ||
In the same year, Yang and colleagues reported a new two-photon excited fluorescence resonance energy transfer (TP-FRET) strategy for the selective detection of HSO3−/SO32−.15 The 2-acetyl-6-dialkylaminonaphthalene (acedan) moiety, which could be used with two-photon excitation, was chosen as the energy donor and hemicyanine as the energy acceptor. As true for 11, the alkene of the hemicyanine was designed to prove the reaction-based response site for HSO3−/SO32−, whereas the positive charge was expected to promote localisation of 12 (Fig. 8) within the mitochondria of living cells. The major advantage of 12 lies in its ability to be excited at a much longer wavelength (i.e. 760 nm) than 11 due to the fact that it can be activated through two-photon excitation. This capability was expected to address the issue of background noise, while reducing cell damage and obviating concerns associated with poor tissue penetration. As expected, endogenous HSO3−/SO32− generated by the reaction between Na2S2O3 and GSH catalysed by thiosulphate sulphurtransferase in HepG2 cells could be tracked using 12via two-photon ratiometric fluorescence imaging. Additionally, endogenous HSO3−/SO32− in fresh rat liver tissues at a depth of 120 μm was imaged using 12. In subsequent research, Yang and co-workers prepared another TP-FRET-based fluorescent probe 13 (Fig. 8), which was used to explore the role of SO2 in febrile seizure-induced brain injury.16 In probe 13, acedan was again used as the donor while anthocyanidin was chosen as the energy acceptor. As previously demonstrated, reaction with HSO3−/SO32− served to break the π-conjugation of the anthocyanidin derivative thus inhibiting the FRET process. Compared to the first-generation probe 12, probe 13 exhibited a much larger FRET efficiency and faster reaction kinetics (a maximum response was observed within 3 seconds). These features allowed 13 to be used to trace endogenous HSO3−/SO32− in febrile seizure-treated rat hippocampus tissues. Due to the fact HSO3−/SO32− and SO2 are in direct equilibrium in aqueous solution, it was suggested that these FRET-based fluorescent probes (11, 12 and 13) could serve as chemical tools that could be used to explore how SO2 acts as a gasotransmitter in various biological processes.
FRET-based sensors for small neutral molecules
In addition to cations and anions, small neutral molecules are important in the regulation of biological processes. A variety of fluorescent probes based on differing design strategies have been reported for detecting small neutral molecules. This section will include some representative FRET-based sensors for biologically important small neutral molecules, including their underlying design strategy, sensing mechanisms and biological applications. The first example is probe 14 that was developed by Sun et al.17 for the detection of nitroxyl (HNO). With this probe, a coumarin/fluorescein dyad was used as the FRET reporter (Fig. 9). Diphenylphosphino benzoate was used as the recognition group for HNO; it was chosen for this purpose due to the expected interaction between the 2-(diphenylphosphino)-benzoate moiety and HNO via formation of an aza-ylide intermediate. In fact, this functional group has been widely applied as a HNO recognition site in many previously developed sensors. Initially, 14 displayed a characteristic coumarin emission band centred at 470 nm, which gradually disappeared upon addition of Angeli's salt (AS), which serves as a source of HNO in aqueous media. Concurrent with the reduction in the intensity of the 470 nm feature, an emission band centred at 517 nm was seen to appear. These spectral changes were interpreted in terms of the proposed FRET process, which was triggered by ring opening of the central lactone via an aza-ylide intermediate and release of the active fluorescein moiety. A linear relationship between the fluorescence intensity ratio (I517/I470) and the AS concentration (0–100 μM) was observed. Using 14, exogenous HNO in HeLa cells could be detected via ratiometric fluorescence imaging.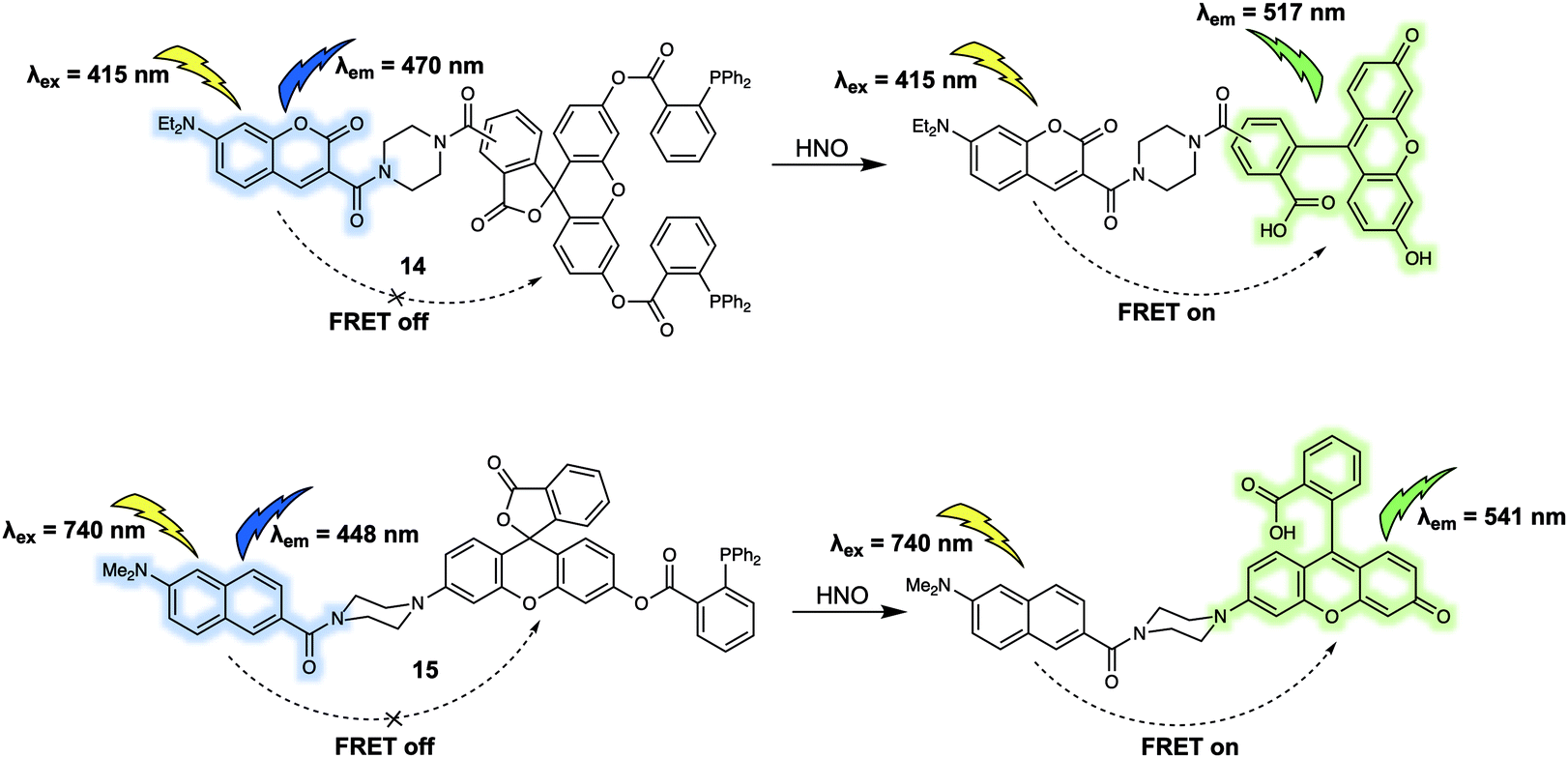 | ||
| Fig. 9 FRET-based probes (14 and 15) developed for the ratiometric detection of HNO. | ||
To facilitate the investigation of HNO concentrations in living cells and live tissues, Tan et al. designed a new FRET-based two-photon fluorescent sensor, 15 (Fig. 9).18 As in the case of 15, the 2-(diphenylphosphino)-benzoate moiety was used as the recognition site for HNO. A naphthalene derivative was chosen as the energy donor since it was expected to facilitate two-photon absorption, while a rhodol fluorophore was chosen as the energy acceptor. The presence of 2-(diphenylphosphino)-benzoate on the phenol group of rhodol stabiliszes the lactone form. Thus, as prepared 15 displays an emission band centred at 448 nm when excited at 740 nm. Addition of AS led to the appearance of the characteristic rhodol emission band centred at 541 nm. A simultaneous decrease in the fluorescence intensity at 448 nm was observed, as expected for an efficient FRET process. Detection of HNO in a frozen rat liver slice was successfully achieved with 15 by means of two-photon ratiometric fluorescent imaging. An imaging depth of 40–150 μm was obtained with the naphthalene channel and 20–130 μm with the rhodol channel.
Nitric oxide (NO) is one of the widely recognised gasotransmitters, playing an important role in numerous signalling events in physiology and pathophysiology. In 2017, Tang and colleagues reported a two-photon FRET-based system for monitoring in situ release of NO.19 Probe 16 was constructed by conjugation of coumarin and naphthalimide cores with NO (Fig. 10). This compound differed from previously discussed probes in this review in that instead of reacting with an analyte to produce a response which can be observed by fluorescence, 16 responded to specific photoirradiation by releasing a small gaseous molecule (NO), which initiated the FRET process and resulted in a change in the fluorescence output. As prepared, 16 displayed a fluorescence emission band centered at around 472 nm when subject to excitation at 408 nm. Upon photoirradiation at 365 nm, the N-nitrosoamine is photocleaved to trigger the release of NO. This, in turn, leads to the production of a new emission at 532 nm that is ascribed to FRET. The release of NO could also be promoted by irradiation under two-photon excitation conditions using a 800 nm laser light with two-photon excitation at 840 nm being used to detect the changes in probe-based fluorescence. This allowed for NO imaging in live HeLa cells.
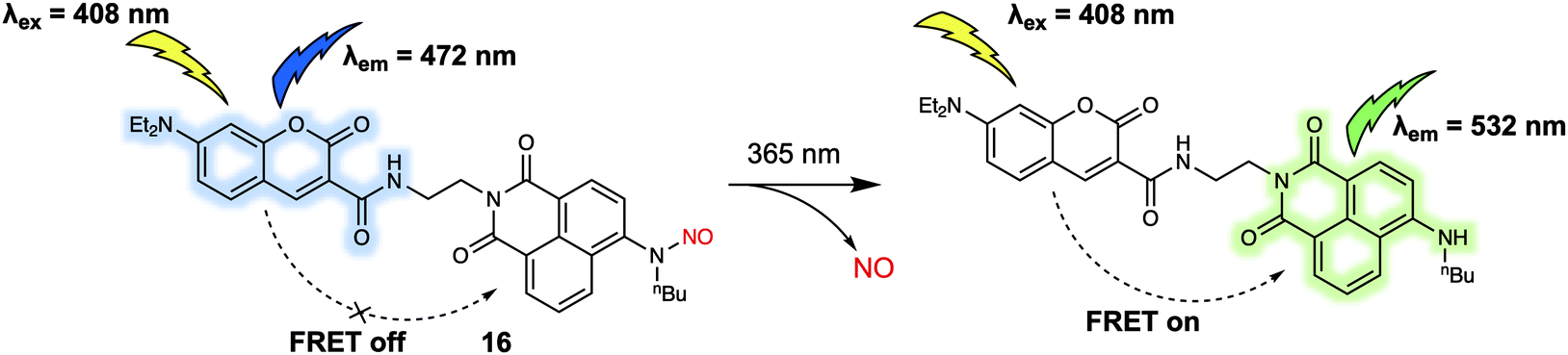 | ||
| Fig. 10 Photoinduced decomposition and fluorescence alteration of probe 16. | ||
Hydrogen sulfide (H2S) is also a key gasotransmitter. Abnormal levels of H2S may induce many types of diseases including Down syndrome. In 2015, Lin et al. reported a new FRET sensor 17 for the detection of H2S.20 In this system, based on a coumarin/naphthalimide dyad, an azide group at the 4-position of naphthalimide was utilised as the specific recognition site due to the well-appreciated fact that it undergoes selective reduction by H2S to form the corresponding naphthyl amine (Fig. 11). The parent probe 17 displayed an emission band centred at around 474 nm corresponding to the coumarin but featured no characteristic naphthalimide emission at around 534 nm, indicating a lack of FRET. The addition of increasing concentrations of H2S led to the gradual appearance of a fluorescence emission band centred at 534 nm. A concomitant decrease in the intensity of the emission at 474 nm was seen, consistent with a FRET process. Importantly, a linear relationship between the fluorescence intensity ratio (I534/I474) and the concentration of H2S (from 1.0 μM to 30 μM) was observed. Consistent with this postulate, it was found that 17 could be used for tracing both exogenous and endogenous H2S in live PC-3 cells. In the same year (2015), Yuan and colleagues reported a FRET platform for sensing H2S.21 In this FRET system, acedan and anthocyanidin type fluorophores were used as the energy donor and acceptor, respectively. Due to the fact that acedan may be photoexcited via either one- or two-photon absorption, 18 could be excited directly at either 378 nm or at 760 nm using a two-photon laser setup (Fig. 11). As prepared, 18 displayed a strong emission at 625 nm under the two-photon excitation (760 nm). This emission was seen to decrease in intensity while that of a new emission band centred at 500 nm was seen to increase upon exposure to H2S. These spectroscopic changes were attributed to the nucleophilic attack of H2S on to the benzopyrylium part of 18, leading to an interruption in the π-conjugated system of the anthocyanidin-analogue, a chemical change that leads to the inhibition of the FRET process. The major advantage of 18 is associated with two well-resolved emission bands that allow for a distinct ratiometric response in both HeLa cells and rat liver slices upon interaction with H2S. As a result, this FRET platform displays minimal cross-talk between dual emission channels when used for ratiometric fluorescence imaging.
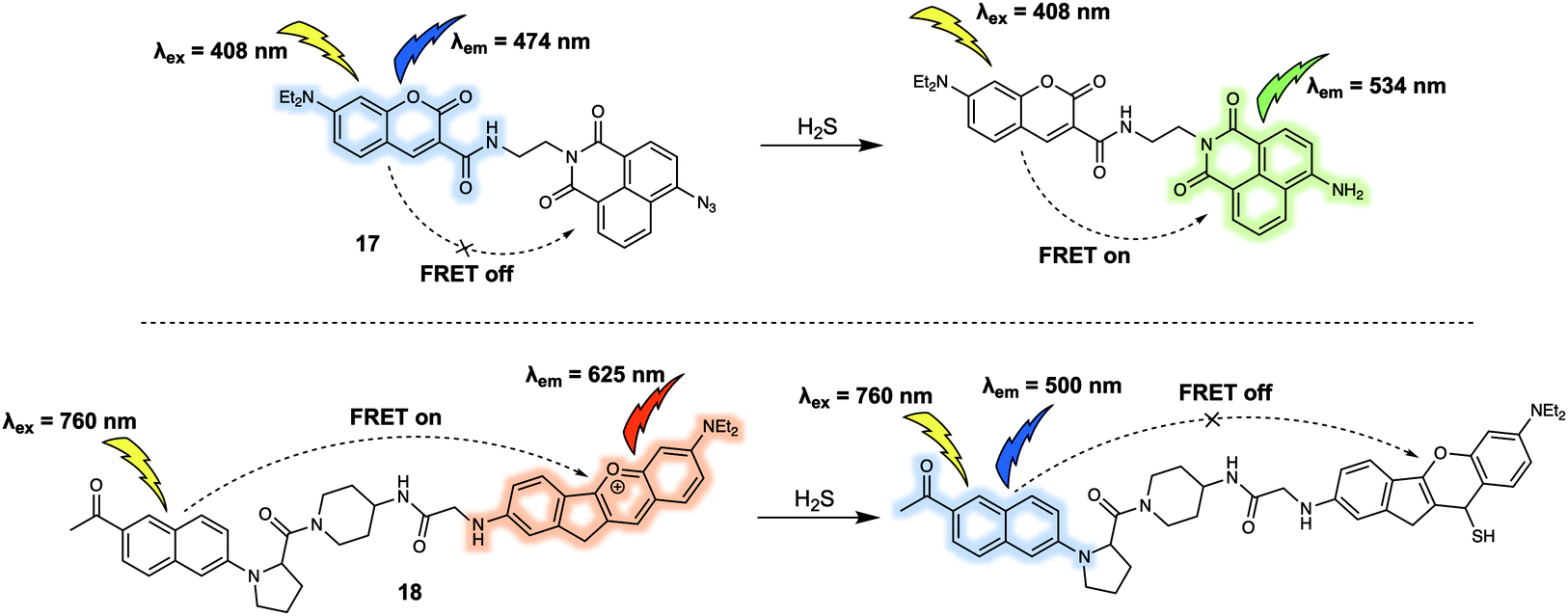 | ||
| Fig. 11 FRET-based probes (17 and 18) developed for the ratiometric detection of H2S. | ||
Hydropolysulfides (R-SnSH, n ≥ 1) are a class of reactive sulphur species that are considered to play a vital role in maintaining redox homeostasis in living systems. In 2018, Urano et al. reported the FRET probe 20 (Fig. 12) that proved effective for the real time detection of RSSHs.22 The design relied on the use of a tetramethylrhodamine methyl ester (TMRM) fluorophore as the energy donor. The acceptor was the silicon-derived rhodamine, 19, which was selected because it displays high inherent selectivity for Na2S2 over GSH and other sulfane sulphur species, including GSSSG and S8. Upon addition of Na2S2, the maximum emission of 20 shifted from 643 nm to 595 nm as a result of nucleophilic attack by the hydropolysulfides on the acceptor, thereby quenching the FRET process. The reaction of the probe with hydropolysulfides proved reversible, thereby enabling real-time tracking of fluctuations in cellular concentrations of RSSHs via fluorescence imaging. For example, using 20 the GSSH-GSSSG redox cycle (an important process in the regulation of oxidative stress within cells) could be visualised in intact cells and in real time.
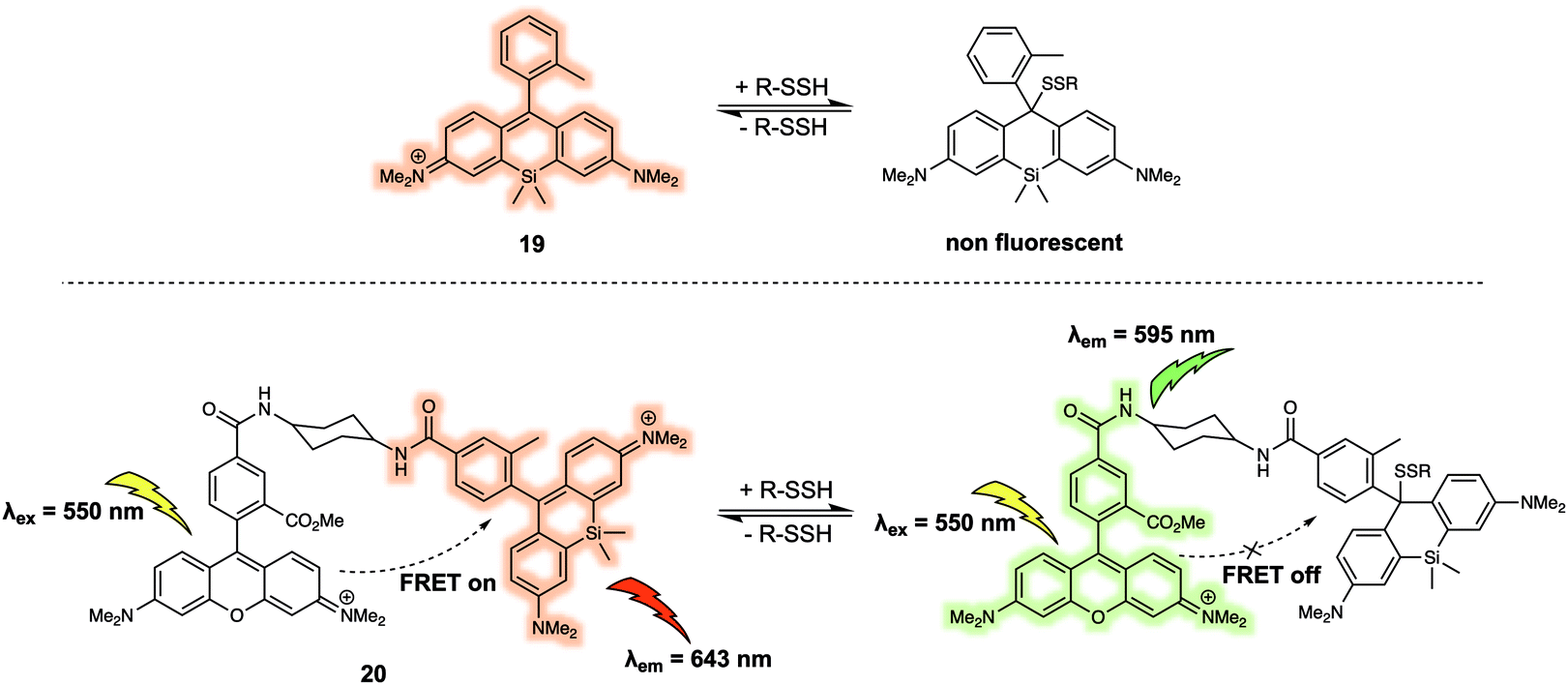 | ||
| Fig. 12 FRET-based reversible probe 20 developed to allow for the ratiometric detection of R-SSH. | ||
Urano et al. developed a similar FRET probe 22 (Fig. 13) for the selective and reversible detection of GSH.23 Nucleophilic attack by GSH on the xanthene ring of 21 occurs rapidly and reversibly. TMRM was used as the energy donor owing to its excellent spectral overlap with 21. It is noteworthy that 22 permitted intracellular GSH concentrations to be successfully determined in different cell lines via ratiometric fluorescence imaging. As part of this study, the authors demonstrated that 22 could be used to monitor changes in the GSH levels in live cells induced by exposure to thiol scavengers or H2O2, or resulting from glucose-deprivation.
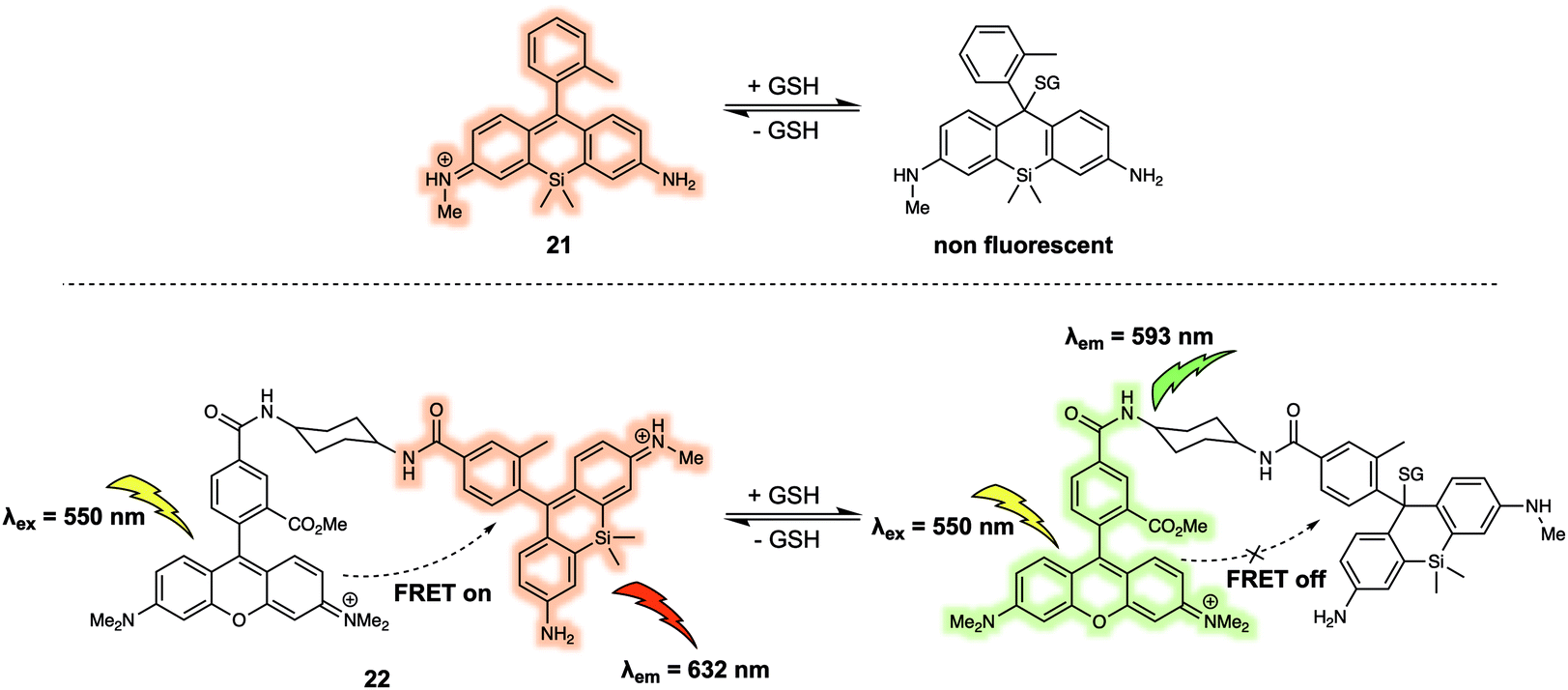 | ||
| Fig. 13 FRET-based probe 22 developed for the ratiometric detection of GSH. | ||
Adenosine triphosphate (ATP) is known to promote many biological processes as the fundamental energy source. It can also be a marker of metabolic activity. However, monitoring of lysosomal ATP is still a challenge due to a lack of efficient techniques for its selective visualisation. In 2018, Ahn et al. developed a FRET-based fluorescent sensor, probe 23 (Fig. 14), that permitted the ratiometric detection of lysosomal ATP in live cells.24 In probe 23, a rhodamine 6G derivative/BODIPY dyad was used as the FRET pair. The probe itself displayed an emission at 454 nm upon excitation at 403 nm. Addition of ATP gradually induced ring opening of the rhodamine lactam because of hydrogen bonding interactions between the tetramine linker and the phosphate groups present in ATP. Additionally, it was suggested that π–π interactions between the adenosine base of ATP and the rhodamine core contributed to the high affinity of 23 towards ATP over other adenosine phosphates, including adenosine diphosphate (ADP) and adenosine monophosphate (AMP). The ATP-induced opening of the rhodamine lactam ring enables a FRET process as evidenced by the appearance of a new fluorescence emission at 557 nm. Since the interaction with ATP, only occurred at acidic pH and not cytosolic pH, the system was suitable for the selective detection of lysosomal ATP in live cells. Using ratiometric fluorescence imaging, lysosomal ATP in live cells and in the corpus callosum of a mouse brain was successfully tracked using 23.
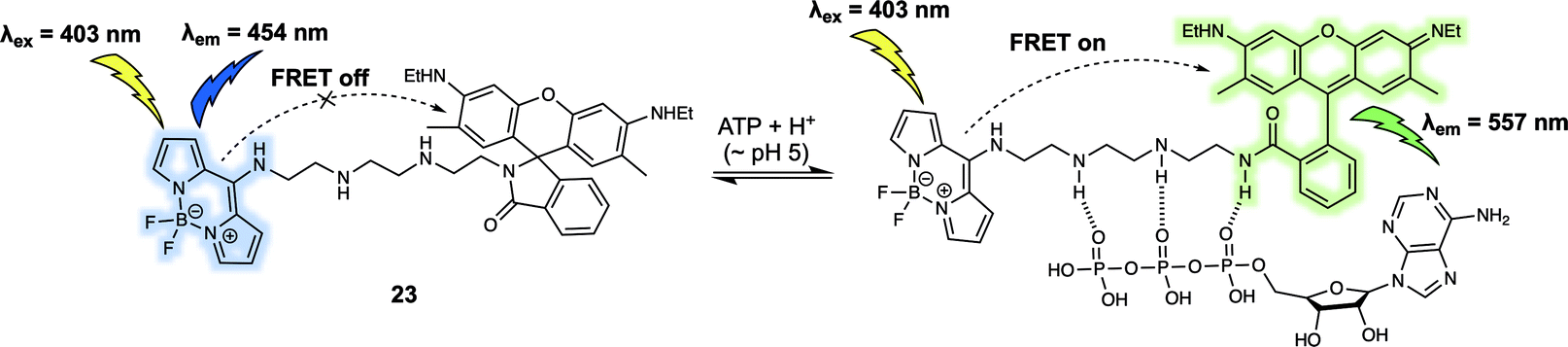 | ||
| Fig. 14 Rhodamine 6G/BODIPY (energy donor/acceptor)-based FRET fluorescent probe 23 developed to permit the ratiometric detection of ATP. | ||
A majority of the small neutral molecules discussed above are endogenously generated or secreted by the living organism itself. However, there is also a need for systems that permit exogenously administered agents, such as prodrugs, to be detected and tracked within cells. In 2019, Ang et al. reported a FRET sensor 24 (Fig. 15) that allowed cisplatin to be detected in live cells and permitted for the mechanism of platinum(IV) (PtIV) prodrug reduction to be explored in vitro.25 Probe 24 relied on the use of a coumarin-466/rhodamine 6G dyad as the FRET pair. As prepared, 24 displayed an emission at 470 nm upon excitation at 400 nm. Addition of cisplatin produced a strong fluorescence emission band centred at 565 nm with concomitant decrease in fluorescence intensity at 470 nm. A sensing mechanism was proposed that involved interaction between the dithiocarbamate recognition motif of the probe and cisplatin, triggered the ring opening of the rhodamine 6G. The resulting fluorescence intensity ratio exhibited a linear dose-dependence response towards cisplatin (0–160 μM). Additionally, they reported that 24 could discriminate PtII complexes from PtIV complexes, allowing for the tracking of PtIV-based prodrug complexes in HeLa cells via ratiometric fluorescence imaging. Using this approach, the authors were able to determine how the relative hydrophobicity of PtIV-based prodrugs modulated their cellular uptake and concordant reduction to the active PtII form. It was proposed that information derived from these studies could be used for the development of more efficacious Pt-based chemotherapy agents.
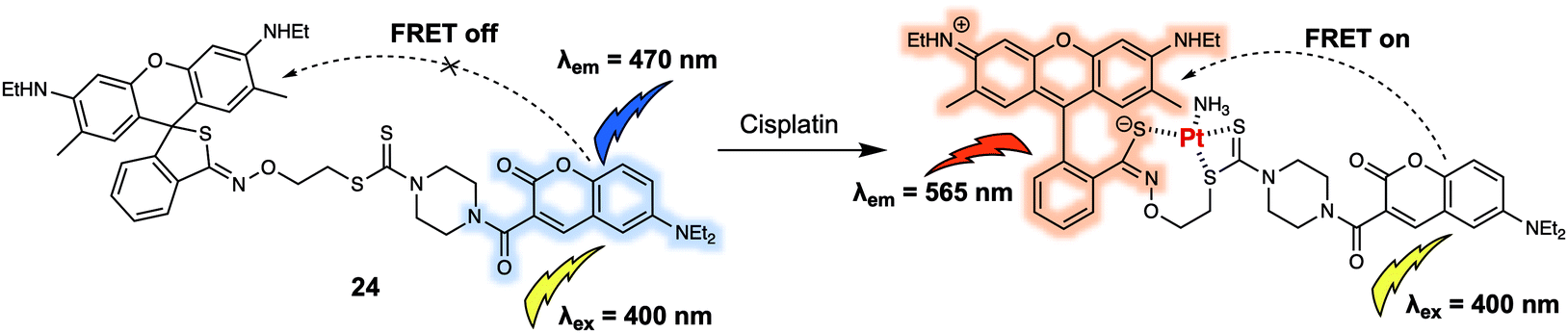 | ||
| Fig. 15 Coumarin-466/rhodamine 6G (energy donor/acceptor)-based FRET fluorescent probe 24 that allows for the ratiometric detection of cisplatin and related PtII species. | ||
FRET-based sensors for biomacromolecules
Biomacromolecules are large molecules that are commonly made up of monomers through covalent conjugation. As a result of their large size and conformational flexibility, most biomolecules, for instance proteins, DNA and RNA, encompass not only primary and secondary structures, but also tertiary structures, all of which play a key role in the regulation of their bioactivity. Being able to sense and monitor biomacromolecules is all-important in terms of exploring their function. One of the time honoured approaches involves the use of green fluorescent protein (GFP)-based fluorescent probes and related systems. Small molecule sensors have some advantages when compared to these nano-sized protein-based fluorescent probes. For instance, bulky fluorescent proteins permit only small ratiometric changes, whereas small-molecular sensors often enjoy high FRET efficiencies due to their relatively small molecular weights and the large analyte-induced changes in structure and optical features that can often be built in as a consequence. Arenz et al. designed the small-molecule based acid sphingomyelinase (ASM) FRET probe 26 (Fig. 16), which bears sphingomyelin analogues for the selective monitoring of the activity of the ASM enzyme in micelles, liposomes, and live cells.26 In a first set of predictive studies, nitrobenzoxadiazole (NBD) and Nile Red (NR) were used as the FRET donor/acceptor pair to develop 25 (Fig. 16). Upon excitation at 430 nm, 25 exhibited exclusively acceptor emission at 624 nm, consistent with high FRET efficiency from the donor NBD to the acceptor NR. The addition of ASM induced a rapid decrease in the acceptors fluorescence. However, no significant increase in the fluorescence expected for the cleaved donor product, NBD, was observed. The authors attributed this finding to the polar NBD dye displaying only very weak fluorescence in aqueous environments. To address this limitation, FRET probe 26 was prepared. In this case an NBD moiety was used as the acceptor rather than donor, while 7-methoxycoumarin-3-carboxylate (MCC) was used as the energy donor. In the case of 26, a high FRET efficiency of 90% was observed. Upon cleavage of the phosphodiester by ASM, 26 exhibited an 87% decrease in the acceptor (NBD) fluorescence that was accompanied by a 900% increase in the donor (MCC) fluorescence intensity.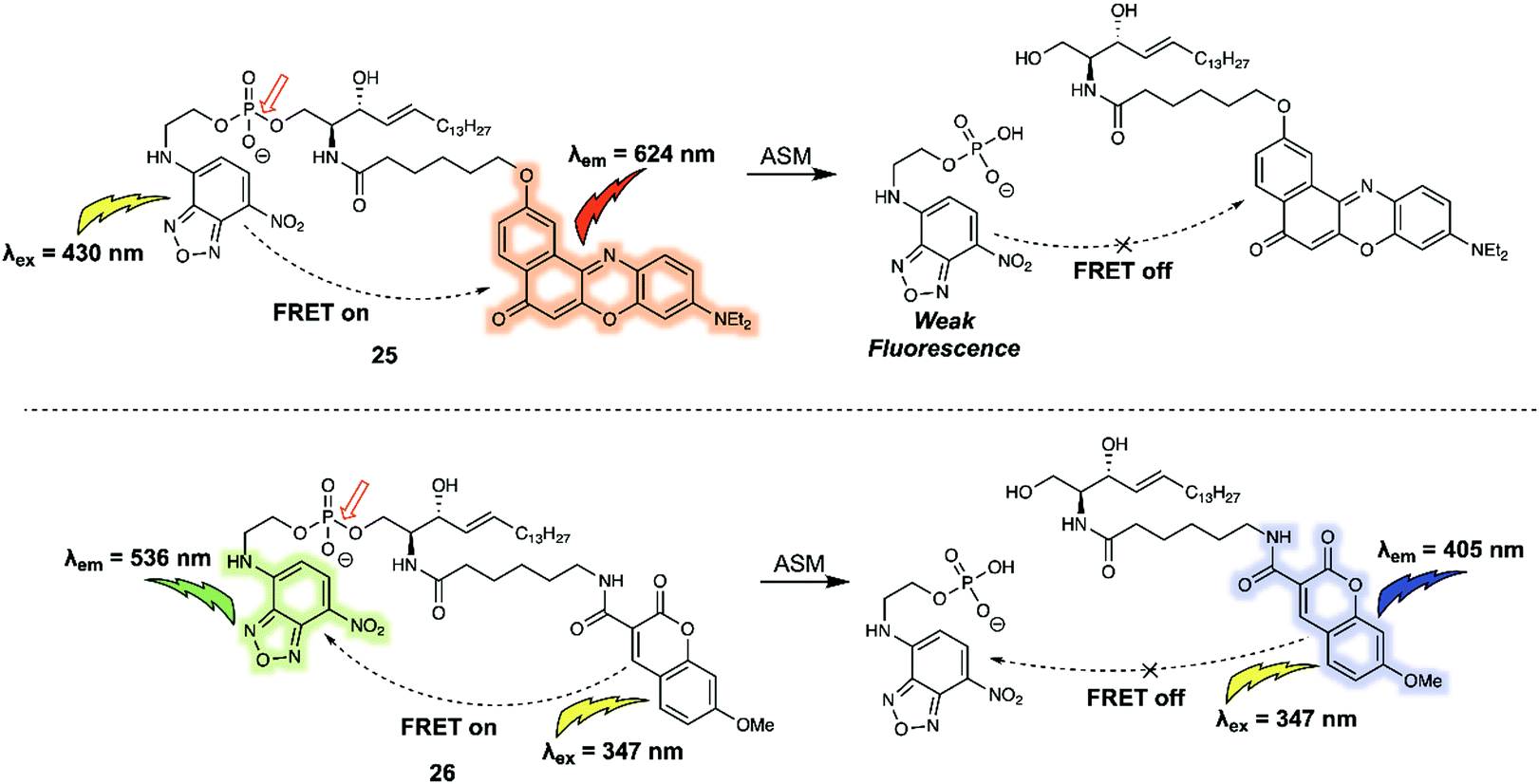 | ||
| Fig. 16 Structures of FRET probe 25 (NBD/NR) and probe 26 (MCC/NBD). The red arrow indicates the site of ASM-induced hydrolysis. | ||
An additional benefit of FRET-based small-molecule probes is their relatively easy design and preparation as compared to their GFP counterparts; this is particularly advantageous when preparing systems that can act as fluorescent substrates for enzymes. A variety of compounds, including short peptides, sugars and other nucleotides, act as specific substrates for a number of enzymes; many of these substrates can be incorporated into FRET reporter systems to afford enzyme-specific FRET sensors. One such example (probe 27) was reported by Schultz et al. who synthesized a FRET-based membrane-targeted reporter to monitor the activity of matrix metalloproteinase 12 (MMP-12).27 Probe 27 (Fig. 17) was created by anchoring a coumarin343/TAMRA FRET pair to a MMP-12 sensitive small peptide, which displayed high selectivity for MMP-12 over other matrix metalloproteases. As prepared, 27 displays an emission at 575 nm, when excited at 450 nm, corresponding to the acceptor (TAMRA) because of FRET. When exposed to MMP-12 in vitro, 27 a new donor emission at 490 nm appeared and a 4.5-fold increase in I490/I575 was observed. MMP-12 activity in RAW264.7 macrophages could then be monitored via real-time ratiometric imaging. Due to lipidation of this probe, once the cleavage event took place, it was internalised into the lipid membrane, enabling the authors to decipher the location of MMP-12 at the cellular level.
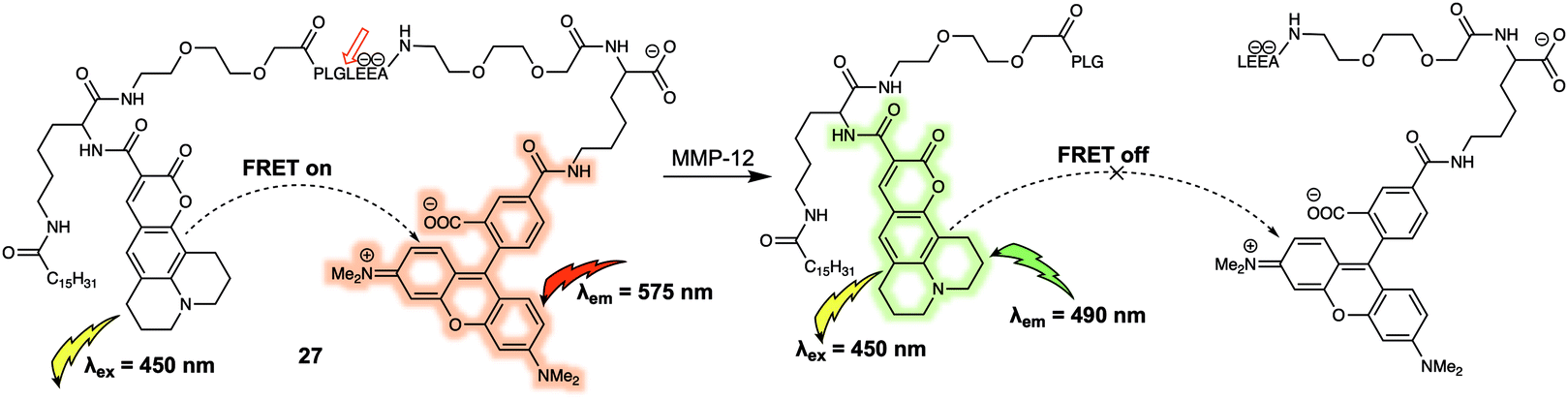 | ||
| Fig. 17 Structure of probe 27 used for the detection of MMP-12. The digestion site is between glycine and leucine. | ||
Using a similar design strategy, the same research team prepared two new small peptide-based fluorescent substrates for monitoring the activity of neutrophil elastase (NE).28 Here, methoxycoumarin/coumarin343 (probe 28) and coumarin343/TAMRA (probe 29) were used as the FRET pairs (Fig. 18). Probe 28 relied on the peptide sequence QPMAVVQSVPQ as the specific recognition site for NE. To avoid diffusion of the intact reporter through the hydrophobic membrane, negative charges were included in the substrate sequence in the form of two glutamic acid residues in the case of 29. Probe 28 and 29 were used to measure NE activity in lung fluid and on neutrophils, respectively.
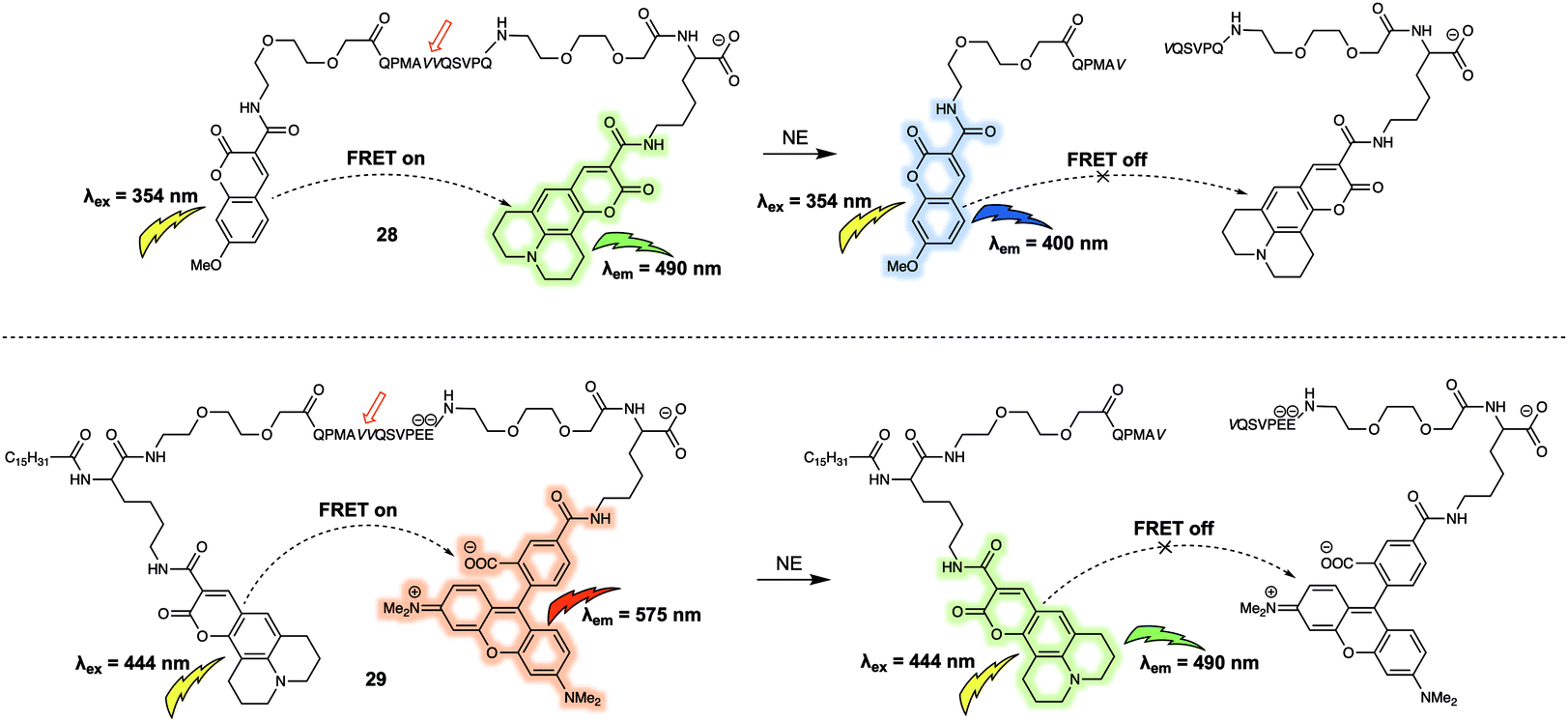 | ||
| Fig. 18 Water soluble 28 and membrane-targeting lipidated 29 FRET probes designed to monitor NE activity. | ||
The FRET-based systems discussed so far in this review have been based on combinations of fluorescent dyes used as energy donors and acceptors, respectively. This has the advantage of affording in favourable cases a ratiometric change in the fluorescence signal upon exposure to the target analyte. An alternative design strategy for FRET-based sensors is an “turn-off” FRET approach, where the acceptor behaves as a quencher. Such sensor combinations are typically used in enzyme-based sensor systems since they can allow for real-time detection, and monitoring of enzyme activity, especially when used in conjunction with live cell imaging. In 2014, Xing et al. developed the FRET-based fluorescent probe 30 (Fig. 19) and showed that it could be used for the real-time visualisation of endogenous cell-surface-associated proteolytic enzymes (furin).29 As with some of the biomolecule probes summarised above, an enzyme-responsive short peptide was used as the specific recognition substrate for the enzyme. 4-(Dimethylaminoazo)benzene-4-carboxamide (Dabcyl) was used as the quencher for the fluorescence donor fluorescein; this pair was chosen due to the spectral overlap between the absorption of the Dabcyl quencher and the emission of the fluorescein donor. When intact, 30 exhibited near baseline levels of fluorescence intensity. This was taken as evidence of efficient FRET between the fluorescein and Dabcyl subunits. After incubation with the enzyme (furin), the fluorescent emission intensity at 525 nm gradually increased with time, a finding interpreted in terms of 30 being degraded by furin to release fluorescein in free, fluorescent state. An optimised lipid moiety was used to anchor 30 to the cell surface, ensuring the response could be correlated with surface activity. The probe was then used for the real-time monitoring of furin expression in the cell lysates of U251 and Lovo cells. Interestingly, time-lapse images of U251 cells treated with 30 allowed the authors to visualise the dynamic enzymatic processes on the cell surface. In addition, interactions between 30 and cell-surface-associated furin enzyme in U251 cells and mouse ear tissue could be visualised using two-photon microscopy.
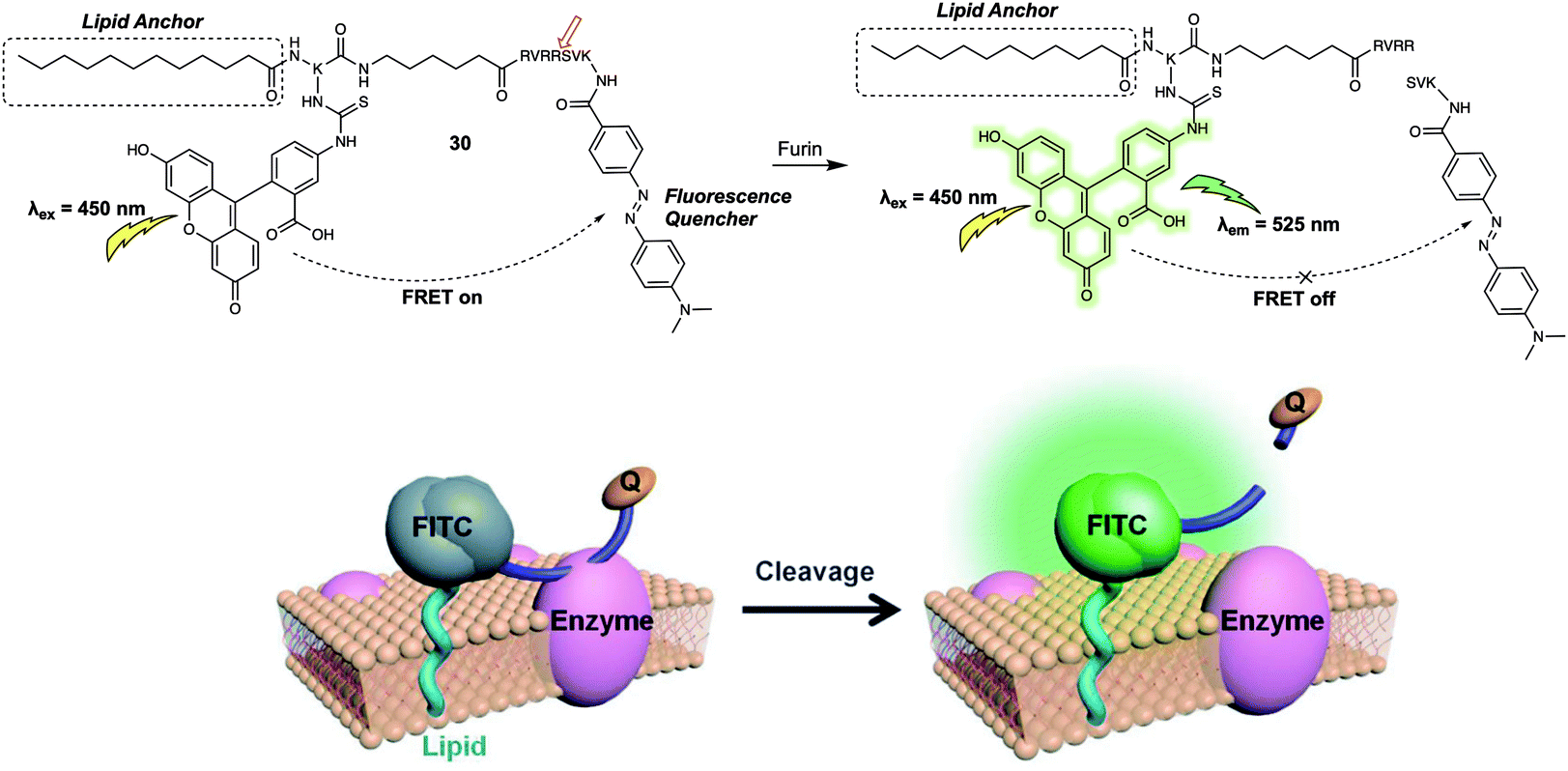 | ||
| Fig. 19 Probe 30 for the detection of cell-surface-associated furin. Reproduced with permission from ref. 29 (Angew. Chem., Int. Ed., 2014, 53, 14357–14362). Copyright (2014) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Membrane bound enzymes play vital roles in various cellular processes, such as membrane transport and cell adhesion. Yao et al. designed a switchable FRET-based fluorescent sensor (probe 31) (Fig. 20) for the real-time imaging of receptor-like protein tyrosine phosphatases (RPTPs).30 In this case, a novel two-photon fluorescent dye (Flu7) was prepared, containing two aliphatic chains. A separate molecule (Q12) containing a “photocaged” phosphorylated phenolic group coupled to a fluorescence quencher (Disperse Red 1), was also synthesized. When anchored to the cell membrane, the positively charged side chains of the Flu7 fluorophore binds with the negatively charged phosphate anion incorporated in Q12. This interaction served to hold the quencher in close proximity to the donor, facilitating FRET-based quenching. As initially prepared, the two components were non-fluorescent, due to this FRET quenching process. Moreover, the photocaged nature of the Q12 quencher resulted in no response being seen in the presence of RPTPs. However, upon irradiation with UV light, the 2-nitrobenzyl group was cleaved from the Q12, allowing RPTPs to dephosphorylate Q12 and release the quencher. This release, in turn, triggered an increase in the fluorescence emission of Flu7. It was found that the FRET system 31 could be used for the real-time imaging of endogenous membrane associated RPTPs in live cells. Due to the fact that it could be photoexcited under two-photon conditions, this FRET system was successfully employed for the real-time imaging of membrane-associated RPTPs in Drosophila brains. The results confirmed that the switchable nature of 31 was both RPTPs and UV dependent. These features allowed for the selective and fast modulation of the fluorescent output under conditions of UV irradiation.
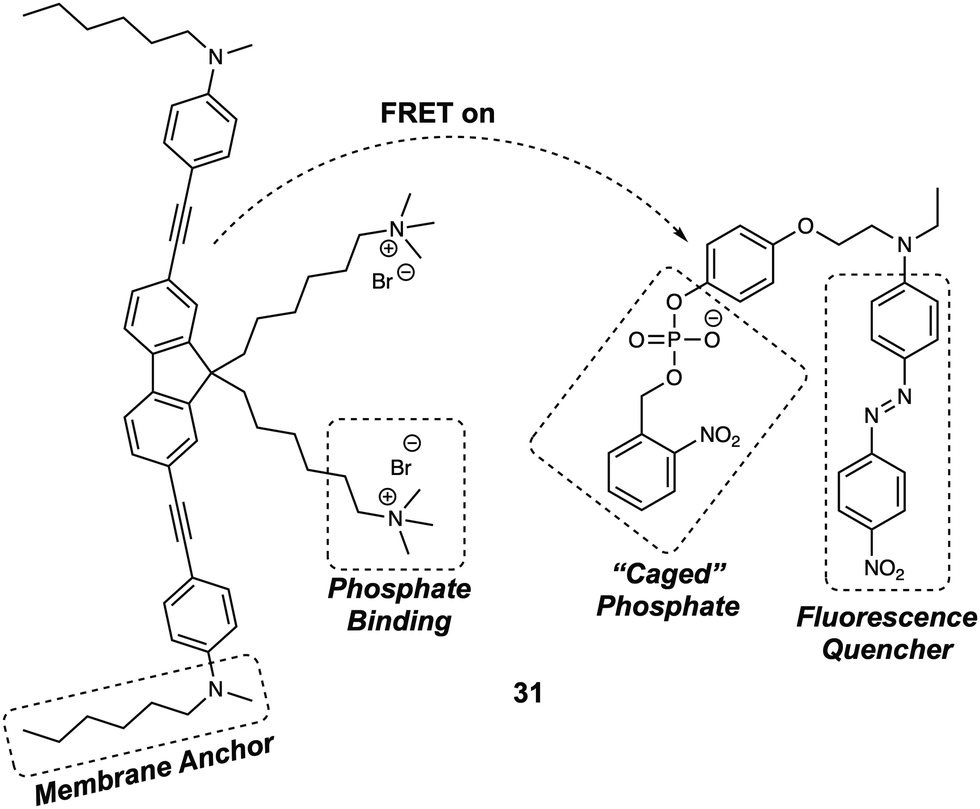 | ||
| Fig. 20 Structure of probe 31 designed to facilitate detection of membrane-associated RPTPs. | ||
Compared to proteases containing cleft-like architectures for cleavage, some enzymes have sterically constrained pocket-shaped active sites, which could hinder the design of FRET-based small-molecular probes. For instance, the design of probes for exo-glycoside hydrolases, these enzymes are characterised by their pocket-shaped active site topologies. Therefore, the designed probes require an enzyme-responsive substrate that is built to interact with the pocket-like active site. In 2015, Vocadlo et al. designed the FRET-based probe 33 (Fig. 21) to monitor the activity of lysosomal human glycoside hydrolase glucocerebrosidase (GCase) in live cells.31 In their initial design, these authors used a 5-[(2-azidoethyl)amino]naphthalene-1-sulfonic acid (EDANS) group as the fluorescence donor and a 4-{[4-(dimethylamino)phenyl]azo}-benzoic acid (DABCYL) derivative as the quencher; to afford 32 (Fig. 21). In probe 32, the quencher moiety was attached to the anomeric centre of the glucose to prevent steric congestion of the active site. Concurrently, the fluorescence donor was installed at the 6-position of glucopyranose to ensure close proximity within the fluorophore (donor)-quencher pair. In fact, 32 exhibited 97.6% quenching efficiency of the EDANS subunit by the linked DABCYL. However, relatively low water solubility precluded the determination of the Km values (a composite rate constant) while short emission wavelengths limited its application in live cell imaging. Therefore, this research team designed and synthesized 33, which relied on a BODIPY fluorophore, a dye with more red-shifted emission than the EDANS subunit in 32, as the fluorescence donor and Black Hole Quencher 2 (BHQ2) as the acceptor. This latter species was chosen due to the fact that its absorption overlaps with the BODIPY emission. Appending the fluorophore (donor) to the non-cleaved side of the glucopyranose ring had the effect of localising the fluorophore in lysosomes after cleavage of the FRET probe by GCase, an effect ascribed to the high polarity of the sugar. Meanwhile, the mildly basic aniline subunit present on 33 also promoted its accumulation in the acidic lysosomes. Fluorescence microscopic studies revealed that the fluorescence of live cells incubated with 33 gradually increased with time. Additionally, the fluorescence signal exhibited a dotted pattern which indicated lysosomal localisation, that was further confirmed by a co-localisation assay with commercially available LysoTracker Green DND-26. These findings led the authors to suggest that dynamic changes in the GCase activity could be monitored using 33. The authors also concluded that 33 could be used for the quantitative detection of the chaperoning of GCase in human fibroblasts.
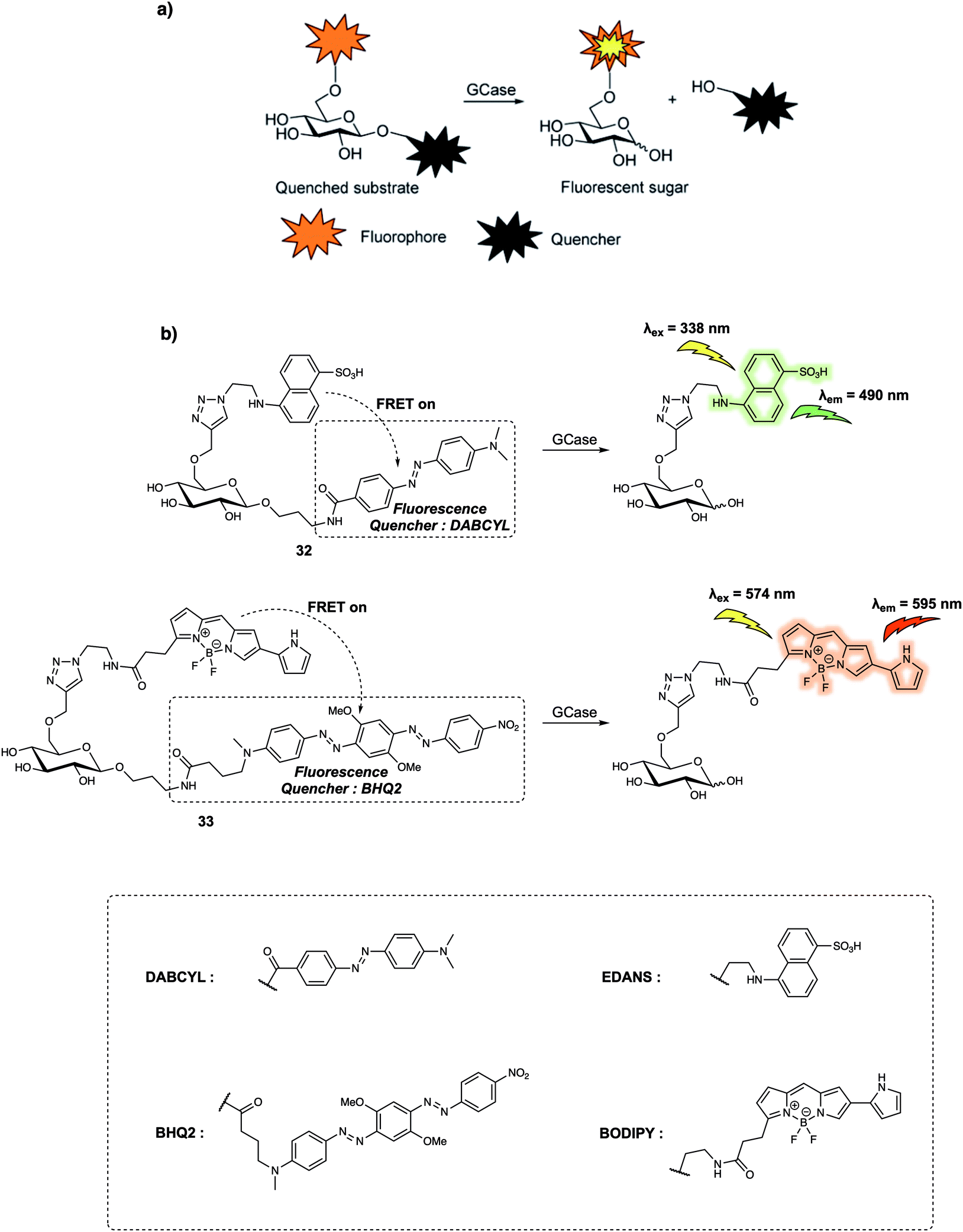 | ||
| Fig. 21 (a) General reaction scheme showing the design of probes that can be used for the fluorescence-quenched monitoring of GCase activity. (b) Sensing mechanism of probes 32 and 33 for the detection of GCase. Reproduced from ref. 31 (J. Am. Chem. Soc., 2015, 137, 1181–1189). Copyright (2015) American Chemical Society. | ||
Although probe 33 proved effective in monitoring GCase activity, attaching a fluorophore to the 6-position hydroxyl group of glucopyranose represents a design strategy that is not generally applicable. In 2017, Vocadlo et al. positioned both the fluorophore and quencher outside of the active site of glucopyranose.32 In this case, the fluorescence donor (EDANS) and quencher (DABCYL) were anchored to the O-linked β-N-acetylglucosamine through a hemiacetal aglycon group. Using this design, the probes preserved the active carbohydrate recognition motif. The FRET quenching efficiency was calculated to be about 99.93% and 99.89% for probes 34 and 35, respectively (Fig. 22). Such high quenching efficiency is thought to reflect the proximity of the FRET partners, presumably enabling contact quenching. It is to be noted that the bis-acetal unit remained stable in solution between pH 2 and 10. Upon addition of exo-acting human O-GlcNAcase (hOGA), the fluorescence intensity of probes 34 and 35 gradually increased with time. Second-order rate constants for the interaction probes 34 and 35 with hOGA were calculated to be 263 and 519 M−1 s−1, respectively; these values are comparable to those seen for the native substrate methyl β-D-N-acetylgluco-pyranoside. Furthermore, 34 could be used to monitor the activity of hOGA in SK-N-SH cell lysate. After cleavage by hOGA, probes 34 and 35 released a hemiacetal, which underwent spontaneous hydrolysis. Resulting in separated fluorescence donor and quencher, giving rise to readily discernible fluorescence changes. It was proposed by the authors that the concept embodied in probes 34 and 35 could be applied to many other types of hydrolases for instance sulfatases.
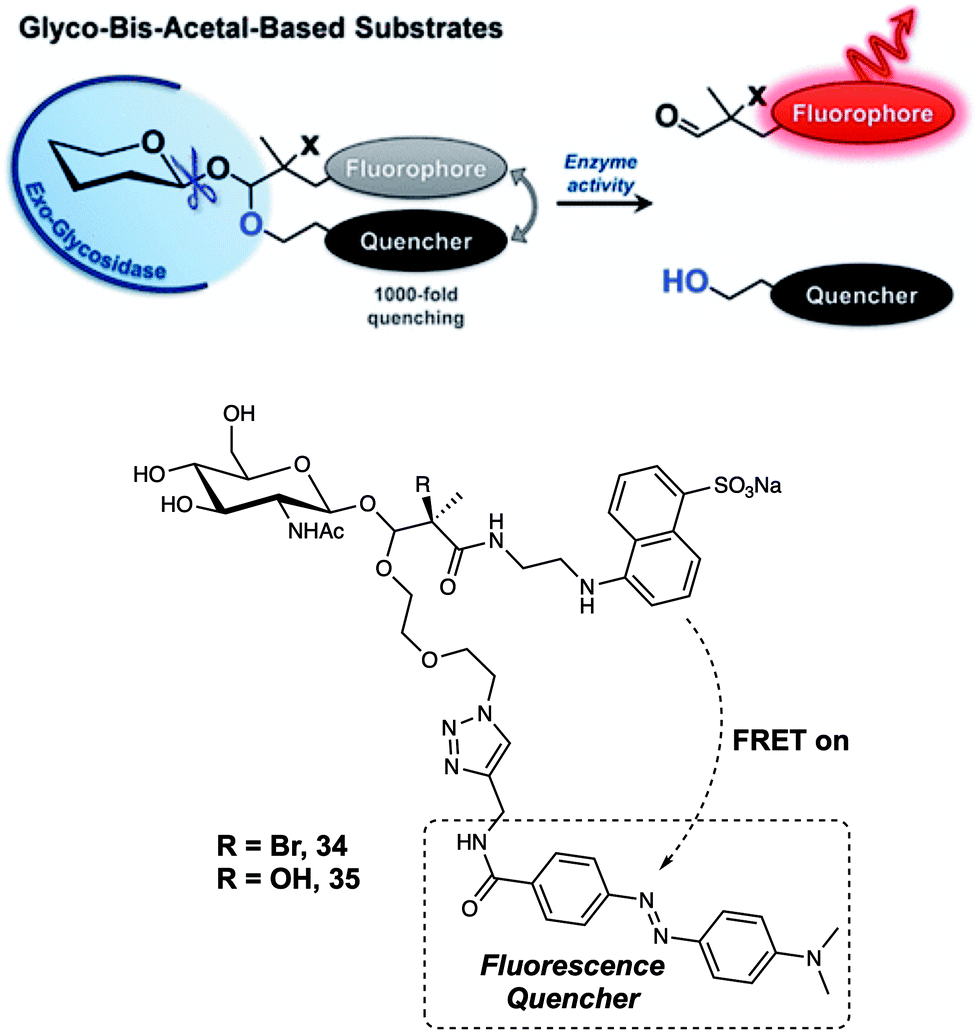 | ||
| Fig. 22 Glycosidase-induced hydrolysis of the fluorescence-quenched probes 34 and 35 serves to increase in fluorescence intensity as the result of releasing a free fluorophore in a form that is not subject to FRET quenching. Reproduced from ref. 32 (J. Am. Chem. Soc., 2017, 139, 8392–8395). Copyright (2017) American Chemical Society. | ||
Another important class of enzymes are the β-lactamases (Blas), which are responsible for the hydrolysis of the β-lactam moiety. In 2012, Xing et al. designed a ruthenium(II) cephalosporin-based FRET fluorescent sensor (probe 36) (Fig. 23) that was designed to permit the intracellular imaging of Blas in bacteria.33 In probe 36, a luminescent Ru(II)(bpy)32+ complex served as the fluorescence donor while a BHQ3 subunit with a broad absorption from 550 nm to 700 nm was used as the quencher. The BHQ3 group was anchored to the 7′-amino group of the cephalosporin core structure through a glycyl linker to form the final FRET system. As prepared, the FRET system displayed weak fluorescence due to quenching of the Ru(II)(bpy)32+ complex by the BHQ3 subunit. After treatment of 36 with TEM-1 Bla, an 8-fold enhancement in fluorescence intensity at 645 nm was observed. This is consistent with the inherent FRET quenching process being inhibited and cleavage of the probe. It was suggested by the authors that 36 had the potential to be used in photodynamic therapy since the constituent Ru(II)(bpy)32+ complex is commonly used as a photosensitizing agent for the generation of reactive oxygen species (such as singlet oxygen, 1O2). In addition, it was thought that the 1O2 generated by the Ru(II)(bpy)32+ photosensitizer could be scavenged by BHQ3 due to the close proximity of these two key elements within 36. Finally, 36 could be used to discriminate several types of bacteria pathogens with varying resistance profiles towards β-lactam antibiotics.
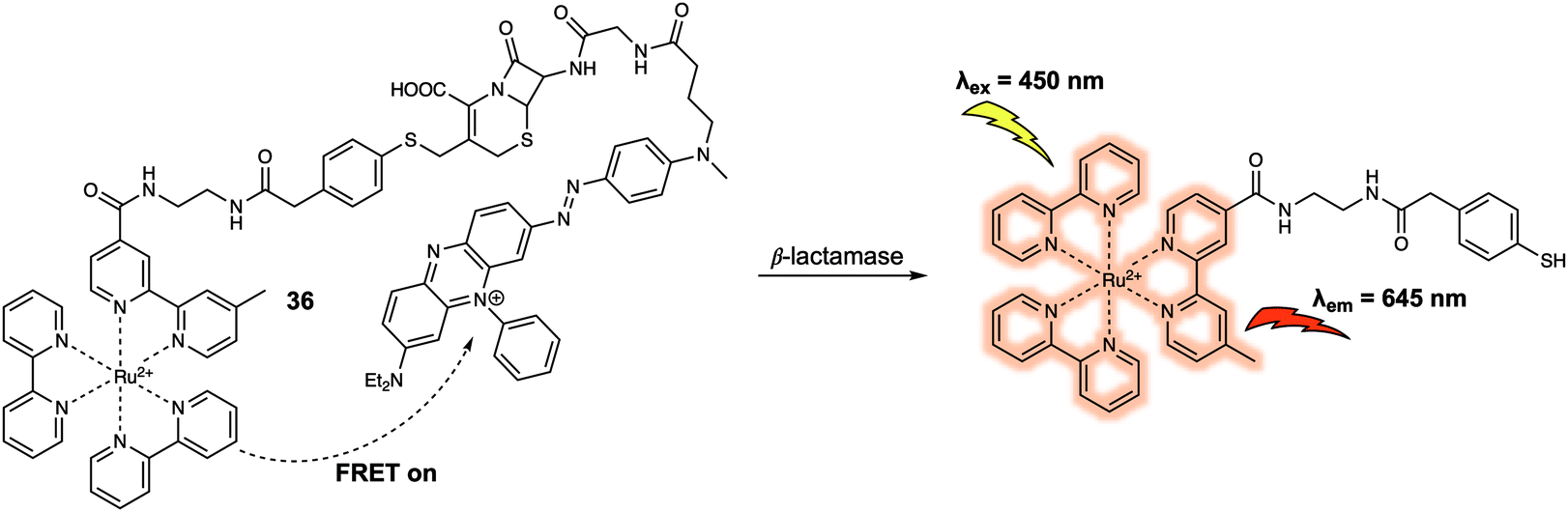 | ||
| Fig. 23 Structures of probe 36 developed for the detection of Blas. | ||
When designing a FRET quenching system, the choice of a suitable quencher is an important consideration. As implied above, an effective quencher should possess an absorption profile that overlaps significantly with the emission band of the donor. To date, several typical dark quenchers, including DABCYL, members of the Black Hole Quencher (BHQ) family, and the QSY series have been studied extensively; a number are now commercially available. Despite these efforts, the development of new quenchers represents a recognised need within the FRET-based sensor community. For instance, quenchers with a near-infrared (NIR) absorption compatible with NIR fluorescence donors would be advantageous in terms of in vivo imaging since they would be subject to lower levels of interference from endogenous fluorophores. In 2015, Urano and colleagues developed a series of silicon-substituted rhodamines (SiRs) for use as NIR quenchers.34 SiRs exhibit a far-red to NIR absorption due to the replacement of the O atom by a Si atom within the xanthene moiety of rhodamine. By attaching aromatic rings directly to the nitrogen atoms of the xanthene skeleton, the fluorescence intensity of the SiRs could be reduced to near background. This reduction was ascribed to fluorescence quenching resulting from free rotation of the bond between the N atom and the xanthene moiety. In the context of this research, both hydrophobic and hydrophilic SiRs were synthesized; this was done by modifying the core structure of the SiRs subunit. This had the additional advantage of enabling their use in a diverse array of solvents. Thus, this series of SiRs could be applied as NIR dark quenchers and used to construct FRET systems. As such, a new FRET system 41, based on SiRs, designed to allow the detection of matrix metalloproteinases (MMPs), could be constructed (Fig. 24). As prepared the fluorescence quantum yield of 41 proved to be less than 0.001. However, a 20-fold fluorescence increase in the emission was seen after interaction with two members of the MMP family (MMP-9 or MT1-MMP). The activity of MMP in tumour tissue could also be followed using this FRET system, thus underscoring the practical utility of SiRs in constructing NIR FRET systems for in vivo imaging.
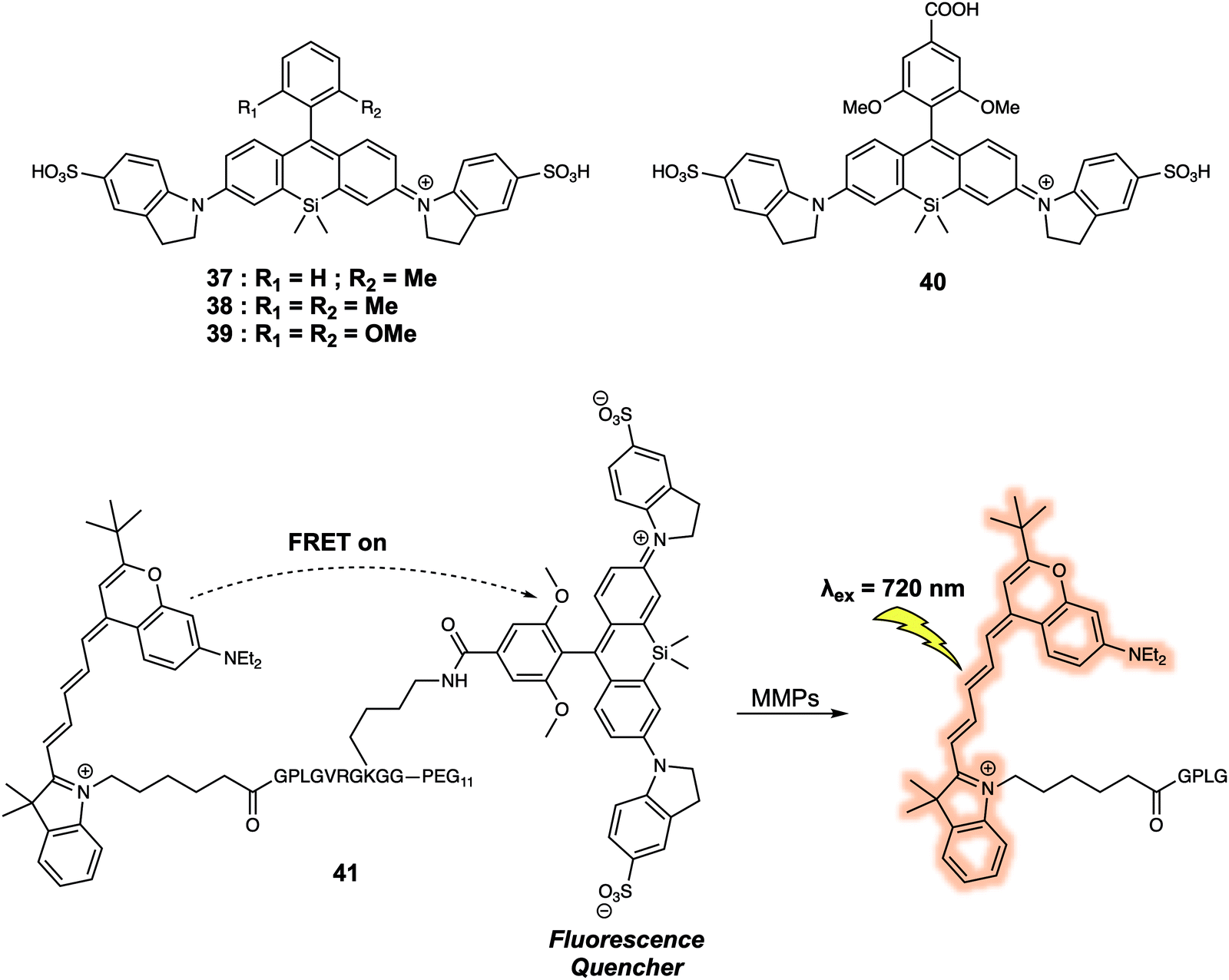 | ||
| Fig. 24 Structures of the water soluble quencher 37–39, and the application of 40 in the construction of FRET probe 41 that allows for the detection of MMPs. | ||
Another set of quenchers that have been used to create FRET-based sensing systems are the so-called aggregation-induced emission luminogens (AIEgens). AIEgens display unique fluorescence properties when compared to traditional dyes due to their ability to exhibit strong emission upon aggregation in solution. Liu et al.35 constructed the FRET system 42 (Fig. 25) using coumarin as the energy donor and an AIEgen as the energy quencher. In 42, the short peptide Asp-Glu-Val-Asp (DEVD) was used as the specific recognition site for the caspase-3 enzyme. Upon excitation at 405 nm, a weak fluorescence signal was observed due to FRET quenching of the donor coumarin by the AIEgen. Addition of caspase-3 induced cleavage of the recognition peptide and release of the coumarin and AIEgen. Subsequently, a fluorescence emission at 465 nm gradually appeared that was ascribed to the recovery of the coumarin fluorescence. In tandem with these spectral changes, a new emission at 665 nm appeared. The presence of this new feature was taken as evidence that upon release from the coumarin moiety, the AIEgen would aggregate in solution thus becoming fluorescent. This FRET probe was then used to monitor the activity of caspase-3 in live HeLa cells by taking advantage of the dual-signal readout. Given the key role of caspase-3 in cancer biology, it was proposed that 42 could have a role to play in screening the apoptosis-inducing capabilities of potential drug candidates.
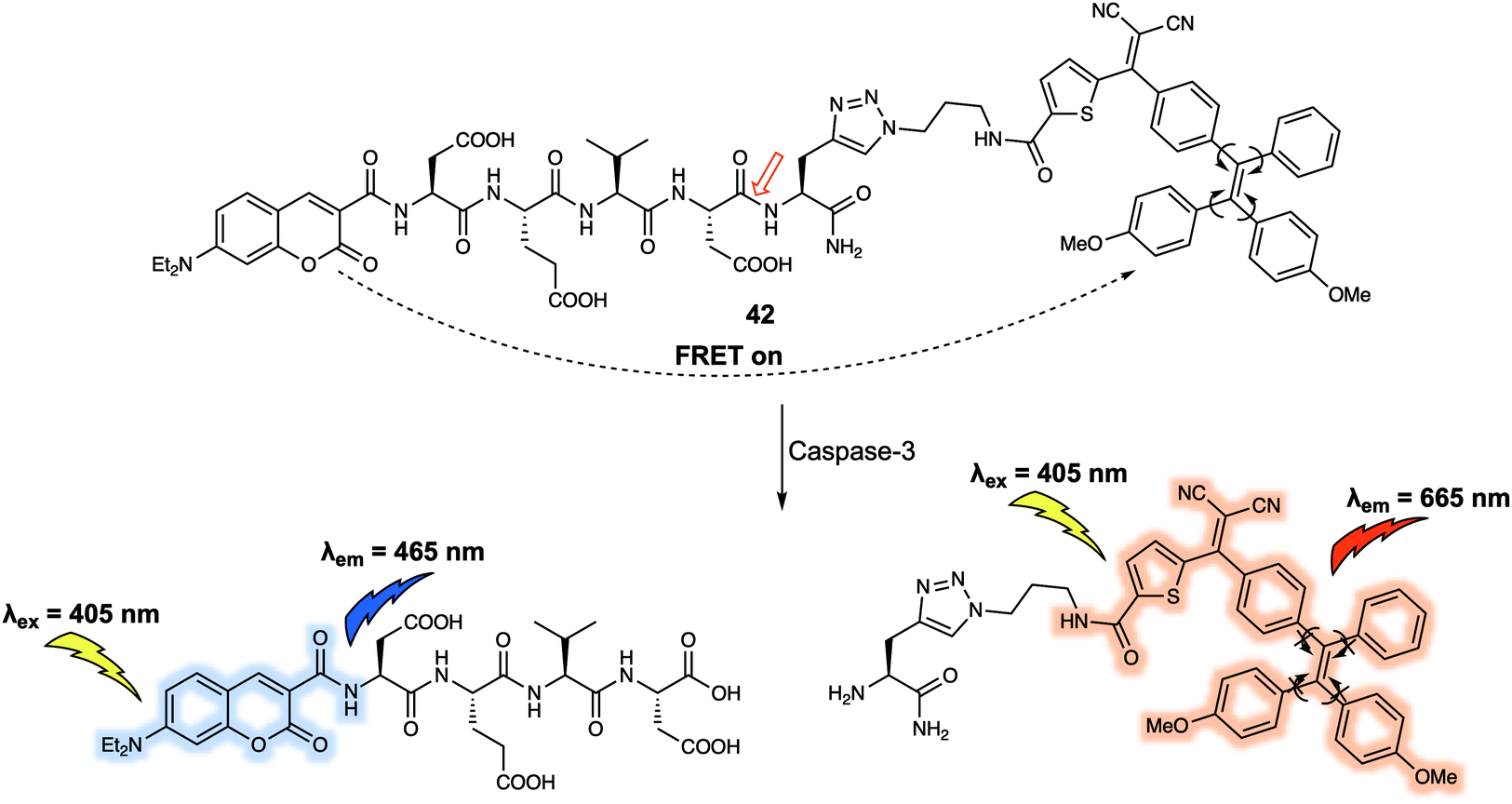 | ||
| Fig. 25 Structure of probe 42 that was developed to detect and monitor caspase-3. | ||
In 2019, a FRET-based strategy for DNA sensing was reported by Nau et al. These researchers prepared a cucurbit[7]uril (CB7)-containing FRET system (43) (Fig. 26).36 Here, the supramolecular CB7 host was covalently attached to a fluorescein dye that was designed to serve as the energy acceptor. A non-covalently bound 4′,6-diamidino-2-phenylindole (DAPI) dye was used as the energy donor. DAPI is well-known to bind strongly to the minor groove of DNA. In the case of 43, where the DAPI is held close to the tethered fluorescein acceptor after encapsulation of DAPI within the hydrophobic environment of the CB7 receptor, therefore, FRET is promoted upon excitation at 360 nm. The incremental additions of DAPI into a fixed concentration of CB7-carboxyfluorescein (CB7-CF) resulted in enhanced emission intensities at both 450 nm and 520 nm. Upon the addition of salmon sperm DNA, the DAPI energy donor is no longer bound appreciably within the CB7 host. Rather, it binds in preference to the minor groove of DNA. This equilibrium-based decomplexation reflects the relatively strong interaction between DNA and DAPI. The loss of DAPI inhibits the FRET process, resulting in a decrease in the fluorescence emission intensity of the fluorescein at 520 nm with concomitant enhancement in the DAPI-derived emission feature centred at 450 nm. This host–guest FRET system was used for the quantitative detection of DNA with an excellent linear dependence of the ratiometric fluorescence signal being observed.
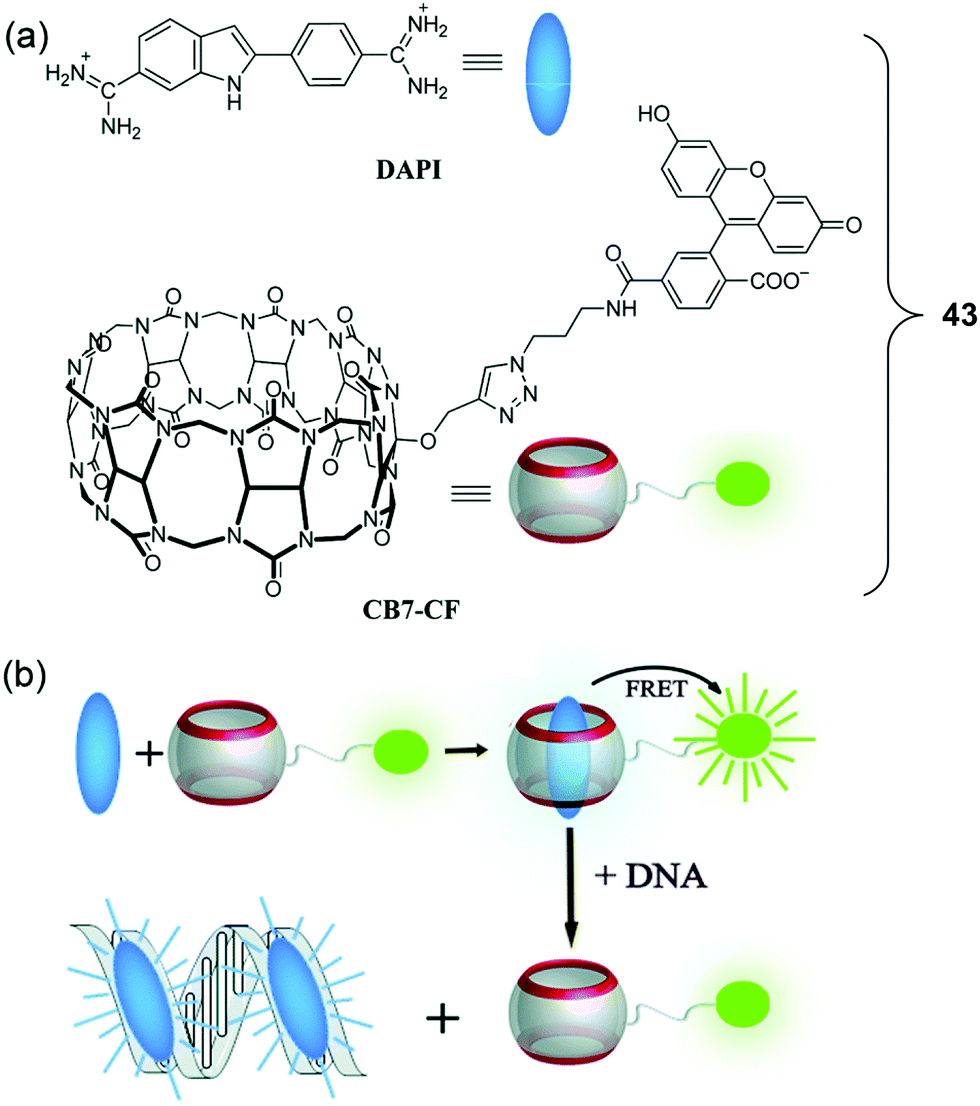 | ||
| Fig. 26 (a) Molecular structures of DAPI and CB7-CF. (b) Schematic illustration of the resulting DNA chemosensing ensemble, which relies on a FRET process between DAPI (donor) and CB7-carboxyfluorescein (acceptor), as well as the relative affinities of the DAPI guest for the CB7 host and double-stranded DNA (dsDNA). Reproduced with permission from ref. 36 (Chem. Commun., 2019, 55, 671–674). Copyright (2019) The Royal Society of Chemistry. | ||
FRET-based systems have also been developed that allow for protein sensing. In 2014, Zhu et al. developed a FRET system (probe 44) (Fig. 27) designed to permit the ratiometric detection of vicinal dithiol-containing proteins (VDPs).37 This FRET probe relies on the fluorescent dyes coumarin and naphthalimide, which serve as the energy donor and acceptor, respectively. An organic arsenic moiety is incorporated into 44 as a highly selective recognition site for the targeted VDPs. Upon excitation at 405 nm (an appropriate excitation wavelength for coumarin), 44 displayed an emission at 540 nm. This is ascribed to the emission of naphthalimide, which was taken as evidence that energy transfer from the coumarin to the naphthalimide occurs. Addition of one equivalent of reduced bovine serum albumin (rBSA), taken as a model VDP induced a remarkable fluorescence enhancement at 470 nm which was assigned to the coumarin emission, reflecting inhibition of the FRET process. A control probe 45 was also prepared. In this case no significant changes in the fluorescence features are seen upon the addition of rBSA. These results led the authors to suggest that the cyclic dithiarsane moiety provides a selective recognition site for rBSA through exchange with the vicinal dithiols of the targeted VDPs. Specifically, it was proposed that the FRET probe 44 was cleaved upon interaction with vicinal dithiols present in the VDPs allowing for attachment of the coumarin. This results in a change in the fluorescent signature that permits ratiometric sensing. In fact, 44 was used for tracing endogenous VDPs in live MCF-7 cells via readout of the ratiometric fluorescence signal. By using this FRET probe the effect of selected redox reagents on cell-surface VDPs could be monitored. The ratiometric features of 44 also permitted the semi-quantitative detection of intracellular VDPs in live cells. Moreover, the distribution of VDPs in live MCF-7 cells could be mapped using this probe. Considering the key roles of VDPs in regulating cellular redox homeostasis, it was suggested by the authors that this FRET-based technique could provide a new and visible chemical tool for the investigation of VDP-related biological processes and may be useful in drug screening assays.
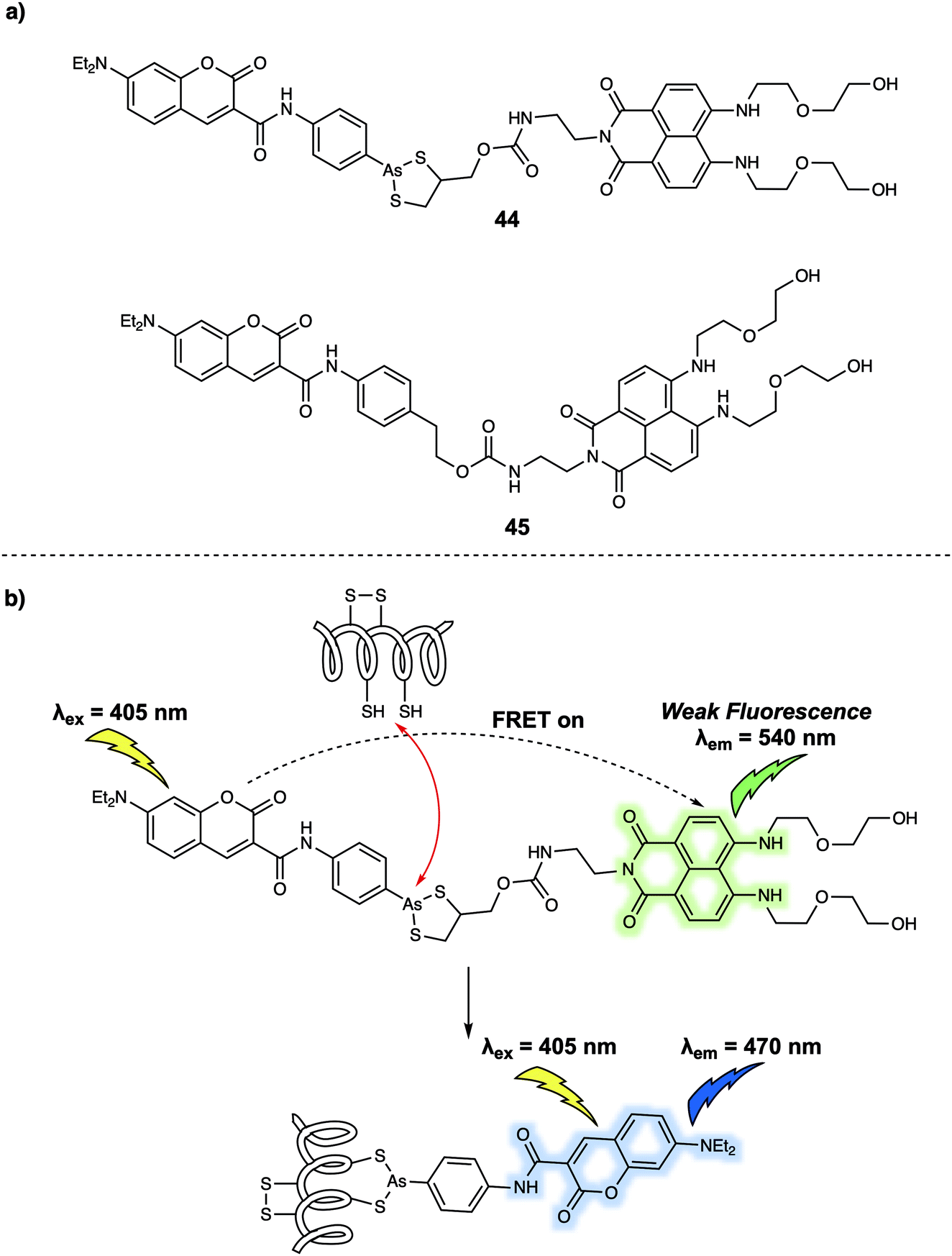 | ||
| Fig. 27 (a) Chemical structures of FRET probes 44 (target probe) and 45 (control probe); (b) design strategy underlying probe 44 and its use for the ratiometric monitoring of VDPs via a FRET process. | ||
FRET-based sensors for the visualisation of cellular microenvironments
Cellular microenvironments refer to the local environment of a cell, which includes both intracellular and extracellular effects that have a direct and indirect effect on cell behaviour. Understanding these environs in detail remains a challenge. FRET-based systems are providing useful tools for this purpose. For the sake of illustration, FRET-based probes designed to explore the effect of pH, hypoxia and mitochondrial membrane potential are discussed in this section. pH is an important factor that is closely related to cellular function and can be used as an indicator for abnormal cell physiology. For instance, the pH of cancer cells is typically lower than that of healthy cells. Meanwhile, the pH of different cellular organelles varies as such lysosomes (below 7.0) and mitochondria (above 7.0). In 2014, Zhang and colleagues designed a new FRET probe 46 for monitoring pH changes in HeLa cells.38 Probe 46 relies on bispyrene and fluorescein subunits as the energy donor and acceptor, respectively (Fig. 28). In acidic solution (pH 3.0), the two pyrene fluorophores form an excimer which resulted in an emission at 459 nm being observed upon excitation at 358 nm. No appreciable fluorescence emission from the fluorescein moiety present in 46 was seen under these conditions, a finding ascribed to the ring closed spirolactam form predominating in acidic media. With increasing pH, the fluorescein spirolactam moiety underwent ring opening and the phenol group becomes subject to deprotonation; this led to an increase in the absorption feature centred around 500 nm. Overlap with pyrenyl excimer emission band allows for energy transfer to the fluorescein subunit via a FRET process; this resulted in an easy-visualised emission at 526 nm. Intracellular pH changes were revealed via ratiometric fluorescence imaging using the probe. By analysing various fluorescence images, it was found that the relative ratio (I510–550nm/I440–480nm) increased from 0.27 to 2.25 as the pH increased from 4.0 to 8.0 in HeLa cells. These differences allowed the use of 46 for the real-time monitoring of pH-related biological processes in live cells.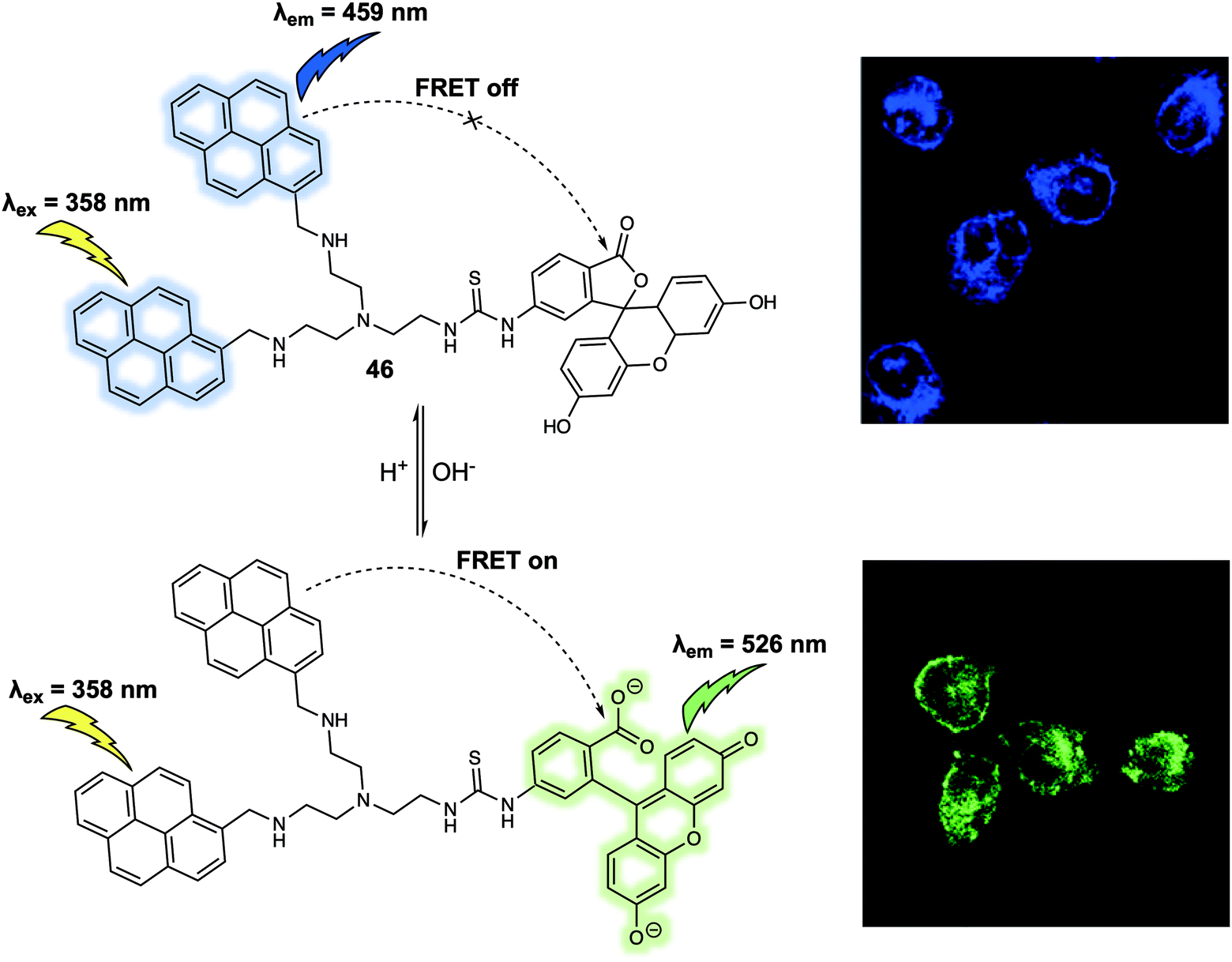 | ||
| Fig. 28 FRET-based probe 46 that permit monitoring of pH changes in HeLa cells. Reproduced with permission from ref. 38 (Anal. Chem., 2014, 86, 10389–10396). Copyright (2014) American Chemical Society. | ||
Another critical cellular environmental factor is hypoxia, which can trigger production of the hypoxia-inducible factor-1 (HIF-1) and accelerate bioreductive reactions. Hypoxia has been associated with a number of diseases, including cancer, cardiovascular disease, and stroke. In 2012, Hanaoka and colleagues designed and synthesized a new FRET-based fluorescence probe 47 and demonstrated that it could be used to detect hypoxia.39 Probe 47 was constructed using Cy5 as the fluorescence donor and QSY-21 as the fluorescence quencher (Fig. 29). QSY-21 is appreciated as being an efficient quencher for the fluorescence emission in the red to NIR region. Hence, under normoxic conditions, no fluorescence response was observed at an excitation wavelength of 650 nm, an observation attributed to the emission of Cy5 being quenched by QSY-21 via a FRET process. In contrast, hypoxic conditions result in the reduction of QSY-21. This caused the absorbance maximum to shift from 660 nm to 470 nm precluding overlap with the Cy5 emission and inhibiting the FRET process. In principle, the radical of QSY-21 generated from the reduction could quench the fluorescence of linked Cy5 via electron transfer. However, no evidence for this quenching mode was seen, a finding ascribed to the relative long linker (above five methylenes) used to construct 47. In operational terms, the salient feature of 47 is that the QSY-21 moiety undergoes reversible reduction/oxidation when subject to hypoxia–normoxia cycles. This proved true when it was tested with live A549 cells. Here, by placing a cover glass over the cells and then removing it, hypoxic–normoxic conditions could be generated. Probe 47 was then dosed into the cells, resulting in the observation of a reversible “off–on” and “on–off” fluorescence signal under these hypoxia–normoxia cycling conditions. The authors stated that 47 was a FRET-based system that could be used to follow repeated hypoxia–normoxia cycles in live cells. As such it offered a crucial visual tool for exploring the response of live cells to hypoxia.
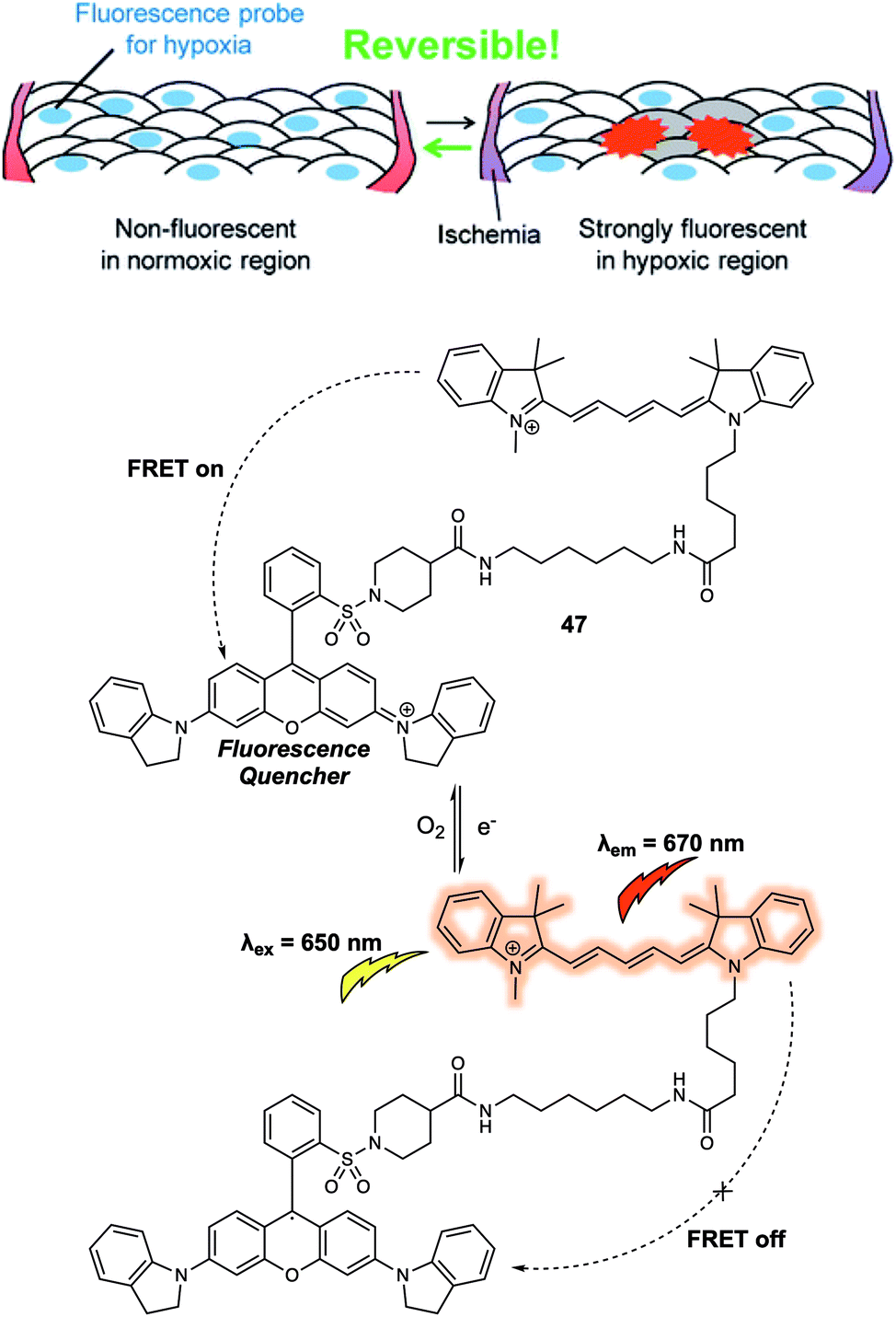 | ||
| Fig. 29 Design of the FRET-based probe 47 that allowed hypoxia in live cells to be monitored by visual means. Reproduced with permission from ref. 39 (J. Am. Chem. Soc., 2012, 134, 19588–19591). Copyright (2012) American Chemical Society. | ||
The mitochondrial membrane potential (ΔΨm) has been proposed as an indicator of apoptosis and necrosis. Polarized mitochondria are characterised by high ΔΨm values, while depolarized mitochondria exhibit low ΔΨm. Fluorescent probes that provide a visual tool for tracking dynamic changes in ΔΨm are thus of interest in the context of fundamental mechanistic research and as tools for assessing the effects of potential drug candidates. Most probes reported exhibit a fluorescence signal readout based on ΔΨm-induced changes in the fluorescence intensity. More effective systems would be those that permit ratiometric sensing. With this desire in mind, Yu et al. reported a new FRET system in 2019 that could be used for monitoring ΔΨm levels through readout of a ratiometric fluorescence signal.40 The FRET system was constructed using a carbazole linked to a benzyl chloride functionalized pyridinium group (48) as the fluorescent donor and a quinoline derivative providing for an extended D–π–A structure (49) as the fluorescence acceptor (Fig. 30). Initially, the fluorescent donor (48) was immobilized to the mitochondria by means of covalent bonds formed between the benzyl chloride moiety of 48 and endogenous protein thiol groups localised in the mitochondria. In this state, 48 displayed a fluorescence signal (green channel, 510–550 nm) under excitation at 405 nm, irrespective of changes in the ΔΨm. The acceptor (49) was then added and found to localise selectively to the mitochondria. In this two-component system, an organelle localised fluorescence emission (DR channel, 665–735 nm) was observed upon excitation at 405 nm. When the ΔΨm decreased by the exogenous addition of carbonyl cyanide 3-chlorophenylhydrazone (CCCP), the FRET signal decreased. This was ascribed to the acceptor 49 no longer binding well to the mitochondrial membrane leading to breakup of the FRET pair 48/49. This FRET system was used to monitor decreases in the ΔΨm induced by CCCP in live A549 cells. In addition, this FRET system could be used to monitor cell damage induced by H2O2 and chloral hydrate reflecting the close relationship between ΔΨm and cell viability.
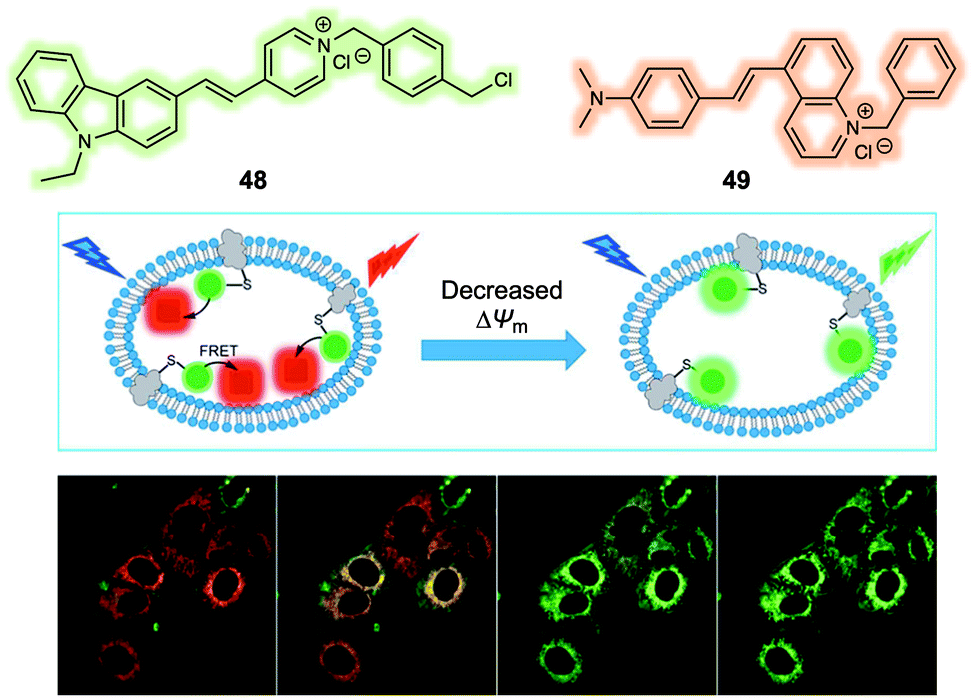 | ||
| Fig. 30 Design of a FRET system (48/49) for the ratiometric detection of mitochondria membrane potential (ΔΨm). Reproduced with permission from ref. 40 (Anal. Chem., 2019, 91, 3704–3709). Copyright (2019) American Chemical Society. | ||
One limitation of the above 48/49-based FRET system is that it relies on the non-specific intracellular targeting of 49 to the mitochondria and as such may still target depolarized mitochondria due to its hydrophobic nature and limiting its sensitivity to ΔΨm. To address this issue, the Lin group designed and synthesized a FRET system that relies on 50 and 51 as the FRET reporter pair (Fig. 31).41 Both 50 and 51 were fluorescent and found to localise in the mitochondria at high ΔΨm. In addition, the overlap of the emission of 50 (maximum at around 540 nm) and the absorption of 51 (maximum at around 530 nm in most solvents) was observed. FRET from 50 to 51 was found when the ΔΨm was high as evidenced by a strong red emission being observed. Conversely, both 50 and 51 were released from the mitochondria under conditions of a reduced ΔΨm. This resulted in a reduction of the FRET effect and as a consequence the cells were found to display a strong green emission and weak red emission. This change in fluorescence features allowed monitoring of ΔΨm in live cells. After release, probe 50 was found to relocalise in the cytoplasm, while 51 moved to the nucleolus because of its relatively high affinity towards RNA. The FRET system 50/51 was used by the authors to monitor cell oxidative damage induced by H2O2via ratiometric fluorescence imaging.
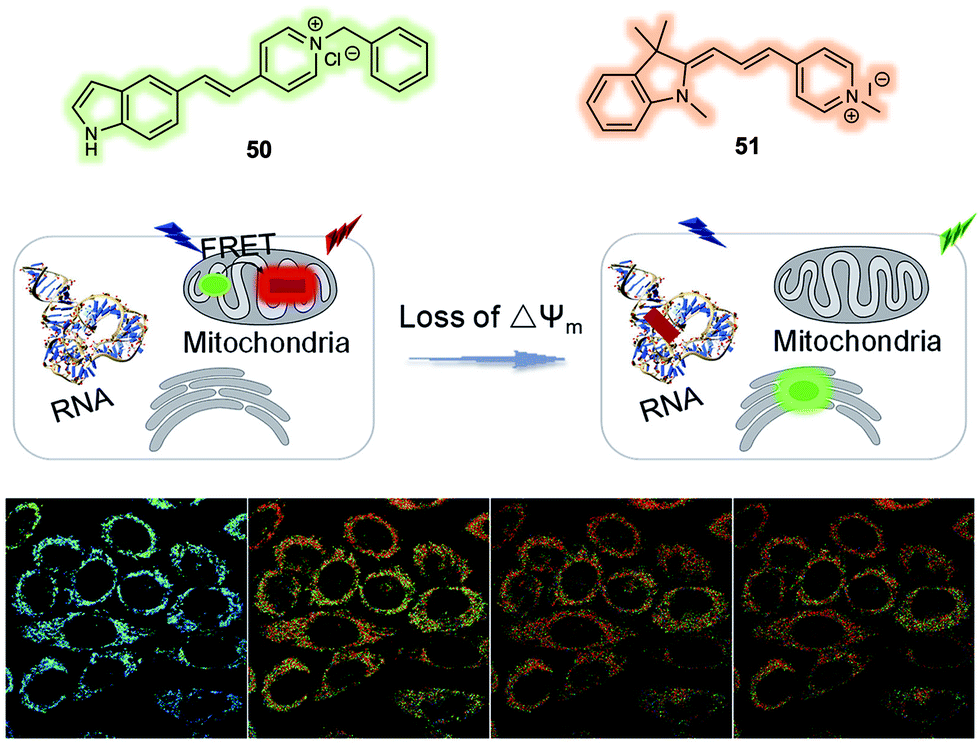 | ||
| Fig. 31 FRET system (50/51) that allows for the ratiometric detection of changes in the mitochondrial membrane potential (ΔΨm). Reproduced with permission from ref. 41 (Anal. Chim. Acta, 2019, 1097, 196–203). Copyright (2019) Elsevier B.V. | ||
Dual/multiple-responsive FRET-based probes
Most small-molecule sensors for use in biological systems are designed to detect a single analyte. This emphasis has afforded robust techniques that permit the role of single species to be studied with minimal interference. However, the ability to monitor simultaneously two or more analytes within the same biological sample is allowing the study of more complex biological processes and has important ramifications for clinical applications, where two or more species may be intimately responsible for a given phenotypic response.42 A common approach to the problem of detecting multiple analytes involves the synchronous use of several single species sensors within one homogeneous solution or in a given single type of cell. While typically easy to implement, this approach is subject to inherent drawbacks. This has motivated efforts to prepare more advanced sensing systems that allow for the simultaneous detection of two or more analytes using a single sensor. The design of such multi-analyte sensor systems is made challenging by the fact that they need incorporate multiple recognition sites and fluorescence reporter subunits within a single molecule while permitting reasonable signal communication (i.e. energy transfer) between the constituent moieties or between the target analytes and the reporter subunits. FRET-based systems containing various fluorescent dyads and suitable linkers have been explored in an effort to address these challenges. For instance, oxidative stress is another disease-correlated complication related to redox imbalance. Its determinants and effects are currently being actively explored in the hopes of understanding a range of disorders from cancer to neurodegenerative disease, as well as in the context of drug discovery efforts. Classic probes for oxidative stress have been intensity-based. In an effort to create a more effective system for quantifying oxidative stress in live cells, New et al. designed an intriguing FRET system (probe 52) based on a coumarin/flavin dyad.43 In its oxidized state, 52 emits a characteristic flavin emission at 520 nm upon excitation at 405 nm, presumably via a FRET process (Fig. 32). In contrast, 52 only displayed coumarin emission at 470 nm in its reduced state as determined from model studies carried out in HEPES buffer using NaBH3CN as the reductant. A linear response in the fluorescence intensity ratio (I520/I470) was observed. This FRET platform was then used for tracing oxidative stress in HeLa cells via ratiometric fluorescence imaging. Probe 52 permitted visualisation by means of three different detection modalities: confocal microscopy, fluorescence lifetime imaging and flow cytometry.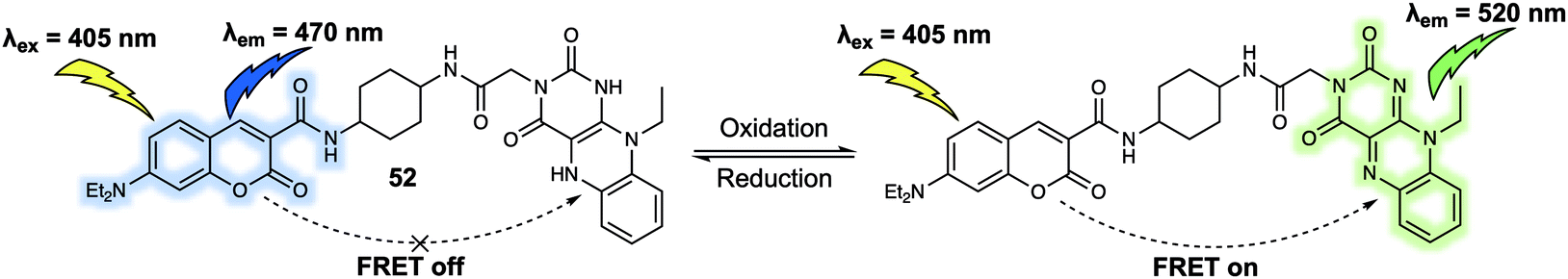 | ||
| Fig. 32 FRET-based probe 52 that facilitates the monitoring of oxidative stress. | ||
Lin et al. reported a FRET-based fluorescent sensor (probe 53) (Fig. 33) that permits the simultaneous monitoring of endogenously produced NO and H2O2 in living macrophage cells.44 Probe 53 contains a coumarin/rhodamine dyad, to which an H2O2-responsive boronate group and an NO-responsive phenylenediamine group were anchored. As prepared, 53 exhibited no fluorescence emission upon excitation at 400 nm or 550 nm. Upon addition of NO, followed by excitation at 550 nm, a strong emission at 580 nm was observed, whilst little emission was observed upon excitation at 400 nm. Subsequent addition of H2O2 led to the appearance of a new strong emission feature at 580 nm upon excitation at 400 nm, as would be expected for a FRET process from the coumarin to the rhodamine moiety. Similarly, just addition of H2O2 resulted in a fluorescence emission increase at 460 nm upon excitation at 400 nm, whilst no fluorescence emission was observed upon excitation at 550 nm. However, a strong fluorescence emission at 580 nm was observed upon excitation at 400 nm after the sequential addition of NO. This system thus displayed logic gate-like sensing, leading the authors to propose that this single FRET probe could emerge as a unique chemical tool that might allow the interplay of H2O2 and NO in various complex biological processes to be explored in greater detail.
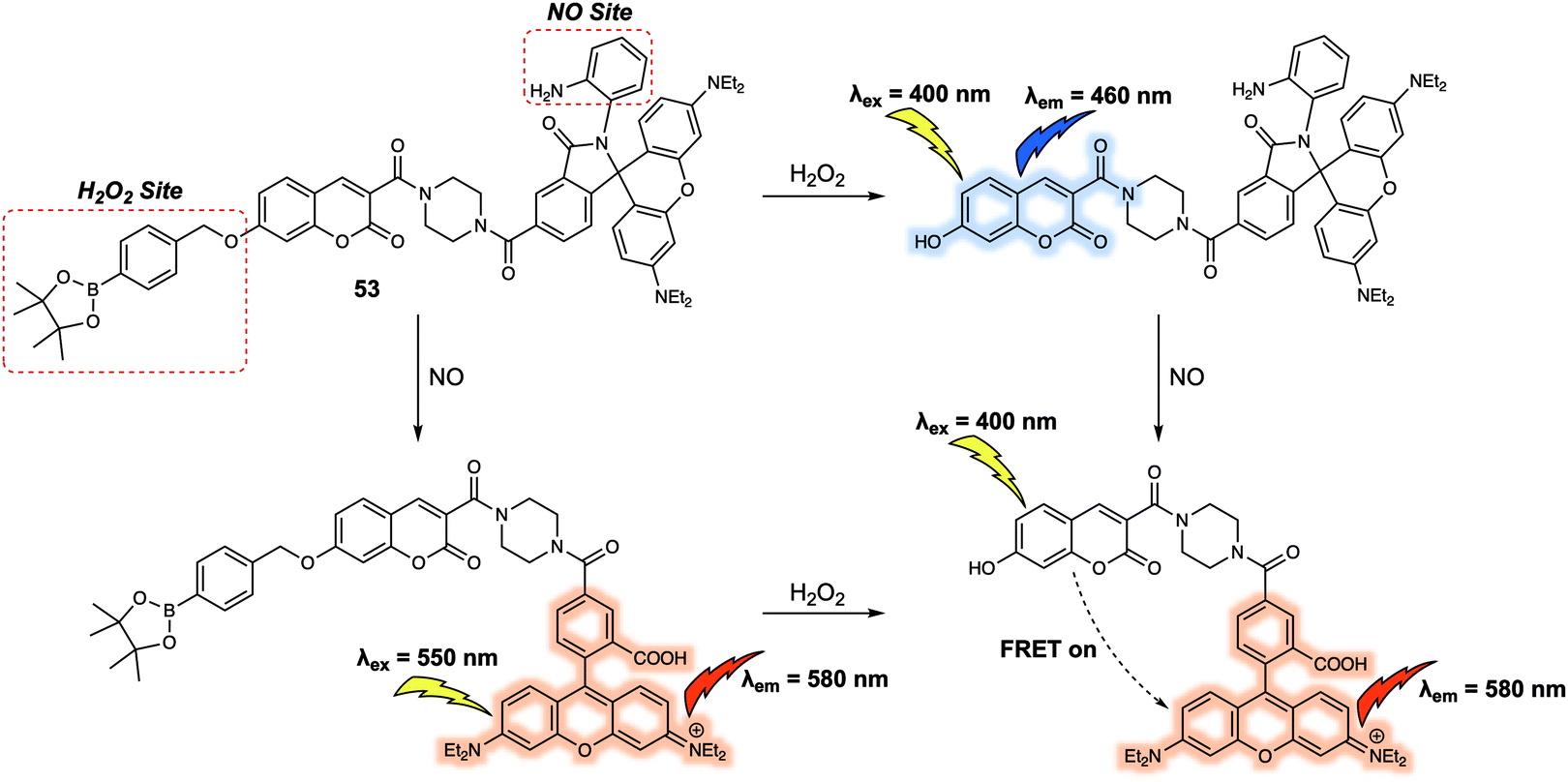 | ||
| Fig. 33 Chemical structure of the dual-responsive probe 53 and its proposed mechanism action when used as a sensor for H2O2 and NO. | ||
Understanding the cross-talk between H2S and NO in biological systems is of particular importance since they could have interlinked roles in a number of prevalent medical disorders, including cardiovascular diseases (ischemia reperfusion) and inflammation. Wu and colleagues reported a FRET-based chemosensor 54 (Fig. 34) that permitted the simultaneous detection of H2S and NO.45 In their design, an H2S-responsive azide group was anchored to a naphthalimide core while an NO-responsive phenylenediamine was anchored to a rhodamine. The two subunits were connected via a linker to create the final FRET-based system. In the absence of H2S and NO, 54 produced a very weak fluorescence signal upon excitation, a finding rationalised in terms of the azide acting as a quenching group for the photoexcited states of the naphthalimide and spirocyclic form of the rhodamine. When exposed to H2S and excited at 425 nm, 54 was found to give rise to an emission centred at 539 nm. Separate addition of NO to 54 led to an emission at 570 nm upon excitation at 550 nm. Simultaneous addition of the two analytes resulted in a fluorescence emission at 570 nm when excited at 425 nm or 550 nm a result of the FRET process from the naphthalimide to rhodamine unit. The authors then demonstrated that 54 could be used to monitor H2S and/or NO in L929 cells.
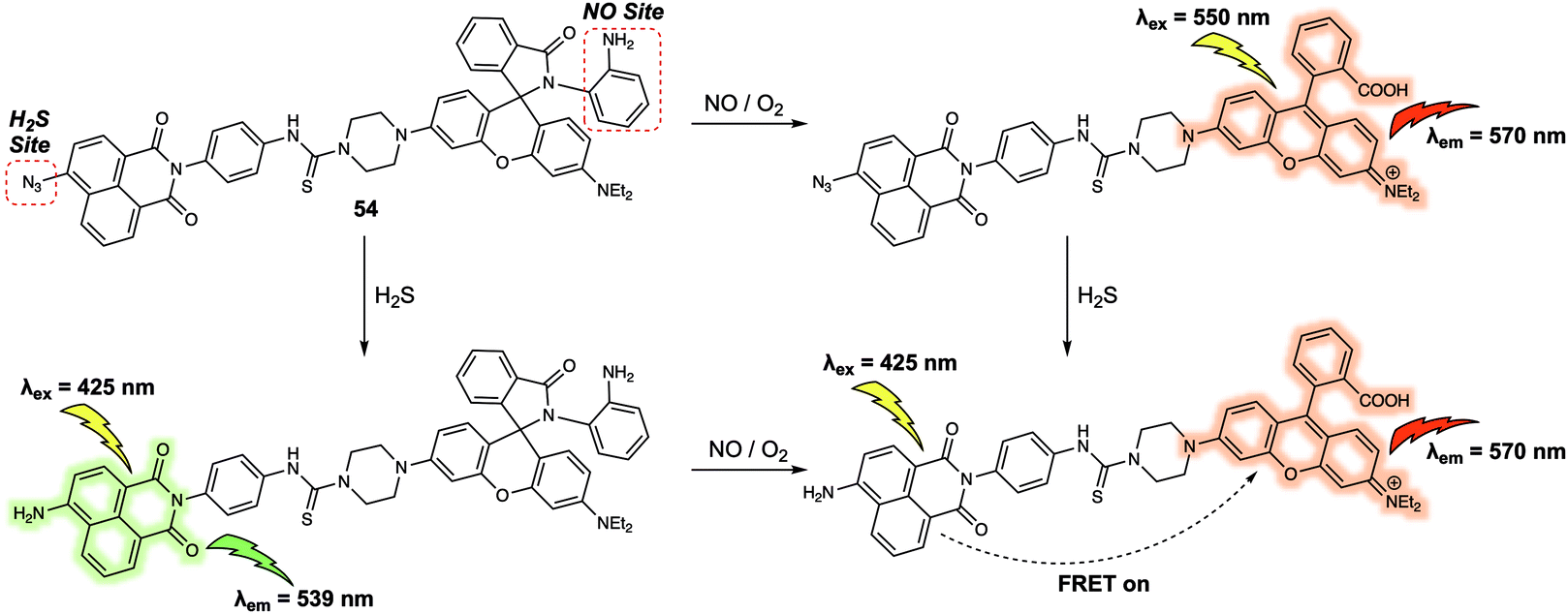 | ||
| Fig. 34 Chemical structure of the dual-responsive probe 54 and its proposed mechanism action when used to detect NO and H2S. | ||
Another dual-responsive FRET system (probe 55) (Fig. 35) was reported by Xian et al.46 This probe, which relies on a coumarin/rhodol dyad as the FRET pair, proved effective for measuring both H2S and hydrogen polysulfides (H2Sn, n > 1). In terms of design, both an H2S-responsive azide and an H2Sn-response phenyl 2-(benzoylthio)benzoate were positioned on the FRET dyad. As expected, the addition of Na2S2 (commonly used for generation of H2Sn) induced a strong fluorescence emission at 542 nm, corresponding to the emission of the rhodol. In contrast, the addition of Na2S (commonly used for generation of H2S) induced characteristic coumarin fluorescence emission at 452 nm as the result of azide reduction. Unfortunately, the reaction of the azide subunit in 55 with H2S also generates a small amount of H2Sn, which led to a peak appearing at 542 nm, corresponding to the fluorescence emission of rhodol. In spite of this complication, 55 could be used to discriminate between H2S and H2Sn both in solution and in living cells by taking advantage of the dual-channel fluorescence signal readout it permits. Moreover, the fluorescence intensity ratio could be used to detect the relative concentrations of H2S and H2Sn in aqueous solution under conditions where both analytes are present.
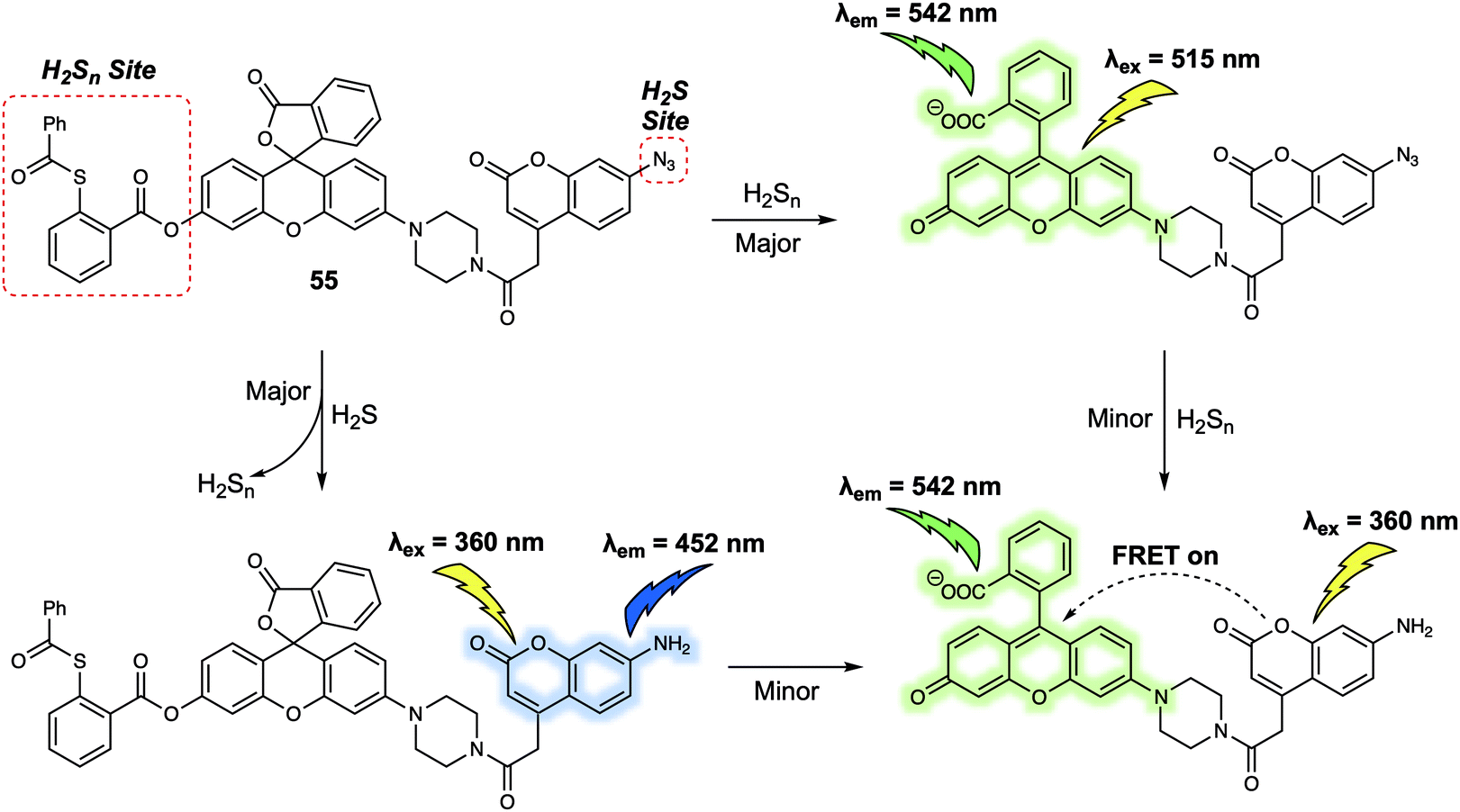 | ||
| Fig. 35 Chemical structure of the dual-responsive probe 55 and the proposed chemical transformations that allow it to be used as a sensor for H2S and H2Sn. | ||
Hlaváč et al. constructed a FRET system (probe 56) with three signal readouts as a ratiometric chemosensor for the simultaneous detection of chymotrypsin and trypsin. Probe 56 (Fig. 36) was constructed by attaching three fluorescent dyes 7-diethylaminocoumarin-3-carboxylic acid (DEAC), fluorescein (Fl) and rhodamine B (RhB) to a short peptide.47 Upon excitation of the donor (DEAC) at 435 nm, FRET to Fl occurs, followed by FRET from Fl to RhB. The system thus exhibited a cascade-like energy transfer process. The two cleavage sites (phenylalanine (specific for chymotrypsin) and lysine (specific for trypsin)) of this short peptide-based fluorescent probe allowed it to discriminate chymotrypsin and trypsin through the cascade-like FRET process. Specifically, it was found to display three discrete fluorescence signals when excited at one single wavelength (435 nm). The disparate signal readouts provided a means to quantify the activity of the target protease.
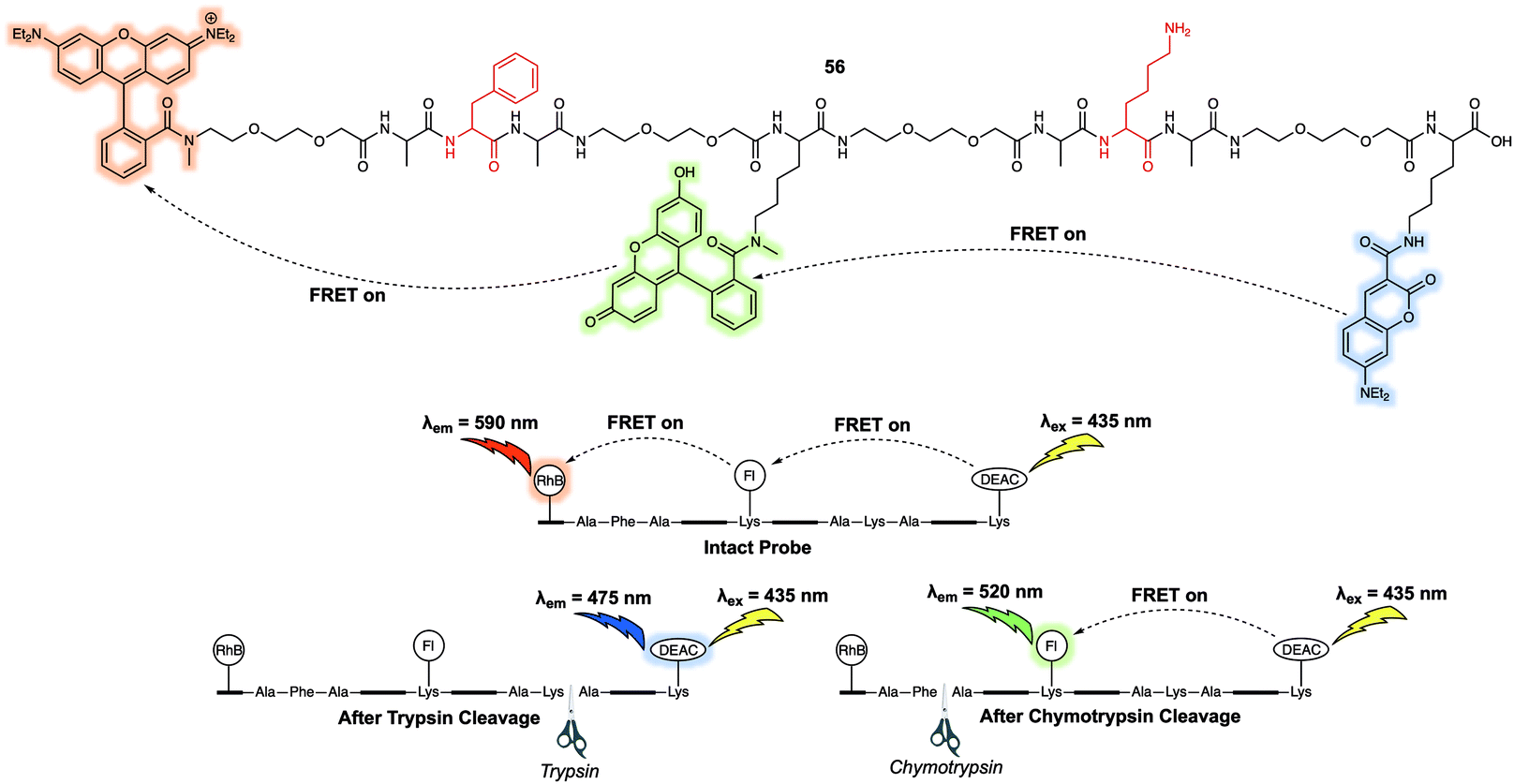 | ||
| Fig. 36 Chemical structure of the dual-responsive, three-fluorophore probe 56 and the working mechanism proposed that allows for the detection of chymotrypsin and trypsin. | ||
In 2019, Zhang and colleagues reported a dual-responsive FRET-based fluorescent sensor (probe 57) (Fig. 37) that was designed to allow investigations of how phosphatase activity in cells depend on H2S levels.48 In analogy to other dual-responsive FRET systems, a coumarin/rhodol dyad exists at the core of this FRET system. Probe 57 incorporated an azide group as the recognition unit for H2S. It also contains a phosphate group that was expected to serve as a recognition site for the phosphatase enzyme being targeted for study. Probe 57 itself exhibited relatively weak fluorescence upon excitation at 360 nm or 510 nm. However, the separate addition of either H2S or phosphatase resulted in the appearance of the characteristic coumarin or rhodol fluorescence emission at 445 nm and 545 nm, respectively. Addition of a fixed concentration of H2S followed by increasing phosphatase additions, enhancement of the emission at 545 nm and concurrent decrease of the emission at 445 nm were observed. These findings were ascribed to a FRET process between donor with the reduced amine and the ring-opened fluorescent acceptor. Importantly, 57 exhibited a high selectivity for H2S and phosphatase over polysulfides and other enzymes. Using this system and a three-channel fluorescence imaging setup it proved possible to detect H2S and phosphatase simultaneously in HeLa cells. Using 57 as a chemosensor, this research team was able to confirm that in vitro the intracellular concentrations of H2S played a vital role in regulating the activity of phosphatase. In particular, it was discovered that cells regulate their H2S concentrations so as to maintain an optimal levels of phosphatase activity.
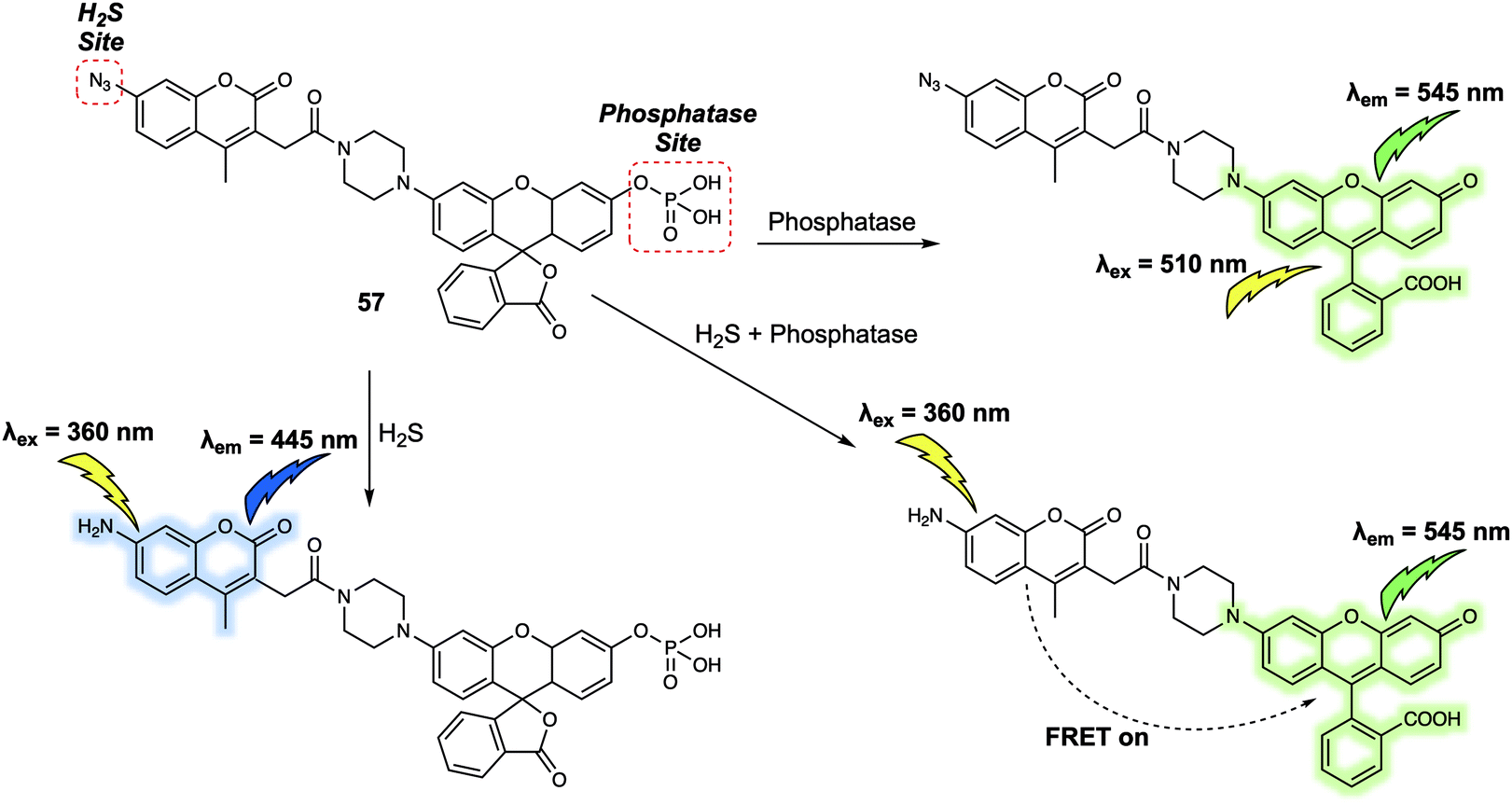 | ||
| Fig. 37 Structure of probe 57 and its proposed fluorescent response mechanism when exposed to H2S, phosphatase, and an H2S/phosphatase mixture. | ||
A dual-responsive FRET system (probe 58) (Fig. 38) was developed by Yang et al. to permit the monitoring of β-D-glucosidase (GCD) and phosphodiesterase I (PDE).49 The proposed sensing mechanism is depicted in Fig. 36. In the absence of either target enzyme probe 58 emits at 656 nm due to FRET from the 7-β-D-glucopyranosyloxycoumarin donor to the meso-tetraphenylporphyrin (TPP) acceptor. Addition of GCD cleaves the β-glycosidic bond to release the 7-hydroxycoumarin, which emits at 460 nm. A slight decrease in the emission of the TPP acceptor at 656 nm was observed, presumably due to a somewhat weakened FRET process. In contrast, sole addition of PDE induced fluorescence emission at 390 nm because the FRET process from 7-β-D-glucopyranosyloxycoumarin to TPP was fully inhibited. Finally, addition of both GCD and PDE resulted in strong emission at 460 nm but very weak emission at 656 nm. This FRET system could be used to detect GCD and PDE levels in Huh7 cells.
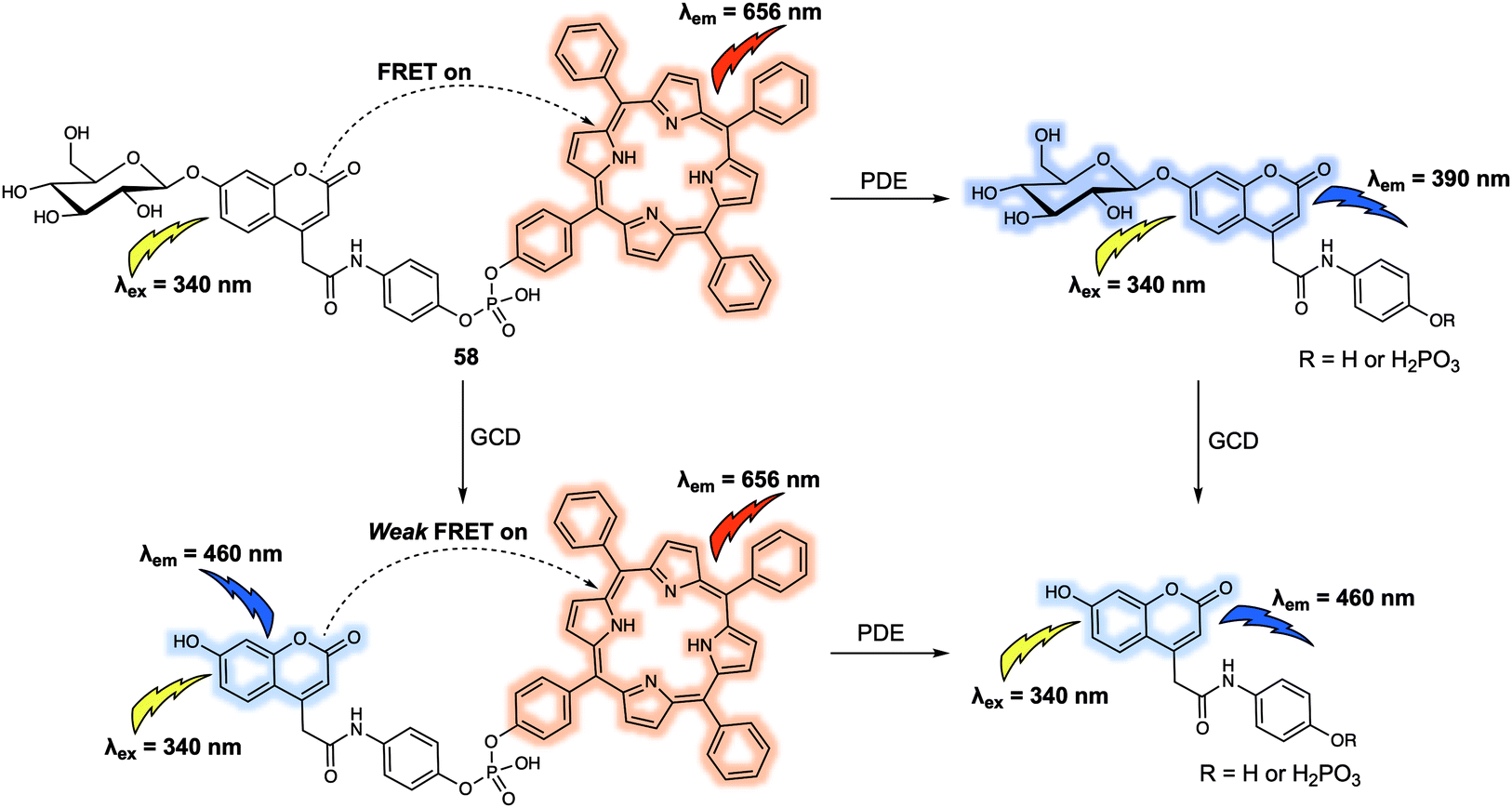 | ||
| Fig. 38 Chemical structure of the dual-responsive probe 58 and the mechanism proposed for the simultaneous detection of GCD and PDE. | ||
Gangliosides, functionalized oligosaccharides critical to cell signal transduction, have also been used to construct FRET-based probes for enzymes. For instance, in 2015 Withers et al. designed and synthesized a FRET system (probe 59) (Fig. 39) for monitoring ganglioside-processing enzyme activity.50 Within probe 59, a 7-hydroxycoumarin served as the FRET donor and a BODIPY as the acceptor. These subunits were attached to the C9 position of the sialic acid and the terminus of the constituent fatty acid alkyl chain, respectively, as shown in Fig. 39. Probe 59 could be cleaved by three ganglioside-degrading enzymes, which are sphingolipid ceramide N-deacylase (Enz1), endoglycoceramidase (Enz2), and neuraminidase (Enz3). In the absence of one of these enzymes, excitation of the 7-hydroxycoumarin donor at 360 nm gave rise to the BODIPY-derived emission at around 518 nm. The FRET efficiency was calculated to be >95%, indicating a highly efficient energy transfer system. Addition of ENz1, ENz2 or ENz3, respectively, induced a substantial increase in the fluorescence intensity at 450 nm corresponding to 7-hydroxcoumarin-based emission. A decrease in the fluorescence intensity at 518 nm was also observed. The change in the ratio I450/I518 was up to 70-fold for both ENz2 and ENz3; however, a relatively low (7.3-fold) change was observed for ENz1, a finding ascribed to an equilibrium being established between hydrolysis and enzymatic synthesis. The ratiometric signal readout from the donor and acceptor of 59 permitted quantification of ENz2 and ENz3 levels in lysate expressed from E. coli cells. Moreover, 59 enabled the fluorescence imaging of ENz3 activity in human lymphoma (Jurkat) cells.
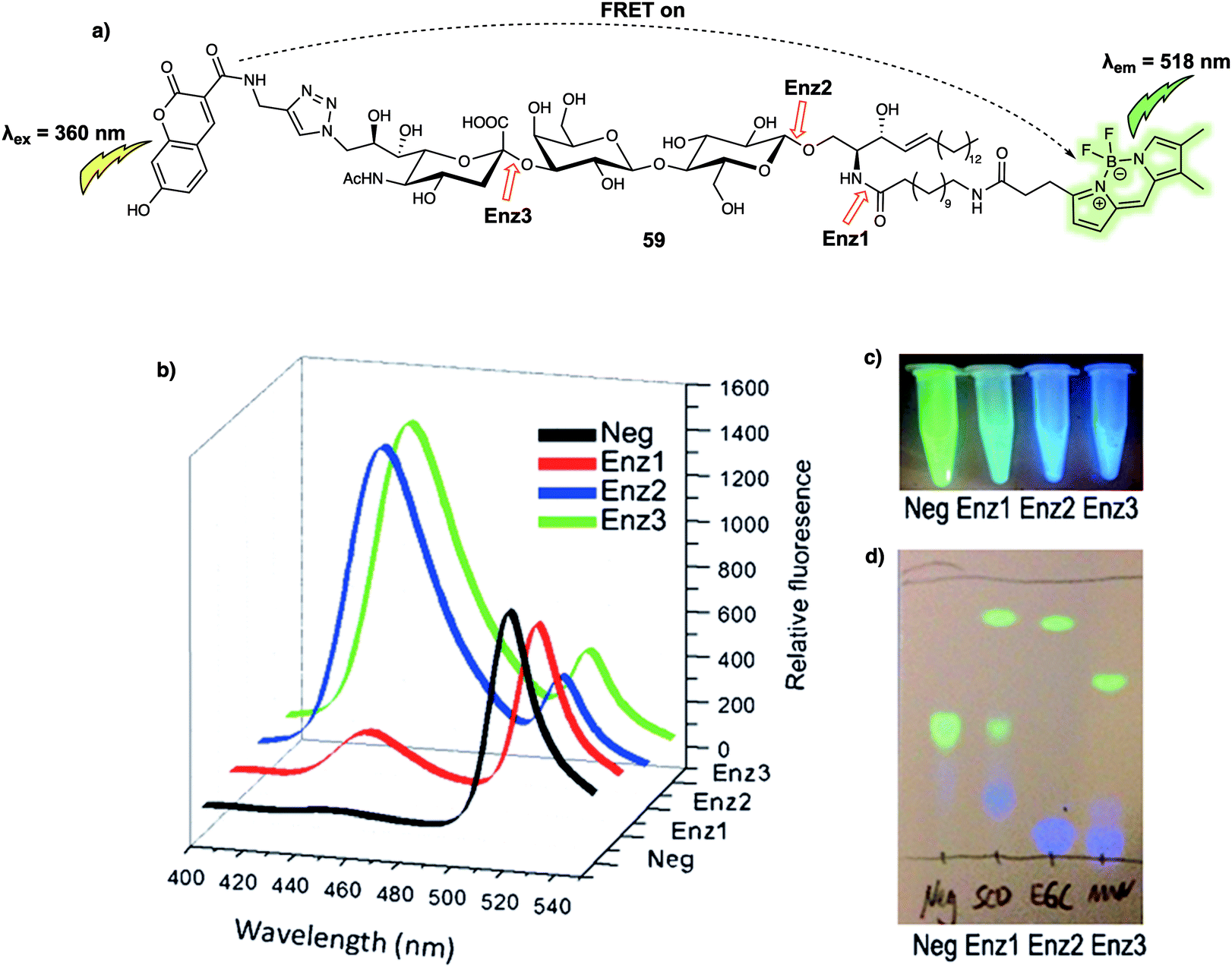 | ||
| Fig. 39 (a) Structure of probe 59 and proposed mechanism for the detection of Enz1, Enz2, and Enz3. Red arrows represent the position where each enzyme cleaved the substrate. (b) Fluorescence spectra recorded before and after incubation with each test enzyme (30 min). (c) Visual detection of the reactions under a UV lamp. (d) Different hydrolysis patterns of probe 59 after incubating with Enz1, Enz2, and Enz3. Neg: unreacted probe 59 as a negative control. Reproduced with permission from ref. 50 (Angew. Chem., Int. Ed., 2015, 54, 5389–5393). Copyright (2015) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Conclusions and perspectives
Förster resonance energy transfer (FRET) is a photochemical process that relies on energy transfer from the donor to the acceptor. This process can be regulated by the distance between the donor and acceptor, as well as the overlap between the emission of the donor and absorption of the acceptor. The unique features of FRET have resulted in the approach being widely applied to the design of various fluorescent probes for the detection of specific analytes. In particular, many small-molecule FRET-based probes exhibit properties that make them ideal for biological applications, including fast uptake by cells, non-invasive imaging, in situ detection with minimal biological interference, and easy structural variability. In this review, we have aimed to summarise recent progress towards the development of small-molecule FRET-based chemosensors and imaging agents. We began with the design principles of donor–acceptor interactions and how they are modulated to create FRET-based sensors. A number of recently developed small-molecule FRET-based chemosensors were then presented so as to illustrate a range of working mechanisms and selected biological applications. In the final section, design strategies used to create dual/multiple-responsive FRET probes were discussed. We then highlight the potential of selected systems to monitor two or more analytes within the same biological sample. The ability to detect multiple targets is expected to allow a greater understanding of complex biological processes since in many cases two or more species may be intimately responsible for a given biological response. Therefore, we strongly believe that future efforts should focus in part on designing FRET-based probes that can detect multiple analytes and provide signal outputs that allow for their easy discrimination. Such systems are expected to follow the rules of logic gates and the associated complexity is expected to aid research in a range of areas, including cell biology and laboratory tests designed to enable faster clinical diagnoses. Another notable area for development is in the area of NIR fluorescent FRET-based probes and their quenchers. Since they permit relatively deep tissue penetration, NIR fluorescent probes are particularly desirable, especially for in vivo analysis. One major challenge associated with the design of FRET-based NIR fluorescent probes involves the synthesis of the constituent NIR fluorescent donors and acceptors. This will require the development of NIR fluorophores with high photostability and significant water solubility, as well as quenchers.Given the combination of challenges and opportunities associated with FRET-based small-molecule probes, we believe that the area remains open for continued development. Unsurprisingly, at this time the need for improved systems is even greater than ever. At the same time, the possibilities to contribute to advances in a wide range of fields is almost limitless. We hope, therefore, that with this review we have inspired the reader to contribute to the further development of the area while providing a knowledge base that will facilitate this task. In this way, we believe the best is yet to come for those working to prepare and exploit new FRET-based small-molecule probes as research tools.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
LW wishes to thank China Scholarship Council and The University of Bath for supporting his PhD work in the UK. BE, SDB and TDJ would like to thank the EPSRC and The University of Bath for funding. TDJ wishes to thank the Royal Society for a Wolfson Research Merit Award. CH thanks the National Natural Science Foundation of China (Grants 21672150, 21302125), Alexander von Humboldt Foundation (AvH), Shanghai Rising-Star Program (19QA1406400), Shanghai Engineering Research Center of Green Energy Chemical Engineering (18DZ2254200), and Program for Changjiang Scholars and Innovative (IRT_16R49). XPH, HT thank the Natural Science Foundation of China (No. 21788102, 21722801 and 91853201), Shanghai Municipal Science and Technology Major Project (2018SHZDZX03), the National Key Sci-Tech Special Projects of Infection Diseases of China (2018ZX10732202) and the international cooperation program of Shanghai Science and Technology Committee (17520750100). JY acknowledges a grant from the National Creative Research Initiative programs of the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (No. 2012R1A3A2048814). JLS thanks the National Institutes of Health (R01 GM103790) and the Robert A. Welch Foundation (F-0018) for support of the work carried out in Austin.References
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- X. Zhang, Y. Xiao and X. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025–8029 CrossRef CAS PubMed.
- K. Aich, S. Goswami, S. Das, C. D. Mukhopadhyay, C. K. Quah and H.-K. Fun, Inorg. Chem., 2015, 54, 7309–7315 CrossRef CAS PubMed.
- A. T. Aron, M. O. Loehr, J. Bogena and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 14338–14346 CrossRef CAS PubMed.
- C. Y.-S. Chung, J. M. Posimo, S. Lee, T. Tsang, J. M. Davis, D. C. Brady and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18285–18294 CrossRef PubMed.
- Y.-R. Zhang, X.-P. Chen, J. Shao, J.-Y. Zhang, Q. Yuan, J.-Y. Miao and B.-X. Zhao, Chem. Commun., 2014, 50, 14241–14244 CAS.
- S.-L. Shen, X. Zhao, X.-F. Zhang, X.-L. Liu, H. Wang, Y.-Y. Dai, J.-Y. Miao and B.-X. Zhao, J. Mater. Chem. B, 2017, 5, 289–295 RSC.
- L. Wu, A. C. Sedgwick, X. Sun, S. D. Bull, X.-P. He and T. D. James, Acc. Chem. Res., 2019, 52, 2582–2597 CrossRef CAS PubMed.
- X. Jia, Q. Chen, Y. Yang, Y. Tang, R. Wang, Y. Xu, W. Zhu and X. Qian, J. Am. Chem. Soc., 2016, 138, 10778–10781 CrossRef CAS PubMed.
- D. Cheng, Y. Pan, L. Wang, Z. Zeng, L. Yuan, X. Zhang and Y.-T. Chang, J. Am. Chem. Soc., 2017, 139, 285–292 CrossRef CAS PubMed.
- D.-P. Li, Z.-Y. Wang, X.-J. Cao, J. Cui, X. Wang, H.-Z. Cui, J.-Y. Miao and B.-X. Zhao, Chem. Commun., 2016, 52, 2760–2763 RSC.
- X. Yang, Y. Zhou, X. Zhang, S. Yang, Y. Chen, J. Guo, X. Li, Z. Qing and R. Yang, Chem. Commun., 2016, 52, 10289–10292 RSC.
- S. Yang, X. Wen, X. Yang, Y. Li, C. Guo, Y. Zhou, H. Li and R. Yang, Anal. Chem., 2018, 90, 14514–14520 CrossRef CAS PubMed.
- H. Zhang, R. Liu, Y. Tan, W. H. Xie, H. Lei, H.-Y. Cheung and H. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 5438–5443 CrossRef CAS PubMed.
- X. Zhu, M. Xiong, H.-W. Liu, G.-J. Mao, L. Zhou, J. Zhang, X. Hu, X.-B. Zhang and W. Tan, Chem. Commun., 2016, 52, 733–736 RSC.
- X. Xie, J. Fan, M. Liang, Y. Li, X. Jiao, X. Wang and B. Tang, Chem. Commun., 2017, 53, 11941–11944 RSC.
- L. He, W. Lin, Q. Xu and H. Wei, Chem. Commun., 2015, 51, 1510–1513 RSC.
- L. Yuan, F. Jin, Z. Zeng, C. Liu, S. Luo and J. Wu, Chem. Sci., 2015, 6, 2360–2365 RSC.
- K. Umezawa, M. Kamiya and Y. Urano, Angew. Chem., Int. Ed., 2018, 57, 9346–9350 CrossRef CAS PubMed.
- K. Umezawa, M. Yoshida, M. Kamiya, T. Yamasoba and Y. Urano, Nat. Chem., 2017, 9, 279–286 CrossRef CAS PubMed.
- Y. W. Jun, T. Wang, S. Hwang, D. Kim, D. Ma, K. H. Kim, S. Kim, J. Jung and K. H. Ahn, Angew. Chem., Int. Ed., 2018, 57, 10142–10147 CrossRef CAS PubMed.
- J. X. Ong, C. S. Q. Lim, H. V. Le and W. H. Ang, Angew. Chem., Int. Ed., 2019, 131, 170–173 CrossRef.
- T. Pinkert, D. Furkert, T. Korte, A. Herrmann and C. Arenz, Angew. Chem., Int. Ed., 2017, 56, 2790–2794 CrossRef CAS PubMed.
- A. Cobos-Correa, J. B. Trojanek, S. Diemer, M. A. Mall and C. Schultz, Nat. Chem. Biol., 2009, 5, 628–630 CrossRef CAS PubMed.
- S. Gehrig, M. A. Mall and C. Schultz, Angew. Chem., Int. Ed., 2012, 51, 6258–6261 CrossRef CAS PubMed.
- J. Mu, F. Liu, M. S. Rajab, M. Shi, S. Li, C. Goh, L. Lu, Q.-H. Xu, B. Liu, L. G. Ng and B. Xing, Angew. Chem., Int. Ed., 2014, 53, 14357–14362 CrossRef CAS PubMed.
- L. Li, X. Shen, Q.-H. Xu and S. Q. Yao, Angew. Chem., Int. Ed., 2013, 52, 424–428 CrossRef CAS PubMed.
- A. K. Yadav, D. L. Shen, X. Shan, X. He, A. R. Kermode and D. J. Vocadlo, J. Am. Chem. Soc., 2015, 137, 1181–1189 CrossRef CAS PubMed.
- S. Cecioni and D. J. Vocadlo, J. Am. Chem. Soc., 2017, 139, 8392–8395 CrossRef CAS PubMed.
- Q. Shao and B. Xing, Chem. Commun., 2012, 48, 1739–1741 RSC.
- T. Myochin, K. Hanaoka, S. Iwaki, T. Ueno, T. Komatsu, T. Terai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 4759–4765 CrossRef CAS PubMed.
- Y. Yuan, R. Zhang, X. Cheng, S. Xu and B. Liu, Chem. Sci., 2016, 7, 4245–4250 RSC.
- S. Zhang, K. I. Assaf, C. Huang, A. Hennig and W. M. Nau, Chem. Commun., 2019, 55, 671–674 RSC.
- C. Huang, T. Jia, M. Tang, Q. Yin, W. Zhu, C. Zhang, Y. Yang, N. Jia, Y. Xu and X. Qian, J. Am. Chem. Soc., 2014, 136, 14237–14244 CrossRef CAS PubMed.
- Y.-X. Wu, X.-B. Zhang, J.-B. Li, C.-C. Zhang, H. Liang, G.-J. Mao, L.-Y. Zhou, W. Tan and R.-Q. Yu, Anal. Chem., 2014, 86, 10389–10396 CrossRef CAS PubMed.
- S. Takahashi, W. Piao, Y. Matsumura, T. Komatsu, T. Ueno, T. Terai, T. Kamachi, M. Kohno, T. Nagano and K. Hanaoka, J. Am. Chem. Soc., 2012, 134, 19588–19591 CrossRef CAS PubMed.
- R. Feng, L. Guo, J. Fang, Y. Jia, X. Wang, Q. Wei and X. Yu, Anal. Chem., 2019, 91, 3704–3709 CrossRef CAS PubMed.
- J. Sun, M. Tian and W. Lin, Anal. Chim. Acta, 2020, 1097, 196–203 CrossRef CAS PubMed.
- X.-P. He, X.-L. Hu, T. D. James, J. Yoon and H. Tian, Chem. Soc. Rev., 2017, 46, 6687–6696 RSC.
- A. Kaur, M. A. Haghighatbin, C. F. Hogan and E. J. New, Chem. Commun., 2015, 51, 10510–10513 RSC.
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305–1315 CrossRef CAS PubMed.
- P. Zhang, J. Li, B. Li, J. Xu, F. Zeng, J. Lv and S. Wu, Chem. Commun., 2015, 51, 4414–4416 RSC.
- W. Chen, A. Pacheco, Y. Takano, J. J. Day, K. Hanaoka and M. Xian, Angew. Chem., Int. Ed., 2016, 55, 9993–9996 CrossRef CAS PubMed.
- Y. Okorochenkova, M. Porubský, S. Benická and J. Hlaváč, Chem. Commun., 2018, 54, 7589–7592 RSC.
- P. Ou, R. Zhang, Z. Liu, X. Tian, G. Han, B. Liu, Z. Hu and Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 2261–2265 CrossRef CAS PubMed.
- Y. Li, H. Wang, J. Li, J. Zheng, X. Xu and R. Yang, Anal. Chem., 2011, 83, 1268–1274 CrossRef CAS PubMed.
- G.-Y. Yang, C. Li, M. Fischer, C. W. Cairo, Y. Feng and S. G. Withers, Angew. Chem., Int. Ed., 2015, 54, 5389–5393 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |