
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas hydrates in sustainable chemistry
Aliakbar
Hassanpouryouzband
 *abc,
Edris
Joonaki†
ad,
Mehrdad
Vasheghani Farahani†
a,
Satoshi
Takeya
e,
Carolyn
Ruppel
f,
Jinhai
Yang
*a,
Niall J.
English
g,
Judith M.
Schicks
h,
Katriona
Edlmann
b,
Hadi
Mehrabian
c,
Zachary M.
Aman
i and
Bahman
Tohidi
a
*abc,
Edris
Joonaki†
ad,
Mehrdad
Vasheghani Farahani†
a,
Satoshi
Takeya
e,
Carolyn
Ruppel
f,
Jinhai
Yang
*a,
Niall J.
English
g,
Judith M.
Schicks
h,
Katriona
Edlmann
b,
Hadi
Mehrabian
c,
Zachary M.
Aman
i and
Bahman
Tohidi
a
aHydrates, Flow Assurance & Phase Equilibria Research Group, Institute of GeoEnergy Engineering, School of Energy, Geoscience, Infrastructure and Society, Heriot-Watt University, Riccarton, Edinburgh, EH14 4AS, UK. E-mail: Hssnpr@ed.ac.uk; Petjy@hw.ac.uk
bSchool of Geosciences, University of Edinburgh, Grant Institute, West Main Road, Edinburgh, EH9 3JW, UK
cDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
dTÜV SÜD National Engineering Laboratory, Scottish Enterprise Technology Park, East Kilbride, South Lanarkshire, G75 0QF, UK
eNational Institute of Advanced Industrial Science and Technology (AIST), Central 5, 1-1-1 Higashi, Tsukuba 305-8565, Japan
fU.S. Geological Survey, Woods Hole, MA, USA
gSchool of Chemical and Bioprocess Engineering, University College Dublin, Belfield, Dublin 4, Ireland
hGFZ German Research Centre for Geosciences, Telegrafenberg, 14473 Potsdam, Germany
iFluid Science & Resources, School of Engineering, University of Western Australia, 35 Stirling Highway, Perth, WA 6009, Australia
First published on 22nd June 2020
Abstract
Gas hydrates have received considerable attention due to their important role in flow assurance for the oil and gas industry, their extensive natural occurrence on Earth and extraterrestrial planets, and their significant applications in sustainable technologies including but not limited to gas and energy storage, gas separation, and water desalination. Given not only their inherent structural flexibility depending on the type of guest gas molecules and formation conditions, but also the synthetic effects of a wide range of chemical additives on their properties, these variabilities could be exploited to optimise the role of gas hydrates. This includes increasing their industrial applications, understanding and utilising their role in Nature, identifying potential methods for safely extracting natural gases stored in naturally occurring hydrates within the Earth, and for developing green technologies. This review summarizes the different properties of gas hydrates as well as their formation and dissociation kinetics and then reviews the fast-growing literature reporting their role and applications in the aforementioned fields, mainly concentrating on advances during the last decade. Challenges, limitations, and future perspectives of each field are briefly discussed. The overall objective of this review is to provide readers with an extensive overview of gas hydrates that we hope will stimulate further work on this riveting field.
1. Introduction
Gas hydrates are ice-like solid compounds that naturally form (or can be formed) under certain conditions of pressure and temperature (P–T) within a gas/water mixture where water molecules hydrogen bond together forming a crystalline lattice and are known as hydrates or clathrates (terms are used interchangeably). Gas hydrates are at the centre of research within sustainable chemistry because of their innovative applications in a wide range of scientific and industrial contexts, such as permanently storing CO2 present in flue gases by forming gas hydrates under oceans. The early era of gas hydrate-related research was primarily dominated by flow assurance, minimising hydrocarbon/gas pipeline blockage by hydrate formation. However, in recent years the upsurge of research in the field was stimulated by expanding the application of hydrates to energy recovery, CO2 capture and storage, gas separation, water desalination, gas storage and transport, refrigeration, etc. More recently the potential for methane escaping from hydrate-bearing sediments and reaching the atmosphere has received significant attention due to the high greenhouse warming potential (GWP) of methane.Recent experimental results backed by theoretical calculations reveal significant potential not only to continue to improve flow assurance but to dramatically increase the scope of gas hydrate-based applications, which requires enabling technologies and elucidation of a new master plan. This could not be achieved without concerted collaborative effort among researchers from different fields of chemistry, physics, geology, engineering, energy industry, humanities, etc., standing as the key to unlocking the contribution hydrates can make to a cleaner atmosphere and support economic and sustainable development. This paper aims to complete the missing links between recent experimental and theoretical efforts in chemistry, and highlight areas of research that will require multi-disciplinary research and collaboration. The idea of clathrate-based applications in a diverse range of sectors is of interest to all of the scientific community and the GWP is a concern to society as a whole. In particular, scientists studying low-carbon and unconventional energy have much to benefit from advances in gas hydrate technologies which can reduce costs and improve efficiencies within the multibillion-dollar oil industry either through the substitution of conventional fossil fuels or optimising extraction.
This paper reviews a substantial body of the theoretical, experimental, and industrial research, advances and lessons in the gas hydrate field, over the last decade. The review includes the current state of the art understanding and advances in technical developments, which are combined with expert perspectives and analyses. It is important to note that the purpose of this review is not to analyse in detail every contribution but to highlight the latest advancements, focus on the most pressing issues preventing further understanding of clathrate hydrates, and importantly realising the practical applications of hydrate-based technologies for sustainable chemistry. The review assembles the different gas hydrate-related subjects relevant to sustainable chemistry, appealing to an even broader community of readers. There are several excellent detailed reviews on different subsections of gas hydrates in the existing literature. Rather than duplicate these here, these reviews are cited here to provide the reader with guidance about critical information that is readily accessible elsewhere. Since the early pioneering reviews and books on the fundamentals of gas hydrates, the gas hydrate-community has driven significant developments and advances in novel hydrate-based technologies, seeking to improve their efficiencies and applicability. Building on these early studies, the evolving gas hydrate-based applications have led to many advances in various fields, allowing a wider range of scientific community to contribute in this area of science.
As outlined in Fig. 1, this review summarizes different properties of gas hydrates (Section 2) and their formation and dissociation kinetics (Section 3) from chemistry and physics perspectives. It then focuses on strategies for protection and removal of hydrocarbon pipelines from gas hydrates (Section 4), presence of natural gas hydrate reservoirs in the earth and potential strategies for their extraction, as well as extraterrestrial hydrates (Section 5). The role of gas hydrates in CO2 capture and storage (Section 6) is discussed next, followed by a treatment of gas hydrates in sustainable development (Section 7). Throughout the review, each subsection covers the related challenges and directions for future investigations of hydrate-based technologies.
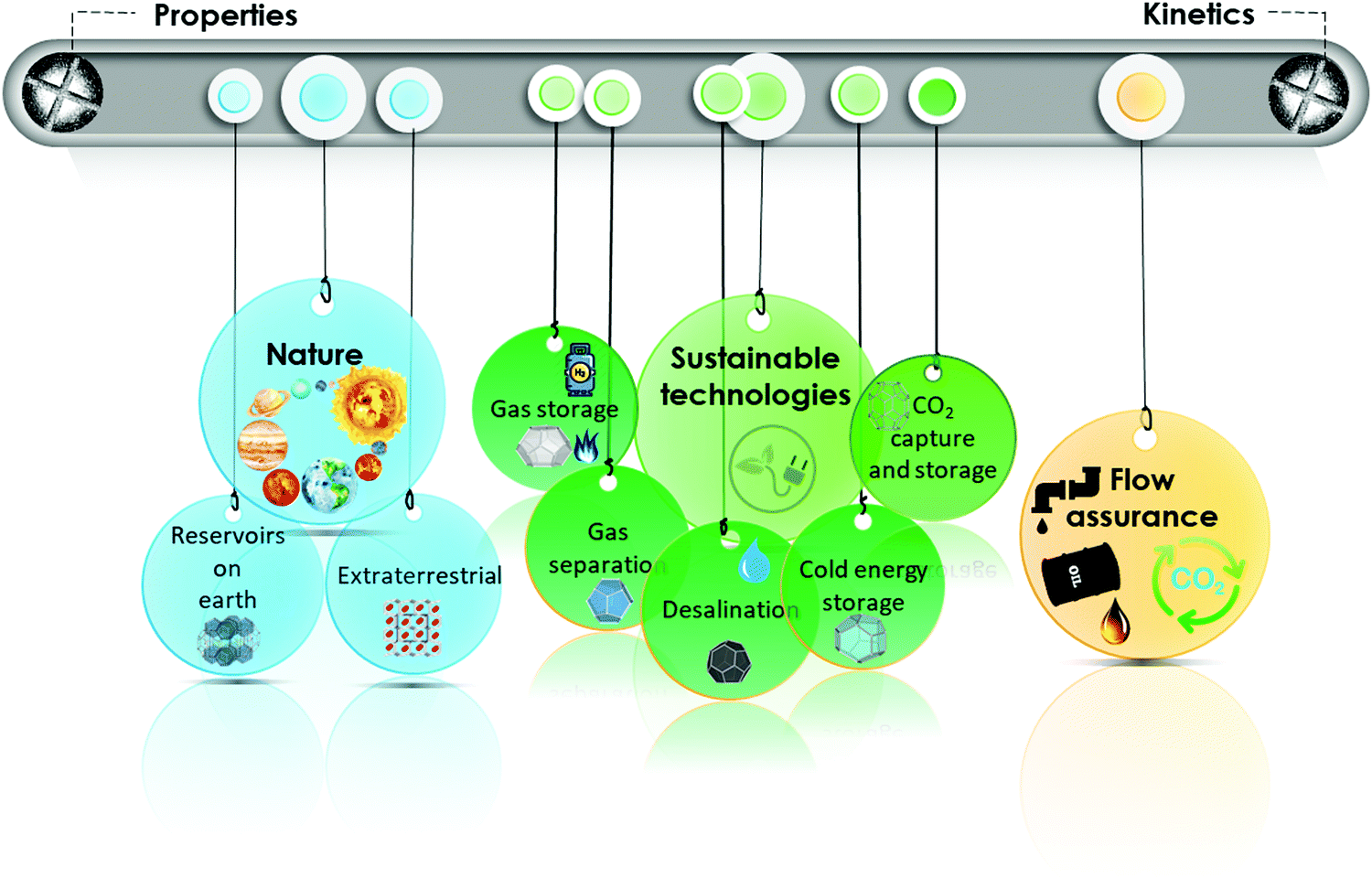 | ||
| Fig. 1 Graphical contents of the review. | ||
2. Gas hydrates’ properties
The unique properties of gas hydrates under various conditions of temperature and pressure have numerous practical applications in science and technology, and they also influence the earth's natural cycles. One example of these natural cycles is the widespread escape of methane from natural reservoirs during certain climate warming events in Earth's history and the resulting changes in ocean chemistry that may have been related to these methane emission events.1 The ability to control the properties of the clathrates using different methods and additives is of great importance to many industrial processes, particularly with regards to reducing the costs and controlling the kinetics of formation/dissociation to maximise the applicability of gas hydrate technologies. Even the multibillion-dollar international oil industry is affected by the properties of gas hydrates, which can form during hydrocarbon production and can plug production facilities and transport pipelines, imposing billions of dollars of financial loses every year. Finally, a fundamental understanding of the properties of gas hydrates is vital to developing hydrate-based technologies.The properties for many types of gas hydrates are well-known within the gas hydrate community, as demonstrated by the many published review papers covering several aspects of this diverse field. This section of the review is focused almost exclusively on studies of gas hydrates properties from the last decade. This section is structured in seven parts, with each part introducing the important work that has provided insights into the different aspects of gas-hydrate properties.
2.1. Hydrate structures
The past few decades have witnessed an impressive body of experimental work devoted to gas hydrate structures and chemistry. For instance, thanks to in situ techniques such as X-ray diffraction (XRD),2 Raman spectroscopy,3 NMR,4,5 and neutron diffraction6 at very low temperatures and high pressures, we are now able to look in real-time into different structures of clathrates, identify cages occupancies, and their evolution over time, providing a deeper insight into the kinetic behaviour of these crystalline structures. The determination and understanding of gas hydrate properties are imperative for controlling their behaviour. For this purpose in situ techniques such as in situ Raman spectroscopy and in situ X-ray diffraction supplemented with calorimetry and/or NMR are helpful tools to determine hydrate formation and dissociation behaviour as well as roles of multiple molecules on structure and cage occupancy, especially for gas hydrates formed from gas mixtures, and as such determining the thermodynamic properties of the resulting hydrate phase. Despite the fact that structures modelled from a range of spectroscopy techniques have gained ground in the field, definitive structural solutions7 by single-crystal XRD (SXRD) are needed mainly because atomic coordination and geometric parameters usually remain uncovered by other spectroscopy methods and complexity emerging from disorder framework is vague. However, the complexity of SXRD measurements requiring restricted size gas-hydrate single crystals mean that SXRD data for gas hydrates remain limited. This section of the review is focused on the discovered structures with particular emphasize on the structures that have not been discussed in previous reviews. Based on guest/host interactions, hydrates are classified into two main groups; (1) clathrate hydrates, crystals with encaged hydrophobic guest molecules in which the interaction between hydrogen-bonded network of water molecules and guest molecules is only by non-directional van der Waals forces. (2) Semi-clathrate hydrates: crystals in which active part of the guest particle molecule physically attach to the water framework and help stabilise hydrophobic guest molecules inside the hydrate lattice.2.1.1.1. Natural gas hydrates. Natural gas hydrates are a common class of clathrate hydrates that have hitherto been identified in natural environments.8 This class of well-known hydrates is classified into three main types: structure I (sI),9 which usually forms by smaller guest molecules (0.4–0.55 nm) and is the most abundant gas hydrate structure on the Earth; structure II (sII),10 which usually forms by larger guest molecules (0.6–0.7 nm) and structure H (sH)8,11 (see Fig. 2), which usually requires both small and large guest molecules for formation. However, there are exceptions; for example, nitrogen, and hydrogen can form sII hydrates, and some intermediate size guest molecules could form both sI and sII depending on the P–T conditions. All three classes consist of a hydrogen-bonded water framework based primarily around a nearly spherical structure unit of pentagonal dodecahedra (small cage) with 12 planar pentagonal faces (512).12 The difference between the structures arises from the way these small cages link; cubic sI unit cells comprise 46 water molecules forming two small cages and six, ellipsoidal-shaped, tetracaidecahedral (large sI cages) with 12 pentagonal and two hexagonal faces (51262) that formed by sharing vertices between 512 blocks without direct face sharing; cubic sII unit cell comprises 136 water molecules located in 16 small cages and 8 hexakaidecahedral (large sII cages) with 12 pentagonal and four hexagonal faces (51264) that formed through sharing faces between small cavities;13 sH unit cell with hexagonal symmetry comprises 34 water molecules arranged in 3 small cavities, two irregular dodecahedron 435663 (medium sH cavities), and one icosahedron 51268 cage (large sH cavities) that is isostructural the hexagonal clathrasil dodecasil-lH.14 The occupancy of each structure depends on the number of cages and each cage could accommodate one or more (such as nitrogen or hydrogen hydrates) guest molecules. The large cages in all structures are reported to commonly have around 100% occupancy,15 but that is not a requirement for the stabilization of the crystals.16 However, the occupancy of smaller cages strongly depends on the type of guest molecules and could be very low or zero in some cases. For example, in sI hydrates, the smaller guest molecules can fill either small or large cavities, whereas, larger guest molecules can only occupy large cages. The P–T of formation conditions strongly affect the occupancy, and as such a stable clathrate could have a range of nonstoichiometric compositions. It has also been reported that under suitable conditions transitions between structures are possible. For example, sI hydrate could transition to sII or sH, upon compression17,18 or addition of appropriate guest molecules.19 More details about the common structures can be found here.12,13,20–24
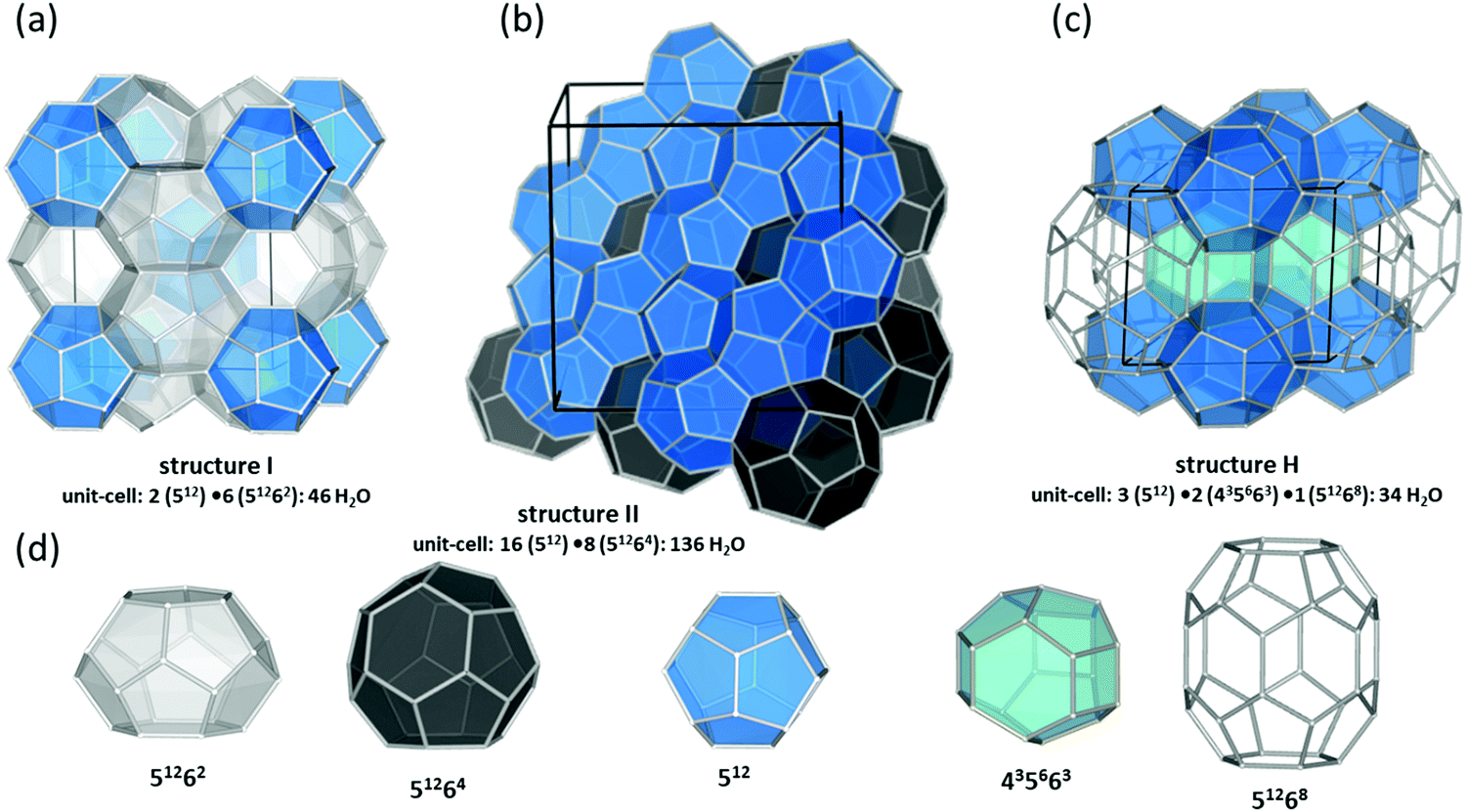 | ||
| Fig. 2 Three natural gas hydrate structures. (a) Structure I, (b) structure II, and (c) structure H, and (d) five different host water cages. The solid lines represent the unit-cell of each hydrate. All the crystal structures through the review were drawn using a visualization software called “Vesta 3”.25 | ||
2.1.1.2. Other structures. The applications of gas hydrates in diverse fields motivate scientists to investigate different hydrate structures. One of the early examples is research by Udachin and Ripmeester,26 which discovered 1.67 choline hydroxide·tetra-n-propylammonium fluoride·30.33H2O, whose structure is characterized by stacks of sH and sII hydrate and which exhibits hydrophobic and hydrophilic (see Section 2.1.2) modes of hydration by guest molecules. Another example is the discovery of trigonal sT hydrate (with dimethyl ether as the guest gas), which lacks polyhedral (512) cages and instead consists of unit cells with 12 small cages (425861) and three types of large cages with different ratios; 12(51263); 12(51262); 24(4151063).27
Traditionally28,29 it was believed that in the pressure range 1–2 GPa, methane liberates from the clathrates and could not be stable at extreme conditions. However, following studies on Titan, the giant moon of Saturn, Voyager 130 located a deep atmosphere made-up of nitrogen with considerable amounts of methane, which may be as high as 21% at the surface,31 and requires a mechanism to keep methane intact over millennia against photochemical processes. Loveday et al.32 was motivated by this finding to investigate methane hydrate above 2 GPa using X-ray and neutron diffraction. They reported methane can form a new hydrate phase (MH-III, known as filled ice33 with an unusual combination of 4-, 6-, and 8-membered water molecules’ rings) at higher pressures with a higher gas to water ratio and structural transition at about 1–2 GPa, remaining stable at least up to 10 GPa. Considering the reversibility of MH-I to MH-II and MH-II to MH-III, the authors suggested that the source of Titan's atmospheric methane is a layer of MH-I that is been formed because of gravitational differentiation from core methane in MH-III at the end of accretion and producing methane via convective processes. This discovery has prompted considerable research into clathrate hydrates at high pressure to investigate their existence in extraterrestrial settings.33–43 Recently, it has been computationally supported that methane could form MH-IV (with 6-membered rings, similar to the ordinary ice (ice Ih))41,44 at even higher pressures, which has been confirmed through experimental research37 by Raman spectroscopy measurements using diamond anvil cells. The authors reported that MH-IV (methane to water ratio ∼ 0.5) forms beyond 40 GPa and is stable up to 150 GPa at room temperature. The behaviour of methane hydrate beyond 150 GPa and the effect of temperature at pressures over greater than 10 GPa needs to be investigated through future research (see Fig. 3).
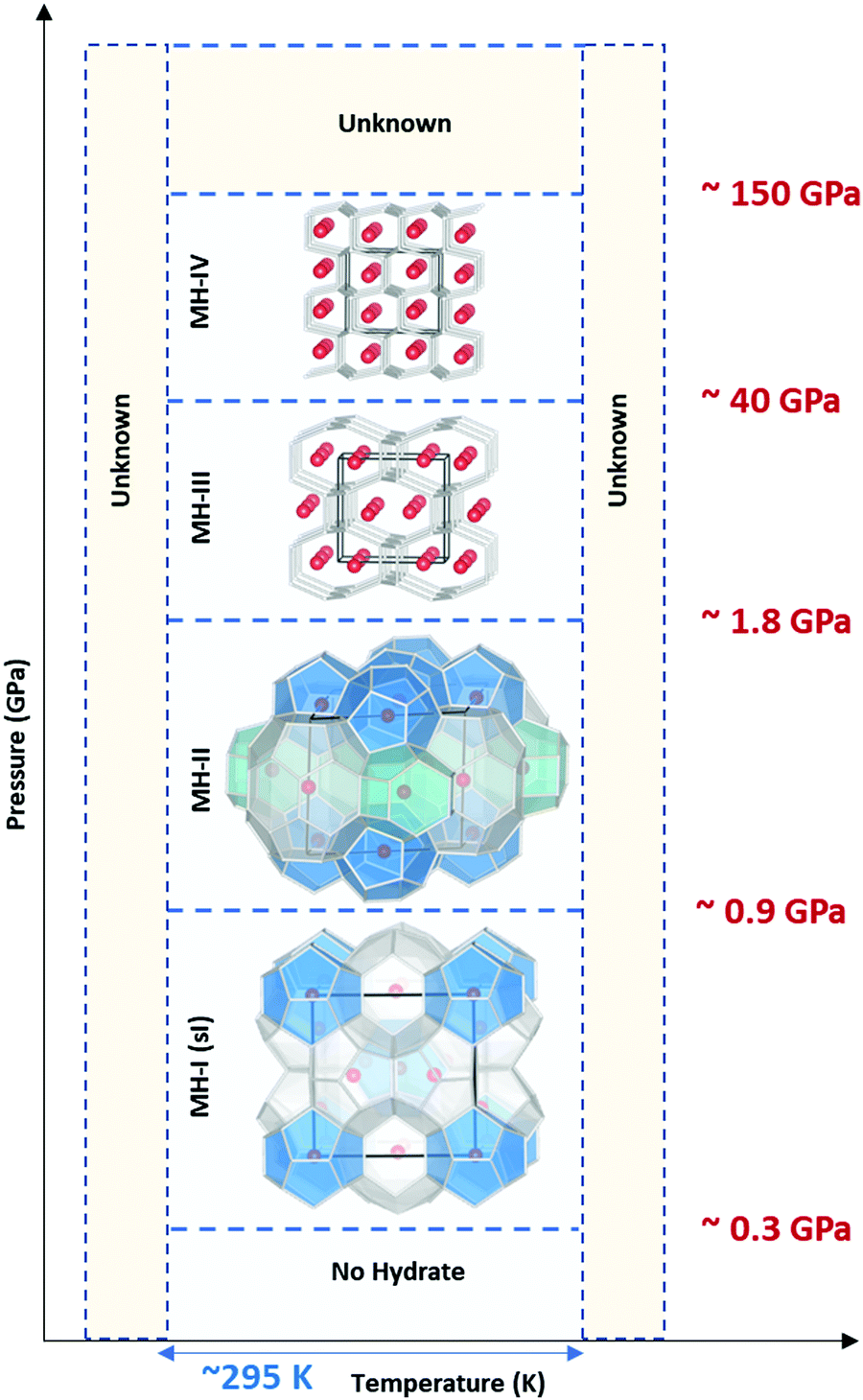 | ||
| Fig. 3 Structural transition of methane hydrate upon compression up to 150 GPa at room temperature. MH-I is the known sI hydrate. MH-II is similar to sH hydrate and may be closely related to it. However, there are challenging reports about whether MH-II has sH structure or not.17,32 | ||
There are also studies that show that unusual clathrates of other gases could also form under specific conditions. It has been found that helium molecules could refill empty neon clathrates (ice XVI) and could enter into ice Ih and ice II structures.45,46 Similarly, it has been shown that hydrogen also could fill ice by forming a structure similar to ice Ic.47 Another study suggested that Ne and O2 could fill ice XVII.48 These findings suggest that, by choosing appropriate sized guest molecules and suitable P–T conditions, it is possible to fill open, low density phases of ice.38 Still, significant further research will be required to characterize clathrates under extreme conditions for a wide range of compositions, temperatures, and pressures. Promising technologies for hydrate research could be advanced further using the developments from a variety of research fields that can contribute to investigating the existence of clathrates at unusual/extreme conditions.
2.1.2.1. The quaternary ammonium salt hydrates. These class of lesser-known clathrate hydrates, namely the semi-clathrate hydrates of ammonium/phosphonium salts, received little attention after their initial description in 1940.49 However, after examples of the separation50 and storage51 of various gases using these types of clathrates were presented, interest in semi-clathrate hydrates increased exponentially. One of the key features of these crystals compared to other classes of clathrate hydrates (sI, sII, and H) is their thermal stability at atmospheric pressure. For example, solutions of tetra-n-butylammonium fluoride under atmospheric conditions form hydrates with a melting point of 310 K.49 The higher thermal stability of semi-clathrate compared to other hydrates is attributed to their structural variety, including, but not limited to, the ways in which the dodecahedra associate. This association can be either by sharing faces or by bonding among vertices to build a range of interstitial multifaceted polyhedrals for hosting guest gas molecules or the ion pairs without immensely disordering the hydrogen-bonding pattern of the water framework.52
In contrast to clathrate hydrates in which gas molecules are encaged with stability provided to cavities by van der Waals interactions, guest molecules in semi-clathrates are both physically attached to the water network by hydrogen bonding and occupy cages. The walls of some of these cages are partially removed or replaced with the active part of the guest particle.53,54 In the case of tetra-n-butylammonium bromide (TBAB) (see Fig. 4) salt semi-clathrates, for example, negatively charged anions (Br−) construct a cage structure with the water molecules. This behaviour makes this class of hydrates ionic rather than molecular inclusion compounds. Hydrophobic cations, however, takes a cage filling role by disordering water molecules and occupying the centre of four cages (namely two tetrakaidecahedra and two pentakaidecahedra)55 without H-bonding with the neighbouring water molecules (i.e. hydrophobic inclusion). This, in turn, leads to shaping of a merged cavity composed of several simple cavities, each of which has a hydrocarbon radical that connects to others by water molecules missing from the vertices.20 All the dodecahedral cages are empty and are potential vacancies to be occupied by another guest molecules. Non-volatility in case of exposure to gases such as H2 and CO2, in addition to typical stability of these clathrates at ambient conditions, is a critical feature in the use of these salts in gas storage, as purity of gas phase is not affected after dissociation of semi-clathrate hydrates. Application of these hydrates in different sectors is reviewed below in relevant chapters.
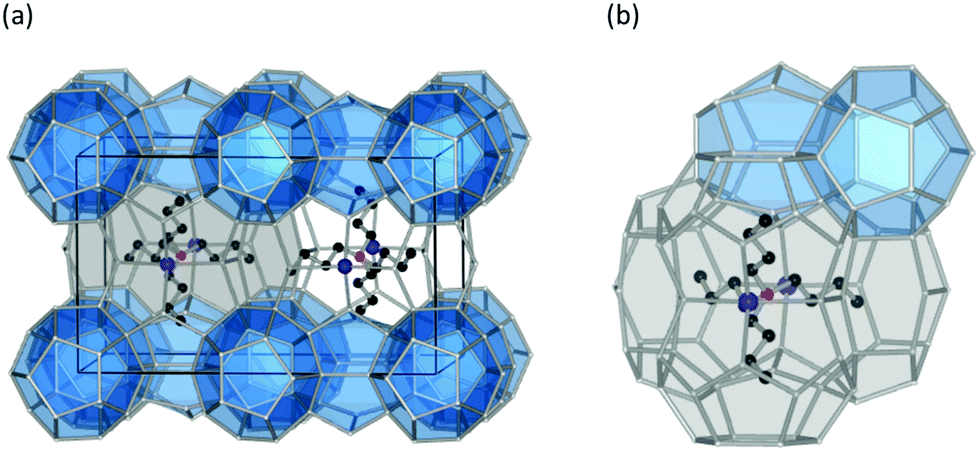 | ||
| Fig. 4 (a) The structure of TBAB hydrate ((C4H9)4N+·Br−·38H2O). The solid lines represent the unit-cell of each hydrate. (b) Magnification of TBAB hydrate structure near tetra-n-butylammonium cation. The blue shaded dodecahedral cages are empty, and as such are candidates for encaging small molecules. | ||
2.1.2.2. The alkylamine hydrates. Amines are organic compounds consisting of a basic N atom with a lone pair of electrons in which one or more of the H atoms is replaced with an aryl or alkyl group, making it different from ammonia. Such amines and amine-related compounds are relevant in a variety of the broad range of anthropogenic uses, including industrial, pharmaceutical, scientific and commercial contexts.56 Of particular importance are the associated risk for release of these compounds into the aquatic system57,58 and potential for amine-based CO2 capture.59,60 Therefore, it is important to understand the processes related to amines in the aquatic environment and the formation of alkylamine hydrate in the presence of water and alkylamines is a key process.
The alkylamine hydrates are another class of little-known stoichiometric semi-clathrate hydrates, except for tert-butylamine,53 which is a clathrate hydrate (see Section 2.1.1). In semi-clathrate alkylamine hydrates, the amine molecule is hydrogen-bonded to the water network, retaining the cage like structure. In contrast to clathrate hydrates, every amine has shown to create different and more complex water framework structures with greater distortions from equal edges and tetrahedral coordination at the vertices compared to that of clathrate hydrates. In the amine hydrate structures, similar to the quaternary ammonium salt hydrates, alkyl chains take the cage-filling role, while the functional group is hydrogen bonded with water latticework.61
Since the early studies of the existence of amine hydrates by Pickering were disclosed in 1893, several workers discovered different physical/chemical properties of these hydrates.62 However, this class of hydrates sees little attention by scientists and there have been limited works on this topic in the last decade. In 2009, Ogata et al.63 detailed the cage occupancy and phase equilibrium relations of mixed H2-trimethylamine clathrates under varying conditions. Following this, several workers investigated various amine hydrate structure transitions when exposed to pressurised CH464–67 or H268 providing significant information about guest–host interactions.
An important, imaginative, and very recent new general trend in hydrate molecular simulation lies in assessing the effects of externally-applied magnetic fields on hydrates by non-equilibrium molecular dynamics (NEMD), including their kinetic properties – which may have profound geophysical implications.80 English and Allen performed such NEMD simulation to show that magnetic fields, including their direction reversals, have important effects on gas-release dynamics from methane hydrates. For a field-polarity switch, there is a sudden increase in the gas release, from effects on rotating water molecules in the hydrate cages due to shifting Lorentz forces.80 Intriguingly, and boldly, it was conjectured that these NEMD-based findings, especially involving switches in field direction, may have a causal link with superchron-related swaps in the Earth's magnetic-field polarity leading to increased methane release into the geosphere.80
2.2. Thermodynamics
As we learned from the previous chapter, gas hydrates are composed of two different kinds of molecules, the host molecules (water), forming the cavities and the guest molecules which are encased into the cavities and stabilize them. Depending on the P–T conditions (see Fig. 5), these components coexist in different phases at equilibrium state. Therefore, development of an efficient thermodynamic route is of significant importance for determining hydrate formation conditions and hydrate compositions of different systems and has a key role in contributing to the varied gas hydrate related industries. For instance, predicting the optimum amount of thermodynamic inhibitor that needs to be added to the gas production stream to stop pipeline blockage by hydrate formation would have been extremely costly without using thermodynamic models. Accordingly, scientific efforts in this area have led to significant advances in thermodynamic modelling.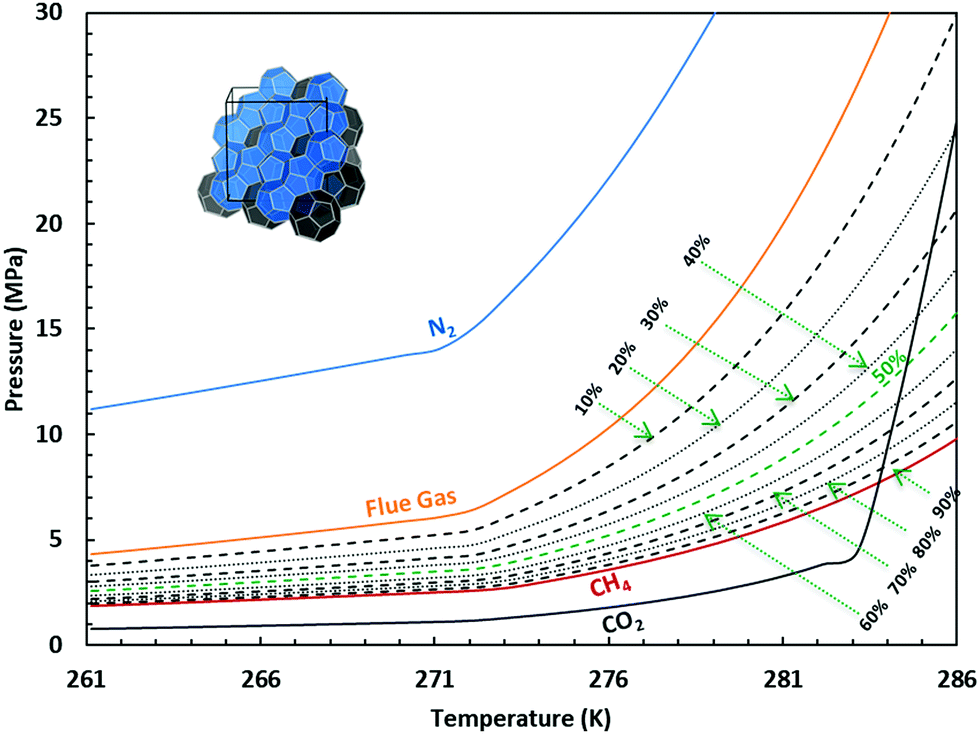 | ||
| Fig. 5 Hydrate stability zones of CO2, N2, CH4, flue gas (14.6% CO2 and 85.4% N2) and mixtures of flue gas with CH4 (dotted lines) as a function of temperature. Increasing the pressure of the system above the predicated phase boundaries could initiate the hydrate formation. Adding gases with higher hydrate stability pressure zones to gases with lower hydrate stability pressure zones, shifts the system phase boundaries to the left (adapted with permission from Hassanpouryouzband et al.,81 Copyright 2019 Springer Nature). | ||
In 1988, Englezos and Bishnoi93 proposed the use of Gibbs minimization of closed system methods to calculate the hydrate fraction and composition in the same way Michelsen94 had done for multiphase flash for liquids and vapour systems. They used the van der Waals and Platteeuw model to calculate the chemical potential of water in hydrate in a similar way to the previously mentioned model. This model was then extended95,96 to multi-hydrate former systems and computationally strengthened. Ballard and Sloan97 proposed another adaptation of general hydrate flash based on Gibbs minimization considering non-ideality about effect of gas adsorption on lattice size. The Gibbs-minimization method has been further improved by such modifications as increasing the speed and robustness of calculations,98–100 implementing different equations of states and activity coefficient models for involving passes,101–108 and etc.86,109–111 to address further limitations of the method. As discussed in the previous section, the presence of ammonium/phosphonium salts also determines the thermodynamic behaviour of the resulting hydrate phase. After the initial neural network approach,112 the first theoretical approach for modelling phase behaviour of semi clathrates was introduced using the statistical associating fluid theory with variable range for electrolytes (SAFT-VRE)113 equation of state for the thermodynamic properties of the liquid phase and (vdW-P) theory combined with the new model for salt hydrates and applied the Gibbs-minimization method under stoichiometric constraints. Following this approach, many workers adjusted tuning parameters of SAFT-VRE for various semi clathrate compositions.114–116 Other approaches such as electrolyte non-random two-liquid (e-NRTL),117 modified Patel–Teja (MPT) EOS118 and electrolyte Cubic-Plus-Association (e-CPA)119 were also later applied for predicting semi clathrates phase equilibria.
In addition to the Gibbs minimization model based on modified van der Waals and Platteeuw (VDW-P), Chen and Guo followed a different approach to predict hydrate dissociation conditions that consists of a new two-step model; (1) quasi-chemical reaction for formation of basic hydrate and (2) adsorption of guest molecules. They considered local stability as well as kinetic mechanisms, which is absent in VDW-P theory.120 In addition to their achievement in extended testing, they obtained more accurate results than VDW-P model in some cases for temperature between 259 & 304 K. However, this model lacks required accuracy over a wide range of pressure, temperature and compositions due to several factors: firstly the authors assumed that all large cavities were completely occupied, which is not in agreement with empirical data and secondly, they tuned the model parameters with the Antoine equation based fugacity functions that proved inaccurate for vapour pressures outside of tuned conditions.121 To address these problems, Klauda and Sandler122 in 2000, applied Quantum mechanical calculations to reduce the number of fitted parameters and remove the assumption of a constant crystal lattice for various guests inside a structure. They also used the quasi-polynomial, QL1123 instead of the Antoine equation for calculation of Langmuir constants, which is accurate beyond the tuned temperature range. Subsequently, they applied their method to single and multi-hydrate former gas mixtures.124 Similar to Gibbs minimization method, there were further modifications on the Chen and Guo method to increase the accuracy and remove the limitations.125–127 Alongside the aforementioned approaches, various neural network algorithms have been reported for phase equilibria of gas hydrates.128–130
In summary, there have been significant advances in predicting hydrate phase equilibria modelling, and there enable us to predict hydrate stability zones, hydrate fractions and compositions plus other derived thermodynamic properties. Despite the major progress in experimental and simulation studies, a series of challenges remain unresolved which offer the opportunity to explore new directions from a thermodynamic modelling perspective. Firstly, current thermodynamic models work reasonably well for those hydrate systems with no or low concentrations of inhibitor; however, when high concentrations of inhibitors were used the errors in prediction increase significantly. Secondly, there are significant errors when calculating hydrate equilibria at high pressures or with very small guest molecules. Thirdly, for systems containing CO2 or H2S in the presence of second guest molecules, uncertainties increase significantly.86
More broadly, in terms of recent sophisticated thermodynamics modelling of cage occupancies, which depends strongly on modelling the subtleties of guest–guest and guest–lattice interactions, Brumby et al. have made impressive progress with Monte Carlo simulations of hydrogen hydrates in the isothermal–isobaric Gibbs ensemble, performing a detailed analysis of cage-occupation distributions.135 In accordance with previous experimental and theoretical studies, they found evidence of very limited double occupancy of small cages, where approximately 0.1% of small cages were doubly occupied at 300 MPa between 225 and 250 K.135
Outside of hydrogen hydrates, where nuclear quantum effects can often be very important, as just discussed, molecular simulation has of course expanded our insights into thermodynamics for other guests. Here, the work of Kvamme et al.136 is important in highlighting how residual thermodynamics and chemical potentials of a variety of guests (with particular focus on methane and CO2, motivated by gas production in marine-hydrate-sediment contexts) and the water-lattice framework can be evaluated from (biased) molecular simulation. This offers the possibility of using molecular simulation as an important prototyping tool for evaluating thermodynamic hydrate-formation and dissociation propensities in a variety of marine-sediment and pipeline settings.
2.3. Thermal properties
A detailed understanding of the underlying mechanisms governing the thermal behaviour of clathrate hydrates, together with accessibility to accurate and reliable thermal property data, makes it possible to enhance simulation scenarios designed to achieve long-term resource recovery and determine the impact of hydrates on climate change.137 The response of clathrate hydrates to a changing thermal environment is governed by the thermal conductivity, thermal diffusivity, specific heat, and enthalpy of formation/dissociation.1382.3.1.1. Thermal conductivity. Distinguished from other molecular crystals, clathrate hydrates generally exhibit an anomalous thermal behaviour in natural systems with a glass-like temperature dependence (positive slope) resembling amorphous solids.139–142 Hydrates have been observed to show a glass-like temperature dependence in thermal conductivity above the Debye temperature, similar to some clathrate-like compounds,143–145 and a crystal-like behaviour below.146,147 This intriguing behaviour is attributed to the interactions between localized low-frequency vibrations of the guest molecules with the acoustic phonons of the host lattice.148–156 Pressure dependence of the thermal conductivity has also been studied by researchers, suggesting a weak direct proportionality.157–162 Despite similarities on the molecular level and the other physical properties, thermal conductivity of clathrates hydrates has been found to be markedly lower than ice Ih.23,163–165 Such behaviour arises also from larger anharmonicities in the intermolecular interactions when compared with ice.148 Thermal conductivity measurements recently conducted on some semi-clathrate hydrates, however, revealed a weak negative temperature dependence, demonstrating the crystal heat transmission characteristics in semi-clathrate hydrates.166,167
Transient hot-wire140,158,159,161,166,168,169 and transient plane source162,164,167,170–174 techniques are the most widely used methods for measurement of the thermal conductivity of clathrate hydrates. Some other techniques, such as the steady-state potentiometric method141,146,147,175 and guarded hot-plate method,163,176 are also sometimes used to measure the thermal conductivity of hydrates. The main challenge associated with the laboratory measurement of the gas hydrates thermal conductivity is that the gas hydrate samples are usually porous and have free water/ice and gas which could impact the quality of the experimental data as the measured thermal conductivity includes the thermal contact resistance (TCR) due to the unavoidable imperfect contact.146,147,164 Recently, however, an experimental system was introduced using the modified freestanding 3ω method which is able to reconstruct the intrinsic thermal properties (thermal conductivity and diffusivity) of clathrate hydrates.177
2.3.1.2. Thermal diffusivity. While thermal conductivity is a measure of the ability of a material to conduct heat thermal diffusivity is the thermal inertia of the material.161 A new experimental configuration based on the approximation solution of the Navier–Stokes heat equation was first developed by Turner et al. in order to measure the thermal diffusivity of clathrate hydrates.178,179 Waite et al. used the infinite line source formulation of Carslaw and Jaeger for simultaneous determination of thermal conductivity and thermal diffusivity using the transient hot-wire method.161,168 Transient place source technique has also used to measure the thermal diffusivity of methane hydrate.162,173
2.3.1.3. Calorimetric studies. The heat stored in or extracted from a material due to a temperature change can be quantified by the heat capacity.138 Clathrate hydrates heat capacity data can provide some information about the motion of encaged guest molecules or reordering of the guest and host.180 Enthalpy data is also necessary as a key thermal property for a realistic evaluation of the recovery schemes proposed for exploitation of natural gas hydrate-bearing sediments.181 Several experimental studies have been conducted to measure the heat capacity142,180–189 and enthalpies of formation/dissociation180,181,183,184,186,188,190–198 of clathrate hydrates, the majority of which use the heat-flow calorimeter180,181,183,184,186,191,192,194 and differential-scanning calorimeter.188,190,193,195,197–199
Experimental measurement of gas hydrate heat capacity is always associated with two major challenges: (1) due to strong dependence of the vapour pressure of gas hydrates on the temperature, increasing the system temperature results in dissociation of the hydrates and consequently renders the apparent heat capacity much higher than the actual value; (2) the presence of free hydrate former species (especially free water) could greatly influence the measured heat capacity of the hydrate.188 Enthalpies of formation/dissociation are also determined indirectly by using the P–T diagram of the system and Clapeyron equation or its Calussius–Clapeyron approximation.198,200 However, this method has limited application when used for semi-clathrate hydrates.199,201 A detailed overview of the calculation of the dissociation enthalpy of methane hydrates can be found elsewhere.202
2.3.1.4. Perspectives on molecular modelling. Underlying mechanisms of the heat conduction in clathrate hydrates can now be explored via molecular simulation thanks to recent advances in computational power. In molecular simulation, thermal conductivity can be estimated by both equilibrium and non-equilibrium Molecular Dynamics (MD)69 (see Fig. 6). A review on the contribution of molecular simulations to our detailed theoretical understanding of mechanisms of thermal conduction in clathrate hydrates can be found elsewhere.69,137 Recently, MD was employed to further investigate the thermal behaviour of the clathrate hydrates,203–206 particularly to evaluate the influence of guest occupancy ratios on the thermal performance of gas hydrates, suggesting an improved thermal conduction by the inclusion of more guest molecules in the cage.207–209 The MD approach was also utilised to study the calorimetric properties of clathrate hydrates including the heat capacity210 and endothermic dissociation process of clathrate hydrates.202,211–213 However, the dependence of the experimental results on the samples’ nature and quality together with the quality of the potential models used in molecular simulation and the system size and electrostatics render difficult a direct and quantitative comparison of theoretical and experimental values.69 This clearly makes it necessary to seek novel and more suitable macroscopic experimental techniques to minimize the effect of the external/internal factors (confining pressure, temperature, residual water, gas or ice, presence of micropores,…) to obtain a well-defined sample whose thermal properties reflect those of pure hydrates.214 Apart from challenges encountered in macroscale studies, there are still a number of technical issues that need to be addressed associated with the molecular modelling required to identify key microscopic mechanisms controlling the thermal properties. This would lead to improved reproducibility of the thermal properties already measured precisely in the laboratory. Integrating the experimental and theoretical studies would enable us to elucidate the thermal behaviour of hydrates at different conditions, especially those difficult or even impossible to achieve experimentally such as extremely low temperature or high pressure conditions. Such integration can be of particular interest when investigating the evolution of the thermal properties of hydrates during various processes such as formation and dissociation.215 The importance of the thermal properties in various hydrate-based applications such as CCS, hydrogen storage, desalination and gas separation, and the key role of the molecular-level mechanisms in controlling the thermal conductivity of hydrates necessitate further experimental studies integrated with molecular simulations to shed light on the thermal behaviour of hydrates.
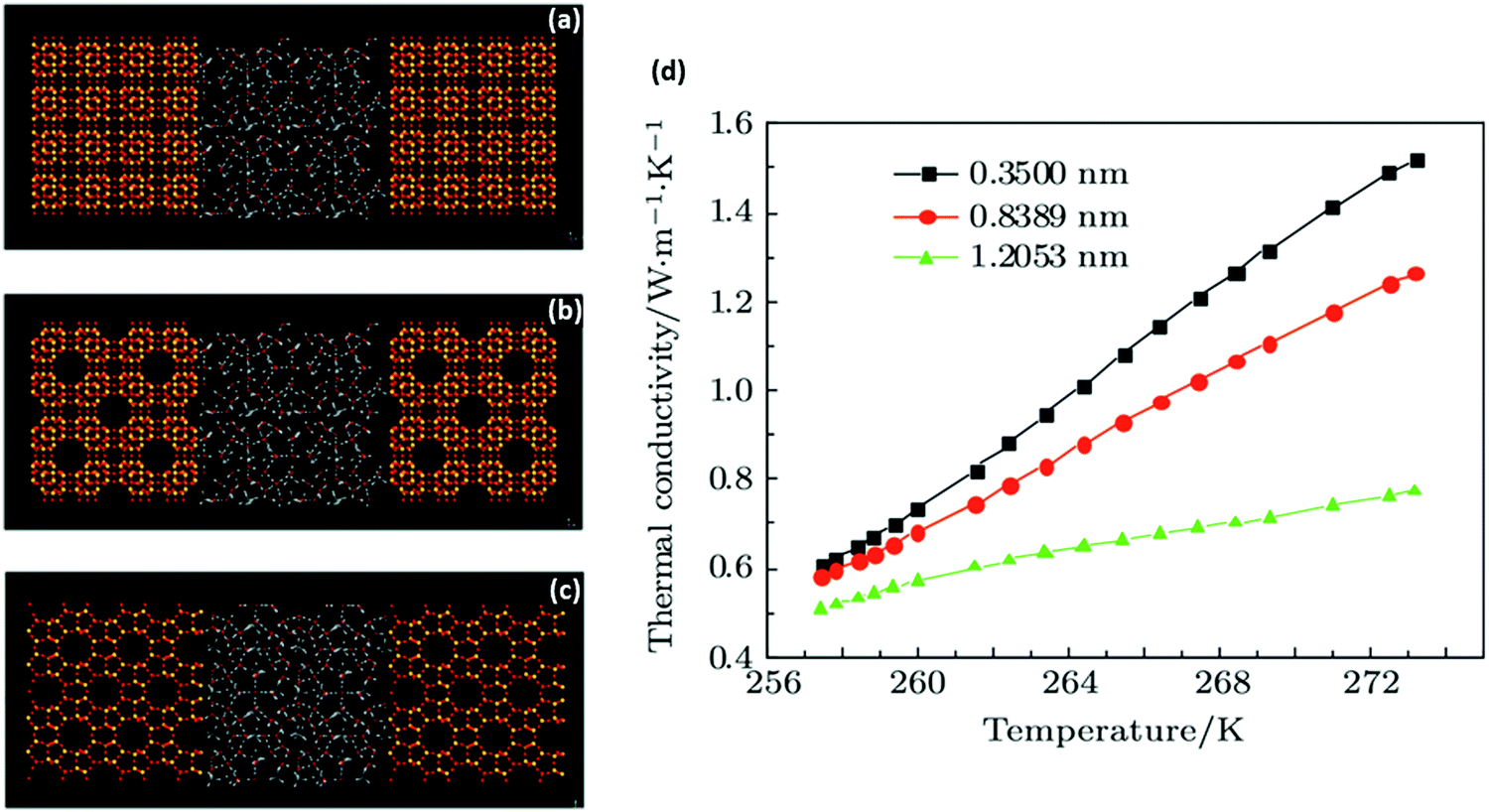 | ||
| Fig. 6 Molecular-dynamics simulation of thermal conduction in methane hydrate–SiO2 porous media: (a–c) snapshot of the final structure of hydrate + porous media simulation system with different pore sizes (a – 0.35, b – 0.8389, c – 1.2053 nm); (d) simulated thermal conductivity values vs. temperature. As observed, the thermal conductivity of the system increases with temperature increase. At a certain temperature, the thermal conductivity further increases as the pore size reduces due to improvement of the SiO2 surface (with higher thermal conductivity) and the micro-energy transfer associated with hydrate–SiO2 at lower pore sizes (adapted with permission from Guo et al.,216 Copyright 2017 Institute of Physics). | ||
Laboratory measurements of the effective thermal conductivity of hydrate-bearing sediments are usually conducted using transient hot-wire169,219,226,227 and transient plane source172,174,220,228–232 techniques. Transient hot-wire technique, however, is not easily adapted for in situ measurement where sample penetration is difficult. This makes the transient plane source technique more conducive towards adaptation for field use.137 Recently, a thermistor-based method combined with Micro-CT observations was employed to further investigate the effect of saturation and spatial distribution of co-existing phases on the effective thermal conductivity of HBS.233–236
Development of accurate predictive models for the effective thermal conductivity of composite materials such as HBS comprises an important portion of the literature about heat transfer in porous media. However, a unified model or prediction procedure with universal applicability has not been found yet.237 Based on their principles, the existing predictive models can be categorized into Mixing Models, Empirical Models, Mathematical Models, Volume Fraction Models, Packing Structure Models, and Pressure-dependent Models. A detailed review of the existing models can be found elsewhere.238
There are few published data on the effective thermal diffusivity of gas hydrate-sand/sediment mixtures.161,178,228 Gas hydrate-bearing sediments can change temperature more rapidly than hydrate-free sediments as the thermal diffusivity of methane hydrate is more than twice that of water.138 Hydrate should therefore be accounted for in transient heat flow applications such as safety assessments for drilling into or through hydrate-bearing sediments.
Unlike the effective thermal diffusivity and conductivity, the heat capacity of HBS depends only on the mass fractions of sediment, hydrate, and pore fluids rather than on their pore-scale distribution and interfacial effects.138 Therefore, hydrate formation can significantly lower the specific heat of sediments.
2.4. Electromagnetic properties
The electromagnetic characteristics of a material including the steady state charge migration under an electric field, polarization and magnetization can be quantitatively expressed by the Electrical conductivity (σ), magnetic permittivity (κ*) and magnetic permeability (μ*).138 Electrical measurements have been widely used as a primary method to detect the spatial distribution of hydrate in natural sediments.239–245 They can also be employed to monitor the formation/dissociation of clathrate hydrates or even to evaluate the performance of hydrate inhibitors.240,246,247 When transformed from an aqueous phase system to pure hydrate, electrical conductivity decreases, meaning that this property can be used to detect hydrate formation/dissociation.247,248 The ionic concentration in solution has a primary effect on resistivity and a second-order effect on magnetic permittivity, hence permittivity can be used as a more reliable parameter to estimate water saturation and extract volumetric hydrate saturation in a multiphase hydrate bearing system as well. For instance, the electrical resistivity logs acquired from natural gas hydrate bearing sediments confirm the presence of less conductive hydrate bearing zones relative to the water saturated regions.249–255 The magnetic permeability, however, is usually considered to be unity for pure hydrates and hydrate bearing sediments as these materials are generally non-ferromagnetic.138 In addition to the electromagnetic properties, dielectric measurements have also been employed to quantify gas hydrate saturation in both laboratory and field studies.256–258Several fields and laboratory studies have been conducted to measure the electrical conductivity of hydrate bearing sediments.283–290 Given that various factors such as the ionic concentration of the aqueous solution, gas exchange and fluid-filling porosity of the pores affect the electromagnetic properties of gas hydrate bearing sediments,291 the electrical conductivity of porous polycrystalline methane hydrates in mixtures with brine and sand was recently studied via in situ impedance measurement to gain insights regarding the petrophysical relations between methane hydrates, brine salinity and the host sediment.292,293 Several possible conduction mechanisms were also determined to correlate resistivity data with methane hydrate saturations, information that can be used to improve the reliability of existing and new electrical models. The models available in the literature have been mainly developed according to the rock-physics models.294–296 As such models cannot sufficiently account for the spatial arrangement of hydrates, effort focuses on addressing this shortcoming using the finite-element method in order to simulate the electrical characteristics of hydrate bearing specimens reconstructed by different methods such as the diffusion limited aggregation (DLA) model.297–299 In addition, electrical resistivity tomography (ERT) has been shown as promising in characterizing electrical properties of hydrate bearing sediments.300,301
The magnetic permittivity is expressed as a complex number to account for its magnitude as well as its phase relative to the excitation. In hydrate studies, however, the real component of the permittivity is mainly investigated given the typical small contribution due to polarization losses (represented by the imaginary component) in the operating frequencies ranging between Hz and kHz.138 As discussed earlier, the magnetic permittivity is influenced by geometric and spatial effects. Some models proposed for estimation of the effective magnetic permittivity of hydrate bearing sediments include the volumetric linear and quadratic methods.
2.5. Mechanical properties
Understanding of the mechanical behaviour of clathrate hydrates and the internal mechanisms of their deformation as well as their interaction with the host sediment is essential in gas production from natural gas-hydrate-bearing sediments, environmental and climate impact studies, hydrogen storage and hydrate technology applications. Similar to the thermal properties, it has been revealed that the mechanical properties of clathrate hydrates are different from those of ice Ih.302,3032.5.1.1. Indirect determination of the mechanical properties. Indirect methods such as acoustic measurements were initially used to infer the mechanical properties of pure clathrate hydrates. For these studies, elastic wave (compressional and shear wave) velocities were measured using different techniques such as the ultrasonic pulse transmission method302,303,308,312–316 and Brillouin spectroscopy method;317–321 the results were then used to calculate the parameters related to elasticity mechanics such as elastic moduli (bulk, shear and Young's moduli) and Poisson's ratio.214 X-ray diffraction, neutron diffraction and Raman spectroscopy techniques are also utilised to calculate the isothermal bulk modulus by measuring the unit cell volume as a function of pressure.322–329
Experimental studies suggest that the elastic properties of pure clathrate hydrates depend upon the hydrate composition and structure, the guest molecule, and cage occupancy.214,312,318 Increasing the system temperature results in a reduced bulk modulus and Poisson's ratio for clathrate hydrates and consequently, a lower compressional wave velocity, similar to observations for ice Ih. However, the Young's and shear moduli were observed to vary depending on the guest molecule and even show anomalous behaviour.214,217,302,303,313,319,330 For different pressures, bulk modulus and Poisson's ratio of clathrate hydrates increase with an increase in the pressure while the Young's, and shear moduli variations strongly depend on the guest molecule.214,302,323,330,331
2.5.1.2. Direct determination of the mechanical properties. Influenced by the aforementioned sources of uncertainty, the strength of pure clathrate hydrates was initially believed to be similar to ice Ih.214,332 However, laboratory constant-strain-rate experiments conducted on poly-crystalline methane hydrate specimens suggested a different stress–strain behaviour with an extraordinary strength (20 to 40-fold higher) compared with ice, which could be attributed to dislocation movement and molecular diffusion.214,306,333–335 Unlike ice Ih which typically displays an ultimate yield strength followed by relaxation to steady-state behaviour, methane hydrate exhibits an extensive strain hardening followed by strain softening during compression.306,335,336 In fact, the methane hydrate stress–strain curve can be divided into two stages: (i) the rapid structural damage stage and (ii) the complete structural damage stage.337 Interestingly, methane hydrates undergo partial decomposition during deformation due to a solid state disproportionation or exsolution process, even well within the stability zone.307,334 Studies of the creep behaviour of methane hydrates suggest direct proportionality of axial and creep strains with external load.335,338
The mechanical strength of pure clathrate hydrates was experimentally shown to be influenced by the system temperature, confining pressure, strain rate and density.214 More specifically, the compressive and shear strength of methane hydrates increase with increasing confining pressure, strain rate and density.339–342 The deviatoric stress increases with an increase in the strain rate at confining pressures less than 10 MPa, while at higher values there is no obvious change in the deviatoric stress.343 Methane hydrate strength was also observed to be very sensitive to temperature, with lower temperatures leading to higher the strength.341,342 When well within the stability zone, the compressive strength of hydrates is higher than that of ice Ih; however, the strengths become closer in value when hydrate is less supercooled (relative to the hydrate phase boundary).340,341 The essential mechanisms causing the difference in the mechanical properties of clathrate hydrates and ice is underpinned by the special hydrate lattice structure and the host, guest and host–guest interactions.214 In the small strain regions (<1.5%), the stress–strain behaviour is generally not influenced by the confining pressure and temperature; however, within at the whole strain region, the mechanical behaviour is markedly influenced by the strain rate.344
2.5.1.3. Theoretical studies. The stringent high pressure–low temperature conditions required for hydrate formation and stability make the direct measurement of the mechanical properties and deformation mechanisms of pure hydrates difficult using common experimental techniques. This in turn, results in poor accuracy for values for the mechanical properties, and the extrapolation of these values can be controversial controversial.214 The rapid progress of modern computer technology together with the unavoidable uncertainties associated with the experimental studies means models are the most promising alternative for providing insights into the mechanical behaviour of pure clathrate hydrates.214
Theoretical approaches such as Density-Functional Theory (DFT),345–352 Molecular Dynamics (MD)336,353–363 and Lattice Dynamics (LD) simulations364–367 have been used to probe the mechanical behaviour of clathrate hydrates, with links to macroscopic phenomena. They have also been employed to deliver insights into problems not well understood by experiments and to validate the experimental results. These models also help elucidate the underlying mechanisms controlling the mechanical properties of clathrate hydrates and to explore why the mechanical behaviour of the clathrate hydrates differ from ice Ih.358
Recently, DFT was successfully employed to investigate the ideal strength of methane hydrates under uniaxial, triaxial, and shear deformation modes and to compare its mechanical behaviour with that of ice Ih.366 The effect of guest molecule size on the mechanical properties of a number of hydrates was also investigated using DFT, suggesting a close relationship between the shear modulus, wave velocity and the level of anisotropy in the hydrate lattice, which itself is a function of guest size.346,347,368 More recently, a DFT study compared the mechanical and vibrational properties of tetrahydrofuran (THF) hydrates with natural gas hydrates, providing significant insights into the accuracy of using THF hydrates as an analogue for natural gas hydrates.349
MD simulations have assisted with the construction of theoretical stress–strain curves for pure clathrate hydrates for different structures and at different pressures and temperatures and led to the determination of such mechanical properties as Poisson's ratio, elastic moduli and strength and identification of fracture initiation process.353,354,357,359,361 The origin of strain hardening in methane hydrates under compressive deformation was also investigated using MD simulations. The simulation results highlight the role of the guest molecules as non-deformable units preventing the failure of hydrate structures and thus leading to the strain-hardening phenomenon.336 MD simulations also revealed that the temperature dependence of the elastic moduli of pure hydrates is dominated by the guest molecule, a phenomenon of particular importance in hydrate-based applications such as carbon capture and storage where CH4 is replaced by CO2.354 The role of guest molecules on the mechanical properties of hydrates was further explored with the aid of tension MD simulations.363 The results indicated that the tensile strength and Young's modulus of hydrates are influenced not only by the type, size and shape of guest molecules but also on its polarity. Of particular interest, MD simulations helped with shedding light on the mechanical instability of monocrystalline and polycrystalline methane hydrates, providing molecular insights regarding destabilising mechanisms of gas hydrates under mechanical loading and their grain-boundary structures353 (see Fig. 7). Moreover, it was shown that polycrystalline hydrates under compression and tension exhibit grain size strengthening at low grain sizes and grain size weakening at larger grain sizes. The intrinsic differences in the mechanical properties of monocrystalline methane hydrate monocrystalline ice Ih were also explored using MD simulations. The simulation results suggest these differences could be due to the host–guest molecule interactions and relative angles which tetrahedral hydrogen bonds make to the loading direction.358 Microscopic insights offered by the molecular modelling efforts could establish the fundamental understanding of the mechanical responses of naturally occurring and artificial synthetic gas hydrates. However, there is a gap in the knowledge as most of the studies have focused on qualitative behaviours and mechanisms and are yet to fit these findings to laws to achieve upscaling.
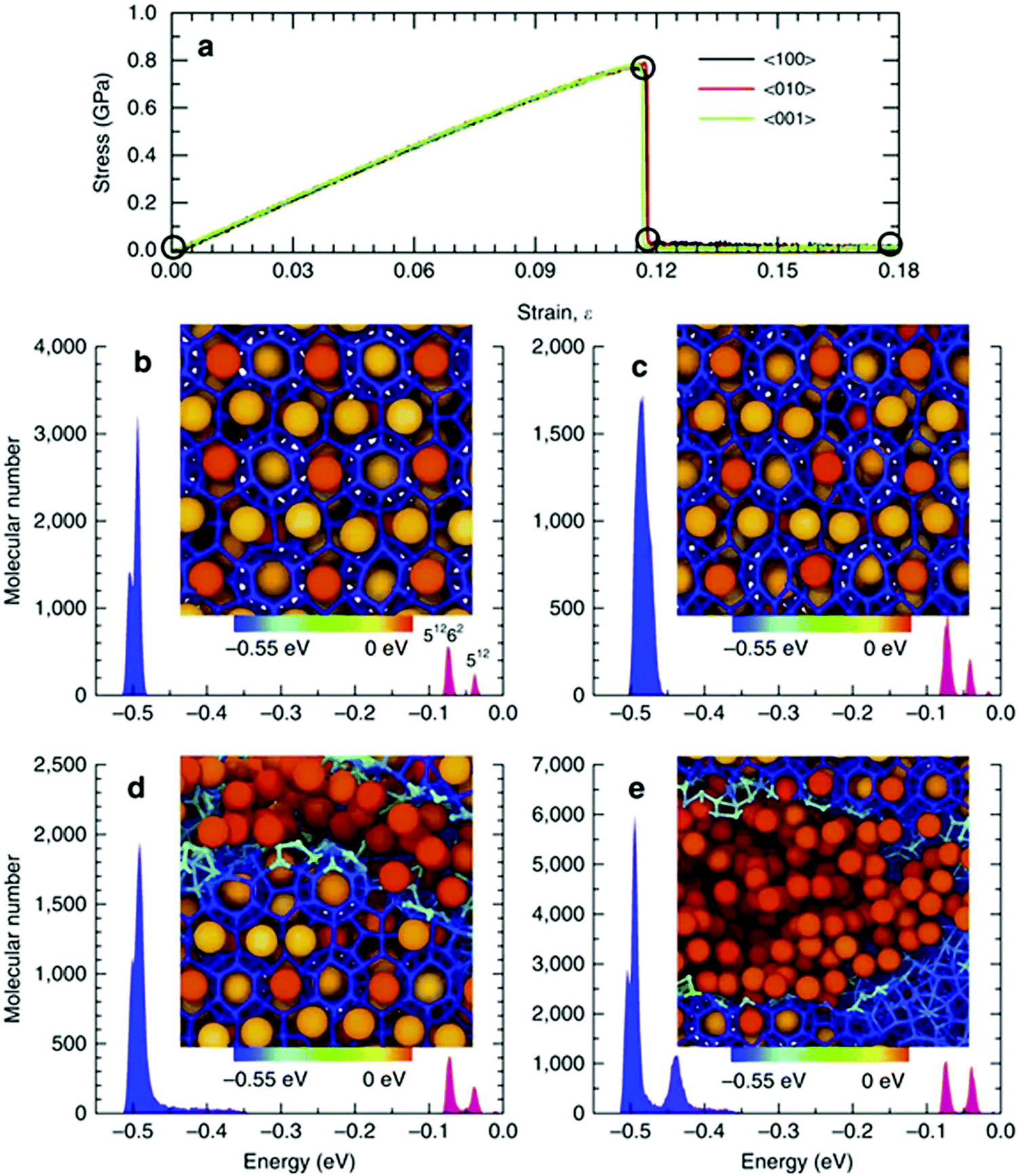 | ||
| Fig. 7 (a) Stress–strain relationships for single-crystal methane hydrate. The molecular cohesive energy distributions and corresponding localized molecular structures (b) initially, (c) at the onset of fracture, (d) immediately after initial fracture and (e) at a strain of 0.18 (reprinted with permission from Wu et al.,353 Copyright 2015 Springer Nature). | ||
The effects of hydrates on the mechanical properties of the host sediment properties strongly depend on where hydrates nucleate and grow in pore space. The pore-scale habit of hydrates is determined primarily by the state of effective stress and host sediment grain size (Fig. 8).400 For coarse-grained natural HBS, when excess water presents in the system, the main pore-scale habit is preferred to be pore-filling,401 whereby hydrates nucleate on the sediment grain boundaries and grow freely into the pore spaces without bridging two or more particles together. Moreover, since gas hydrate is suspended in pore fluid, it primarily alters the pore fluid bulk stiffness, fluid conduction properties and bulk density of the sediment.402 At saturations generally more than 40%, pore-filling hydrates turn into load-bearing, where hydrates bridge neighbouring grains and become a part of the skeleton and accordingly, contribute to the mechanical stability of the sediment. For these two pore-scale habits, there is always a water film remaining on the grain surface. When excess gas presents in the system, grain-coating habit is preferred by hydrates, whereby they form a coating around grains. When there is enough free gas in the system, the grain coatings can coalesce to cement intergranular contacts and even use up remaining water.403 Pore-scale habit for fine-grained HBS is different from coarse-grained sediments, mainly due to smaller pore-size and higher specific surface. While capillarity acts as a hindrance for hydrate nucleation, hydrate growth can displace sediment grains, leading to particle-displacive segregated morphology, e.g. lenses, nodules, chunks and veins.369 A comprehensive discussion regarding the pore-scale habit of HBS can be found elsewhere (Fig. 8).403
It must be noted that laboratory-formed HBS samples are more often used to study properties of HBS;404 however, they are not necessarily representative of naturally occurring HBS samples, particularly in pore-scale habits and physical properties. In recent years, the advancement of pressure core acquisition and analysis technology has enabled scholars to image and interrogate pressure cores to study hydrate habits of nearly intact naturally occurring HBS preserved at their in situ pore pressures.405,406 Post-recovery analytical capabilities for pressure core samples have also allowed for reliable and systematic measurement of physical properties.407
 | ||
| Fig. 8 Core-scale hydrate morphology in HBS samples and conceptual pore-scale habits for fine-grained and coarse-grained sediments (reprinted with permission from Ren et al.,403 Copyright 2020 Elsevier Ltd). | ||
2.5.2.1. Fluid flow and permeability characteristics. Permeability is a measure of the ability of a porous medium to allow fluids to pass through it, and the relationship between fluid flow and permeability controls both fluid-flow pathways and the accumulation, distribution and saturation of gas hydrates.138 This is particularly important for gas transport, production or migration into the oceanic environment.408 Apart from the mineralogy, shape and packing arrangement of the grains and the size (specific surface area) and interconnectivity (tortuosity) of the pores, the presence of gas hydrates adds additional complexities because the spatial distribution of hydrates can alter the pore size, shape and interconnectivity and accordingly the permeability of sediments.138 To date, numerous macro- and micro-scale experimental studies have been conducted to investigate fluid flow through hydrate bearing porous sediments and explore the link between the permeability evolution and hydrodynamics of gas-hydrate systems.401,409–424 A review regarding the experimental techniques for investigating the permeability properties of hydrate-bearing sediments can be found elsewhere.425
Furthermore, several numerical simulation studies have been conducted to elucidate the geological processes associated with hydrate formation and dissociation in porous media under a wide range of conditions.426–441 Studies also to predict the behaviour of gas hydrate bearing sediments during gas production,442–449 a process for which the absolute and relative permeability values and relationships are crucial given their influence on gas and water production rates. A review regarding recently developed relative permeability models can be found elsewhere.381In situ observations of pore structures using X-ray microCT and Nuclear Magnetic Resonance Imaging (MRI) can significantly improve our pore-scale understanding of the permeability characteristics of gas hydrate bearing sediments as a function of hydrate saturation.406,450–458 A review regarding the analytical and empirical correlations used for determination of permeability of hydrate bearing sediments has been published by Joseph et al.459
Fines, which commonly coexist with sediments that host natural gas hydrates, can be readily mobilized by water and/or gas flow during gas production.460 Fines migration inherently involves the permeability impairment associated with the generation, movement, and retention of sub 100 μm solid particles in porous media.461 Several experimental and numerical studies have recently been conducted to further understanding of the contribution to permeability evolution due to fines that are mobilized during gas production.460,462–467
To date, an accountable number of studies have been undertaken to characterize fluid flow in sediments containing hydrates given their vital role in exploration and exploitation of natural gas hydrate as well as assessment of the impacts due to the hydrate dissociation on submarine instabilities, marine ecosystem and global climate change. However, there exist several challenges mainly due to the difference between the simulated HBS samples, core samples and natural ones, leading to the discrepancies between the measured/estimated permeability and the real values.403 The main challenges in lab-scale studies include difficulties associated with the HBS sample size and methods followed to form hydrates within the host sediment and maintain the equilibrium conditions during the test process (to avoid hydrate formation or dissociation).401,462,468 On the other hand, the flow test method (Steady State/Unsteady State) adds more uncertainties since these methods are usually time-consuming and inherently rely on assumptions far from the real conditions governing HBS occurrences.469 This makes it essential to address the lab challenges in accordance with the flow test methods. Of particular importance is the pore-sale habit of hydrates in artificial samples which is predominantly controlled by the hydrate formation method. The pore-scale habit in conjunction with the pore-structure, anisotropy and heterogeneity influence the permeability characteristics of HBS,470 which makes it challenging to synthesize artificial samples imitating natural ones. Thus, to bridge the gap of measured permeability between laboratory samples and natural sediments, it is of utmost priority to upgrade the existing methods and apparatuses or even develop new measurement methods. Challenges encountered in theoretical analysis and numerical simulations are mainly due to several simplifications such as assuming homogeneous and isotropic reservoir/porous medium where hydrates are uniformly distributed with a predetermined pore-scale habit. Insights provided by the lab scale studies are required to be considered in numerical simulations to be able to make reliable permeability predictions. Further discussion in this regard can be found elsewhere.403
2.5.2.2. Geophysical properties. Geophysical studies are essential in assessing natural gas hydrate bearing sediments and are extensively used for detection, mapping and characterization of gas hydrates occurrence471 and tracking their formation/dissociation process within sediments472 given that hydrates have much higher elastic moduli than the pore fluids and alter the stiffness of the pore fluid and host sediment.473 Hydrate formation has an impact on the skeletal stiffness of host sediment, enlarging contacts between grains, which in turn, results in the reinforcement of the bulk and shear moduli. Accordingly, hydrate bearing sediments show elevated elastic wave velocities depending on the saturation and pore-scale habit of the hydrates within the sediment framework.265 However, the strong dependence of the elastic moduli of hydrate bearing sediments on the pore-scale habit of the hydrates may cause ambiguity when inferring the hydrate saturations from measured velocities.474,475 A review in this regard can be found elsewhere.138
A tremendous number of field-scale geophysical studies have been undertaken in North America,476–482 Asia,245,250,254,483–490 Europe,491,492 Africa493–496 and Oceania497–502 in order to understand the natural occurrences of gas hydrates and quantify the extent and distribution of gas hydrates within the sediments for exploration purposes. Seismic techniques are the most commonly used methods for detecting gas hydrate occurrences, particularly in the marine environment, where they are employed to identify Bottom-Simulating Reflections (BSR).503 BSRs can be observed at a depths of up to several hundred meters below the seafloor in continental margin sedimentary sections and are a seismic reflection likely to be caused by the elastic velocity contrast between the overlying gas hydrate-bearing sediments and the underlying gas saturated sediments.504 The BSR is the main identifier for the presence of gas hydrates, corresponding to the deepest level at which natural gas hydrates are stable. BSRs can be either continuous, discontinuous, or plumbing.505,506 However, sediments can contain gas hydrates without having a BSR, particularly if hydrate is not present near the phase boundary due to the nature of the sediments there or insufficient methane for hydrate to form directly above the free gas phase.507 Elastic wave velocities combined with amplitude variation with offset (AVO) and amplitude variation with angle (AVA) data from BSRs have been used to estimate associated gas hydrate and free-gas concentrations as well as infer the distribution of natural gas hydrate bearing sediments.508–516 Vertical seismic profiling surveys (VSP) are also used along with downhole log data to evaluate the effect of gas hydrates on the elastic velocity of hydrate bearing sediments.517–519
Laboratory analyses of natural or artificial gas hydrate bearing specimens have been conducted to study the impact of hydrates on the elastic wave velocities of different types of sediments. Laboratory studies confirm the significant impact of hydrates where increasing gas hydrates within the host sediments result in elevation of the elastic wave velocities.284,520–528 However, the method of synthesizing gas hydrates in sediment substantially affects the pore-scale habit of hydrates.138,284,529 Given the recent interest in permanent hydrate-based storage of CO2 in geological formations, seismic survey methods could be used to remotely monitor the CH4–CO2 replacement process and evaluate the stability of the host sediment.530–532
Several models have also been developed to establish a relationship between the elastic moduli and the gas hydrate saturation in order to quantify the amount of gas hydrates accumulated in the sediments and predict HBS physical properties.533 These models consider the elastic properties of sediments, pore fluid(s) and gas hydrates534 and generally can be categorized into Time-average equations, Weighted-average equations, Cementation theory-based models, and Effective Medium Theory (EMT) models. Detailed reviews of these can be found elsewhere.402,535–537 A number of theoretical and numerical approaches have also been developed to detect and quantify gas hydrates in submarine and permafrost regions to give more insights into elastic wave attenuation mechanisms.538–540
2.5.2.3. Geomechanical characteristics. The strength of a hydrate bearing sediment subjected to a principal effective stresses (axial (σ1′) and confining stress (σ3′)) at a specific pore P–T is expressed by the cohesive resistance (c) and frictional resistance and described by the friction angle (φ). These properties can be obtained by applying the Coulomb failure criterion which relates the shear stress at failure (τf) to the normal effective stress acting on the failure plane (σn′) in the Mohr–Coulomb failure diagram.138 The strength parameters together with the other geomechanical properties such as Young's modulus (E), Poisson's ratio (ν) and dilatancy angle (ψ) are measured using the triaxial compression tests.381 Recently, numerous triaxial experiments have been carried out to determine the geomechanical properties of natural and synthetic gas hydrate bearing sediment samples at high pressure and low temperature conditions.386,389,391,541–558 Shear strength of specimens at specific pore pressures and temperatures with a given hydrate saturation could be tested under undrained (CU) or drained (CD) modes after consolidation, a detailed discussion of this can be found elsewhere.138 Given the fact that the geomechanical properties of sediment predominantly depends upon the grain type, shape, packing, fines content and degree of consolidation,559,560 the presence of hydrates within sediment generally results in bridging/binding of sediment grains and consequently a higher stiffness, pre-failure dilation, and strength whereas the friction angle has been interestingly observed to remain constant with increasing the hydrate saturation.381
The pore-scale habit of hydrates can markedly affect the strength evolution of the host sediment, particularly at lower saturations (<30.0%).561 According to recent experimental studies, there is a critical saturation at which the local growth of hydrates and/or their extension across adjacent grains result in creating hydrate networks or frame structures throughout the specimens and accordingly substantial enhancement of the cohesive resistance, normalized stiffness and volumetric dilation as well as switchover of the stress–strain response from strain-hardening to strain-softening.387,393,542,562–564 This critical saturation ranging from 15.0 to 50.0% is essentially affected by the spatial distribution of hydrates in the sediment matrix and requires to be taken into consideration to avoid the formation of geomechanical instabilities associated with the hydrate dissociation when predicting the response of gas hydrate bearing sediments to different external stimuli such as mechanical loading and P–T variations during exploitation. At higher saturations (>80.0%), however, the sediment grains will be cemented by hydrates exhibiting a monolithic system with minimal host sediment characteristics.381 The presence of occluded pores within the sediment makes the measurement of the pore pressure difficult, hence interpretations at these saturations are usually based on the total stress, not the effective stress. Further details of this have been provided elsewhere.381
Confining stress (σ3) and system temperature also affect the geomechanical behaviour of hydrate bearing sediments. At a given hydrate saturation, higher confining stresses result in higher interlocking and crushing of grains and accordingly overall increase in shear strength and stiffness, reduction in Poisson's ratio and dilatancy angle and switchover of the stress–strain response from strain-hardening to strain-softening.565–567 Temperature reduction results in higher stability of hydrates within the host sediment and a higher shear strength. Several experimental studies have been conducted recently to explore the stress–strain response of hydrate bearing sediments as a function of hydrate dissociation. This process is associated with significant changes in the matrix of gas hydrate bearing sediments such as an increase in pore pressure under undrained condition or fluid flow under drained condition, secondary hydrate/ice formation and loosening of grains.568–577 Creep tests have recently been conducted on hydrate bearing specimens which are particularly relevant given the importance of predicting the long-term stability of gas hydrate bearing sediments in Arctic and permafrost regions.578–580 More recently, the geomechanical characteristics of gas hydrate bearing frozen sediments have been experimentally studied to investigate the influence of gas hydrates and ice coexistence on the geomechanical strength of the sediment.581–583
The geomechanical behaviour of gas hydrate bearing sediments has also been simulated mainly via applying the Mohr–Coulomb, Cam-clay, Duncan–Chang and critical-state models. These geomechanical models available in the literature have been employed to capture the effect of hydrate saturation and spatial distribution on the sediment strength, stiffness and dilation characteristics.584–590 The geomechanical models are also increasingly included in numerical reservoir simulators to investigate the coupled geomechanical response of hydrate reservoirs to different external stimuli.382,575,591–597 Additionally, the Discrete Element Method (DEM) has been extensively utilised for the simulation of the mechanical behaviour of hydrate bearing sediments under triaxial compression.598–609 More recently, some micromechanical models have been proposed for hydrate bearing sediments whereby the hydrates are represented as solid particles positioned between sand particles and contributing to the skeleton response even for small strains.610,611
2.6. Rheological properties of hydrate slurries
Rheology studies on hydrates have provided insights into the rheological properties of hydrate-laden suspensions, which may be found in oil and gas transportation pipeline networks612,613 during and after hydrate formation, in order to improve hydrate flow assurance strategies.614,615 Investigation of the viscosity alteration of gas hydrate slurries due to presence of hydrate particles and aggregates in pipelines is essential in flow assurance studies. This enables the development of a reliable model capable of describing hydrate-laden flow characteristics in pipelines to reliably determine the extent to which hydrates may cause flow hindrance.616,617 Recently there has been a paradigm shift in flow assurance from “complete hydrate avoidance” – where large quantities of hydrate inhibitors are injected into flowlines to prevent gas hydrate formation – to “hydrate management” – where gas hydrates are allowed to form in the flowline, but the hydrate slurry properties are controlled. This shift renders it imperative to gain knowledge of the rheological behaviour of the gas hydrate slurries in order to find the key factors influencing the transportability of gas hydrates in flowlines.618 Clathrate hydrate slurries also have applications in refrigeration where hydrate slurries are used as a two-phase (solid–liquid) secondary refrigerant (TPSR)619–623 to provide chilling.To date, several efforts, including large-scale flow loop-based619,620,624–637 and small-scale benchtop rheometer-based638–653 studies, have been undertaken to obtain experimental evidence on the rheological properties of hydrates and investigate the effect of hydrate agglomeration and deposition on hydrate slurry viscosity. However, it has always been difficult to reproducibly control the hydrate formation under high pressures for hydrate slurry characterisation.614,640 In flow loop-based studies, the turbulent behaviour of industrial pipelines is approximated, and the apparent slurry viscosity estimated from measured pressure drops by applying the Hagen–Poiseuille and Rabinowitsh–Mooney equations under a number of assumptions.654 Flow loop experiments provide a practical way to study the effects of flow regimes and patterns on hydrate slurry rheological behaviour.635 In benchtop rheometer-based studies, the shear rate, shear stress and accordingly the viscosity of a hydrate slurry sample are directly measured using high-pressure rheometers and/or autoclaves.639,655,656 These studies allow for an intuitive approach when studying the effect of different factors such as water fraction, hydrate volume fraction and shear rate on the rheological behaviour of hydrate slurries.635 However, since each measurement technique has its own limitations, comparison of different types of experimental studies is not straightforward. Moreover, most studies have been conducted under different conditions (temperature, pressure, composition) causing complexities when attempting to compare the experimental data with model predictions.657
To predict hydrate blockage in multiphase flowlines, the experimental studies are incorporated into the development of an appropriate formation/agglomeration model and coupled with transport and kinetic models. To date, a number of empirical and physical models have been developed to predict the rheological properties (viscosity and yield stress) of hydrate slurries formed from water-in-oil emulsions.626,658,659 The rheological models currently available in the literature have been reviewed elsewhere.657
Further experimental and modelling studies are necessary to achieve reliable relative viscosity for a given hydrate slurry as a function of the hydrate volume fraction. In addition, as initial work on understanding the viscosity of gas hydrate slurries focused on understanding the behaviour of ice slurries, comparing the rheological properties of ice slurries to those of a gas hydrate slurries could help with understanding the influence of particle agglomeration on viscosity and yield stress. An advanced knowledge of the rheological properties of gas hydrate slurries is also necessary for the other hydrate-based applications such as desalination, gas separation and energy storage, where hydrate-laden slurry flows in purpose-built flowlines with configurations different from oil and gas transportation pipeline networks. Literature survey shows that the experimental and modelling studies on rheology properties of gas hydrate slurries have been mainly concerned with energy applications. This makes it necessary to extend rheological studies in both experimental and modelling aspects in compliance with the other hydrate-based applications given the transportability of the hydrates in flowlines is controlled by the relative viscosity of hydrate slurry.
2.7 Interfacial phenomena
Interfacial systems consisting of fluids and surface-active materials play a critical role in gas hydrate nucleation and growth phenomena (Fig. 9). Thermodynamics of interfacial areas containing the alterations to the chemical potential, interfacial tension (IFT), entropy, and Gibbs free energy throughout the gas hydrate formation process are of enormous interest and require a fundamental understanding of the gas hydrates interfacial characteristics.660 Due to the significant surface district created in the hydrate system, IFT can directly influence the total free energy.661,662 The potential molecular interactions at the particle–particle and particle–surface interfaces will change based on the IFT, contact angle, and Gibbs free energy alterations,663 when hydrate crystals exist in their various continuous phases including gaseous, oleic, and aqueous phases.The interface free energy can be explained using an IFT per unit area, to exchange the pressure term in the Gibbs–Duhem equation as follows:660,664
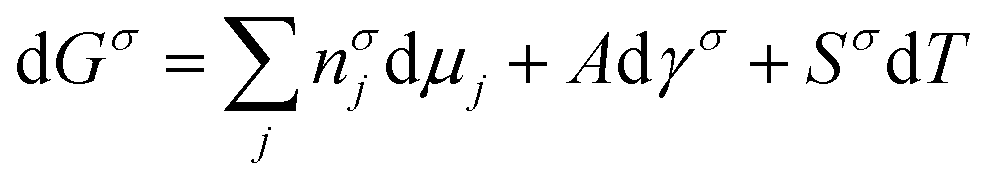
Surface active chemicals or surfactants could affect the clathrates crystallization rate through various roles.682,683 Surfactants could facilitate the hydrate nucleation phenomenon by reducing the clathrate–aqueous phase surface free energy, since they adsorb onto that interface.684 Additionally, they could boost the mass transfer process by increasing the blending of water and methane which results in the acceleration of hydrate crystal growth. Surfactant adsorption onto the interface surface, however, could abate the hydrate crystals growth rate.685 Various adsorption drivers have been suggested for surfactants at the hydrate crystals surfaces in contact with the aqueous phase.682,686–691 Realisation of the drivers of the amphiphilic molecular interaction with the clathrate surface is critical for their utilisation and optimization for suppressing the hydrate crystallisation rate. In this regard, sodium dodecyl sulfate (SDS) was recognised as one of the most effectual surface active species to boost the nucleation and growth of gas hydrates crystals.682,687,692
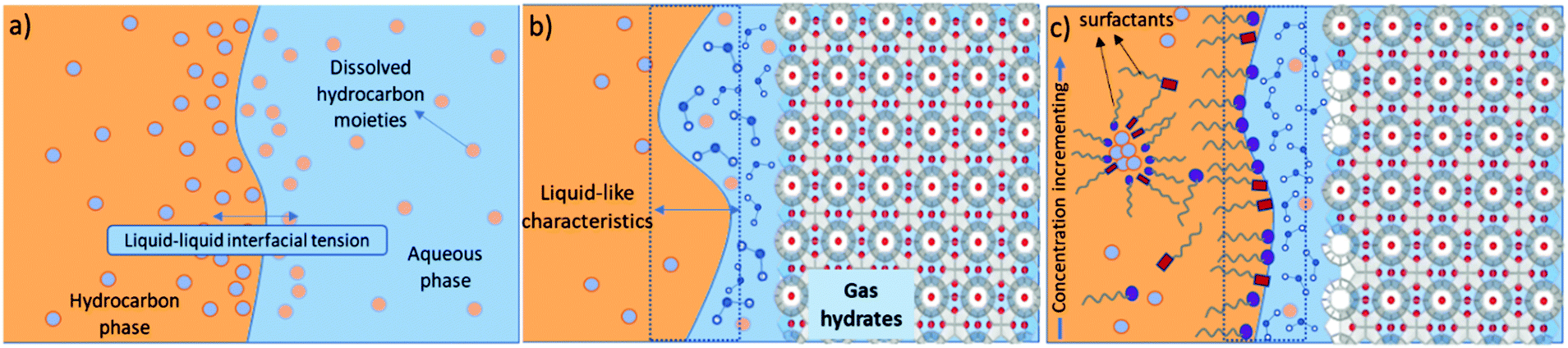 | ||
| Fig. 9 The various solid hydrate and fluid–fluid interactions which play crucial roles in hydrates crystals formation and inhibitions phenomena; (a) formation of water in oleic phase and hydrocarbon in aqueous phase emulsions; (b) hydrate crystals (solid phase) formation, growth, and their interaction with peripheral water and hydrocarbon molecules at interface; (c) hydrate crystals-water-hydrocarbon molecular interactions in the presence of different surface-active chemical species. | ||
By conducting zeta potential measurements,686 it has been shown that SDS adsorbs to the hydrate surface through hydrogen bonding of the anionic head group of SDS to the water molecules on the hydrate surface, leading to an arrangement of the active moieties at the interface where the hydrocarbon tail stays in the aqueous phase.686 Lo et al.690 realised that SDS adsorbs to a cyclopentane hydrate through the Langmuir isotherm and noted that the first saturation point related to the generation of a monolayer surfactant via anionic hydrogen bonding to the hydrate surface. The second saturation point related to the creation of a bilayer with the ionic functional groups of the second surfactant stratum adjusted towards the water phase.
The influence of surfactants on the cyclopentane clathrate hydrate crystallization have been investigated.693 At low surfactant volumes, reduced interfacial tension owing to the shrinking district of the water–cyclopentane interface was reported, resulting in formation of a planar clathrate hydrate crystal. At higher surfactant volumes, the interfacial tension fluctuated during the crystallisation phenomenon, which led to crystal morphology alteration towards the conical shape. The interfacial tension fluctuation behaviour is due to the growth and liberation of the hydrate crystals from the droplet surface. The prevalent interfacial behaviour presents a microscopic point of view, which can be employed to realise the cold flow operating strategy for hydrate slurries,694 where the probable capillary-aggregation phenomenon between hydrate crystals is profoundly underrated within the gas hydrate stability region.
3. Kinetics of gas-hydrates formation and dissociation
Developing further potential applications of hydrates are primarily hindered by the problems associated with hydrate formation and dissociation. Understanding the kinetic process of clathrate hydrate remains a challenging aspect of hydrate-related problems. For instance, increasing the rate of gas hydrate formation is critical in gas storage and gas separation operations that utilise hydrates. Whereas depressing the rate of hydrate formation and rapid dissociation is important for hydrate risk management in the area of flow assurance. Controlling the rate of hydrate dissociation is important for natural gas storage and environmental issues. In this section, we review growth and dissociation kinetics of clathrate hydrates.3.1. Crystallization
Crystallization occurs in two major steps (see Fig. 10). The first step is known as nucleation, which is the appearance of a crystalline phase from disordered or liquid-state molecules. The second step is known as crystal growth, which is the subsequent increase in the size of particles and leads to a crystalline form. Three canonical hydrate structures (sI, sII, and sH) are known and guest–host interactions play a crucial role as hydrates are thermodynamically stable only when a minimum number of cages are filled with the guest molecules. It has been found that the number ratio of cages filled with guest molecules (cage occupancy) of the largest cage is almost 100% and that the cage occupancy of smaller cages depends on the formation conditions. Thus, the stoichiometry of a hydrate depends on the formation conditions, such as pressure and temperature, and the type of guest molecule. Also, the kinetics of hydrates depend on temperatures above and below the melting point of ice (273 K) due to the phase change of water (solid, liquid, or vapour).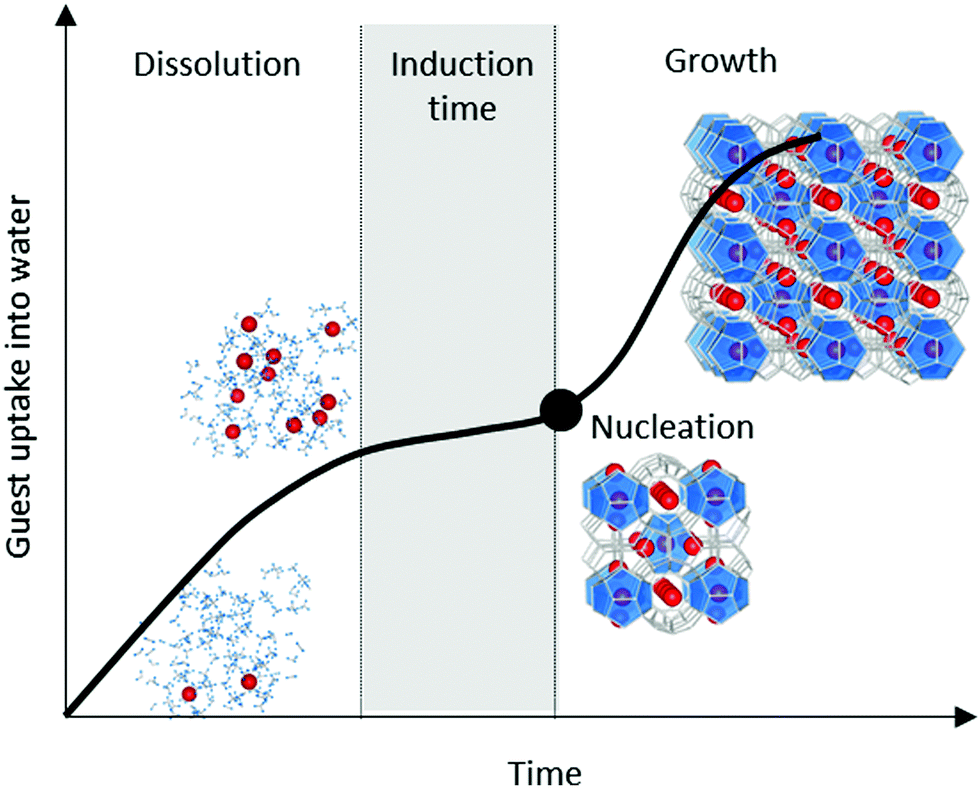 | ||
| Fig. 10 Typical time dependence of the hydrate crystallization process. | ||
The period between the times when the system temperature is lowered, or pressure is increased, to the equilibrium P–T condition at the time of hydrate nucleation is called the induction time. The induction time has been confirmed experimentally in various studies and the period basically depends on the driving force.696 The induction time data for hydrates fits the probability function to the theoretical probability distribution function describing the behaviour of stochastic processes697–702 based on classical nucleation theory (CNT). Crystalline hydrate growth occurs only after nuclei larger than critical radius, which is the minimum size that must be formed before crystalline hydrate growth, is attained. A molecular-level hypothesis of the induction time in homogeneous nucleation of hydrate is that cage-like water clusters surrounding guests combine to form the initial amorphous hydrate like structures.703 This is called labile-cluster hypothesis (see Fig. 11), and several other mechanisms for homogeneous nucleation have been proposed by recent molecular dynamic simulations: local-structuring hypothesis,704 blob hypothesis,705 and cage-absorption hypothesis.706 The reader is referred further to Section 3.1.3 of the present work. From experimental studies, Raman spectroscopic measurements of methane hydrates suggest that water clusters, which are similar to small cages of structure I hydrate, are readily formed and the formation of large cages of structure I hydrate from liquid water may be rate-determining during the hydrate formation.707 However, no significant difference between the structure of water before methane hydrate formation and that after the hydrate dissociation was observed by neutron diffraction method.708 Also, no significant difference between that of the liquid mixture of THF at room temperature and supercooled temperature was observed by X-ray Raman scattering measurements.709
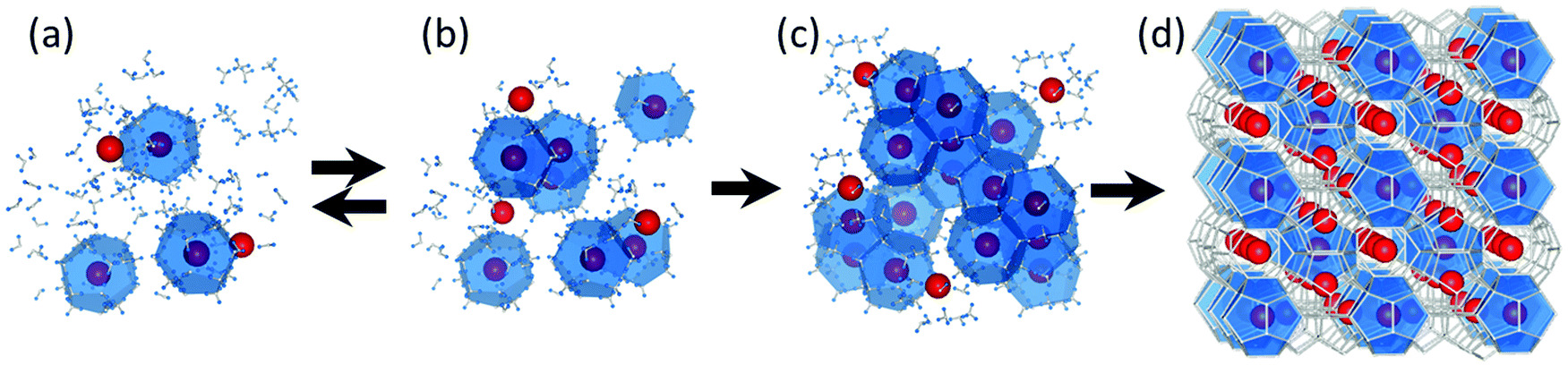 | ||
| Fig. 11 Labile-cluster model of hydrate nucleation: (a) labile-clusters, (b) agglomeration of clusters, (c) primary nucleolus, and (d) hydrate crystal. | ||
A significant effect on shortening induction time of hydrate nucleation has been observed by using dissociated hydrate water or ice melted water.710 This phenomenon is called a memory effect. The occurrence of residual structure is suggested by the difference in water viscosity between that before hydrate formation and that after hydrate dissociation.711 The higher viscosity of water after the hydrate dissociation returns only after heating to a temperature well above hydrate equilibrium. The stochastic nature of these waters has been confirmed by statistical methods.697,712,713 However, no significant structural difference of water after hydrate dissociation was found in different thermal histories by neutron diffraction.714 Recently, the formation of submicron-sized bubbles, so called nanobubbles, due to hydrate dissociation was reported.715,716 A significant number of nanobubbles remain dispersed in the dissociated water for more than 1 day, and the water has memory effect. MD simulations of methane hydrate dissociation also suggest the formation of methane nanobubbles in water.717 The guest–water interfaces of these nanobubbles may act as nucleation sites for heterogeneous nucleation of hydrate without the need for residual structures. Detection and measurements of the sudden nucleation event may be difficult because the nucleation sites are themselves a stochastic process forming the crystalline hydrate phase at a certain point suddenly. Although previous investigations have not thoroughly examined the stochastic hydrate nucleation process, these hypotheses listed above as well as CNT may give insights into fully understanding the complex mechanism.
(i) Guest–water system. At guest–water interfaces, hydrate films (layers) grow laterally along the interface after the nucleation. Investigations of hydrate crystals grown from the guest–water interface are important for the overall understanding of the kinetics of hydrate formation. Experimental studies on hydrate growth at the guest–water interface have been conducted.718–720 The important considerations for modelling the lateral growth kinetics are mass or/and heat transfer at the growing front of hydrate film.721,722 After the growth of hydrate film to around 10 μm in thickness,723 the guest and water phases are separated, and further hydrate formation is suppressed. Polycrystalline film covering the interface and the morphologies of hydrate crystals have a significant dependence on subcooling and gas composition. With large subcooling, the shapes of hydrate crystals are generally sword-like or triangular, with the shape changing to a polygon and a large polygon when the subcooling is smaller.724–727 Hydrate films grow in thickness though the films formed at guest–water interfaces by subsequent mass-transfer resulting in further hydrate formation. A detailed review of the hydrate growth models have been reported,22 but it is still debated how to diffuse molecules through hydrate films to grow the thickness of hydrate film.728
(ii) Guest–ice system. At guest–ice interface, hydrate also grows although their formation rate may be smaller than that of liquid water. In the initial step of xenon hydrate formation, hydrate film with thickness less than 0.1 μm is formed before the reaction becomes too slow. The number ratio of small and large cage is about 1 during the early part of the reaction, and the ratio changes to its equilibrium value of 3–4 when nucleation processes were finished, and rapid growth commenced.729 Moreover, the formation of transient hydrate structures has been observed in the initial step for pure methane,730 carbon dioxide,310 or carbon monoxide731 hydrates. These experiments suggest that a precursor phase or initial structure are different from the equilibrium hydrate structure. MD simulations support the occurrence of transient structures.732,733
A shrinking-core model accounts for the growth of hydrate film in thickness.310,734–737 In this model, the reaction consisted of three steps: (a) the initial reaction of guest with ice; (b) the growth of hydrate film and growth in thickness; (c) the mass diffusion of water/guest through the hydrate film. The activation energy of the processes was obtained from the kinetic rate constant as a function of temperature using the Arrhenius equation. Transport of guest molecules by a series of jumps between neighbouring hydrate cages was demonstrated and the rate-determining process may change in different temperature ranges.738,739 Other mathematical diffusion models have been proposed,740–742 and other factors affecting the growth of hydrates may need to be considered.743 For a careful examination of the kinetic process proposed by the model, further comparative researches between experiment and theoretical simulations are required.
(i) Labile-cluster hypothesis (LCH):703 where unstable entities featuring a guest surrounded by water molecules in their first coordination shells diffuse in the liquid phase as a single entity, and the clusters of varying sizes combine to form larger stable clusters which grow into the hydrate crystal.704,744
(ii) Local-structuring hypothesis (LSH):745 where well-solvated guests meet, forming a cluster with the guests in a configuration consistent with the clathrate crystal, the water molecules rearrange to form a hydrate framework.
(iii) Blob Hypothesis (BH):705 a modified form of LSH, where stochastic local-density fluctuations of guests in water favour ‘blob’ formation as amorphous clusters of solvent-separated guests at high solvated-guest concentrations close to that in the hydrate.746
(iv) Cage-adsorption hypothesis (CAH):706 where guest molecules increase their local concentration to induce hydrate formation by spontaneous cage formation, followed by guest “adsorption” to cages and stabilisation thereof.704,706,747–749
Although not at guest–water interfaces, homogeneous methane-hydrate nucleation studies have observed formation of methane–water clusters which combine to form a critical nucleus,750–752 supporting the concept that locales with high guest concentration, although not highly structured (arising perhaps from either local-density fluctuations and/or cage adsorption), can result in hydrate nucleation.
Recent molecular-simulation studies have indicated typical hydrate-nucleation timescales of the order of microseconds,665,705,706,745,746,750–753 and this is discussed further in Lauricella et al.752 Often, a hydrate nucleates initially into phases inconsistent with common bulk crystal structures, but containing a variety of structural units,665,750–753 as well as ‘amorphous’ units.753,754 In relation to the Blob Hypothesis, English et al. studied the size of the water–methane interface at realistic solvated-methane concentrations, finding not-dissimilar early-stage nucleation mechanisms.69 In addition, solvated-methane-concentration effect on nucleation rate have been assessed,752,755 with innate challenges in MD simulation for methane-hydrate nucleation being discussed with some acuity756 while Hall et al. have placed hydrate nucleation within the wider context of the funnel model, in view of the panoply of competing amenable pathways.757,758 Recently, biased-simulation methods have offered much microscopic insight: Lauricella et al. have examined free-energy landscapes for hydrate nucleation from metadynamics,747,759 and restrained MD.752 Małolepsza et al. have developed an enhanced-sampling technique and applied to hydrate nucleation.760 Bi et al. applied the Forward-Flux Sampling method to methane-hydrate nucleation,761 computing nucleation-pathway free-energy landscapes.762
Since the pioneering gas-hydrate molecular simulations of Tse et al. over 35 years ago,74 simulation has led to a great number of microscopic insights into hydrate behaviour and properties, from thermal-conduction mechanisms to hydrogen storage and carbon capture. In this brief foray into some very recent advances in molecular simulation since the review by English et al.,69 it has been understandably impossible to be truly exhaustive in these ‘cataloguing’ exercises, such has been the recent surge in this specific research activity; rather, some important contemporary trends have been highlighted. One area not highlighted so far has been to remark on the (relative lack of) research ‘crossover’ between the clathrate-hydrate and inorganic-clathrates (e.g., clathrasils) communities, which has been a feature to a lesser extent than, say, between the clathrate-hydrate and semi-clathrate arena. In the area of inorganic clathrates, important insights into cage and lattice behaviour, thermodynamics and dynamical properties (e.g., thermal conductivity763) are being made, with important parallels to current and previous work in clathrate hydrates. Clearly, the power of molecular simulation to ‘leverage’ and transfer this body of experience and insight into transformational work in inorganic clathrates is an important contemporary trend in clathrate modelling; in this regard, the important contributions of Li, Stroebel and co-workers are to be welcomed.764
In the realm of clathrate hydrates, in terms of outlook for achieving greater levels of quantitative agreement relative to experiment for physical properties, the ‘acid test’ of spectroscopy (e.g., Raman data) remains a formidable challenge. For instance, Futera et al.765 discusses measures to potentially increase further the fidelity of H2-water potential tailored for hydrogen hydrates. The use of force-matching to high-quality ab initio MD simulation134 to achieve better-accuracy potentials, beyond the pairwise approximation for classic empirical potentials, is a future important area, and it is to be hoped that neural-network-tuned potentials will offer important progress here in the decades ahead, especially in hydrate-nucleation and formation simulations, with a large variety and heterogeneity in local environments – where model parameterisation in one phase alone will be less likely to succeed.
Since the effect of inhibitors and surfactants depend upon the guest species, the search for environmentally friendly and more effective materials have been conducted. For example, experimental results of methane + propane mixed gas hydrate suggest that porous hydrate film, but not an impermeable film, can be grown at the guest–water interface in the presence of SDS. The porous hydrate film then allows efficient water-to-gas contact for further crystallization.683 The formation of smoke-like hydrate from gas bubbles, which are partially covered by hydrate film, in water with SDS is also suggested.774 In the case of methane hydrate, the amount of crystalized hydrate increased in the presence of the SDS as the hydrate crystals detached from the interface due to increased wettability. The size of individual hydrate crystals with SDS become smaller than those formed from pure water systems due to increased hydrate nucleation. With higher subcooling temperature, the hydrates grow upright crystal fibres because of large capillary forces due to smaller pores between hydrate crystals.775 Based on these experimental results, the crystal growth and morphology in the system with surfactant can be controllable by changing concentration of surfactant. A detailed review of the hydrate promoters including surfactants coupled with other promoters has been reported.776,777
3.2. Dissociation
Hydrate dissociation is an endothermic process as with ice melting. Hydrates separate into water and guest molecules by breaking up hydrogen bonding networks of water molecules and the van der Waals interaction forces between guest and host water molecules. Dissociation occurs in two major steps; (a) the initial destruction of the hydrate crystal and (b) the release of water and guest molecules. Dissociation kinetics of hydrates also largely depend on the temperature above and below 273 K.The dissociation rate is controlled by three factors: the intrinsic dissociation reaction rate, the rate of guest flow produced from the crystalline hydrate, and the rate of heat transfer to the hydrate for endothermic reaction. The hydrate dissociation kinetic models presented so far are based on either one of these factors or multiple factors to couple these effects. The effect of multicomponent hydrate-forming gas,788 additives in the water or type of surrounding solutions789–791 and the form and size of hydrate samples792 may change the rate-determining stage of hydrate dissociation. A detailed review of the hydrate dissociation models has been reported.793
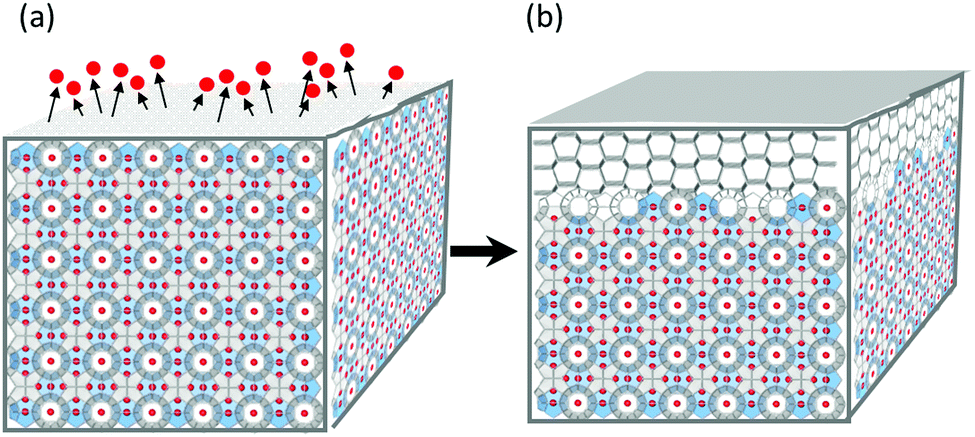 | ||
| Fig. 12 Schematic model of the self-preservation or anomalous preservation of gas hydrate: (a) methane hydrate during dissociation and (b) the hydrate being covered by ice layer. | ||
Methane hydrate dissociation by rapid pressure drop under isothermal conditions is unexpectedly slow and occurs only for temperature ranges between 240 K and 270 K. The dissociation rates vary by several orders of magnitude with two minima at around 250 and 268 K. This phenomenon is termed as “anomalous preservation”.800 Experimental studies suggest that this anomalously slow dissociation is related to ice morphology or structures formed at the surface of the dissociating hydrate.801–807 It has been revealed that the preserved methane hydrate is tightly covered by an ice layer with a thickness of about 100 μm or less.808–810
From a practical application perspective, hydrate pelletizing technology for natural gas storage and transportation have been proposed and evaluated.811–813 To date, methane or natural gas have been successfully stored within hydrate pellets for more than three months at 253 K under atmospheric pressure.814 Recently, the phenomena under more complex systems coexisting with other materials such as surfactant,815 hydrophilic silica beads,816 crude oil,817 dry water,818 and saline water819–821 have been reported. The dissociation reactions largely depend on the type of guest species.822,823
3.3. Kinetics in sediments
The morphologies of naturally occurring hydrate are varied depending on hydrate kinetics as well as type of sediments: e.g. pore-filling hydrate in coarse sands or veined or nodule-type hydrate in fine sediments. Several hydrate habits in sediments have been known which were explained in Section 2.5.2. All these habits depend on hydrate nucleation and growth processes in the pore, where the physical properties of the hydrate sediments are affected by the hydrate formed in the pore space.400,824 Accordingly, understanding of kinetics and microstructure of hydrate at the pore scale is important to advance our knowledge of hydrate sediments.From experimental studies on hydrate sediments, mechanisms of hydrate formation and dissociation processes at the pore scale have been studied and incorporated into continuum scale simulations. Observations of hydrate nucleation, growth, and dissociation in 2D configuration glass micromodels were conducted by optical microscopy.825–828 Morphology changes of hydrate mixed with fine glass beads inside of thin layer were observed as a function of growth rate.829,830 Nondestructive imaging methods, such as MRI and X-ray Computed Tomography (CT), have been applied to study the distribution of hydrates in frozen sediments831,832 and other attempts by means of microfocus X-ray CT contributed to continued studies of the sedimentary matrices at pore scale.833,834 In the last decade, MRI and X-ray CT measurements have extended hydrate kinetic studies to 3D configurations of the microstructure of hydrate sediments. MRI measurements, which are suitable for detecting hydrate and water but suffer from insufficient spatial resolution for measurement at the pore scale, revealed multiple nucleation events at different times and locations within the sediment during hydrate formation processes.835 X-ray CT with micro or higher spatial resolution has also been used for kinetic studies of hydrates in sediments with the aid of contrast agents or heavy guest substances such as Xe due to insufficient image contrast of X-ray CT for distinguishing hydrate and water or ice.715,836–839 In the X-ray CT images of hydrate formation processes, nucleation starts at the water–gas interface from partially saturated water and that starts in the bulk of the liquid which is water saturated with guest substances. Occurrence of fluid phase films between hydrates and the sediment particles were also observed. In the hydrate dissociation images, the decomposition starts at the hydrate–gas interface with melt water accumulating at dissociating hydrate surfaces. Then inhomogeneous dissociation facilitates redistribution of water and local hydrate reformation in the pore space. From a basic science point of view, guest substances such as THF or CO2 have been widely used as model samples for laboratory investigations. Because of the complete miscibility or high water solubility of these guests, growth rates of these hydrates from water are faster than methane. Also, to improve visibility of X-ray CT image, a contrast agent or Xe are used as a contrast medium even though they may change the hydrate kinetics. Ultimately, experiments using methane or natural gas with pure water or sea water are preferred for incorporating into the continuum scale simulations of geological systems. In addition to abovementioned imaging techniques, neutron scattering technique have also been employed840 to investigate gas hydrate kinetics in the presence of solid surfaces. Cox et al.840 utilised time-resolved neutron scattering experiments and molecular dynamics simulations to identify that the methane hydrate formation is insensitive with respect to the presence of various clay and silica nanoparticle surfaces.
Recently, numerical models of pore-scale hydrate dissociation have been proposed to estimate the dissociation rate of methane hydrates. The numerical models suggest that the water layer prevents guest gas from escaping into the gas phase due to the mass-transfer-limitation, which ultimately affects the driving force of hydrate dissociation by depressurization.841–843 A numerical model of methane hydrate formation was also proposed for determining the locations of the initial hydrate nucleation and for estimating the morphological distribution of methane hydrate in porous media using classical nucleation theory.844
Understanding the formation and dissociation kinetics of natural gas hydrates in sediment is important for the efficient and economic development of natural gas hydrates as unconventional natural gas resources in permafrost or marine sediments. On the other hand, dissociation of hydrates and subsequent outgassing could affect the atmosphere of solar planets845,846 as well as the Earth.1 The self-preservation phenomenon is also true in shallow permafrost locations. Gases releasing from the permafrost may contain natural gas produced by the dissociation of hydrates in the pore space of sediments, which can exist in permafrost due to self-preservation.847,848 Accordingly, more detailed knowledge of the kinetic model of hydrates in complex system of sediments is needed.
3.4. Influence of additives
Gas hydrate formation is very susceptible to contamination owing to the presence of any foreign additives other than water and the guest gas moieties, which can result in critical influences on the thermodynamics and kinetics of gas hydrate formation. The foreign species, such as salts, bio-surfactants, alcohols, etc., may be deliberately augmented to water–gas systems to influence the kinetics of hydrate formation. Additionally, gas hydrate nucleation and growth are strongly related to the hydrophobicity of gaseous species and chemical additives (e.g. nanoparticles) which can either be hydrophilic or hydrophobic and when in the aqueous phase might boost gas hydrate formation,849,850 however, they could also prevent hydrate growth if they engaged at the aqueous–oleic phase interface.851 While some results illustrated that nanoparticles might promote hydrate formation at lesser concentrations,852,853 however, the authors did not denote whether the nanoparticles in their tests were hydrophilic or hydrophobic. A macroscopic experimental study conducted by Wang et al.854 proposed that hydrophilic nanoparticles could influence a hydrate inhibition efficiency which could be used to decrease the hydrate reformation and aggregation throughout the gas hydrate or deep-water drilling if their contents could appropriately be controlled to avoid instability of the well borehole. Generally, some additives can introduce hydrophobicity influence855 which results in organising the peripheral water molecules into a clathrate like hydrophobic hydration shell, creating an incremented local gas content surrounding the hydrophobe,856,857 and increase the hydrate crystal formation rate accordingly. The creation of semiclathrate hydrates in the presence of partial hydrophobic additives (e.g. THF858,859) and pure hydrophobes (e.g. cyclopentane860,861) augments the hydrophobicity associated hydrates formation kinetics. Typically, the addition of surfactants which can be cationic (e.g. TBAB,201,862–866 dodecyltrimethylammonium (DTAC),862,867 dodecyl amine hydrochloride (DAH),868N-dodecylpropane-1,3-diamine hydrochloride, (DN2Cl)868), anionic (e.g. SDS,682,867,869–871 sodium tetradecyl sulfate (STS),871 sodium hexadecyl sulfate (SHS),871 sodium oleate871), and nonionic (e.g. THF,867,870,872,873 ethoxylated nonylphenol,874 Tween 80,875 cyclopentane861,876), at specific concentration could facilitate gas hydrate formation. Solid species can also have a hydrophobic effect and induced gas hydrate formation. Glass surfaces with various coatings including partial hydrophobic N,N-dimethyl-noctadecyl-3-aminopropyl trimethoxysilyl and hydrophobic octadecyltrichlorosilane877 boosted hydrate crystallisation and incremented kinetics of hydrate formation.878In the following subsections, the influences of typical additives, salts and alcohols, on gas hydrate formation have been reviewed and the existing hypothesis for their efficacies have been discussed.
3.5. Growth acceleration
Many researchers have employed non-surfactant-based methodologies to accelerate gas hydrate formation including sand packs, nanoparticles, hydrogels, and porous compounds such as activated carbon and porous nano-silica to enlarge the solid–liquid interface.899–906 Nano-metal particles have been utilised to boost gas hydrate formation, which include nano-Cu,907 synthesized silver nanoparticles,908,909 spherical nano-copper oxide,853,910 nano-Al2O3,911 and nano-ZnO,911,912 plus the use of advanced nanofluids consisting of carbon nanotubes913–916 to improve heat and mass transfer phenomena throughout the gas hydrate formation process.917 In the subsections below, we detail the effect of different techniques for facilitating the formation of gas hydrates and their corresponding kinetics.A theoretical model of incorporated hydrate dissolution and formation when applying the shock wave along with diffusion in gas bubbles containing aqueous phase and consideration of heat influences throughout the process, was developed.932 Integrated carbon dioxide hydrate formation and dissociation owing to the presence of the shock wave front in carbon dioxide + nitrogen mixture bubbles containing water in presence of a surfactant at various preliminary static pressures and moderate was illustrated experimentally and theoretically by Chernov and Dontsov.934 They have developed a theoretical model of the studied multi-component gas mixture system taking into consideration heat influences, which is not considered in the generalised model.932 They have observed that gas bubbles fragment into the tinier inclusions in the wave front which results in facilitation of hydrate formation owing to the increment increase of the water–gas interface area in addition to the movement of gas bubbles with a higher velocity in respect to the aqueous phase which leads to strengthening the heat and mass transfer phenomenon. It can be implied that the hydrate formation happens due to the hydrate film sorption progression on the surface of small gas inclusions, and the acceleration of this phenomenon is attained by utilising the kinetic parameters in addition to the consideration of heat and mass transfer processes at the aqueous–gas phase interface, which are significant. The usual time of the gas hydration process in the presence of the shock wave is multiple orders lower when compared to the duration of hydrate formation process using the existing methods of hydration. Higher shock wave amplitudes cause higher rates of gas hydrate formation and dissociation, and the presence of surfactants seem to have no notable influence on the hydration and dissociation phenomena in a system facing shock waves.934 While applying the physical fields has a promoting influence on gas hydrate formation and decomposition, it is typically assumed that physical fields demonstrate their positive effects within a particular span of frequency, and they have no influence on the kinetics of hydrate formation beyond that domain.
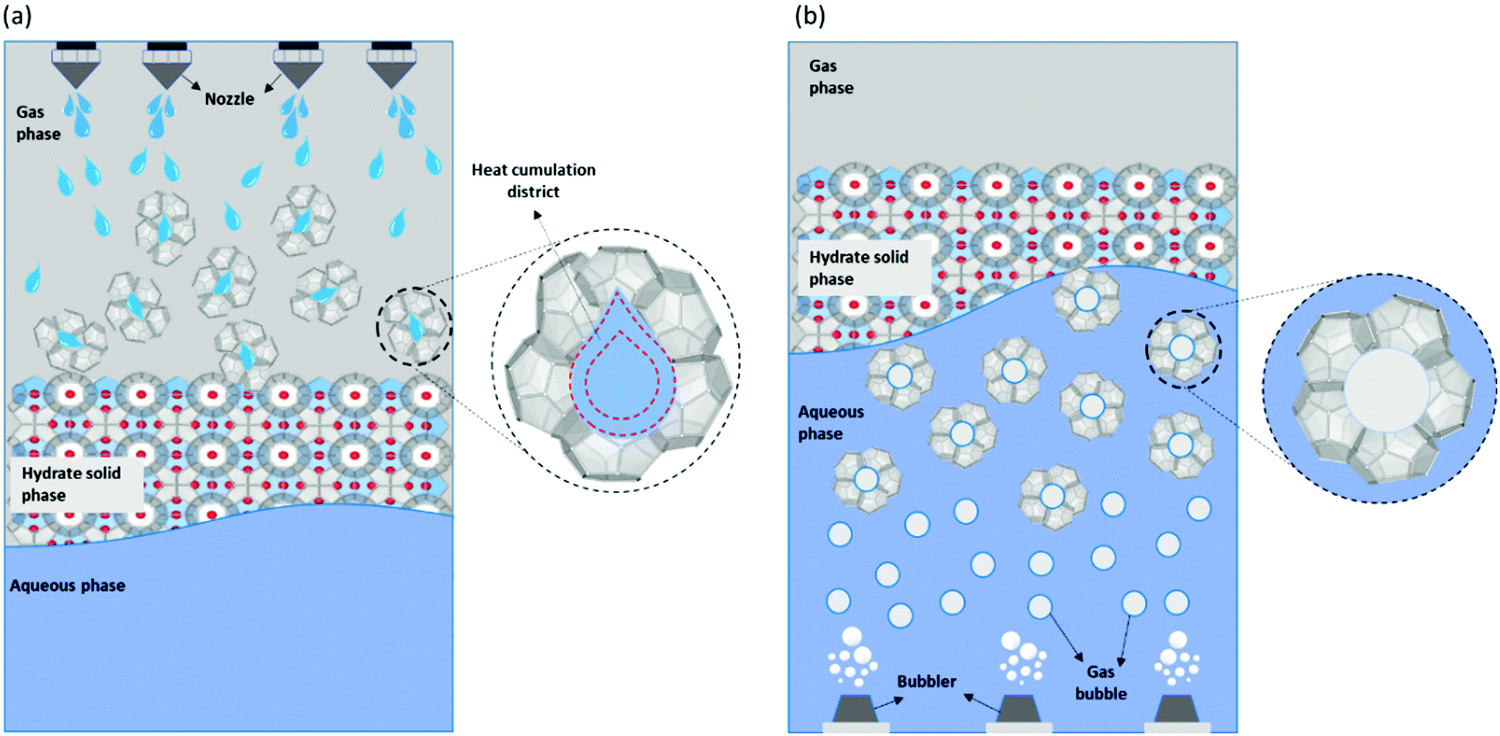 | ||
| Fig. 13 (a) Formation of gas hydrate using the spraying technique (b) schematic diagram of hydrate formation as gas bubbles rise through the continuous aqueous phase. | ||
The bubbling technique949 where gas is injected at high pressure into the aqueous phase via a distributor at the underneath of the reactor results in movement of the gas bubbles in the aqueous phase due to the buoyancy forces, increasing the vapour–aqueous phase disturbance and kinetics of hydrate formation at the periphery of the bubbles accordingly (see Fig. 13b).
4. Chemistry in flow assurance
The challenging hydrate nucleation and growth phenomena can occur at high pressure and low temperature conditions895 as a result of unpremeditated shut-ins,955 poor performance of chemicals injection pumps,956 and incremental increase of aqueous phase in the stream,957 which could result in occluded pipes and halted flow. Gas hydrate associated flow assurance challenges can occur not only in hydrocarbon pipeline systems but also within CO2 and hydrogen transportation pipelines where trace amounts of water impurities could exist in the stream. Flow assurance problems need to be addressed using a wide range of adequate methodologies, otherwise they could also negatively affect the determination of quantities of valuable cargo fluids in custody transfer and allocation systems. In the last two decades, gas hydrate associated flow assurance research studies have been undertaken directed towards identifying optimised mitigation strategies to attain more appropriate management of gas hydrate blockages which can happen in pipeline systems. This section aims to summarise the current state of the art progress of the understanding of gas hydrate associated flow assurance challenges in the oil & gas, and carbon capture & storage (CCS) sectors and to provide an assessment of the current designs of gas hydrate inhibitors for remediation purposes. It also challenges the gas hydrate community to develop new experimental facilities, methodologies and mathematical models for improving the early diagnosis and treatment of complex gas hydrate induced pipeline blockage in a cost effective and influential manner. Some review articles127,955,958–960 on gas hydrate associated flow assurance challenges and induced hydrate risks demonstrate different aspects of this important issue in the oil & gas industry. Fig. 14 shows that in harsh conditions, hydrate growth could lead to thorough transportation pipeline blockage through a mainly three-step process after the water in hydrocarbon emulsification phenomenon: (i) hydrate nucleation; (ii) hydrate-crystal growth and agglomeration; (iii) hydrate-plug formation from aggregates and blocking the flow line.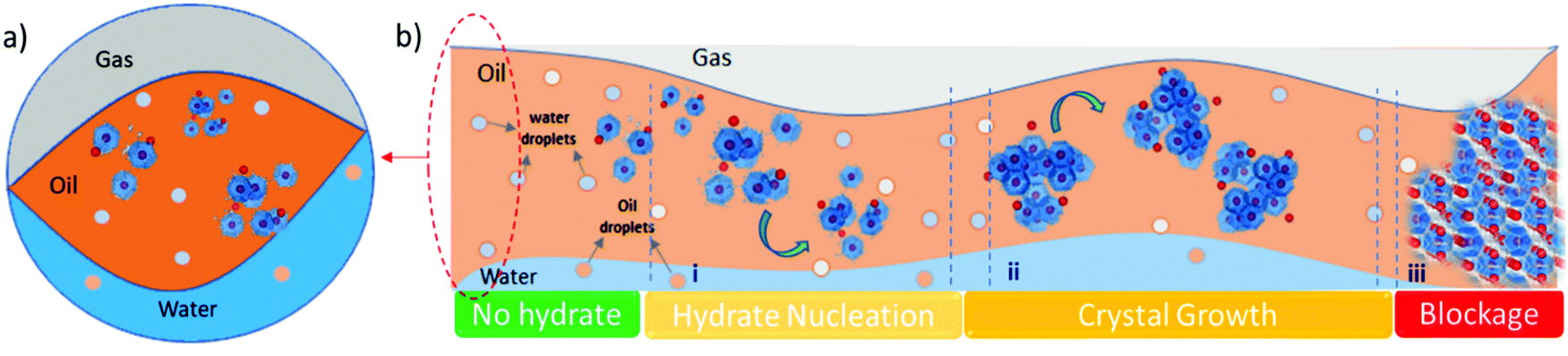 | ||
| Fig. 14 (a) Schematic model of multiphase flow in pipeline, where hydrate crystal aggregates could flow in gaseous, oleic, and aqueous phases, and/or could deposit onto the pipe solid surface; (b) schematic diagram of the drivers of hydrate crystals formation and aggregation in hydrocarbon transportation pipelines and three-step process of (i) hydrate nucleation, (ii) crystal growth, and (iii) blockage. | ||
4.1. Inhibition methods
Different mitigation and remediation strategies are utilised to handle hydrate blockage within pipes such as depressurization,616,961 chemical,962 mechanical,963 and thermal961,964 methodologies. Owing to the risks associated with some of the aforementioned methods such as hydrate plug movements,965 intensive research efforts have focused on potential applications of three types of chemistries (see Fig. 15) namely thermodynamic966–968 and kinetic hydrate inhibitors, THIs and KHIs respectively, and anti-agglomerates (AAs) to manage these risks. This section is structured in three subsections in which the detailed discussion of each class of hydrate inhibitors and their respective inhibition drivers have been addressed. Various reviews660,957,969–971 regarding the fundamentals of chemical inhibition, interfacial and physiochemical phenomena of gas hydrates for hydrate risk management cover most of the significant research works on chemical injection scenarios. In the following subsections, we present the most recent findings out of research studies on chemical-hydrate inhibition.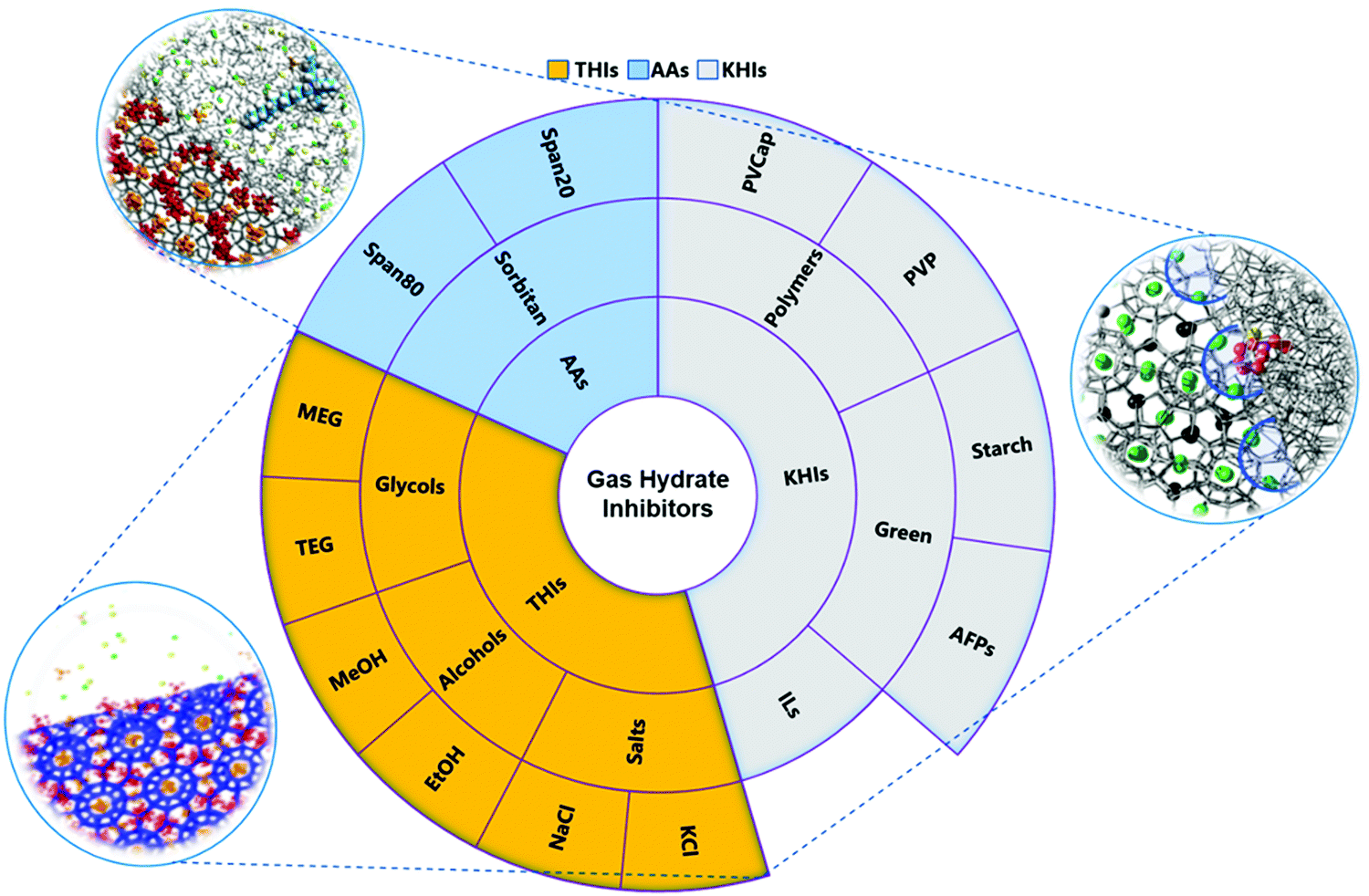 | ||
| Fig. 15 Schematic examples of various types of gas hydrate inhibitors: THIs, KHIs, and AAs, and corresponding molecular interactions with clathrate hydrates. | ||
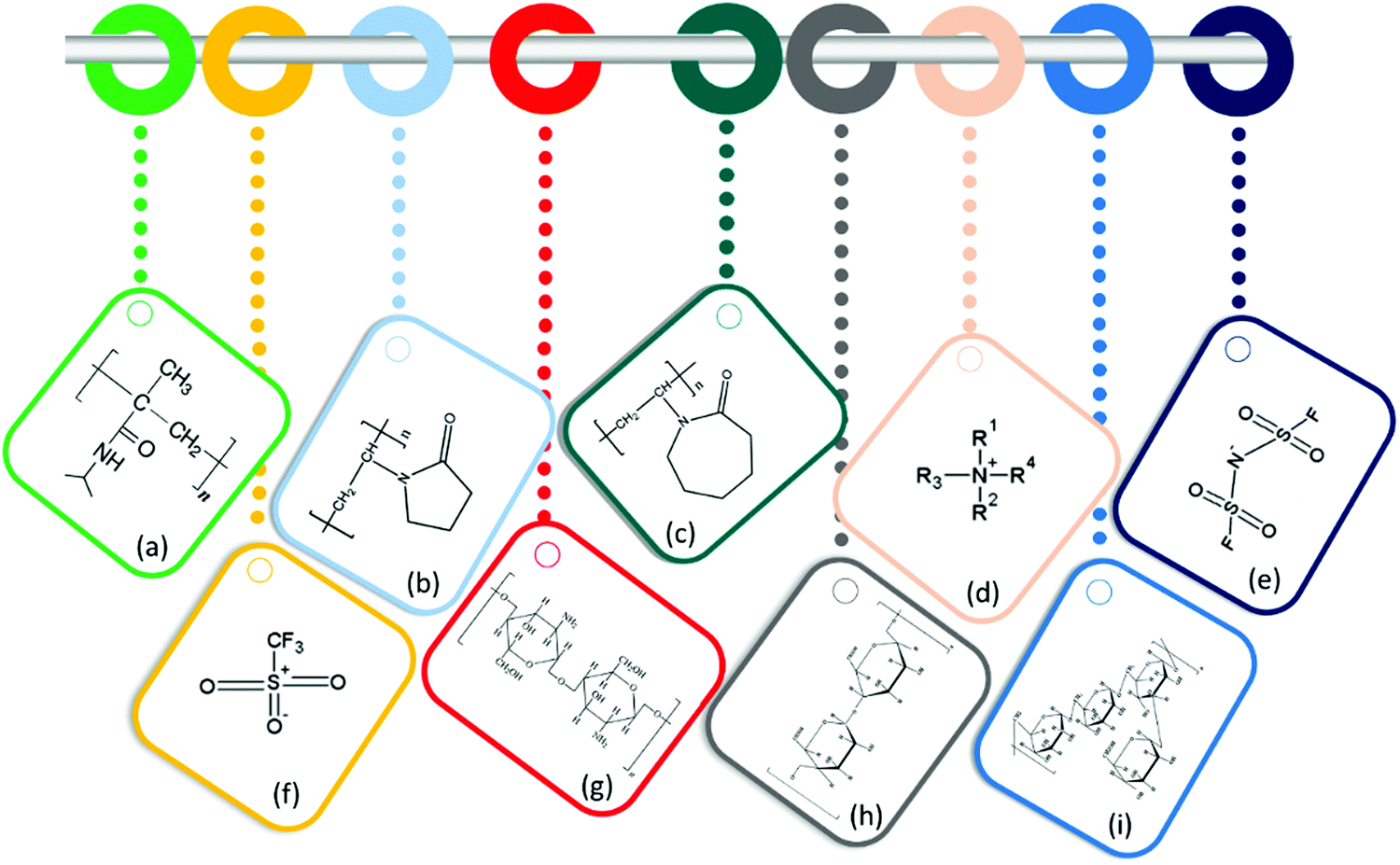 | ||
| Fig. 16 (a) Molecular structure of poly(N-isopropylmethacrylamide); typical poly(N-vinyllactam) polymers with incrementing lactam ring sizes: (b) PVP (five-ring); (c) PVCap (seven-ring); some fundamental structures of ionic liquids (ILs) utilised as kinetic hydrate inhibitors in the literature. Cations: (d) tetraalkylammonium [Nn1,n2,n3,n4]; anions: (e) bis(fluorosulfonyl)imide [f2N]; (f) triflate [OTf]; the chemical structures of green inhibitors: (g) chitosan; (h) tapioca starch; and (i) dextran. | ||
The AFPs biomolecules or ice structuring proteins, which are found in specific cold-adapted organisms,1072 could bind to germinal hydrate crystals and inhibit their growth within a given temperature range. Xu et al.1073 found that pectin, which contains linear regions of 1,4-linked-α-D-galacturonosyl units and methyl esters with hydroxyl functional groups, could form H-bonds with water molecules and rupture water structures, hence, significantly lessen the hydrate crystal growth rate and hinder hydrate formation. In addition to aforementioned biochemistries of KHIs, the hydrophilic waterborne polyurea/urethanes (WPUUs)1074–1076 with hydroxyl functional groups have recently gained interest from researchers as one of the greenest inhibitors owing to their eco-friendly/non-toxic nature.1077–1079 Different researchers have investigated the compatibility of KHIs with other flow assurance chemicals such as corrosion inhibitors to measure potential suppressed KHIs efficiency. Recent studies1080,1081 suggest newly designed KHIs for simultaneous inhibition of gas-hydrate formation and steel pipelines’ corrosion.
A widely utilised type of AAs contains quaternary ammonium surfactants1092 which are usually bonded to n-butyl or n-pentyl functional groups along with long alkyl chains and form a well-structured AA hindrance thin layer at the clathrate–oleic phase interface.1085 It has been illustrated that the AA thin film could hinder further hydrate aggregation and crystal growth processes through the formation of an obstacle for methane transfer to the hydrate surface.694,1085 The creation of a thin interfacial layer of AAs at the clathrate–oleic phase interface could be crucial for minimising the agglomeration of hydrate aggregates1087,1093 which consist of a combination of surfactants and alkanes from the oleic phase, as interfacial tests depict that linear alkanes would incorporate with the surface active species to create an ordered interfacial monolayer at the aqueous–oleic phase and/or the hydrate–oil interfaces.1094,1095 Due to adequate adsorption of AAs onto the gas hydrate aggregates surface through head binding, tail binding, and head & tail binding, the contact angle of the hydrate surface would be incremented resulting in the wettability transition from water wet to oil wet,1082,1084 reducing adhesion force between hydrate aggregates,1096 and prevention of agglomeration phenomenon accordingly. Sometimes Sorbitan type surfactants, such as Span20 to Span80,1086 are employed as AAs to inhibit hydrate particle agglomeration instead of ionic chemicals.1097 It has been shown that AAs can augment the inhibition efficiency of alcoholic thermodynamic inhibitors including MeOH,1098 and in presence of salts in the aqueous phase.1099 Different research studies reported that the inhibition yield of ionic AAs are increased due to the salinity increment of the aqueous phase1100,1101 which leads to higher stability of the emulsion,1102 separating of the AA moieties away from the aqueous phase,1014 and hindering of hydrate aggregates agglomeration processes. Recently, Mehrabian et al.1103 developed a fundamental understanding of the influence of salt on quaternary ammonium cationic AA adsorption phenomenon onto the sII methane–propane hydrate surface in aqueous environment providing insights into the hydrate–salt and AA–salt molecular interactions on an energetic level. Their molecular dynamic simulation results noted that the AA molecule first adsorbs onto the hydrate surface through interactions with either its head or tail (Fig. 17a and b) and then, the other section of the molecule interacts as the second step, forming the head and tail binding configuration at the same time (Fig. 17c).
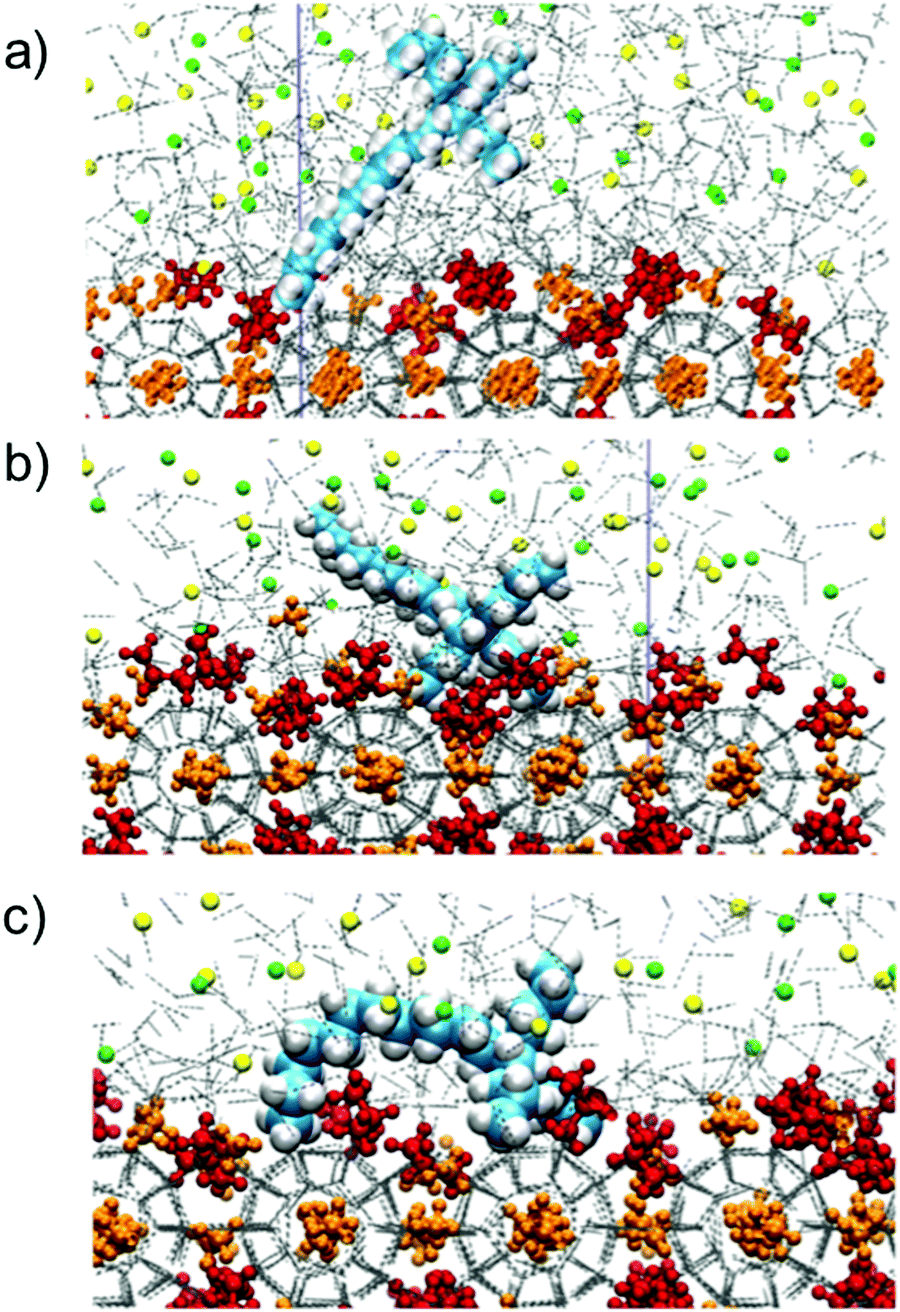 | ||
| Fig. 17 Snapshots of molecular-dynamics simulation of AA molecule adsorption phenomenon onto the gas hydrate surface for three different kinds of molecular interactions with surface active species at the interface: (a) solely the long hydrocarbon tail of the AA molecule interacts with the hydrate surface, (b) solely the ionic head of the AA molecule has binding affinity to the hydrate surface, and (c) the AA molecule interacts with hydrate surface through simultaneously head and tail bindings to the surface (adapted with permission from Mehrabian et al.1103 Copyright 2018 American Chemical Society). | ||
The salt-content augmentation in the aqueous phase can facilitate these interactions through AA solubility reduction in the aqueous phase and strong interactions of AA cationic head groups with chloride anions at the interface which result in an AA efficiency increase. In some cases, AAs boost the formation of gas hydrates1104 which can catalyse hydrate growth which facilitates novel applications in areas such as the storage of natural gas7,1105,1106 and the desalination of water1107,1108 amongst others.1109,1110 Further research studies would be required through affiliating molecular level data using micromechanical force measurements, new molecular dynamic simulations665,745,747,1111–1114 and macroscopic experiments including rocking cell to lead to the establishment of an appropriate synergism between modelling and tests results at all relative length and timescales that is required to handle the gas hydrate associated flow assurance challenges within the pipelines.
Moreover, some experimental studies using sum frequency generation or other spectroscopic techniques would also be needed to probe the molecular structure of AAs at different interfaces to build a fundamental understanding on how a variety of AAs perform in different systems. Such experimental data could assist scientists to realise whether the attained data from numerous simulation investigations are reliable or not in addition to determining potential applications of the studied systems in CO2 hydrate-based CCS processes (see to Section 6.1.2.2).
4.2. Experimental test methods
Over the past decade, researchers have spent an enormous deal of effort on the development and application of various experimental facilities which have created new trajectories for hydrate mitigation and control in intricate pipeline systems.Different flow loops with a variety of dimensions and operating conditions have been widely utilised to investigate the character of hydrate/ice plugging, deposition mechanisms and induced flow patterns under a series of flow rates and water cuts1115–1117 for water dissolved in a liquid condensate system1118 and multiphase systems (e.g. natural gas + diesel oil + water1116) with/out inhibitors,1119 w/o emulsion systems.1120 A high-pressure visual cell1121 and stainless-steel hydrate equilibrium cell1122,1123 were utilised to determine the kinetics of hydrate formation and methane hydrate film growth within a cold tube. Douïeb et al.1124 illustrated the hydrate induction time using a stress/strain-controlled rheometer. They performed the tests at various flow shear rates and realised that higher shear rates could result in shorter hydrate formation induction times probably owing to the greater mixing severity.
A newly designed 1.5 m long cylindrical windowed experimental rig965 was also employed to visually investigate the efficiency of some thermodynamic hydrate inhibitors1125 and combinations of them, with different densities at removing hydrate blockage in vertical pipes. Although the accuracy and quantitative ranking capability of an automated lag-time apparatus (ALTA)667,699 has been validated for hydrate nucleation and KHI chemical development, rocking cells1126 have been widely utilised for the development and validation of KHIs as well. The high-pressure differential scanning calorimetry (DSC)1053,1102,1127 is another experimental facility which has been employed to investigate the effects of AAs, KHIs and NaCl salt and their mixtures on water-in-oil emulsions and gas hydrate formation characteristics. In the last decade, various state of the art analytical techniques have been utilised to identify gas hydrate structures and respective noncovalent interactions at aqueous–vapour phase interface. Raman spectroscopy and synchrotron powder X-ray diffraction (PXRD)1012 have been used for the characterisation of methane and natural gas hydrate crystal structures and corresponding hydrate cage occupation characteristics. In order to optimise gas hydrate inhibitor injection rate, numerous different analytical techniques have been used to determine the concentration of THIs, KHIs, and salt in the aqueous phase. Some experiments were conducted with an acoustic multisensory for determination of the MeOH and ethanol concentration in aqueous solutions without the presence of salts. A combination of near-infrared (NIR) and chemometric methods were applied to measure alcohol contents in gasoline1128,1129 and ppm level concentration of dissolved chloride salts of a variety of alkali and earth alkaline cations in water.1130 Additionally, tests using an ultraviolet (UV)-visible spectrometer was performed to monitor the concentration of PVCAp1131 and different types of polymers1132 in water. Recently, Haghi et al.1133 developed a novel methodology by combining the UV and NIR spectra associated with the partial least-squares (PLS) method to estimate the concentration of MEG, MeOH, and PVCap with/out presence of NaCl in water samples mainly for inhibitor injection rate optimisation purposes. A sealed pipe on a rocking surface called a rock-flow cell1134 is a newly developed testing flow assurance tool to understand the formation and deposition of solids particles including gas hydrates under suitable field conditions including transient conditions, appropriate shear stress and involved flow regimes. Additionally, a new unidirectional growth technique1021 has been employed to understand the interfacial phenomenon of tetrahydrofuran (THF) clathrate hydrate crystal growth from PVP containing an aqueous phase to study the influence of KHIs on hydrate crystal growth inhibition. For more rapid and accurate evaluation of KHIs, a new crystal-growth inhibition (CGI) based technique1135,1136 has been developed with principles similar to the semi-industrial hydrate flow loop. Fig. 18 depicts the experimental CGI regions identified for 0.5 wt% PVCap with methane in both the autoclave setup and the rocking cell which exhibit that all the determined CGI regions are identical in both experimental facilities with minimal discrepancy within the ±0.5 °C experimental error.
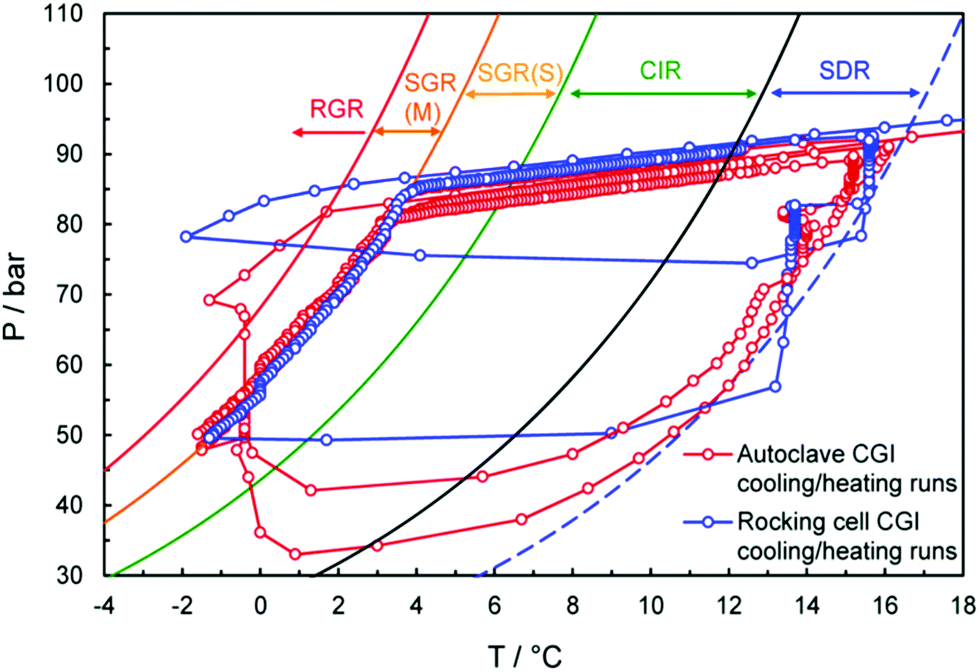 | ||
| Fig. 18 Experimentally identified crystal growth inhibition (CGI) regions for 0.5 wt% PVCap in aqueous phase + methane; SDR: slow dissociation region, CIR: complete inhibition region, SGR(S), slow growth region (slow), SGR(M), slow growth region (medium), and RGR: rapid growth region. CGI cooling and heating tests for 0.5 wt% PVCap in aqueous phase + methane in the rocking cell and standard autoclave setups (adapted with permission from Mozaffar et al.,1135 Copyright 2016 American Chemical Society). | ||
Generally, it is well understood that gas hydrates are formed either in the bulk aqueous phase or the interspersed liquid phase. Most of the techniques which have been employed to form gas hydrates in the interspersed aqueous phase in which the vapour–aqueous phase interface area and mass transfer between phases are increased accordingly.
Numerous research studies on the effects of nanoparticles on gas hydrate formation have been conducted in an agitated vessel902,915 instead of a packed bed849 in the attendance of different chemical compounds including hydrogels,1137 porous polymers,1138 and foams.1044 The utilisation of nanoparticles in solution has a positive influence on the enhancement of hydrate formation kinetics. Through the same drivers of surfactants’ effectiveness, the affirmative influence of nanoparticles can be lessened as their concentration increases, which is occurred due to the extreme heat generation and a raise in the viscosity of the mixture.899 Detailed discussion in this regard are presented in the growth acceleration section (Section 3.5).
4.3. Advancement
Focused beam reflectance measurements (FBRM)1142 are often utilised to investigate the dispersed hydrate particle size. The FBRM probe determines both the number and size of chord lengths at a specific location in space, which result in the creation of a chord length distribution (CLD). The FBRM experiments have been successfully conducted for emulsions characterisation and droplet size distributions in oil,1143,1144 illustration of hydrate nucleation and growth behaviour,1145 evaluation of anti-agglomerate additives performance1146,1147 and estimation of hydrate formation and dissociation rate in gas–water solutions.1148 Moreover, the FBRM experimental data has been combined with resistance-to-flow determinations1142 to unravel the connection between hydrate particle distribution and bed formation and determine the onset of hydrate bed formation.
5. Gas hydrates in nature
5.1. Energy recovery
Naturally occurring gas hydrates that have accumulated in submarine continental margins (>97%), and permafrost regions (3%>) over millennia with methane as the predominant guest molecule, are considered as a low-carbon energy source and a CO2 sink. The uncertainty in estimating total hydrate resources introduces considerable variability into the resulting projections of the potential contribution of these reserves. However, estimation of the amount of energy stored in the hydrate reservoirs consistently outweighs that of the other conventional energy sources combined. For a more exhaustive examination of studies into gas hydrate reservoirs, the reader is referred to available reviews.1156–1161 The purpose of this part of the review is to offer a brief overview of gas hydrates in nature and present the most recent advancement in strategies that have considered the potential options for energy recovery from hydrate reservoirs. A graphical summary of this section is provided in Fig. 19. | ||
| Fig. 19 Potential methods for extracting gas from hydrate reservoirs: (1) thermal stimulation: raising temperature of the reservoir by various techniques (yellow dot) to move the system condition outside of the Hydrate Stability Zone (HSZ) (red dot). (2) Depressurization: reducing the pressures of the reservoir by removing part of the gas phase to move the system outside of HSZ stimulates the hydrate dissociation. (3) Chemical potential shift – (i) inhibitor injection and (ii) smaller guest gas injection: shifting the HSZ above reservoir condition triggering the dissociation of hydrates. (iii) CO2 replacement: destabilizing the existing gas in the hydrate phase by injection of more favourable guest molecules. | ||
Although thermal-stimulation methods have been considered as a promising candidate for fulfilling the gas-hydrate production goals from hydrate reservoirs, it is not an economically viable solution unless the hydrate layers exhibit sufficient thickness and saturation. To conquer this drawback, some strategies have attempted to combine thermal stimulation with other methods that are discussed in following sections.
As the gas is produced following the depressurization, the reservoirs can exhibit improved permeability when compared to its initial status. Thus, the availability of methane gas near the wellbore is expected to improve in long-term operations, resulting in higher production rates. This factor plays the main role in the majority of the numerical simulations. However, there are still economic barriers regarding the challenging access to gas hydrate reserves, uncertainty on predicting the recoverable amounts,1169 and more importantly, significant environmental hazards associated with the stability of the geological structures after gas production, all of which are the subject of ongoing discussions. The endothermic dissociation of gas hydrates,1188 which would require an external energy source to transfer heat to the reservoirs, poses additional challenges and stands as the main challenge in the experimental modelling. It has also been reported that depressurization, apart from being a slow method, also has the possibility of ice formation1189 when rapidly done, thereby causing a reduction or complete blockage of permeability which in turn affects the volume of gas produced. Accordingly, it is been widely suggested in the literature to combine the depressurisation technique with others to remove the aforementioned barriers. While thermal stimulation or inhibitor injection are expensive methods, depressurization usually requires the addition of heat to ensure that the heat consumed by decomposition of the gas hydrate will not reduce the production rate.1190,1191
5.1.3.1. Inhibitor injection. Inhibitor injection is an established subject initiated decades ago1192 and mostly applied to flow assurance related topics. However, we found only a limited number of studies dealing with application of this methodology in hydrate bearing sediments. Research interest has decreased in recent years, mostly driven by increasing concerns of the effect of injected inhibitors on the environment and the high cost of the method when considering amount of inhibitor that is required to be injected into the reservoir. Inhibitor injection is another strategy to shift the chemical potential of the hydrates by depressing the activity of water in coexisting phases to overcome the hydrogen bonding of water framework. The freezing-point depression of water by thermodynamic inhibitors is analogous to this method. The main inhibitors used in this method are salts and alcohols, the effect of which are reviewed in previous chapter. The conventional workflow in the preparation of methane recovery strategies with inhibitor injection begins with the selection and injection of inhibitors, which can be heated before injection, followed by depressurization. In the most recent studies, two groups of workers investigated the efficiency of methane production under MeOH1193 and polyethylene glycol (PEG) polymer1194 injection with various concentration and rates, and defined the optimum rate and concentration for inhibitor injection. Beside inhibitor injection concentration and rate, reservoir temperature, pressure and hydrate surface area in contact with inhibitor are important parameters for improving the efficiency.1192 The possibility of combining this method with other methods will be discussed in following sections.
5.1.3.2. Heavier guest gas injection. The key approach here to produce methane is to use the higher stability of CO2 in hydrates (causing the chemical disequilibrium) for replacement with methane, storing CO2 and producing methane. A similar approach can be used to separate CO2 from different gases streams. A notable research in the development of this method has been conducted over the past 10 years.1195,1196 Hence, more detailed review about the application of gas hydrate related technology in CO2 research is provided in Section 6.
5.1.3.3. Smaller guest gas injection. One of the established methods of gas exploitation from gas hydrate reserves is injecting smaller guest gases with a higher pressure hydrate stability zone (HSZ), releasing gases from the cages owing to chemical potential instability. This could address the safety concerns regarding the high-pressure gas pockets trapped between isolated regions and allow production of methane hydrate at higher pressures within the methane HSZ. Several gas mixtures candidates have been suggested in the past decade, mainly N2 mixed gases. Injection of N2, in this context, changes the fugacity of gas phases, making it easier for the CH4 molecules encaged in water framework to be released, which may also be used as an alternative way for flow assurance.1197 In a recent study, Okwananke et al.1198 experimentally investigated the kinetics of methane recovery by N2 and compressed air injection followed by step-wise depressurization over range of temperature and concluded that pure N2 is a more efficient agent for methane recovery. Zhang et al.1199 focused on N2 injection methods by using a MRI system in order to characterize the phenomena and found that initial fast dissociation of CH4 is followed by the slower rate limited N2 diffusion. Both the mentioned studies suggest that this method is more useful for the dispersed hydrates with higher contact areas to increase the rate of dissociation. The rest of the studies combined with CO2 mixed gases which will be reviewed in Section 6.
5.2. Environmental aspects
Gas hydrate dynamics (see Fig. 20) and past climate events have been linked in the literature for decades, and researchers have investigated the possible synergies especially for the Paleocene-Eocene Thermal Maximum hyperthermal event1212–1214 and the Late Quaternary “clathrate gun hypothesis” of Kennett et al.,1215,1216 which was re-examined by Cannariato and Stott.1217 Recent findings about hydrate dynamics during these time periods are summarized by Ruppel and Kessler.1 For the contemporary Earth, gas hydrates in certain geological settings could already be breaking down due to warming ocean waters1205 and the residual effects of deglaciation since the Last Glacial Maximum,1218 which are mostly relevant at high northern latitudes. The feedback component, which requires methane formerly bound in hydrates to reach the atmosphere, is not operating over the largest (deepwater marine) component of global reservoirs1219–1221 or the shallow-water setting linked to degrading subsea permafrost.1222 Elevated sea-air methane fluxes observed at other high-latitude sites1223,1224 cannot presently be linked to methane released by gas hydrate degradation.
 | ||
| Fig. 20 Schematic shows various terrestrial and marine settings for gas hydrate occurrences and the impact of warming climate on gas hydrates. The base and top of the gas hydrate stability zone are denoted by dashed black and dashed violet curves, respectively. Note that the top of the gas hydrate stability zone in the ocean is within the water column, meaning that the seafloor lies well within the stability zone at depths greater than the upper continental slope. Permafrost is denoted as blue shading beneath the ice sheet (cold-based glacier) on the far left and beneath the land (terrestrial permafrost) and high-latitude continental shelf (subsea permafrost). Bubbles are shown to indicate possible gas emissions associated with hydrate breakdown and are color-coded depending on their fate. White bubbles are emitted as methane at the seafloor, but the methane is rapidly stripped from the bubbles so that no methane remains by the time these bubbles reach the sea surface. Methane that persists through the near-seafloor zone of anaerobic methane oxidation (shown in red) is usually oxidized aerobically once it reaches ocean waters. Blue bubbles are produced by gas hydrate degradation at the base of the stability zone, which will be the first locus of dissociation in the deep marine environment in response to ocean warming. In permafrost areas, gas hydrate can simultaneously dissociate at the top (purple bubbles) and bottom (blue bubbles) of the stability zone. The blue bubbles indicate gas that might re-form as new gas hydrate as the gas migrates upward to the stability zone after release. Purple bubbles have a better chance of migrating through the sediments towards the surface if they are not stopped by low permeability ice-bearing permafrost. This methane may be oxidized by microbes within sediments or remain as methane and continue migrating upward if pathways exist. Bubbles within no colour and outlined in black are those that may emit methane to the atmosphere. Locations experiencing deglaciation or subsea permafrost thaw since the last glacial maximum are the most prone to releasing gas hydrate methane that can reach the atmosphere. Upper continental slopes on many marine continental margins are likely experiencing net hydrate dissociation now, but this methane is not reaching the atmosphere based on the studies completed to date (adapted with permission from Ruppel and Kessler,1 Copyright 2017 John Wiley and Sons). | ||
There are several characteristics of gas hydrates that motivate interest in possible synergies between gas hydrate dynamics and climate change. Firstly, 15% of global methane, corresponding to ∼1500 Gt carbon, is estimated to be trapped in methane hydrate deposits,1160 and methane is a greenhouse gas that is 25 times more potent (over a century) and 84 times more potent (over two decades) than CO2 on a unit mass basis.1225 Secondly, most gas hydrates exist at shallow depths (up to a few hundreds of meters) beneath the seafloor, even shallower depths in deglaciated terrains, and deeper depths, but still closer to the surface than conventional reservoirs, in permafrost areas. The proximity of these deposits to the surface/seafloor means that migration pathways are relatively short compared to those for gas migration from conventional reservoirs. Thirdly, because gas hydrates are stable only in a specific P–T range, climate change and sea level perturbations can perturb hydrate stability. For plausible changes in sea level and temperature over the next few centuries, the effect of rising temperatures on the gas hydrate reservoir will likely outstrip the stabilizing effect of sea level rise nearly everywhere in the oceans.1
An exhaustive review by Ruppel and Kessler1 provides deep background on climate–hydrate interactions. They discuss the major geologic settings for gas hydrates and possible synergies with contemporary climate change; assess how the Intergovernmental Panel on Climate Change (IPCC) reports have incorporated hydrate sources;1225 and detail the challenges for accurately tracking methane released from gas hydrate through the geosphere, ocean, or atmosphere. A review by James et al.1226 provides additional information focused on the Arctic Ocean.
For the contemporary Earth, research mostly focuses on the response of gas hydrate reservoirs to warming ocean waters or atmospheric temperatures.1,1227 Such perturbations can drive hydrate dissolution in pore waters within the stability zone and cause hydrate dissociation where gas hydrates are close to the stability boundary, which is a relatively narrow zone within the formation. For most marine hydrates, warming-induced dissociation would occur only at the base of the reservoir, while permafrost-associated and deglacial hydrates can experience dissociation simultaneously at both the top and base of the hydrate zone.1228
Dissociation of gas hydrate releases methane into the formation, where the methane mostly remains trapped. If the gas migrates to shallower depths, it often encounters the hydrate stability zone, where the gas may re-form hydrate with available free water, or the permafrost zone, where water ice may block flow conduits and prevent methane from reaching the surface. Methane released by hydrate dissociation in marine environments also encounters a biochemical barrier to reaching the seafloor since more than 90% of methane may be consumed by AOM processes within the sulfate reduction zone (SRZ) in the shallowest part of the seafloor.1229
If methane survives transport through the SRZ1230 and reaches the seafloor, emitted bubbles usually do not retain their methane for long as they ascend in the water column due to dissolution into the seawater.1231 Nominally, gas bubbles emitted at water depths greater than 100 m (the shallowest deepwater marine hydrate zone is ∼350 m in high-latitude waters) will be stripped of their methane before the bubbles reach the sea surface.1231 Bubbles emitted at seafloor that lies within the hydrate stability may form hydrate rinds as they ascend through the water column,1232,1233 but the methane still escapes from the bubble,1234 especially after the hydrate shell disappears. Methane dissolved in the oceans is rapidly consumed by bacterial aerobic oxidation, producing CO2 as a byproduct.1235,1236 This process can acidify deep waters1237 but does prevent methane from reaching the atmosphere. An increasingly number of studies demonstrate that, even over seafloor seeps, sea-air methane flux in deep ocean environments is very low, partially because of the consumption of methane in the water column. Some studies also show that methane dissolved in near surface waters is dominated by young methane produced by planktonic processes, not the fossil methane typically trapped in hydrates.1221,1222 The details of methane release for hydrate settings other than the deepwater marine environment are provided later in this section.
Where are contemporary gas hydrates most susceptible to climate change processes? At water depths greater than ∼1000 m, seafloor conditions are supercooled relative to the hydrate stability, and bottom water temperature is expected to remain within the stability zone for hundreds to thousands of years into the future.1,1227 The bulk of the global gas hydrate reservoir is therefore unlikely to be noticeably perturbed by warming climate over these time scales. Many studies of climate-driven gas hydrate degradation focus on upper continental slopes at the landward or “feather” edge1227,1238 of gas hydrate stability,1,431,1239–1242 which corresponds to 450 to 700 m water depth in temperate regions. Ruppel1204 estimated that ∼3.5% of the gas hydrate reservoirs could be within this depth range although several factors could contribute to gas hydrates being rarer in these locations than elsewhere in deepwater reservoirs. At upper continental slope water depths, seafloor P–T are close to the stability boundary. Ocean warming on seasonal, decadal, or longer time scales could lead to dissociation and release of methane, which does not need to migrate far through the sediments to reach the seafloor. The discovery of upper continental slope methane seeps1243–1246 on numerous continental margins in the past few years has led to speculation that some of these features are sourced from gas hydrate dissociation. The connection is difficult to prove, particularly where the leaking gas does not have a special composition that can be linked to that of local gas-hydrate deposits.1247
A special case of climate-induced hydrate dissociation applies to high-latitude regions, where onshore permafrost has been thawing rapidly, even in the past decade. Intrapermafrost gas hydrate (Dallimore et al., 2015), which is thought to be relatively rare, could be experiencing dissociation due to such thawing. Permafrost that developed sub-aerially (i.e., not below ice sheets) is unlikely to host methane hydrates shallower than ∼200 m deep, and contemporary methane releases in permafrost areas are probably attributable almost exclusively to non-hydrate sources.1
Subsea permafrost was formerly subaerial permafrost before it was inundated during deglacial sea level rise.1248–1255 Degradation of subsea permafrost and any associated hydrates has been rapid since the Last glacial Maximum (LGM). However, water column methane does not carry a strong fossil methane signature, as would be expected if the methane were derived from gas hydrate degradation.1222
Formerly subglacial hydrates are a special case since hydrate can form at shallow depths in the ground beneath thick ice sheets (e.g., Fig. 11 of Ruppel and Kessler1). Deglaciation raises the temperature and releases the pressure on these deposits, which then dissociate. Some of the gas released from the seafloor into shallow seas that cover such areas may reach the atmosphere1256–1259 although more research is required to link methane releases directly to gas hydrate dissociation. Deglaciation could also be a factor in future methane releases from gas hydrates currently beneath ice sheets in Antarctica1260 and Greenland.1261
In fact, part of the released methane migrates upwards into the sulfate methane transition zone (SMTZ), where sulfate is diffusing from upper layers, causing anaerobic oxidation of methane (AOM) by syntrophic partnership between anaerobic meth-anotrophs (ANME) and sulfate-reducing bacteria.1230,1278 Here, methane acts as a fuel for the growth of anaerobic microorganisms.1279 This oceanic methane biogeochemistry process, serve as a major sink, causing considerable increase in dissolved inorganic carbon, alkalinity, and hydrogen sulphide. Promotion of the precipitation of authigenic carbonates and iron sulphide is another effect of this process. This in turn, influences the sea floor morphology, and landscape.1229
The spatial association between hydrate-related features (e.g., BSRs, gas-charged sediments) and submarine slope failures has been well-established on the margins of the Atlantic Ocean, northern Gulf of Mexico, the western Beaufort Sea, South China Sea, and Norwegian part of the Arctic Ocean, as well as on the New Zealand and Cascadia margins.380,1286–1291 Among the many slides investigated for their potential interaction with gas hydrates, two mega-slides, the Storegga Slide offshore Norway379,1292,1293 and the Cape Fear slide offshore the Southeastern United States,1281 have been the best studied and experienced some failure stages large enough to have triggered tsunami. Neither of these mega-slides are now interpreted to have been triggered by processes related to gas hydrate dynamics: Haflidison et al.1294 show that the Storegga slide started at the toe, where gas hydrate would have been actively forming in deepwater. A recent interpretation of the Cape Fear Slide highlights the role of inherited structure, sediment loading, and fluid flow in the initiation of the seafloor failure.1282
Many seafloor landslides originate on upper continental slopes in the depth range where gas hydrates are relatively ubiquitous on marine continental margins, and thus the spatial association between slides and hydrate-related features is not surprising. While the evidence for a link between gas hydrate dynamics and some other seafloor destabilization features1258,1295,1296 (e.g., pingos, pockmarks) is strong, the connection between gas hydrates and submarine slides remains more tenuous. Controversy persists about whether the existence of gas hydrates and/or free gas in the sedimentary section pre-conditions slopes to fail when triggered externally (e.g., by an earthquake) or whether the hydrate-related features directly provoke slope failures.
Addressing this controversy requires reconstructing conditions in the hydrate reservoir prior to slope failure, a task rendered difficult by the fact that seafloor deformation and mass transport accompanying landslide events destroy many of the pre-existing features. The mechanical state of the slope immediately before failure depends on a combination of factors unrelated to gas hydrate dynamics (e.g., bulk lithology, sedimentary layering, rate of sedimentation/erosion, fluid advection rates, and pore pressure, which can also be modulated by hydrates and gas) and factors related to the hydrate system itself (e.g., hydrate and gas saturation, whether hydrate forms in unconsolidated or already consolidated sediments1297). As noted in Section 2.5, gas hydrates are rheologically much stronger than saturated, unconsolidated marine sediments,334 but hydrates only provide cohesion to sediments when its saturation exceeds ∼40% of pore volume.138,1298 At lower saturations, gas hydrate accumulations are strong components set in a weaker matrix of saturated marine sediments. Even low saturations of gas hydrate can clog permeability and lead to increased pore pressure, which in turn enhances the likelihood of hydraulic fracturing, gas migration, formation of pipe structures, and seafloor failure1285,1299–1301 even for relatively gas hydrate deposits well within the stability zone. Stronger, hydrate-bearing sediments1302,1303 are also typically arrayed above rheologically weak, gas-charged sediments that are below BSRs. The contrast in mechanical properties between hydrate-bearing sediments above and gas-charged sediments below could produce an unstable condition that leaves slopes poised for failure when triggered by external (e.g., earthquakes, tides) or internal (hydraulic fracturing) events.
Although some researchers have postulated that depressurization of upper continental slope gas hydrates during periods of lowered sea level triggered widespread slope failures,1304,1305 slide timing does not correlate well with sea level low-stands. Hydrate dissociation caused by warming of intermediate-depth ocean waters impinging on upper continental slopes therefore receives more attention in contemporary studies.1240,1241,1246,1306,1307 Unless the gas and water released by hydrate dissociation at the base of the stability zone are removed by advection, the pore pressure can increase, weakening slope sediments through liquefaction or fracturing. Whether these changes drive slopes to failure or merely condition slopes for later failure returns to the crux of the controversy surrounding the interplay of gas hydrate dynamics and seafloor instability.
To place hydrate dissociation in context, it is important to consider that hydrate saturations are probably less than a few percent of pore space in most marine sediments; that hydrate dissociation is endothermic and therefore self-limiting, meaning that runaway dissociation driven by only a single factor (e.g., ocean warming) is unlikely;1 and that hydrate dissociation in response to natural driving forces is a relatively slow process and already occurs continuously at the base of the stability zone in response to sedimentation. The best-studied submarine slide events occurred over several stages and thousands of years,1282,1294 and it is possible that none, only one, or a few of the stages of these complex slope failures were preceded by hydrate-related processes, including dissociation.
Studies have attempted to link the timing of submarine slope failures to climate warming events such as Late Quaternary Dansgaard/Oeschger (D/O) cycles.1308 However, within the limitations of available age data, large-scale slope failures in the North Atlantic coincide more closely with Heinrich events,1309,1310 which are characterized by episodes of ice rafting and injection of cold meltwaters.
As noted by Maslin et al.,380 much better dating of submarine slide events will be required before the temporal correlation between seafloor destabilization episodes and climate perturbations can be unraveled. Maslin et al.380 also posit that future submarine slide events may be connected not to warming-induced dissociation in gas hydrate reservoirs, but rather depressurization-induced dissociation caused by melting of ice sheets in Greenland and Antarctica. Deglacial episodes since the Last Glacial Maximum are already implicated in some smaller-scale seafloor destabilizations like crater, pockmark, and marine pingo development.1258,1311,1312
Anthropogenic activities that affect hydrate-bearing sediments may also induce seafloor failure. For example, extraction of deep, warm fluids through a conductor that crosses hydrate-bearing layers could drive dissociation near a borehole, weaken seafloor sediments, and possibly threaten seafloor infrastructure.1313,1314 In a case documented by Hadley et al.,1315 the oil production strategy was altered to avoid the hazards associated with attendant hydrate dissociation in the reservoir. Seafloor stability is also a consideration in deepwater marine production testing that is conducted to analyze the efficiency of methane extraction during controlled gas hydrate dissociation.1316,1317 Depending on the depth and lithology of the target hydrate reservoir and its hydrate saturation, the rate of dissociation and gas extraction, and the pre-existing shear stress in the sediments, seafloor collapse could be a potential hazard.
 | ||
| Fig. 21 The schematic depiction of the solar system shows the planets and other celestial bodies at their approximate orbital positions relative to the sun. The sizes of Pluto, Ceres, the moons of some gaseous planets, and the comet have been increased to make them visible. Bodies portrayed in colour are known or postulated to have clathrates at the surface, in their interiors, or in their atmospheres. For Jupiter, Saturn, and Uranus, only the moons with possible clathrates are shown. Comets move through the solar system, and one is portrayed here schematically to highlight that clathrates likely occur in some of these icy bodies. | ||
As recognized by Mille1324 more than 50 years ago, P–T conditions on the surface of or within the interiors of some planets, moons, and comets and within some planetary atmospheres are suitable for the formation and long-term stability of clathrates, including complex, multi-gas hydrates or hydrate structures that would be considered exotic on Earth.34,323,1320,1325 Water, the critical building block for clathrate cages, is confirmed or inferred to exist in liquid or frozen form on Mars, Europa and Ganymede (moons of Jupiter), Titan and Enceladus (moons of Saturn), Pluto, and the dwarf planet Ceres, and water ice is a common constituent of comets.1326
On the surface or within the interiors of Mars, Saturn, Uranus, Neptune, and Pluto and on moons of Jupiter, Saturn, and Uranus, methane, ethane, CO2, ammonia, and/or other gases coexist with water phases in varying amounts and are abundant enough to form clathrate. CO2 and methane hydrate could form at or near the surface of Mars, particularly at the polar ice caps.846,1327–1330 Methane and ethane hydrates are postulated to comprise the outer icy shell of Titan,1320,1321,1331,1332 and methane hydrate may contribute to the strength of Ceres’ outer crust.1333 On Pluto, methane hydrate is postulated to form an insulating layer that protects the near-surface water ice from the deeper and warmer liquid-water layer.1334 On Enceladus, the dynamics of water plumes detected at the surface may be affected by the presence of ammonia hydrate.1335
Clathrates also occur in comets,1336,1337 which has led to the inference that hydrates may form within the interstellar medium,1338 a view challenged by some laboratory experiments.1339 The existence of clathrates on highly mobile comets implies that these objects could be important in delivering compounds like methane to other extraterrestrial bodies.1340,1341
The recognition of extraterrestrial clathrates containing gases sometimes produced on Earth by biological processes (e.g., methane, ethane) has provoked substantial interest among astrobiologists. Outstanding questions include the source of the hydrocarbon compounds trapped in some extraterrestrial hydrates and the potential for other bodies in the solar system to host the type of hydrate-associated microbial communities found beneath the ocean floor on Earth.1326,1342,1343
6. Gas hydrate-based CO2 capture, transport and storage
The IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels suggests that, to limit the possible impacts and risks on the people, economies and ecosystems to moderate levels, net global emissions of anthropogenic CO2 must be reduced by about 45% by 2030 and to zero by 2050 compared to the 2010 levels.1344 In addition to the use of low-carbon energy and improvements to energy efficiency, CCS has also been considered as one of the most effective approaches to meet the stringent reduction target for CO2 emissions.1345 CCS operation is a chain of CO2 capture or separation from flue gas, transport of the captured CO2 to a storage site and finally, storage of the CO2 in a desired geological formation. The current CO2 capture technologies such as physical adsorption, chemical absorption, cryogenic separation, and membranes are technically applicable but not economically viable for large-scale applications in the short to medium term.1346,1347 Gas hydrate-based CCS technologies have been extensively studied in the past decades as a route towards significant improvements to the economic feasibility of large-scale CCS operations.13486.1. CO2 capture
CO2, N2 (nitrogen) and O2 (oxygen); H2 (hydrogen) and CO2; and CH4 (methane) and CO2 are the major gas components for a flue gas, fuel gas, and biogas, respectively. All these gases can form hydrates at certain thermodynamic conditions. Experimental data and thermodynamic prediction of gas hydrate phase equilibria show that, for temperatures from 273 to 283 K, the phase equilibrium pressure of CO2 hydrate is more than 10 MPa lower than those of N2 hydrate and O2 hydrate, and more than 200 MPa lower than that of H2 hydrate. The significantly lower formation pressure of CO2 hydrate leads to a higher affinity to occupancy of the suitable cages in hydrate crystals compared to the other gases, which is the fundamental basis of a hydrate-based gas separation process (HBSP). Chazallon and Pirim quantitatively found that the flue gas containing 2 to 70 mol% CO2 will form structure I hydrate, while structure II hydrate was thermodynamically stable for the flue gas with 1 mol% CO2.1350 Hassanpouryouzband et al. observed formation of sII hydrates in the presence of CO2 + N2 at higher pressures with up to 14.6 mol% CO2 in the system.1351 Furthermore, it was found that statically constant P–T conditions are not signs of thermodynamic equilibrium for the CO2–N2 mixtures and a significant fraction of CO2 captured in the hydrate phase could occur at the final constant pressure.1352 In short, flue gas or fuel gas can form CO2 hydrate or CO2-mixed hydrates in which CO2 occupies the majority of the guest molecules at certain temperature and pressure conditions; decomposition of the hydrates can produce CO2-rich gas; the CO2-rich gas could be further processed through a series of HBSP cycles to achieve the desired purity of CO2 gas.
6.1.2.1. Thermodynamic promoters. Thermodynamic promoters are chemical additives that are added to the system to reduce the hydrate formation pressure.1060,1353–1357 These additives themselves can form hydrates in which some cages are left to accommodate small molecules such CO2, N2, CH4 and H2. The most-studied thermodynamic promoters include tetrahydrofuran (THF), cyclopentane (CP), propane (C3H8) and tetra-n-butyl quaternary ammonium along with phosphonium salts such as TBAB, tetra-n-butyl ammonium chloride (TBAC), TBAF, tetra-n-butyl ammonium nitrate (TBANO3), tetra-n-butyl phosphonium bromide (TBPB), and tetra-n-butyl phosphonium chloride (TBPC),54,1348,1358–1360 some of which are discussed in Section 2.1.2.
The THF, CP and C3H8 are structure II hydrate formers. Given that their molecules will occupy some of the large cavities of the structure II hydrate, CO2 molecules can only fill the rest of the large cavities or some of the small cavities and the occupancy depends on the gas composition and thermodynamic conditions.1361–1363 The tetra-n-butyl ammonium and phosphonium salts form semiclathrate hydrates.54,1364,1365 X-ray diffraction and Raman spectroscopic analyses indicated that there are different structures of the semiclathrate hydrates, such as tetragonal hydrate structure (TBAC), orthorhombic hydrate structure (TBPB and TBPC), and polymorphic phases with both orthorhombic and tetragonal hydrate structures (TBAB).1366,1367 In such semiclathrate hydrate structures, water molecules incorporated with the cations form cage-like structures through hydrogen-bonds, whilst tetra-n-butyl ammonium (TBA) cations and tetra-n-butyl phosphonium (TBP) cations (i.e., the butyl chains) occupy the large cages. The small pentagonal dodecahedral cages (512) are usually left empty for small gas molecules such as CO2, N2, CH4 and O2. Fig. 4 illustrates the structure of TBAB hydrate with a hydration number of 38 ((C4H9)4N+·Br−·38H2O).1366 Br− anions and water molecules build the clathrate cages indicated in solid lines, two TBA cations fill four large cages, six dodecahedra are empty, the dashed lines indicate the broken part of the cage structure (H atoms are not indicated for simplicity).
The presence of low concentrations of the thermodynamic promoters leads to a large shift of the hydrate phase equilibrium conditions towards lower pressure or higher temperature, which is crucial to reduce the energy penalty of the gas hydrate-based CO2 capture process. The shift in the hydrate formation pressures and temperatures is related to both the concentration of the promoter and the composition of the gas mixture. In general, the reduction in the hydrate formation pressure is relatively smaller for the structure II hydrate promoters than for the semiclathrate hydrate promoters. Among the aforementioned semiclathrate hydrate promoters TBAF has the lowest formation pressure, hence the highest formation temperature.1365 Two parameters, namely split fraction and separation factor, are introduced for a quantitative assessment of HBSP:1368 split fraction is defined as the ratio of the amount of CO2 in the hydrate phase and the feed gas mixture, indicating the capability of CO2 capture with hydrate formation; the separation factor is defined as a ratio of the relative amount of CO2 and the second gas component in the hydrate phase and gas phase after hydrate formation, indicating the capability of separating CO2 from the other gas component. Furthermore, low formation pressures and high formation rates are essential to facilitate a HBSP for CO2 capture at industrial scales.
Kim et al. investigated the effect of three typical types of thermodynamic hydrate promoters on CO2 capture from a simulated flue gas that was composed of 20 mol% CO2 and 80 mol% N2, including THF (soluble in water, sII clathrate former), CP (insoluble in water, sII clathrate former), and TBAC (water-soluble, semiclathrate former).1362 They set the experiments at the same pressure (3.1 MPa) and the same degree of subcooling (5.0 K) in the presence of the same volume of the aqueous phase. For 1.0 mol% of each promoter, the TBAC hydrate had the highest CO2 concentration in the hydrate phase and the highest CO2 split fraction. This is attributed to the fact that THF molecules and CP molecules will form structure II hydrates and occupy a certain portion of the large cages (51264) while CO2 molecules also prefer the large cages. This competition considerably reduces the availability of the large cages for CO2 molecules.1369 Hashimoto et al. reported dependence of CO2 selectivity on semiclathrate hydrate structures of TBAB, TBAC, TBPB and TBPC for a gas mixture of 15 mol% CO2 and 85 mol% N2. Their results showed that as the initial pressure increases, the gas uptake in the hydrate phase linearly increases and the mole fraction of CO2 in the hydrate phase does not change much for TBAC, TBPB and TBPC. The CO2 fraction irregularly responds to the increase in the initial pressure at 1 MPa. Given that, the TBAC has the lowest gas uptake and the highest CO2 fraction in its hydrate phase, which was also reported for CO2–CH4 gas mixtures (i.e. fuel gas),1370 it is a plausible explanation that the orthorhombic structure of the TBPB and TBPC hydrates has the largest gas capacity,54 the tetragonal structure of the TBAC hydrates has the highest selectivity for CO2, and the TBAB hydrates with both orthorhombic and tetragonal hydrate structures show the highest gas capacity and high selectivity for CO2 capture. Rodriguez et al. recently reported that the separation factor of TBAB hydrates depends slightly on the cooling rate and strongly on the types of TBAB hydrates.1371
The CO2 split fraction and separation factor are relative to a number of parameters such as the composition of feed gases, the concentration of the chemical additive, and the driving force (i.e., the difference in the temperature and pressure between the experimental conditions and the hydrate equilibrium conditions). The CO2 split fraction also strongly depends on the ratio of the feed gas to the aqueous solution. As a typical example, both CO2 split fraction and separation factor are much higher for a fuel gas containing 40 mol% CO2 and 60 mol% H2 than for a flue gas containing 15 mol% CO2 and 85 mol% N2. This can be understood given that there is a much higher CO2 content in the fuel gas and H2 molecules require a significantly higher pressure to stay in the hydrate cages compared to N2 molecules in the flue gas.1360 Moreover, the presence of thermodynamic promotion additives usually reduces the separation factor and split fraction for fuel gases but not for flue gases.1348
6.1.2.2. Kinetic promoters. Some surfactants in the aqueous phase can act as kinetic promoters to shorten the induction time and promote hydrate growth,776 while some other surfactants are used to prevent gas hydrate agglomeration for flow assurance (Section 4.1.3) and control of hydrate morphology (Section 3.1.4). There are three types of surfactants including anionic, cationic, and non-ionic surfactant. Experimental studies showed that anionic surfactants such as SDS, sodium tetradecyl sulfate (STS) and sodium hexadecyl sulfate (SHS)871 are the most effective kinetic promoters692,1372 when compared to non-ionic and cationic surfactants such as Tween-40, Twenn-80, DTACl875 and amino acids.1373,1374 The molecules of such a surfactant have a hydrophilic head and a lipophilic (hydrophobic) end that are amphiphilic to both polar and nonpolar substances. At a concentration above the critical micellar concentration,686,1375 the molecules of a hydrate former form some kind of molecular clusters with the surfactant molecules through the hydrophobic interactions. Raman spectra indicate that these molecular clusters may have a structure similar to the hydrate cages.1376 The presence of surfactants also enhances dissolution of hydrate forming gases in water, supplying gas molecules for fast hydrate growth. These enable hydrate structures to form more easily and faster and thus reduce the induction time. Furthermore, surfactants reduce the interfacial tension between the surfaces of hydrate crystals and water, resulting in a significant influence on the hydrate morphology. Lo et al. investigated the adsorption of SDS on CP hydrates and THF hydrates by measurement of zeta potential and pyrene fluorescence. As schematically illustrated in Fig. 22, the dissociated DS− anions adsorb on the CP hydrate surface and then the electrostatic repulsion forces push the hydrate particles apart from each other.686,1375 As a result, the SDS adsorption leads to the formation of small hydrate particles and hence good contact between gas and water, promoting the kinetics of gas hydrate formation.
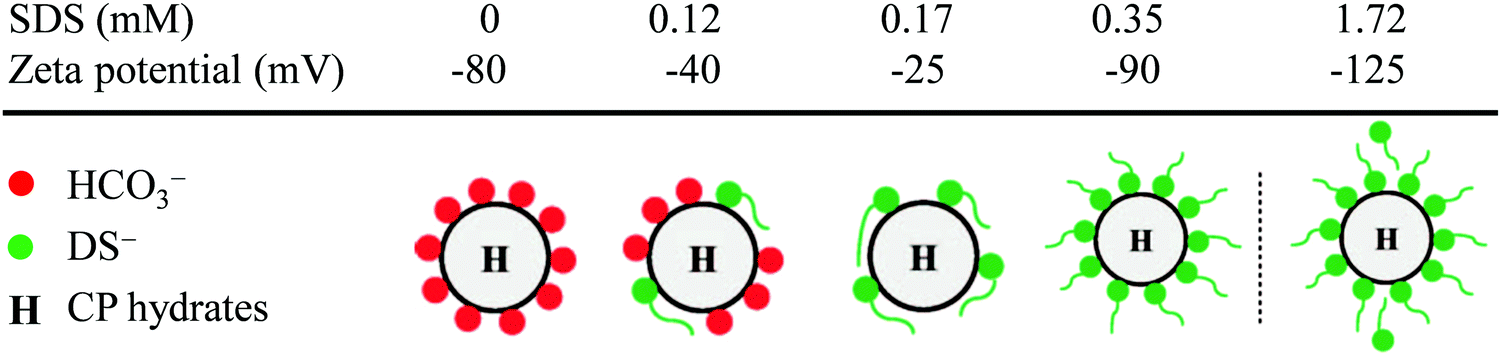 | ||
| Fig. 22 SDS adsorption at the interface between CP hydrates and water. It was assumed that the bicarbonate anions (HCO3−) came from dissolved CO2 from air. A monolayer of DS− is completed at 0.17 mM of SDS, at which the induction time sharply decreased (adapted with permission from Lo et al.,1375 Copyright 2008 American Chemical Society). | ||
Zhang et al.1377 studied the effect of SDS on the kinetics of hydrate formation for CO2–CH4 mixtures simulating biogas in a glass bead pack at 274.2 K and 6 MPa with 288 ppm of SDS in water. Their results showed that 288 ppm of SDS in water promoted hydrate formation for CO2–CH4 mixtures and the presence of CO2 deteriorated the promotion performance of SDS because of the competing adsorption of the HCO3− and SD− at the surface of the hydrate crystals. It was reported that, for a biogas with 40% CO2 and 60% CH4, the presence of SDS reduces the separation factor and the addition of some ligin derivatives improves the CO2 separation.1378,1379 It was recently found that for ternary gas mixtures of CO2, CH4 and N2, SDS provided a much stronger promotion of CO2 hydrate formation, compared to CH4 and N2.1377 Ricaurte et al. also found that mixing 3000 ppm of SDS with 4.0 wt% of THF significantly improved both the split separation and separation factor for a gas mixture of 75 mol% CO2 and 25 mol% CH4 at a target temperature of 275 K and a load pressure of 4.0 MP.1380
The kinetics of hydrate formation can also be improved in some physical materials by increasing the hydrate forming gas–water contact area and enhancing the heat transfer process during hydrate formation,899 more details of which can be found in Section 3.5. The materials that were investigated for CO2 capture include silica sand,1108,1381 silica gels,1382,1383 foams,1384 nanoparticles,1385,1386 and hydroquinone hydrate-bearing porous particles.1387 Additionally materials were experimentally tested for storage of methane and hydrogen as clathrate hydrates, such as nano-hollow silica,1388 metal foams,1389 emulsions,1138 hydrogel,1390 dry water,951,1391 and porous carbon in nanotubes.854,1392 Li et al. found that the addition of nano-Al2O3 particles in TBAB significantly improved the hydrate formation process for a syngas with 39.8 mol% CO2 and 60.2 mol% H2.1393 A graphene–SO3−–Ag nanoparticle material was also found to be a few times more efficient than SDS to promote CO2 hydrate formation.1394 A comparison of silica sand, polyurethane foam, silica gel, and stirred reactors indicated that silica sand leads to the highest hydrate conversion rate in 120 minutes at 274.2 K and 6.0 MPa for a gas mixture with 38.1 mol% CO2, 59.4 mol% hydrogen, and 2.5 mol% propane.1348
Thermodynamic modelling and molecular dynamic simulation show that, among these impure components, SO2 and H2S are stronger hydrate formers and more stable in clathrate structures than CO2.1396–1398 Experimental results using conventional autoclaves1399–1401 and Raman spectroscopy1402 confirmed the model prediction of the hydrate phase equilibrium. For the other impure components that are less stable in clathrate structures, the presence of these impurities leads to a shift of the phase boundary of CO2 hydrate toward higher pressure and lower temperature before the appearance of liquid CO2, as shown in Fig. 23.1403,1404 Equilibrium and non-equilibrium thermodynamic modelling suggests that H2S not only shifts CO2 hydrate formation conditions but also improves the kinetics of CO2 hydrate growth due to its high solubility in water, while CH4, Ar and N2 that are not very soluble in water do not measurably affect the kinetics of CO2 hydrate formation.1405 Similar experimental results were reported for a synthetic flue gas with 1 mol% of SO2.1399
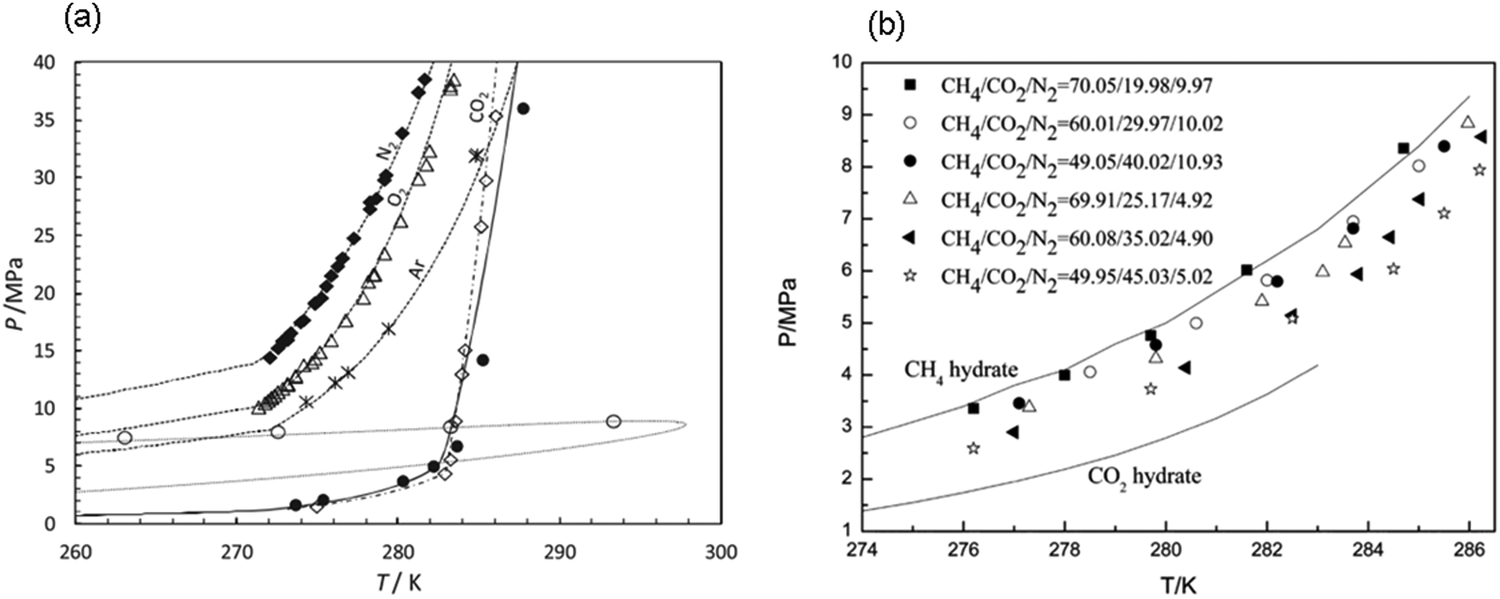 | ||
| Fig. 23 Effect of common impurities on the phase equilibrium of CO2 hydrate. (a) ● (○ phase envelope) CO2 with 5.05 mol% O2, 2.05 mol% Ar and 3.07 mol% N2; ⋄ pure CO2; ♦ pure N2; △ pure O2; * pure Ar; ○ (adapted with permission from Chapoy et al.,1403 Copyright 2013 Elsevier Ltd). (b) CH4–CO2–N2 mixtures (adapted with permission from Zang et al.,1404 Copyright 2018 American Chemical Society). | ||
Chapoy et al. experimentally investigated the effect of thermodynamic properties of CO2-rich systems containing impurities such as O2, N2 and Ar. Their results showed that the presence of 5.05 mol% O2, 2.05 mol% Ar and 3.07 mol% N2 in the CO2-rich system results in a water content 10–30% less and a density 35% lower compared to that of pure CO2.1403 Lang and Servio measured the solubility of CO2–N2 gas mixtures under hydrate–liquid–vapour equilibrium and found that CO2 solubility decreases and N2 solubility increases when the system pressure increases at a constant temperature.1406 More recently, Hassanpouryouzband et al. measured solubility parameters for various molar combinations of CO2–N2 mixtures over wide range of pressures, temperatures, and water salinities. The authors then used the measured data to calibrate available thermodynamic models.1407
Burnol et al. conducted a sensitivity analysis of the capacity of CO2 storage in the deep saline aquifers of the Paris Basin.1408 Their results indicated that a few molar percent of N2 or/and CH4 in the simulated CO2 streams leads to one order of magnitude reduction in the storage capacity of CO2, which was attributed to the effect of N2 and CH4 on the negative buoyancy zone and the gas hydrate stability zone. The negative change in the Gibbs free energy calculated from molecular dynamics suggested that SO2 molecules have higher tendency to occupy the large cages in structure I hydrate that are preferred by CO2 molecules, while H2S molecules will compete with CO2 molecules for the small cages,1397 which was observed using Raman spectroscopy.1402 As a result, a high concentration of these impure components present in the CO2 stream will also reduce the amount of CO2 in the hydrate phase and in turn the storage capacity of CO2 as gas hydrates.
6.2. CO2 storage
Hydrate storage of CO2 has been studied through theoretical simulations and laboratory experiments, mainly focused on the feasibility of CO2 storage as hydrate in depleted oil and gas reservoirs. This is because their geological characteristics are well known, the desired seal integrity is proven, and the necessary infrastructures are already in place. Reservoir simulations show that a number of depleted gas reservoirs in Northern Alberta (Canada) are suitable for CO2 hydrate formation and that the storage capacity will be influenced by the initial reservoir conditions including pressure, temperature and porosity, the residual hydrocarbon gas, temperature rise due to heat release from the exothermic reaction of CO2 hydrate formation, and the operational conditions of CO2 injection such as injection rate and pressure.1420,1421 Experimental measurements of the interfacial tension between CO2 hydrate and water is 18 times smaller than that between ice and water.1422 The low interfacial tension allows porous media and other foreign particles to help the adsorption of CO2 molecules on hydrate crystals, and thus to promote CO2 hydrate nucleation and formation kinetics,904,1423–1425 which is also in agreement with the outcome of molecular dynamic simulations.1426 The presence of salts in pore water enhances CO2 hydrate nucleation but leads to less CO2 hydrate formation.904,1424,1425,1427 Englezos and his colleagues carried out a series of experiments using different injection methods to simulate in situ process of CO2 injection into depleted gas and oil reservoirs in Northern Alberta (Canada), such as vertical injection,1428 horizontal injection,1429 and two spiral tubes at both top and bottom of the simulated sediments.1430 They concluded that the storage of CO2 as gas hydrate in depleted oil or gas reservoirs is feasible in terms of both hydrate formation kinetics and the storage capacity.1431 More recently, the formation of flue gas hydrate (CO2 + N2) in permafrost regions and marine sediments is been suggested as an option for CO2 storage and secondary sealing of hydrate reservoirs.1351 It was found that more than 92% of the CO2 present in the injection gas could be stored at certain conditions. In addition, the presence of N2 in the injection phase increases the safety of storage in case of a temperature rise, as N2 releases first with continued retention of CO2 in the hydrate phase.1351
In sediments the CO2 replacement process is constrained by heat and mass transfer due to fine pores and secondary hydrate formation. Experimental studies showed that the presence of excess water and clays resulted in slow CO2–CH4 exchange rate1449 and that formation of CO2 hydrate crusts wrapping on the methane hydrate crystals can act as a barrier to prevent CO2 molecule from diffusing into the methane hydrate crystals.1440,1450,1451 Thermal stimulation can destabilize methane hydrates and thus enhance the process of CO2 replacement reaction.1452–1454 It was reported that the injection of CO2–N2 mixtures, compressed air, or flue gas can enhance methane recovery from methane hydrate because the N2 molecules help to move some of the methane molecules out of the small cages, while CO2 molecules replace the methane molecules in the large cages,1455–1457 and was confirmed by thermodynamic modelling.1458 Cha et al. measured the composition of the methane hydrate by 13C nuclear magnetic resonance analysis and found that the methane composition formula changed from (1.6S)·(6.0L)·46H2O and (1.0S)·(4.1L)·46H2O before and after contact with a gas mixture of 20 mol% CO2 and 80 mol% N2 at 273 K and 10 MPa, respectively.4 The first field trial of the CO2 replacement technique was successfully completed in the North Slope of Alaska.1459 77% N2 was added to the CO2 stream to prevent secondary hydrate formation and promote CO2–CH4 replacement. After about 6 weeks of gas production, about 54% of the injected CO2 was stored underground and more than 50% of the produced methane was retained in the hydrate reservoir until the well pressure was further reduced below the methane hydrate dissociation pressure.
Direct injection of flue gas into methane hydrate-bearing sediments was experimentally investigated to reduce the cost of CO2 capture.81,1407,1460,1461 The nitrogen present in the injected flue gas shifted the hydrate equilibrium conditions, leading to decomposition of a large portion of the methane hydrate. The CO2 can the form CO2 hydrate or CO2–CH4 or CO2–CH4–N2 hydrates from the dissociated methane hydrate or by CO2 replacement in the methane hydrate-bearing sediments.1462,1463 It was further reported that 81.9% CO2 present in the injected flue gas was captured and stored in methane hydrate-bearing frozen sediments at 261.2 K.81Fig. 24 illustrates the principal mechanism of the flue gas injection method.
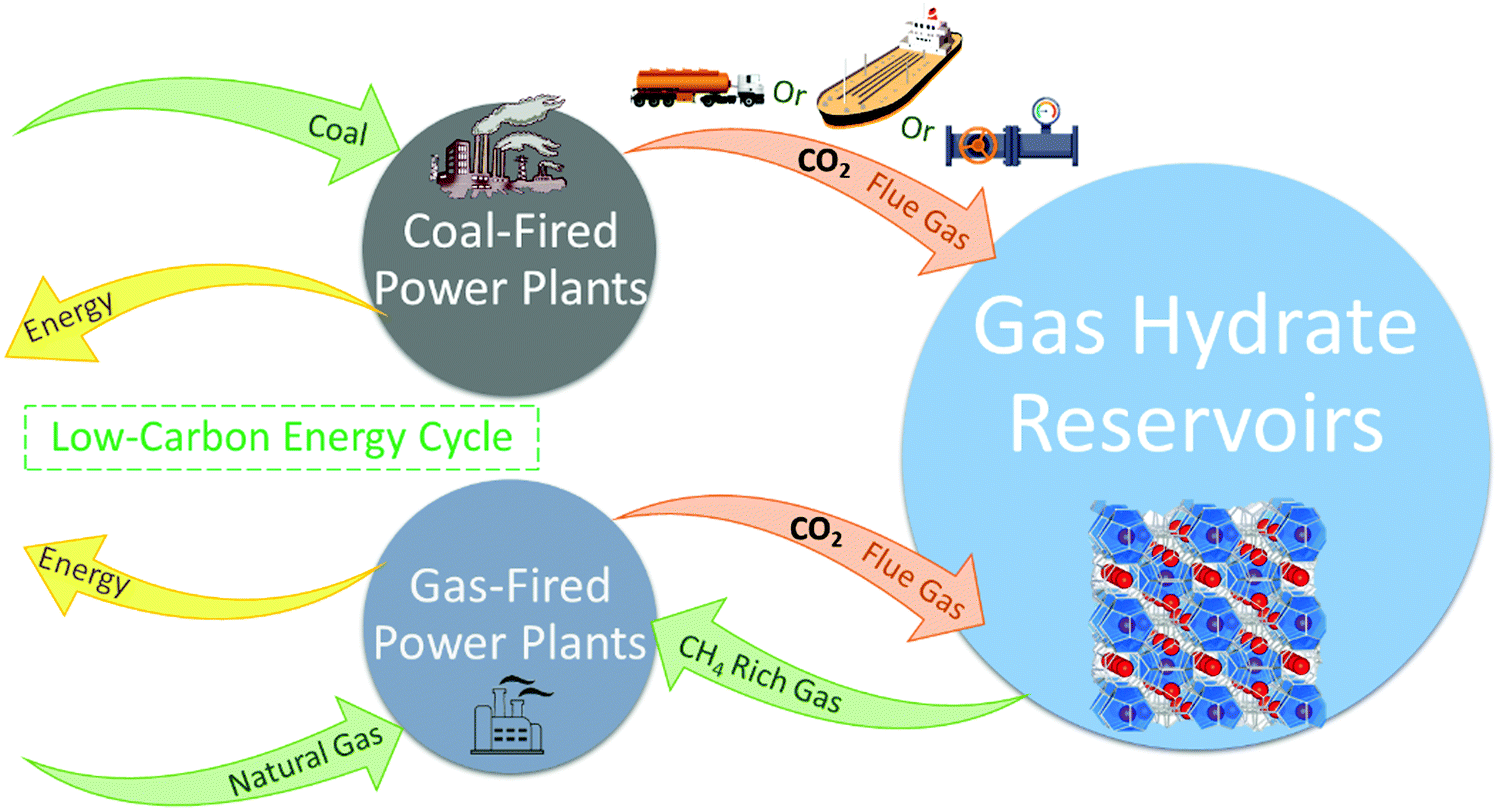 | ||
| Fig. 24 Illustration of the principal mechanism of the direct injection of flue gas for methane recovery from gas hydrate reservoirs and CO2 capture and storage simultaneously (adapted with permission from Hassanpouryouzband et al.,81 Copyright 2019 Springer Nature). | ||
There is not a clear picture how CO2 molecules replace the methane molecules that are entrapped in the methane hydrate. Early molecular dynamic simulations indicated that methane hydrate dissociates first after contacting with CO2 and then reform CO2 hydrate or CO2-mixed hydrates,1443,1464 while some simulation work suggested that direct swapping of methane and CO2 molecules or a transient co-occupation of one hydrate cage could occur in the outer lay next to the hydrate surface, which is buffered by the fluctuation of water molecules.1465 The recent molecular dynamic simulation by Wu et al. suggested that adsorption of CO2 molecules on methane hydrate surfaces can help to stabilize the methane hydrate and that both direct swapping and co-growth of CO2-mixed hydrates could occur during a CO2 replacement process.1466 However, most of the experiments showed that methane hydrate dissociation was not observed using a range of different methods including magnetic resonance imaging,1436,1437 differential scanning calorimeter,1467 electrical resistance.1451 In contrast, observations using in situ Raman spectroscopy found that free water phase appeared during CO2 replacement.1450 The physical models of CO2–CH4 molecular exchange were developed based on an assumption of a solid-to-solid transition process.1468,1469
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 gigatonnes of CO2, or about 40–1000 years of total global CO2 emissions, depending on the in situ density of CO2, sediment porosity, and sweep efficiency.1474,1475 Tohidi et al. conducted several time-lasting experiments demonstrating that the upward migrating CO2 tends to form hydrates at the base of the hydrate stability zone and that the CO2 hydrate formation process and the formed CO2 hydrate layer slowed down the CO2 diffusion rate by several times to 3 orders of magnitude.1476 In addition to CO2 hydrate formation, molecular dynamic simulations suggested that intercalation of CO2 within smectite minerals in brine aquifers can induce clay swelling to restrict CO2 migration.1477 The recent experimental results from The University of Bergen showed that a self-sealing layer could form in hours with injection of CO2 into cool aquifers and that the formation of the CO2 hydrate seal strongly depends on the rock properties.1416,1478
500 gigatonnes of CO2, or about 40–1000 years of total global CO2 emissions, depending on the in situ density of CO2, sediment porosity, and sweep efficiency.1474,1475 Tohidi et al. conducted several time-lasting experiments demonstrating that the upward migrating CO2 tends to form hydrates at the base of the hydrate stability zone and that the CO2 hydrate formation process and the formed CO2 hydrate layer slowed down the CO2 diffusion rate by several times to 3 orders of magnitude.1476 In addition to CO2 hydrate formation, molecular dynamic simulations suggested that intercalation of CO2 within smectite minerals in brine aquifers can induce clay swelling to restrict CO2 migration.1477 The recent experimental results from The University of Bergen showed that a self-sealing layer could form in hours with injection of CO2 into cool aquifers and that the formation of the CO2 hydrate seal strongly depends on the rock properties.1416,1478
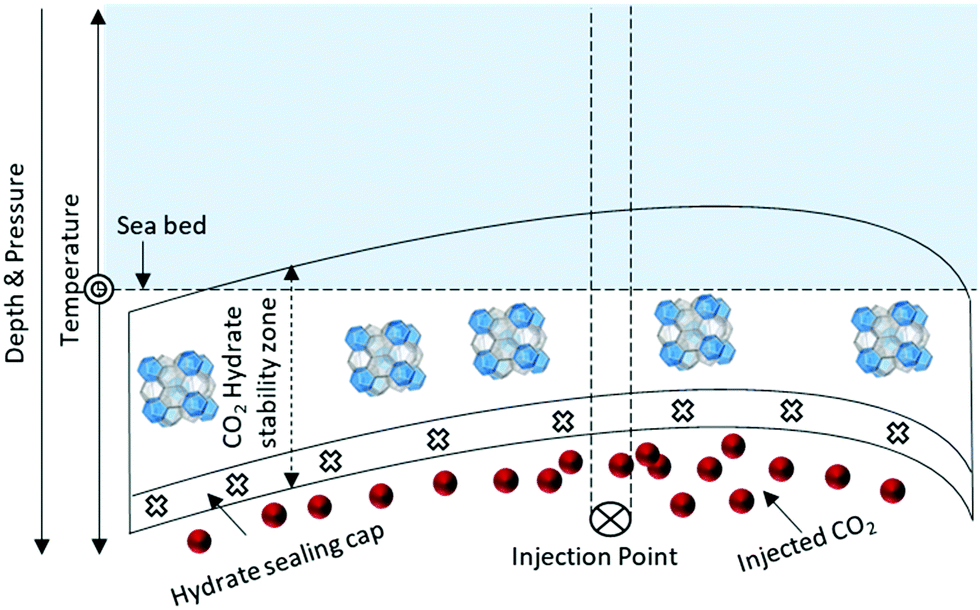 | ||
| Fig. 25 Schematic diagram of the CO2 self-sealing mechanism. | ||
6.3. Transport of CO2
Gas hydrate slurries (i.e., mixtures of CO2 hydrate and water) were initially proposed as an alternative to liquefied natural gas to reduce the cost of natural gas transport for long distances.1479,1480 It was further developed and investigated as a cold flow technology to prevent hydrate blockage in hydrocarbon transport pipelines.626,1481,1482 Further developments in the cold flow technology are reviewed in Section 7.2. The feasibility of CO2 hydrate slurries for CO2 transport was investigated recently.1483,1484 The main advantage of this concept is that it is expected to reduce the energy required for compression or liquefaction because the CO2 hydrate can be produced through the gas hydrate-based separation process. Similar to the transport of natural gas as solid hydrates,1485 CO2 hydrate can be made into pellets and transported using trucks or ships.332 The self-preservation and stability at relatively low pressures of CO2 hydrates make it feasible to carry the CO2 hydrate pellets for long distance using existing cargo ships that can provide chilled tanks at 1.5–2 MPa and around 243 K.814,1486 However, it should be noted that the transport of CO2 as hydrate forms is still a new concept in its early development stage.6.4. Further development toward large-scale deployments
In the past decades extensive experimental studies have successfully led to significant reductions in the hydrate formation temperature and pressure for CO2 separation from flue gas, syngas and fuel gas. Meanwhile, a variety of thermodynamic models, molecular dynamic models, and other physical models have been developed to describe the separation phenomena and processes. The results of lab-based research have demonstrated that gas hydrate-based CO2 capture technology is an energy efficient, environmentally friendly, and simple process compared to the other conventional technologies.1346,1359,1487 So far no industrial usage of gas hydrate-based CO2 capture technology has been reported. Therefore, further development is expected to be directed toward trials and implementation of large-scale deployments, such as the study of suitability and performance of combined thermodynamic and kinetic promoters and the design and optimisation of continuous separation processes,1487,1488 hybrid processes,1489 and integration with renewable energy (e.g., solar energy).1490 For transport of CO2 in the form of CO2 hydrate, there is still lack of fundamental understanding of the characteristics and rheological properties of CO2 hydrate slurries. For geological storage of CO2 as hydrates, systematic investigation is needed to provide a better understanding of the effect of CO2 hydrate formation on the integrity of CO2 injection wells, and the geomechanical, geophysical and geothermal properties of the storage formations.1491–1493 After the first field trial of the CO2 replacement method in the North Slope of Alaska,1459 more scientific test trials are ultimately demanded to undertake in situ evaluations of the different approaches that have proved feasible in lab experiments, for example, the CO2 replacement method and flue gas injection method. Moreover, comprehensive economic assessments of an entire chain of the hydrate-based CO2 capture and storage process are needed to provide the policymakers and the CCS industry with sufficient confidence for large-scale deployments of hydrate-based CCS technologies.7. Other industries
7.1. Gas storage
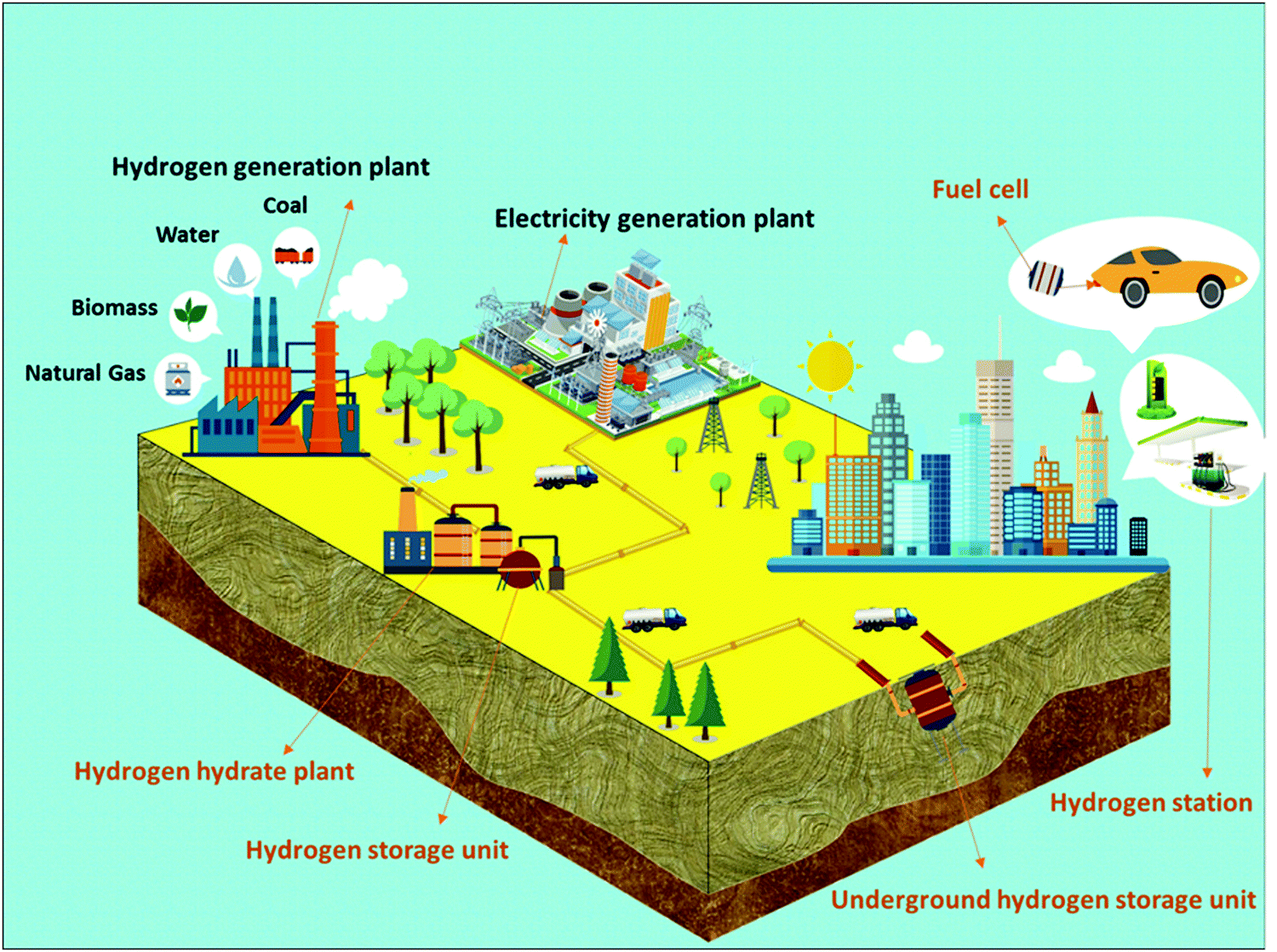 | ||
| Fig. 26 A graphical illustration of an entire clathrate hydrate-based hydrogen storage system that consists of three parts: hydrate synthetic plant, storage unit, and release unit in addition to transportation facilities. Hydrogen gas from a hydrogen production plant is converted into hydrogen hydrate at a hydrate plant. The hydrogen hydrate can be in a form of transportable slurries through pipelines. The hydrate slurries are then transported to a storage unit such as storage tanks or underground silos. | ||
Considering the molecular diameter of hydrogen, in the past it was considered too small to stabilize the water framework either as a single guest or in a gas mixture. However, after several successive reports about H2 hydrate formation,47,1318 and H2–THF hydrate formation1509 at extreme P–T conditions, interest in the potential of this field increased. Since then, numerous studies1510,1511 have characterized binary H2 hydrates at different condition as sII clathrates, having a maximum reported1512 storage capacity of 11.2 wt% at 500 MPa and 77 K. These forms of storage are not suitable for widespread commercial application as there are significant energy costs in storing and transporting sII hydrogen clathrates. The research developed to add THF859 to form hydrogen-THF clathrates at low pressures with around 1 wt% H2. Following that, a study1513 claimed that it is possible to store hydrogen up to around 4 wt% at low pressures by “tuning THF contents”. In response, several studies1514,1515 challenged the capacities reported in the previous study, claiming the maximum storage of hydrogen couldn’t be more than 1 wt% in the investigated conditions. Although the maximum hydrogen capacity is a matter of conflict, the success of the tuning effect for mixed guest hydrates was developed and reported in later studies.1516,1517 THF is a volatile organic compound and contamination of hydrogen after dissociation with these compounds would be harmful to fuel cells or turbines. To circumvent this problem, it is been suggested to store hydrogen in semi-clathrates of quaternary ammonium compounds with a maximum reported storage capacity of about 1 wt% H2.51 In addition to the different studies on THF-mixed1518–1520 hydrates, quaternary ammonium-mixed hydrates,866,1521,1522 or other sII promoters-mixed (e.g. cyclopentane,1523 cyclohexanone,1524 furan and tetrahydrothiophene1525) hydrates, attempts been made to use other promoters for storing hydrogen in different structures such as sH,1526 alkylamine hydrates,63 and etc.1505,1508 However, all of the abovementioned promoters suffer from lower hydrogen storage capacity when compared to pure hydrogen hydrates, as the promoters occupy major1527 fractions of the large cavities, reducing the hydrogen occupancy in the hydrate phase.
The kinetics of hydrogen hydrate formation and dissociation is another parameter that plays a crucial role in the commercialization potential of clathrate based hydrogen storage. The time required for the formation of hydrogen hydrate could be in the order of a day, resulting in an extreme energy penalty for keeping the system at low temperatures and high pressures for that duration. Therefore, methods for increasing the rate of hydrogen hydrate formation have been widely studied and include formation of hydrate from reactive forms of ice1528 or quenching the seeds of clathrates in the system.1529 More recently, it is been suggested to mix hydrogen with natural gas components, mainly, methane,1530–1533 and propane1534,1535 to enhance the formation kinetics. Here methane and propane could serve a dual purpose, i.e. as they will be used as a promoter to form hydrogen hydrate at lower pressures and also, they could be burnt to produce energy, increasing the energy density of the clathrates.
The research into the application of clathrate hydrates in hydrogen storage has experienced a remarkable growth in the last few years. Several milestones have been achieved: the storage of hydrogen at low pressures, the increase of hydrogen hydrate formation rate, and strategies to improve the capacity of hydrogen storage. Important efforts are also being driven toward improving the understanding of the physical properties of hydrogen inside clathrates.1536–1538 These advances envisage a promising future for hydrate-based hydrogen storage, yet important limitations must be addressed. The capacity of hydrogen storage needs to be further increased under moderate conditions to ensure progress. The kinetics of hydrate formation and dissociation required to be further improved to make the hydrate-based technologies competitive with other energy storage sectors. Reaching a few seconds time-scale for formation could be the next milestone for kinetics improvement. Full chain economic analysis for this method is also missing in the literature. The future of the field will not only depend on how the scientists circumvent the mentioned and forthcoming problems but also on how they tackle the technical challenges experienced in the other hydrogen storage methods.
After proposing the possibility of natural gas storage in hydrates,1548 similar to that of hydrogen storage, development of natural gas storage in hydrates has followed the same pathways. Numerous works have been done on characterizing methane (as the major component of natural gas) hydrates,767,1549–1551 followed by attempts for moderating the formation P–T conditions and increasing the storage capacity of clathrates such as using Ice powder,1552,1553 THF,1554–1556 semi clathrates,1557,1558 and etc.1559 Various methods890,1560–1566 have also been applied to increase the kinetics of natural gas/methane hydrate formation. The formation pressure of methane hydrate in comparison with hydrogen is significantly lower at similar temperatures, and even lower when mixed with other heavier natural gas components that is the main reason for further development of clathrate based natural gas storage. During the last decade, global research activity into natural gas storage in hydrates has increased to the point where several demonstration phase projects have been undertaken, such as the natural gas hydrates pellet reactor814,1567 which didn’t encounter any fundamental technical barriers and has led to several commercialization analysis report812,1568 being published. A more detailed review and list of the patents in the commercialization pathway of this technology can be found in another review paper.1485 As gas hydrate based natural gas storage approaches the commercial stages of development, some technical uncertainties remain, such as those related to gas capacity and the kinetics of hydrate formation and dissociation/energy recovery mainly due to limited heat transfer and self-preservation effect; however success of the demonstration projects provided experiences and practical data to push the technology forward to commercial stage.
To solve this problem, Mcturk and Waller1576 reported a method for continuous storage of O3 in the form of molecular clathrates to isolate them, preserving the O3 for a longer time. To reduce the hydrate formation pressure, they used carbon tetrachloride (CCl4) as the ozone solvent and help guest due to its stability to Ozone. Nevertheless, they were unable to store O3 in more than 30% of the small cages and the remaining small cages were left free. Hence, they suggested the use of ozone clathrates at higher pressures for a higher storage capacity. Following this, Vysokikh et al.,1577 experimentally investigated the possibility of hydrate formation under the Earth's stratosphere conditions by forming hydrate with O3-ice and O3-hydrogen chloride. Subsequently Muromachi et al.,1578 investigated the formation of O3–O2, and O3–CCl4 hydrates. The authors found that O3 could be preserved for more than 20 days after hydrate formation under atmospheric pressure. These hydrates have three orders of magnitude more O3 than a typical “ozonated water” and has been used for disinfection. Nakajima et al.1579 investigated the formation hydrate from mixtures of O3 + O2 + CO2 and concluded that the ozone could be preserved at atmospheric pressure for more than four weeks by forming clathrates of the mentioned mixture. These findings have sparked considerable interest in clathrate hydrates as potential ozone storage materials and many workers have assessed the formation of O3 mixed hydrates,1580–1585 in an attempt to increase the capacity of O3 in the clathrates formed from the O3 + O2 + CO2 mixture.1586,1587 In 2018, Subbotin et al.1588 studied the storage capacity of O3 in O3 + O2 + N2 + CO2 mixed gas hydrates. They found that the storage capacity of such hydrates could be several times higher than O3 + O2 + CO2 mixed hydrates, providing more favourable thermobaric conditions for O3 storage. More recently, Watanabe et al.1589 conducted a more detailed study on the kinetics of continuous O3 + O2 + CO2 mixed hydrate formation by detailing the effect various influencing parameters on the storage capacity, kinetics of O3 mixed hydrate formation, and O3 decomposition to O2 in the gas phase.
7.2. Cold energy storage
The need for reducing hydrofluorocarbons in refrigeration according to the Kyoto Protocol and Montreal Protocol1590 has directed considerable efforts toward the development of more environmentally friendly refrigerants1591–1593 and the construction of more efficient refrigerant systems as well as the wider applications of cost-effective cold storage systems which have been proposed as another solution for storing intermittent renewable energies.1594,1595 Among different working media for cold storage systems such as water, ice, and eutectic salt gas hydrates are attractive and versatile because of their high temperature zone, suitable latent heat of fusion (more than Ice in CO2 hydrate), and better heat transfer efficiency has and have been described and compared in detail in several reviews.920,1596–1600 The conventional workflow for hydrate cold storage begins with the reduction of the system temperature to hydrate formation point, followed by exothermic hydrate formation that is the main cold storage part. Finally, stored cold energy could be consumed through endothermic hydrate dissociation either through depressurization, receiving heat from the environment (see Fig. 27), or a combination of both. Based on this, many lab-scale hydrate-based cold storage systems1601,1602 have been developed including direct contact type, and indirect contact type, also known as secondary refrigeration, where the primary circuit (cold generation place) and secondary system (the place to be cooled) are physically separated and the generated cold is transferred by the fluid/phase change material (PCM)1603 between two places.920,1596,1604,1605 The longstanding interest in investigating the hydrate phase equilibria and their formation/dissociation kinetics due to their importance in different sectors, removes the initial barriers. The potential of using different hydrate formers such as CO2,1604,1606,1607 CH4 and various refrigerants1608–1613 or their combinations1614–1618 (e.g. CO2–THF,1619,1620 and a refrigerant + methane1621/CO21622) as guest molecules in hydrate-based cold storage is well-demonstrated. Among different candidates, CO2 or CO2-mixed (additives such as THF to reduce formation pressure) has received greater attention due to the higher solubility of CO2, higher latent heat of fusion,1623 availability, lower environmental impact (if captured from CO2 production industry) and moderate formation conditions. However, CO2 is a corrosive gas and will cost extra for building and maintaining the CO2-resistive cold storage systems.1624 Therefore, choosing a suitable guest molecule/s for hydrate-based cold storage is an ongoing topic. Following the choosing of a satisfactory guest molecule, characterizing hydrate formation/dissociation in the relevant environments1151,1625,1626 and applying a suitable growth acceleration method, the principal understanding of hydrate slurries properties become important (see Section 2.6) and are the main step for all the hydrate-based applications. Since continued advancements in hydrate-based cold storage systems highly depend on the further improvement of the devices for efficient cold storage systems, concerns about the possibility of gas leakage and the energy penalty for gas compression could be significant. Another critical point we would like to highlight is that a detailed economic assessment for a large-scale hydrate-based cold storage method is required to reach the next milestone. It is expected that the research in this field will further develop by fusion of various fields such as process engineering and metallurgy. We hope that the remaining barriers for applying this technology will be removed in the near future.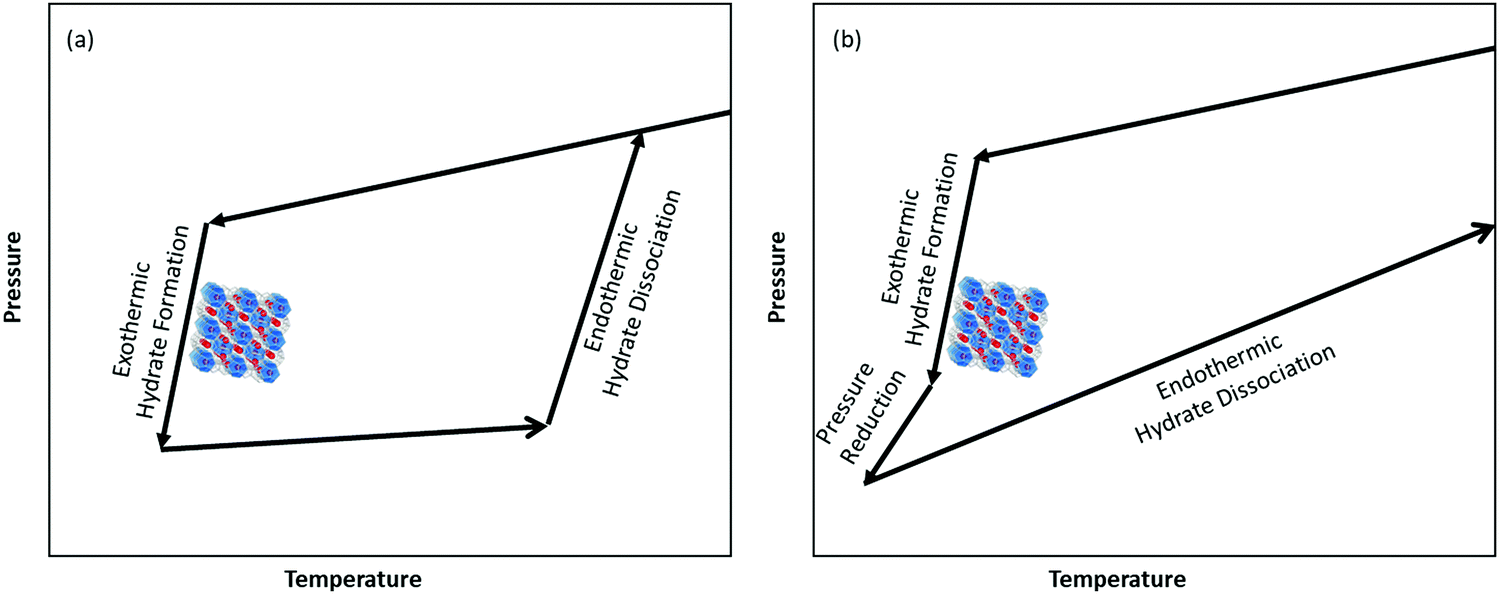 | ||
| Fig. 27 Two modes of hydrate cold storage technology, namely heating (a) and (b) depressurising for cooling (based on Cheng et al.920). | ||
7.3. Clathrate hydrate-based desalination
The escalating freshwater demand by the growing global population and industrial/agricultural activities is putting unprecedented pressure on the world's freshwater resources. The current pace of human population growth, industrialization, urbanization and the over-exploitation of the freshwater resources are contributing substantially to the global water scarcity.1627,1628 To address the declining trend in global freshwater availability1629 a radical rethink of the global water management strategies and policies is necessary to ensure human welfare,1630 and ensure the long-term viability of the Earth's freshwater resources.1631 Given the limited total usable freshwater supply, a variety of seawater desalination technologies have been developed over the last several decades to meet the rapid growth of the worldwide water demand and augment the supply of water, particularly in arid regions such as the Middle East.1632 Desalination is the process of removing salt from seawater or brackish water to make it usable for drinking or irrigation.1633 Traditional thermal-based, membrane-based and chemical-based desalination technologies, such as Multi-Stage Flashing (MSF), Multi-Effect Desalination (MED) and Reverse Osmosis (RO), are energy-intensive processes normally using fossil fuels, which in turn contribute to global warming.1632,1634,1635 Given ice crystals are made up of essentially pure water, desalination technologies based on indirect and direct freezing were also introduced, yet are not widely applied commercially due to high costs.1636 Thus, to make a real impact, innovative desalination technologies must be developed to secure safe sufficient clean water, while reducing the cost and energy use.1637 The emerging desalination technologies for water treatment have been critically reviewed in the literature.1633Clathrate hydrate-based desalination (see Fig. 28), classified as a freezing or crystallization approach, was proposed more than 70 years ago as a potential technology for the desalination of seawater.1638 In this process, an electrolyte solution such as seawater or brackish water is contacted with a hydrate forming agent at a favourable P–T (which could be above the freezing point of water). The water molecules encage the hydrate former molecules and form the clathrate hydrate, thereby salts and other impurities become excluded from the crystalline structure.1639 The salt does not disturb the morphology of the hydrate crystal and acts as a thermodynamic inhibitor, shifting the hydrate phase equilibrium to a higher pressure at a given temperature and accordingly, reducing the driving force for the hydrate formation (see Sections 3.4.1 and 4.1.1). The hydrate crystal can then be mechanically separated and removed from the brine and decomposed to potable water and hydrate former which is then recovered and recycled.1638 The volumetric efficiency of the process is represented by the water recovery, which is defined as the volume of freshwater recovered from the feed solution, and depends upon the kinetics of the hydrate formation and the volume of water converted to hydrates along with the recoverable amount of hydrate crystals at the separation step from brine.1640 It must be noted that the eutectic composition of the feed solution constrains the maximum recoverable water. Another metric is salt rejection, which represents the efficiency of the salt removal in the process and is defined as the salt concentration evolution relative to its initial value.1638
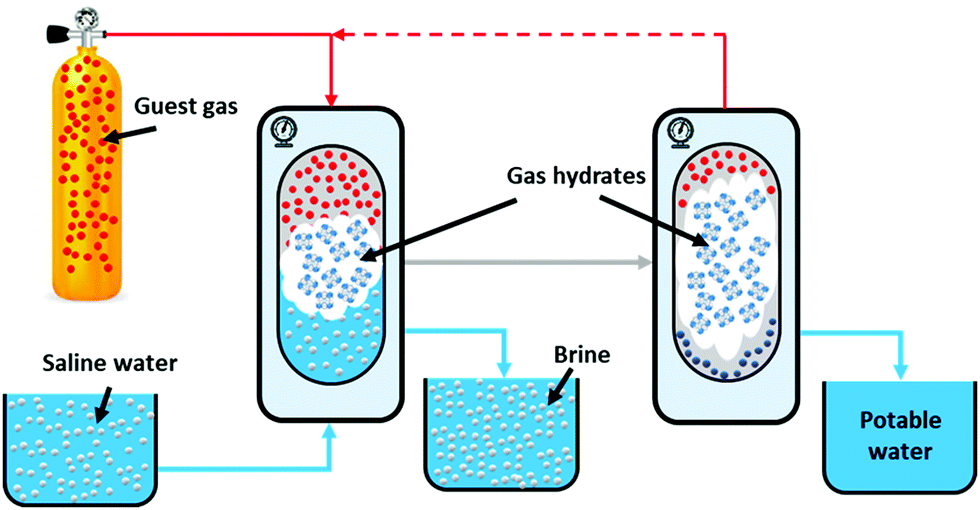 | ||
| Fig. 28 Schematic diagram of the clathrate hydrate-based desalination process. Seawater is contacted with an appropriate hydrate former such as CO2 at desired thermodynamic conditions to form clathrate hydrates while excluding salts from the crystalline structure. The hydrate crystal can then be removed from the brine and dissociated to drinking water and hydrate former which could be recycled. | ||
The application of hydrates in the field of seawater desalination has been happening since the 1940s. Since then, a great deal of research has been carried out worldwide to find suitable hydrate formers considering the important factors of eco-friendliness, non-toxicity, stability, availability and economic viability.1641 To date the application of a variety of hydrate forming agents such as propane (C3H8),1642,1643 cyclopentane (C5H10),1639,1644–1649 carbon dioxide (CO2),900,1107,1650,1651 refrigerant gases (HFC, HCFC and CFC)1652–1655 and sulphur hexafluoride (SF6)1656 have been applied to the hydrate-based desalination process. Numerous studies and pilot plant scale tests have also been conducted to determine phase equilibrium975,1657–1662 and evaluate/improve the kinetics or alleviate the temperature requirements for hydrate formation (e.g. by using secondary hydrate guest gases).1663–1669 Given the main hindrance for the early commercialization of hydrate-based desalination was the challenges associated with the separation of the hydrate phase out of the brine, studies have been conducted to investigate the salt removal efficiency and enhance it via secondary treatment processes after hydrate formation.1670–1677 Moreover, there have been a number of inventions focused on crystal separation from the saline water, salt removal from the hydrate slurry and reduction in induction time, a detailed review of which can be found elsewhere.1638
The criteria for a suitable hydrate former together with other technical issues have made it challenging to propose a commercial hydrate-based desalination scheme. For instance, SF6, CFC, HFC and HCFC have been found to be inappropriate as they have an adverse impact on ozone depletion even though they are atmospheric hydrate formers and alkanes and cycloalkanes are flammable fluids which pose safety issues at large-scale applications. Ethane and propane, however, have been proposed as favourable hydrate formers according to an integrated thermodynamic approach.1641 Moreover, a comprehensive and systematic assessment conducted on cyclopentane revealed its viability as an atmospheric hydrate forming agent.1649 Despite all the efforts and inventions developed attempting to commercialize the hydrate-based desalination process by addressing the technical challenges together with energy efficiency and environmental concerns, its application on a commercial scale has not yet been successful. Economic feasibility must be accounted for as the operation cost depends upon a variety of factors such as brine temperature, favourable thermodynamic conditions, salt content, mobility of salt and yield.1678–1680 Therefore, there is still a need to develop eco-friendly and energy-efficient hydrate-based desalination methods that are viable to be deployed at the industrial scale.
Recently, an optimal design approach of the RO-hydrate hybrid system for seawater desalination was proposed.1681 Moreover, a novel hydrate-based desalination process utilizing LNG waste cold energy as a heat sink has been modelled, simulated and its economic feasibility evaluated.1682–1684 The results show that LNG appropriately replaces the external refrigeration cycle, hence the process is able to desalinate high concentration brines with low energy consumption, and hence this coupling could be economically favourable at higher plant capacity. A multifunctional desalination apparatus via gas hydrate with various operation modes and separation methods were also developed and its application was tested for a continuous desalination process with multi-time injection, separation and purging.1685 The ultimate desalination efficiency was observed to be higher than 80% with a water recovery of above 30%. As a form of CO2-hydrate, CO2 can act with dual character extracting potable water from brine while being collected from gas stream. Therefore, it could be considered as a potential working medium for the developing technology of coupled CO2 capture-clathrate hydrate-based desalination, whereby CO2 is captured as CO2-hydrate in the presence of seawater, then decomposed to CO2 and desalinated water where both are desired.1686 Although few investigations have been conducted in this regard so far,1108,1687–1690 such capability undoubtedly could provide a huge potential opportunity for more active research in this field, particularly on the technical aspects such as the favourable formation pressure and rate along with cost minimization. Recent advances in both technical and economic aspects continue progress toward commercial and viable clathrate-hydrate based desalination technologies. However, further experimental studies are required to enhance the desalination efficiency via reducing the amount of salts trapped among hydrate crystals in each cycle as well as optimizing key operating parameters such as the working gas (hydrate formers), usage of different additives, and finally operating pressure and temperature. At molecular level, MD simulation could also be undertaken to provide fundamental insights regarding the role of crystal nucleation, growth and separation for applications in hydrate-based desalination.
7.4. Gas separation
Gas separation refers to an operation that is being used to either purify or separate certain component/s from the feed gas. Gas separation is necessary for different sectors including natural gas purification,1691 hydrogen separation,1692 CO2 seperation,1693 biogas separations,1694 and etc.1695 This technique is commonly implemented in multiphase system. Since, for separating a certain group of molecules in a homogeneous gas mixture, molecular recognition is mandatory. Here, each phase can be described as mechanically separable and a discrete homogenous part of the system. The three most common gas separation methods are solvent/sorbent based separation,1696 cryogenic distillation,1697 and membranes.1698 Recently, hydrate-based gas separation has emerged as a potential technique. The applicability of this method lies in the strong affinity of some of gas molecules to the hydrate phase. In fact, the main challenge in the separation field is the variation in phase affinity of different molecules during phase change, increasing the difficulty of generalizing the separation method.One remarkable property of gas hydrates is their various occupancies for different gases which can be used to capture and separate less desirable species from the feed gas by forming hydrates followed by dissociating of the formed hydrates after separating from the gas phase. In such a process, the composition of both remaining gas phase and dissociated gas phase will be significantly different from the feed gas. Variation in solubility of different gases (e.g. CO2 and N21407) helps this process, since gases with higher solubility usually have higher cage occupancy at the hydrate phase. In some cases (e.g. CO2 + N21351), the purity of the captured species could be more than 90% percent after the first attempt. Repeating this process will increase the purity of the separated gases to the desired value (see Fig. 29). Another attractive point of using gas hydrate for gas separation is the endothermic nature of hydrate formation which reduces the required energy for cooling the system during the process. In terms of temperature and pressure effect, the cage occupancy of gases depends on both of these factors, which in turn affects the selectivity of gases in the hydrate phase.
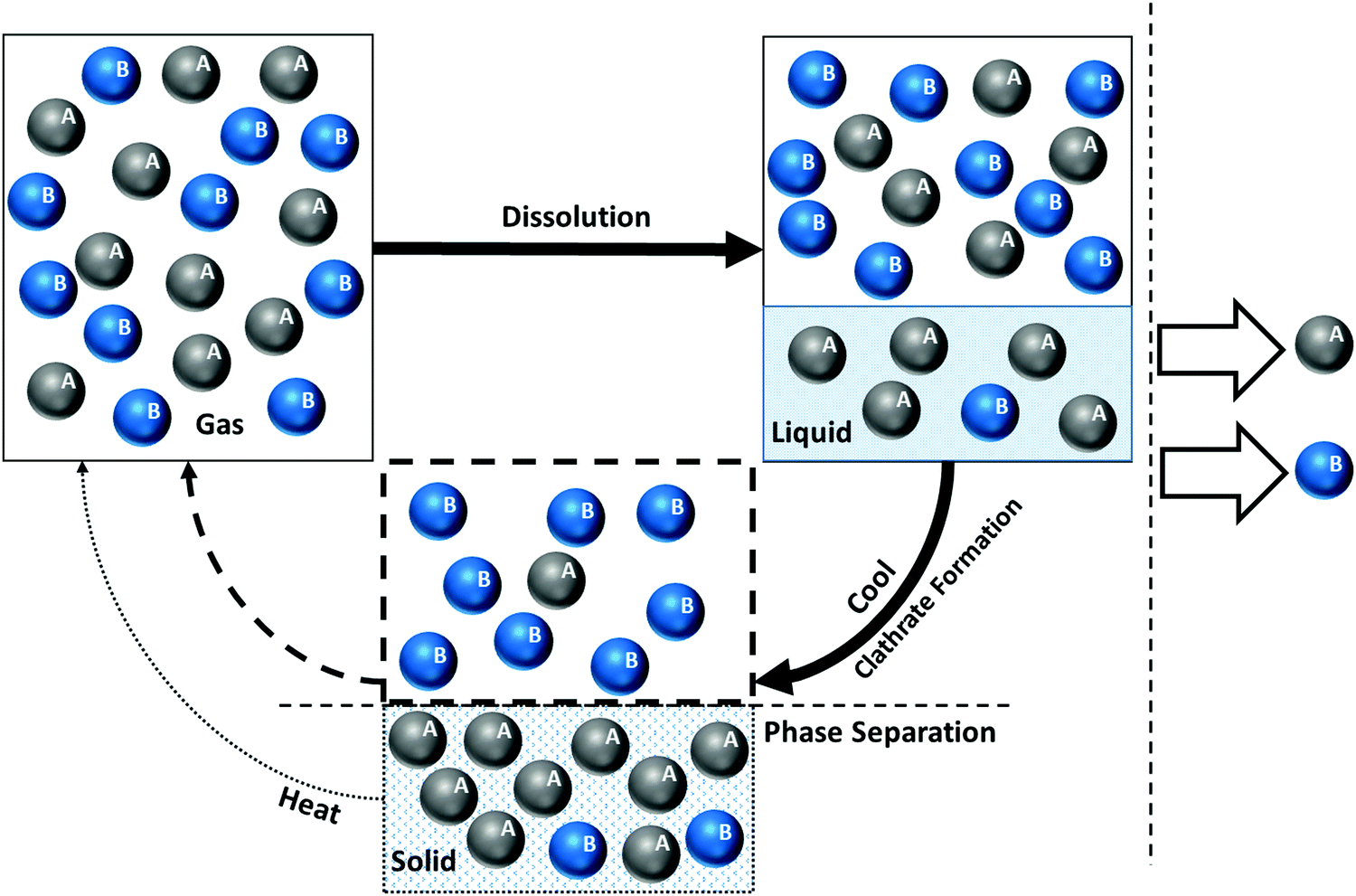 | ||
| Fig. 29 Schematic diagram of hydrate-based gas separation. | ||
Although it looks quite simple to employ this separation process, in practice it demands complicated operations to separate the gas phase from the hydrate phase efficiently and keep the process continuous, as plugging different parts of the separation apparatus by hydrate formation is a predictable problem.1110,1360,1699–1702 Accordingly, different types of reactor configurations have been suggested, one of which is SIMTECHE, which has reached the commercial demonstration stage. Other reactors exist including but not limited to stirred reactors, unstirred reactors, fixed bed1703 reactors, a detailed review of which can be found elsewhere.1348 Despite the fact that this method is based on the differences in cage occupancies, the key limitation of this method is the cage occupancies of the gases, as it can’t be used to separate gases with similar cage occupancies. Accordingly hydrate based separation is mostly associated with CO2 capture and separation (see Section 6) that has considerably lower HSZ and consequently higher cage occupancies. However, it is also used to separate other gases such as N2,1352 H2,1704 H2S,1705 CH4,1706 SF6,1707–1710 N2O,1711 CHF3,1712 and other refrigerant gases.1713–1715 In the majority of hydrate-based gas separation studies, the authors studied the equilibrium condition of mixed gases, the effect of additives for enhancing gas uptake rate or the neutralising separation P–T conditions that have already been reviewed in the relevant sections of this article. Recently, it is been suggested to combine this method with membranes1716,1717 or chemical absorption1489 to enhance the efficiency of gas separation. The results show that when combined with chemical absorption methods it has about 30% lower energy when compared with cryogenic separation only.1489
Although there has been substantial research into hydrate based gas separation since the introduction of the field and the advantages of this method over conventional techniques in terms of being the most environmentally benign approach, there are still numerous issues to be addressed to persuade industries and societies to accept the developed hydrate-based gas separation methods. The fact that establishing new separation plants is capital-intensive makes it harder to persuade the industry, especially when economic studies for most of the proposed method are missing, making it a very high-risk investment for the industry. It is however important and necessary to compare the efficiency of the method with existing separation methods in detail from both the scientific and commercial point of view. In the field of hydrate-based gas separation, the challenge is to design more efficient reactors beside other common challenges such as kinetics and neutralised P–T conditions. On the basis of the promising development of hydrate-based gas separation in recent years, and the considerable growth potential, hydrate-based gas separation is an excellent candidate technique to make a significant shift in gas separation processes in various industrial applications.
8. Final remarks
Over the past decade, significant progress has been achieved in characterising gas hydrates’ behaviour, understanding their role in Nature, and developing gas hydrate-based applications for sustainable technologies. In this review, the main properties of gas hydrates and their kinetic behaviour have been summarized, including those related to pure hydrates and those that can exist in Nature within sediments. This was followed by reviewing the role of gas hydrates in conventional flow assurance that appears to have reached a satisfying level of maturity. However, there is always the need for further optimisation of the hydrate-inhibition process by reducing the costs associated with buying substances or services. The discovery of a vast number of natural gas hydrates reservoirs has enabled the establishment of a new and fruitful area within natural gas recovery, offering different methods for recovering the gas hydrates. The potential interaction of these reservoirs with the atmosphere, however, is of increasing concern particularly with regards to their greenhouse warming potential and their potential role in marine stability. In addition, the stability of gas hydrates at extremely high pressures has gathered interest in the field from various branches of astronomy and planetary research. This review has summarized the different potential interactions of gas hydrates and their existence in nature as well as developing methods for extracting gas hydrates. The development of a broad range of applications for gas-hydrate-based technologies in sustainable development including CO2 capture and storage, water desalination, energy storage, etc., has also increased research attention in this field. Numerous diverse methods have been designed to demonstrate and enhance the capability of gas-hydrate-based technologies. The ongoing investigation in designing new apparatus, processes, or promotional compounds for increasing hydrates’ capacity or enhancing their formation/dissociation rate has provided many successful approaches for the application of gas hydrates in sustainable development. This review also compiles the potential application of gas hydrates in sustainable industries, focusing on the general findings in these fields. Considering the extended coverage of the review article, when relevant, the related outlook was provided at the end of each subsection. Although the application of gas hydrates in sustainable industries is in its infancy, intensive research for characterising gas hydrates has already provided important advances, enhancing technologies at the conceptual level. Therefore, we anticipate that the emergence of new gas-hydrate-based applications in sustainable industries will be delivered in the near future, allowing the gas-hydrate community to continue to take an active role in the transition to a lower-carbon economy. Accordingly, we hope the current review will help to motivate scientists working in this exciting field to achieve this commendable goal.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
A. H. and K. E. were partially supported by funding from UKRI-EPSRC (grant number EP/S027815/1). C. R. was partially supported by DOE-USGS Interagency agreement DE-FE0023495. C. R. thanks L. Stern and W. Waite for insights that improved her contributions. E. J. is partially supported by Flow Programme project sponsored by Department for Business, Energy and Industrial Strategy (BEIS), UK. Any use of trade, firm or product name is for descriptive purposes only and does not imply endorsement by the U.S. Government.References
- C. D. Ruppel and J. D. Kessler, Rev. Geophys., 2017, 55, 126–168 CrossRef.
- M. T. Kirchner, R. Boese, W. E. Billups and L. R. Norman, J. Am. Chem. Soc., 2004, 126, 9407–9412 CrossRef CAS PubMed.
- C. Petuya, F. Damay, D. Talaga and A. Desmedt, J. Phys. Chem. C, 2017, 121, 13798–13802 CrossRef CAS.
- M. Cha, K. Shin, H. Lee, I. L. Moudrakovski, J. A. Ripmeester and Y. Seo, Environ. Sci. Technol., 2015, 49, 1964–1971 CrossRef CAS PubMed.
- A. Haber, M. Akhfash, C. K. Loh, Z. M. Aman, E. O. Fridjonsson, E. F. May and M. L. Johns, Langmuir, 2015, 31, 8786–8794 CrossRef CAS PubMed.
- A. Klapproth, R. O. Piltz, S. J. Kennedy and K. A. Kozielski, J. Phys. Chem. C, 2019, 123, 2703–2715 CrossRef CAS.
- C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 237–257 CrossRef CAS PubMed.
- H. Lu, Y. T. Seo, J. W. Lee, I. Moudrakovski, J. A. Ripmeester, N. R. Chapman, R. B. Coffin, G. Gardner and J. Pohlman, Nature, 2007, 445, 303–306 CrossRef CAS PubMed.
- R. K. McMullan and G. A. Jeffrey, J. Chem. Phys., 1965, 42, 2725–2732 CrossRef CAS.
- T. C. W. Mak and R. K. McMullan, J. Chem. Phys., 1965, 42, 2732–2737 CrossRef.
- J. A. Ripmeester, J. S. Tse, C. I. Ratcliffe and B. M. Powell, Nature, 1987, 325, 135–136 CrossRef CAS.
- D. W. Davidson, Water in crystalline hydrates aqueous solutions of simple nonelectrolytes, Springer, 1973, pp. 115–234 Search PubMed.
- D. Hornby, Hydrogen bonding in biological structures, Springer Science & Business Media, 1993, vol. 323 Search PubMed.
- S. Alavi, J. A. Ripmeester and D. D. Klug, J. Chem. Phys., 2006, 125, 104501 CrossRef PubMed.
- Y. A. Dyadin and K. A. Udachin, J. Struct. Chem., 1987, 28, 394–432 CrossRef.
- J. A. Ripmeester and D. W. Davidson, J. Mol. Struct., 1981, 75, 67–72 CrossRef CAS.
- I. M. Chou, A. Sharma, R. C. Burruss, J. Shu, H. K. Mao, R. J. Hemley, A. F. Goncharov, L. A. Stern and S. H. Kirby, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13484–13487 CrossRef CAS PubMed.
- L. Yang, C. A. Tulk, D. D. Klug, I. L. Moudrakovski, C. I. Ratcliffe, J. A. Ripmeester, B. C. Chakoumakos, L. Ehmd, C. D. Martin and J. B. Parise, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6060–6064 CrossRef CAS PubMed.
- J. A. Ripmeester, C. I. Ratcliffe and J. S. Tse, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 3731–3745 RSC.
- Y. A. Dyadin, I. V. Bondaryuk and L. S. Aladko, J. Struct. Chem., 1995, 36, 995–1045 CrossRef.
- C. A. Koh, Chem. Soc. Rev., 2002, 31, 157–167 RSC.
- E. D. Sloan Jr., C. A. Koh and C. A. Koh, Clathrate Hydrates of Natural Gases, CRC Press, 2007, vol. 20074156 Search PubMed.
- E. D. Sloan, Nature, 2003, 426, 353–359 CrossRef CAS PubMed.
- S. Alavi, K. Udachin, C. I. Ratcliffe and J. A. Ripmeester, Supramolecular Chemistry: From Molecules to Nanomaterials, 2012 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- K. A. Udachin and J. A. Ripmeester, Nature, 1999, 397, 420–423 CrossRef CAS PubMed.
- K. A. Udachin, C. I. Ratcliffe and J. A. Ripmeester, Angew. Chem., Int. Ed., 2001, 40, 1303–1305 CrossRef CAS PubMed.
- H. Hirai, T. Kondo, M. Hasegawa, T. Yagi, Y. Yamamoto, T. Komai, K. Nagashima, M. Sakashita, H. Fujihisa and K. Aoki, J. Phys. Chem. B, 2000, 104, 1429–1433 CrossRef CAS.
- O. Grasset, C. Sotin and F. Deschamps, Planet. Space Sci., 2000, 48, 617–636 CrossRef CAS.
- O. B. Toon, C. P. McKay, R. Courtin and T. P. Ackerman, Icarus, 1988, 75, 255–284 CrossRef CAS.
- E. Lellouch, A. Coustenis, D. Gautier, F. Raulin, N. Dubouloz and C. Frère, Icarus, 1989, 79, 328–349 CrossRef CAS.
- J. S. Loveday, R. J. Nelmes, M. Guthrie, S. A. Belmonte, D. R. Allan, D. D. Klug, J. S. Tse and Y. P. Handa, Nature, 2001, 410, 661–663 CrossRef CAS PubMed.
- A. Levi and R. E. Cohen, Astrophys. J., 2019, 882, 71 CrossRef CAS.
- J. S. Loveday and R. J. Nelmes, Phys. Chem. Chem. Phys., 2008, 10, 937–950 RSC.
- H. Hirai, T. Tanaka, T. Kawamura, Y. Yamamoto and T. Yagi, J. Phys. Chem. Solids, 2004, 65, 1555–1559 CrossRef CAS.
- L. E. Bove and U. Ranieri, Philos. Trans. R. Soc., A, 2019, 377, 20180262 CrossRef CAS PubMed.
- S. Schaack, U. Ranieri, P. Depondt, R. Gaal, W. F. Kuhs, P. Gillet, F. Finocchi and L. E. Bove, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16204–16209 CrossRef CAS PubMed.
- C. G. Salzmann, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16164–16166 CrossRef CAS PubMed.
- H. Kadobayashi, H. Hirai, H. Ohfuji, M. Ohtake and Y. Yamamoto, J. Chem. Phys., 2018, 148, 164503 CrossRef PubMed.
- T. Tanaka, H. Hirai, T. Matsuoka, Y. Ohishi, T. Yagi, M. Ohtake, Y. Yamamoto, S. Nakano and T. Irifune, J. Chem. Phys., 2013, 139, 104701 CrossRef PubMed.
- X. Cao, Y. Huang, X. Jiang, Y. Su and J. Zhao, Phys. Chem. Chem. Phys., 2017, 19, 15996–16002 RSC.
- J. E. Proctor, H. E. Maynard-Casely, M. A. Hakeem and D. Cantiah, J. Raman Spectrosc., 2017, 48, 1777–1782 CrossRef CAS.
- L. Bezacier, E. Le Menn, O. Grasset, O. Bollengier, A. Oancea, M. Mezouar and G. Tobie, Phys. Earth Planet. Inter., 2014, 229, 144–152 CrossRef CAS.
- Y. Huang, K. Li, X. Jiang, Y. Su, X. Cao and J. Zhao, J. Phys. Chem. A, 2018, 122, 6007–6013 CAS.
- D. Londono, W. F. Kuhs and J. L. Finney, Nature, 1988, 332, 141 CrossRef CAS.
- W. F. Kuhs, T. C. Hansen and A. Falenty, J. Phys. Chem. Lett., 2018, 9, 3194–3198 CrossRef CAS PubMed.
- W. L. Vos, L. W. Finger, R. J. Hemley and H. K. Mao, Phys. Rev. Lett., 1993, 71, 3150–3153 CrossRef CAS PubMed.
- M. Catti, L. del Rosso, L. Ulivi, M. Celli, F. Grazzi and T. Hansen, Phys. Chem. Chem. Phys., 2019, 21, 14671–14677 RSC.
- D. L. Fowler, W. V. Loebenstein, D. B. Pall and C. A. Kraus, J. Am. Chem. Soc., 1940, 62, 1140–1142 CrossRef CAS.
- Y. Kamata, H. Oyama, W. Shimada, T. Ebinuma, S. Takeya, T. Uchida, J. Nagao and H. Narita, Jpn. J. Appl. Phys., Part 1, 2004, 43, 362–365 CrossRef CAS.
- A. Chapoy, R. Anderson and B. Tohidi, J. Am. Chem. Soc., 2007, 129, 746–747 CrossRef CAS PubMed.
- R. K. McMullan, M. Bonamico and G. A. Jeffrey, J. Chem. Phys., 1963, 39, 3295–3310 CrossRef CAS.
- D. W. Devidson, Water Compr. Treatise, 1973, 115–234 Search PubMed.
- G. A. Jeffrey, J. Inclusion Phenom., 1984, 1, 211–222 CrossRef CAS.
- W. Shimada, M. Shiro, H. Kondo, S. Takeya, H. Oyama, T. Ebinuma and H. Narita, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o65–o66 CrossRef PubMed.
- S. A. Lawrence, Amines: synthesis, properties and applications, Cambridge University Press, 2004 Search PubMed.
- F. Ma, Y. Wan, G. Yuan, L. Meng, Z. Dong and J. Hu, Environ. Sci. Technol., 2012, 46, 3236–3243 CrossRef CAS PubMed.
- X. Ge, A. S. Wexler and S. L. Clegg, Atmos. Environ., 2011, 45, 524–546 CrossRef CAS.
- O. G. Brakstad, L. Sørensen, K. Zahlsen, K. Bonaunet, A. Hyldbakk and A. M. Booth, Int. J. Greenhouse Gas Control, 2018, 70, 157–163 CrossRef CAS.
- H. Guo, C. Li, X. Shi, H. Li and S. Shen, Appl. Energy, 2019, 239, 725–734 CrossRef CAS.
- G. A. Jeffrey and R. K. M. Ullan, Prog. Inorg. Chem., 2009, 16, 43 Search PubMed.
- M. D. Gregory, Hydration of Several Aliphatic Amines in Some Non-Polar Solvents, The University of Oklahoma, 1968 Search PubMed.
- K. Ogata, T. Tsuda, S. Amano, S. Hashimoto, T. Sugahara and K. Ohgaki, Chem. Eng. Sci., 2010, 65, 1616–1620 CrossRef CAS.
- W. Shin, S. Park, J. W. Lee, Y. Seo, D. Y. Koh, J. Seol and H. Lee, J. Phys. Chem. C, 2012, 116, 16352–16357 CrossRef CAS.
- S. Lee, Y. Lee, S. Park and Y. Seo, J. Phys. Chem. B, 2012, 116, 13476–13480 CrossRef CAS PubMed.
- Y. Youn, J. Seol, M. Cha, Y. H. Ahn and H. Lee, J. Chem. Eng. Data, 2014, 59, 2004–2012 CrossRef CAS.
- Y. Youn, M. Cha and H. Lee, Fluid Phase Equilib., 2016, 413, 123–128 CrossRef CAS.
- S. Park, H. Kang, K. Shin, Y. Seo and H. Lee, Phys. Chem. Chem. Phys., 2015, 17, 1949–1956 RSC.
- N. J. English and J. M. D. MacElroy, Chem. Eng. Sci., 2015, 121, 133–156 CrossRef CAS.
- B. C. Barnes and A. K. Sum, Curr. Opin. Chem. Eng., 2013, 2, 184–190 CrossRef.
- N. J. English and J. S. Tse, J. Phys. Chem. A, 2011, 115, 6226–6232 CrossRef CAS PubMed.
- A. Falenty, T. C. Hansen and W. F. Kuhs, Nature, 2014, 516, 231 CrossRef CAS PubMed.
- Y. Krishnan, M. R. Ghaani and N. J. English, J. Phys. Chem. C, 2019, 123, 27554–27560 CrossRef CAS.
- J. S. Tse, M. L. Klein and I. R. McDonald, J. Phys. Chem., 1983, 87, 4198–4203 CrossRef CAS.
- A. Wallqvist, J. Chem. Phys., 1992, 96, 5377–5382 CrossRef CAS.
- N. J. English and G. M. Phelan, J. Chem. Phys., 2009, 131, 74704 CrossRef PubMed.
- N. J. English and S. T. John, Phys. Rev. Lett., 2009, 103, 15901 CrossRef PubMed.
- N. J. English, S. T. John and D. J. Carey, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 134306 CrossRef.
- P. Warrier, M. N. Khan, V. Srivastava, C. M. Maupin and C. A. Koh, J. Chem. Phys., 2016, 145, 211705 CrossRef PubMed.
- N. J. English and C. C. R. Allen, Sci. Total Environ, 2019, 661, 664–669 CrossRef CAS PubMed.
- A. Hassanpouryouzband, J. Yang, A. Okwananke, R. Burgass, B. Tohidi, E. Chuvilin, V. Istomin and B. Bukhanov, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- T. E. Chnology, J. Pet. Technol., 1941, 43, 1–414 Search PubMed.
- D. L. Katz, Trans. AIME, 1945, 160, 140–149 CrossRef.
- J. H. van der Waals and J. C. Platteeuw, Adv. Chem. Phys., 1958, 1–57 Search PubMed.
- J. H. van der Waals, Trans. Faraday Soc., 1956, 52, 184–193 RSC.
- M. N. Khan, P. Warrier, C. J. Peters and C. A. Koh, Fluid Phase Equilib., 2018, 463, 48–61 CrossRef CAS.
- J. E. Lennard-Jones and A. F. Devonshire, Proc. R. Soc. London, Ser. A, 1937, 163, 53–70 CAS.
- V. McKoy and O. Sinanoǧlu, J. Chem. Phys., 1963, 38, 2946–2956 CrossRef CAS.
- W. R. Parrish and J. M. Prausnitz, Ind. Eng. Chem. Process Des. Dev., 1972, 11, 26–35 CrossRef CAS.
- D. R. Hafemann and S. L. Miller, J. Phys. Chem., 1969, 73, 1392–1397 CrossRef CAS.
- H. J. Ng and D. B. Robinson, Ind. Eng. Chem. Fundam., 1976, 15, 293–298 CrossRef CAS.
- G. D. Holder, G. Corbin and K. D. Papadopoulos, Ind. Eng. Chem. Fundam., 1980, 19, 282–286 CrossRef CAS.
- P. Englezos and P. R. Bishnoi, Fluid Phase Equilib., 1988, 42, 129–140 CrossRef CAS.
- M. L. Michelsen, Fluid Phase Equilib., 1982, 9, 1–19 CrossRef CAS.
- P. R. Bishnoi, A. K. Gupta, P. Englezos and N. Kalogerakis, Fluid Phase Equilib., 1989, 53, 97–104 CrossRef CAS.
- A. K. Gupta, P. Raj Bishnoi and N. Kalogerakis, Fluid Phase Equilib., 1991, 63, 65–89 CrossRef CAS.
- A. L. Ballard and E. D. Sloan, Fluid Phase Equilib., 2002, 194–197, 371–383 CrossRef CAS.
- I. S. V. Segtovich, A. G. Barreto and F. W. Tavares, Fluid Phase Equilib., 2016, 413, 196–208 CrossRef CAS.
- T. H. Sirino, M. A. M. Neto, D. Bertoldi, R. E. M. Morales and A. K. Sum, Fluid Phase Equilib., 2018, 475, 45–63 CrossRef CAS.
- S. Hielscher, V. Vinš, A. Jäger, J. Hrubý, C. Breitkopf and R. Span, Fluid Phase Equilib., 2018, 459, 170–185 CrossRef CAS.
- M. A. Mahabadian, A. Chapoy, R. Burgass and B. Tohidi, Fluid Phase Equilib., 2016, 414, 117–132 CrossRef CAS.
- N. M. Alsaifi and P. Englezos, Fluid Phase Equilib., 2011, 302, 169–178 CrossRef CAS.
- H. Haghighi, A. Chapoy, R. Burgess and B. Tohidi, Fluid Phase Equilib., 2009, 276, 24–30 CrossRef CAS.
- L. Li, L. Zhu and J. Fan, Fluid Phase Equilib., 2016, 409, 291–300 CrossRef CAS.
- A. H. S. Dehaghani and M. H. Badizad, Fluid Phase Equilib., 2016, 427, 328–339 CrossRef.
- J. Kondori, S. Zendehboudi and L. James, Ind. Eng. Chem. Res., 2018, 57, 13833–13855 CrossRef CAS.
- A. M. Palma, A. J. Queimada and J. A. P. Coutinho, Ind. Eng. Chem. Res., 2019, 58, 19239–19250 CrossRef CAS.
- M. S. Waseem and N. M. Alsaifi, J. Chem. Thermodyn., 2018, 117, 223–235 CrossRef CAS.
- M. N. Khan, P. Warrier, C. J. Peters and C. A. Koh, J. Nat. Gas Sci. Eng., 2016, 35, 1388–1404 CrossRef CAS.
- M. N. Khan, P. Warrier, C. J. Peters and C. A. Koh, J. Nat. Gas Sci. Eng., 2016, 35, 1355–1361 CrossRef CAS.
- M. Connolly, H. Pan and H. Tchelepi, Ind. Eng. Chem. Res., 2019, 58, 14954–14974 CrossRef CAS.
- A. H. Mohammadi, V. Belandria and D. Richon, Chem. Eng. Sci., 2010, 65, 4302–4305 CrossRef CAS.
- A. Galindo, A. Gil-Villegas, G. Jackson and A. N. Burgess, J. Phys. Chem. B, 1999, 103, 10272–10281 CrossRef CAS.
- A. Fukumoto, L. P. S. Silva, P. Paricaud, D. Dalmazzone and W. Fürst, Int. J. Hydrogen Energy, 2015, 40, 9254–9266 CrossRef CAS.
- P. Babu, P. Paricaud and P. Linga, Fluid Phase Equilib., 2016, 413, 80–85 CrossRef CAS.
- A. Fukumoto, P. Paricaud, D. Dalmazzone, W. Bouchafaa, T. T.-S. Ho and W. Fürst, J. Chem. Eng. Data, 2014, 59, 3193–3204 CrossRef CAS.
- H. Najibi, K. Momeni, M. T. Sadeghi and A. H. Mohammadi, J. Chem. Thermodyn., 2015, 87, 122–128 CrossRef CAS.
- Q.-L. Ma, J.-L. Qi, G.-J. Chen and C.-Y. Sun, Fluid Phase Equilib., 2016, 430, 178–187 CrossRef CAS.
- L. Sun, X. Liang, N. Von Solms and G. M. Kontogeorgis, Fluid Phase Equilib., 2019, 486, 37–47 CrossRef CAS.
- G.-J. Chen and T.-M. Guo, Fluid Phase Equilib., 2002, 122, 43–65 CrossRef.
- K. Růržička and V. Majer, AIChE J., 2004, 42, 1723–1740 CrossRef.
- J. B. Klauda and S. I. Sandler, Ind. Eng. Chem. Res., 2000, 39, 3377–3386 CrossRef CAS.
- K. Růržička and V. Majer, AIChE J., 1996, 42, 1723–1740 CrossRef.
- J. B. Klauda and S. I. Sandler, Chem. Eng. Sci., 2003, 58, 27–41 CrossRef CAS.
- A. Martin and C. J. Peters, J. Phys. Chem. C, 2008, 113, 422–430 CrossRef.
- H. Delavar and A. Haghtalab, Fluid Phase Equilib., 2015, 394, 101–117 CrossRef CAS.
- S. Shahnazar and N. Hasan, Fluid Phase Equilib., 2014, 379, 72–85 CrossRef CAS.
- E. Khamehchi, E. Shamohammadi and S. H. Yousefi, Gas Process. J., 2013, 1, 41–50 Search PubMed.
- A. Chapoy, A. H. Mohammadi and D. Richon, Oil Gas Sci. Technol., 2007, 62, 701–706 CrossRef CAS.
- A. A. Ibrahim, T. A. Lemma, M. L. Kean and M. G. Zewge, Appl. Mech. Mater., 2016, 819, 569–574 Search PubMed.
- C. J. Burnham and N. J. English, J. Phys. Chem. C, 2016, 120, 16561–16567 CrossRef CAS.
- C. J. Burnham, Z. Futera and N. J. English, Phys. Chem. Chem. Phys., 2017, 19, 717–728 RSC.
- M. Pérez-Rodríguez, J. Otero-Fernández, A. Comesaña, Á. M. Fernández-Fernández and M. M. Piñeiro, ACS Omega, 2018, 3, 18771–18782 CrossRef PubMed.
- C. J. Burnham, Z. Futera and N. J. English, J. Chem. Phys., 2018, 148, 102323, DOI:10.1063/1.4999909.
- P. E. Brumby, D. Yuhara, T. Hasegawa, D. T. Wu, A. K. Sum and K. Yasuoka, J. Chem. Phys., 2019, 150, 134503, DOI:10.1063/1.5084785.
- B. Kvamme, J. Zhao, N. Wei and N. Saeidi, Energies, 2020, 13, 880 CrossRef.
- N. J. English and J. S. Tse, Energies, 2010, 3 Search PubMed.
- W. F. Waite, J. C. Santamarina, D. D. Cortes, B. Dugan, D. N. Espinoza, J. Germaine, J. Jang, J. W. Jung, T. J. Kneafsey, H. Shin, K. Soga, W. J. Winters and T. Yun, Rev. Geophys., 2009, 47, 1–38 CrossRef.
- R. G. Ross, P. Andersson and G. Bäckström, Nature, 1981, 290, 322–323 CrossRef CAS.
- P. Andersson and R. G. Ross, J. Phys. C: Solid State Phys., 1983, 16, 1423–1432 CrossRef CAS.
- J. S. Tse and M. A. White, J. Phys. Chem., 1988, 92, 5006–5011 CrossRef CAS.
- R. G. Ross and P. Andersson, Can. J. Chem., 1982, 60, 881–892 CrossRef CAS.
- G. A. Slack, MRS Proc., 1997, 478, 47 CrossRef CAS.
- G. S. Nolas, D. T. Morelli and T. M. Tritt, Annu. Rev. Mater. Sci., 1999, 29, 89–116 CrossRef CAS.
- J. L. Cohn, G. S. Nolas, V. Fessatidis, T. H. Metcalf and G. A. Slack, Phys. Rev. Lett., 1999, 82, 779–782 CrossRef CAS.
- A. I. Krivchikov, B. Y. Gorodilov, O. A. Korolyuk, V. G. Manzhelii, H. Conrad and W. Press, J. Low Temp. Phys., 2005, 139, 693–702 CrossRef CAS.
- A. I. Krivchikov, B. Y. Gorodilov, O. A. Korolyuk, V. G. Manzhelii, O. O. Romantsova, H. Conrad, W. Press, J. S. Tse and D. D. Klug, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 64203 CrossRef.
- J. S. Tse, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 17, 259–266 CrossRef CAS.
- R. Inoue, H. Tanaka and K. Nakanishi, J. Chem. Phys., 1996, 104, 9569–9577 CrossRef CAS.
- J. S. Tse, V. P. Shpakov, V. V. Murashov and V. R. Belosludov, J. Chem. Phys., 1997, 107, 9271–9274 CrossRef CAS.
- J. S. Tse, C. I. Ratcliffe, B. M. Powell, V. F. Sears and Y. P. Handa, J. Phys. Chem. A, 1997, 101, 4491–4495 CrossRef CAS.
- J. Baumert, C. Gutt, V. P. Shpakov, J. S. Tse, M. Krisch, M. Müller, H. Requardt, D. D. Klug, S. Janssen and W. Press, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 174301 CrossRef.
- A. I. Krivchikov, A. N. Yushchenko, O. A. Korolyuk, F. J. Bermejo, R. Fernandez-Perea, I. Bustinduy and M. A. González, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 24202 CrossRef.
- T. Takabatake, K. Suekuni, T. Nakayama and E. Kaneshita, Rev. Mod. Phys., 2014, 86, 669–716 CrossRef CAS.
- A. Desmedt, L. Bedouret, E. Pefoute, M. Pouvreau, S. Say-Liang-Fat and M. Alvarez, Eur. Phys. J.-Spec. Top., 2012, 213, 103–127 CrossRef CAS.
- R. P. Warzinski, I. K. Gamwo, E. J. Rosenbaum, E. M. Myshakin, H. Jiang, K. D. Jordan, N. J. English and D. W. Shaw, Alternative Energy and Shale Gas Encyclopedia, 2016, pp. 680–686 Search PubMed.
- M. M. Safarov and M. A. Zaripova, Meas. Tech., 1993, 36, 435–438 CrossRef.
- O. Andersson and H. Suga, J. Phys. Chem. Solids, 1996, 57, 125–132 CrossRef CAS.
- O. Andersson and G. P. Johari, J. Chem. Phys., 2009, 131, 114503 CrossRef PubMed.
- N. J. English, J. S. Tse and R. Gallagher, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 92201 CrossRef.
- W. F. Waite, L. A. Stern, S. H. Kirby, W. J. Winters and D. H. Mason, Geophys. J. Int., 2007, 169, 767–774 CrossRef.
- J. Zhao, S. Sun, C. Liu and Q. Meng, Heat Mass Transfer, 2018, 54, 3287–3295 CrossRef CAS.
- J. G. Cook and D. G. Leaist, Geophys. Res. Lett., 1983, 10, 397–399 CrossRef CAS.
- D. Huang and S. Fan, J. Chem. Eng. Data, 2004, 49, 1479–1482 CrossRef CAS.
- J. F. Gabitto and C. Tsouris, Physical properties of gas hydrates: A review, J. Thermodyn., 2010271291, http://downloads.hindawi.com/journals/jtd/2010/271291.pdf Search PubMed.
- K. Fujiura, Y. Nakamoto, Y. Taguchi, R. Ohmura and Y. Nagasaka, Fluid Phase Equilib., 2016, 413, 129–136 CrossRef CAS.
- D. Li, D. Liang, H. Peng and L. Wan, J. Therm. Anal. Calorim., 2016, 123, 1391–1397 CrossRef CAS.
- W. F. Waite, L. Y. Gilbert, W. J. Winters and D. H. Mason, Rev. Sci. Instrum., 2006, 77, 44904 CrossRef.
- W. F. Waite, Preliminary Laboratory Thermal Conductivity Measurements in Pure Methane Hydrate and Methane Hydrate-Sediment Mixtures: A Progress Report, 2011 Search PubMed.
- D. Z. Huang, S. S. Fan, D. Q. Liang and Z. P. Feng, Chin. J. Geophys., 2005, 48, 1201–1207 CrossRef.
- D. Li, J. Du, S. He, D. Liang, X. Zhao and X. Yang, Sci. China: Chem., 2012, 55, 373–379 CrossRef CAS.
- S. Sun, X. Jin, C. Liu, Q. Meng and Y. Zhang, Int. J. Heat Mass Transfer, 2018, 127, 88–96 CrossRef CAS.
- E. J. Rosenbaum, N. J. English, J. K. Johnson, D. W. Shaw and R. P. Warzinski, J. Phys. Chem. B, 2007, 111, 13194–13205 CrossRef CAS PubMed.
- S. Sun, J. Zhao, J. Zhao, Y. Hao and J. Yang, J. Chem. Thermodyn., 2019, 132, 423–431 CrossRef CAS.
- A. I. Krivchikov, V. G. Manzhelii, O. A. Korolyuk, B. Y. Gorodilov and O. O. Romantsova, Phys. Chem. Chem. Phys., 2005, 7, 728–730 RSC.
- Y. Handa and J. G. Cook, J. Phys. Chem., 1987, 91, 6327–6328 CrossRef CAS.
- J. Li, Z.-L. Wang and G.-C. Yao, Chin. Phys. Lett., 2018, 35, 70502 CrossRef.
- P. Kumar, D. Turner and E. D. Sloan, J. Geophys. Res. Solid Earth, 2004, 109, B01207, DOI:10.1029/2003JB002763.
- D. J. Turner, P. Kumar and E. D. Sloan, Int. J. Thermophys., 2005, 26, 1681–1691 CrossRef CAS.
- Y. P. Handa, R. E. Hawkins and J. J. Murray, J. Chem. Thermodyn., 1984, 16, 623–632 CrossRef CAS.
- D. G. Leaist, J. J. Murray, M. L. Post and D. W. Davidson, J. Phys. Chem., 1982, 86, 4175–4178 CrossRef CAS.
- Y. P. Handa, J. Chem. Thermodyn., 1985, 17, 201–208 CrossRef CAS.
- Y. P. Handa, J. Chem. Thermodyn., 1986, 18, 891–902 CrossRef CAS.
- Y. P. Handa, J. Chem. Thermodyn., 1986, 18, 915–921 CrossRef CAS.
- O. Yamamuro, M. Oguni, T. Matsuo and H. Suga, Solid State Commun., 1987, 62, 289–292 CrossRef CAS.
- Y. Paul Handa, Ind. Eng. Chem. Res., 1988, 27, 872–874 CrossRef CAS.
- O. Yamamuro, M. Oguni, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1988, 49, 425–434 CrossRef CAS.
- R. M. Rueff, E. Dendy Sloan and V. F. Yesavage, AIChE J., 1988, 34, 1468–1476 CrossRef CAS.
- E. Tombari, S. Presto, G. Salvetti and G. P. Johari, J. Chem. Phys., 2006, 124, 154507, DOI:10.1063/1.2188944.
- J. Gao and N. M. Kenneth, Chin. J. Chem. Eng., 2003, 11, 276–279 CAS.
- J. S. Lievois, R. Perkins, R. J. Martin and R. Kobayashi, Fluid Phase Equilib., 1990, 59, 73–97 CrossRef CAS.
- S.-P. Kang, H. Lee and B.-J. Ryu, J. Chem. Thermodyn., 2001, 33, 513–521 CrossRef CAS.
- Y. Zhang, P. G. Debenedetti, R. K. PruD’Homme and B. A. Pethica, J. Phys. Chem. B, 2004, 108, 16717–16722 CrossRef CAS.
- M. B. Rydzy, J. M. Schicks, R. Naumann and J. Erzinger, J. Phys. Chem. B, 2007, 111, 9539–9545 CrossRef CAS PubMed.
- G. Li, D. Liu and Y. Xie, J. Therm. Anal. Calorim., 2010, 102, 819–826 CrossRef CAS.
- T.-H. Kwon, T. J. Kneafsey and E. V. L. Rees, J. Phys. Chem. B, 2011, 115, 8169–8175 CrossRef CAS PubMed.
- Y. Lee, S. Lee, J. Lee and Y. Seo, Chem. Eng. J., 2014, 246, 20–26 CrossRef CAS.
- L. Mu and N. von Solms, J. Chem. Thermodyn., 2018, 117, 33–42 CrossRef CAS.
- J. Deschamps and D. Dalmazzone, J. Therm. Anal. Calorim., 2009, 98, 113 CrossRef CAS.
- S. Sun, J. Zhao and D. Yu, Fluid Phase Equilib., 2018, 456, 92–97 CrossRef CAS.
- W. Lin, A. Delahaye and L. Fournaison, Fluid Phase Equilib., 2008, 264, 220–227 CrossRef CAS.
- I. N. Tsimpanogiannis, V. K. Michalis and I. G. Economou, Fluid Phase Equilib., 2019, 489, 30–40 CrossRef CAS.
- Z.-L. Wang, K.-P. Yuan and D.-W. Tang, Chin. Phys. Lett., 2015, 32, 104401 CrossRef.
- Q.-B. Li and C. Liu, K. Cheng Je Wu Li Hsueh Pao/Journal Eng. Thermophys., 2015, 36, 714–719.
- D. Yang, Y. Liu, Y. Xiu, H. Xu, K. Yuan and Z. Xu, Zhongguo Shiyou Daxue Xuebao (Ziran Kexue Ban)/Journal China Univ. Pet. (Edition Nat. Sci.), 2016, 40, 141–145.
- H. Ghafari and H. Mohammadi-Manesh, Mol. Simul., 2019, 45, 614–622 CrossRef CAS.
- L. Wan, D. Liang, N. Wu and J. Guan, Huagong Xuebao, 2012, 63, 382–386 CAS.
- L. Wan, D. Liang, N. Wu and J. Guan, Sci. China: Chem., 2012, 55, 167–174 CrossRef CAS.
- L. H. Wan, D. Q. Liang and J. A. Guan, Adv. Mater. Res., 2014, 1008–1009, 861–872 Search PubMed.
- F. L. Ning, K. Glavatskiy, Z. Ji, S. Kjelstrup and T. J. H. Vlugt, Phys. Chem. Chem. Phys., 2015, 17, 2869–2883 RSC.
- V. S. Baghel, R. Kumar and S. Roy, J. Phys. Chem. C, 2013, 117, 12172–12182 CrossRef CAS.
- S. Alavi and R. Ohmura, J. Chem. Phys., 2016, 145, 154708, DOI:10.1063/1.4964673.
- Y. Hu, B. R. Lee and A. K. Sum, Fluid Phase Equilib., 2017, 450, 24–29 CrossRef CAS.
- F. Ning, Y. Yu, S. Kjelstrup, T. J. H. Vlugt and K. Glavatskiy, Energy Environ. Sci., 2012, 5, 6779–6795 RSC.
- J. Kondori, S. Zendehboudi and M. E. Hossain, J. Pet. Sci. Eng., 2017, 159, 754–772 CrossRef CAS.
- P. Guo, Y.-K. Pan, L.-L. Li and B. Tang, Chin. Phys. B, 2017, 26, 73101 CrossRef.
- C. Ruppel, Thermal State of the Gas Hydrate Reservoir, in Natural Gas Hydrate, ed. M. D. Max, Springer Netherlands, Dordrecht, 2000, pp. 29–42 Search PubMed.
- Q. Chen, S. Diao and Y. Ye, Thermophysical Properties of Gas Hydrate in Porous Media, in Natural Gas Hydrates, ed. Y. Ye and C. Liu, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 141–167 Search PubMed.
- D. D. Cortes, A. I. Martin, T. S. Yun, F. M. Francisca, J. C. Santamarina and C. Ruppel, J. Geophys. Res. Solid Earth, 2009, 114, B11103, DOI:10.1029/2008JB006235.
- D. Huang and S. Fan, J. Geophys. Res. Solid Earth, 2005, 110, B01311, DOI:10.1029/2004JB003314.
- J. Zhao, C. Cheng, Y. Song, W. Liu, Y. Liu, K. Xue, Z. Zhu, Z. Yang, D. Wang and M. Yang, Energies, 2012, 5 Search PubMed.
- S. Dangayach, D. N. Singh, P. Kumar, S. K. Dewri, B. Roy, C. Tandi and J. Singh, Mar. Pet. Geol., 2015, 67, 653–662 CrossRef CAS.
- Z. R. Chong, S. H. B. Yang, P. Babu, P. Linga and X.-S. Li, Appl. Energy, 2016, 162, 1633–1652 CrossRef.
- E. Chuvilin and B. Bukhanov, Energy Fuels, 2017, 31, 5246–5254 CrossRef CAS.
- E. Chuvilin, Effect of Ice and Hydrate Formation on Thermal Conductivity of Sediments, in Impact of Thermal Conductivity on Energy Technologies, ed. B. Bukhanov, Intech Open, Rijeka, 2018, ch. 7 Search PubMed.
- R. D. Stoll and G. M. Bryan, J. Geophys. Res., 1979, 84, 1629–1634 CrossRef CAS.
- W. F. Waite, B. J. deMartin, S. H. Kirby, J. Pinkston and C. D. Ruppel, Geophys. Res. Lett., 2002, 29, 82–84 CrossRef.
- M. Muraoka, M. Ohtake, N. Susuki, Y. Yamamoto, K. Suzuki and T. Tsuji, J. Geophys. Res. Solid Earth, 2014, 119, 8021–8033 CrossRef CAS.
- S. Dai, J.-H. Cha, E. J. Rosenbaum, W. Zhang and Y. Seol, Geophys. Res. Lett., 2015, 42, 6295–6305 CrossRef.
- M. Muraoka, N. Susuki, H. Yamaguchi, T. Tsuji and Y. Yamamoto, Energy Fuels, 2015, 29, 1345–1351 CrossRef CAS.
- D. Li and D. Liang, Int. J. Heat Mass Transfer, 2016, 92, 8–14 CrossRef CAS.
- L. Dongliang, P. Hao and L. Deqing, Int. J. Heat Mass Transfer, 2017, 104, 566–573 CrossRef.
- J. Zhao, B. Wang, L. Yang, C. Cheng and Y. Song, Rev. Sci. Instrum., 2015, 86, 85110 CrossRef PubMed.
- L. Yang, J. Zhao, W. Liu, M. Yang and Y. Song, Energy, 2015, 79, 203–211 CrossRef.
- L. Yang, J. Zhao, B. Wang, W. Liu, M. Yang and Y. Song, Fuel, 2016, 179, 87–96 CrossRef CAS.
- B. Wang, Z. Fan, P. Lv, J. Zhao and Y. Song, Int. J. Heat Mass Transfer, 2017, 110, 142–150 CrossRef CAS.
- Y. Dong, J. S. McCartney and N. Lu, Geotech. Geol. Eng., 2015, 33, 207–221 CrossRef.
- J. Mo and H. Ban, Case Stud. Therm. Eng., 2017, 10, 423–433 CrossRef.
- R. D. Hyndman, T. Yuan and K. Moran, Earth Planet. Sci. Lett., 1999, 172, 167–177 CrossRef CAS.
- X. Zhou, S. Fan, D. Liang, D. Wang and N. Huang, J. Nat. Gas Chem., 2007, 16, 399–403 CrossRef CAS.
- A. E. Cook, B. I. Anderson, A. Malinverno, S. Mrozewski and D. S. Goldberg, Geophysics, 2010, 75, F173–F185 CrossRef.
- G. Y. Kim, B. Y. Yi, D. G. Yoo, B. J. Ryu and M. Riedel, Mar. Pet. Geol., 2011, 28, 1979–1985 CrossRef.
- M. W. Lee and T. S. Collett, Mar. Pet. Geol., 2011, 28, 439–449 CrossRef.
- A. E. Cook, B. I. Anderson, J. Rasmus, K. Sun, Q. Li, T. S. Collett and D. S. Goldberg, Mar. Pet. Geol., 2012, 34, 72–84 CrossRef.
- M. W. Lee and T. S. Collett, Mar. Pet. Geol., 2013, 47, 195–203 CrossRef.
- M. Priegnitz, J. Thaler, E. Spangenberg, C. Rücker and J. M. Schicks, Rev. Sci. Instrum., 2013, 84, 104502 CrossRef PubMed.
- M. Karamoddin and F. Varaminian, J. Ind. Eng. Chem., 2014, 20, 3815–3820 CrossRef CAS.
- B. Tohidi, J. Yang, M. Salehabadi, R. Anderson and A. Chapoy, Environ. Sci. Technol., 2010, 44, 1509–1514 CrossRef CAS PubMed.
- K. Schwalenberg, E. Willoughby, R. Mir and R. N. Edwards, Marine Gas Hydrate Electromagnetic Signatures in Cascadia and Their Correlation with Seismic Blank Zones, First Break, 2005, 23, 57–63 Search PubMed.
- U. Shankar and M. Riedel, Mar. Pet. Geol., 2011, 28, 1768–1778 CrossRef.
- X. Wang, S. Wu, M. Lee, Y. Guo, S. Yang and J. Liang, Mar. Pet. Geol., 2011, 28, 1625–1633 CrossRef.
- X. Wang, M. Lee, T. Collett, S. Yang, Y. Guo and S. Wu, Mar. Pet. Geol., 2014, 51, 298–306 CrossRef.
- X. Wang, T. S. Collett, M. W. Lee, S. Yang, Y. Guo and S. Wu, Mar. Geol., 2014, 357, 272–292 CrossRef.
- J. Liu, J. Zhang, F. Ma, M. Wang and Y. Sun, Mar. Pet. Geol., 2017, 88, 225–234 CrossRef.
- J. Wei, Y. Fang, H. Lu, H. Lu, J. Lu, J. Liang and S. Yang, Mar. Pet. Geol., 2018, 98, 622–628 CrossRef CAS.
- J. R. Kliner and J. L. H. Grozic, Can. Geotech. J., 2006, 43, 551–562 CrossRef CAS.
- Y. F. Sun and D. Goldberg, Geophys. Res. Lett., 2005, 32, L04313, DOI:10.1029/2004GL021976.
- Y. Sun, D. Goldberg, T. Collett and R. Hunter, Mar. Pet. Geol., 2011, 28, 450–459 CrossRef.
- R. E. Grimm, D. E. Stillman, S. F. Dec and M. A. Bullock, J. Phys. Chem. B, 2008, 112, 15382–15390 CrossRef CAS PubMed.
- W. L. Du Frane, L. A. Stern, K. A. Weitemeyer, S. Constable, J. C. Pinkston and J. J. Roberts, Geophys. Res. Lett., 2011, 38, L09313, DOI:10.1029/2011GL047243.
- W. L. Du Frane, L. A. Stern, K. A. Weitemeyer, S. Constable, J. C. Pinkston and J. J. Roberts, Electrical conductivity of laboratory-synthesized methane hydrate, Lawrence Livermore National Lab (LLNL), Livermore, CA, United States, 2011 Search PubMed.
- T. M. Nahir, J. Am. Chem. Soc., 2005, 127, 12431 CrossRef CAS.
- L. Stern, W. L. Du Frane, K. A. Weitemeyer, S. Constable and J. J. Roberts, AGU Fall Meeting Abstracts, 2012.
- R. Lu, L. A. Stern, W. L. Du Frane, J. C. Pinkston, J. J. Roberts and S. Constable, AGU Fall Meeting Abstracts, 2018 Search PubMed.
- J. Lee, Hydrate-Bearing Sediments: Formation and Geophysical Properties, Georgia Institute of Technology, 2007 Search PubMed.
- K. A. Weitemeyer, S. C. Constable, K. W. Key and J. P. Behrens, Geophys. Res. Lett., 2006, 33, L03304, DOI:10.1029/2005GL024896.
- K. Schwalenberg, M. Haeckel, J. Poort and M. Jegen, Mar. Geol., 2010, 272, 79–88 CrossRef CAS.
- K. A. Weitemeyer, S. Constable and A. M. Tréhu, Geophys. J. Int., 2011, 187, 45–62 CrossRef CAS.
- S.-K. Hsu, C.-W. Chiang, R. L. Evans, C.-S. Chen, S.-D. Chiu, Y.-F. Ma, S.-C. Chen, C.-H. Tsai, S.-S. Lin and Y. Wang, J. Asian Earth Sci., 2014, 92, 224–232 CrossRef.
- B. K. Goswami, K. A. Weitemeyer, T. A. Minshull, M. C. Sinha, G. K. Westbrook, A. Chabert, T. J. Henstock and S. Ker, J. Geophys. Res. Solid Earth, 2015, 120, 6797–6822 CrossRef CAS.
- E. Attias, K. Weitemeyer, T. A. Minshull, A. I. Best, M. Sinha, M. Jegen-Kulcsar, S. Hölz and C. Berndt, Geophys. J. Int., 2016, 206, 1093–1110 CrossRef CAS.
- D. Lim, H. Ro, Y. J. Seo, J. Y. Lee, J. Lee, S. J. Kim, Y. Park and H. Lee, Energy Fuels, 2017, 31, 708–713 CrossRef CAS.
- K. Schwalenberg, D. Rippe, S. Koch and C. Scholl, J. Geophys. Res. Solid Earth, 2017, 122, 3334–3350 CrossRef CAS.
- J. Jing, K. Chen, M. Deng, Q. Zhao, X. Luo, G. Tu and M. Wang, J. Asian Earth Sci., 2019, 171, 201–212 CrossRef.
- R. Tharimela, A. Augustin, M. Ketzer, J. Cupertino, D. Miller, A. Viana and K. Senger, Interpretation, 2019, 7, SH111–SH131 CrossRef.
- M. Riedel, T. S. Collett, H.-S. Kim, J.-J. Bahk, J.-H. Kim, B.-J. Ryu and G. Y. Kim, Mar. Pet. Geol., 2013, 47, 222–235 CrossRef.
- U. Shankar and M. Riedel, Mar. Pet. Geol., 2014, 58, 265–277 CrossRef.
- A. E. Cook and W. F. Waite, J. Geophys. Res. Solid Earth, 2018, 123, 2069–2089 CrossRef CAS.
- E. Attias, K. Weitemeyer, S. Hölz, S. Naif, T. A. Minshull, A. I. Best, A. Haroon, M. Jegen-Kulcsar and C. Berndt, Geophys. J. Int., 2018, 214, 1701–1714 CrossRef CAS.
- Z. Weidong, L. Yongjun, R. Shaoran and W. Ruihe, 2011 International Conference on Materials for Renewable Energy & Environment, IEEE, 2011, vol. 2, pp. 1954–1958.
- Y. Chen, D. Li, D. Liang, X. Zhou and N. Wu, Acta Pet. Sin., 2013, 34, 507–512 CrossRef CAS.
- J. Cai, W. Wei, X. Hu and D. A. Wood, Earth-Sci. Rev., 2017, 171, 419–433 CrossRef.
- J. C. Santamarina, F. Francisca, T. S. Yun, J. Y. Lee, A. I. Martin and C. Ruppel, AAPG Hedberg Conference: Gas hydrates: Energy resource potential and associated geological hazards. AAPG, Vancouver, BC, 2004.
- J. Y. Lee, J. C. Santamarina and C. Ruppel, J. Geophys. Res. Solid Earth, 2010, 115, B11104, DOI:10.1029/2009JB006669.
- J. C. Santamarina and C. Ruppel, Geophys. Charact. Gas Hydrates, Geophys. Dev. Ser., 2010, 14, 373–384 Search PubMed.
- E. Spangenberg and J. Kulenkampff, Geophys. Res. Lett., 2006, 33, L24315, DOI:10.1029/2006GL028188.
- S. R. Ren, Y. Liu, Y. Liu and W. Zhang, J. Pet. Sci. Eng., 2010, 70, 52–56 CrossRef CAS.
- Y. F. Chen, D. Q. Liang and N. Y. Wu, Appl. Mech. Mater., 2013, 432, 104–108 Search PubMed.
- M. E. Permyakov, N. A. Manchenko, A. D. Duchkov, A. Y. Manakov, A. N. Drobchik and A. K. Manshtein, Russ. Geol. Geophys., 2017, 58, 642–649 CrossRef.
- M. Pohl, Ultrasonic and Electrical Properties of Hydrate-Bearing Sediments, Colorado School of Mine, Arthur Lakes Library, 2018 Search PubMed.
- F.-G. Li, C.-Y. Sun, S.-L. Li, G.-J. Chen, X.-Q. Guo, L.-Y. Yang, H. Pan, S. Li and K. Zhang, Energy Fuels, 2012, 26, 6210–6217 CrossRef CAS.
- W. L. Du Frane, L. A. Stern, S. Constable, K. A. Weitemeyer, M. M. Smith and J. J. Roberts, J. Geophys. Res. Solid Earth, 2015, 120, 4773–4783 CrossRef CAS.
- R. Lu, L. A. Stern, W. L. Du Frane, J. C. Pinkston, J. J. Roberts and S. Constable, J. Geophys. Res. Solid Earth, 2019, 124, 10877–10892, DOI:10.1029/2019JB018364.
- X. Hu, C. Zou, Z. Lu, C. Yu, C. Peng, W. Li, Y. Tang, A. Liu and K. S. Kouamelan, J. Geophys. Eng., 2019, 16, 215–228 CrossRef.
- H. Pan, H. Li, D. Grana, Y. Zhang, T. Liu and C. Geng, Mar. Pet. Geol., 2019, 105, 273–283 CrossRef.
- T. Liu, X. Liu and T. Zhu, Mar. Pet. Geol., 2019, 104036 Search PubMed.
- D. Huaimin, S. Jianmeng, C. Likai, S. Liyuan, Y. Weichao, L. Yafen, L. Zhenzhou and F. Hui, J. Geophys. Eng., 2018, 15, 1399–1406 CrossRef.
- H. Dong, J. Sun, Z. Lin, H. Fang, Y. Li, L. Cui and W. Yan, J. Geophys. Eng., 2018, 15, 275–285 CrossRef.
- H. Dong, J. Sun, J. Zhu, L. Liu, Z. Lin, N. Golsanami, L. Cui and W. Yan, Fuel, 2019, 248, 27–37 CrossRef CAS.
- M. Priegnitz, J. Thaler, E. Spangenberg, J. M. Schicks, J. Schrötter and S. Abendroth, Geophys. J. Int., 2015, 202, 1599–1612 CrossRef CAS.
- T. Kremer, C. Vieira and A. Maineult, J. Appl. Geophys., 2018, 158, 11–28 CrossRef.
- M. B. Helgerud, Wave speeds in gas hydrate and sediments containing gas hydrate: A laboratory and modeling study, Stanford University, Palo Alto, 2001 Search PubMed.
- M. B. Helgerud, W. F. Waite, S. H. Kirby and A. Nur, J. Geophys. Res. Solid Earth, 2009, 114, B02212, DOI:10.1029/2008JB006132.
- Y. Li, Y. Song, W. Liu and F. Yu, Energies, 2012, 5 Search PubMed.
- L. Yanghui, Z. Honghua, Y. Feng, S. Yongchen, L. Weiguo, L. Qingping and Y. Haiyuan, J. Cold Reg. Eng., 2012, 26, 149–159 CrossRef.
- L. A. Stern, S. H. Kirby and W. B. Durham, Science, 1996, 273, 1843–1848 CrossRef CAS.
- L. A. Stern, S. H. Kirby and W. B. Durham, Energy Fuels, 1998, 12, 201–211 CrossRef CAS.
- W. F. Waite, M. B. Helgerud, A. Nur, J. C. Pinkston, L. A. Stern, S. H. Kirby and W. B. Durham, Ann. N. Y. Acad. Sci., 2000, 912, 1003–1010 CrossRef CAS.
- W. F. Kuhs, A. Klapproth, F. Gotthardt, K. Techmer and T. Heinrichs, Geophys. Res. Lett., 2000, 27, 2929–2932 CrossRef CAS.
- D. K. Staykova, W. F. Kuhs, A. N. Salamatin and T. Hansen, J. Phys. Chem. B, 2003, 107, 10299–10311 CrossRef CAS.
- W. F. Kuhs, G. Genov, E. Goreshnik, A. Zeller, K. S. Techmer and G. Bohrmann, Int. J. Offshore Polar Eng., 2004, 14, ISOPE-04-14-4-305 Search PubMed.
- B. I. Pandit and M. S. King, Proceedings of the 4th Canadian Permafrost Conference, 1982, pp. 335–342.
- M. Bathe, S. Vagle, G. A. Saunders and E. F. Lambson, J. Mater. Sci. Lett., 1984, 3, 904–906 CrossRef CAS.
- L. I. Berge, K. A. Jacobsen and A. Solstad, J. Geophys. Res. Solid Earth, 1999, 104, 15415–15424 CrossRef CAS.
- M. B. Helgerud, W. F. Waite, S. H. Kirby and A. Nur, Can. J. Phys., 2003, 81, 47–53 CrossRef CAS.
- M. Pohl, M. Prasad and M. L. Batzle, Geophys. Prospect., 2018, 66, 1349–1357 CrossRef.
- B. L. Whiffen, H. Kiefte and M. J. Clouter, Geophys. Res. Lett., 1982, 9, 645–648 CrossRef CAS.
- H. Kiefte, M. J. Clouter and R. E. Gagnon, J. Phys. Chem., 1985, 89, 3103–3108 CrossRef CAS.
- H. Shimizu, T. Kumazaki, T. Kume and S. Sasaki, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 212102 CrossRef.
- S. Sasaki, T. Kumazaki, I. Suwa, T. Kume and H. Shimizu, J. Phys.: Condens. Matter, 2002, 14, 10445 CrossRef CAS.
- J. Beam, J. Yang, J. Liu, C. Liu and J.-F. Lin, J. Chem. Phys., 2016, 144, 154501 CrossRef.
- W. F. Kuhs, B. Chazallon, P. G. Radaelli and F. Pauer, J. Inclusion Phenom. Mol. Recognit. Chem., 1997, 29, 65–77 CrossRef CAS.
- J. S. Loveday, R. J. Nelmes, M. Guthrie, S. A. Belmonte, D. R. Allan, D. D. Klug, J. S. Tse and Y. P. Handa, Nature, 2001, 410, 661–663 CrossRef CAS PubMed.
- B. Chazallon and W. F. Kuhs, J. Chem. Phys., 2002, 117, 308–320 CrossRef CAS.
- C. Sanloup, H. Mao and R. J. Hemley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 25–28 CrossRef CAS PubMed.
- A. Klapproth, E. Goreshnik, D. Staykova, H. Klein and W. F. Kuhs, Can. J. Phys., 2003, 81, 503–518 CrossRef CAS.
- Z. M. Jendi, A. D. Rey and P. Servio, Mol. Simul., 2015, 41, 572–579 CrossRef CAS.
- J. Baumert, C. Gutt, M. Krisch, H. Requardt, M. Müller, J. S. Tse, D. D. Klug and W. Press, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 54302 CrossRef.
- A. Y. Manakov, A. Y. Likhacheva, V. A. Potemkin, A. G. Ogienko, A. V. Kurnosov and A. I. Ancharov, ChemPhysChem, 2011, 12, 2476–2484 CrossRef CAS PubMed.
- D. Wang, D. Li, H. Zhang, S. Fan and H. Zhao, Sci. China, Ser. G: Phys., Mech. Astron., 2008, 51, 1905–1913 CrossRef CAS.
- H. Hirai, Y. Uchihara, H. Fujihisa, M. Sakashita, E. Katoh, K. Aoki, K. Nagashima, Y. Yamamoto and T. Yagi, J. Chem. Phys., 2001, 115, 7066–7070 CrossRef CAS.
- S. Tanaka, F. Maruyama, O. Takano, K. Uchida and N. Oya, Int. Confer. Gas Hydrate, Trondheim, Norw.
- W. Zhang, Experimental Deformation of Methane Hydrate: New Results, National Energy Technology Laboratory, 1999 Search PubMed.
- W. B. Durham, S. H. Kirby, L. A. Stern and W. Zhang, J. Geophys. Res. Solid Earth, 2003, 108, DOI:10.1029/2002JB001872.
- W. B. Durham, L. A. Stern and S. H. Kirby, Can. J. Phys., 2003, 81, 373–380 CrossRef CAS.
- J. Jia, Y. Liang, T. Tsuji, S. Murata and T. Matsuoka, Sci. Rep., 2016, 6, 23548 CrossRef CAS PubMed.
- F. Yu, Y. Song, W. Liu, Y. Li and W. Lam, J. Pet. Sci. Eng., 2011, 77, 183–188 CrossRef CAS.
- Y. Li, W. Liu, Y. Song, M. Yang and J. Zhao, J. Nat. Gas Sci. Eng., 2016, 33, 347–354 CrossRef CAS.
- M. Hyodo, Y. Nakata, N. Yoshimoto, M. Fukunaga, K. Kubo, Y. Nanjo, T. Matsuo, A. F. L. Hyde and K. Nakamura, The Twelfth International Offshore and Polar Engineering Conference, International Society of Offshore and Polar Engineers, 2002.
- Y. Nabeshima and T. T. Matsui, Static Shear Behaviors of Methane Hydrate and Ice, Proc. ISOPE Ocean Min. Symp., 2003, 156–159 CAS.
- Y. Nabeshima and T. Matsui, Fifth ISOPE Ocean Min. Symp., 2003, 4.
- Y. Nabeshima, Y. Takai and T. Komai, Sixth ISOPE Ocean Min. Symp., 2005, 4.
- Y. Song, F. Yu, Y. Li, W. Liu and J. Zhao, J. Nat. Gas Chem., 2010, 19, 246–250 CrossRef CAS.
- F. Yu, Y. Song, W. Liu, Y. Li and J. Zhao, Proc. Int. Conf. Offshore Mech. Arct. Eng., 2010, 705–710, DOI:10.1115/OMAE2010-21174.
- C. R. Miranda and T. Matsuoka, Proceedings of the 6th International Conference on Gas Hydrates, 2008.
- T. M. Vlasic, P. D. Servio and A. D. Rey, Cryst. Growth Des., 2017, 17, 6407–6416 CrossRef CAS.
- X. Zong, G. Cheng, N. Qiu, Q. Huang, J. He, S. Du and Y. Li, Chem. Lett., 2017, 46, 1141–1144 CrossRef CAS.
- Z. M. Jendi, Multiscale Modelling of Gas Hydrate Mechanical and Thermal Properties, McGill University, 2015 Search PubMed.
- T. M. Vlasic, P. D. Servio and A. D. Rey, Ind. Eng. Chem. Res., 2019, 58, 16588–16596 CrossRef CAS.
- H. Huo, Y. Liu, Z. Zheng, J. Zhao, C. Jin and T. Lv, J. Renewable Sustainable Energy, 2011, 3, 63110 CrossRef.
- T. M. Vlasic, P. Servio and A. D. Rey, AIP Adv., 2016, 6, 85317 CrossRef.
- Z. M. Jendi, P. Servio and A. D. Rey, Phys. Chem. Chem. Phys., 2016, 18, 10320–10328 RSC.
- J. Wu, F. Ning, T. T. Trinh, S. Kjelstrup, T. J. H. Vlugt, J. He, B. H. Skallerud and Z. Zhang, Nat. Commun., 2015, 6, 8743 CrossRef CAS PubMed.
- J. Jia, Y. Liang, T. Tsuji, S. Murata and T. Matsuoka, Sci. Rep., 2017, 7, 1290 CrossRef PubMed.
- F. Ning, K. Glavatskiy, T. Vlugt and S. Kjelstrup, Prepr. Pap. – Am. Chem. Soc., Div. Fuel Chem., 2010, 55, 1 Search PubMed.
- H. Jiang and K. D. Jordan, J. Phys. Chem. C, 2010, 114, 5555–5564 CrossRef CAS.
- H. A. Sveinsson, Molecular modeling of fracture in methane hydrates, MS thesis, 2015 Search PubMed.
- P. Cao, J. Wu, Z. Zhang, B. Fang and F. Ning, J. Phys. Chem. C, 2018, 122, 29081–29093 CrossRef CAS.
- H. A. Sveinsson and A. Malthe-Sørenssen, Phys. Chem. Chem. Phys., 2019, 21, 13539–13544 RSC.
- S. V. Gudkovskikh and M. V. Kirov, J. Mol. Model., 2019, 25, 32 CrossRef.
- S. Cai, Q. Tang, S. Tian, Y. Lu and X. Gao, Int. J. Mol. Sci., 2019, 20 Search PubMed.
- J. Wu, B. Skallerud, J. He and Z. Zhang, Procedia IUTAM, 2017, 21, 11–16 CrossRef.
- Q. Shi, P. Cao, Z. Han, F. Ning, H. Gong, Y. Xin, Z. Zhang and J. Wu, Cryst. Growth Des., 2018, 18, 6729–6741 CrossRef CAS.
- V. P. Shpakov, J. S. Tse, C. A. Tulk, B. Kvamme and V. R. Belosludov, Chem. Phys. Lett., 1998, 282, 107–114 CrossRef CAS.
- V. R. Belosludov, V. P. Shpakov, J. S. Tse, R. V. Belosludov and Y. Kawazoe, Ann. N. Y. Acad. Sci., 2000, 912, 993–1002 CrossRef CAS.
- Z. M. Jendi, P. Servio and A. D. Rey, Cryst. Growth Des., 2015, 15, 5301–5309 CrossRef CAS.
- A. O. Akheramka, Molecular Dynamics Simulations to Study the Effect of Fracturing on the Efficiency Of CH4–CO2 Replacement in Hydrates, University of Alaska Fairbanks, 2018 Search PubMed.
- S. M. Daghash, P. Servio and A. D. Rey, Mol. Simul., 2019, 1–14 Search PubMed.
- L. Lei and J. C. Santamarina, Mar. Pet. Geol., 2019, 109, 899–911 CrossRef.
- S. Goto, O. Matsubayashi and S. Nagakubo, J. Geophys. Res. Solid Earth, 2016, 121, 3200–3219 CrossRef CAS.
- J. Rutqvist, G. J. Moridis, T. Grover and T. Collett, J. Pet. Sci. Eng., 2009, 67, 1–12 CrossRef CAS.
- J. Yoneda, A. Masui, Y. Konno, Y. Jin, K. Egawa, M. Kida, T. Ito, J. Nagao and N. Tenma, Mar. Pet. Geol., 2014, 66, 471–486 CrossRef.
- H. Lee, Y. Seo, Y. T. Seo, I. L. Moudrakovski and J. A. Ripmeester, Angew. Chem., Int. Ed., 2003, 42, 5048–5051 CrossRef CAS PubMed.
- T. S. Collett, Proc. 4th Int. Conf. Gas Hydrate, 2002, 2002.
- J. Rutqvist and G. J. Moridis, Offshore Technol. Conf., 2008, 17 Search PubMed.
- F. Ning, N. Wu, Y. Yu, K. Zhang, G. Jiang, L. Zhang, J. Sun and M. Zheng, Geophys. J. Int., 2013, 193, 1385–1398 CrossRef.
- J. Sun, F. Ning, H. Lei, X. Gai, M. Sánchez, J. Lu, Y. Li, L. Liu, C. Liu, N. Wu, Y. He and M. Wu, J. Pet. Sci. Eng., 2018, 170, 345–367 CrossRef CAS.
- H. N. Wang, X. P. Chen, M. J. Jiang and Z. Y. Guo, J. Nat. Gas Sci. Eng., 2019, 68, 102885 CrossRef CAS.
- N. Sultan, P. Cochonat, J.-P. Foucher and J. Mienert, Mar. Geol., 2004, 213, 379–401 CrossRef CAS.
- M. Maslin, M. Owen, R. Betts, S. Day, T. D. Jones and A. Ridgwell, Philos. Trans. R. Soc., A, 2010, 368, 2369–2393 CrossRef CAS PubMed.
- K. P. Lijith, B. R. C. Malagar and D. N. Singh, Mar. Pet. Geol., 2019, 104, 270–285 CrossRef.
- B. Song, Y. Cheng, C. Yan, Y. Lyu, J. Wei, J. Ding and Y. Li, J. Nat. Gas Sci. Eng., 2019, 65, 197–211 CrossRef.
- A. L. Handwerger, A. W. Rempel and R. M. Skarbek, Geochem., Geophys., Geosyst., 2017, 18, 2429–2445 CrossRef.
- A. Li, R. J. Davies and J. Yang, Mar. Geol., 2016, 380, 264–271 CrossRef.
- J. A. Priest, C. R. I. Clayton and E. V. L. Rees, Mar. Pet. Geol., 2014, 58, 187–198 CrossRef CAS.
- J. Yoneda, A. Masui, Y. Konno, Y. Jin, K. Egawa, M. Kida, T. Ito, J. Nagao and N. Tenma, Mar. Pet. Geol., 2015, 66, 451–459 CrossRef.
- K. Miyazaki, A. Masui, Y. Sakamoto, K. Aoki, N. Tenma and T. Yamaguchi, J. Geophys. Res. Solid Earth, 2011, 116, B06102, DOI:10.1029/2010JB008049.
- Y. Li, Y. Song, F. Yu, W. Liu and R. Wang, Pet. Explor. Dev., 2011, 38, 637–640 CrossRef CAS.
- M. Hyodo, Y. Li, J. Yoneda, Y. Nakata, N. Yoshimoto, A. Nishimura and Y. Song, J. Geophys. Res. Solid Earth, 2013, 118, 5185–5194 CrossRef.
- Z. Liu, H. Wei, L. Peng, C. Wei and F. Ning, J. Pet. Sci. Eng., 2017, 149, 56–64 CrossRef CAS.
- B. Wang, P. Huo, T. Luo, Z. Fan, F. Liu, B. Xiao, M. Yang, J. Zhao and Y. Song, Energies, 2017, 10, 531 CrossRef.
- T. S. Yun, F. M. Francisca, J. C. Santamarina and C. Ruppel, Geophys. Res. Lett., 2005, 32, L10609, DOI:10.1029/2005GL022607.
- T. S. Yun, J. C. Santamarina and C. Ruppel, J. Geophys. Res. Solid Earth, 2007, 112, B04106, DOI:10.1029/2006JB004484.
- J. Jang and J. C. Santamarina, Mar. Pet. Geol., 2016, 77, 235–246 CrossRef CAS.
- M. Ben Clennell, M. Hovland, J. S. Booth, P. Henry and W. J. Winters, J. Geophys. Res. Solid Earth, 1999, 104, 22985–23003 CrossRef.
- C. R. I. Clayton, J. A. Priest and E. V. L. Rees, Geotechnique, 2010, 60, 435 CrossRef.
- L. Lei and J. C. Santamarina, J. Geophys. Res. Solid Earth, 2018, 123, 2583–2596 CrossRef CAS.
- W. Lingyun and J. L. Grozic, J. Geotech. Geoenviron. Eng., 2008, 134, 547–550 CrossRef.
- Y. Xu, Y. Seol, J. Jang and S. Dai, Geotechnical Frontiers 2017, 2017, pp. 766–772 Search PubMed.
- S. Dai, J. C. Santamarina, W. F. Waite and T. J. Kneafsey, J. Geophys. Res. Solid Earth, 2012, 117, B11205, DOI:10.1029/2012JB009667.
- N. Mahabadi, S. Dai, Y. Seol and J. Jang, J. Pet. Sci. Eng., 2019, 174, 696–703 CrossRef CAS.
- W. F. Waite, J. C. Santamarina, D. D. Cortes, B. Dugan, D. N. Espinoza, J. Germaine, J. Jang, J. W. Jung, T. J. Kneafsey and H. Shin, Rev. Geophys., 2009, 47(4) DOI:10.1029/2008RG000279.
- X. Ren, Z. Guo, F. Ning and S. Ma, Earth-Sci. Rev., 2020, 103100 CrossRef.
- L. Lei, Z. Liu, Y. Seol, R. Boswell and S. Dai, J. Geophys. Res. Solid Earth, 2019, 124, 3335–3349 CrossRef CAS.
- S. Dai, R. Boswell, W. F. Waite, J. Jang, J. Y. Lee and Y. Seol, The 9th International Conference on Gas Hydrates, 2017, pp. 25–30.
- C. D. Ruppel, J. Y. Lee and I. Pecher, J. Geophys. Res. Solid Earth, 2019, 124, 7525–7537 CrossRef.
- J. Jang, S. Dai, J. Yoneda, W. F. Waite, L. A. Stern, L.-G. Boze, T. S. Collett and P. Kumar, Mar. Pet. Geol., 2019, 108, 537–550 CrossRef CAS.
- M. T. Reagan and G. J. Moridis, Geophys. Res. Lett., 2007, 34, L22709, DOI:10.1029/2007GL031671.
- H. Minagawa, R. Ohmura, Y. Kamata, T. Ebinuma, H. Narita and Y. Masuda, Fifth International Conference on Gas Hydrates, Tapir Acad. Trondheim, Norway, 2005, p. 1058.
- A. Kumar, B. Maini, P. R. Bishnoi, M. Clarke, O. Zatsepina and S. Srinivasan, J. Pet. Sci. Eng., 2010, 70, 114–122 CrossRef.
- H. Liang, Y. Song, Y. Chen and Y. Liu, Pet. Sci. Technol., 2011, 29, 79–87 CrossRef CAS.
- A. Johnson, S. Patil and A. Dandekar, Mar. Pet. Geol., 2011, 28, 419–426 CrossRef CAS.
- B. Li, X.-S. Li, G. Li, J.-L. Jia and J.-C. Feng, Energies, 2013, 6, 3622–3636 CrossRef CAS.
- M. L. Delli and J. L. H. Grozic, J. Pet. Sci. Eng., 2014, 120, 1–9 CrossRef CAS.
- C.-H. Li, Q. Zhao, H.-J. Xu, K. Feng and X.-W. Liu, Appl. Geophys., 2014, 11, 207–214 CrossRef.
- Y. Konno, J. Yoneda, K. Egawa, T. Ito, Y. Jin, M. Kida, K. Suzuki, T. Fujii and J. Nagao, Mar. Pet. Geol., 2014, 66, 487–495 CrossRef.
- W. Liu, Z. Wu, Y. Li, Y. Song, Z. Ling, J. Zhao and Q. Lv, J. Nat. Gas Sci. Eng., 2016, 36, 378–384 CrossRef CAS.
- G. Li, D.-M. Wu, X.-S. Li, Q.-N. Lv, C. Li and Y. Zhang, Appl. Energy, 2017, 202, 282–292 CrossRef CAS.
- Z. Wu, Y. Li, X. Sun, P. Wu and J. Zheng, Appl. Energy, 2018, 230, 1304–1310 CrossRef CAS.
- S. Dai, J. Kim, Y. Xu, W. F. Waite, J. Jang, J. Yoneda, T. S. Collett and P. Kumar, Mar. Pet. Geol., 2019, 108, 705–713, DOI:10.1016/j.marpetgeo.2018.08.016.
- D. W. Meyer, P. B. Flemings, D. DiCarlo, K. You, S. C. Phillips and T. J. Kneafsey, J. Geophys. Res. Solid Earth, 2018, 123, 5350–5371 CrossRef.
- J. Yoneda, M. Oshima, M. Kida, A. Kato, Y. Konno, Y. Jin, J. Jang, W. F. Waite, P. Kumar and N. Tenma, Mar. Pet. Geol., 2019, 108, 524–536 CrossRef.
- Z. Wu, Y. Li, X. Sun, M. Li and R. Jia, Mar. Pet. Geol., 2018, 91, 373–380 CrossRef CAS.
- A. Okwananke, A. Hassanpouryouzband, M. Vasheghani Farahani, J. Yang, B. Tohidi, E. Chuvilin, V. Istomin and B. Bukhanov, J. Pet. Sci. Eng., 2019, 180, 435–444 CrossRef CAS.
- J. Zhang, Y. Ye, G. Hu and S. Diao, Experimental Techniques for Permeability and Mechanical Properties of Hydrate-Bearing Sediments, in Natural Gas Hydrates, ed. Y. Ye and C. Liu, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 169–192 Search PubMed.
- S. K. Garg, J. W. Pritchett, A. Katoh, K. Baba and T. Fujii, J. Geophys. Res. Solid Earth, 2008, 113, B01201, DOI:10.1029/2006JB004768.
- Q. Zhao, K.-J. Dunn and X.-W. Liu, Appl. Geophys., 2011, 8, 101–109 CrossRef.
- M. L. Delli and J. L. H. Grozic, SPE J., 2013, 18, 274–284 CrossRef CAS.
- S. Chatterjee, G. Bhatnagar, B. Dugan, G. R. Dickens, W. G. Chapman and G. J. Hirasaki, J. Geophys. Res. Solid Earth, 2014, 119, 6705–6732 CrossRef CAS.
- S. Dai and Y. Seol, Geophys. Res. Lett., 2014, 41, 4176–4184 CrossRef CAS.
- K. N. Darnell and P. B. Flemings, Geophys. Res. Lett., 2015, 42, 10–765 CrossRef.
- D. H. Kang, T. S. Yun, K. Y. Kim and J. Jang, Geophys. Res. Lett., 2016, 43, 9018–9025 CrossRef.
- H. Daigle, J. Pet. Sci. Eng., 2016, 146, 526–535 CrossRef CAS.
- J. Katagiri, Y. Konno, J. Yoneda and N. Tenma, J. Nat. Gas Sci. Eng., 2017, 45, 537–551 CrossRef.
- J. Behseresht and S. L. Bryant, J. Geophys. Res. Solid Earth, 2017, 122, 3585–3623 CrossRef.
- K. You and P. B. Flemings, J. Geophys. Res. Solid Earth, 2018, 123, 4582–4600 CrossRef CAS.
- P. Mohammadmoradi and A. Kantzas, Mar. Pet. Geol., 2018, 89, 786–798 CrossRef CAS.
- J. Liu, M. Haeckel, J. Rutqvist, S. Wang and W. Yan, J. Geophys. Res. Solid Earth, 2019, 124, 4399–4427 CrossRef CAS.
- L. Wei, A. Cook, H. Daigle, A. Malinverno, M. Nole and K. You, Geochem., Geophys., Geosyst., 2019, 20, 3985–4000 CrossRef CAS.
- H. Singh, E. M. Myshakin and Y. Seol, SPE J., 2019, 24, 547–562 CrossRef CAS.
- J. Nimblett and C. Ruppel, J. Geophys. Res. Solid Earth, 2003, 108(B9) DOI:10.1029/2001jb001650.
- M. Kurihara, K. Funatsu, H. Ouchi, Y. Masuda, M. Yasuda, K. Yamamoto, M. Numasawa, T. Fujii, H. Narita and S. R. Dallimore, Proceedings of the 6th International Conference on Gas Hydrates (ICGH 2008), Vancouver, British Columbia, 2008.
- E. M. Myshakin, B. J. Anderson, K. Rose and R. Boswell, Energy Fuels, 2011, 25, 1077–1091 CrossRef CAS.
- E. M. Myshakin, M. Gaddipati, K. Rose and B. J. Anderson, Mar. Pet. Geol., 2012, 34, 169–185 CrossRef.
- N. Mahabadi and J. Jang, Geochem., Geophys., Geosyst., 2014, 15, 2346–2353 CrossRef CAS.
- J. Sun, F. Ning, S. Li, K. Zhang, T. Liu, L. Zhang, G. Jiang and N. Wu, J. Unconv. Oil Gas Resour., 2015, 12, 23–33 CrossRef.
- N. Mahabadi, S. Dai, Y. Seol, T. Sup Yun and J. Jang, Geochem., Geophys., Geosyst., 2016, 17, 3099–3110 CrossRef CAS.
- N. Mahabadi, X. Zheng and J. Jang, Geophys. Res. Lett., 2016, 43, 4279–4287 CrossRef CAS.
- B. P. VanderBeek and A. W. Rempel, J. Geophys. Res. Solid Earth, 2018, 123, 5394–5411 CrossRef CAS.
- Y. Jin, J. Hayashi, J. Nagao, K. Suzuki, H. Minagawa, T. Ebinuma and H. Narita, Jpn. J. Appl. Phys., 2007, 46, 3159–3162 CrossRef CAS.
- Y. Seol and T. J. Kneafsey, J. Geophys. Res. Solid Earth, 2011, 116, B08102 Search PubMed.
- T. J. Kneafsey, Y. Seol, A. Gupta and L. Tomutsa, SPE J., 2011, 16, 78–94 CrossRef CAS.
- Y. Konno, Y. Jin, T. Uchiumi and J. Nagao, Rev. Sci. Instrum., 2013, 84, 64501 CrossRef PubMed.
- J.-Q. Wang, J.-F. Zhao, M.-J. Yang, Y.-H. Li, W.-G. Liu and Y.-C. Song, Fuel, 2015, 145, 170–179 CrossRef CAS.
- X. Chen, R. Verma, D. N. Espinoza and M. Prodanović, Water Resour. Res., 2018, 54, 600–608 CrossRef.
- E. Kossel, C. Deusner, N. Bigalke and M. Haeckel, J. Geophys. Res. Solid Earth, 2018, 123, 1235–1251 CrossRef CAS.
- J. Wang, L. Zhang, J. Zhao, L. Ai and L. Yang, J. Nat. Gas Sci. Eng., 2018, 51, 141–146 CrossRef.
- S. Chanda and R. P. Singh, Therm. Sci. Eng. Prog., 2019, 11, 380–391 CrossRef.
- J. Joseph, D. N. Singh, P. Kumar, S. K. Dewri, C. Tandi and J. Singh, Mar. Georesour. Geotechnol., 2016, 34, 450–464 CrossRef CAS.
- S. C. Cao, J. Jang, J. Jung, W. F. Waite, T. S. Collett and P. Kumar, Mar. Pet. Geol., 2019, 108, 714–730, DOI:10.1016/j.marpetgeo.2018.09.010.
- M. Vasheghani Farahani, S. Foroughi, S. Norouzi and S. Jamshidi, J. Energy Resour. Technol., 2019, 141(12) DOI:10.1115/1.4044976.
- J. W. Jung, J. Jang, J. C. Santamarina, C. Tsouris, T. J. Phelps and C. J. Rawn, Energy Fuels, 2012, 26, 480–487 CrossRef CAS.
- G. Han, T. Kwon, J. Y. Lee and T. J. Kneafsey, J. Geophys. Res. Solid Earth, 2018, 123, 2539–2558 CrossRef.
- Z. A. Jarrar, R. I. Al-Raoush, J. A. Hannun, K. A. Alshibli and J. Jung, Geomech. Energy Environ., 2018, 100105 Search PubMed.
- G. Han, T.-H. Kwon, J. Y. Lee and J. Jung, Geomech. Energy Environ., 2019, 100131 Search PubMed.
- C. Shuang, J. Jung and T. Kwon, The Characteristics of Fines Migration and Clogging of Sediments Recovered from the Gas Hydrate Deposits from the Ulleng Basin, East Sea, Korea, Proceedings of the XVII ECSMGE, 2019 Search PubMed.
- J. Jung, H. Kang, S. C. Cao, R. I. Al-Raoush, K. Alshibli and J. Y. Lee, Geofluids, 2019 DOI:10.1155/2019/5061216.
- G. Moridis, T. S. Collett, M. Pooladi-Darvish, S. H. Hancock, C. Santamarina, R. Boswell, T. J. Kneafsey, J. Rutqvist, M. B. Kowalsky, M. T. Reagan, E. D. Sloan, A. Sum and C. Koh, SPE Reserv. Eval. Eng., 2011, 14, 76–112 CrossRef CAS.
- W. M. Mahmud, S. Elmabrouk and H. K. Ben Mahmud, Int. J. Pet. Petrochemical Eng. Vol., 2017, 3, 54–64 Search PubMed.
- J. Choi, S. Dai, J. Cha and Y. Seol, Geochem., Geophys., Geosyst., 2014, 15, 1648–1656 CrossRef CAS.
- J. Wang, P. Jaiswal, S. S. Haines, P. E. Hart and S. Wu, Geophysics, 2018, 83, B167–B181 CrossRef.
- Q. T. Bu, G. W. Hu, Y. G. Ye, C. L. Liu, C. F. Li, A. I. Best and J. S. Wang, J. Geophys. Eng., 2017, 14, 555–569 CrossRef.
- M. Riedel, E. C. Willoughby and S. Chopra, Geophysical characterization of gas hydrates, Society of Exploration Geophysicists, 2010 Search PubMed.
- A. I. Best, J. A. Priest, C. R. I. Clayton and E. V. L. Rees, Earth Planet. Sci. Lett., 2013, 368, 78–87 CrossRef CAS.
- Y. Luo, J. Peng, P. Li, J. He and L. Li, J. Nat. Gas Sci. Eng., 2015, 22, 90–97 CrossRef.
- R. Ghosh, K. Sain and M. Ojha, Mar. Geophys. Res., 2010, 31, 29–37 CrossRef.
- Z. Zhang, D. H. Han and Q. Yao, Geophysics, 2011, 76, B139–B150 CrossRef.
- R. Dash and G. Spence, Geophys. J. Int., 2011, 187, 1363–1377 CrossRef CAS.
- A. Schlesinger, J. Cullen, G. Spence, R. Hyndman, K. Louden and D. Mosher, Mar. Pet. Geol., 2012, 35, 105–115 CrossRef.
- Z. Zhang, D. R. McConnell and D. H. Han, Mar. Pet. Geol., 2012, 34, 119–133 CrossRef.
- J. I. T. Hillman, A. E. Cook, H. Daigle, M. Nole, A. Malinverno, K. Meazell and P. B. Flemings, Mar. Pet. Geol., 2017, 86, 1357–1373 CrossRef.
- J. Wang, P. Jaiswal, S. S. Haines, Y. Yang, P. E. Hart and S. Wu, Interpretation, 2019, 1–56 CrossRef.
- N. Satyavani and K. Sain, Mar. Georesources Geotechnol., 2015, 33, 191–201 CrossRef CAS.
- Y. Konno, Y. Jin, J. Yoneda, M. Kida, K. Egawa, T. Ito, K. Suzuki and J. Nagao, Mar. Pet. Geol., 2014, 66, 425–433 CrossRef.
- R. Chatterjee, D. K. Singha, M. Ojha, M. K. Sen and K. Sain, J. Nat. Gas Sci. Eng., 2016, 33, 562–572 CrossRef.
- J. Qian, X. Wang, T. S. Collett, D. Dong, Y. Guo, P. Su and J. Liang, Interpretation, 2017, 5, SM33–SM48 CrossRef.
- X. Wang, B. Liu, J. Qian, X. Zhang, Y. Guo, P. Su, J. Liang, J. Jin, Z. Luan, D. Chen, S. Xi and C. Li, J. Asian Earth Sci., 2018, 168, 27–37 CrossRef.
- J. Guan and D. Liang, Mar. Pet. Geol., 2018, 91, 225–235 CrossRef CAS.
- S. Gullapalli, P. Dewangan, A. Kumar, G. Dakara and C. K. Mishra, Mar. Pet. Geol., 2019, 110, 695–705 CrossRef CAS.
- C. K. Mishra, P. Dewangan, G. Sriram, A. Kumar and G. Dakara, Mar. Pet. Geol., 2019, 104037 Search PubMed.
- S. Singhroha, S. Chand and S. Bünz, J. Geophys. Res. Solid Earth, 2019, 124, 4343–4364, DOI:10.1029/2018JB016574.
- T. A. Minshull, H. Marín-Moreno, P. Betlem, J. Bialas, S. Bünz, E. Burwicz, A. L. Cameselle, G. Cifci, M. Giustiniani, J. I. T. Hillman, S. Hölz, J. R. Hopper, G. Ion, R. León, V. Magalhaes, Y. Makovsky, M.-P. Mata, M. D. Max, T. Nielsen, S. Okay, I. Ostrovsky, N. O’Neill, L. M. Pinheiro, A. A. Plaza-Faverola, D. Rey, S. Roy, K. Schwalenberg, K. Senger, S. Vadakkepuliyambatta, A. Vasilev and J.-T. Vázquez, Mar. Pet. Geol., 2020, 111, 735–764 CrossRef CAS.
- M. Nyamapfumba and G. A. McMechan, Geophysics, 2012, 77, O55–O63 CrossRef.
- J. Wei, T. Pape, N. Sultan, J. L. Colliat, T. Himmler, L. Ruffine, A. de Prunelé, B. Dennielou, S. Garziglia, T. Marsset, C. A. Peters, A. Rabiu and G. Bohrmann, Mar. Pet. Geol., 2015, 59, 359–370 CrossRef CAS.
- A. N. Le, M. Huuse, J. Redfern, R. L. Gawthorpe and D. Irving, Mar. Pet. Geol., 2015, 68, 629–647 CrossRef.
- O. T. Akinsanpe, A. A. Adepelumi, U. K. Benjamin and D. E. Falebita, J. Ocean Univ. China, 2017, 16, 1027–1034 CrossRef CAS.
- G. J. Crutchley, D. R. A. Fraser, I. A. Pecher, A. R. Gorman, G. Maslen and S. A. Henrys, J. Geophys. Res. Solid Earth, 2015, 120, 725–743 CrossRef CAS.
- G. J. Crutchley, G. Maslen, I. A. Pecher and J. J. Mountjoy, Interpretation, 2016, 4, SA1–SA12 CrossRef.
- D. R. A. Fraser, A. R. Gorman, I. A. Pecher, G. J. Crutchley and S. A. Henrys, Mar. Pet. Geol., 2016, 77, 399–408 CrossRef.
- S. Koch, H. Schroeder, M. Haeckel, C. Berndt, J. Bialas, C. Papenberg, D. Klaeschen and A. Plaza-Faverola, Geo-Mar. Lett., 2016, 36, 187–196 CrossRef CAS.
- H. Wang, G. J. Crutchley and T. Stern, Mar. Pet. Geol., 2017, 88, 69–80 CrossRef.
- A. R. Gorman, P. T. Fletcher, D. Baker, D. R. A. Fraser, G. J. Crutchley and S. A. Henrys, ASEG Ext. Abstr., 2018, 2018, 1–4 Search PubMed.
- U. Majumdar, A. E. Cook, W. Shedd and M. Frye, Geophys. Res. Lett., 2016, 43, 7044–7051 CrossRef.
- J. A. Priest, A. I. Best and C. R. I. Clayton, J. Geophys. Res. Solid Earth, 2005, 110, 1–13 Search PubMed.
- J. I. T. Hillman, A. E. Cook, D. E. Sawyer, H. M. Küçük and D. S. Goldberg, Earth Planet. Sci. Lett., 2017, 459, 157–169 CrossRef CAS.
- P. Aird, in Deepwater Drilling, ed. P. B. T.-D. D. Aird, Gulf Professional Publishing, 2019, pp. 17–68 Search PubMed.
- W. Xu and C. Ruppel, J. Geophys. Res. Solid Earth, 1999, 104, 5081–5095 CrossRef CAS.
- C. Ecker, J. Dvorkin and A. Nur, Geophysics, 1998, 63, 1659–1669 CrossRef.
- D. C. Mosher, Mar. Pet. Geol., 2011, 28, 1540–1553 CrossRef.
- W. Shedd, R. Boswell, M. Frye, P. Godfriaux and K. Kramer, Mar. Pet. Geol., 2012, 34, 31–40 CrossRef.
- G. Sriram, P. Dewangan, T. Ramprasad and P. Rama Rao, J. Geophys. Res. Solid Earth, 2013, 118, 2258–2274 CrossRef.
- J. Qian, X.-J. Wang, S.-G. Wu, Z. Wang and S.-X. Yang, Mar. Geophys. Res., 2014, 35, 125–140 CrossRef.
- B. Hosseini Shoar, A. Javaherian, N. Keshavarz Farajkhah and M. Seddigh Arabani, Mar. Pet. Geol., 2014, 51, 184–196 CrossRef.
- R.-W. Zhang, H.-Q. Li, B.-J. Zhang, H.-D. Huang and P.-F. Wen, Appl. Geophys., 2015, 12, 453–464 CrossRef.
- T. Liu and X. Liu, Geophysics, 2018, 83, B143–B154 CrossRef.
- K. P. Arun, K. Sain and J. Kumar, J. Nat. Gas Sci. Eng., 2018, 50, 90–100 CrossRef.
- Y. Mi, A. Sakai, R. Walia, R. D. Hyndman and S. R. Dallimore, CREWES Res. Rep.
- H. Tak, J. Byun, S. J. Seol and D. G. Yoo, Mar. Pet. Geol., 2013, 47, 204–213 CrossRef CAS.
- U. S. Yadav, K. M. Shukla, M. Ojha, P. Kumar and U. Shankar, Mar. Pet. Geol., 2019, 108, 551–561, DOI:10.1016/j.marpetgeo.2019.02.001.
- G. W. Hu, Y. G. Ye, J. Zhang, C. L. Liu, S. B. Diao and J. S. Wang, J. Geophys. Res. Solid Earth, 2010, 115, B02102, DOI:10.1029/2008JB006160.
- Q. Zhang, F.-G. Li, C.-Y. Sun, Q.-P. Li, X.-Y. Wu, B. Liu and G.-J. Chen, Am. Mineral., 2011, 96, 1425–1432 CrossRef CAS.
- G. W. Hu, Y. G. Ye, J. Zhang, S. B. Diao and C. L. Liu, Chin. J. Geophys., 2012, 55, 635–647, DOI:10.1002/cjg2.1758.
- G. Hu, Y. Ye, J. Zhang, C. Liu and Q. Li, Mar. Pet. Geol., 2014, 52, 1–8 CrossRef.
- M. Schindler, M. L. Batzle and M. Prasad, Geophys. Prospect., 2017, 65, 1025–1036 CrossRef.
- D. Sadeq, K. Alef, S. Iglauer, M. Lebedev and A. Barifcani, Int. J. Hydrogen Energy, 2018, 43, 23193–23200 CrossRef CAS.
- X. Li, Y. Liu, H. Zhang, B. Xiao, X. Lv, H. Yao, W. Pang, Q. Li, L. Yang, Y. Song and J. Zhao, Energies, 2019, 12(10), 1997 CrossRef CAS.
- Z. Liu, J. Kim, L. Lei, F. Ning and S. Dai, J. Geophys. Res. Solid Earth, 2019, 124, 3307–3319 CrossRef CAS.
- T. S. Yun, Mechanical and Thermal Study of Hydrate Bearing Sediments, Georgia Institute of Technology, 2005 Search PubMed.
- W. J. Winters, W. F. Waite, D. H. Mason, L. Y. Gilbert and I. A. Pecher, J. Pet. Sci. Eng., 2007, 56, 127–135 CrossRef CAS.
- D. N. Espinoza and J. C. Santamarina, Int. J. Greenhouse Gas Control, 2011, 5, 1031–1038 CrossRef CAS.
- H.-S. Kim, G.-C. Cho and T.-H. Kwon, Geochem., Geophys., Geosyst., 2013, 14, 1787–1799 CrossRef CAS.
- X.-H. Wang, F.-G. Li, Y.-X. Xu, C.-Y. Sun, H. Pan, B. Liu, L.-Y. Yang, G.-J. Chen and Q.-P. Li, Energy Convers. Manage., 2015, 99, 274–281 CrossRef CAS.
- S. Chand, T. A. Minshull, D. Gei and J. M. Carcione, Geophys. J. Int., 2004, 159, 573–590 CrossRef.
- X. Liu, X. Yin and X. Luan, Sci. China: Earth Sci., 2018, 61, 1261–1278 CrossRef CAS.
- D. Sava and B. Hardage, Rock-Physics Models for Gas-Hydrate Systems Associated with Unconsolidated Marine Sediments, in Natural Gas Hydrates–Energy Resource Potential and Associated Geologic Hazards, ed. T. Collett, A. Johnson, C. Knapp and R. Boswell, AAPG Memoir 89, 2009, pp. 505–524 Search PubMed.
- H. Pan, H. Li, J. Chen, Y. Zhang, X. Liu, S. Cai and C. Cao, J. Pet. Sci. Eng., 2019, 182, 106268 CrossRef CAS.
- G. A. Dugarov, A. A. Duchkov, A. D. Duchkov and A. N. Drobchik, J. Nat. Gas Sci. Eng., 2019, 63, 38–46 CrossRef CAS.
- S. Chand, T. A. Minshull, J. A. Priest, A. I. Best, C. R. I. Clayton and W. F. Waite, Geophys. J. Int., 2006, 166, 543–552 CrossRef.
- F. I. Zyserman, P. M. Gauzellino and J. E. Santos, J. Appl. Geophys., 2012, 86, 98–108 CrossRef.
- H. Marín-Moreno, S. K. Sahoo and A. I. Best, J. Geophys. Res. Solid Earth, 2017, 122, 1835–1847 Search PubMed.
- W. Liu, J. Zhao, Y. Luo, Y. Song, Y. Li, M. Yang, Y. Zhang, Y. Liu and D. Wang, Mar. Pet. Geol., 2013, 46, 201–209 CrossRef CAS.
- Z. M. Sun, J. Zhang, C. L. Liu, S. J. Zhao and Y. G. Ye, Appl. Mech. Mater., 2013, 275–277, 326–331 Search PubMed.
- H. Ghiassian and J. L. H. Grozic, Mar. Pet. Geol., 2013, 43, 310–319 CrossRef.
- S. Pinkert and J. L. H. Grozic, J. Geophys. Res. Solid Earth, 2016, 121, 4147–4155 CrossRef.
- W. Liu, T. Luo, Y. Li, Y. Song, Y. Zhu, Y. Liu, J. Zhao, Z. Wu and X. Xu, J. Nat. Gas Sci. Eng., 2016, 32, 20–27 CrossRef CAS.
- T. Luo, Y. Song, Y. Zhu, W. Liu, Y. Liu, Y. Li and Z. Wu, Mar. Pet. Geol., 2016, 77, 507–514 CrossRef CAS.
- S. Gupta, C. Deusner, M. Haeckel, R. Helmig and B. Wohlmuth, Geochem., Geophys., Geosyst., 2017, 18, 3419–3437 CrossRef CAS.
- S. Kajiyama, Y. Wu, M. Hyodo, Y. Nakata, K. Nakashima and N. Yoshimoto, J. Nat. Gas Sci. Eng., 2017, 45, 96–107 CrossRef CAS.
- T. Luo, Y. Li, W. Liu, X. Sun and S. Shen, Energies, 2017, 10 Search PubMed.
- Z. Liu, S. Dai, F. Ning, L. Peng, H. Wei and C. Wei, Geophys. Res. Lett., 2018, 45, 715–723 CrossRef CAS.
- T. Luo, Y. Li, X. Sun, S. Shen and P. Wu, J. Pet. Sci. Eng., 2018, 171, 302–314 CrossRef CAS.
- T. Hirose, W. Tanikawa, Y. Hamada, W. Lin, K. Hatakeda, O. Tadai, H. Y. Wu, S. Nomura, N. Abe, L. P. Gupta, T. Sugihara, Y. Masaki, M. Kinoshita and Y. Yamada, Mar. Pet. Geol., 2019, 108, 348–355, DOI:10.1016/j.marpetgeo.2018.08.017.
- Y. Kuang, L. Yang, Q. Li, X. Lv, Y. Li, B. Yu, S. Leng, Y. Song and J. Zhao, J. Pet. Sci. Eng., 2019, 181, 106173 CrossRef CAS.
- D. Li, Z. Wang, D. Liang and X. Wu, Energies, 2019, 12 CrossRef.
- Y. Li, T. Luo, X. Sun, W. Liu, Q. Li, Y. Li and Y. Song, Energies, 2019, 12 CrossRef.
- B. Gong, Y. Jiang and L. Chen, J. Nat. Gas Sci. Eng., 2019, 69, 102915 CrossRef CAS.
- Y. Li, P. Wu, W. Liu, X. Sun, Z. Cui and Y. Song, Rev. Sci. Instrum., 2019, 90, 55106 CrossRef PubMed.
- L. Dong, Y. Li, H. Liao, C. Liu, Q. Chen, G. Hu, L. Liu and Q. Meng, J. Pet. Sci. Eng., 2020, 184, 106478 CrossRef CAS.
- M. Hyodo, Y. Wu, K. Nakashima, S. Kajiyama and Y. Nakata, J. Geophys. Res. Solid Earth, 2017, 122, 7511–7524 CrossRef CAS.
- B. Loret, Fluid Injection in Deformable Geological Formations, Springer, 2019, pp. 305–367 Search PubMed.
- B. N. Madhusudhan, C. R. I. Clayton and J. A. Priest, J. Geophys. Res. Solid Earth, 2019, 124, 65–75 CrossRef CAS.
- X. H. Zhang, X. B. Lu, Y. H. Shi and Z. Xia, Mar. Pet. Geol., 2015, 67, 72–80 CrossRef CAS.
- J.-H. Choi, S. Dai, J.-S. Lin and Y. Seol, J. Geophys. Res. Solid Earth, 2018, 123, 3347–3357 CrossRef CAS.
- X. H. Zhang, D. S. Luo, X. B. Lu, L. L. Liu and C. L. Liu, Acta Mech. Sin., 2018, 34, 266–274 CrossRef CAS.
- K. Miyazaki, N. Tenma, K. Aoki and T. Yamaguchi, Energies, 2012, 5 Search PubMed.
- M. Hyodo, Y. Li, J. Yoneda, Y. Nakata, N. Yoshimoto and A. Nishimura, Mar. Pet. Geol., 2014, 51, 52–62 CrossRef CAS.
- C. Yan, Y. Cheng, M. Li, Z. Han, H. Zhang, Q. Li, F. Teng and J. Ding, Int. J. Hydrogen Energy, 2017, 42, 19810–19818 CrossRef CAS.
- M. Hyodo, J. Yoneda, N. Yoshimoto and Y. Nakata, Soils Found., 2013, 53, 299–314 CrossRef.
- Y. Song, Y. Zhu, W. Liu, J. Zhao, Y. Li, Y. Chen, Z. Shen, Y. Lu and C. Ji, Mar. Pet. Geol., 2014, 51, 70–78 CrossRef CAS.
- S. Uchida, A. Klar and K. Yamamoto, Second EAGE Workshop on Geomechanics and Energy, 2015.
- M. Hyodo, Y. Nakata and N. Yoshimoto, Japanese Geotech. Soc. Spec. Publ., 2016, 2, 62–75 Search PubMed.
- H. Han, Y. Wang, X.-S. Li, J.-X. Yu, J.-C. Feng and Y. Zhang, Fuel, 2016, 182, 446–453 CrossRef CAS.
- Y. Song, Y. Zhu, W. Liu, Y. Li, Y. Lu and Z. Shen, J. Pet. Sci. Eng., 2016, 147, 77–86 CrossRef CAS.
- H. Iwai, Y. Konishi, K. Saimyou, S. Kimoto and F. Oka, Soils Found., 2018, 58, 1113–1132 CrossRef.
- M. Zhou, K. Soga, K. Yamamoto and H. Huang, Geomech. Energy Environ., 2018, 100111 Search PubMed.
- B. Song, Y. Cheng, C. Yan, Z. Han and Q. Li, 53rd US Rock Mechanics/Geomechanics Symposium, American Rock Mechanics Association, 2019.
- Y. Song, T. Luo, B. N. Madhusudhan, X. Sun, Y. Liu, X. Kong and Y. Li, J. Nat. Gas Sci. Eng., 2019, 72, 103031 CrossRef CAS.
- R. Wang, W. Liu, Y. Li, F. Yu, X. Nie and Y. Song, Proc. Int. Conf. Offshore Mech. Arct. Eng., 2012, 703–708, DOI:10.1115/OMAE2012-84080.
- K. Miyazaki, N. Tenma and T. Yamaguchi, Energies, 2017, 10 Search PubMed.
- Y. Li, P. Wu, X. Sun, W. Liu, Y. Song and J. Zhao, Energies, 2019, 12 CrossRef.
- Y. Li, W. Liu, Y. Zhu, Y. Chen, Y. Song and Q. Li, Appl. Energy, 2016, 162, 1627–1632 CrossRef CAS.
- E. M. Chuvilin, B. A. Bukhanov, S. I. Grebenkin, V. V. Doroshin and A. V. Iospa, Cold Reg. Sci. Technol., 2018, 153, 101–105 CrossRef.
- J. Yang, A. Hassanpouryouzband, B. Tohidi, E. Chuvilin, B. Bukhanov, V. Istomin and A. Cheremisin, J. Geophys. Res. Solid Earth, 2019, 124, 2551–2563 CrossRef CAS.
- S. Uchida, K. Soga and K. Yamamoto, J. Geophys. Res. Solid Earth, 2012, 117, B03209, DOI:10.1029/2011JB008661.
- M. Jiang, F. Zhu, F. Liu and S. Utili, Int. J. Numer. Anal. Methods Geomech., 2014, 38, 1823–1854 CrossRef CAS.
- S. Uchida, X.-G. Xie and Y. F. Leung, J. Geophys. Res. Solid Earth, 2016, 121, 5580–5595 CrossRef CAS.
- X. Gai and M. Sánchez, Gas, 2017, 1, 2 Search PubMed.
- X. Zhang, J. Lin, X. Lu, L. Liu, C. Liu, M. Li and Y. Su, Int. J. Numer. Anal. Methods Geomech., 2018, 42, 931–942 CrossRef CAS.
- K. Li, R. Liu, L. Kong and X. Zhao, Geotech. Geol. Eng., 2019, 37, 2893–2902 CrossRef.
- J. Liu and M. Jiang, Environ. Geotech., 2019, 1–12 Search PubMed.
- S. Gupta, R. Helmig and B. Wohlmuth, Comput. Geosci., 2015, 19, 1063–1088 CrossRef.
- X. Chen, X. Zhang, X. Lu, W. Wei and Y. Shi, Acta Mech. Sin., 2016, 32, 905–914 CrossRef CAS.
- Q. Li, Y. Cheng, H. Zhang, C. Yan and Y. Liu, J. Ocean Univ. China, 2018, 17, 35–45 CrossRef.
- G. Jin, H. Lei, T. Xu, X. Xin, Y. Yuan, Y. Xia and J. Juo, Mar. Pet. Geol., 2018, 92, 424–436 CrossRef CAS.
- M. Sánchez, C. Santamarina, M. Teymouri and X. Gai, J. Geophys. Res. Solid Earth, 2018, 123, 10326–10348 CrossRef.
- Y. Sohn, J. Y. Lee, K.-I. Song and T.-H. Kwon, Offshore Technol. Conf., 2019, 17 Search PubMed.
- X. Sun, L. Wang, H. Luo, Y. Song and Y. Li, J. Pet. Sci. Eng., 2019, 177, 971–982 CrossRef CAS.
- J.-W. Jung, J. C. Santamarina and K. Soga, J. Geophys. Res. Solid Earth, 2012, 117, B04202, DOI:10.1029/2011JB009040.
- M. Jiang, F. Zhu and S. Utili, Comput. Geotech., 2015, 69, 551–563 CrossRef.
- Z. Shen and M. Jiang, Comput. Geotech., 2016, 75, 225–243 CrossRef.
- M. Xu, E. Song, H. Jiang and J. Hong, Granul. Matter, 2016, 18, 79 CrossRef.
- J. Mingjing, H. Jie, W. Jianfeng, C. Bruno and Z. Fangyuan, Int. J. Geomech., 2016, 16, 4015087 CrossRef.
- M. Jiang, J. He, J. Wang, Y. Zhou and F. Zhu, C. R. Mec., 2017, 345, 868–889 CrossRef.
- M. Jiang, D. Peng and J. Y. Ooi, Eng. Geol., 2017, 223, 92–109 CrossRef.
- M. Jiang, Z. Shen and D. Wu, Landslides, 2018, 15, 2227–2241 CrossRef.
- M. Jiang, Z. Shen, W. Zhou, M. Arroyo and W. Zhang, Granul. Matter, 2018, 20, 63 CrossRef.
- M. Jiang, J. Liu, C. Y. Kwok and Z. Shen, C. R. Mec., 2018, 346, 815–832 CrossRef.
- M. Jiang, J. Liu and Z. Shen, Eng. Geol., 2019, 261, 105280 CrossRef.
- Y. Jiang and B. Gong, Mar. Georesources Geotechnol., 2019, 1–15 Search PubMed.
- E. Cohen and A. Klar, Granul. Matter, 2019, 21, 36 CrossRef.
- E. Cohen, A. Klar and K. Yamamoto, Energies, 2019, 12, 2131 CrossRef CAS.
- A. Sinquin, T. Palermo and Y. Peysson, Oil Gas Sci. Technol., 2004, 59, 41–57 CrossRef.
- A. Fidel-Dufour, F. Gruy and J.-M. Herri, Chem. Eng. Sci., 2006, 61, 505–515 CrossRef CAS.
- A. K. Sum, C. A. Koh and E. D. Sloan, Ind. Eng. Chem. Res., 2009, 48, 7457–7465 CrossRef CAS.
- A. Ahuja, Hydrate Forming Emulsion: Rheology and Morphology Analysis for Flow Assurance, The City College of New York, 2015 Search PubMed.
- E. D. Sloan, Natural gas hydrates in flow assurance, Gulf Professional Publishing, 2010 Search PubMed.
- J. G. Delgado-Linares, A. A. A. Majid, E. D. Sloan, C. A. Koh and A. K. Sum, Energy Fuels, 2013, 27, 4564–4573 CrossRef CAS.
- K.-L. Yan, K. Guo, C.-Y. Sun, S.-S. Niu, B. Liu, D.-J. Shen, J. Chen, R.-Q. Zhong, G.-J. Chen and Q.-P. Li, Fluid Phase Equilib., 2014, 377, 9–15 CrossRef CAS.
- M. Darbouret, M. Cournil and J.-M. Herri, Int. J. Refrig., 2005, 28, 663–671 CrossRef CAS.
- A. Delahaye, L. Fournaison, S. Marinhas and M. C. Martínez, Chem. Eng. Sci., 2008, 63, 3551–3559 CrossRef CAS.
- Y. Salehy, P. Clain, A. Boufares, V. Osswald, A. Delahaye and L. Fournaison, Energetika, 2017, 63(3) DOI:10.6001/energetika.v63i3.3561.
- J. Oignet, A. Delahaye, J.-P. Torré, C. Dicharry, H. M. Hoang, P. Clain, V. Osswald, Z. Youssef and L. Fournaison, Chem. Eng. Sci., 2017, 158, 294–303 CrossRef CAS.
- N. Liu, J. Zhou, M. Gao and P. Cheng, Int. Commun. Heat Mass Transfer, 2019, 102, 34–41 CrossRef CAS.
- S. Jerbi, A. Delahaye, L. Fournaison and P. Haberschill, Int. J. Refrig., 2010, 33, 1625–1631 CrossRef CAS.
- Z. W. Ma, P. Zhang, R. Z. Wang, S. Furui and G. N. Xi, Int. J. Heat Mass Transfer, 2010, 53, 3745–3757 CrossRef CAS.
- A. Delahaye, L. Fournaison, S. Jerbi and N. Mayoufi, Ind. Eng. Chem. Res., 2011, 50, 8344–8353 CrossRef CAS.
- P. Clain, A. Delahaye, L. Fournaison, N. Mayoufi, D. Dalmazzone and W. Fürst, Chem. Eng. J., 2012, 193–194, 112–122 CrossRef CAS.
- B.-Z. Peng, J. Chen, C.-Y. Sun, A. Dandekar, S.-H. Guo, B. Liu, L. Mu, L.-Y. Yang, W.-Z. Li and G.-J. Chen, Chem. Eng. Sci., 2012, 84, 333–344 CrossRef CAS.
- X. Lv, B. Shi, Y. Wang and J. Gong, Energy Fuels, 2013, 27, 7294–7302 CrossRef CAS.
- S. Jerbi, A. Delahaye, J. Oignet, L. Fournaison and P. Haberschill, Int. J. Refrig., 2013, 36, 1294–1301 CrossRef CAS.
- K.-L. Yan, C.-Y. Sun, J. Chen, L.-T. Chen, D.-J. Shen, B. Liu, M.-L. Jia, M. Niu, Y.-N. Lv, N. Li, Z.-Y. Song, S.-S. Niu and G.-J. Chen, Chem. Eng. Sci., 2014, 106, 99–108 CrossRef CAS.
- L. Ding, B. Shi, X. Lv, Y. Liu, H. Wu, W. Wang and J. Gong, Chem. Eng. Sci., 2016, 146, 199–206 CrossRef CAS.
- Z. Rehman, K. Seong, S. Lee and M. H. Song, Chem. Eng. Commun., 2018, 205, 822–832 CrossRef CAS.
- B.-H. Shi, L. Ding, W.-Q. Li, X.-F. Lv, Y. Liu, S.-F. Song, C.-Y. Ruan, H.-H. Wu, W. Wang and J. Gong, Asia-Pac. J. Chem. Eng., 2018, 13, e2193 CrossRef.
- L. Ding, B. Shi, Y. Liu, S. Song, W. Wang, H. Wu and J. Gong, Fuel, 2019, 239, 126–137 CrossRef CAS.
- X. Lv, J. Zuo, Y. Liu, S.-D. Zhou, D. Lu, B. Shi and H. Zhao, RSC Adv., 2019, 9, 32873–32888 RSC.
- W. Fu, Z. Wang, J. Zhang, Y. Cao and B. Sun, J. Pet. Sci. Eng., 2020, 184, 106504 CrossRef CAS.
- J. Peixinho, P. U. Karanjkar, J. W. Lee and J. F. Morris, Langmuir, 2010, 26, 11699–11704 CrossRef CAS.
- E. B. Webb, P. J. Rensing, C. A. Koh, E. Dendy Sloan, A. K. Sum and M. W. Liberatore, Rev. Sci. Instrum., 2012, 83, 15106 CrossRef PubMed.
- E. B. Webb, P. J. Rensing, C. A. Koh, E. D. Sloan, A. K. Sum and M. W. Liberatore, Energy Fuels, 2012, 26, 3504–3509 CrossRef CAS.
- E. B. Webb, C. A. Koh and M. W. Liberatore, Langmuir, 2013, 29, 10997–11004 CrossRef CAS.
- G. Zylyftari, J. W. Lee and J. F. Morris, Chem. Eng. Sci., 2013, 95, 148–160 CrossRef CAS.
- E. B. Webb, C. A. Koh and M. W. Liberatore, Ind. Eng. Chem. Res., 2014, 53, 6998–7007 CrossRef CAS.
- A. Ahuja, G. Zylyftari and J. F. Morris, J. Chem. Eng. Data, 2015, 60, 362–368 CrossRef CAS.
- A. Memon and H.-J. Ng, J. Chem. Eng. Data, 2015, 60, 293–298 CrossRef CAS.
- A. Ahuja, G. Zylyftari and J. F. Morris, J. Nonnewton. Fluid Mech., 2015, 220, 116–125 CrossRef CAS.
- A. K. Yegya Raman, S. Koteeswaran, D. Venkataramani, P. Clark, S. Bhagwat and C. P. Aichele, Fuel, 2016, 179, 141–149 CrossRef CAS.
- P. U. Karanjkar, A. Ahuja, G. Zylyftari, J. W. Lee and J. F. Morris, Rheol. Acta, 2016, 55, 235–243 CrossRef CAS.
- A. A. A. Majid, D. T. Wu and C. A. Koh, Langmuir, 2017, 33, 11436–11445 CrossRef CAS PubMed.
- B.-H. Shi, S. Chai, L. Ding, Y.-C. Chen, Y. Liu, S.-F. Song, H.-Y. Yao, H.-H. Wu, W. Wang and J. Gong, AIChE J., 2018, 64, 3502–3518 CrossRef CAS.
- G. A. B. Sandoval, E. J. Soares, R. L. Thompson, R. do Nascimento Siqueira, R. M. de Andrade, F. Campos and A. Teixeira, Energy Fuels, 2018, 32, 2733–2741 CrossRef CAS.
- A. Ahuja, A. Iqbal, M. Iqbal, J. W. Lee and J. F. Morris, Energy Fuels, 2018, 32, 5877–5884 CrossRef CAS.
- Y. Qin, P. F. Pickering, M. L. Johns, E. F. May and Z. M. Aman, Energy Fuels, 2020 DOI:10.1021/acs.energyfuels.9b00395.
- B.-H. Shi, S. Chai, L.-Y. Wang, X. Lv, H.-S. Liu, H.-H. Wu, W. Wang, D. Yu and J. Gong, Fuel, 2016, 185, 323–338 CrossRef CAS.
- Y. Qin, Z. M. Aman, P. F. Pickering, M. L. Johns and E. F. May, J. Nonnewton. Fluid Mech., 2017, 248, 40–49 CrossRef CAS.
- Y. Hoon Sohn, J. Kim, K. Shin, D. Chang, Y. Seo, Z. M. Aman and E. F. May, Chem. Eng. Sci., 2015, 126, 711–718 CrossRef.
- A. A. A. Majid, D. T. Wu and C. A. Koh, Engineering, 2018, 4, 321–329 CrossRef CAS.
- R. Camargo and T. Palermo, Proceedings of the 4th International Conference on Gas Hydrates, 2002, vol. 1, pp. 880–885.
- H. Moradpour, A. Chapoy and B. Tohidi, Fuel, 2011, 90, 3343–3351 CrossRef CAS.
- Z. M. Aman and C. A. Koh, Chem. Soc. Rev., 2016, 45, 1678–1690 RSC.
- R. H. Fowler, Proc. R. Soc. London, Ser. A, 1937, 159, 229–246 CAS.
- S. R. H. Fowler and E. A. Guggenheim, Statistical Thermodynamics. A Version of Statistical Mechanics [by RH Fowler] for Students of Physics and Chemistry, Cambridge, 1939 Search PubMed.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Press, 2015 Search PubMed.
- E. B. Nauman, Chemical reactor design, optimization, and scaleup, John Wiley & Sons, 2008 Search PubMed.
- M. R. Walsh, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Science, 2009, 326, 1095–1098 CrossRef CAS PubMed.
- E. F. May and R. Wu, Chem. Eng. Sci., 2014, 107, 1–12 CrossRef CAS.
- R. Wu, K. A. Kozielski, P. G. Hartley, E. F. May, J. Boxall and N. Maeda, AIChE J., 2013, 59, 2640–2646 CrossRef CAS.
- B. C. Barnes, G. T. Beckham, D. T. Wu and A. K. Sum, J. Chem. Phys., 2014, 140, 164506 CrossRef PubMed.
- B. C. Barnes, B. C. Knott, G. T. Beckham, D. T. Wu and A. K. Sum, J. Phys. Chem. B, 2014, 118, 13236–13243 CrossRef CAS PubMed.
- K. Kinnari, J. Hundseid, X. Li and K. M. Askvik, J. Chem. Eng. Data, 2015, 60, 437–446 CrossRef CAS.
- M. Kharrat and D. Dalmazzone, J. Chem. Thermodyn., 2003, 35, 1489–1505 CrossRef CAS.
- L.-K. Wang, G.-J. Chen, G.-H. Han, X.-Q. Guo and T.-M. Guo, Fluid Phase Equilib., 2003, 207, 143–154 CrossRef CAS.
- D. Liang, K. Guo, R. Wang and S. Fan, Fluid Phase Equilib., 2001, 187, 61–70 CrossRef.
- C. J. Taylor, K. T. Miller, C. A. Koh and E. D. Sloan Jr, Chem. Eng. Sci., 2007, 62, 6524–6533 CrossRef CAS.
- S. Li, C. Sun, B. Liu, X. Feng, F. Li, L. Chen and G. Chen, AIChE J., 2013, 59, 2145–2154 CrossRef CAS.
- J. Chen, C. Sun, B. Liu, B. Peng, X. Wang, G. Chen, J. Y. Zuo and H. Ng, AIChE J., 2012, 58, 2216–2225 CrossRef CAS.
- I. E. Dzyaloshinskii, E. M. Lifshitz and L. P. Pitaevskii, Adv. Phys., 1961, 10, 165–209 CrossRef.
- M. Bienfait, Surf. Sci., 1992, 272, 1–9 CrossRef CAS.
- Z. M. Aman, E. P. Brown, E. D. Sloan, A. K. Sum and C. A. Koh, Phys. Chem. Chem. Phys., 2011, 13, 19796–19806 RSC.
- D. Nenow and A. Trayanov, J. Cryst. Growth, 1986, 79, 801–805 CrossRef.
- H.-J. Butt and M. Kappl, Surface and interfacial forces, Wiley Online Library, 2010 Search PubMed.
- J. S. Zhang, S. Lee and J. W. Lee, Ind. Eng. Chem. Res., 2007, 46, 6353–6359 CrossRef CAS.
- J. Yoslim, P. Linga and P. Englezos, J. Cryst. Growth, 2010, 313, 68–80 CrossRef CAS.
- T. Yagasaki, M. Matsumoto, Y. Andoh, S. Okazaki and H. Tanaka, J. Phys. Chem. B, 2014, 118, 1900–1906 CrossRef CAS PubMed.
- P. M. Naullage, Y. Qiu and V. Molinero, J. Phys. Chem. Lett., 2018, 9, 1712–1720 CrossRef CAS PubMed.
- J. S. Zhang, C. Lo, P. Somasundaran, S. Lu, A. Couzis and J. W. Lee, J. Phys. Chem. C, 2008, 112, 12381–12385 CrossRef CAS.
- J. Zhang and J. W. Lee, Energy Fuels, 2009, 23, 3045–3047 CrossRef CAS.
- P. U. Karanjkar, J. W. Lee and J. F. Morris, Cryst. Growth Des., 2012, 12, 3817–3824 CrossRef CAS.
- O. Salako, C. Lo, A. Couzis, P. Somasundaran and J. W. Lee, J. Colloid Interface Sci., 2013, 412, 1–6 CrossRef CAS PubMed.
- C. Lo, J. S. Zhang, A. Couzis, P. Somasundaran and J. W. Lee, J. Phys. Chem. C, 2010, 114, 13385–13389 CrossRef CAS.
- C. Lo, J. Zhang, P. Somasundaran and J. W. Lee, J. Colloid Interface Sci., 2012, 376, 173–176 CrossRef CAS PubMed.
- Y. Zhong and R. E. Rogers, Chem. Eng. Sci., 2000, 55, 4175–4187 CrossRef CAS.
- K. Dann and L. Rosenfeld, Langmuir, 2018, 34, 6085–6094 CrossRef CAS PubMed.
- F. Sicard, T. Bui, D. Monteiro, Q. Lan, M. Ceglio, C. Burress and A. Striolo, Langmuir, 2018, 34, 9701–9710 CrossRef CAS PubMed.
- M. Khurana, Z. Yin and P. Linga, ACS Sustainable Chem. Eng., 2017, 5, 11176–11203 CrossRef CAS.
- V. Natarajan, P. R. Bishnoi and N. Kalogerakis, Chem. Eng. Sci., 1994, 49, 2075–2087 CrossRef CAS.
- S. Takeya, A. Hori, T. Hondoh and T. Uchida, J. Phys. Chem. B, 2000, 104, 4164–4168 CrossRef CAS.
- I. L. Moudrakovski, G. E. McLaurin, C. I. Ratcliffe and J. A. Ripmeester, J. Phys. Chem. B, 2004, 108, 17591–17595 CrossRef CAS.
- N. Maeda, Rev. Sci. Instrum., 2014, 85, 65115 CrossRef PubMed.
- T. M. Svartaas, W. Ke, S. Tantciura and A. U. Bratland, Energy Fuels, 2015, 29, 8195–8207 CrossRef CAS.
- S. Sun, X. Peng, Y. Zhang, J. Zhao and Y. Kong, J. Chem. Thermodyn., 2017, 107, 141–152 CrossRef CAS.
- P. J. Metaxas, V. W. S. Lim, C. Booth, J. Zhen, P. L. Stanwix, M. L. Johns, Z. M. Aman, G. Haandrikman, D. Crosby and E. F. May, Fuel, 2019, 252, 448–457 CrossRef CAS.
- E. D. Sloan and F. Fleyfel, AIChE J., 1991, 37, 1281–1292 CrossRef CAS.
- G.-J. Guo, Y.-G. Zhang, M. Li and C.-H. Wu, J. Chem. Phys., 2008, 128, 194504 CrossRef PubMed.
- N. J. English, M. Lauricella and S. Meloni, J. Chem. Phys., 2014, 140, 204714 CrossRef PubMed.
- G.-J. Guo and P. M. Rodger, J. Phys. Chem. B, 2013, 117, 6498–6504 CrossRef CAS PubMed.
- S. Subramanian and E. D. Sloan Jr, Fluid Phase Equilib., 1999, 158, 813–820 CrossRef.
- P. Buchanan, A. K. Soper, H. Thompson, R. E. Westacott, J. L. Creek, G. Hobson and C. A. Koh, J. Chem. Phys., 2005, 123, 164507 CrossRef PubMed.
- H. Conrad, F. Lehmkühler, C. Sternemann, A. Sakko, D. Paschek, L. Simonelli, S. Huotari, O. Feroughi, M. Tolan and K. Hämäläinen, Phys. Rev. Lett., 2009, 103, 218301 CrossRef PubMed.
- A. Vysniauskas and P. R. Bishnoi, Chem. Eng. Sci., 1983, 38, 1061–1072 CrossRef CAS.
- E. D. Sloan, S. Subramanian, P. N. Matthews, J. P. Lederhos and A. A. Khokhar, Ind. Eng. Chem. Res., 1998, 37, 3124–3132 CrossRef CAS.
- R. Ohmura, M. Ogawa, K. Yasuoka and Y. H. Mori, J. Phys. Chem. B, 2003, 107, 5289–5293 CrossRef CAS.
- B. Sowa and N. Maeda, J. Phys. Chem. A, 2015, 119, 10784–10790 CrossRef CAS PubMed.
- C. J. Benmore and A. K. Soper, J. Chem. Phys., 1998, 108, 6558–6560 CrossRef CAS.
- M. Chaouachi, A. Falenty, K. Sell, F. Enzmann, M. Kersten, D. Haberthür and W. F. Kuhs, Geochem., Geophys., Geosyst., 2015, 16, 1711–1722 CrossRef CAS.
- T. Uchida, K. Yamazaki and K. Gohara, J. Phys. Chem. C, 2016, 120, 26620–26629 CrossRef CAS.
- S. A. Bagherzadeh, S. Alavi, J. Ripmeester and P. Englezos, J. Chem. Phys., 2015, 142, 214701 CrossRef PubMed.
- M. Sugaya and Y. H. Mori, Chem. Eng. Sci., 1996, 51, 3505–3517 CrossRef CAS.
- T. Uchida, T. Ebinuma, J. Kawabata and H. Narita, J. Cryst. Growth, 1999, 204, 348–356 CrossRef CAS.
- P. Servio and P. Englezos, AIChE J., 2003, 49, 269–276 CrossRef CAS.
- E. M. Freer, M. S. Selim and E. D. Sloan Jr, Fluid Phase Equilib., 2001, 185, 65–75 CrossRef CAS.
- T. Mochizuki and Y. H. Mori, Chem. Eng. Sci., 2017, 171, 61–75 CrossRef CAS.
- R. Ohmura, S. Kashiwazaki and Y. H. Mori, J. Cryst. Growth, 2000, 218, 372–380 CrossRef CAS.
- R. Tanaka, R. Sakemoto and R. Ohmura, Cryst. Growth Des., 2009, 9, 2529–2536 CrossRef CAS.
- K. Saito, M. Kishimoto, R. Tanaka and R. Ohmura, Cryst. Growth Des., 2010, 11, 295–301 CrossRef.
- S.-L. Li, C.-Y. Sun, B. Liu, Z.-Y. Li, G.-J. Chen and A. K. Sum, Sci. Rep., 2014, 4, 4129 CrossRef PubMed.
- X. Y. Zeng, G. Wu, J. R. Zhong, D. Y. Chen, C. Y. Sun and G. J. Chen, Cryst. Growth Des., 2019, 19, 3158–3165 CrossRef CAS.
- S.-L. Li, Y.-F. Wang, C.-Y. Sun, G.-J. Chen, B. Liu, Z.-Y. Li and Q.-L. Ma, Chem. Eng. Sci., 2015, 135, 412–420 CrossRef CAS.
- I. L. Moudrakovski, A. A. Sanchez, C. I. Ratcliffe and J. A. Ripmeester, J. Phys. Chem. B, 2001, 105, 12338–12347 CrossRef CAS.
- J. M. Schicks and J. A. Ripmeester, Angew. Chem., Int. Ed., 2004, 43, 3310–3313 CrossRef CAS PubMed.
- J. Zhu, S. Du, X. Yu, J. Zhang, H. Xu, S. C. Vogel, T. C. Germann, J. S. Francisco, F. Izumi and K. Momma, Nat. Commun., 2014, 5, 4128 CrossRef CAS PubMed.
- L. C. Jacobson, M. Matsumoto and V. Molinero, J. Chem. Phys., 2011, 135, 74501 CrossRef PubMed.
- S. Liang and P. G. Kusalik, J. Chem. Phys., 2015, 143 DOI:10.1063/1.4923465.
- S. Takeya, T. Hondoh and T. Uchida, Ann. N. Y. Acad. Sci., 2000, 912, 973–982 CrossRef CAS.
- R. W. Henning, A. J. Schultz, V. Thieu and Y. Halpern, J. Phys. Chem. A, 2000, 104, 5066–5071 CrossRef CAS.
- X. Wang, A. J. Schultz and Y. Halpern, J. Phys. Chem. A, 2002, 106, 7304–7309 CrossRef CAS.
- W. F. Kuhs, D. K. Staykova and A. N. Salamatin, J. Phys. Chem. B, 2006, 110, 13283–13295 CrossRef CAS PubMed.
- A. Falenty, A. N. Salamatin and W. F. Kuhs, J. Phys. Chem. C, 2013, 117, 8443–8457 CrossRef CAS.
- A. N. Salamatin, A. Falenty, T. C. Hansen and W. F. Kuhs, Energy Fuels, 2015, 29, 5681–5691 CrossRef CAS.
- V. A. Vlasov, Theor. Found. Chem. Eng., 2012, 46, 576–582 CrossRef CAS.
- V. S. Shagapov, A. S. Chiglintseva and G. R. Rafikova, Theor. Found. Chem. Eng., 2018, 52, 560–567 CrossRef CAS.
- W. Liu, Y. Li, L. Zhang, S. Shen, M. Yang, J. Zhao and Y. Song, Theor. Found. Chem. Eng., 2019, 53, 305–317 CrossRef CAS.
- M. T. Nguyen, J. Amtawong, K. Smoll, A. Chanez, M. Yamano, G.-B. H. Dinh, S. Sengupta, R. W. Martin and K. C. Janda, J. Phys. Chem. C, 2016, 120, 8482–8489 CrossRef CAS.
- G.-J. Guo, Y.-G. Zhang, Y.-J. Zhao, K. Refson and G.-H. Shan, J. Chem. Phys., 2004, 121, 1542–1547 CrossRef CAS PubMed.
- R. Radhakrishnan and B. L. Trout, J. Chem. Phys., 2002, 117, 1786–1796 CrossRef CAS.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Am. Chem. Soc., 2010, 132, 11806–11811 CrossRef CAS PubMed.
- G.-J. Guo, M. Li, Y.-G. Zhang and C.-H. Wu, Phys. Chem. Chem. Phys., 2009, 11, 10427–10437 RSC.
- G.-J. Guo, Y.-G. Zhang and H. Liu, J. Phys. Chem. C, 2007, 111, 2595–2606 CrossRef CAS.
- G.-J. Guo, Y.-G. Zhang, C.-J. Liu and K.-H. Li, Phys. Chem. Chem. Phys., 2011, 13, 12048–12057 RSC.
- S. Sarupria and P. G. Debenedetti, J. Phys. Chem. Lett., 2012, 3, 2942–2947 CrossRef CAS PubMed.
- M. Lauricella, S. Meloni, N. J. English, B. Peters and G. Ciccotti, J. Phys. Chem. C, 2014, 118, 22847–22857 CrossRef CAS.
- M. Lauricella, G. Ciccotti, N. J. English, B. Peters and S. Meloni, J. Phys. Chem. C, 2017, 121, 24223–24234 CrossRef CAS.
- R. W. Hawtin, D. Quigley and P. M. Rodger, Phys. Chem. Chem. Phys., 2008, 10, 4853–4864 RSC.
- L. C. Jacobson and V. Molinero, J. Am. Chem. Soc., 2011, 133, 6458–6463 CrossRef CAS PubMed.
- M. R. Walsh, G. T. Beckham, C. A. Koh, E. D. Sloan, D. T. Wu and A. K. Sum, J. Phys. Chem. C, 2011, 115, 21241–21248 CrossRef CAS.
- J. A. Ripmeester and S. Alavi, ChemPhysChem, 2010, 11, 978–980 CrossRef CAS PubMed.
- K. W. Hall, S. Carpendale and P. G. Kusalik, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12041–12046 CrossRef CAS PubMed.
- K. W. Hall, Z. Zhang, C. J. Burnham, G.-J. Guo, S. Carpendale, N. J. English and P. G. Kusalik, J. Phys. Chem. Lett., 2018, 9, 6991–6998 CrossRef CAS PubMed.
- M. Lauricella, S. Meloni, S. Liang, N. J. English, P. G. Kusalik and G. Ciccotti, J. Chem. Phys., 2015, 142, 244503 CrossRef PubMed.
- E. Małolepsza and T. Keyes, J. Phys. Chem. B, 2015, 119, 15857–15865 CrossRef PubMed.
- Y. Bi and T. Li, J. Phys. Chem. B, 2014, 118, 13324–13332 CrossRef CAS PubMed.
- Y. Bi, A. Porras and T. Li, J. Chem. Phys., 2016, 145, 211909 CrossRef PubMed.
- N. J. English and S. T. John, Comput. Mater. Sci., 2017, 126, 1–6 CrossRef CAS.
- Y. Bi, E. Xu, T. A. Strobel and T. Li, Commun. Chem., 2018, 1, 15 CrossRef.
- Z. Futera, M. Celli, L. del Rosso, C. J. Burnham, L. Ulivi and N. J. English, J. Phys. Chem. C, 2017, 121, 3690–3696 CrossRef CAS.
- R. Ohmura, W. Shimada, T. Uchida, Y. H. Mori, S. Takeya, J. Nagao, H. Minagawa, T. Ebinuma and H. Narita, Philos. Mag., 2004, 84, 1–16 CrossRef CAS.
- R. Ohmura, S. Matsuda, T. Uchida, T. Ebinuma and H. Narita, Cryst. Growth Des., 2005, 5, 953–957 CrossRef CAS.
- J. D. Lee, M. Song, R. Susilo and P. Englezos, Cryst. Growth Des., 2006, 6, 1428–1439 CrossRef CAS.
- R. Larsen, C. A. Knight and E. D. Sloan Jr, Fluid Phase Equilib., 1998, 150, 353–360 CrossRef.
- P. Gayet, C. Dicharry, G. Marion, A. Graciaa, J. Lachaise and A. Nesterov, Chem. Eng. Sci., 2005, 60, 5751–5758 CrossRef CAS.
- M. T. Storr, P. C. Taylor, J.-P. Monfort and P. M. Rodger, J. Am. Chem. Soc., 2004, 126, 1569–1576 CrossRef CAS PubMed.
- B. J. Anderson, J. W. Tester, G. P. Borghi and B. L. Trout, J. Am. Chem. Soc., 2005, 127, 17852–17862 CrossRef CAS PubMed.
- B. Kvamme, T. Kuznetsova and K. Aasoldsen, J. Mol. Graphics Modell., 2005, 23, 524–536 CrossRef CAS PubMed.
- S. Y. Lee, H. C. Kim and J. D. Lee, J. Cryst. Growth, 2014, 402, 249–259 CrossRef CAS.
- H. Hayama, M. Mitarai, H. Mori, J. Verrett, P. Servio and R. Ohmura, Cryst. Growth Des., 2016, 16, 6084–6088 CrossRef CAS.
- A. Kumar, G. Bhattacharjee, B. D. Kulkarni and R. Kumar, Ind. Eng. Chem. Res., 2015, 54, 12217–12232 CrossRef CAS.
- Y. He, M.-T. Sun, C. Chen, G.-D. Zhang, K. Chao, Y. Lin and F. Wang, J. Mater. Chem. A, 2019, 7, 21634–21661 RSC.
- A. Gupta, S. F. Dec, C. A. Koh and E. D. Sloan, J. Phys. Chem. C, 2007, 111, 2341–2346 CrossRef CAS.
- S. F. Dec, K. E. Bowler, L. L. Stadterman, C. A. Koh and E. D. Sloan, J. Phys. Chem. A, 2007, 111, 4297–4303 CrossRef CAS PubMed.
- M. Kida, Y. Jin, N. Takahashi, J. Nagao and H. Narita, J. Phys. Chem. A, 2010, 114, 9456–9461 CrossRef CAS PubMed.
- S. F. Dec, J. Phys. Chem. C, 2012, 116, 9660–9665 CrossRef CAS.
- J.-R. Zhong, Y.-F. Sun, W.-Z. Li, Y. Xie, G.-J. Chen, C.-Y. Sun, L.-Y. Yang, H.-B. Qin, W.-X. Pang and Q.-P. Li, Appl. Energy, 2019, 250, 873–881 CrossRef CAS.
- N. J. English, J. K. Johnson and C. E. Taylor, J. Chem. Phys., 2005, 123, 244503 CrossRef PubMed.
- N. J. English and E. T. Clarke, J. Chem. Phys., 2013, 139, 94701 CrossRef PubMed.
- Z. Sun, H. Wang, J. Yao, Z. Sun, K. Bongole, X. Zhu, L. Liu and J. Wang, J. Nat. Gas Sci. Eng., 2018, 55, 235–242 CrossRef CAS.
- J. Kondori, S. Zendehboudi and L. James, Fuel, 2019, 249, 264–276 CrossRef CAS.
- H. C. Kim, P. R. Bishnoi, R. A. Heidemann and S. S. H. Rizvi, Chem. Eng. Sci., 1987, 42, 1645–1653 CrossRef CAS.
- M. A. Clarke and P. R. Bishnoi, Chem. Eng. Sci., 2001, 56, 4715–4724 CrossRef CAS.
- T. Kawamura, K. Ohga, K. Higuchi, J. H. Yoon, Y. Yamamoto, T. Komai and H. Haneda, Energy Fuels, 2003, 17, 614–618 CrossRef CAS.
- T. Kawamura, Y. Sakamoto, M. Ohtake, Y. Yamamoto, T. Komai, H. Haneda and J.-H. Yoon, Ind. Eng. Chem. Res., 2006, 45, 360–364 CrossRef CAS.
- S. Fan, Y. Zhang, G. Tian, D. Liang and D. Li, Energy Fuels, 2006, 20, 324–326 CrossRef CAS.
- S. Yao, Y. Li, W. Wang, G. Song, Z. Shi, X. Wang and S. Liu, Energy Fuels, 2019, 33(6), 5208–5215, DOI:10.1021/acs.energyfuels.9b01003.
- Z. Yin, Z. R. Chong, H. K. Tan and P. Linga, J. Nat. Gas Sci. Eng., 2016, 35, 1362–1387 CrossRef CAS.
- V. A. Istomin and V. S. Yakushev, Gas-Hydrates Self-Preservation Effect, in Physics and Chemistry of Ice, ed. N. Maeno and T. Hondoh, Hokkaido University Press, Sapporo, Japan, 1992, pp. 136–140 Search PubMed.
- S. Takeya, W. Shimada, Y. Kamata, T. Ebinuma, T. Uchida, J. Nagao and H. Narita, J. Phys. Chem. A, 2001, 105, 9756–9759 CrossRef CAS.
- S. Takeya, T. Ebinuma, T. Uchida, J. Nagao and H. Narita, J. Cryst. Growth, 2002, 237, 379–382 CrossRef.
- V. R. Belosludov, O. S. Subbotin, D. S. Krupskii, T. Ikeshoji, R. V. Belosludov, Y. Kawazoe and J. Kudoh, J. Phys.: Conf. Ser., 2006, 29, 198 CrossRef CAS.
- D. Bai, D. Zhang, X. Zhang and G. Chen, Sci. Rep., 2015, 5, 14599 CrossRef CAS PubMed.
- V. A. Vlasov, Int. J. Heat Mass Transfer, 2016, 102, 631–636 CrossRef CAS.
- L. A. Stern, S. Circone, S. H. Kirby and W. B. Durham, J. Phys. Chem. B, 2001, 105, 1756–1762 CrossRef CAS.
- W. F. Kuhs, G. Genov, D. K. Staykova and T. Hansen, Phys. Chem. Chem. Phys., 2004, 6, 4917–4920 RSC.
- K. Takeya, K. Nango, T. Sugahara, K. Ohgaki and A. Tani, J. Phys. Chem. B, 2005, 109, 21086–21088 CrossRef CAS PubMed.
- W. Shimada, S. Takeya, Y. Kamata, T. Uchida, J. Nagao, T. Ebinuma and H. Narita, J. Phys. Chem. B, 2005, 109, 5802–5807 CrossRef CAS PubMed.
- S. Takeya and J. A. Ripmeester, ChemPhysChem, 2010, 11, 70–73 CrossRef CAS PubMed.
- V. P. Melnikov, A. N. Nesterov, A. M. Reshetnikov and V. A. Istomin, Chem. Eng. Sci., 2011, 66, 73–77 CrossRef CAS.
- M. Kida, M. Watanabe, Y. Jin and J. Nagao, Jpn. J. Appl. Phys., 2015, 54, 65502 CrossRef.
- S. Y. Misyura, Sci. Rep., 2016, 6, 30324 CrossRef CAS PubMed.
- S. Takeya, A. Yoneyama, K. Ueda, K. Hyodo, T. Takeda, H. Mimachi, M. Takahashi, T. Iwasaki, K. Sano and H. Yamawaki, J. Phys. Chem. C, 2011, 115, 16193–16199 CrossRef CAS.
- S. Takeya, A. Yoneyama, K. Ueda, H. Mimachi, M. Takahashi, K. Sano, K. Hyodo, T. Takeda and Y. Gotoh, J. Phys. Chem. C, 2012, 116(26), 13842–13848, DOI:10.1021/jp302269v.
- A. Falenty, W. F. Kuhs, M. Glockzin and G. Rehder, Energy Fuels, 2014, 28(10), 6275–6283, DOI:10.1021/ef501409g.
- T. Takaoki, 4th International Conference on Natural Gas Hydrate, Japan, 2002, 2002.
- G. Rehder, R. Eckl, M. Elfgen, A. Falenty, R. Hamann, N. Kähler, W. F. Kuhs, H. Osterkamp and C. Windmeier, Energies, 2012, 5, 2499–2523 CrossRef CAS.
- H. J. Kang, Y. Yang, M. S. Ki, M. S. Shin, J. Choi, J. H. Cha and D. Lee, Ocean Eng., 2016, 113, 162–173, DOI:10.1016/j.oceaneng.2015.12.052.
- H. Mimachi, M. Takahashi, S. Takeya, Y. Gotoh, A. Yoneyama, K. Hyodo, T. Takeda and T. Murayama, Energy Fuels, 2015, 29, 4827–4834 CrossRef CAS.
- G. Zhang and R. E. Rogers, Chem. Eng. Sci., 2008, 63, 2066–2074 CrossRef CAS.
- A. Hachikubo, S. Takeya, E. Chuvilin and V. Istomin, Phys. Chem. Chem. Phys., 2011, 13, 17449–17452, 10.1039/c1cp22353d.
- A. S. Stoporev, A. Y. Manakov, L. K. Altunina, A. V. Bogoslovsky, L. A. Strelets and E. Y. Aladko, Energy Fuels, 2014, 28(2), 794–802, DOI:10.1021/ef401779d.
- V. P. Mel’nikov, L. S. Podenko, A. N. Nesterov, A. O. Drachuk, N. S. Molokitina and A. M. Reshetnikov, Dokl. Chem., 2016, 466, 53–56, DOI:10.1134/S0012500816020038.
- H. Sato, H. Sakamoto, S. Ogino, H. Mimachi, T. Kinoshita, T. Iwasaki, K. Sano and K. Ohgaki, Chem. Eng. Sci., 2013, 91, 86–89, DOI:10.1016/j.ces.2013.01.014.
- S. Takeya, H. Mimachi and T. Murayama, Appl. Energy, 2018, 230, 86–93, DOI:10.1016/j.apenergy.2018.08.015.
- P. S. R. Prasad and B. S. Kiran, Sci. Rep., 2019, 9, 5860 CrossRef PubMed.
- S. Takeya and J. A. Ripmeester, Angew. Chem., Int. Ed., 2008, 47(7), 1276–1279, DOI:10.1002/anie.200703718.
- H. Ohno, H. Narita and J. Nagao, J. Phys. Chem. Lett., 2011, 2(3), 201–205, DOI:10.1021/jz101595t.
- J. A. Priest, E. V. L. Rees and C. R. I. Clayton, J. Geophys. Res. Solid Earth, 2009, 114, B11205, DOI:10.1029/2009JB006284.
- B. Tohidi, R. Anderson, M. Ben Clennell, R. W. Burgass and A. B. Biderkab, Geology, 2001, 21(9), 867–870, DOI:10.1130/0091-7613(2001)029<0867:VOOGHF>2.0.CO;2.
- D. Katsuki, R. Ohmura, T. Ebinuma and H. Narita, Philos. Mag., 2006, 86(12), 1753–1761, DOI:10.1080/14786430500509062.
- D. Katsuki, R. Ohmura, T. Ebinuma and H. Narita, Philos. Mag., 2007, 87(7), 1057–1069, DOI:10.1080/14786430601021652.
- D. Katsuki, R. Ohmura, T. Ebinuma and H. Narita, J. Appl. Phys., 2008, 104, 083514, DOI:10.1063/1.3000622.
- K. Nagashima, T. Suzuki, M. Nagamoto and T. Shimizu, J. Phys. Chem. B, 2008, 112(32), 9876–9882, DOI:10.1021/jp802487d.
- M. Muraoka and K. Nagashima, J. Phys. Chem. C, 2012, 116(44), 23342–23350, DOI:10.1021/jp306224w.
- M. Mork, G. Schei and R. Larsen, Ann. N. Y. Acad. Sci., 2000, 912(1), 897–905, DOI:10.1111/j.1749-6632.2000.tb06843.x.
- T. Uchida, S. Dallimore and J. Mikami, Ann. N. Y. Acad. Sci., 2000, 912(1), 1021–1033, DOI:10.1111/j.1749-6632.2000.tb06857.x.
- S. Jin, S. Takeya, J. Hayashi, J. Nagao, Y. Kamata, T. Ebinuma and H. Narita, Jpn. J. Appl. Phys., Part 1, 2004, 43(8A) DOI:10.1143/JJAP.43.5673.
- S. Jin, J. Nagao, S. Takeya, Y. Jin, J. Hayashi, Y. Kamata, T. Ebinuma and H. Narita, Jpn. J. Appl. Phys., Part 2, 2006, 45, 24–28, DOI:10.1143/JJAP.45.L714.
- S. A. Bagherzadeh, I. L. Moudrakovski, J. A. Ripmeester and P. Englezos, Energy Fuels, 2011, 25(7), 3083–3092 CrossRef CAS.
- P. Kerkar, K. W. Jones, R. Kleinberg, W. B. Lindquist, S. Tomov, H. Feng and D. Mahajan, Appl. Phys. Lett., 2009, 95, 024102, DOI:10.1063/1.3120544.
- P. B. Kerkar, K. Horvat, K. W. Jones and D. Mahajan, Geochem., Geophys., Geosyst., 2014, 15, 4759–4768, DOI:10.1002/2014GC005373.
- L. Yang, A. Falenty, M. Chaouachi, D. Haberthür and W. F. Kuhs, Geochem., Geophys., Geosyst., 2016, 17(9), 3717–3732, DOI:10.1002/2016GC006521.
- L. Lei, Y. Seol, J. H. Choi and T. J. Kneafsey, Mar. Pet. Geol., 2019, 104, 451–467, DOI:10.1016/j.marpetgeo.2019.04.004.
- S. J. Cox, D. J. F. Taylor, T. G. A. Youngs, A. K. Soper, T. S. Totton, R. G. Chapman, M. Arjmandi, M. G. Hodges, N. T. Skipper and A. Michaelides, J. Am. Chem. Soc., 2018, 140, 3277–3284 CrossRef CAS PubMed.
- P. Y. Yu, W. Y. Sean, R. Y. Yeh, L. H. Chiang Hsieh, R. Q. Hsu and T. Sato, Int. J. Heat Mass Transfer, 2017, 113, 176–183, DOI:10.1016/j.ijheatmasstransfer.2017.05.053.
- X. Wang, B. Dong, W. Li, M. Yu and Y. Song, Int. J. Heat Mass Transfer, 2018, 122, 1182–1197 CrossRef CAS.
- X. Wang, B. Dong, C. Chen, W. Li and Y. Song, Int. J. Heat Mass Transfer, 2019, 144, 118656 CrossRef CAS.
- A. Fukumoto, K. Kamada, T. Sato, H. Oyama, H. Torii, F. Kiyono, J. Nagao, N. Temma and H. Narita, J. Nat. Gas Sci. Eng., 2018, 50, 269–281, DOI:10.1016/j.jngse.2017.12.016.
- S. R. Gainey and M. E. Elwood Madden, Icarus, 2012, 218(1), 513–524, DOI:10.1016/j.icarus.2011.12.019.
- D. Ambuehl and M. Elwood Madden, Icarus, 2014, 234, 45–52, DOI:10.1016/j.icarus.2014.01.037.
- V. S. Yakushev and E. M. Chuvilin, Cold Reg. Sci. Technol., 2000, 31(3), 189–197, DOI:10.1016/S0165-232X(00)00012-4.
- E. Chuvilin, B. Bukhanov, D. Davletshina, S. Grebenkin and V. Istomin, Geosciences, 2018, 8, 431 CrossRef CAS.
- V. D. Chari, D. V. Sharma, P. S. R. Prasad and S. R. Murthy, J. Nat. Gas Sci. Eng., 2013, 11, 7–11 CrossRef CAS.
- F. Farhang, A. V. Nguyen and K. B. Sewell, Energy Fuels, 2014, 28, 7025–7037 CrossRef CAS.
- M. Cha, S. Baek, J. Morris and J. W. Lee, Chem. – Asian J., 2014, 9, 261–267 CrossRef CAS PubMed.
- S. Arjang, M. Manteghian and A. Mohammadi, Chem. Eng. Res. Des., 2013, 91, 1050–1054 CrossRef CAS.
- M. Aliabadi, A. Rasoolzadeh, F. Esmaeilzadeh and A. Alamdari, J. Nat. Gas Sci. Eng., 2015, 27, 1518–1522 CrossRef CAS.
- R. Wang, T. Liu, F. Ning, W. Ou, L. Zhang, Z. Wang, L. Peng, J. Sun, Z. Liu, T. Li, H. Sun and G. Jiang, J. Energy Chem., 2019, 30, 90–100 CrossRef.
- J. Grdadolnik, F. Merzel and F. Avbelj, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 322–327 CrossRef CAS PubMed.
- D. Chandler, Nature, 2005, 437(7059), 640–647 CrossRef CAS PubMed.
- W. Kauzmann, Advances in protein chemistry, Elsevier, 1959, vol. 14, pp. 1–63 Search PubMed.
- H. Lee, J. Lee, D. Y. Kim, J. Park, Y.-T. Seo, H. Zeng, I. L. Moudrakovski, C. I. Ratcliffe and J. A. Ripmeester, Materials For Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World Scientific, 2011, pp. 285–288 Search PubMed.
- L. J. Florusse, C. J. Peters, J. Schoonman, K. C. Hester, C. A. Koh, S. F. Dec, K. N. Marsh and E. D. Sloan, Science, 2004, 306, 469–471 CrossRef CAS PubMed.
- B. Tohidi, A. Danesh, A. C. Todd, R. W. Burgass and K. K. Østergaard, Fluid Phase Equilib., 1997, 138, 241–250 CrossRef CAS.
- L. C. Ho, P. Babu, R. Kumar and P. Linga, Energy, 2013, 63, 252–259 CrossRef CAS.
- X.-S. Li, Z.-M. Xia, Z.-Y. Chen and H.-J. Wu, Energy Fuels, 2011, 25, 1302–1309 CrossRef CAS.
- N. H. Duc, F. Chauvy and J.-M. Herri, Energy Convers. Manage., 2007, 48, 1313–1322 CrossRef CAS.
- X.-S. Li, C.-G. Xu, Z.-Y. Chen and H.-J. Wu, Energy, 2010, 35, 3902–3908 CrossRef CAS.
- S. M. Kim, J. D. Lee, H. J. Lee, E. K. Lee and Y. Kim, Int. J. Hydrogen Energy, 2011, 36, 1115–1121 CrossRef CAS.
- M. Arjmandi, A. Chapoy and B. Tohidi, J. Chem. Eng. Data, 2007, 52, 2153–2158 CrossRef CAS.
- M. Ricaurte, C. Dicharry, X. Renaud and J.-P. Torré, Fuel, 2014, 122, 206–217 CrossRef CAS.
- J. Du, H. Li and L. Wang, Adv. Powder Technol., 2014, 25, 1227–1233 CrossRef CAS.
- M. Ricaurte, C. Dicharry, D. Broseta, X. Renaud and J.-P. Torré, Ind. Eng. Chem. Res., 2012, 52, 899–910 CrossRef.
- C. F. da Silva Lirio, F. L. P. Pessoa and A. M. C. Uller, Chem. Eng. Sci., 2013, 96, 118–123 CrossRef.
- K. Okutani, Y. Kuwabara and Y. H. Mori, Chem. Eng. Sci., 2008, 63, 183–194 CrossRef CAS.
- H. J. Lee, J. D. Lee, P. Linga, P. Englezos, Y. S. Kim, M. S. Lee and Y. Do Kim, Energy, 2010, 35, 2729–2733 CrossRef CAS.
- J.-P. Torré, C. Dicharry, M. Ricaurte, D. Daniel-David and D. Broseta, Energy Procedia, 2011, 4, 621–628 CrossRef.
- H. Ganji, M. Manteghian, M. R. Omidkhah and H. R. Mofrad, Fuel, 2007, 86, 434–441 CrossRef CAS.
- A. Kumar, T. Sakpal, P. Linga and R. Kumar, Fuel, 2013, 105, 664–671 CrossRef CAS.
- J. Zhang, P. Yedlapalli and J. W. Lee, Chem. Eng. Sci., 2009, 64, 4732–4736 CrossRef CAS.
- H. Li, P. Stanwix, Z. Aman, M. Johns, E. May and L. Wang, J. Phys. Chem. A, 2016, 120, 417–424 CrossRef CAS PubMed.
- N. N. Nguyen, A. V. Nguyen, K. M. Steel, L. X. Dang and M. Galib, J. Phys. Chem. C, 2017, 121, 3830–3840 CrossRef CAS.
- S. Dong, M. Li and A. Firoozabadi, Fuel, 2017, 210, 713–720 CrossRef CAS.
- S. Dong and A. Firoozabadi, J. Chem. Thermodyn., 2018, 117, 214–222 CrossRef CAS.
- F. Jiménez-Ángeles and A. Firoozabadi, ACS Cent. Sci., 2018, 4, 820–831 CrossRef PubMed.
- N. N. Nguyen and A. V. Nguyen, Fuel, 2015, 156, 87–95 CrossRef CAS.
- F. Farhang, A. V. Nguyen and M. A. Hampton, Energy Fuels, 2014, 28, 1220–1229 CrossRef CAS.
- B. Sowa, X. H. Zhang, P. G. Hartley, D. E. Dunstan, K. A. Kozielski and N. Maeda, Energy Fuels, 2014, 28, 6877–6888 CrossRef CAS.
- B. Lukanov and A. Firoozabadi, Langmuir, 2014, 30, 6373–6383 CrossRef CAS PubMed.
- L. Piatkowski, Z. Zhang, E. H. G. Backus, H. J. Bakker and M. Bonn, Nat. Commun., 2014, 5, 4083 CrossRef CAS.
- P. Jungwirth and D. J. Tobias, J. Phys. Chem. B, 2002, 106, 6361–6373 CrossRef CAS.
- S. Pal and F. Müller-Plathe, J. Phys. Chem. B, 2005, 109, 6405–6415 CrossRef CAS PubMed.
- A. Kumar, S. S. Vedula, R. Kumar and P. Linga, J. Chem. Thermodyn., 2018, 117, 2–8 CrossRef CAS.
- A. Kumar, H. P. Veluswamy, R. Kumar and P. Linga, Appl. Energy, 2019, 235, 21–30 CrossRef CAS.
- A. V. Palodkar and A. K. Jana, Energy Fuels, 2019, 33, 1433–1443 CrossRef CAS.
- P. Thoutam, S. Rezaei Gomari, A. Chapoy, F. Ahmad and M. Islam, ACS Omega, 2019, 4(19), 18210–18218 CrossRef CAS PubMed.
- G. C. Nihous, C. K. Kinoshita and S. M. Masutani, Chem. Eng. Sci., 2009, 64, 2767–2771 CrossRef CAS.
- F. E. Anderson and J. M. Prausnitz, AIChE J., 1986, 32, 1321–1333 CrossRef CAS.
- E. D. Sloan and C. A. Koh, Clathrate hydrates of natural gases, third edition, Chem. IndThen Boca Raton-Marcel Dekker Then CRC Press, York, 2008 Search PubMed.
- S. Bobev and K. T. Tait, Am. Mineral., 2004, 89, 1208–1214 CrossRef CAS.
- H. K. Abay and T. M. Svartaas, Energy Fuels, 2009, 24, 752–757 CrossRef.
- J. Amtawong, J. Guo, J. S. Hale, S. Sengupta, E. B. Fleischer, R. W. Martin and K. C. Janda, J. Phys. Chem. Lett., 2016, 7, 2346–2349 CrossRef CAS PubMed.
- P. Linga and M. A. Clarke, Energy Fuels, 2017, 31, 1–13 CrossRef CAS.
- M. Yang, Y. Song, L. Jiang, W. Liu, B. Dou and W. Jing, Appl. Energy, 2014, 135, 504–511 CrossRef CAS.
- A. Siangsai, P. Rangsunvigit, B. Kitiyanan, S. Kulprathipanja and P. Linga, Chem. Eng. Sci., 2015, 126, 383–389 CrossRef CAS.
- V. Govindaraj, D. Mech, G. Pandey, R. Nagarajan and J. S. Sangwai, J. Nat. Gas Sci. Eng., 2015, 26, 810–818 CrossRef CAS.
- Y. Zhang, X.-S. Li, Z.-Y. Chen, G. Li and Y. Wang, J. Nat. Gas Sci. Eng., 2016, 35, 1463–1471 CrossRef CAS.
- S. H. B. Yang, P. Babu, S. F. S. Chua and P. Linga, Appl. Energy, 2016, 162, 1131–1140 CrossRef CAS.
- S. Wang, M. Yang, W. Liu, J. Zhao and Y. Song, J. Pet. Sci. Eng., 2016, 145, 565–572 CrossRef CAS.
- B.-H. Shi, L. Yang, S.-S. Fan and X. Lou, Fuel, 2017, 194, 395–405 CrossRef CAS.
- J. Li, D. Liang, K. Guo, R. Wang and S. Fan, Energy Convers. Manage., 2006, 47, 201–210 CrossRef CAS.
- A. Mohammadi, M. Manteghian, A. Haghtalab, A. H. Mohammadi and M. Rahmati-Abkenar, Chem. Eng. J., 2014, 237, 387–395 CrossRef CAS.
- F. Wang, Y.-M. Song, G.-Q. Liu, G. Guo, S.-J. Luo and R.-B. Guo, Appl. Energy, 2018, 213, 227–234 CrossRef CAS.
- H. Najibi, M. M. Shayegan and H. Heidary, J. Nat. Gas Sci. Eng., 2015, 23, 315–323 CrossRef CAS.
- H. Kakati, A. Mandal and S. Laik, J. Ind. Eng. Chem., 2016, 35, 357–368 CrossRef CAS.
- M. Mohammadi, A. Haghtalab and Z. Fakhroueian, J. Chem. Thermodyn., 2016, 96, 24–33 CrossRef CAS.
- A. Ghozatloo, N. M. Shariaty and M. Hassanisadi, Iran. J. Chem. Eng., 2014, 11, 67–73 Search PubMed.
- N.-J. Kim, S.-S. Park, H. T. Kim and W. Chun, Int. Commun. Heat Mass Transfer, 2011, 38, 31–36 CrossRef CAS.
- J. Pasieka, S. Coulombe and P. Servio, Chem. Eng. Sci., 2013, 104, 998–1002 CrossRef CAS.
- Y.-M. Song, F. Wang, G. Guo, S.-J. Luo and R.-B. Guo, Appl. Energy, 2018, 224, 175–183 CrossRef CAS.
- Y. Song, F. Wang, G. Liu, S. Luo and R. Guo, Energy Fuels, 2017, 31, 1850–1857 CrossRef CAS.
- I. S. Gudmundsson, RF Pat.
- D. Liang, S. He and D. Li, Chin. Sci. Bull., 2009, 54, 965–971 CAS.
- C. Cheng, F. Wang, Y. Tian, X. Wu, J. Zheng, J. Zhang, L. Li, P. Yang and J. Zhao, Renewable Sustainable Energy Rev., 2020, 117, 109492 CrossRef CAS.
- S.-S. Park and N.-J. Kim, J. Ind. Eng. Chem., 2013, 19, 1668–1672 CrossRef CAS.
- H. J. Hong, C. H. Ko, M. H. Song, S. Lee and K. Seong, J. Ind. Eng. Chem., 2016, 41, 183–189 CrossRef CAS.
- Y. Kozo, F. Tetsuro, K. Takahiro and K. Yuichi, Production method for gas hydrates and device for producing same, Pat.GB, 2000, 2347938 Search PubMed.
- K. Carpenter and V. Bahadur, J. Phys. Chem. Lett., 2016, 7, 2465–2469 CrossRef CAS.
- H. Kumano, T. Hirata, K. Mitsuishi and K. Ueno, Int. J. Refrig., 2012, 35, 1266–1274 CrossRef CAS.
- A. A. Chernov, A. A. Pil’Nik, D. S. Elistratov, I. V. Mezentsev, A. V. Meleshkin, M. V. Bartashevich and M. G. Vlasenko, Sci. Rep., 2017, 7, 40809 CrossRef CAS PubMed.
- V. E. Dontsov, V. E. Nakoryakov and A. A. Chernov, J. Appl. Mech. Tech. Phys., 2007, 48, 346–360 CrossRef CAS.
- V. E. Dontsov, V. E. Nakoryakov and A. A. Chernov, Dokl. Ross. Akad. Nauk, 2006, vol. 411, pp. 190–193.
- V. S. Shagapov, S. A. Lepikhin and I. A. Chiglintsev, Thermophys. Aeromechanics, 2010, 17, 229–241 CrossRef.
- V. E. Dontsov, A. A. Chernov and E. V. Dontsov, Thermophys. Aeromechanics, 2007, 14, 21–35 CrossRef.
- V. E. Dontsov and A. A. Chernov, Int. J. Heat Mass Transfer, 2009, 52, 4919–4928 CrossRef CAS.
- V. E. Dontsov, V. E. Nakoryakov and A. A. Chernov, J. Eng. Thermophys., 2009, 18, 1–7 CrossRef.
- Y. Zhang, L. Zhao, S. Deng, R. Zhao, X. Nie and Y. Liu, J. Therm. Sci., 2019, 28, 948–961 CrossRef CAS.
- A. A. Chernov and V. E. Dontsov, Int. J. Heat Mass Transfer, 2011, 54, 4307–4316 CrossRef CAS.
- J. Park, K. Shin, J. Kim, H. Lee, Y. Seo, N. Maeda, W. Tian and C. D. Wood, J. Phys. Chem. C, 2015, 119, 1690–1699 CrossRef CAS.
- S. Fan, L. Yang, Y. Wang, X. Lang, Y. Wen and X. Lou, Chem. Eng. Sci., 2014, 106, 53–59 CrossRef CAS.
- P. Linga, N. Daraboina, J. A. Ripmeester and P. Englezos, Chem. Eng. Sci., 2012, 68, 617–623 CrossRef CAS.
- D. J. Turner, K. T. Miller and E. D. Sloan, Chem. Eng. Sci., 2009, 64, 3996–4004 CrossRef CAS.
- X. Zhao, Z. Qiu and W. Huang, J. Nat. Gas Sci. Eng., 2015, 22, 270–278 CrossRef CAS.
- M. Mork, Formation rate of natural gas hydrate-reactor experiments and models, 2002 Search PubMed.
- P. Skovborg, H. J. Ng, P. Rasmussen and U. Mohn, Chem. Eng. Sci., 1993, 48, 445–453 CrossRef CAS.
- J. S. Parent and P. R. Bishnoi, Chem. Eng. Commun., 1996, 144, 51–64 CrossRef CAS.
- P. Englezos, N. Kalogerakis, P. D. Dholabhai and P. R. Bishnoi, Chem. Eng. Sci., 1987, 42, 2647–2658 CrossRef CAS.
- J. D. Lee and P. Englezos, Chem. Eng. Sci., 2006, 61, 1368–1376 CrossRef CAS.
- J. Yang and B. Tohidi, Chem. Eng. Sci., 2011, 66, 278–283 CrossRef CAS.
- M. F. Qureshi, M. Atilhan, T. Altamash, S. Aparicio, M. Aminnaji and B. Tohidi, J. Nat. Gas Sci. Eng., 2017, 38, 50–58 CrossRef CAS.
- S. Fujita, K. Watanabe and Y. H. Mori, AIChE J., 2009, 55, 1056–1064 CrossRef CAS.
- R. Ohmura, S. Kashiwazaki, S. Shiota, H. Tsuji and Y. H. Mori, Energy Fuels, 2002, 16, 1141–1147 CrossRef CAS.
- M. Takahashi, T. Kawamura, Y. Yamamoto, H. Ohnari, S. Himuro and H. Shakutsui, J. Phys. Chem. B, 2003, 107, 2171–2173 CrossRef CAS.
- B. P. Binks and R. Murakami, Nat. Mater., 2006, 5, 865 CrossRef CAS PubMed.
- W. Wang, C. L. Bray, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 11608–11609 CrossRef CAS PubMed.
- G. Hu, Y. Ye, C. Liu, Q. Meng, J. Zhang and S. Diao, Fuel Process. Technol., 2011, 92, 1617–1622 CrossRef CAS.
- Q. Zhang, C. Li, Q. Wu and B. Zhang, RSC Adv., 2018, 8, 27171–27180 RSC.
- E. D. Sloan Jr, Method for Controlling Clathrate Hydrates in Fluid Systems, US Pat., 5432292, 1995 Search PubMed.
- S. Mokhatab, R. J. Wilkens and K. J. Leontaritis, Energy Sources, Part A, 2007, 29, 39–45 CrossRef CAS.
- C. Vörös, V. Füvesi and Á. Pintér, Hung. J. Ind. Chem., 2013, 41, 7–10 Search PubMed.
- M. Tariq, D. Rooney, E. Othman, S. Aparicio, M. Atilhan and M. Khraisheh, Ind. Eng. Chem. Res., 2014, 53, 17855–17868 CrossRef CAS.
- L. E. Zerpa, J.-L. Salager, C. A. Koh, E. D. Sloan and A. K. Sum, Ind. Eng. Chem. Res., 2010, 50, 188–197 CrossRef.
- A. Qasim, M. S. Khan, B. Lal and A. M. Shariff, J. Pet. Sci. Eng., 2019, 106418 CrossRef CAS.
- A. Striolo, Mol. Phys., 2019, 1–13 Search PubMed.
- S. R. Davies, M. S. Selim, E. D. Sloan, P. Bollavaram and D. J. Peters, AIChE J., 2006, 52, 4016–4027 CrossRef CAS.
- J.-H. Cha, C. Ha, S.-P. Kang, J. W. Kang and K.-S. Kim, Fluid Phase Equilib., 2016, 413, 75–79 CrossRef CAS.
- M. Nepomiluev and V. Streletskaya, SPE Russian Oil and Gas Exploration & Production Technical Conference and Exhibition, Society of Petroleum Engineers, 2014.
- E. M. Reyna and S. R. Stewart, SPE/IADC drilling conference, Society of Petroleum Engineers, 2001.
- M. Aminnaji, B. Tohidi, R. Burgass and M. Atilhan, J. Nat. Gas Sci. Eng., 2017, 45, 840–847 CrossRef CAS.
- N. Daraboina, P. Linga, J. Ripmeester, V. K. Walker and P. Englezos, Energy Fuels, 2011, 25, 4384–4391 CrossRef CAS.
- M. Cha, K. Shin, Y. Seo, J.-Y. Shin and S.-P. Kang, J. Phys. Chem. A, 2013, 117, 13988–13995 CrossRef CAS PubMed.
- J. Kim, K. Shin, Y. Seo, S. J. Cho and J. D. Lee, J. Phys. Chem. B, 2014, 118, 9065–9075 CrossRef CAS PubMed.
- M. A. Kelland, Energy Fuels, 2018, 32, 12001–12012 CrossRef CAS.
- M. S. Kamal, I. A. Hussein, A. S. Sultan and N. von Solms, Renewable Sustainable Energy Rev., 2016, 60, 206–225 CrossRef CAS.
- M. A. Kelland, Energy Fuels, 2006, 20, 825–847 CrossRef CAS.
- T. Yagasaki, M. Matsumoto and H. Tanaka, Phys. Chem. Chem. Phys., 2015, 17, 32347–32357 RSC.
- X. Sun, G. Zhou, J. Zhu, H. Wu, G. Lu and D. Bai, ChemPhysChem, 2019, 20, 2553–2565 CrossRef CAS PubMed.
- T. Y. Sylva, C. K. Kinoshita and S. M. Masutani, Chem. Eng. Sci., 2016, 155, 10–15 CrossRef CAS.
- Q. Lv, X. Zang, X. Li and G. Li, Fluid Phase Equilib., 2018, 458, 272–277 CrossRef CAS.
- D. Bai, Z. Wu, C. Lin and D. Zhou, Fluid Phase Equilib., 2019, 487, 76–82 CrossRef CAS.
- H. Piramoon, M. K. Moraveji, A. Parvareh and A. Azimi, Pet. Sci. Technol., 2019, 37, 1924–1930 CrossRef CAS.
- R. J. Kirkpatrick, Am. Mineral. J. Earth Planet. Mater., 1975, 60, 798–814 CAS.
- T. Yagasaki, M. Matsumoto, Y. Andoh, S. Okazaki and H. Tanaka, J. Phys. Chem. B, 2014, 118, 11797–11804 CrossRef CAS PubMed.
- K. S. Sujith and C. N. Ramachandran, J. Phys. Chem. B, 2017, 121, 153–163 CrossRef CAS PubMed.
- H. Kim, H. P. Veluswamy, Y. Seo and P. Linga, Cryst. Growth Des., 2018, 18, 6984–6994 CrossRef CAS.
- A. Majumdar, E. Mahmoodaghdam and P. R. Bishnoi, J. Chem. Eng. Data, 2000, 45, 20–22 CrossRef CAS.
- A. H. Mohammadi and D. Richon, J. Chem. Thermodyn., 2012, 44, 26–30 CrossRef CAS.
- H. Najibi, Z. Kamali and A. H. Mohammadi, Fluid Phase Equilib., 2013, 342, 71–74 CrossRef CAS.
- N. A. Sami, K. Das, J. S. Sangwai and N. Balasubramanian, J. Chem. Thermodyn., 2013, 65, 198–203 CrossRef CAS.
- H. Najibi, E. Amiri and A. H. Mohammadi, Fluid Phase Equilib., 2014, 363, 70–73 CrossRef CAS.
- M. Dastanian, A. A. Izadpanah and M. Mofarahi, J. Chem. Eng. Data, 2017, 62, 1701–1707 CrossRef CAS.
- M. Dastanian, A. A. Izadpanah and M. Mofarahi, J. Chem. Eng. Data, 2018, 63, 1675–1681 CrossRef CAS.
- K. N. Mahadev and P. R. Bishnoi, Can. J. Chem. Eng., 1999, 77, 718–722 CrossRef CAS.
- M. D. Jager, C. J. Peters and E. D. Sloan, Fluid Phase Equilib., 2002, 193, 17–28 CrossRef CAS.
- C. Eichholz, A. Majumdar, M. A. Clarke, L. R. Oellrich and P. R. Bishnoi, J. Chem. Eng. Data, 2004, 49, 847–851 CrossRef CAS.
- R. Masoudi, B. Tohidi, A. Danesh, A. C. Todd, R. Anderson, R. W. Burgass and J. Yang, Chem. Eng. Sci., 2005, 60, 4213–4224 CrossRef CAS.
- A. H. Mohammadi and D. Richon, J. Chem. Thermodyn., 2009, 41, 1374–1377 CrossRef CAS.
- H. Najibi, A. Chapoy, H. Haghighi and B. Tohidi, Fluid Phase Equilib., 2009, 275, 127–131 CrossRef CAS.
- G.-H. Kwak, K.-H. Lee, S. Y. Hong, S. D. Seo, J. D. Lee, B. R. Lee and A. K. Sum, J. Chem. Eng. Data, 2018, 63, 2179–2184 CrossRef CAS.
- R. Masoudi, B. Tohidi, R. Anderson, R. W. Burgass and J. Yang, Fluid Phase Equilib., 2004, 219, 157–163 CrossRef CAS.
- P. D. Dholabhai, J. S. Parent and P. R. Bishnoi, Fluid Phase Equilib., 1997, 141, 235–246 CrossRef CAS.
- Q. Nasir, K. K. Lau, B. Lal and K. M. Sabil, J. Chem. Eng. Data, 2014, 59, 3920–3926 CrossRef CAS.
- Z.-G. Sun, S.-S. Fan, L. Shi, Y.-K. Guo and K.-H. Guo, J. Chem. Eng. Data, 2001, 46, 927–929 CrossRef CAS.
- A. P. Semenov, A. S. Stoporev, R. I. Mendgaziev, P. A. Gushchin, V. N. Khlebnikov, V. S. Yakushev, V. A. Istomin, D. V. Sergeeva and V. A. Vinokurov, J. Chem. Thermodyn., 2019, 137, 119–130 CrossRef CAS.
- J.-W. Lee and S.-P. Kang, Ind. Eng. Chem. Res., 2011, 50, 8750–8755 CrossRef CAS.
- Y. Seo, H. Kim and J. Park, ASME 2016 35th International Conference on Ocean, Offshore and Arctic Engineering, American Society of Mechanical Engineers Digital Collection, 2016.
- H. Kim, J. Park, Y. Seo and M. Ko, Chem. Eng. Sci., 2017, 158, 172–180 CrossRef CAS.
- S. Xu, S. Fan, H. Yao, Y. Wang, X. Lang, P. Lv and S. Fang, J. Chem. Thermodyn., 2017, 104, 212–217 CrossRef CAS.
- J.-H. Sa, G.-H. Kwak, B. R. Lee, D.-H. Park, K. Han and K.-H. Lee, Sci. Rep., 2013, 3, 2428 CrossRef PubMed.
- C. B. Bavoh, O. Nashed, M. S. Khan, B. Partoon, B. Lal and A. M. Sharif, J. Chem. Thermodyn., 2018, 117, 48–53 CrossRef CAS.
- Y. Liu, B. Chen, Y. Chen, S. Zhang, W. Guo, Y. Cai, B. Tan and W. Wang, Energy Technol., 2015, 3, 815–819 CrossRef CAS.
- H. P. Veluswamy, Q. W. Hong and P. Linga, Cryst. Growth Des., 2016, 16, 5932–5945 CrossRef CAS.
- J.-H. Sa, B. R. Lee, D.-H. Park, K. Han, H. D. Chun and K.-H. Lee, Environ. Sci. Technol., 2011, 45, 5885–5891 CrossRef CAS PubMed.
- H. Roosta, A. Dashti, S. H. Mazloumi and F. Varaminian, J. Mol. Liq., 2016, 215, 656–663 CrossRef CAS.
- C. B. Bavoh, B. Partoon, B. Lal, G. Gonfa, S. F. Khor and A. M. Sharif, Chem. Eng. Sci., 2017, 171, 331–339 CrossRef CAS.
- J.-H. Sa, G.-H. Kwak, K. Han, D. Ahn, S. J. Cho, J. D. Lee and K.-H. Lee, Sci. Rep., 2016, 6, 31582 CrossRef CAS PubMed.
- J.-H. Sa, G.-H. Kwak, K. Han, D. Ahn and K.-H. Lee, Sci. Rep., 2015, 5, 11526 CrossRef PubMed.
- S. K. Nagappayya, R. M. Lucente-Schultz, V. M. Nace and V. M. Ho, J. Chem. Eng. Data, 2014, 60, 351–355 CrossRef.
- K.-S. Kim, J. W. Kang and S.-P. Kang, Chem. Commun., 2011, 47, 6341–6343 RSC.
- A. Perrin, O. M. Musa and J. W. Steed, Chem. Soc. Rev., 2013, 42, 1996–2015 RSC.
- M. A. Kelland, Energy Fuels, 2017, 31, 5046–5054 CrossRef CAS.
- E. Luna-Ortiz, M. Healey, R. Anderson and E. Sørhaug, Energy Fuels, 2014, 28, 2902–2913 CrossRef CAS.
- O. Nashed, K. M. Sabil, L. Ismail, A. Japper-Jaafar and B. Lal, J. Chem. Thermodyn., 2018, 117, 147–154 CrossRef CAS.
- C. Magnusson, E. Abrahamsen, M. A. Kelland, A. Cely, K. Kinnari, X. Li and K. M. Askvik, Energy Fuels, 2018, 32, 5772–5778 CrossRef CAS.
- M. Muraoka, N. Susuki and Y. Yamamoto, RSC Adv., 2016, 6, 63880–63885 RSC.
- P. C. Chua and M. A. Kelland, Energy Fuels, 2012, 26, 4481–4485 CrossRef CAS.
- L. Cheng, K. Liao, Z. Li, J. Cui, B. Liu, F. Li, G. Chen and C. Sun, Chem. Eng. Sci., 2019, 207, 305–316, DOI:10.1016/j.ces.2019.06.032.
- E. D. Sloan Jr, Method for Controlling Clathrate Hydrates in Fluid Systems, US Pat., 5880319, 1999 Search PubMed.
- F. T. Reyes and M. A. Kelland, Energy Fuels, 2013, 27, 3730–3735 CrossRef CAS.
- H.-B. Qin, Z.-Y. Zhang, C.-Y. Sun, G.-J. Chen, Q.-L. Ma and Z.-F. Ning, J. Chem. Eng. Data, 2017, 62, 2770–2775 CrossRef CAS.
- Q. Zhang, X. Shen, X. Zhou and D. Liang, Energy Fuels, 2016, 31, 839–846 CrossRef.
- J. Xu, L. Li, J. Liu, X. Wang, Y. Yan and J. Zhang, Phys. Chem. Chem. Phys., 2018, 20, 8326–8332 RSC.
- J. Park, S. S. Lee, H. Kim, S.-H. Kim and Y. Seo, Energy Fuels, 2018, 32, 9001–9009 CrossRef CAS.
- A. Perrin, M. J. Goodwin, O. M. Musa, D. J. Berry, P. Corner, K. Edkins, D. S. Yufit and J. W. Steed, Cryst. Growth Des., 2017, 17, 3236–3249 CrossRef CAS.
- T. Yagasaki, M. Matsumoto and H. Tanaka, J. Am. Chem. Soc., 2015, 137, 12079–12085 CrossRef CAS PubMed.
- Q. Wang, C. Wang, S. Ma, P. Lu and J. Dong, ACS Sustainable Chem. Eng., 2018, 6, 13532–13542 CrossRef CAS.
- P. C. Chua, M. A. Kelland, T. Hirano and H. Yamamoto, Energy Fuels, 2012, 26, 4961–4967 CrossRef CAS.
- P. C. Chua, M. A. Kelland, K. Ishitake, K. Satoh, M. Kamigaito and Y. Okamoto, Energy Fuels, 2012, 26, 3577–3585 CrossRef CAS.
- L. S. Ree, E. Opsahl and M. A. Kelland, Energy Fuels, 2010, 24(4), 2554–2562 CrossRef.
- H. Roosta, A. Dashti, S. H. Mazloumi and F. Varaminian, J. Mol. Liq., 2018, 253, 259–269 CrossRef CAS.
- M. A. Kelland, T. M. Svartaas, J. Øvsthus and T. Namba, Ann. N. Y. Acad. Sci., 2000, 912, 281–293 CrossRef CAS.
- L. Del Villano, M. A. Kelland, G. M. Miyake and E. Y.-X. Chen, Energy Fuels, 2010, 24, 2554–2562 CrossRef CAS.
- H. Ajiro, Y. Takemoto, M. Akashi, P. C. Chua and M. A. Kelland, Energy Fuels, 2010, 24, 6400–6410 CrossRef CAS.
- A. A. Bertolazzo, P. M. Naullage, B. Peters and V. Molinero, J. Phys. Chem. Lett., 2018, 9, 3224–3231 CrossRef CAS PubMed.
- B. Widom, P. Bhimalapuram and K. Koga, Phys. Chem. Chem. Phys., 2003, 5, 3085–3093 RSC.
- M. A. Kelland, Production chemicals for the oil and gas industry, CRC Press, 2014 Search PubMed.
- C. D. Magnusson and M. A. Kelland, Energy Fuels, 2015, 29, 6347–6354 CrossRef CAS.
- J. Wang, R. Wang, R.-H. Yoon and Y. Seol, J. Chem. Eng. Data, 2014, 60, 383–388 CrossRef.
- M. A. Kelland, Production Chemicals for the Oil and Gas Industry, 2010, vol. 72 Search PubMed.
- L. Wan, D.-Q. Liang, Q. Ding and G. Hou, Fuel, 2019, 239, 173–179 CrossRef CAS.
- A. M. P. S. G. da Silva, S. I. C. Lopes, P. Brogueira, T. J. V. Prazeres, M. Beija and J. M. G. Martinho, J. Colloid Interface Sci., 2008, 327, 129–137 CrossRef PubMed.
- I. Idziak, D. Avoce, D. Lessard, D. Gravel and X. X. Zhu, Macromolecules, 1999, 32, 1260–1263 CrossRef CAS.
- E. G. Dirdal and M. A. Kelland, Energy Fuels, 2019, 33, 7127–7137 CrossRef CAS.
- L. H. S. Ree and M. A. Kelland, Energy Fuels, 2018, 32, 10639–10648 CrossRef CAS.
- Q. Zhang, I. M. Heyns, R. Pfukwa, B. Klumperman and M. A. Kelland, Energy Fuels, 2018, 32, 12337–12344 CrossRef CAS.
- M. S. Khan, B. Lal, A. M. Shariff and H. Mukhtar, J. Mol. Liq., 2019, 274, 33–44 CrossRef CAS.
- C. Xiao, N. Wibisono and H. Adidharma, Chem. Eng. Sci., 2010, 65, 3080–3087 CrossRef CAS.
- C. Xiao and H. Adidharma, Chem. Eng. Sci., 2009, 64, 1522–1527 CrossRef CAS.
- K. Kim and S.-P. Kang, Proceedings of the 7th International Conference on Gas Hydrates (ICGH 2011), 2011, pp. 17–21.
- T. Kitajima, N. Ohtsubo, S. Hashimoto, T. Makino, D. Kodama and K. Ohgaki, Am. Chem. Sci. J., 2012, 2, 100–110 CrossRef CAS.
- L. K. Chun and A. Jaafar, Asian J. Sci. Res., 2013, 6, 374–380 CrossRef CAS.
- R. K. McMullan, T. C. Mak and G. A. Jeffrey, J. Chem. Phys., 1966, 44, 2338–2345 CrossRef CAS.
- Q. Chen, Y. Yu, P. Zeng, W. Yang, Q. Liang, X. Peng, Y. Liu and Y. Hu, J. Nat. Gas Chem., 2008, 17, 264–267 CrossRef CAS.
- X.-S. Li, Y.-J. Liu, Z.-Y. Zeng, Z.-Y. Chen, G. Li and H.-J. Wu, J. Chem. Eng. Data, 2010, 56, 119–123 CrossRef.
- K. Tumba, P. Reddy, P. Naidoo, D. Ramjugernath, A. Eslamimanesh, A. H. Mohammadi and D. Richon, J. Chem. Eng. Data, 2011, 56, 3620–3629 CrossRef CAS.
- H. Zeng, L. D. Wilson, V. K. Walker and J. A. Ripmeester, J. Am. Chem. Soc., 2006, 128, 2844–2850 CrossRef CAS.
- X. Guo, W. Zhao, X. Pang, X. Liao, X. Hu and J. Wu, Food Hydrocolloids, 2014, 35, 217–225 CrossRef CAS.
- D. Le Corre, J. Bras and A. Dufresne, Biomacromolecules, 2010, 11, 1139–1153 CrossRef CAS PubMed.
- M. R. Talaghat, J. Nat. Gas Sci. Eng., 2014, 18, 7–12 CrossRef CAS.
- L. Wan, N. Zhang and D.-Q. Liang, J. Mol. Liq., 2019, 292, 111435 CrossRef CAS.
- S. Yaqub and L. K. Keong, J. Ind. Eng. Chem., 2019, 79, 131–145 CrossRef CAS.
- S. Yaqub, A. bin Mohd Shariff and N. B. Mellon, J. Nat. Gas Sci. Eng., 2019, 65, 68–81 CrossRef CAS.
- Y. Xu, M. Yang and X. Yang, J. Nat. Gas Chem., 2010, 19, 431–435 CrossRef CAS.
- S. Yaqub, B. Lal, B. Partoon and N. B. Mellon, Fluid Phase Equilib., 2018, 477, 40–57 CrossRef CAS.
- V. Srivastava, D. S. Chauhan, P. G. Joshi, V. Maruthapandian, A. A. Sorour and M. A. Quraishi, ChemistrySelect, 2018, 3, 1990–1998 CrossRef CAS.
- P. L. Davies, J. Baardsnes, M. J. Kuiper and V. K. Walker, Philos. Trans. R. Soc., B, 2002, 357, 927–935 CrossRef CAS PubMed.
- S. Xu, S. Fan, S. Fang, X. Lang, Y. Wang and J. Chen, Sci. Rep., 2016, 6, 23220 CrossRef CAS.
- A. Farhadian, M. A. Varfolomeev, A. Kudbanov and S. R. Gallyamov, Chem. Eng. Sci., 2019, 206, 507–517 CrossRef CAS.
- A. Farkhadian, M. A. Varfolomeev, Y. F. Zaripova and V. V. Yarkovoi, Chem. Technol. Fuels Oils, 2019, 55, 159–164 CrossRef CAS.
- A. Farhadian, A. Kudbanov, M. A. Varfolomeev and D. Dalmazzone, Sci. Rep., 2019, 9, 1–10 CrossRef CAS PubMed.
- X. Zhou, Y. Li, C. Fang, S. Li, Y. Cheng, W. Lei and X. Meng, J. Mater. Sci. Technol., 2015, 31, 708–722 CrossRef.
- J. Hu, K. Peng, J. Guo, D. Shan, G. B. Kim, Q. Li, E. Gerhard, L. Zhu, W. Tu and W. Lv, ACS Appl. Mater. Interfaces, 2016, 8, 17499–17510 CrossRef CAS PubMed.
- H. K. Shendi, I. Omrani, A. Ahmadi, A. Farhadian, N. Babanejad and M. R. Nabid, Prog. Org. Coat., 2017, 105, 303–309 CrossRef CAS.
- Q. Sheng, K. C. da Silveira, W. Tian, C. Fong, N. Maeda, R. Gubner and C. D. Wood, Energy Fuels, 2017, 31, 6724–6731 CrossRef CAS.
- J. Park, H. Kim, Q. Sheng, C. D. Wood and Y. Seo, Energy Fuels, 2017, 31, 9363–9373 CrossRef CAS.
- Z. M. Aman, K. Olcott, K. Pfeiffer, E. D. Sloan, A. K. Sum and C. A. Koh, Langmuir, 2013, 29, 2676–2682 CrossRef CAS.
- C. Liu, M. Li, C. Liu, K. Geng and Y. Li, Energy Fuels, 2016, 30, 6240–6248 CrossRef CAS.
- E. P. Brown, S. Hu, J. Wells, X. Wang and C. A. Koh, Energy Fuels, 2018, 32, 6619–6626 CrossRef CAS.
- T. Bui, A. Phan, D. Monteiro, Q. Lan, M. Ceglio, E. Acosta, P. Krishnamurthy and A. Striolo, Langmuir, 2017, 33, 2263–2274 CrossRef CAS PubMed.
- Z. Huo, E. Freer, M. Lamar, B. Sannigrahi, D. M. Knauss and E. D. Sloan Jr, Chem. Eng. Sci., 2001, 56, 4979–4991 CrossRef CAS.
- A. Phan, T. Bui, E. Acosta, P. Krishnamurthy and A. Striolo, Phys. Chem. Chem. Phys., 2016, 18, 24859–24871 RSC.
- M. A. Kelland, T. M. Svartaas, J. Øvsthus, T. Tomita and K. Mizuta, Chem. Eng. Sci., 2006, 61, 4290–4298 CrossRef CAS.
- M. R. Anklam, J. D. York, L. Helmerich and A. Firoozabadi, AIChE J., 2008, 54, 565–574 CrossRef CAS.
- E. P. Brown and C. A. Koh, Energy Fuels, 2016, 30, 8065–8071 CrossRef CAS.
- L. M. Frostman, SPE Annual Technical Conference and Exhibition, Society of Petroleum Engineers, 2000.
- P. C. Chua and M. A. Kelland, Energy Fuels, 2018, 32, 1674–1684 CrossRef CAS.
- T. Bui, F. Sicard, D. Monteiro, Q. Lan, M. Ceglio, C. Burress and A. Striolo, J. Phys. Chem. Lett., 2018, 9, 3491–3496 CrossRef CAS PubMed.
- Y. Tokiwa, H. Sakamoto, T. Takiue, M. Aratono and H. Matsubara, J. Phys. Chem. B, 2015, 119, 6235–6241 CrossRef CAS PubMed.
- Q. Lei and C. D. Bain, Phys. Rev. Lett., 2004, 92, 176103 CrossRef PubMed.
- R. B. Asserson, A. C. Hoffmann, S. Høiland and K. M. Asvik, J. Pet. Sci. Eng., 2009, 68, 209–217 CrossRef CAS.
- Z. M. Aman, L. E. Dieker, G. Aspenes, A. K. Sum, E. D. Sloan and C. A. Koh, Energy Fuels, 2010, 24, 5441–5445 CrossRef CAS.
- J. D. York and A. Firoozabadi, J. Phys. Chem. B, 2008, 112, 10455–10465 CrossRef CAS PubMed.
- J. D. York and A. Firoozabadi, Energy Fuels, 2009, 23, 2937–2946 CrossRef CAS.
- S. Gao, Energy Fuels, 2009, 23, 2118–2121 CrossRef CAS.
- Z. M. Aman, A. Haber, N. N. A. Ling, A. Thornton, M. L. Johns and E. F. May, Energy Fuels, 2015, 29, 7948–7955 CrossRef CAS.
- C. Liu, M. Li, V. K. Srivastava and C. A. Koh, Energy Fuels, 2016, 30, 2555–2562 CrossRef CAS.
- H. Mehrabian, M. A. Bellucci, M. R. Walsh and B. L. Trout, J. Phys. Chem. C, 2018, 122, 12839–12849 CrossRef CAS.
- A. Striolo, A. Phan and M. R. Walsh, Curr. Opin. Chem. Eng., 2019, 25, 57–66 CrossRef.
- J. S. Gudmundsson, Method for Production of Gas Hydrates for Transportation and Storage, US Pat., 5536893, 1996 Search PubMed.
- Y. Katoh, K. Horiguchi, T. Iwasaki and S. Nagamori, Process for Producing Gas Hydrate Pellet, US Pat., 7999141, 2011 Search PubMed.
- K. Park, S. Y. Hong, J. W. Lee, K. C. Kang, Y. C. Lee, M.-G. Ha and J. D. Lee, Desalination, 2011, 274, 91–96 CrossRef CAS.
- P. Babu, R. Kumar and P. Linga, Chem. Eng. Sci., 2014, 117, 342–351 CrossRef CAS.
- X. J. Shi and P. Zhang, Int. J. Refrig., 2014, 42, 77–89 CrossRef CAS.
- A. Eslamimanesh, A. H. Mohammadi, D. Richon, P. Naidoo and D. Ramjugernath, J. Chem. Thermodyn., 2012, 46, 62–71 CrossRef CAS.
- P. Pirzadeh and P. G. Kusalik, J. Am. Chem. Soc., 2013, 135, 7278–7287 CrossRef CAS PubMed.
- B. C. Knott, V. Molinero, M. F. Doherty and B. Peters, J. Am. Chem. Soc., 2012, 134, 19544–19547 CrossRef CAS PubMed.
- J. Zhang, R. W. Hawtin, Y. Yang, E. Nakagava, M. Rivero, S. K. Choi and P. M. Rodger, J. Phys. Chem. B, 2008, 112, 10608–10618 CrossRef CAS PubMed.
- F. Jiménez-Ángeles and A. Firoozabadi, J. Phys. Chem. C, 2015, 119, 8798–8804 CrossRef PubMed.
- S. V. Joshi, G. A. Grasso, P. G. Lafond, I. Rao, E. Webb, L. E. Zerpa, E. D. Sloan, C. A. Koh and A. K. Sum, Chem. Eng. Sci., 2013, 97, 198–209 CrossRef CAS.
- G. Song, Y. Li, W. Wang, K. Jiang, X. Ye and P. Zhao, Chem. Eng. Sci., 2017, 158, 480–489 CrossRef CAS.
- C. Ruan, L. Ding, B. Shi, Q. Huang and J. Gong, RSC Adv., 2017, 7, 48127–48135 RSC.
- J. W. Nicholas, C. A. Koh, E. D. Sloan, L. Nuebling, H. He and B. Horn, AIChE J., 2009, 55, 1882–1888 CrossRef CAS.
- J. Chen, K.-L. Yan, G.-J. Chen, C.-Y. Sun, B. Liu, N. Ren, D.-J. Shen, M. Niu, Y.-N. Lv and N. Li, Chem. Eng. Sci., 2015, 122, 284–290 CrossRef CAS.
- X. F. Lv, B. H. Shi, Y. Wang, Y. X. Tang, L. Y. Wang and J. Gong, Oil Gas Sci. Technol., 2015, 70, 1111–1124 CrossRef CAS.
- I. Rao, C. A. Koh, E. D. Sloan and A. K. Sum, Ind. Eng. Chem. Res., 2013, 52, 6262–6269 CrossRef CAS.
- L. Jensen, K. Thomsen and N. von Solms, Chem. Eng. Sci., 2008, 63, 3069–3080 CrossRef CAS.
- X. Li, C. Chen, Y. Chen, Y. Li and H. Li, Energy Fuels, 2015, 29, 2277–2288 CrossRef CAS.
- S. Douïeb, S. Archambault, L. Fradette, F. Bertrand and B. Haut, Can. J. Chem. Eng., 2017, 95, 187–198 CrossRef.
- M. Aminnaji, B. Tohidi, R. Burgass and M. Atilhan, J. Nat. Gas Sci. Eng., 2017, 40, 17–23 CrossRef CAS.
- A. Lone and M. A. Kelland, Energy Fuels, 2013, 27, 2536–2547 CrossRef CAS.
- N. Maeda, M. A. Kelland and C. D. Wood, Chem. Eng. Sci., 2018, 183, 30–36 CrossRef CAS.
- Q. Guo and G. W. Small, Appl. Spectrosc., 2013, 67, 913–923 CrossRef CAS PubMed.
- A. Ouyang and J. Liu, Meas. Sci. Technol., 2013, 24, 25502 CrossRef.
- A. A. Gowen, F. Marini, Y. Tsuchisaka, S. De Luca, M. Bevilacqua, C. O’Donnell, G. Downey and R. Tsenkova, Talanta, 2015, 131, 609–618 CrossRef CAS PubMed.
- R. Anderson, F. Tohidi, H. Mozaffar and B. Tohidi, J. Pet. Sci. Eng., 2016, 145, 520–526 CrossRef CAS.
- M. K. Gibbons and B. Örmeci, J. Water Supply Res. Technol., 2013, 62, 205–213 CrossRef CAS.
- R. K. Haghi, J. Yang and B. Tohidi, Ind. Eng. Chem. Res., 2018, 57, 11728–11737 CrossRef.
- J.-H. Sa, A. Melchuna, X. Zhang, R. Morales, A. Cameirao, J.-M. Herri and A. K. Sum, Ind. Eng. Chem. Res., 2019, 58, 8544–8552 CrossRef CAS.
- H. Mozaffar, R. Anderson and B. Tohidi, Energy Fuels, 2016, 30, 10055–10063 CrossRef CAS.
- B. Tohidi, R. Anderson, H. Mozaffar and F. Tohidi, Energy Fuels, 2015, 29, 8254–8260 CrossRef CAS.
- A. Ding, L. Yang, S. Fan and X. Lou, Chem. Eng. Sci., 2013, 96, 124–130 CrossRef CAS.
- F. Su, C. L. Bray, B. Tan and A. I. Cooper, Adv. Mater., 2008, 20, 2663–2666 CrossRef CAS PubMed.
- B. Tohidi, R. Anderson, A. Chapoy, J. Yang and R. W. Burgass, Energy Fuels, 2012, 26, 4053–4058 CrossRef CAS.
- J. Yang, A. Chapoy, S. Mazloum and B. Tohidi, SPE Prod. Oper., 2012, 27, 376–381 CAS.
- B. Tohidi, A. Chapoy, J. Yang, F. Ahmadloo, I. Valko and Z. M. Zain, Offshore Technology Conference, Offshore Technology Conference, 2008.
- M. Akhfash, Z. M. Aman, J. Du, P. F. Pickering, M. L. Johns, C. A. Koh and E. F. May, Energy Fuels, 2017, 31, 4875–4885 CrossRef CAS.
- J. A. Boxall, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Langmuir, 2011, 28, 104–110 CrossRef PubMed.
- J. A. Boxall, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Ind. Eng. Chem. Res., 2009, 49, 1412–1418 CrossRef.
- D. J. Turner, K. T. Miller and E. D. Sloan, Chem. Eng. Sci., 2009, 64, 5066–5072 CrossRef CAS.
- J. Chen, Y.-F. Wang, C.-Y. Sun, F.-G. Li, N. Ren, M.-L. Jia, K.-L. Yan, Y.-N. Lv, B. Liu and G.-J. Chen, Energy Fuels, 2014, 29, 122–129 CrossRef CAS.
- A. Cameirão, H. Le Ba, M. Darbouret, J.-M. Herri, J.-L. Peytavy and P. Glénat, J. Cryst. Growth, 2012, 342, 65–71 CrossRef.
- M. A. Clarke and P. R. Bishnoi, Chem. Eng. Sci., 2005, 60, 695–709 CrossRef CAS.
- K. AlHarooni, A. Barifcani, D. Pack, R. Gubner and V. Ghodkay, J. Pet. Sci. Eng., 2015, 135, 608–617 CrossRef CAS.
- T. M. Latta, M. E. Seiersten and S. A. Bufton, Offshore Technology Conference, Offshore Technology Conference, 2013.
- K. Alef, C. Smith, S. Iglauer, R. Gubner and A. Barifcani, Fuel, 2018, 222, 638–647 CrossRef CAS.
- A. Hussain, S. Gharfeh and S. S. Adham, SPE International Production and Operations Conference & Exhibition, Society of Petroleum Engineers, 2012.
- S. Xu, S. Fan, Y. Wang and X. Lang, Chem. Eng. Sci., 2017, 171, 293–302 CrossRef CAS.
- J. W. Bartels, R. A. Jones and R. M. Lucente-Schultz, Removal of Hydrate Inhibitors from Waste Streams, US Pat., 15/423,797, 2017 Search PubMed.
- J. Minier-Matar, S. Gharfeh, A. Hussain and S. Adham, Separation of Kinetic Hydrate Inhibitors from an Aqueous Solution, US Pat., 10131551, 2016 Search PubMed.
- Z. R. Chong, S. H. B. Yang, P. Babu, P. Linga and X. Sen Li, Appl. Energy, 2016, 162, 1633–1652 CrossRef.
- K. A. Kvenvolden and T. D. Lorenson, Geophysical Monograph Series, 2000, vol. 124, pp. 3–18 Search PubMed.
- K. A. Kvenvolden and M. A. McMenamin, Geolocial Surv. Circ., 1980, 825, 1–11 Search PubMed.
- J. H. Hand, D. L. Katz and V. K. Verma, Natural Gases in Marine Sediments, Springer US, MA, 1974, pp. 179–194 Search PubMed.
- R. Boswell and T. S. Collett, Energy Environ. Sci., 2011, 4, 1206–1215 RSC.
- X. Sen Li, C. G. Xu, Y. Zhang, X. K. Ruan, G. Li and Y. Wang, Appl. Energy, 2016, 172, 286–322 CrossRef.
- L. G. Tang, R. Xiao, G. Li, Z. P. Feng, X. Sen Li and S. S. Fan, Guocheng Gongcheng Xuebao, 2006, 6, 548–553 CAS.
- G. Li, X. Sen Li and Y. Wang, Energy Procedia, 2014, 61, 803–807 CrossRef CAS.
- D. L. Li, D. Q. Liang, S. S. Fan, X. Sen Li, L. G. Tang and N. S. Huang, Energy Convers. Manage., 2008, 49, 2207–2213 CrossRef CAS.
- C. Cranganu, J. Pet. Sci. Eng., 2009, 65, 76–80 CrossRef CAS.
- J. M. Schicks, E. Spangenberg, R. Giese, M. MLuzi-Helbin, M. Priegnitz and B. Beeskow-Strauch, Energies, 2013, 6, 3002–3016 CrossRef CAS.
- Y. Song, Y. Kuang, Z. Fan, Y. Zhao and J. Zhao, Int. J. Heat Mass Transfer, 2018, 121, 207–214 CrossRef CAS.
- Z. Yin, G. Moridis, Z. R. Chong, H. K. Tan and P. Linga, Ind. Eng. Chem. Res., 2018, 57, 5776–5791 CrossRef CAS.
- M. Terzariol, G. Goldsztein and J. C. Santamarina, Energy, 2017, 141, 1622–1628 CrossRef CAS.
- Ş. Merey and S. N. Longinos, J. Nat. Gas Sci. Eng., 2018, 52, 248–266 CrossRef.
- Z. Yin, G. Moridis, Z. R. Chong, H. K. Tan and P. Linga, Appl. Energy, 2018, 230, 444–459 CrossRef CAS.
- K. Yamamoto, Y. Terao, T. Fujii, T. Ikawa, M. Seki, M. Matsuzawa and T. Kanno, Proceedings of the Annual Offshore Technology Conference, Offshore Technology Conference, 2014, vol. 3, pp. 1802–1812.
- Z. R. Chong, Z. Yin and P. Linga, Energy Procedia, 2017, 105, 4963–4969 CrossRef CAS.
- R. Sun, Z. Fan, M. Yang, W. Pang, Y. Li and Y. Song, J. Nat. Gas Sci. Eng., 2019, 65, 125–134 CrossRef CAS.
- S. Circone, L. A. Stern, S. H. Kirby, J. C. Pinkston and W. B. Durham, Ann. N. Y. Acad. Sci., 2006, 912, 544–555 CrossRef.
- M. Yang, Y. Gao, H. Zhou, B. Chen and Y. Li, Int. J. Energy Res., 2019, 43(10), 5493–5505, DOI:10.1002/er.4669.
- Y. Gao, M. Yang, J. Nan Zheng and B. Chen, Fuel, 2018, 232, 99–107 CrossRef CAS.
- K. U. Heeschen, S. Abendroth, M. Priegnitz, E. Spangenberg, J. Thaler and J. M. Schicks, Energy Fuels, 2016, 30, 6210–6219 CrossRef CAS.
- Y. Wang, J. C. Feng, X. Sen Li, L. Zhan and X. Y. Li, Energy, 2018, 160, 835–844 CrossRef CAS.
- T. Lv, X. Li, Z. Chen, D. Sun, Y. Zhang, K. Yan and J. Cai, Energy Technol., 2018, 6, 2501–2511 CrossRef CAS.
- Z. R. Chong, J. W. R. Moh, Z. Yin, J. Zhao and P. Linga, Appl. Energy, 2018, 229, 637–647 CrossRef.
- Z. R. Chong, J. Zhao, J. H. R. Chan, Z. Yin and P. Linga, Appl. Energy, 2018, 214, 117–130 CrossRef.
- H. Shin and J. C. Santamarina, Acta Geotech., 2017, 12, 883–895 CrossRef.
- B. R. C. Malagar, K. P. Lijith and D. N. Singh, J. Nat. Gas Sci. Eng., 2019, 168–184 CrossRef CAS.
- L. Yang, Y. Liu, H. Zhang, B. Xiao, X. Guo, R. Wei, L. Xu, L. Sun, B. Yu, S. Leng and Y. Li, Chin. J. Chem. Eng., 2019, 27(9), 2133–2147, DOI:10.1016/j.cjche.2019.02.028.
- Z. R. Chong, S. H. B. Yang, P. Babu, P. Linga, X.-S. Sen Li, Z. R. Chong, P. Babu and X.-S. Sen-Li, Appl. Energy, 2015, 162, 1633–1652 CrossRef.
- Z. Yin and P. Linga, Chin. J. Chem. Eng., 2019, 27(9), 2026–2036, DOI:10.1016/j.cjche.2019.01.005.
- K. Yamamoto, T. Kanno, X. X. Wang, M. Tamaki, T. Fujii, S. S. Chee, X. W. Wang, V. Pimenov and V. Shako, RSC Adv., 2017, 7, 5554–5577 RSC.
- Y. Bai and Q. Li, Sci. China: Technol. Sci., 2010, 53, 2469–2476 CrossRef CAS.
- V. C. Nair, S. K. Prasad, R. Kumar and J. S. Sangwai, Appl. Energy, 2018, 225, 755–768 CrossRef CAS.
- Y. Wang, X. Sen Li, G. Li, N. S. Huang and J. C. Feng, Fuel, 2014, 117, 688–696 CrossRef CAS.
- J. H. Sira, S. L. Patil and V. A. Kamath, in Proceedings - SPE Annual Technical Conference and Exhibition, Society of Petroleum Engineers, 1990, vol. Pi, pp. 977–984 Search PubMed.
- G. Li, D. Wu, X. Li, Y. Zhang, Q. Lv and Y. Wang, Energy Fuels, 2017, 31, 5411–5418 CrossRef CAS.
- V. Chandrasekharan Nair, D. Mech, P. Gupta and J. S. Sangwai, Energy Fuels, 2018, 32, 6657–6668 CrossRef CAS.
- S. Merey, R. I. Al-Raoush, J. Jung and K. A. Alshibli, J. Pet. Sci. Eng., 2018, 171, 48–62 CrossRef CAS.
- A. V. Palodkar and A. K. Jana, Sci. Rep., 2018, 8, 16563 CrossRef PubMed.
- J. L. Panter, A. L. Ballard, A. K. Sum, E. D. Sloan and C. A. Koh, Energy Fuels, 2011, 25, 2572–2578 CrossRef CAS.
- A. Okwananke, J. Yang, B. Tohidi, E. Chuvilin, V. Istomin, B. Bukhanov and A. Cheremisin, J. Chem. Thermodyn., 2018, 117, 138–146 CrossRef CAS.
- L. Zhang, Y. Kuang, X. Zhang, Y. Song, Y. Liu and J. Zhao, Ind. Eng. Chem. Res., 2017, 56, 7585–7592 CrossRef CAS.
- K. Rose, R. Boswell and T. Collett, Mar. Pet. Geol., 2011, 28, 311–331 CrossRef.
- J. Ye, X. Qin, H. Qiu, N. Wu, H. Lu, W. Xie, J. Lu, C. Lu, Z. Kuang, Q. Liang, H. Lu, B. Kou and J. Wei, China Geol., 2018, 1, 1–5 Search PubMed.
- R. Boswell, D. Schoderbek, T. S. Collett, S. Ohtsuki, M. White and B. J. Anderson, Energy Fuels, 2017, 31, 140–153 CrossRef CAS.
- S. H. Hancock, T. S. Collett, S. R. Dallimore, T. Satoh, T. Inoue, E. Huenges, J. Henninges and B. Weatherill, Geol. Surv. Can., Bull., 2005, 585, 1–15 Search PubMed.
- C. Ruppel, MITEI Nat. gas Report, Suppl. Pap. Methane Hydrates 4, 2011, 4, 1–25.
- K. A. Kvenvolden, Global Biogeochem. Cycles, 1988, 2, 221–229 CrossRef CAS.
- K. A. Kvenvolden, Rev. Geophys., 1993, 31, 173–187 CrossRef.
- G. J. F. MacDonald, Clim. Change, 1990, 16, 247 CrossRef.
- J. Bohannon, Science, 2008, 319(5871), 1753 CrossRef CAS PubMed.
- V. Krey, J. G. Canadell, N. Nakicenovic, Y. Abe, H. Andruleit, D. Archer, A. Grubler, N. T. M. Hamilton, A. Johnson, V. Kostov, J.-F. Lamarque, N. Langhorne, E. G. Nisbet, B. O’Neill, K. Riahi, M. Riedel, W. Wang and V. Yakushev, Environ. Res. Lett., 2009, 4, 34007 CrossRef.
- A. Mascarelli, A sleeping giant?, Nat. Clim. Change, 2009, 46–49 CrossRef.
- G. Whiteman, C. Hope and P. Wadhams, Nature, 2013, 499, 401–403 CrossRef CAS PubMed.
- J. P. Kennett and L. D. Stott, Nature, 1991, 353, 225–229 CrossRef.
- G. R. Dickens, J. R. O’Neil, D. K. Rea and R. M. Owen, Paleoceanography, 1995, 10, 965–971 CrossRef.
- G. R. Dickens, M. M. Castillo and J. C. G. Walker, Geology, 1997, 25, 259–262 CrossRef CAS.
- J. P. Kennett, K. G. Cannariato, I. L. Hendy and R. J. Behl, Science, 2000, 288, 128–133 CrossRef CAS PubMed.
- J. P. Kennett, K. G. Cannariato, I. L. Hendy and R. J. Behl, Methane hydrates in Quaternary climate change: The clathrate gun hypothesis, American Geophysical Union, Washington, D.C., 2002 Search PubMed.
- K. G. Cannariato and L. D. Stott, Geochem., Geophys., Geosyst., 2004, 5(5) DOI:10.1029/2003GC000600.
- P. Serov, S. Vadakkepuliyambatta, J. Mienert, H. Patton, A. Portnov, A. Silyakova, G. Panieri, M. L. Carroll, J. Carroll and K. Andreassen, Proc. Natl. Acad. Sci. U. S. A., 2017, 201619288 Search PubMed.
- R. E. Fisher, S. Sriskantharajah, D. Lowry, M. Lanoisellé, C. M. R. Fowler, R. H. James, O. Hermansen, C. Lund Myhre, A. Stohl, J. Greinert, P. B. R. Nisbet-Jones, J. Mienert and E. G. Nisbet, Geophys. Res. Lett., 2011, 38, L21803, DOI:10.1029/2011GL049319.
- C. A. Graves, L. Steinle, G. Rehder, H. Niemann, D. P. Connelly, D. Lowry, R. E. Fisher, A. W. Stott, H. Sahling and R. H. James, J. Geophys. Res.: Oceans, 2015, 120, 6185–6201 Search PubMed.
- J. W. Pohlman, J. Greinert, C. Ruppel, A. Silyakova, L. Vielstädte, M. Casso, J. Mienert and S. Bünz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5355–5360 CrossRef CAS PubMed.
- K. J. Sparrow, J. D. Kessler, J. R. Southon, F. Garcia-Tigreros, K. M. Schreiner, C. D. Ruppel, J. B. Miller, S. J. Lehman and X. Xu, Sci. Adv., 2018, 4, eaao4842 CrossRef PubMed.
- B. F. Thornton, M. C. Geibel, P. M. Crill, C. Humborg and C.-M. Mörth, Geophys. Res. Lett., 2016, 43, 5869–5877 CrossRef CAS.
- N. Shakhova, I. Semiletov, I. Leifer, V. Sergienko, A. Salyuk, D. Kosmach, D. Chernykh, C. Stubbs, D. Nicolsky and V. Tumskoy, Nat. Geosci., 2014, 7, 64 CrossRef CAS.
- T. F. Stocker, D. Qin, G. K. Plattner, M. M. B. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Climate change 2013 the physical science basis: Working Group I contribution to the fifth assessment report of the intergovernmental panel on climate change, Cambridge University Press, 2013, vol. 9781107057 Search PubMed.
- R. H. James, P. Bousquet, I. Bussmann, M. Haeckel, R. Kipfer, I. Leifer, H. Niemann, I. Ostrovsky, J. Piskozub, G. Rehder, T. Treude, L. Vielstädte and J. Greinert, Limnol. Oceanogr., 2016, 61, S283–S299 CrossRef CAS.
- C. D. Ruppel, Nat. Educ. Knowl., 2011, 3, 29 Search PubMed.
- A. E. Taylor, S. R. Dallimore, P. R. Hill, D. R. Issler, S. Blasco and F. Wright, J. Geophys. Res.: Earth Surf., 2013, 118, 2365–2379 CrossRef.
- W. S. Reeburgh, Chem. Rev., 2007, 107, 486–513 CrossRef CAS PubMed.
- K. Knittel and A. Boetius, Annu. Rev. Microbiol., 2009, 63, 311–334 CrossRef CAS PubMed.
- D. F. McGinnis, J. Greinert, Y. Artemov, S. E. Beaubien and A. Wuest, J. Geophys. Res., 2006, 111, C09007 CrossRef.
- G. Rehder, P. W. Brewer, E. T. Peltzer and G. Friederich, Geophys. Res. Lett., 2002, 29, 21–24 CrossRef.
- G. Rehder, I. Leifer, P. G. Brewer, G. Friederich and E. T. Peltzer, Mar. Chem., 2009, 114, 19–30 CrossRef CAS.
- B. Wang, S. A. Socolofsky, J. A. Breier and J. S. Seewald, J. Geophys. Res.: Oceans, 2016, 121, 2203–2230 Search PubMed.
- B. B. Ward, K. A. Kilpatrick, P. C. Novelli and M. I. Scranton, Nature, 1987, 327, 226–229 CrossRef CAS.
- S. Mau, D. L. Valentine, J. F. Clark, J. Reed, R. Camilli and L. Washburn, Geophys. Res. Lett., 2007, 34(22), L22603, DOI:10.1029/2007GL031344.
- F. Garcia-Tigreros and J. D. Kessler, J. Geophys. Res.: Biogeosci., 2018, 123, 2135–2144 CrossRef CAS.
- R. J. Davies, J. Yang, A. Li, S. Mathias and R. Hobbs, Earth Planet. Sci. Lett., 2015, 423, 202–209 CrossRef CAS.
- A. R. Gorman and K. Senger, J. Geophys. Res.: Solid Earth, 2010, 115, B07105, DOI:10.1029/2009JB006680.
- B. J. Phrampus and M. J. Hornbach, Nature, 2012, 490, 527–530 CrossRef CAS PubMed.
- B. J. Phrampus, M. J. Hornbach, C. D. Ruppel and P. E. Hart, J. Geophys. Res.: Solid Earth, 2014, 119, 8594–8609 CrossRef CAS.
- S. L. Hautala, E. A. Solomon, H. P. Johnson, R. N. Harris and U. K. Miller, Geophys. Res. Lett., 2014, 41, 8486–8494 CrossRef CAS.
- H. P. Johnson, U. K. Miller, M. S. Salmi and E. A. Solomon, Geochem., Geophys., Geosyst., 2015, 16, 3825–3839 CrossRef CAS.
- I. R. MacDonald, N. L. Guinasso, R. Sassen, J. M. Brooks, L. Lee and K. T. Scott, Geology, 1994, 22, 699–702 CrossRef CAS.
- G. K. Westbrook, K. E. Thatcher, E. J. Rohling, A. M. Piotrowski, H. Pälike, A. H. Osborne, E. G. Nisbet, T. A. Minshull, M. Lanoisellé, R. H. James, V. Hühnerbach, D. Green, R. E. Fisher, A. J. Crocker, A. Chabert, C. Bolton, A. Beszczynska-Möller, C. Berndt and A. Aquilina, Geophys. Res. Lett., 2009, 36, L15608 CrossRef.
- A. Skarke, C. Ruppel, M. Kodis, D. Brothers and E. Lobecker, Nat. Geosci., 2014, 7, 657 CrossRef CAS.
- C. Berndt, T. Feseker, T. Treude, S. Krastel, V. Liebetrau, H. Niemann, V. J. Bertics, I. Dumke, K. Dünnbier, B. Ferré, C. Graves, F. Gross, K. Hissmann, V. Hühnerbach, S. Krause, K. Lieser, J. Schauer and L. Steinle, Science, 2014, 343, 284–287 CrossRef CAS PubMed.
- L. L. Brothers, P. E. Hart and C. D. Ruppel, Geophys. Res. Lett., 2012, 39, L15501, DOI:10.1029/2012GL052222.
- L. L. Brothers, B. M. Herman, P. E. Hart and C. D. Ruppel, Geochem., Geophys., Geosyst., 2016, 17(11), 4354–4365, DOI:10.1002/2016GC006584.
- P. P. Overduin, T. Schneider von Deimling, F. Miesner, M. N. Grigoriev, C. Ruppel, A. Vasiliev, H. Lantuit, B. Juhls and S. Westermann, J. Geophys. Res.: Oceans, 2019, 124, 3490–3507 Search PubMed.
- V. Rachold, D. Y. Bolshiyanov, M. N. Grigoriev, H.-W. Hubberten, R. Junker, V. V. Kunitsky, F. Merker, P. P. Overduin and W. Schneider, EOS, Trans. Am. Geophys. Union, 2007, 88, 149 CrossRef.
- P. Rekant, H. A. Bauch, T. Schwenk, A. Portnov, E. Gusev, V. Spiess, G. Cherkashov and H. Kassens, Arktos, 2015, 1, 11 CrossRef.
- P. Serov, A. Portnov, J. Mienert, P. Semenov and P. Ilatovskaya, J. Geophys. Res.: Earth Surf., 2015, 120, 1515–1529 CrossRef.
- A. Portnov, A. J. Smith, J. Mienert, G. Cherkashov, P. Rekant, P. Semenov, P. Serov and B. Vanshtein, Geophys. Res. Lett., 2013, 40, 3962–3967 CrossRef CAS.
- C. D. Ruppel, B. M. Herman, L. L. Brothers and P. E. Hart, Geochem., Geophys., Geosyst., 2016, 17, 4333–4353 CrossRef.
- K. Andreassen, A. Hubbard, M. Winsborrow, H. Patton, S. Vadakkepuliyambatta, A. Plaza-Faverola, E. Gudlaugsson, P. Serov, A. Deryabin and R. Mattingsdal, Science, 2017, 356, 948–953 CrossRef CAS PubMed.
- A. Portnov, S. Vadakkepuliyambatta, J. Mienert and A. Hubbard, Nat. Commun., 2016, 7, 10314, DOI:10.1038/ncomms10314.
- P. Serov, S. Vadakkepuliyambatta, J. Mienert, H. Patton, A. Portnov, A. Silyakova, G. Panieri, M. L. Carroll, J. L. Carroll, K. Andreassen and A. Hubbard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6215–6220 CrossRef CAS PubMed.
- M. Waage, A. Portnov, P. Serov, S. Bünz, K. A. Waghorn, S. Vadakkepuliyambatta, J. Mienert and K. Andreassen, Geochem., Geophys., Geosyst., 2019, 20, 630–650 CrossRef CAS.
- J. L. Wadham, S. Arndt, S. Tulaczyk, M. Stibal, M. Tranter, J. Telling, G. P. Lis, E. Lawson, A. Ridgwell, A. Dubnick, M. J. Sharp, A. M. Anesio and C. E. H. Butler, Nature, 2012, 488, 633–637 CrossRef CAS PubMed.
- G. Lamarche-Gagnon, J. L. Wadham, B. S. Lollar, S. Arndt, P. Fietzek, A. D. Beaton, A. J. Tedstone, J. Telling, E. A. Bagshaw and J. R. Hawkings, Nature, 2019, 565, 73 CrossRef CAS PubMed.
- R. John Parkes, B. A. Cragg and P. Wellsbury, Hydrogeol. J., 2000, 8, 11–28 CrossRef.
- B. D. Lanoil, R. Sassen, M. T. La Duc, S. T. Sweet and K. H. Nealson, Appl. Environ. Microbiol., 2001, 67, 5143–5153 CrossRef CAS PubMed.
- H. J. Mills, R. J. Martinez, S. Story and P. A. Sobecky, Appl. Environ. Microbiol., 2005, 71, 3235–3247 CrossRef CAS PubMed.
- J. R. Marchesi, A. J. Weightman, B. A. Cragg, R. John Parkes and J. C. Fry, FEMS Microbiol. Ecol., 2001, 34, 221–228 CrossRef CAS PubMed.
- F. Inagaki, T. Nunoura, S. Nakagawa, A. Teske, M. Lever, A. Lauer, M. Suzuki, K. Takai, M. Delwiche, F. S. Colwell, K. H. Nealson, K. Horikoshi, S. D’Hondt and B. B. Jørgensen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2815–2820 CrossRef CAS PubMed.
- R. J. Parkes, G. Sellek, G. Webster, D. Martin, E. Anders, A. J. Weightman and H. Sass, Environ. Microbiol., 2009, 11, 3140–3153 CrossRef CAS PubMed.
- L. J. Hamdan, M. Sikaroodi and P. M. Gillevet, Geomicrobiol. J., 2012, 29, 340–351 CrossRef.
- B. R. Briggs, F. Inagaki, Y. Morono, T. Futagami, C. Huguet, A. Rosell-Mele, T. D. Lorenson and F. S. Colwell, FEMS Microbiol. Ecol., 2012, 81, 88–98 CrossRef CAS PubMed.
- J. C. Fry, G. Webster, B. A. Cragg, A. J. Weightman and R. J. Parkes, FEMS Microbiol. Ecol., 2006, 58, 86–98 CrossRef CAS PubMed.
- J.-W. Lee, K. K. Kwon, A. Azizi, H.-M. Oh, W. Kim, J.-J. Bahk, D.-H. Lee and J.-H. Lee, Mar. Pet. Geol., 2013, 47, 136–146 CrossRef CAS.
- L. Jiao, X. Su, Y. Wang, H. Jiang, Y. Zhang and F. Chen, Geosci. Front., 2015, 6, 627–633 CrossRef.
- H. Lei, Y. Yang, S. Kandasamy and C. Shi, Acta Geol. Sin. Ed., 2018, 92, 333–341 CrossRef CAS.
- H. Cui, X. Su, F. Chen, M. Holland, S. Yang, J. Liang, P. Su, H. Dong and W. Hou, Mar. Environ. Res., 2019, 144, 230–239 CrossRef CAS PubMed.
- M. D. Max, Natural gas hydrate in oceanic and permafrost environments, Springer Science & Business Media, 2003, vol. 5 Search PubMed.
- R. K. Thauer, A. K. Kaster, H. Seedorf, W. Buckel and R. Hedderich, Nat. Rev. Microbiol., 2008, 6, 579–591 CrossRef CAS PubMed.
- K. U. Hinrichs, J. M. Hayes, W. Bach, A. J. Spivackl, L. R. Hmelo, N. G. Holm, C. G. Johnson and S. P. Sylva, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14684–14689 CrossRef CAS PubMed.
- R. Conrad, Environ. Microbiol. Rep., 2009, 1, 285–292 CrossRef CAS PubMed.
- K. Knittel, G. Wegener and A. Boetius, Microbial Communities Utilizing Hydrocarbons and Lipids: Members, Metagenomics and Ecophysiology, 2019, pp. 1–21 Search PubMed.
- P. J. Talling, M. L. Clare, M. Urlaub, E. Pope, J. E. Hunt and S. F. L. Watt, Oceanography, 2014, 27, 32–45 CrossRef.
- M. J. Hornbach, L. L. Lavier and C. D. Ruppel, Geochem., Geophys., Geosyst., 2007, 8(12) DOI:10.1029/2007GC001722.
- J. C. Hill, D. S. Brothers, M. J. Hornbach, D. E. Sawyer, D. J. Shillington and A. Bécel, Geol. Soc. London, Spec. Publ., 2019, 477, 169–181 CrossRef.
- D. E. Smith, S. Harrison and J. T. Jordan, Quat. Sci. Rev., 2013, 82, 93–103 CrossRef.
- E. L. Soutter, I. A. Kane and M. Huuse, Geology, 2018, 46, 511–514 CrossRef CAS.
- B. Dugan and P. B. Flemings, Science, 2000, 289, 288–291 CrossRef CAS PubMed.
- R. E. Kayen and H. J. Lee, Mar. Geotechnol., 1991, 10, 125–141 CrossRef.
- J. Mienert, J. Posewang and M. Baumann, Geol. Soc. London, Spec. Publ., 1998, 137, 275–291 CrossRef CAS.
- J. J. Mountjoy, I. Pecher, S. Henrys, G. Crutchley, P. M. Barnes and A. Plaza-Faverola, Geochem., Geophys., Geosyst., 2014, 15, 4137–4156 CrossRef CAS.
- S. Yelisetti, G. D. Spence and M. Riedel, Geophys. J. Int., 2014, 199, 441–458 CrossRef.
- D. E. Sawyer, R. A. Mason, A. E. Cook and A. Portnov, Sci. Rep., 2019, 9, 128 CrossRef PubMed.
- X. Wu, Q. Liang, Y. Ma, Y. Shi, Z. Xia, L. Liu and M. Haeckel, Energies, 2018, 11 CAS.
- S. Bünz, J. Mienert, M. Vanneste and K. Andreassen, Geophysics, 2005, 70, B19–B34 CrossRef.
- J. Mienert, M. Vanneste, S. Bünz, K. Andreassen, H. Haflidason and H. P. Sejrup, Mar. Pet. Geol., 2005, 22, 233–244 CrossRef CAS.
- H. Haflidason, R. Lien, H. Sejrup, C. Forsberg and P. Bryn, Mar. Pet. Geol., 2005, 22, 123–136 CrossRef.
- M. Hovland, J. V. Gardner and A. G. Judd, Geofluids, 2002, 2, 127–136 CrossRef.
- N. Sultan, G. Bohrmann, L. Ruffine, T. Pape, V. Riboulot, J.-L. Colliat, A. De Prunelé, B. Dennielou, S. Garziglia, T. Himmler, T. Marsset, C. A. Peters, A. Rabiu and J. Wei, J. Geophys. Res. Solid Earth, 2014, 119, 2679–2694 CrossRef CAS.
- W. J. Winters, I. A. Pecher, W. F. Waite and D. H. Mason, Am. Mineral., 2004, 89, 1221–1227 CrossRef CAS.
- T. S. Yun, C. J. Santamarina and C. Ruppel, J. Geophys. Res. Solid Earth, 2007, 12, B04106, DOI:10.1029/2006JB004484.
- H. Daigle and B. Dugan, J. Geophys. Res. Solid Earth, 2010, 115, B11103 CrossRef.
- J. Elger, C. Berndt, L. Rüpke, S. Krastel, F. Gross and W. H. Geissler, Nat. Commun., 2018, 9, 715 CrossRef PubMed.
- W. Xu and L. N. Germanovich, J. Geophys. Res. Solid Earth, 2006, 111, B01104, DOI:10.1029/2004JB003600.
- J. L. H. Grozic, Submarine mass movements and their consequences, Springer, 2010, pp. 11–30 Search PubMed.
- J. L. H. Grozic and H. Ghiassian, in Proc., 63rd Canadian Geotechnical Conf, 2010, pp. 459–466.
- J. S. Booth, D. W. O’leary, P. Popenoe, W. W. Danforth and W. C. Schwab, in Submarine Landslides Selected Studies in the US Exclusive Economic Zone, ed. W. C. Schwab, H. J. Lee and D. C. Twichell, 1993, pp. 14–22 Search PubMed.
- P. Popenoe, E. A. Schmuck, W. P. Dillon, W. C. Schwab, H. J. Lee and D. C. Twichell, Submarine Landslides Selected Studies in the US Exclusive Economic Zone, 1993, pp. 40–53 Search PubMed.
- M. F. Nixon and J. L. H. Grozic, Can. Geotech. J., 2007, 44, 314–325 CrossRef CAS.
- T.-H. Kwon and G.-C. Cho, Energies, 2012, 5 Search PubMed.
- J. P. Kennett, K. G. Cannariato, I. L. Hendy and R. J. Behl, Methane hydrates in quaternary climate change: the clathrate gun hypothesis, 2003, vol. 54, pp. 1–9 Search PubMed.
- A. Dawson, S. Bondevik and J. T. Teller, The Holocene, 2011, 21, 1167–1171 CrossRef.
- M. Maslin, M. Owen, S. Day and D. Long, Geology, 2004, 32, 53–56 CrossRef CAS.
- K. Andreassen, A. Hubbard, M. Winsborrow, H. Patton, S. Vadakkepuliyambatta, A. Plaza-Faverola, E. Gudlaugsson, P. Serov, A. Deryabin, R. Mattingsdal, J. Mienert and S. Bünz, Science, 2017, 356, 948–953 CrossRef CAS PubMed.
- M. Hovland and H. Svensen, Mar. Geol., 2006, 228, 15–23 CrossRef.
- R. Freij-Ayoub, C. Tan, B. Clennell, B. Tohidi and J. Yang, J. Pet. Sci. Eng., 2007, 57, 209–220 CrossRef CAS.
- M. Hovland and O. T. Gudmestad, Nat. Gas Hydrates Occur. Distrib. Detect., 2001, 307–315 Search PubMed.
- C. Hadley, D. Peters, A. Vaughan and D. Bean, in International Petroleum Technology Conference, International Petroleum Technology Conference, 2008.
- T. Kvalstad, K. Yamamoto, S. Noguchi, S. Uchida and K. Soga, Effect of gas hydrate production on seabed stability in the Eastern Nankai Trough area, 2011 Search PubMed.
- H. Zhang, X. Luo, J. Bi, G. He and Z. Guo, Mar. Georesour. Geotechnol., 2019, 37, 467–476 CrossRef CAS.
- J. I. Lunine and D. J. Stevenson, Astrophys. J. Suppl. Ser., 1985, 58, 493 CrossRef CAS.
- J. S. Kargel and J. I. Lunine, Clathrate Hydrates on Earth and in the Solar System, in Solar System Ices, ed. B. Schmitt, C. De Bergh and M. Festou, Springer Netherlands, Dordrecht, 1998, pp. 97–117 Search PubMed.
- O. Mousis, J. I. Lunine, C. Thomas, M. Pasek, U. Marbœuf, Y. Alibert, V. Ballenegger, D. Cordier, Y. Ellinger, F. Pauzat and S. Picaud, Astrophys. J., 2009, 691, 1780–1786 CrossRef CAS.
- O. Mousis, E. Chassefière, N. G. Holm, A. Bouquet, J. H. Waite, W. D. Geppert, S. Picaud, Y. Aikawa, M. Ali-Dib, J. L. Charlou and P. Rousselot, Astrobiology, 2015, 15, 308–326 CrossRef CAS PubMed.
- R. W. Carlson, M. S. Anderson, R. Mehlman and R. E. Johnson, Icarus, 2005, 177, 461–471 CrossRef CAS.
- S. H. Royle, W. Montgomery, S. P. Kounaves and M. A. Sephton, J. Geophys. Res.: Planets, 2017, 122, 2793–2802 CAS.
- S. L. Miller, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1798–1808 CrossRef CAS PubMed.
- I.-M. Chou, A. Sharma, R. C. Burruss, R. J. Hemley, A. F. Goncharov, L. A. Stern and S. H. Kirby, J. Phys. Chem. A, 2001, 105, 4664–4668 CrossRef CAS.
- M. Choukroun, S. W. Kieffer, X. Lu and G. Tobie, Clathrate Hydrates: Implications for Exchange Processes in the Outer Solar System, in The Science of Solar System Ices, ed. M. S. Gudipati and J. Castillo-Rogez, Astrophysics and Space Science Library, Springer New York, New York, NY, 2013, pp. 409–454 Search PubMed.
- A. Dobrovolskis and A. P. Ingersoll, Icarus, 1975, 26, 353–357 CrossRef CAS.
- S. L. Miller and W. D. Smythe, Science, 1970, 170, 531–533 CrossRef CAS PubMed.
- O. Mousis, E. Chassefière, J. Lasue, V. Chevrier, M. E. Elwood Madden, A. Lakhlifi, J. I. Lunine, F. Montmessin, S. Picaud, F. Schmidt and T. D. Swindle, Space Sci. Rev., 2013, 174, 213–250 CrossRef CAS.
- R. E. Pellenbarg, M. D. Max and S. M. Clifford, J. Geophys. Res.: Planets, 2003, 108(E4), 8042, DOI:10.1029/2002JE001901.
- A. G. Davies, C. Sotin, M. Choukroun, D. L. Matson and T. V. Johnson, Icarus, 2016, 274, 23–32 CrossRef CAS.
- J. P. Osegovic and M. D. Max, J. Geophys. Res.: Planets, 2005, 110, E08004, DOI:10.1029/2005JE002435.
- R. R. Fu, A. I. Ermakov, S. Marchi, J. C. Castillo-Rogez, C. A. Raymond, B. H. Hager, M. T. Zuber, S. D. King, M. T. Bland, M. Cristina De Sanctis, F. Preusker, R. S. Park and C. T. Russell, Earth Planet. Sci. Lett., 2017, 476, 153–164 CrossRef CAS.
- S. Kamata, F. Nimmo, Y. Sekine, K. Kuramoto, N. Noguchi, J. Kimura and A. Tani, Nat. Geosci., 2019, 12, 407–410 CrossRef CAS.
- K. Shin, R. Kumar, K. A. Udachin, S. Alavi and J. A. Ripmeester, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14785–14790 CrossRef CAS PubMed.
- U. Marboeuf, O. Mousis, J.-M. Petit, B. Schmitt, A. L. Cochran and H. A. Weaver, Astron. Astrophys., 2010, 525 DOI:10.1051/0004-6361/201015438.
- A. Luspay-Kuti, O. Mousis, M. Hässig, S. A. Fuselier, J. I. Lunine, B. Marty, K. E. Mandt, P. Wurz and M. Rubin, Sci. Adv., 2016, 2, e1501781 CrossRef PubMed.
- J. Ghosh, R. R. J. Methikkalam, R. G. Bhuin, G. Ragupathy, N. Choudhary, R. Kumar and T. Pradeep, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1526–1531 CrossRef CAS PubMed.
- M. Choukroun, T. H. Vu and E. C. Fayolle, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14407–14408 CrossRef CAS PubMed.
- G. J. Flynn, F. Horz, S. Bajt and S. R. Sutton, Lunar and Planetary Science Conference, 1996, vol. 27.
- M. Fries, A. Christou, D. Archer, P. Conrad, W. Cooke, J. Eigenbrode, I. L. ten Kate, M. Matney, P. Niles, M. Sykes, A. Steele and A. Treiman, Geochemical Perspect. Lett., 2016, 2, 10–23 CrossRef.
- M. J. Russell, A. E. Murray and K. P. Hand, Astrobiology, 2017, 17, 1265–1273 CrossRef CAS PubMed.
- K. P. Hand, C. F. Chyba, J. C. Priscu, R. W. Carlson and K. H. Nealson, Europa, 2009, 589–629 Search PubMed.
- IPCC, IPCC report Global Warming of 1.5 C: Summary for Policymakers, 2018.
- J. Rogelj, A. Popp, K. V. Calvin, G. Luderer, J. Emmerling, D. Gernaat, S. Fujimori, J. Strefler, T. Hasegawa, G. Marangoni, V. Krey, E. Kriegler, K. Riahi, D. P. Van Vuuren, J. Doelman, L. Drouet, J. Edmonds, O. Fricko, M. Harmsen, P. Havlík, F. Humpenöder, E. Stehfest and M. Tavoni, Nat. Clim. Change, 2018, 8, 325–332 CrossRef CAS.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- S. Budinis, S. Krevor, N. Mac Dowell, N. Brandon and A. Hawkes, Energy Strateg. Rev., 2018, 22, 61–81 CrossRef.
- P. Babu, P. Linga, R. Kumar and P. Englezos, Energy, 2015, 85, 261–279 CrossRef CAS.
- E. D. Sloan Jr. and C. A. Koh, Clathrate Hydrates of Natural Gases, CRC Press, 3rd edn, 2007 Search PubMed.
- B. Chazallon and C. Pirim, Chem. Eng. J., 2018, 342, 171–183 CrossRef CAS.
- A. Hassanpouryouzband, J. Yang, B. Tohidi, E. Chuvilin, V. Istomin and B. Bukhanov, ACS Sustainable Chem. Eng., 2019, 7, 5338–5345 CrossRef CAS.
- A. Hassanpouryouzband, J. Yang, B. Tohidi, E. Chuvilin, V. Istomin, B. Bukhanov and A. Cheremisin, ACS Sustainable Chem. Eng., 2018, 6, 5732–5736 CrossRef CAS.
- S. Adisasmito, R. J. Frank and E. D. Sloan, J. Chem. Eng. Data, 1991, 36, 68–71 CrossRef CAS.
- H. Oyama, W. Shimada, T. Ebinuma, Y. Kamata, S. Takeya, T. Uchida, J. Nagao and H. Narita, Fluid Phase Equilib., 2005, 234, 131–135 CrossRef CAS.
- N. Mayoufi, D. Dalmazzone, A. Delahaye, P. Clain, L. Fournaison and W. Fürst, J. Chem. Eng. Data, 2011, 56, 2987–2993 CrossRef CAS.
- Y. Zhang, S. M. Sheng, X. D. Shen, X. B. Zhou, W. Z. Wu, X. P. Wu and D. Q. Liang, J. Chem. Eng. Data, 2017, 62, 2461–2465 CrossRef CAS.
- J. N. Zheng and M. Yang, J. Chem. Eng. Data, 2019, 64, 372–379 CrossRef CAS.
- R. Mcmullan and G. A. Jeffrey, J. Chem. Phys., 1959, 31, 1231–1234 CrossRef CAS.
- H. Dashti, L. Zhehao Yew and X. Lou, J. Nat. Gas Sci. Eng., 2015, 23, 195–207 CrossRef CAS.
- Z. W. Ma, P. Zhang, H. S. Bao and S. Deng, Renewable Sustainable Energy Rev., 2016, 53, 1273–1302 CrossRef CAS.
- R. Kumar, P. Englezos, I. Moudrakovski and J. A. Ripmeester, AIChE J., 2009, 55, 1548–1594 CrossRef.
- S. Kim, S. D. Choi and Y. Seo, Energy, 2017, 118, 950–956 CrossRef CAS.
- Z. Xia, Z. Li, X. Li, Z. Chen, G. Li and Y. Wang, Energy Procedia, 2019, 158, 5850–5855 CrossRef CAS.
- S. Muromachi, S. Takeya, Y. Yamamoto and R. Ohmura, CrystEngComm, 2014, 16, 2056–2060 RSC.
- T. Kobori, S. Muromachi, T. Yamasaki, S. Takeya, Y. Yamamoto, S. Alavi and R. Ohmura, Cryst. Growth Des., 2015, 15, 3862–3867 CrossRef CAS.
- W. Shimada, M. Shiro, H. Kondo, S. Takeya, H. Oyama, T. Ebinuma and H. Marita, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, 65–66 CrossRef.
- H. Hashimoto, T. Yamaguchi, H. Ozeki and S. Muromachi, Sci. Rep., 2017, 7, 17216 CrossRef PubMed.
- P. Linga, R. Kumar and P. Englezos, Chem. Eng. Sci., 2007, 62, 4268–4276 CrossRef CAS.
- H. P. Veluswamy, K. P. Premasinghe and P. Linga, Energy Procedia, 2017, 105, 5048–5054 CrossRef.
- J. Lim, W. Choi, J. Mok and Y. Seo, Chem. Eng. J., 2019, 369, 686–693 CrossRef CAS.
- C. T. Rodriguez, Q. Du Le, C. Focsa, C. Pirim and B. Chazallon, Chem. Eng. J., 2020, 382, 122867 CrossRef.
- U. Karaaslan and M. Parlaktuna, Energy Fuels, 2000, 14, 1103–1107 CrossRef CAS.
- H. P. Veluswamy, P. Y. Lee, K. Premasinghe and P. Linga, Ind. Eng. Chem. Res., 2017, 56, 6145–6154 CrossRef CAS.
- J. Zheng, K. Bhatnagar, M. Khurana, P. Zhang, B. Y. Zhang and P. Linga, Appl. Energy, 2018, 217, 377–389 CrossRef CAS.
- C. Lo, J. S. ZHang, P. Somasundaran, S. Lu, A. Couzis and J. W. Lee, Langmuir, 2008, 24, 12723–12726 CrossRef CAS PubMed.
- C. Lo, J. Zhang, P. Somasundaran and J. W. Lee, J. Phys. Chem. Lett., 2010, 1, 2676–2679 CrossRef CAS.
- L. Zhang, Y. Kuang, S. Dai, J. Wang, J. Zhao and Y. Song, Chem. Eng. J., 2020, 379, 122357 CrossRef CAS.
- P. Di Profio, V. Canale, N. D’Alessandro, R. Germani, A. Di Crescenzo and A. Fontana, ACS Sustainable Chem. Eng., 2017, 5, 1990–1997 CrossRef CAS.
- J. Yi, D. L. Zhong, J. Yan and Y. Y. Lu, Energy, 2019, 171, 61–68 CrossRef CAS.
- M. Ricaurte, C. Dicharry, D. Broseta, X. Renaud and J. P. Torré, Ind. Eng. Chem. Res., 2013, 52, 899–910 CrossRef CAS.
- T. J. Kneafsey, L. Tomutsa, G. J. Moridis, Y. Seol, B. M. Freifeld, C. E. Taylor and A. Gupta, J. Pet. Sci. Eng., 2007, 56, 108–126 CrossRef CAS.
- A. Adeyemo, R. Kumar, P. Linga, J. Ripmeester and P. Englezos, Int. J. Greenhouse Gas Control, 2010, 4, 478–485 CrossRef CAS.
- Y. T. Seo, I. L. Moudrakovski, J. A. Ripmeester, J. W. Lee and H. Lee, Environ. Sci. Technol., 2005, 39, 2315–2319 CrossRef CAS.
- A. Nambiar, P. Babu and P. Linga, Can. J. Chem., 2015, 93, 808–814 CrossRef CAS.
- I. Kim, M. Nole, S. Jang, S. Ko, H. Daigle, G. A. Pope and C. Huh, RSC Adv., 2017, 7, 9545–9550 RSC.
- S. Said, V. Govindaraj, J. M. Herri, Y. Ouabbas, M. Khodja, M. Belloum, J. S. Sangwai and R. Nagarajan, J. Nat. Gas Sci. Eng., 2016, 32, 95–108 CrossRef CAS.
- R. Coupan, F. Plantier, J. P. Torré, C. Dicharry, P. Sénéchal, F. Guerton, P. Moonen, A. Khoukh, S. A. Kessas and M. Hemati, Chem. Eng. J., 2017, 325, 35–48 CrossRef CAS.
- P. S. R. Prasad, Y. Sowjanya and V. Dhanunjana Chari, J. Phys. Chem. C, 2014, 118, 7759–7764 CrossRef CAS.
- L. Yang, S. Fan, Y. Wang, X. Lang and D. Xie, Ind. Eng. Chem. Res., 2011, 50, 11563–11569 CrossRef CAS.
- F. Su, C. L. Bray, B. O. Carter, G. Overend, C. Cropper, J. A. Iggo, Y. Z. Khimyak, A. M. Fogg and A. I. Cooper, Adv. Mater., 2009, 21, 2382–2386 CrossRef CAS.
- B. O. Carter, W. Wang, D. J. Adams and A. I. Cooper, Langmuir, 2010, 26, 3186–3193 CrossRef CAS.
- W. Zhao, J. Bai, J. S. Francisco and X. C. Zeng, J. Phys. Chem. C, 2018, 122, 7951–7958 CrossRef CAS.
- Z. Y. Li, Z. M. Xia, X. Sen Li, Z. Y. Chen, J. Cai, G. Li and T. Lv, Energy Fuels, 2018, 32, 2064–2072 CrossRef CAS.
- Y. He and F. Wang, J. Mater. Chem. A, 2018, 6, 22619–22625 RSC.
- R. T. J. Porter, M. Fairweather, M. Pourkashanian and R. M. Woolley, Int. J. Greenhouse Gas Control, 2015, 36, 161–174 CrossRef CAS.
- A. Chapoy, R. Burgass, B. Tohidi and I. Alsiyabi, J. Chem. Eng. Data, 2015, 60, 447–463 CrossRef CAS.
- M. Nohra, T. K. Woo, S. Alavi and J. A. Ripmeester, J. Chem. Thermodyn., 2012, 44, 5–12 CrossRef CAS.
- B. Kvamme, E. Iden, J. Tveit, V. Veland, M. Zarifi and K. Qorbani, J. Chem. Eng. Data, 2017, 62, 1645–1658 CrossRef CAS.
- N. Daraboina, J. Ripmeester and P. Englezos, Int. J. Greenhouse Gas Control, 2013, 15, 97–103 CrossRef CAS.
- C. Y. Sun, G. J. Chen, W. Lin and T. M. Guo, J. Chem. Eng. Data, 2003, 48, 600–602 CrossRef CAS.
- Z. T. Ward, C. E. Deering, R. A. Marriott, A. K. Sum, E. D. Sloan and C. A. Koh, J. Chem. Eng. Data, 2015, 60, 403–408 CrossRef CAS.
- B. Beeskow-Strauch, J. M. Schicks, E. Spangenberg and J. Erzinger, Chem. – Eur. J., 2011, 17, 4376–4384 CrossRef CAS PubMed.
- A. Chapoy, M. Nazeri, M. Kapateh, R. Burgass, C. Coquelet and B. Tohidi, Int. J. Greenhouse Gas Control, 2013, 19, 92–100 CrossRef CAS.
- X. Zang and D. Liang, J. Chem. Eng. Data, 2018, 63, 197–201 CrossRef CAS.
- B. Kvamme, T. Kuznetsova, B. Jensen, S. Stensholt, J. Bauman, S. Sjøblom and K. Nes Lervik, Phys. Chem. Chem. Phys., 2014, 16, 8623–8638 RSC.
- F. Lang and P. Servio, J. Chem. Eng. Data, 2014, 59, 2547–2550 CrossRef CAS.
- A. Hassanpouryouzband, M. V. Farahani, J. Yang, B. Tohidi, E. Chuvilin, V. Istomin and B. Bukhanov, Ind. Eng. Chem. Res., 2019, 58, 3377–3394 CrossRef CAS.
- A. Burnol, I. Thinon, L. Ruffine and J. M. Herri, Int. J. Greenhouse Gas Control, 2015, 35, 96–109 CrossRef CAS.
- F. M. Orr, JPT, J. Pet. Technol., 2004, 56, 90–97 CrossRef CAS.
- F. M. Orr Jr., Science, 2009, 325, 1656–1658 CrossRef PubMed.
- D. P. Schrag, Science, 2009, 325, 1658–1659 CrossRef CAS PubMed.
- Y. Teng and D. Zhang, Sci. Adv., 2018, 4, eaao6588 CrossRef PubMed.
- H. Koide, M. Takahashi, Y. Shindo, Y. Tazaki, M. Iijima, K. Ito, N. Kimura and K. Omata, Energy, 1997, 22, 279–283 CrossRef CAS.
- B. A. Remepel and A. W. Buffett, J. Geophys. Res., 1997, 102, 10151–10164 CrossRef.
- S. Almenningen, J. Gauteplass, L. P. Hauge, T. Barth, M. A. Fernø and G. Ersland, J. Pet. Sci. Eng., 2019, 177, 880–888 CrossRef CAS.
- J. Gauteplass, S. Almenningen, G. Ersland and T. Barth, Int. J. Greenhouse Gas Control, 2018, 78, 21–26 CrossRef CAS.
- X. H. Ta, T. S. Yun, B. Muhunthan and T.-H. Kwon, Geochem., Geophys., Geosyst., 2015, 16, 912–924 CrossRef CAS.
- J. W. Jung and J. C. Santamarina, Geochem., Geophys., Geosyst., 2011, 12, 1–9 CrossRef.
- K. Miyazaki, Y. Oikawa, H. Haneda and T. Yamaguchi, Int. J. Offshore Polar Eng., 2016, 26, 315–320 CrossRef CAS.
- M. Uddin, D. Coombe and F. Wright, J. Energy Resour. Technol., 2008, 130, 032502 CrossRef.
- O. Y. Zatsepina and M. Pooladi-Darvish, SPE Reserv. Eval. Eng., 2012, 15, 98–108 CrossRef CAS.
- O. Y. Zatsepina and B. A. Buffett, Fluid Phase Equilib., 2002, 200, 263–275 CrossRef CAS.
- D. Y. Koh, H. Kang, D. O. Kim, J. Park, M. Cha and H. Lee, ChemSusChem, 2012, 5, 1443–1448 CrossRef CAS PubMed.
- P. Mekala, M. Busch, D. Mech, R. S. Patel and J. S. Sangwai, J. Pet. Sci. Eng., 2014, 122, 1–9 CrossRef CAS.
- K. Lee, S. H. Lee and W. Lee, Int. J. Greenhouse Gas Control, 2013, 14, 15–24 CrossRef CAS.
- Z. He, P. Linga and J. Jiang, Phys. Chem. Chem. Phys., 2017, 19, 15657–15661 RSC.
- C. Holzammer, J. M. Schicks, S. Will and A. S. Braeuer, J. Phys. Chem. B, 2017, 121, 8330–8337 CrossRef CAS PubMed.
- D. Sun and P. Englezos, J. Nat. Gas Sci. Eng., 2016, 35, 1416–1425 CrossRef CAS.
- M. Massah, D. Sun, H. Sharifi and P. Englezos, J. Chem. Thermodyn., 2018, 117, 106–112 CrossRef CAS.
- D. Sun and P. Englezos, Int. J. Greenhouse Gas Control, 2014, 25, 1–8 CrossRef CAS.
- D. Sun and P. Englezos, Can. J. Chem. Eng., 2017, 95, 69–76 CrossRef CAS.
- K. Ohgaki, K. Takano, H. Sangawa, T. Matsubara and S. Nakano, J. Chem. Eng. Jpn., 1996, 29, 478–483 CrossRef CAS.
- Y. T. Seo and H. Lee, J. Phys. Chem. B, 2001, 105, 10084–10090 CrossRef CAS.
- E. M. Yezdimer, P. T. Cummings and A. A. Chialvo, J. Phys. Chem. A, 2002, 106, 7982–7987 CrossRef CAS.
- T. Uchida, I. Y. Ikeda, S. Takeya, Y. Kamata, R. Ohmura, J. Nagao, O. Y. Zatsepina and B. A. Buffett, ChemPhysChem, 2005, 6, 646–654 CrossRef CAS.
- G. Ersland, J. Husebø, A. Graue, B. A. Baldwin, J. Howard and J. Stevens, Chem. Eng. J., 2010, 158, 25–31 CrossRef CAS.
- A. Graue, B. Kvamme, B. Baldwin, J. Stevens, J. J. Howard, E. Aspenes, G. Ersland, J. Husebo and D. Zornes, SPE J., 2008, 13, 146–152 CrossRef CAS.
- M. Ota, Y. Abe, M. Watanabe, R. L. Smith and H. Inomata, Fluid Phase Equilib., 2005, 228–229, 553–559 CrossRef CAS.
- S. Lee, S. Park, Y. Lee and Y. Seo, Chem. Eng. J., 2013, 225, 636–640 CrossRef CAS.
- Q. Yuan, C. Y. Sun, B. Liu, X. Wang, Z. W. Ma, Q. L. Ma, L. Y. Yang, G. J. Chen, Q. P. Li, S. Li and K. Zhang, Energy Convers. Manage., 2013, 67, 257–264 CrossRef CAS.
- M. Ota, T. Saito, T. Aida, M. Watanabe, Y. Sato, R. L. Smith and H. Inomata, AIChE J., 2007, 53, 2715–2721 CrossRef CAS.
- M. Ota, K. Morohashi, Y. Abe, M. Watanabe, R. Lee Smith and H. Inomata, Energy Convers. Manage., 2005, 46, 1680–1691 CrossRef CAS.
- Y. Qi, M. Ota and H. Zhang, Energy Convers. Manage., 2011, 52, 2682–2687 CrossRef CAS.
- N. I. Papadimitriou, I. N. Tsimpanogiannis, I. G. Economou and A. K. Stubos, Phys. Chem. Chem. Phys., 2018, 20, 28026–28038 RSC.
- K. Shin, Y. Park, M. Cha, K. P. Park, D. G. Huh, J. Lee, S. J. Kim and H. Lee, Energy Fuels, 2008, 22, 3160–3163 CrossRef CAS.
- Y. Lee, W. Choi, K. Shin and Y. Seo, Energy Convers. Manage., 2017, 150, 356–364 CrossRef CAS.
- Y. Lee, Y. Ju Seo, T. Ahn, J. Lee, J. Y. Lee, S. J. Kim and Y. Seo, Chem. Eng. J., 2017, 308, 50–58 CrossRef CAS.
- J. M. Schicks, M. Luzi and B. Beeskow-Strauch, J. Phys. Chem. A, 2011, 115, 13324–13331 CrossRef CAS PubMed.
- J. Yang, A. Chapoy, B. Tohidi, P. S. Jadhawar and J. Lee, OTC, 2008.
- J. H. Yoon, T. Kawamura, Y. Yamamoto and T. Komai, J. Phys. Chem. A, 2004, 108, 5057–5059 CrossRef CAS.
- J. W. Jung and J. C. Santamarina, Geochem., Geophys., Geosyst., 2010, 11, 1–8 CrossRef.
- L. Zhang, L. Yang, J. Wang, J. Zhao, H. Dong, M. Yang, Y. Liu and Y. Song, Chem. Eng. J., 2017, 308, 40–49 CrossRef CAS.
- P. L. Stanwix, N. M. Rathnayake, F. P. P. De Obanos, M. L. Johns, Z. M. Aman and E. F. May, Energy Environ. Sci., 2018, 11, 1828–1840 RSC.
- A. M. Gambelli and F. Rossi, Energy, 2019, 172, 423–434 CrossRef CAS.
- H. Kang, D. Y. Koh and H. Lee, Sci. Rep., 2014, 4, 1–8 Search PubMed.
- D. Lim, H. Ro, Y. Seo, Y. Ju Seo, J. Y. Lee, S. J. Kim, J. Lee and H. Lee, J. Chem. Thermodyn., 2017, 106, 16–21 CrossRef CAS.
- S. S. Tupsakhare and M. J. Castaldi, Appl. Energy, 2019, 236, 825–836 CrossRef CAS.
- D. R. Bhawangirkar and J. S. Sangwai, Ind. Eng. Chem. Res., 2019, 58, 14462–14475 CrossRef CAS.
- D. Schoderbek and K. L. Martin, J. Howard, S. Silpngarmlert and K. Hester, in Society of Petroleum Engineers - Arctic Technology Conference 2012, Offshore Technology Conference, 2012, vol. 1, pp. 155–171.
- J. Yang, A. Okwananke, B. Tohidi, E. Chuvilin, K. Maerle, V. Istomin, B. Bukhanov and A. Cheremisin, Energy Convers. Manage., 2017, 136, 431–438 CrossRef CAS.
- A. Hassanpouryouzband, J. Yang, B. Tohidi, E. Chuvilin, V. Istomin, B. Bukhanov and A. Cheremisin, Environ. Sci. Technol., 2018, 52, 4324–4330 CrossRef CAS PubMed.
- D. Sun, J. Ripmeester and P. Englezos, J. Chem. Eng. Data, 2016, 61, 4061–4067 CrossRef CAS.
- J. M. Schicks, B. Strauch, K. U. Heeschen, E. Spangenberg and M. Luzi-Helbing, J. Geophys. Res. Solid Earth, 2018, 123, 3608–3620 CrossRef CAS.
- D. Bai, X. Zhang, G. Chen and W. Wang, Energy Environ. Sci., 2012, 5, 7033–7041 RSC.
- Y. T. Tung, L. J. Chen, Y. P. Chen and S. T. Lin, J. Phys. Chem. B, 2011, 115, 15295–15302 CrossRef CAS PubMed.
- G. Wu, L. Tian, D. Chen, M. Niu and H. Ji, J. Phys. Chem. C, 2019, 123, 13401–13409 CrossRef CAS.
- S. Lee, Y. Lee, J. Lee, H. Lee and Y. Seo, Environ. Sci. Technol., 2013, 47, 13184–13190 CrossRef CAS PubMed.
- A. N. Salamatin, A. Falenty and W. F. Kuhs, J. Phys. Chem. C, 2017, 121, 17603–17616 CrossRef CAS.
- T. Yasunami, K. Sasaki and Y. Sugai, J. Can. Pet. Technol., 2010, 49, 44–50 CrossRef CAS.
- J. A. Jimenez and R. J. Chalaturnyk, Proc. SPE/ISRM Rock Mech. Pet. Eng. Conf., 2002, 340–352 Search PubMed.
- K. Edlmann, S. Haszeldine and C. I. McDermott, Environ. Earth Sci., 2013, 70, 3393–3409 CrossRef CAS.
- H. Koide, M. Takahashi, H. Tsukamoto and Y. Shindo, Energy Convers. Manage., 1995, 36, 505–508 CrossRef CAS.
- C. A. Rochelle, A. P. Camps, D. Long, A. Milodowski, K. Bateman, D. Gunn, P. Jackson, M. A. Lovell and J. Rees, Geol. Soc. Spec. Publ., 2009, 319, 171–183 CrossRef CAS.
- J. S. Levine, J. M. Matter, D. Goldberg, A. Cook and K. S. Lackner, Geophys. Res. Lett., 2007, 34, 1–5 CrossRef.
- J. K. Eccles and L. Pratson, Geophys. Res. Lett., 2012, 39, 1–7 Search PubMed.
- B. Tohidi, J. Yang, M. Salehabadi, R. Anderson and A. Chapoy, Environ. Sci. Technol., 2010, 44, 1509–1514, DOI:10.1021/es902450j.
- E. M. Myshakin, W. A. Saidi, V. N. Romanov, R. T. Cygan and K. D. Jordan, J. Phys. Chem. C, 2013, 117, 11028–11039 CrossRef CAS.
- J. Gauteplass, S. Almenningen, G. Ersland, T. Barth, J. Yang and A. Chapoy, Chem. Eng. J., 2020, 381, 122646 CrossRef CAS.
- J. S. Gudmundsson, F. Hveding and A. Berrehaug, Proc. Int. Offshore Polar Eng. Conf., 1995, 282–288 Search PubMed.
- V. Andersson and J. Ó. N. S. Gudmundsson, Ann. N. Y. Acad. Sci., 2000, 912, 322–329 CrossRef CAS.
- R. Larsen, A. Lund, V. Andersson and K. W. Hjarbo, Proc. - SPE Annu. Tech. Conf. Exhib., 2001, 1905–1909.
- R. Azarinezhad, A. Chapoy, R. Anderson and B. Tohidi, SPE Proj. Facil. Constr., 2010, 5, 58–64 CrossRef.
- S. Matsuo, H. Umeda, S. Takeya and T. Fujita, Energies, 2017, 10, 1–13 CrossRef.
- B. Prah and R. Yun, Appl. Therm. Eng., 2018, 128, 653–661 CrossRef CAS.
- H. P. Veluswamy, A. Kumar, Y. Seo, J. D. Lee and P. Linga, Appl. Energy, 2018, 216, 262–285 CrossRef CAS.
- C. Han, U. Zahid, J. An, K. Kim and C. Kim, Curr. Opin. Chem. Eng., 2015, 10, 42–48 CrossRef.
- S. P. Kang and H. Lee, Environ. Sci. Technol., 2000, 34, 4397–4400 CrossRef CAS.
- G. Deppe, A. Lee, O. Han, B. Nguyen-Phuoc and D. Spencer, 3rd Annu. Conf. Carbon Sequestration, Washint. DC, 2004.
- C.-G. Xu, Y.-S. Yu, W.-J. Xie, Z.-M. Xia, Z.-Y. Chen and X.-S. Li, Appl. Energy, 2019, 255, 113791 CrossRef CAS.
- Y. Liu, S. Deng, R. Zhao, J. He and L. Zhao, Renewable Sustainable Energy Rev., 2017, 77, 652–669 CrossRef CAS.
- M. Bai, Z. Zhang and X. Fu, Renewable Sustainable Energy Rev., 2016, 59, 920–926 CrossRef CAS.
- V. Vilarrasa and J. Rutqvist, Earth-Sci. Rev., 2017, 165, 245–256 CrossRef CAS.
- R. Shukla, P. Ranjith, A. Haque and X. Choi, Fuel, 2010, 89, 2651–2664 CrossRef CAS.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS.
- R. Moliner, M. J. Lázaro and I. Suelves, Int. J. Hydrogen Energy, 2016, 41, 19500–19508 CrossRef CAS.
- D. Parra, L. Valverde, F. J. Pino and M. K. Patel, Renewable Sustainable Energy Rev., 2019, 101, 279–294 CrossRef CAS.
- P. Di Profio, S. Arca, F. Rossi and M. Filipponi, Int. J. Hydrogen Energy, 2009, 34, 9173–9180 CrossRef CAS.
- P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 CrossRef CAS.
- F. Nadeem, S. M. S. Hussain, P. K. Tiwari, A. K. Goswami and T. S. Ustun, IEEE Access, 2019, 7, 4555–4585 Search PubMed.
- R. Tarkowski, Renewable Sustainable Energy Rev., 2019, 105, 86–94 CrossRef CAS.
- J. Andersson and S. Grönkvist, Int. J. Hydrogen Energy, 2019, 44, 11901–11919 CrossRef CAS.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- V. V. Struzhkin, B. Militzer, W. L. Mao, H. K. Mao and R. J. Hemley, Chem. Rev., 2007, 107, 4133–4151 CrossRef CAS PubMed.
- H. P. Veluswamy, R. Kumar and P. Linga, Appl. Energy, 2014, 122, 112–132 CrossRef CAS.
- S. Alavi and J. A. Ripmeester, Mol. Simul., 2017, 43, 808–820 CrossRef CAS.
- T. A. Strobel, C. A. Koh and E. D. Sloan, Fluid Phase Equilib., 2007, 261, 382–389 CrossRef CAS.
- T. A. Strobel, K. C. Hester, C. A. Koh, A. K. Sum and E. D. Sloan, Chem. Phys. Lett., 2009, 478, 97–109 CrossRef CAS.
- K. A. Udachin, J. Ltpkowski and M. Tkacz, Supramol. Chem., 1994, 3, 181–183 CrossRef CAS.
- W. L. Mao, H. Kwang Mao, A. F. Goncharov, V. V. Struzhkin, Q. Guo, J. Hu, J. Hu, R. J. Hemley, M. Somayazulu and Y. Zhao, Science, 2002, 297, 2247–2249 CrossRef CAS PubMed.
- V. V. Struzhkin, B. Militzer, W. L. Mao, H. K. Mao and R. J. Hemley, Chem. Rev., 2007, 107, 4133–4151 CrossRef CAS PubMed.
- K. A. Lokshin and Y. Zhao, Appl. Phys. Lett., 2006, 88, 131909 CrossRef.
- H. Lee, J. W. Lee, D. Y. Kim, J. Park, Y. T. Seo, H. Zeng, I. L. Moudrakovski, C. I. Ratcliffe and J. A. Ripmeester, Mater. Sustainable Energy Appl., 2010, 434, 285–288 Search PubMed.
- R. Anderson, A. Chapoy and B. Tohidi, Langmuir, 2007, 23, 3440–3444 CrossRef CAS PubMed.
- T. A. Strobel, C. J. Taylor, K. C. Hester, S. F. Dec, C. A. Koh, K. T. Miller and E. D. Sloan, J. Phys. Chem. B, 2006, 110, 17121–17125 CrossRef CAS PubMed.
- D. Y. Kim, J. Park, J. W. Lee, J. A. Ripmeester and H. Lee, J. Am. Chem. Soc., 2006, 128, 15360–15361 CrossRef CAS PubMed.
- D. Y. Koh, H. Kang and H. Lee, Chem. Commun., 2013, 49, 6782–6784 RSC.
- J. Liu, J. Hou, J. Xu, H. Liu, G. Chen and J. Zhang, Int. J. Hydrogen Energy, 2017, 42, 17136–17143 CrossRef CAS.
- J. Cai, R. Yan, C. G. Xu, Z. Y. Chen and X. Sen Li, Energy Procedia, 2019, 158, 5149–5155 CrossRef CAS.
- S. P. Kaur and C. N. Ramachandran, Int. J. Hydrogen Energy, 2018, 43, 19559–19566 CrossRef CAS.
- H. P. Veluswamy, T. Yang and P. Linga, Cryst. Growth Des., 2014, 14, 1950–1960 CrossRef CAS.
- J. Cai, Y. Q. Tao, N. von Solms, C. G. Xu, Z. Y. Chen and X. Sen Li, Appl. Energy, 2019, 243, 1–9 CrossRef CAS.
- D. Jianwei, L. Deqing, L. Dongliang and L. Xinjun, Ind. Eng. Chem. Res., 2010, 49, 11797–11800 CrossRef.
- T. A. Strobel, K. C. Hester, E. D. Sloan and C. A. Koh, J. Am. Chem. Soc., 2007, 129, 9544–9545 CrossRef CAS PubMed.
- T. Tsuda, K. Ogata, S. Hashimoto, T. Sugahara, M. Moritoki and K. Ohgaki, Chem. Eng. Sci., 2009, 64, 4150–4154 CrossRef CAS.
- T. A. Strobel, C. A. Koh and E. D. Sloan, J. Phys. Chem. B, 2008, 112, 1885–1887 CrossRef CAS.
- K. C. Hester, T. A. Strobel, E. D. Sloan, C. A. Koh, A. Huq and A. J. Schultz, J. Phys. Chem. B, 2006, 110, 14024–14027 CrossRef CAS.
- R. Kumar, D. D. Klug, C. I. Ratcliffe, C. A. Tulk and J. A. Ripmeester, Angew. Chem., Int. Ed., 2013, 52, 1531–1534 CrossRef CAS PubMed.
- R. G. Grim, P. B. Kerkar, E. D. Sloan, C. A. Koh and A. K. Sum, J. Chem. Phys., 2012, 136, 234504 CrossRef PubMed.
- Y. Matsumoto, R. G. Grim, N. M. Khan, T. Sugahara, K. Ohgaki, E. D. Sloan, C. A. Koh and A. K. Sum, J. Phys. Chem. C, 2014, 118, 3581–3589 CrossRef.
- R. V. Belosludov, R. K. Zhdanov, O. S. Subbotin, H. Mizuseki, Y. Kawazoe and V. R. Belosludov, J. Renewable Sustainable Energy, 2014, 6, 53132 CrossRef.
- M. N. Khan, L. J. Rovetto, C. J. Peters, E. D. Sloan, A. K. Sum and C. A. Koh, J. Chem. Eng. Data, 2015, 60, 418–423 CrossRef CAS.
- Z. Zhang, P. G. Kusalik and G. J. Guo, J. Phys. Chem. C, 2018, 122, 7771–7778 CrossRef CAS.
- H. P. Veluswamy, J. C. Yew and P. Linga, J. Chem. Eng. Data, 2015, 60, 228–237 CrossRef CAS.
- H. P. Veluswamy, J. Y. Chen and P. Linga, Chem. Eng. Sci., 2015, 126, 488–499 CrossRef CAS.
- S. Alavi and J. A. Ripmeester, Angew. Chem., Int. Ed., 2007, 46, 6102–6105 CrossRef CAS PubMed.
- D. P. Luis, I. E. Romero-Ramirez, A. González-Calderón and J. López-Lemus, J. Chem. Phys., 2018, 148, 114503 CrossRef CAS PubMed.
- Q. Lu, X. He, W. Hu, X. Chen and J. Liu, J. Phys. Chem. C, 2019, 123, 12052–12061 CrossRef CAS.
- A. Celzard and V. Fierro, Energy Fuels, 2005, 19, 573–583 CrossRef CAS.
- V. Rozyyev, D. Thirion, R. Ullah, J. Lee, M. Jung, H. Oh, M. Atilhan and C. T. Yavuz, Nat. Energy, 2019, 4, 604–611 CrossRef CAS.
- D. Lozano-Castelló, J. Alcañiz-Monge, M. A. De La Casa-Lillo, D. Cazorla-Amorós and A. Linares-Solano, Fuel, 2002, 81, 1777–1803 CrossRef.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- J. Silvestre-Albero, Green Energy and Technology, Springer, 2019, pp. 383–403 Search PubMed.
- K. Kim, H. Kang and Y. Kim, Energies, 2015, 8, 3142–3164 CrossRef CAS.
- W. Wang, C. Ma, P. Lin, L. Sun and A. I. Cooper, Energy Environ. Sci., 2013, 6, 105–107 RSC.
- J. G. Speight, Natural Gas: A Basic Handbook, Gulf Professional Publishing, 2007 Search PubMed.
- I. N. Tsimpanogiannis, J. Costandy, P. Kastanidis, S. El Meragawi, V. K. Michalis, N. I. Papadimitriou, S. N. Karozis, N. I. Diamantonis, O. A. Moultos, G. E. Romanos, A. K. Stubos and I. G. Economou, Mol. Phys., 2018, 116, 2041–2060 CrossRef CAS.
- B. Miller and E. R. Strong, Am. Gas Assoc. Mon., 1946, 28, 63–67 Search PubMed.
- S. Song, B. Shi, W. Yu, L. Ding, Y. Chen, Y. Yu, C. Ruan, Y. Liu, W. Wang and J. Gong, Chem. Eng. J., 2019, 361, 1264–1284 CrossRef CAS.
- S. Ma, J. Nan Zheng, M. Tian, D. Tang and M. Yang, Fuel, 2020, 261, 116364 CrossRef.
- A. Gupta, J. Lachance, E. D. Sloan and C. A. Koh, Chem. Eng. Sci., 2008, 63, 5848–5853 CrossRef CAS.
- G. McLaurin, K. Shin, S. Alavi and J. A. Ripmeester, Angew. Chem., Int. Ed., 2014, 53, 10429–10433 CrossRef CAS PubMed.
- Y.-A. Chen, L.-K. Chu, C.-K. Chu, R. Ohmura and L.-J. Chen, Sci. Rep., 2019, 9, 1–11 CrossRef PubMed.
- G. Pandey, H. P. Veluswamy, J. Sangwai and P. Linga, Energy Fuels, 2019, 33, 4865–4876 CrossRef CAS.
- A. Kumar, H. P. Veluswamy, P. Linga and R. Kumar, Fuel, 2019, 236, 1505–1511 CrossRef CAS.
- M. Khurana, H. P. Veluswamy, N. Daraboina and P. Linga, Chem. Eng. J., 2019, 370, 760–771 CrossRef CAS.
- W. Wang, B. O. Carter, C. L. Bray, A. Steiner, J. Bacsa, J. T. A. Jones, C. Cropper, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Mater., 2009, 21, 3810–3815 CrossRef CAS.
- L. Li, S. Wang and W. Wang, IOP Conference Series: Materials Science and Engineering, IOP Publishing, 2018, vol. 301, p. 12033 Search PubMed.
- L. Yang, X. Lan, D. Liu, G. Cui, B. Dou and J. Wang, Chem. Eng. J., 2019, 374, 802–810 CrossRef CAS.
- E. Chaturvedi, N. Prasad and A. Mandal, J. Nat. Gas Sci. Eng., 2018, 56, 246–257 CrossRef CAS.
- Q. Sun, B. Chen, Y. Li, Z. Xu, X. Guo, X. Li, W. Lan and L. Yang, Fluid Phase Equilib., 2018, 475, 95–99 CrossRef CAS.
- Y. M. Song, F. Wang, S. J. Luo, R. B. Guo and D. Xu, Fuel, 2019, 243, 185–191 CrossRef CAS.
- J. Chen, T. Wang, Z. Zeng, J. H. Jiang, B. Deng, C. Z. Chen, J. Y. Li, C. H. Li, L. M. Tao, X. Li and S. X. Xiao, Chem. Eng. J., 2019, 363, 349–355 CrossRef CAS.
- A. Farhadian, M. A. Varfolomeev, Z. Abdelhay, D. Emelianov, A. Delaunay and D. Dalmazzone, Ind. Eng. Chem. Res., 2019, 58, 7752–7760 CrossRef CAS.
- Z. Zhang, P. G. Kusalik and G.-J. Guo, J. Phys. Chem. C, 2019, 123, 20579–20586 CrossRef CAS.
- F. Filarsky, C. Schmuck and H. J. Schultz, Ind. Eng. Chem. Res., 2019, 58, 16687–16695 CrossRef CAS.
- J. D. Lee, J. W. Lee, K. N. Park and J. H. Kim, Double helix type gas hydrate reactor, US Pat., 8,936,759.20, 2015 Search PubMed.
- J. Javanmardi, K. Nasrifar, S. H. Najibi and M. Moshfeghian, Appl. Therm. Eng., 2005, 25, 1708–1723 CrossRef.
- M. J. Molina and F. S. Rowland, Nature, 1974, 249, 810–812 CrossRef CAS.
- P. S. Bailey, CHAPTER XI – The Practical Side, in Ozonation in Organic Chemistry, ed. P. S. Bailey, Academic Press, 1982, pp. 355–370 Search PubMed.
- M. Sillanpää, M. C. Ncibi and A. Matilainen, J. Environ. Manage., 2018, 208, 56–76 CrossRef PubMed.
- F. Zhang, B. Hong, W. Zhao, Y. Yang, J. Bao, C. Gao and S. Sun, Chem. Commun., 2019, 55, 3757–3760 RSC.
- S. Öztekin, Emerging Postharvest Treatment of Fruits and Vegetables, Apple Academic Press, 2018, pp. 213–276 Search PubMed.
- R. Pandiselvam, S. Subhashini, E. P. Banuu Priya, A. Kothakota, S. V. Ramesh and S. Shahir, Ozone Sci. Eng., 2019, 41, 17–34 CrossRef CAS.
- N. F. Gray, Microbiology of Waterborne Diseases, Elsevier, 2014, pp. 617–630 Search PubMed.
- G. McTurk and J. G. Waller, Nature, 1964, 202, 1107 CrossRef CAS.
- T. A. Vysokikh, T. V. Yagodovskaya, S. V. Savilov and V. V. Lunin, Russ. J. Phys. Chem. A, 2008, 82, 686–689 CAS.
- S. Muromachi, K. A. Udachin, S. Alavi, R. Ohmura and J. A. Ripmeester, Chem. Commun., 2016, 52, 5621–5624 RSC.
- T. Nakajima, S. Akatsu, R. Ohmura, S. Takeya and Y. H. Mori, Angew. Chem., Int. Ed., 2011, 50, 10340–10343 CrossRef CAS PubMed.
- S. Muromachi, T. Nakajima, R. Ohmura and Y. H. Mori, Fluid Phase Equilib., 2011, 305, 145–151 CrossRef CAS.
- S. Muromachi, H. D. Nagashima, J.-M. Herri and R. Ohmura, J. Chem. Thermodyn., 2013, 64, 193–197 CrossRef CAS.
- S. Muromachi, R. Ohmura and Y. H. Mori, J. Chem. Thermodyn., 2012, 49, 1–6 CrossRef CAS.
- K. Shishido, S. Muromachi and R. Ohmura, World Acad. Sci. Eng. Technol. Int. J. Chem. Mol. Nucl. Mater. Metall. Eng., 2013, 7, 111–115 Search PubMed.
- O. S. Subbotin, T. P. Adamova, R. V. Belosludov, H. Mizuseki, Y. Kawazoe and V. R. Belosludov, J. Struct. Chem., 2012, 53, 627–633 CrossRef CAS.
- Y. Yu Bozhko, K. V. Gets, R. K. Zhdanov and O. S. Subbotin, J. Phys.: Conf. Ser., 2018, 1128, 12084 CrossRef.
- K. Shishido, S. Muromachi, R. Nakamura, S. Takeya and R. Ohmura, New J. Chem., 2014, 38, 3160–3165 RSC.
- T. Nakajima, T. Kudo, R. Ohmura, S. Takeya and Y. H. Mori, PLoS One, 2012, 7, e48563 CrossRef CAS PubMed.
- O. S. Subbotin, Y. Y. Bozhko, R. K. Zhdanov, K. V. Gets, V. R. Belosludov, R. V. Belosludov and Y. Kawazoe, Phys. Chem. Chem. Phys., 2018, 20, 12637–12641 RSC.
- K. Watanabe, H. D. Nagashima, R. Nakamura and R. Ohmura, ACS Sustainable Chem. Eng., 2018, 6, 11624–11632 CrossRef CAS.
- G. J. M. Velders, A. R. Ravishankara, M. K. Miller, M. J. Molina, J. Alcamo, J. S. Daniel, D. W. Fahey, S. A. Montzka and S. Reimann, Science, 2012, 335, 922–923 CrossRef CAS PubMed.
- N. Abas, A. R. Kalair, N. Khan, A. Haider, Z. Saleem and M. S. Saleem, Renewable Sustainable Energy Rev., 2018, 90, 557–569 CrossRef CAS.
- P. A. Domanski, M. O. McLinden, A. F. Kazakov, J. S. Brown, R. Brignoli, J. Heo, J. Wojtusiak and I. I. R. G. 13th, Review of Refrigerants Evolution, Lorentzen Conference of Natural Refrigerants, 2018 Search PubMed.
- A. Kasaeian, S. M. Hosseini, M. Sheikhpour, O. Mahian, W. M. Yan and S. Wongwises, Renewable Sustainable Energy Rev., 2018, 96, 91–99 CrossRef CAS.
- G. Li, Y. Hwang, R. Radermacher and H. H. Chun, Energy, 2013, 51, 1–17 CrossRef CAS.
- G. Li, Y. Hwang, R. Radermacher and H. H. Chun, Energy, 2013, 51, 1–17 CrossRef CAS.
- T. Ogawa, T. Ito, K. Watanabe, K. Ichi Tahara, R. Hiraoka, J. Ichi Ochiai, R. Ohmura and Y. H. Mori, Appl. Therm. Eng., 2006, 26, 2157–2167 CrossRef CAS.
- X. Wang, M. Dennis and L. Hou, Renewable Sustainable Energy Rev., 2014, 36, 34–51 CrossRef CAS.
- A. Delahaye, L. Fournaison and D. Dalmazzone, Gas Hydrates 2 Geosci. Issues Potential Ind. Appl, 2018, 315–358.
- H. Hashemi, S. Babaee, A. H. Mohammadi, P. Naidoo and D. Ramjugernath, Int. J. Refrig., 2019, 98, 410–427 CrossRef CAS.
- P. Zhang and Z. W. Ma, Renewable Sustainable Energy Rev., 2012, 16, 5021–5058 CrossRef CAS.
- Y. Bi, T. Guo, T. Zhu, S. Fan, D. Liang and L. Zhang, Appl. Energy, 2004, 78, 111–121 CrossRef CAS.
- Y. Xie, G. Li, D. Liu, N. Liu, Y. Qi, D. Liang, K. Guo and S. Fan, Appl. Energy, 2010, 87, 3340–3346 CrossRef CAS.
- J. Chen, Z. Qiu and Y. Zheng, Int. Conf. Power Eng. 2013, ICOPE 2013, 2013, 65, 120–132.
- N. Xie, C. Tan, S. Yang and Z. Liu, ACS Sustainable Chem. Eng., 2019, 7, 1502–1511 CrossRef CAS.
- R. Matsuura, K. Watanabe, Y. Yamauchi, H. Sato and R. Ohmura, Renew. Energy Power Qual. J., 2019, 17, 118–122 Search PubMed.
- Z. Youssef, L. Fournaison, A. Delahaye and M. Pons, Int. J. Refrig., 2019, 98, 202–210 CrossRef CAS.
- Q. Sun and Y. T. Kang, Renewable Sustainable Energy Rev., 2016, 62, 478–494 CrossRef CAS.
- A. Eslamimanesh, A. H. Mohammadi and D. Richon, Chem. Eng. Sci., 2011, 66, 5439–5445 CrossRef CAS.
- H. Hashemi, S. Babaee, P. Naidoo, A. H. Mohammadi and D. Ramjugernath, J. Chem. Eng. Data, 2014, 59, 3907–3911 CrossRef CAS.
- H. Hashemi, S. Babaee, A. H. Mohammadi, P. Naidoo and D. Ramjugernath, J. Chem. Thermodyn., 2015, 82, 47–52 CrossRef CAS.
- F. Nikbakht, A. A. Izadpanah, F. Varaminian and A. H. Mohammadi, Int. J. Refrig., 2012, 35, 1914–1920 CrossRef CAS.
- M. Karamoddin and F. Varaminian, J. Chem. Thermodyn., 2013, 65, 213–219 CrossRef CAS.
- P. T. Ngema, P. Naidoo, A. H. Mohammadi, D. Richon and D. Ramjugernath, J. Chem. Eng. Data, 2016, 61, 827–836 CrossRef CAS.
- P. T. Ngema, P. Naidoo, A. H. Mohammadi and D. Ramjugernath, J. Chem. Eng. Data, 2019, 136, 59–76 CAS.
- S. Zafar, I. Dincer and M. Gadalla, International Communications in Heat and Mass Transfer, Springer, 2017, vol. 89, pp. 165–175 Search PubMed.
- S. Zafar, I. Dincer and M. Gadalla, Green Energy and Technology, Springer, 2018, pp. 1165–1175 Search PubMed.
- M. A. M. Hassan, H. M. Abdel-Hameed and O. E. Mahmoud, J. Energy Resour. Technol., 2019, 141, 42003 CrossRef CAS.
- X. Wang and M. Dennis, Appl. Energy, 2016, 167, 59–69 CrossRef CAS.
- Q. Sun, S. Kim and Y. T. Kang, Appl. Energy, 2017, 190, 249–256 CrossRef CAS.
- S. Kim, S. H. Lee and Y. T. Kang, Energy, 2017, 120, 362–373 CrossRef CAS.
- K. Nemoto, T. Ikeda, H. Mori, S. Alavi, S. Takeya and R. Ohmura, New J. Chem., 2019, 43, 13068–13074 RSC.
- H. Lee, C. Park, E. Lee, J. D. Lee and Y. Kim, J. Chem. Eng. Data, 2017, 62, 4395–4400 CrossRef CAS.
- S. Jerbi, A. Delahaye, L. Fournaison and P. Haberschill, Int. J. Refrig., 2010, 33, 1625–1631 CrossRef CAS.
- Y. S. Choi and S. Nešić, Int. J. Greenhouse Gas Control, 2011, 5, 788–797 CrossRef CAS.
- Y. Bi, T. Guo, L. Zhang, L. Chen and F. Sun, Appl. Energy, 2010, 87, 1149–1157 CrossRef CAS.
- Y. Bi, X. Liu and M. Jiang, Energy, 2014, 73, 908–915 CrossRef.
- C. J. Vörösmarty, A. Y. Hoekstra, S. E. Bunn, D. Conway and J. Gupta, Science, 2015, 349, 478–479 CrossRef PubMed.
- C. J. Vörösmarty, P. B. McIntyre, M. O. Gessner, D. Dudgeon, A. Prusevich, P. Green, S. Glidden, S. E. Bunn, C. A. Sullivan and C. R. Liermann, Nature, 2010, 467, 555 CrossRef PubMed.
- M. Rodell, J. S. Famiglietti, D. N. Wiese, J. T. Reager, H. K. Beaudoing, F. W. Landerer and M.-H. Lo, Nature, 2018, 557(7707), 651–659 CrossRef CAS PubMed.
- A. D. Khawaji, I. K. Kutubkhanah and J.-M. Wie, Desalination, 2008, 221, 47–69 CrossRef CAS.
- M. Kummu, J. H. A. Guillaume, H. De Moel, S. Eisner, M. Flörke, M. Porkka, S. Siebert, T. I. E. Veldkamp and P. J. Ward, Sci. Rep., 2016, 6, 38495 CrossRef CAS PubMed.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nanoscience And Technology: A Collection of Reviews from Nature Journals, World Scientific, 2010, pp. 337–346 Search PubMed.
- A. Subramani and J. G. Jacangelo, Water Res., 2015, 75, 164–187 CrossRef CAS PubMed.
- N. Ghaffour, T. M. Missimer and G. L. Amy, Desalination, 2013, 309, 197–207 CrossRef CAS.
- N. Ghaffour, J. Bundschuh, H. Mahmoudi and M. F. A. Goosen, Desalination, 2015, 356, 94–114 CrossRef CAS.
- J. Zheng, F. Cheng, Y. Li, X. Lü and M. Yang, Chin. J. Chem. Eng., 2019, 27, 2037–2043 CrossRef.
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712–717 CrossRef CAS PubMed.
- P. Babu, A. Nambiar, T. He, I. A. Karimi, J. D. Lee, P. Englezos and P. Linga, ACS Sustainable Chem. Eng., 2018, 6, 8093–8107 CrossRef CAS.
- Y.-N. Lv, S.-S. Wang, C.-Y. Sun, J. Gong and G.-J. Chen, Desalination, 2017, 413, 217–222 CrossRef CAS.
- C. Xu, X. Li, K. Yan, X. Ruan, Z. Chen and Z. Xia, Chin. J. Chem. Eng., 2019, 27, 1998–2013 CrossRef.
- P. Sahu, S. Krishnaswamy, K. Ponnani and N. K. Pande, Desalination, 2018, 436, 144–151 CrossRef CAS.
- W. G. Knox, M. Hess, G. E. Jones and H. B. Smith, Chem. Eng. Prog., 1961, 57, 66–71 CAS.
- Y. T. Ngan and P. Englezos, Ind. Eng. Chem. Res., 1996, 35, 1894–1900 CrossRef CAS.
- M. Nakajima, R. Ohmura and Y. H. Mori, Ind. Eng. Chem. Res., 2008, 47, 8933–8939 CrossRef CAS.
- D. Corak, T. Barth, S. Høiland, T. Skodvin, R. Larsen and T. Skjetne, Desalination, 2011, 278, 268–274 CrossRef CAS.
- L. Cai, B. A. Pethica, P. G. Debenedetti and S. Sundaresan, Chem. Eng. Sci., 2014, 119, 147–157 CrossRef CAS.
- L. Cai, B. A. Pethica, P. G. Debenedetti and S. Sundaresan, Chem. Eng. Sci., 2016, 141, 125–132 CrossRef CAS.
- H. Xu, M. N. Khan, C. J. Peters, E. D. Sloan and C. A. Koh, J. Chem. Eng. Data, 2018, 63, 1081–1087 CrossRef CAS.
- S. Ho-Van, B. Bouillot, J. Douzet, S. M. Babakhani and J. M. Herri, J. Environ. Chem. Eng., 2019, 7, 103359 CrossRef CAS.
- K. C. Kang, P. Linga, K. Park, S.-J. Choi and J. D. Lee, Desalination, 2014, 353, 84–90 CrossRef CAS.
- M. Yang, Y. Song, L. Jiang, Y. Liu and Y. Li, Ind. Eng. Chem. Res., 2014, 53, 10753–10761 CrossRef CAS.
- R. A. McCormack and G. A. Niblock, Investigation of high freezing temperature, zero ozone, and zero global warming potential, clathrate formers for desalination, US Department of the Interior, Bureau of Reclamation, Technical Service, 2000.
- B. A. Simmons, R. W. Bradshaw, D. E. Dedrick, R. T. Cygan, J. A. Greathouse and E. H. Majzoub, Desalination utilizing clathrate hydrates (LDRD final report), Sandia National Laboratories, 2008.
- M. Karamoddin and F. Varaminian, Desalin. Water Treat., 2014, 52, 2450–2456 CrossRef CAS.
- P. T. Ngema, C. Petticrew, P. Naidoo, A. H. Mohammadi and D. Ramjugernath, J. Chem. Eng. Data, 2014, 59, 466–475 CrossRef CAS.
- Y. Seo, D. Moon, C. Lee, J.-W. Park, B.-S. Kim, G.-W. Lee, P. Dotel, J.-W. Lee, M. Cha and J.-H. Yoon, Environ. Sci. Technol., 2015, 49, 6045–6050 CrossRef CAS PubMed.
- L. J. Wang, X. M. Zhang, H. H. Li, L. Shao, D. Zhang and L. Jiao, Adv. Mater. Res., 2013, 616–618, 1202–1207 Search PubMed.
- D. Lee, Y. Lee, S. Lee and Y. Seo, Korean J. Chem. Eng., 2016, 33, 1425–1430 CrossRef CAS.
- M. N. Khan, Phase Equilibria Modeling of Inhibited Gas Hydrate Systems Including Salts: Applications in Flow Assurance, Seawater Desalination and Gas Separation, Colorado School of Mines, Arthur Lakes Library, 2016 Search PubMed.
- Y. Zhang, S.-M. Sheng, X.-D. Shen, X.-B. Zhou, W.-Z. Wu, X.-P. Wu and D.-Q. Liang, J. Chem. Eng. Data, 2017, 62, 2461–2465 CrossRef CAS.
- S. Ho-Van, B. Bouillot, J. Douzet, S. M. Babakhani and J.-M. Herri, Ind. Eng. Chem. Res., 2018, 57, 14774–14783 CrossRef CAS.
- S. Maghsoodloo Babakhani, S. Ho-Van, B. Bouillot, J. Douzet and J.-M. Herri, Chem. Eng. Sci., 2020, 214, 115442 CrossRef.
- J.-H. Cha and Y. Seol, ACS Sustainable Chem. Eng., 2013, 1, 1218–1224 CrossRef CAS.
- Q. Lv, L. Li, X. Li and Z. Chen, Energy Fuels, 2015, 29, 6104–6110 CrossRef CAS.
- Q. Lv, X. Li and G. Li, Energy Procedia, 2019, 158, 5144–5148 CrossRef CAS.
- W. Choi, Y. Lee, J. Mok, S. Lee, J. D. Lee and Y. Seo, Chem. Eng. J., 2019, 358, 598–605 CrossRef CAS.
- S. D. Seo, S. Y. Hong, A. K. Sum, K.-H. Lee, J. D. Lee and B. R. Lee, Chem. Eng. J., 2019, 370, 980–987 CrossRef CAS.
- A. Nambiar, P. Babu and P. Linga, ChemEngineering, 2019, 3, 31 CrossRef CAS.
- M. N. Khan, C. J. Peters and C. A. Koh, Desalination, 2019, 468, 114049 CrossRef.
- S. Han, J.-Y. Shin, Y.-W. Rhee and S.-P. Kang, Desalination, 2014, 354, 17–22 CrossRef CAS.
- M. Karamoddin and F. Varaminian, J. Mol. Liq., 2016, 223, 1021–1031 CrossRef CAS.
- S. Han, Y.-W. Rhee and S.-P. Kang, Desalination, 2017, 404, 132–137 CrossRef CAS.
- H. Fakharian, H. Ganji and A. Naderifar, J. Environ. Chem. Eng., 2017, 5, 4269–4273 CrossRef CAS.
- H. Fakharian, H. Ganji and A. Naderifar, J. Taiwan Inst. Chem. Eng., 2017, 72, 157–162 CrossRef CAS.
- K. C. Kang, S. Y. Hong, S. J. Cho, D. H. Kim and J. D. Lee, J. Nanosci. Nanotechnol., 2017, 17, 4059–4062 CrossRef CAS.
- H. Lee, J. D. Lee and Y. Kim, Ind. Eng. Chem. Res., 2018, 57, 12980–12986 CrossRef CAS.
- H. Fakharian, H. Ganji, A. Naderifar, H. R. Mofrad and M. Kakavand, J. Water Reuse Desalin., 2019, 9, 396–404 CrossRef.
- J. Javanmardi and M. Moshfeghian, Appl. Therm. Eng., 2003, 23, 845–857 CrossRef CAS.
- Y. Ghalavand, M. S. Hatamipour and A. Rahimi, Desalin. Water Treat., 2015, 54, 1526–1541 CAS.
- S.-H. Yun and D.-S. Woo, J. Korean Soc. Water Wastewater, 2019, 33, 281–289 CrossRef.
- H. Lee, H. Ryu, J.-H. Lim, J.-O. Kim, J. Dong Lee and S. Kim, Desalin. Water Treat., 2016, 57, 9009–9017 CrossRef CAS.
- T. He, S. K. Nair, P. Babu, P. Linga and I. A. Karimi, Appl. Energy, 2018, 222, 13–24 CrossRef CAS.
- Z. R. Chong, T. He, P. Babu, J. Zheng and P. Linga, Desalination, 2019, 463, 69–80 CrossRef CAS.
- T. He, Z. R. Chong, J. Zheng, Y. Ju and P. Linga, Energy, 2019, 170, 557–568 CrossRef.
- J. Zheng and M. Yang, Desalination, 2020, 478, 114284 CrossRef.
- M. Sarshar and A. H. Sharafi, Desalin. Water Treat., 2011, 28, 59–64 CrossRef CAS.
- M. Yang, J. Zheng, W. Liu, Y. Liu and Y. Song, Energy, 2015, 93, 1971–1979 CrossRef CAS.
- J. Zheng, M. Yang, Y. Liu, D. Wang and Y. Song, J. Chem. Thermodyn., 2017, 104, 9–15 CrossRef CAS.
- J. Lee, K.-S. Kim and Y. Seo, Chem. Eng. J., 2019, 375, 121974 CrossRef CAS.
- S. Hong, S. Moon, Y. Lee, S. Lee and Y. Park, Chem. Eng. J., 2019, 363, 99–106 CrossRef CAS.
- D. Saha, H. A. Grappe, A. Chakraborty and G. Orkoulas, Chem. Rev., 2016, 116, 11436–11499 CrossRef CAS PubMed.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef CAS PubMed.
- L. C. Tome and I. M. Marrucho, Chem. Soc. Rev., 2016, 45, 2785–2824 RSC.
- S. Basu, A. L. Khan, A. Cano-Odena, C. Liu and I. F. J. Vankelecom, Chem. Soc. Rev., 2010, 39, 750–768 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- T. W. Jagger, A. E. Van Brunt and N. P. Van Brunt, Pressure Swing Adsorption Gas Separation Method and Apparatus, US Pat., 6641644, 2003 Search PubMed.
- A. S. Holmes and J. M. Ryan, Cryogenic distillative separation of acid gases from methane, US Pat., 4318723, 1982 Search PubMed.
- J. Yao and H. Wang, Chem. Soc. Rev., 2014, 43, 4470–4493 RSC.
- C.-G. Xu and X.-S. Li, RSC Adv., 2014, 4, 18301–18316 RSC.
- Y. Wang, X. Lang and S. Fan, J. Energy Chem., 2013, 22, 39–47 CrossRef CAS.
- A. Li, J. Wang and B. Bao, Greenhouse Gases: Sci. Technol., 2019, 9, 175–193 CrossRef CAS.
- P. Kastanidis, I. N. Tsimpanogiannis, G. E. Romanos, A. K. Stubos and I. G. Economou, J. Chem. Eng. Data., 2019, 64(12), 4991–5016, DOI:10.1021/acs.jced.9b00630.
- D.-L. Zhong, J.-L. Wang, Y.-Y. Lu, Z. Li and J. Yan, Energy, 2016, 102, 621–629 CrossRef CAS.
- P. Linga, R. Kumar and P. Englezos, Chem. Eng. Sci., 2007, 62, 4268–4276 CrossRef CAS.
- Z. Xia, X. Li, Z. Chen, G. Li, J. Cai, Y. Wang, K. Yan and C. Xu, Appl. Energy, 2017, 207, 584–593 CrossRef CAS.
- J. Zhao, Y. Zhao and W. Liang, Energy Technol., 2016, 4, 864–869 CrossRef CAS.
- E. K. Lee, J. D. Lee, H. J. Lee, B. R. Lee, Y. S. Lee, S. M. Kim, H. O. Park, Y. S. Kim, Y.-D. Park and Y. Do Kim, Environ. Sci. Technol., 2009, 43, 7723–7727 CrossRef CAS PubMed.
- I. Cha, S. Lee, J. D. Lee, G. Lee and Y. Seo, Environ. Sci. Technol., 2010, 44, 6117–6122 CrossRef CAS PubMed.
- C. K. Ahn, G.-H. Kim, J. E. Lee, K. S. Kim and K. Kim, Sep. Purif. Technol., 2019, 217, 221–228 CrossRef CAS.
- G. Ko and Y. Seo, Environ. Sci. Technol., 2019, 53, 12945–12952 CrossRef CAS PubMed.
- Y.-H. Ahn, D. Lim, J. Min, J. Kim, B. Lee, J. W. Lee and K. Shin, Chem. Eng. J., 2019, 359, 1629–1634 CrossRef CAS.
- E. Kim, G. Ko and Y. Seo, ACS Sustainable Chem. Eng., 2017, 5, 5485–5492 CrossRef CAS.
- H. Tajima, T. Nagata, Y. Abe, A. Yamasaki, F. Kiyono and K. Yamagiwa, Ind. Eng. Chem. Res., 2010, 49, 2525–2532 CrossRef CAS.
- E. Kim, W. Choi and Y. Seo, Fluid Phase Equilib., 2018, 471, 55–60 CrossRef CAS.
- J. Wang, W. Han, S. Wang, H. Tang, W. Liu, Y. Li, C. Lu, J. Zhang, E. M. Kennedy and X. Li, Catal. Sci. Technol., 2019, 9, 1338–1348 RSC.
- P. Warrier, M. Naveed Khan, M. A. Carreon, C. J. Peters and C. A. Koh, J. Renewable Sustainable Energy, 2018, 10, 34701 CrossRef.
- R. Kumar, P. Linga, J. A. Ripmeester and P. Englezos, J. Environ. Eng., 2009, 135, 411–417 CrossRef CAS.
Footnote |
| † E. J. and M. V. F. have equally contributed to the review article. |
| This journal is © The Royal Society of Chemistry 2020 |