
Hybrid polyoxometalates as post-functionalization platforms: from fundamentals to emerging applications
Alexander V.
Anyushin†
 ,
Aleksandar
Kondinski†
and
Tatjana N.
Parac-Vogt
*
,
Aleksandar
Kondinski†
and
Tatjana N.
Parac-Vogt
*
KU Leuven, Department of Chemistry, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: tatjana.vogt@kuleuven.be
First published on 3rd December 2019
Abstract
Polyoxometalates (POMs) represent an important group of metal–oxo nanoclusters, typically comprised of early transition metals in high oxidation states (mainly V, Mo and W). Many plenary POMs exhibit good pH, solvent, thermal and redox stability, which makes them attractive components for the design of covalently integrated hybrid organic–inorganic molecules, herein referred to as hybrid-POMs. Until now, thousands of organic hybrid-POMs have been reported; however, only a small fraction can be further functionalized using other organic molecules or metal cations. This emerging class of ‘post-functionalizable’ hybrid-POMs constitute a valuable modular platform that permits coupling of POM properties with different organic and metal cation functionalities, thereby expanding the key physicochemical properties that are relevant for application in (photo)catalysis, bioinorganic chemistry and materials science. The post-functionalizable hybrid-POM platforms offer an opportunity to covalently link multi-electron redox responsive POM cores with virtually any (bio)organic molecule or metal cation, generating a wide range of materials with tailored properties. Over the past few years, these materials have been showcased in the preparation of framework materials, functional surfaces, surfactants, homogeneous and heterogeneous catalysts and light harvesting materials, among others. This review article provides an overview on the state of the art in POM post-functionalization and highlights the key design and structural features that permit the discovery of new hybrid-POM platforms. In doing so, we aim to make the subject more comprehensible, both for chemists and for scientists with different materials science backgrounds interested in the applications of hybrid (POM) materials. The review article goes beyond the realms of polyoxometalate chemistry and encompasses emerging research domains such as reticular materials, surfactants, surface functionalization, light harvesting materials, non-linear optics, charge storing materials, and homogeneous acid–base catalysis among others.
 Alexander V. Anyushin | Dr Anyushin obtained his PhD from Nikolaev Institute of Inorganic Chemistry, Siberian Branch of the Russian Academy of Sciences. During his postdoctoral studies at Nikolaev Institute, Dr Anyushin investigated novel synthetic approaches in noble metal chemistry and in the chemistry of hybrid organic–inorganic compounds, including transition metal clusters and polyoxometalates. He is currently a post-doc in the group of Prof. Parac-Vogt at KU Leuven where he is focusing on developing functional biopolymer–metal–oxo hybrid materials. Dr Anyushin has an enduring interest in the areas of organic–inorganic hybrid molecules, polyoxometalate-based functional materials for catalysis and bio-inorganic hybrid systems. |
 Aleksandar Kondinski | Aleksandar Kondinski graduated from Jacobs University Bremen in 2010 with a BSc in Chemistry. He completed his PhD under the supervision of Prof. Thomas Heine at the same university in 2016. After a postdoc at RWTH Aachen University under the supervision of Prof. Paul Kögerler, he joined Prof. Parac-Vogt to study hybrid organic–inorganic materials. His research interests include development of advanced polyoxometalates for nanoelectronics and catalytical applications. |
 Tatjana N. Parac-Vogt | Tatjana N. Parac-Vogt is a full professor of chemistry at KU Leuven, Belgium. She studied chemistry at the University of Belgrade, former Yugoslavia, and obtained her PhD from Iowa State University in Ames, USA. Following her PhD, she performed post-doctoral research at the University of California at Berkeley in the group of Professor Ken Raymond. After that she spent two years in Germany as Alexander von Humboldt Fellow, before moving to KU Leuven in 2000, where she is currently the head of the Laboratory of Bioinorganic Chemistry. She has an active interest in promoting gender and cultural diversity in the working environment, and is also active as a coach and mentor to younger female scientists. |
1. Introduction
Metal oxides represent the most dominant materials in the Earth's crust and as such have been shaping technological and civilizational developments for millennia.1,2 In the current era of rapid socio-technological breakthroughs, the demand for discovery of novel, better performing and more sustainable advanced materials is greater than ever before.3–5 These circumstances fortify the wide interest in the further exploitation of Earth's abundant metal oxides and their confinement to the nanoscale with the aim of unlocking a plethora of new remarkable properties that aid in solving urgent global challenges relating to clean energy production,6–9 electrocatalysis,10,11 photocatalysis,12,13 electron storage14,15 and others. Over the past century, atomic-precision synthesis of discrete nanoscale molecular metal-oxides16,17 (i.e. nanomolecular species) formed during self-assembly processes has enabled the preparation of a vast number of transition metal–oxo nanoclusters which often exhibit similar skeletal topologies.18 Among these, one of the largest families are the nanomolecular metal-oxides made of VA and VIA group metals in high oxidation state (mainly V, Mo and W, less frequently Nb and Ta) to which we commonly refer to as polyoxometalates (POMs).1,19–22All-inorganic POMs are commonly formed in condensation processes of simple anions in solution (typically water) under various pH and temperature conditions.23,24 Owing to their all-inorganic nature and robust shell topologies, many POMs exhibit remarkable solution, redox and thermal stability, making them interesting for many actual and potential applications in catalysis,25,26 molecular magnetism,27–29 and life science.30–32 However, in some cases the overall negative charge and all-inorganic nature of POMs may limit their application scope. As a result, there has been a growing interest in diversifying the physicochemical properties of metal-oxide clusters by creating hybrid organic/bioinorganic POM architectures.21
Although the definition of hybrid materials is very broad, inorganic–(bio)organic hybrid materials are often classified and grouped depending on how the inorganic and organic counterparts are integrated in the structure.33 Depending on the nature of interaction with the (bio)organic counterpart, i.e. whether the interaction is ionic or covalent in nature, one distinguishes between two different hybrid POM classes.34 Class I hybrid POMs exhibit predominantly ionic interactions with various (bio)organic molecules. The molybdenum storage proteins35,36 are an ultimate example where such interactions exist in nature, while the interaction of POMs with biological materials and their packing in protein crystals have ultimately aided the elucidation of the ribosome structure by the Yonath Group37 which effectively led to the Nobel Prize in Chemistry in 2009. Besides these remarkable achievements, many other POM-hybrid composites such as those formed with polymers are also known.38–40 Over the past years, encapsulation of catalytically active POMs in metal organic frameworks (MOFs) which is typically based on electrostatic interactions has been a leading concept for performing selective catalytic oxidation reactions.41
Class II hybrid POMs have a covalent link with their corresponding (bio)organic counterparts.34 The covalent bond often provides stronger integration of two parts, resulting in improved stability and atomistic homogeneity, thus allowing for fine-tuning of the electronic and spectroscopic properties.42 The formation of hybrid covalently/coordinatively integrated (bio)organic functionalities with POMs dates back to the beginning of the 20th century when the groups of Miolatti, Rosenhein and co-workers attempted the synthesis of organo-functionalized polyoxomolybdates.43,44 Using single crystal X-ray diffraction experiments in 1975 Pope and co-workers revealed these structures to be [((CH3)2AsO2)Mo4O13H]2− polyanions.45 This discovery provoked a remarkable interest in the study of hybrid POMs, which just in a few years afterwards resulted in a number of new and well-characterized structures.21 To date, there have been a number of reviews written by different groups that address a large number of known hybrid POM structures.18,45–48 Many of these covalently integrated materials have been of interest to different communities due to the potential to devise metallopolymers49 and photo(electro-)chemical catalysts,50 and also due to the potential to graft them onto metal and graphite surfaces, which aids the preparation of surface-functionalized electrodes and nanoparticles.34,51
Despite the rich structural versatility of POMs, only four “classical” all-inorganic structural POM architypes have been effectively studied and further derivatized.52 These include Lindqvist [M6O19]q−, Anderson–Evans [XM6O24]q−, Keggin [XM12O40]q−, and Wells–Dawson [X2M18O62]q− (Fig. 1) which bare the names of the scientists who deduced and resolved their structures.52 All these topologies can be prepared by using MoVI or WVI addenda metals (M) in the presence of p-block heteroelements (X). These classical POM architypes exhibit one or two terminal oxo ligands per addenda unit which largely defines their redox stability.53 Using the bonding parameters and the electron density distribution,54–56 the classical POMs can be described as clathrate models where neutral [MO3]n hosts with n = 6, 12, or 18 encapsulate oxo or hetero-group guests.18
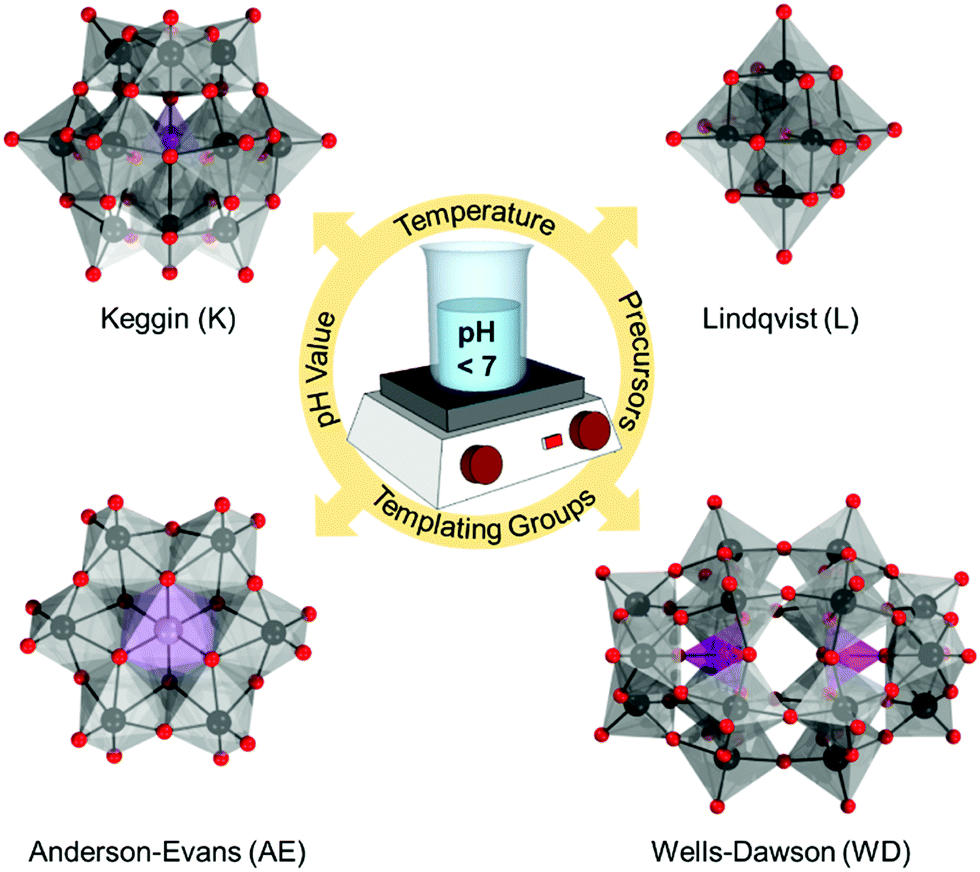 | ||
| Fig. 1 Combined ball-and-stick and polyhedral representation of the four classical POM architypes formed typically in aqueous media by condensation of simple mononuclear metal sources and hetero-groups under varying experimental conditions. Colour code: M = black balls, O = red balls, {MO6} = grey octahedra, {XO6}/{XO4} = violet octahedra/tetrahedra. | ||
Typically, all-inorganic POMs are produced following solution condensation of simple anionic building blocks (e.g. VO43−, MoO42−, WO42−) as a function of pH, concentration, temperature, etc. The hybrid structures of classical POMs can be obtained by creating unsaturated POM structures and reacting them with organic/organometallic compounds. Alternatively, an in situ reaction of organic, typically O-donating, ligands with metal-based building blocks during the course of synthesis can be performed. In general, the in situ self-assembly of hybrid POM is often very sensitive to the applied conditions, and therefore strict control is necessary as changing one parameter can lead to very different POM products.57 This can be of course turned into advantage and exploited for discovering novel hybrid POM structures; however it is often not an efficient strategy towards hybrid POM diversification. The integration of two new desired building blocks by such an approach is highly uncertain, and heavily relies on a trial and error approach for finding appropriate reaction conditions to minimize side reactions, rather than on a comprehensive and rational synthetic design.
Hybrid POMs having reactive groups or ligand-like sites which can be further covalently linked to other organic molecules or which make coordinative bonds with metal cations or metal-containing clusters are typically referred to as hybrid POM platforms or hybrid POM scaffolds. In essence, these hybrid POM platforms act as building blocks in designing other discrete, oligomer or network materials, which distinguishes them from the other hybrid plenary POMs that do not afford such diversification (see the scheme in Fig. 2). Following this, over the past decades there has been an increasing interest in developing hybrid POM platforms that afford versatile post-functionalization, while minimizing the undesired reactions between the POM framework and the added (bio)organic moieties. Many of the post-functionalization reactions are based on amide, ester, thioester, and sulfonamide bond formation, Huisgen coupling, Sonogashira coupling, Heck reaction, Suzuki reaction, metal coordination, polymerization, nucleophilic substitution reactions and others.
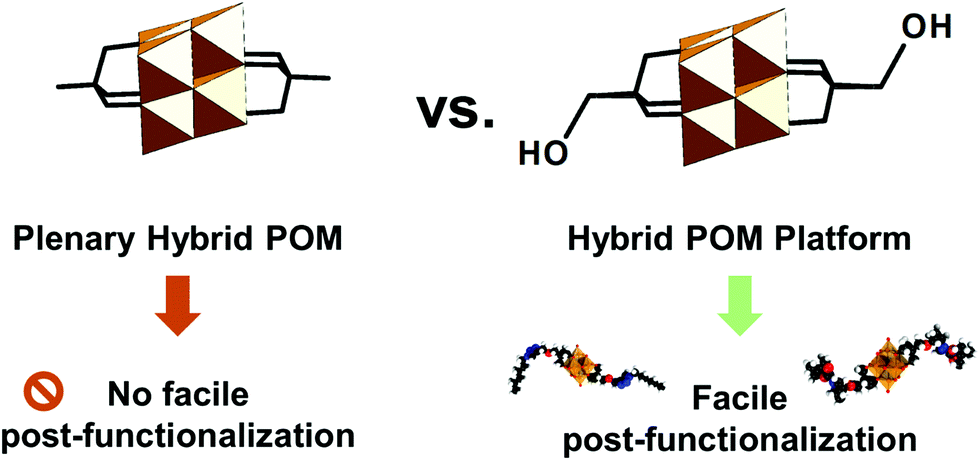 | ||
| Fig. 2 Schematic depiction of the difference between plenary hybrid POMs and hybrid POM platforms, exemplified using hybrid Lindqvist type hexavanadates. Facile post-functionalization is possible using terminal anchoring groups that can react under mild and environmentally benign conditions. | ||
Due to their relatively facile post-functionalization by using well-known methods in organic chemistry, such hybrid POM platforms represent a special subsection of POM compounds which provide the most efficient way for matching POMs with desired (bio)organic functionalities. In this work we review and describe the constitution of the hybrid POM platforms based on their different compositions, topologies and binding modes. This will allow a deeper understanding of each individual subclass and will provide insights into the reactivity of each individual platform. Furthermore, the typical applications that have been achieved over the past years, with relevance to communities in materials science, catalysis, magnetism, surface science, electrochemistry and others, will be highlighted for each platform.
2. On the constitution of hybrid POM platforms
The hybrid POM platforms consist of four building blocks, namely, (i) the POM core, which represents a plenary POM or a fragment of a plenary POM structure; (ii) the bridging unit which can be a metalorganic moiety, tris-alkoxo unit or other functionality; (iii) a spacer which may be a single bond or an organic moiety; (iv) an anchoring group or an anchoring site that can undergo facile post-functionalization with other organic molecules or coordination to metal cations (see Fig. 3). The constituents ii–iv are organic/metalorganic in character and their number and properties define the modularity of the hybrid platform, and thus one can jointly refer to ii–iv as the linker unit.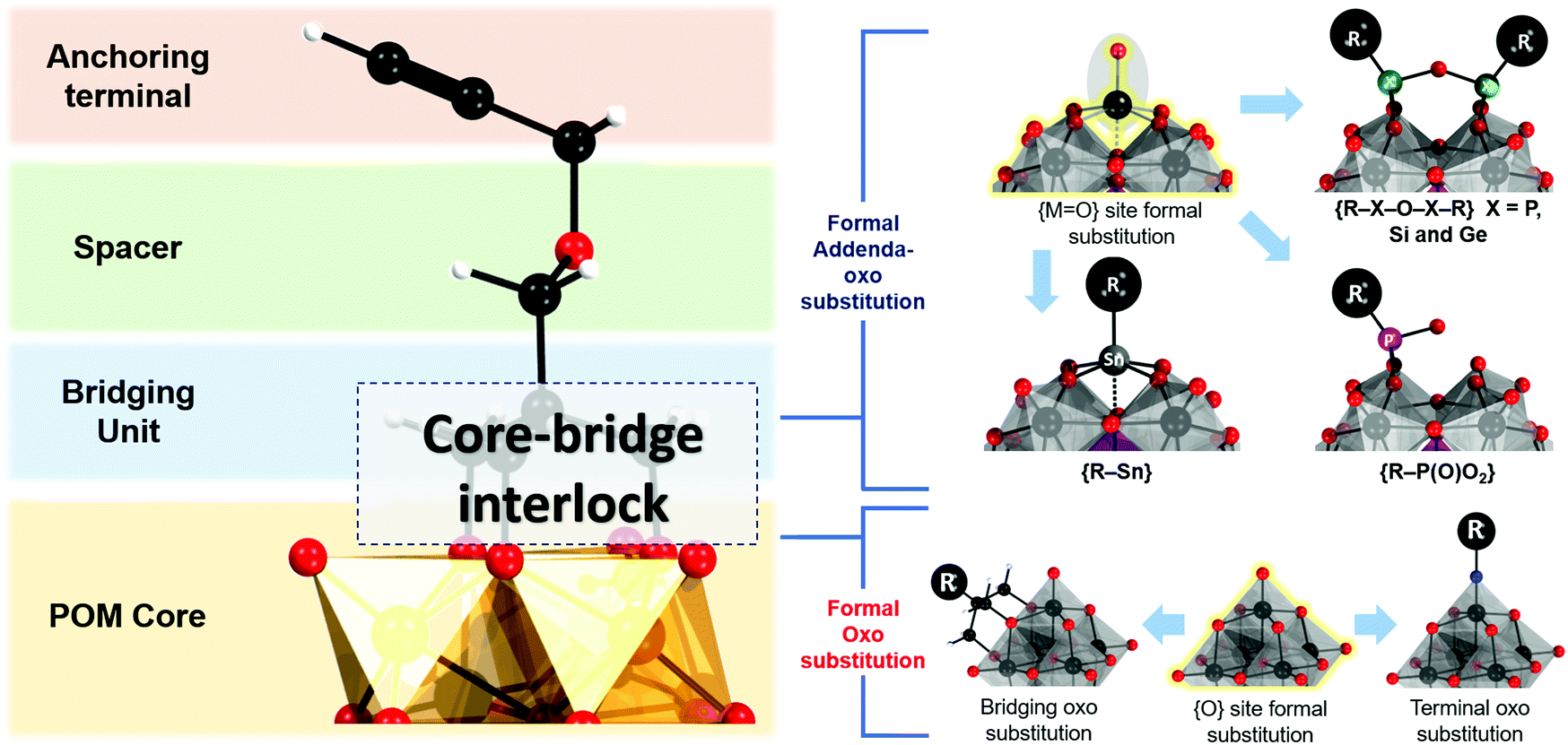 | ||
| Fig. 3 Schematic representation of the four different components comprising a hybrid POM architecture (left) and the different interlocking routes for interlocking the bridging and the POM core units following addenda-oxo and only oxo substitution. Colour code: {MO6} = grey/orange octahedra, C/R = black balls, O = red balls, N = blue balls, H = white balls, P = purple balls, X = green balls, Sn = grey balls. | ||
The nature of the anchoring group typically determines the type of reaction that can be used for post-functionalization. Although this can ultimately define the reactivity of the platform (see Section 2.4), the physicochemical properties of the overall hybrid POM platform and the possible binding modes are also influenced by the modularity and the electronic structure of the core POM moiety and the bridging moiety (Section 2.5). However, for a successful hybrid POM platform design, it is essential that the selected POM core and bridging unit are compatible with each other, and this relationship will be described in detail.
The strategy for integration of the POM core and the bridging unit relies on two parameters which are closely related to the properties of the POM core unit. These are (a) the possibility to form so-called lacunary or unsaturated POM structures; (b) the solution lability of the terminal and the μ2-oxo ligands. Lacunary POM structures typically imply topologies obtained by the formal loss of a single {MO} moiety or multiple {MxO2x} moieties, leading to monolacunary or polylacunary POM structures, respectively. The lacunary species thus exhibit highly nucleophilic terminal oxo sites which can interact with various cations. P-block based organo-bridges such as {R-Sn}, handle-like {R-X-O-X-R} (where X = P, Si and Ge) and {R-P(O)O2} can be interlocked at the lacunary sites (see Fig. 3). However, lacunary species are mainly limited to polyoxotungstates such as Keggin and Wells Dawson structures, while lacunary POMs are uncommon among polyoxomolybdates and polyoxovanadates. Consequently, Keggin and Wells–Dawson-type hybrid platforms are mainly W-based. On the other hand, the high lability of the oxo ligands enables substitution with tridentate trisalkoxide ligands or so-called triolates {(OCH2)3CR}3−. Tris-alkoxide functionalized POMs are dominant among the vanadium Lindqvist and the Anderson–Evans polyoxomolybdates, where predominantly the oxo ligands from the central {XO6} group undergo substitution. Finally, formal substitution of terminal oxo groups of the hexamolybdate Lindqvist is possible with nitrogen-based ligands.
In this context, this review covers V, Mo and W based hybrid POM platforms distributed among the four general POM archetypes (see Tables 1–5). To the best of our knowledge, polyoxoniobate and polyoxotantalate based hybrid POM platforms remain largely unknown, which may be due to the difficulty of these POMs to form lacunary species or to promote formal oxo exchanges with other hetero-ligands. Further on, the type of the binding modes of the hybrid POM platform is dependent on the type and the number of bridging units, and is based on the capacity of the core POM unit to accommodate bridging. These aspects are not of a simple nature and are thus further discussed in Sections 2.1–2.4.
| Modularity | Platform compound formula | Ref. | Method/reacting group | Ref. |
|---|---|---|---|---|
| [A-α-PW9O34{(Si(tBu)-R′)3}] | Cat3[α-PW9O34{(Si(tBu)-OH)3}] | Cat = TBA,88,89 Cat = Hex4N90 | Cl3Si-R, Cat = TBA | 88 and 89 |
| Cl3Ge-CH2CH2COOH, Cat = TBA | 91 | |||
| Coordination of transition metal ion; Ti(OiPr)4 | 90 | |||
| Coordination of transition metal ion; (iPrO)3VO | 92 | |||
| HO-surface, TiO2 or ZnO | 93 | |||
| TBA3[α-PW9O34{(Si((CH2)3SH)-OH)3}] | 94 | (MeO)3Si-(CH2)3SH | 94 | |
| [A-α-XW9O34{(OSi-R)3(E-R′)}], X = P, Si; E = Si, Ge | TBA3H[α-SiW9O34{(OSi(CH2)6-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)3(Si(CH2)6-CHCH2)}] CH2)3(Si(CH2)6-CHCH2)}] |
62 | Copolymerization; H2CC(Me)C(O)Me |
95 |
| TBA3[α-PW9O34{(OSi-CHCH2)3(Si-CHCH2)}] |
96 | Not reported yet | ||
| TBA3[α-PW9O34{(Osi(CH2)3OC(O)C(Me)-CHCH2)3(Si(CH2)3OC(O)C(Me)-CHCH2)}] |
97 | Copolymerization; H2CCH-Ph |
97 | |
| TBA3[α-PW9O34{(OSitBu)3(Ge(CH2)2–COOH)}] | 91 | Amidation; NH2-CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
91 | |
| [A-α-PW9O34{P(O)-R}2] | TBA3Na2[α-PW9O34{P(O)CH2-CHCH2}2] |
98 | Not reported yet | |
| TBA3Na2[α-PW9O34{(P(O)-tBu)}2] | 98 | Coordination of transition metal ion; Ca(NO3)2, [Ti(thf)Cl4], ZrOCl2, La(NO3)3 | 99 and 100 | |
| [B-α-AsW9O33{P(O)-R}2] | TBA3NaH[α-AsW9O33{P(O)(CH2)2–COOH}2] | 101 | Amide bond formation; NH2-R | 101 and 102 |
| Coordination of transition metal ion; La(NO3)3, VOCl3 | 99 | |||
| [B-α-AsW9O33{(Si(tBu)-R′)3}] | TBA3[α-AsW9O34{Si(tBu)-OH}3] | 88 | Cl3Si-H | 88 |
| [B-α-SbW9O34{(Si(tBu)-R′)3}] | (Hex4N)3[α-SbW9O34{Si(tBu)-OH}3] | 90,92 | Coordination of transition metal ion; Ti(OiPr)4 | 90 |
| Coordination of transition metal ion; (iPrO)3VO | 92 | |||
| [A-α-SiW9O28{(OCH2)2C(CH2-OH)2}3] | TBA4[α-SiW9O28{(OCH2)2C(CH2-OH)2}3] | 62 | Dimerization with TBA4[α-A-XW9O28{OMe}6], X = Si, Ge | 62 |
| [γ-SiW10O36{P(O)-R}2], X = Si, Ge | TBA4[γ-SiW10O36{P(O)CH2-COOH}2] | 103 | Amidation; H2N-R | 103 |
| [γ-SiW10O36{O(Si-R)2}] | Cat4[γ-SiW10O36{O(Si(Me)-OH)2}], Cat = TBA, Cs | 104 | Formation of Si-O bond, Cl–Si surface | 104 |
| TBA4[γ-SiW10O36{O(Si(CH2)3OC(O)-C(Me)CH2)2}] |
105 | Copolymerization; H2CC(Me)C(O)OMe |
106 | |
| TBA4[γ-SiW10O36{O(Si(CH2)3-SH2)2}] | 107 | Grafting on Au NP | 107 | |
| TBA4[γ-SiW10O36{O(Si(CH2)3-NH2)2}] | 108 | Sulfonamidation; ClSO2-R | 109–111 | |
| Polymerization; PMDA + ODA | 112 | |||
| TBA3H[γ-SiW10O36{O(Si(CH2)6-CHCH2)2}] |
109 | Copolymerization; H2CC(Me)C(O)OMe |
109 | |
| TBA4[γ-H2SiW10O36{Pd2(OAc)2}] | 113 | Exchange of the acetate ligands to HOOC(CH2)nCOOH (n = 1, 3, 5) | 114 | |
| [γ-SiW10O36{OSi-R}4] | Cat4[γ-SiW10O36{OSi(CH2)3OC(O)-C(Me)CH2}4] |
Cat = TBA1.1Na2.9,115 Cat = TBA3H116 | Copolymerization; H2CCHC(O)NH2 |
115 |
| [γ-PW10O36{O(Si-R)2}] | TBA3[γ-PW10O36{Si(tBu)-OH}2] | 117 | Cl2SiMe2 | 117 |
| Coordination of transition metal ion; (iPrO)3VO | 92 | |||
| [γ-GeW10O36{P(O)-R}2] | TBA4[γ-GeW10O36{P(O)CH2-COOH}2] | 103 | Amidation; H2N-R | 103 |
| [α-XW11O39{O(Si-R)2}], X = P, Si | TBA3[α-PW11O39{O(SiPh-4-NH2)2}] | 118 | Amide bond formation; NH2-R | 119 |
| TBA3[α-PW11O39{O(Si-CHCH2)2}] |
120 | Hydrosilylation; H-Si-R3 | 120 | |
| Copolymerization; butyl acrylate | 121 | |||
| (Me4N)3[α-PW11O39{O(Si(CH2)3-NH2)2}] | 122 | Coordination of transition metal ion; cis-[PtII(NH3)2Cl4] | 122 | |
| TBA3[α-PW11O39{O(Si-Ph-4-I)2}] | 118 and 123 | Sonogashira coupling; HCC-Ar-R |
118 and 123–125 | |
| TBA3[α-PW11O39{O(Si-Ph-4-CC-4′-Py)2}] |
126 | Coordination of transition metal ion; trans-[PdII(MeCN)2Cl2] | 126 | |
| TBA3[α-PW11O39{O(Si(CH2)3-SH)2}] | 127 | Hydrosulfuration; maleimide-R | 128 | |
| TBA4[α-SiW11O39{O(Si-OH)2}] | 129 | HO–Si-surface | 129 | |
| TBA4[α-SiW11O39{O(Si-chain-CHCH2)2}] chain = none, CH2, (CH2)3COOC(Me), 4-Ph |
130 | Polymerization with itself | 130 | |
| Cat4[α-SiW11O39{O(Si-CH2-CHCH2)2}] |
Cat = H121 | Copolymerization; H2CCHC(O)OnBu and (H2CCHC(O)O(CH2)3)2 |
121 and 131 | |
| Cat = TBA3H109 | Copolymerization; H2CC(Me)C(O)OMe |
109 | ||
| Cat4[α-SiW11O39{O(Si(CH2)3-NH2)2}] | 132 | Imine bond formation; H(O)C-R, Cat = Hex4N | 132 | |
| Copolyimidation PMDA + ODA, Cat = TBA | 133 | |||
| Amidation; ClC(O)-R, Cat = TBA | 134 and 135 | |||
| HOOC-(azobenzene)n, Cat = TBA | 136 | |||
| HOOC-CNTs, Cat = TBA | 137 | |||
| TBA4[α-SiW11O39{O(Si(CH2)3-SH)2}] | 138 | NP stabilization; Pd(OAc)2 | 138 | |
| TBA4[α-SiW11O39{O(Si(CH2)3NHC(O)(CH2)2OC(O)-CHCH2)2}] |
41 | Polymerization with itself | 41 | |
| (NHex4)4[α-SiW11O39{O(Si(CH2)3-NCH-Ph-2-OH)2}] |
132 | Metal coordination; M(OAc)2; M = MnII, CoII, NiII, PdII | 132 | |
| M = FeII, CuII | 139 | |||
| M = PdII | 140 | |||
| Coordination of transition metal ion; MoO2(acac)2 | 141 and 142 | |||
| Cat4[α-SiW11O39{O(Si(CH2)3-PPh2)2}] Cat = K, {(C8H17)3(CH3)N} | 132 | Metal coordination; [RhI(COD)2Cl]2 | 132 | |
| [α-PMo11O39{Ge-R}] | TBA4[α-PMo11O39{Ge-Ph-4-I}] | 143 | Sonogashira coupling; HCC-Ph-R, HCC-Fc |
143 |
| TBA4[α-PM11O39{Ge-Ph-4-CC-Ph-4′-NN-NEt2}] |
143 | Diazonation + surface grafting; HOOCCF3 | 143 | |
| [α-XW11O39{E-R}] X = P, Si, Ge, Ga, E = Sn, Ge | TBA4[α-PW11O39{Sn–Cl}] | 144 | Amidation; NR′R′′R′′′, NH-R2, NH2-R | 145 |
| TBA4[α-PW11O39{Sn(CH2)2-COOH}] | 128 and 146 | Amidation; H2N-R-biotin | 146 | |
| Amidation; H2N-R | 128 | |||
| TBA4[α-PW11O39{Sn-Ph-4-I}] | 147 | Sonogashira and other coupling; HCC-Ph-R |
123, 124 and 147–149 | |
| ROC(O)-CHCH2; thiophene-Si(Me)2-CHCH2 |
150 | |||
| TBA4[α-PW11O39{Sn-Ph-4-CC-CH2-OH}] |
149 | HS-C(O)-Me | 149 | |
| TBA4[α-PW11O39{Sn-Ph-4-CC-(CH2)n-OH}], n = 1 or 4 |
149 | Grafting on Au(111) | 149 | |
| TBA4[α-PW11O39{Sn-Ph-4-CC-Ph-4′-COOH}] |
151 | Amidation; NH2-R | 151 | |
| TBA4[α-PW11O39{Ge-Ph-4-I}] | 152 and 153 | Sonogashira coupling; HCC-Ph-R |
152–155 | |
| TBA4[α-PW11O39{Ge-Ph-4-CC-Ph-4′-R′}], R′ = NH2, N = N-NEt2 |
153 | Diazonation + surface grafting; HCl or NOBF4 | 152, 153 and 156 | |
| Amidation; HOOC-R | 156 and 157 | |||
| TBA4[α-PW11O39{Ge(CH2)2-COOH}] | 91 | Amidation; NH2-CH2CCH |
91 | |
| TBA4[α-PW11O39{Ge-Ph-4-CC-Ph-4′-NH-C(O)(CH2)4-C3H5S3}] |
157 | Formation of Au–S bond; Au(111) | 157 | |
| TBA5[α-SiW11O39{Sn(CH2)2-COOH}] | 158–160 | Amidation; NH2-R | 128 and 160 | |
| TBA4[α-SiW11O39{Sn(CH2)2-C(O)}] |
160 | Amidation; NH2-R | 161 and 162 | |
| H2N-CH2CC-dNTP |
163 | |||
| TBA5[α-SiW11O39{Sn(CH2)2C(O)NH(CH2)3-N3}] | 159 and 160 | Cu(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
159 and 160 | |
| TBA5[α-SiW11O39{Sn(CH2)2C(O)NHCH2-CCH}] |
159 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; N3-R | 159 | |
| Cat5[α-SiW11O39{Ge(CH2)2-COOH}], Cat = Li+, Cs | 164 | Amidation, esterification; HO-R, NH2-R | 164 | |
| TBA5[α-SiW11O39{Ge-Ph-4-I}] | 153 | Sonogashira coupling; HCC-Ar-R |
153 | |
| TBA5[α-SiW11O39{Ge-Ph-4-CC-Ph-4′-R′}], R′ = NH2, N = N-NEt2 |
153 | Diazonation + surface grafting; HCl or NOBF4 | 152 and 153 | |
| [GeW9V3O37{(OCH2)3C-NH-R}] | TBA4[GeW9V3O37{(OCH2)3C-NH2}] | 75 | Amidation; HOOC-R | 75 |
| TBA4[GeW9V3O37{(OCH2)3CNHC(O)CH(-NH-Boc)CH2-SH}] | 75 | HS-R and SCN-fluorescein | 75 |
| Modularity | Platform compound formula | Ref. | Method/reacting group | Ref. |
|---|---|---|---|---|
| [P2W15V3O59{(OCH2)3CNH-CO-R}] | Cat6[P2W15V3O59{(OCH2)3C-NH2}] | Cat = TBA72 | HOOC-fullerene | 72 and 165 |
| Cat = TBA4H274 | Amidation; Cl–C(O)–porphyrin | 136 | ||
| Cl-(O)C-C(Me)CH2 |
166 | |||
| TBA5H[P2W15V3O59{(OCH2)3CNHC(O)-C(Me)CH2}] |
166 | Copolymerization; H2CC(Me)C(O)OMe, H2CC(Me)C(O)OPh-4-SMe2·SO3CF3 |
166 | |
| TBA5H[P2W15V3O59{(OCH2)3CNHC(O)(CH2)11-6-(3,5-dioxo-4-azatricyclo[5.2.1.02-exo,6-exo]-dec-8-en-4-yl)}] | 167 | Copolymerization with itself; | 167 | |
| 6-(3,5-Dioxo-4-azatricyclo[5.2.1.02-exo,6-exo]-dec-8-en-4-yl)hexanoic acid | 168 | |||
| TBA6[P2W15V3O59{(OCH2)3CNHC(O)CH2-N3}] | 169 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
169 | |
| TBA5H[P2W15V3O59{(OCH2)3CNHC(O)-C(Me2)Br}] | 170 | Copolymerization with styrene; CH2CH–C6H5 |
170 | |
| TBA6[P2W15V3O59{(OCH2)3CNHC(O)CH2O-coumarin}] | 171 | Reversible photodimerization (254/365 nm light) | 171 | |
| TBA5,7H0,3[P2W15V3O59{(OCH2)3CNHC(O)CHCH-Ph-3,5-(CC-3′-Py)2}] |
172 | Electrocatalyzed copolymerization with Zn(II)-porphyrin; Zn(II)-octaethylporphyrin complex | 172 | |
| TBA5H[P2W15V3O59{(OCH2)3CNHC(O)-terpyridyl}] | 173 and 174 | Coordination of transition metal ion; [RuIII(iPrSPh)2(CH3OH)Cl3] | 173 | |
| [ReI(CO)5Br] | 174 | |||
| TBA5H[P2W15V3O59{(OCH2)3CNHC(O)CH2-bipy}] | 174 and 175 | Coordination of transition metal ion; [ReI(CO)5Br] | 174 and 175 | |
| TBA12[{P2W15V3O59{(OCH2)3CNHC(O)}2-{5,5′-(2,2′-bipy)}}] | 174 | Coordination of transition metal ion; [ReI(CO)5Br] | 174 | |
| TBA10H2[(P2V3W15O59{(OCH2)3NHC(O)-C5H3N}-)2] | 176 | Coordination of transition metal ion; ZnCl2 | 177 | |
| [P2W15V3O59{(OCH2)2(R)CNH-C()-R′}] |
TBA5H[P2W15V3O59{(OCH2)2R′CNHC()-4-Py-R′′}] |
178–180 | Coordination of transition metal ion; Pd(OAc)2 | 178 and 179 |
| TBA5[P2W15V3O59{(OCH2)2(C2H5)CNHC()CHCH-Ph-3,5-(CC-n′-Py)2}], n = 3,3, 3,4, and 4,4 |
172 | Electrocatalyzed copolymerization with Zn(II)-porphyrin; Zn(II)-octaethylporphyrin complex | 172 | |
| TBA5[P2W15V3O59{(OCH2)2(C2H5)CNHC()-(Ph-4-Me)-3,5-(CC-n′-Py)2}] |
181 | Electrocatalyzed copolymerization with Zn(II)-porphyrin; Zn(II)-octaethylporphyrin complex | 181 | |
| [P2W17O61{E-R}] E = Ge, Sn | TBA6H[α2-P2W17O61{Sn-Alk}], Alk = nBu | 182 | Addition of electrophile, OCN-aryl |
183 |
| TBA6H[α2-P2W17O61{Sn-Alk}], (CH2)2CONHCH2-CCH, (CH2)2CON(C2H4)2CH2 |
184 | |||
| TBA7[α2-P2W17O61{Sn-Ph-4-I}] | 185 | Sonogashira and other coupling; HCC-Ar-R |
147 and 186–188 | |
| TBA7[P2W17O61{Sn(CH2)2-COOH}] α1- and α2-WD | 184 and 189 | Amidation and esterification; NH2-R, HO-R | 184 and 189 | |
| TBA6[P2W17O61{Sn(CH2)2-C(O)}] α1- and α2-WD |
α1-WD190 | Amidation; NH2-R | 190–193 | |
| α2-WD194 | Amidation; H2N-R | 162, 163 and 195–199 | ||
| α1- and α2-WD TBA7[P2W17O61{Sn(CH2)2C(O)NH(CH2)3-N3}] | 159 and 194 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R (α1- and α2-) |
159 | |
| Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R (α1-) |
200 | |||
| α1- and α2-WD TBA7[P2W17O61{Sn(CH2)2C(O)NHCH2-CCH}] (α1- and α2-) |
159 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; N3-R (α1- and α2-) | 159 | |
| TBA7[α1-P2W17O61{Sn(CH2)2C(O)NH(CH2)3-N3C2-CH2OC(O)-C(Me)2-SC(S)S-C12H25}] | 193 and 200 | RAFT-copolymerization; CH2CH-C(O)NEt2 |
200 | |
| CH2CH-Ph |
193 | |||
| [α2-P2W17O61{O(Si-R)2}] | (Me2NH2)6[α2-P2W17O61{O(Si(CH2)3O(O)C-C(Me)CH2)2}] |
201 and 202 | Copolymerization; H2CC(Me)C(O)OMe |
201 and 202 |
| TBA6[α2-P2W17O61{O(Si-Ph-4-I)2}] | 185 | Sonogashira coupling; HCC-Ar-R |
185, 186 and 188 | |
| [Ru(bpy)2(ppy-H)]PF6 | 147 | |||
| TBA6[α2-P2W17O61{O(Si-Ph-4-CC-Ph-terpyridyl)2}] |
203 | Coordination of transition metal ion; [FeII(H2O)6](ClO4)2 | 203 | |
| TBA6[α2-P2W17O61{O(Si-Ph-4-CC-4′-Py)2}] |
204 | Coordination of transition metal ion; trans-[PdII(MeCN)2Cl2] | 126 and 204 | |
| TBA6[α2-P2W17O61{O(SiPh-4-CH2-N3)2}] | 205 and 206 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
205–208 | |
| (Me2NH2)5H[α2-P2W17O61{O(Si(CH2)3-SH)2}] | 209 and 210 | Cl-CH2-latex nanoparticles | 211 | |
| [α2-P2W17O61{P(O)-R}2] | TBA6[α2-P2W17O61{P(O)Ph-4-CCH}] |
205 and 212 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; N3-R | 205 |
| Sonogashira coupling; I-Ph-R | 213 | |||
| Hagihara dehydrohalogenating coupling; HgCl2 + CuI | 213 | |||
| trans-[PtII(PBu3)2Cl2] + CuI | 214 | |||
| TBA6[α2-P2W17O61{P(O)Ph-4-CH2-N3}2] | 205, 206 and 208 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
205, 206, 208 and 215 | |
| TBA6[α2-P2W17O61{P(O)(CH2)2-N3}2] | 73 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
73 | |
| K6[α2-P2W17O61{P(O)Ph-4-COOH}2] | 216 and 217 | Coordination of transition metal ion; M(NO3)2, M = Zn, Co, Mn | 218 |
| Modularity | Platform compound formula | Ref. | Method/reacting group | Ref. |
|---|---|---|---|---|
| TBA3[MnMo6O18{(OCH2)3C-R}2] | TBA3[MnMo6O18{(OCH2)3C-4-Py}2] | 68 and 299 | Coordination of transition metal ion; CuI | 3 |
| M(NO3)2, M = Zn, Cu | 299 | |||
| [δ-MnMo6O18{(OCH2)3C-NH2}2] | TBA3[MnMo6O18{(OCH2)3C-NH2}2] | 5, 287 and 302 | Imine formation; H(O)C-R | 284–287, 289 and 306 |
| Amidation, HOOC-R | 264, 266, 267, 269, 271, 272, 279, 280, 281 and 307–309 | |||
| Amidation; Cl(O)C-R | 260, 262 and 295 | |||
| Amidation; O(OC-R)2 | 255, 257, 259 and 261 | |||
| Phenylthiourification; SCN-Ph | 282 | |||
| Mo = N imide bond formation; O = Mo-POM | 274–276 | |||
| 273, 275 and 276 | ||||
| TBA3[MnMo6O18{(OCH2)3CNH-Ph-4-I}2] | 290 | Sonogashira coupling; HCC-aryl |
290 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)(CH2)2COOH}2] | 259 and 278 | Amidation; H2N-R | 259 and 278 | |
| TBA3[MnMo6O18{(OCH2)3CNCH-pyrene}{(OCH2)3C-NH2}] |
310 | Amidation; HOOC-R-surface(Au) | 310 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)(CH2)2C(O)-N-succinimide}2] | 259 | Amidation; H2N-peptide | 259 and 263 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-spiropyran-NO2}{(OCH2)3C-NH2}] | 266 | Amidation; HOOC-R | 267, 268 and 293 | |
| Reversible photoisomerization | 267, 268 and 293 | |||
| TBA3[MnMo6O18((OCH2)3CCH3){(OCH2)3C-NH2}] | 260 | Amidation; HOOC-TPP | 260 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-R}2], R = spiropyran-NO2 | 267 | Reversible photoisomerization | 267 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-R}2], R = spironaphthoxazine | 266 | Reversible photoisomerization | 266 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-spiropyran-NO2}{(OCH2)3CNHC(O)-C(Me)CH2} |
261 | Copolymerization; H2CC(Me)-COOMe |
261 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)(CH2)2Ph-4-N3}] | 293 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-BODIPY |
293 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-spiropyran-NO2}{(OCH2)3C3CNHC(O)(CH2)2Ph-4-N3}] | 293 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-BODIPY, reversible photoisomerization |
293 | |
| TBA3[MnMo6O18{(OCH2)3C-NH-Fmoc}{(OCH2)3C-NH2}] | 258 | Ester bond formation; HOOC-R | 258 | |
| Ester bond formation; O(OC-R)2 | 291 | |||
| TBA3[MnMo6O18{(OCH2)3CNHC(O)CH2-Cl}2] | 292 and 311 | Amide and ester bond formation; NH2-R, HN-R2, HS-R and HOOC-R | 311 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)CH2-N3}2] | 292 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
292 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)Ph-N3}2] | 291 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
291 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)Ph-4-N3}{(OCH2)3C-NH2}] | 291 | Amide bond formation and Cu(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R and HOOC-R |
291 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)(CH2)3-CCH}2] |
291 | Cu(I)-Catalyzed azide–alkyne 1,3-dipolar cycloaddition; HCC-R |
291 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)(CH2)3-CCH}{(OCH2)3C-NH2}] |
291 | Amide bond formation and Cu(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition; N3-R and HOOC-R | 291 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)CH2-Base}2], base = adenine or thymidine | 296 | H bond formation; OC-base, N-base and H-N-base |
296 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-aryl}2], aryl = maleimide, furan or anthracene | 291 | Metal-free Diels–Alder Click reaction; aryls | 291 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-C(CMe) = CH2}2] | 255 and 256 | Copolymerization H2CC(Me)C(O)OMe and H2CC(Me)C(O)O-4-Ph-SMe2*CF3SO3 |
255 and 256 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)CH2O-coumarin}2] | 294 | Reversible photodimerization (254/365 nm light) | 294 | |
| TBA3[MnMo6O18{(OCH2)3CNHC-Ph(2-OH)-4-NN-Ph-4′-O-CnH2n+1}2], n = 8, 10, 12 |
287 | Reversible photoisomerization (300–400/>400 nm light) | 287 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-(2,6-bis-(pyrazol-1-yl)-pyridine)}2] | 305 | Coordination of transition metal ion; Fe(ClO4)2 | 305 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-terpyridyl}2] | 173 | Coordination of transition metal ion; [Pd(CH3CN)2Cl2] | 173 | |
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-4-Py}2] | 298 and 299 | Coordination of transition metal ion; [Pd(PhCN)2Cl2] | 298 | |
| M(NO3)2, M = Zn, Cu | 299 | |||
| [Ru(TTP)(CO)(CH3CN)] and [Zn(TTP)] (TPP = meso-tetraphenylporphyrin) | 304 | |||
| Chain H-bonding with HOOC-R-COOH, R = (CH2)n (n = 0–3), 1,2-Ph, 1,3-Ph, 1,4-Ph, cis-HCCH, trans-HCCH, D-H(OH)CC(OH)H, L-H(OH)CC(OH)H |
312 | |||
| Electrocopolymerization; [ZnII(Porph)], Porph = octaethylporphyrin or 5,15-dipyridinium-octaethylporphyrin | 295 | |||
| TBA3[MnMo6O18{(OCH2)3CNCH-4-bipy-4′-Me}2] |
302 | Coordination of transition metal ion; Ir(III) | 302 and 303 | |
| Coordination of transition metal ion; Cu(I) | 313 | |||
| TBA3[MnMo6O18{(OCH2)3CNHC(O)-DibromoAryl}2], dibromoAryl = 3,5-dibromobenzene or 2,5-dibromothiophene | 264 | Sonogashira–Hagihara coupling; (HCC)3-1,3,5-Ph |
264 | |
| [δ-MnMo6O18{(OCH2)3CX}{(OCH2)3CNH-R}2] | TBA3[MnMo6O18((OCH2)3CNO2){(OCH2)3C-NH2}] | 285 | Imide formation; H(O)C-R | 285 |
| [δ-MnMo6O18{(OCH2)3CNH-R}2] | TBA3[MnMo6O18{(OCH2)3CNHC(O)-Ph-4-NN-Ph}2] |
289 | Reversible photoisomerization (300–400/>400 nm light) | 289 |
| [δ-MnMo6O18{(OCH2)3CCH2-OR}2] | TBA3[MnMo6O18{(OCH2)3CCH2-OH}2] | 283 and 314 | Esterification; ε-caprolactone | 283 and 314 |
| [δ-FeMo6O18{(OCH2)3C-R}2] | TBA3[FeMo6O18{(OCH2)3C-NH2}2] | 303 | Imide formation; H(O)C-R | 303 |
| MoN imide bond formation; OMo-POM |
273, 275 and 276 | |||
| TBA3[FeMo6O18{(OCH2)3CNCH-4-bipy-4′-Me}2] |
303 | Coordination of transition metal ion; Ir(III) [Ir(ppy)2(μ2-Cl)]2 | 303 | |
| [Cu(CH3CN)4]BF4 | 313 | |||
| [δ-CoMo6O18{(OCH2)3C-NH2}2] | TBA3[CoMo6O18{(OCH2)3C-NH2}2] | 303 | Imide formation; H(O)C-R | 303 |
| TBA3[CoMo6O18{(OCH2)3CNCH-4-bipy-4′-Me}2] |
303 | Coordination of transition metal ion; Ir(III) [Ir(ppy)2(μ2-Cl)] | 303 | |
| [Cu(CH3CN)4]BF4 | 313 | |||
| [δ-CrMo6O18(OH)3{(OCH2)3C-R}] | TBA4[CrMo6O18(OH)3{(OCH2)3CNH-4-Py}] | 300 | Coordination of transition metal ion; Cu(I) | 300 |
| [δ-AlMo6O18(OH)3{(OCH2)3C-R}] | TBA3[AlMo6O18(OH)3{(OCH2)3C-R′}], R′ = OH, NH2, NHCH2COOH | 315 | R′ = NH2, amidation, HOOC-R | 265, 269 and 270 |
| Modularity | Platform compound formula | Ref. | Method/reacting group | Ref. |
|---|---|---|---|---|
| 1 | (TBA)2[V3Mo3O16((OCH2)3CCH2OH)] | 319 | Esterification; HOOC-R | 319 |
| [V6O7(OCH3)9(OCH2)3C-NH2] | 320 | Imine formation, R-CHO | 320 | |
| [V6O7(OCH3)9(OCH2)3CCH2-N3] | 320 | Catalytic azide–alkyne 1,3-dipolar cycloaddition; R-CC–H |
320 | |
| 2 | TBA2[V6O13{(OCH2)3C-NH2}2] | 321 and 322 | Amide formation; HOOC-R Suc-OC(O)-R | 322–325 |
| Terminal oxo substitution in hexamolybdates | 326 | |||
| TBA2[V6O13{(OCH2)3CCH2-OH}2] | 327 | Esterification; HOOC-R | 70 and 328–332 | |
| Tosylation reaction with Cl-SO3-Ph-4-Me | 333 | |||
| TBA2[V6O13{(OCH2)3CCH2-SO3Ph-4-Me}2] | 333 | Nucleophilic substitution; NaBr or NaN3 | 333 | |
| TBA2[V6O13{(OCH2)3CCH2-N3}2] | 333 | Catalytic azide–alkyne 1,3-dipolar cycloaddition; R–CC–H |
334 | |
| TBA2[V6O13{(OCH2)3CCH2OCH2-CCH}2] |
42 | Catalytic azide–alkyne 1,3-dipolar cycloaddition; R-N3 | 42 | |
| TBA2[V6O13{(OCH2)3CCH2OC(O)(CH2)2-COOH}2] | 70 | Amide formation; H2NH-R | 70 | |
| TBA2[V6O13{(OCH2)3CCH2OC(O)-C6H4-COOH}2] | 329 | Amidation reaction; H2N-R | 335 | |
| M2[V6O13{(OCH2)3C-COOH}2], M = Na+, TBA+ | 336 | Ester bond formation; HO-R | 337 | |
| Coordination of transition metal ion; Zn(NO3)2 | 337 | |||
| TBA2[V6O13{(OCH2)3CNHC(O)-4-Py}2] | 338 | Coordination to Zn(II)-porphyrins | 135 | |
| Coordination to Zn(II), Co(II), Mn(II), Ni(II) | 338 | |||
| Coordination to RuII-meso-tetraphenylporphyrin | 304 | |||
| Coordination to Tb3+ | 339 | |||
| TBA2[V6O13{(OCH2)3CNHC(O)-3-Py}2] | 340 | Coordination to Pd2+ | 340 | |
| TBA2[V6O13{(OCH2)3CNHCH2-Ph-4-COOH}2] | 339 and 341 | Coordination to Co2+ | 341 | |
| 3 | Na2[VIV3VV3O10{(OCH2)3C-NH2}3] | 67 | Amidation reaction; HOOC-R | 67 |
| Modularity | Platform compound formula | Ref. | Method/reacting group | Ref. |
|---|---|---|---|---|
| 1 | TBA2[Mo6O18{N-Ph(2,6-Me)-4-I}] |
360 | Sonogashira coupling; HCC-Ar-R, HCC-Fc |
361–366 |
| TBA2[Mo6O18{N-Ph(2,6-Me)-4-CCH}] |
360 | Sonogashira coupling; I-Ar-R | 363 and 367 | |
| TBA2[Mo6O18{N-Ph(2,6-R′)-4-X}], R′ = Me, Et, X = Br, I |
368 | Heck coupling; H2CC-R2 |
368 | |
| Sonogashira coupling; HCC-Ar-R |
363 | |||
| TBA2[Mo6O18{N-Ph-4-I}] |
369 | Sonogashira coupling; HCC-Ar-R |
357 and 369 | |
| TBA2[Mo6O18{N-Ph(2,6-Me)-4-CC-Ph-2′,5′-(CCH)2}] |
370 | Sonogashira coupling; I-Ar-R | 370 | |
| TBA2[Mo6O18{N-Ph-4-CHCH2}] |
371 | Copolymerization; H2CCH-Ph-4-Me |
371 | |
| TBA2[Mo6O18{N-Ar-OH}], Ar-OH = Ph-2-Me-4-OH |
372 | Esterification; Ar-OH = Ph-2-Me-4-OH, HOOC-R | 372 and 373 | |
| O(C(O)-R)2 | 374 | |||
| TBA2[Mo6O18{N-n-Py}], n = 3 or 4 |
375 | Alkylation; Me-I, Et-I | 375 | |
| TBA2[Mo6O18{N-Ph(2,6-Me)-4-CH2-N(C2H4)2O}] |
358 | Alkylation; allyl-I, benzyl-I | 358 | |
| TBA2[Mo6O18{N Ph-4-CH2-N3C2H2}] |
376 | Coordination of transition metal ion; fac-[ReI(phen)(CO)3(H2O)](CF3SO3) | 376 | |
| TBA2[Mo6O18{N-Ph-4-COOH}] |
377 | Coordination of transition metal ion; Cu(OAc)2 | 377 | |
| TBA2[Mo6O18{NPh(2,6-Me)-4-CC-terpyridyl}] |
378 | Coordination of transition metal ion; Zn(OAc)2, Ru(terpyridine)Cl3 | 378 | |
| TBA2[Mo6O18{NCH2CH2CH2Cl}] Top of Form |
379 | Nucleophilic substitution; NaI, AgNO3 | 379 | |
| TBA2[Mo6O18{NPh(2,6-Me)-4-CC-Ph-N3Et2}] |
365 | Diazonation + surface grafting S(111) or Si(100); HBF4 | 365 | |
| 2 | TBA2[Mo6O17{N-Ph(2,6-iPr)-4-I}2] |
380 | Sonogashira and other couplings; X = I, HCC-Ar-R |
364 and 380 |
| (HCC)-Ar-(CCH) |
366 | |||
| TBA2[Mo6O17{N-Ph(2,6-iPr)-4-Br}2] |
297 | Mixture of thiophene-based Br-substituted monomers | 359 and 381 | |
| TBA2[Mo6O17{N-Ph(2,6-iPr)-4-CCH}2] |
367 | Sonogashira coupling; Ar-I | 367 | |
| TBA2[Mo6O17{N-Ph(2,6-iPr)-4-CC-terpyridyl}2] |
361 | Coordination of transition metal ion; FeCl2 | 361 | |
| TBA2[Mo6O17{N-Ph(2,6-iPr)-4-(CH2)5O-Ph(4′-Me)-2′,5′-I}2] |
370 | Sonogashira coupling; HCC-Ar-R |
370 |
2.1. Keggin (K) and Wells–Dawson (WD) structures
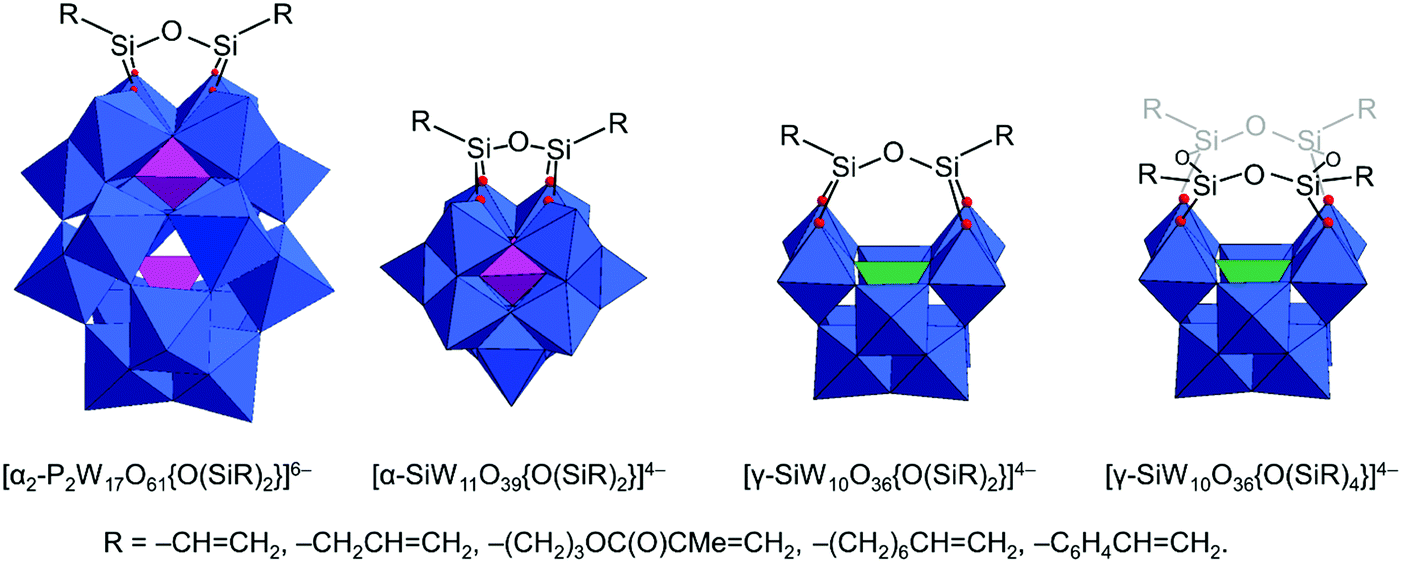
The original Keggin structure refers to {M12O36} topology which upon encapsulation of a single tetrahedral guest unit collapses to the Td symmetry point group.18 The structure can virtually further isomerize by stepwise rotation of each of the four triads, leading to α-, β-, γ-, δ-, ε-Keggin isomers.58 Among the lacunary POMs, density functional calculations and experimental validations reveal that the lacunary α-{XW11O39} Keggin structure exhibits the highest inherent and solution stability59 and as such it is a common POM core fragment that will be discussed throughout this review. Another rotational isomer is the γ-Keggin structure; however, this structure has two W centers in an edge-sharing fashion. High positive charges in close proximity lead to repulsions, so that dilacunary structures such as γ-{SiW10O36} and γ-{GeW10O36} are commonly isolated (see Fig. 4).60,61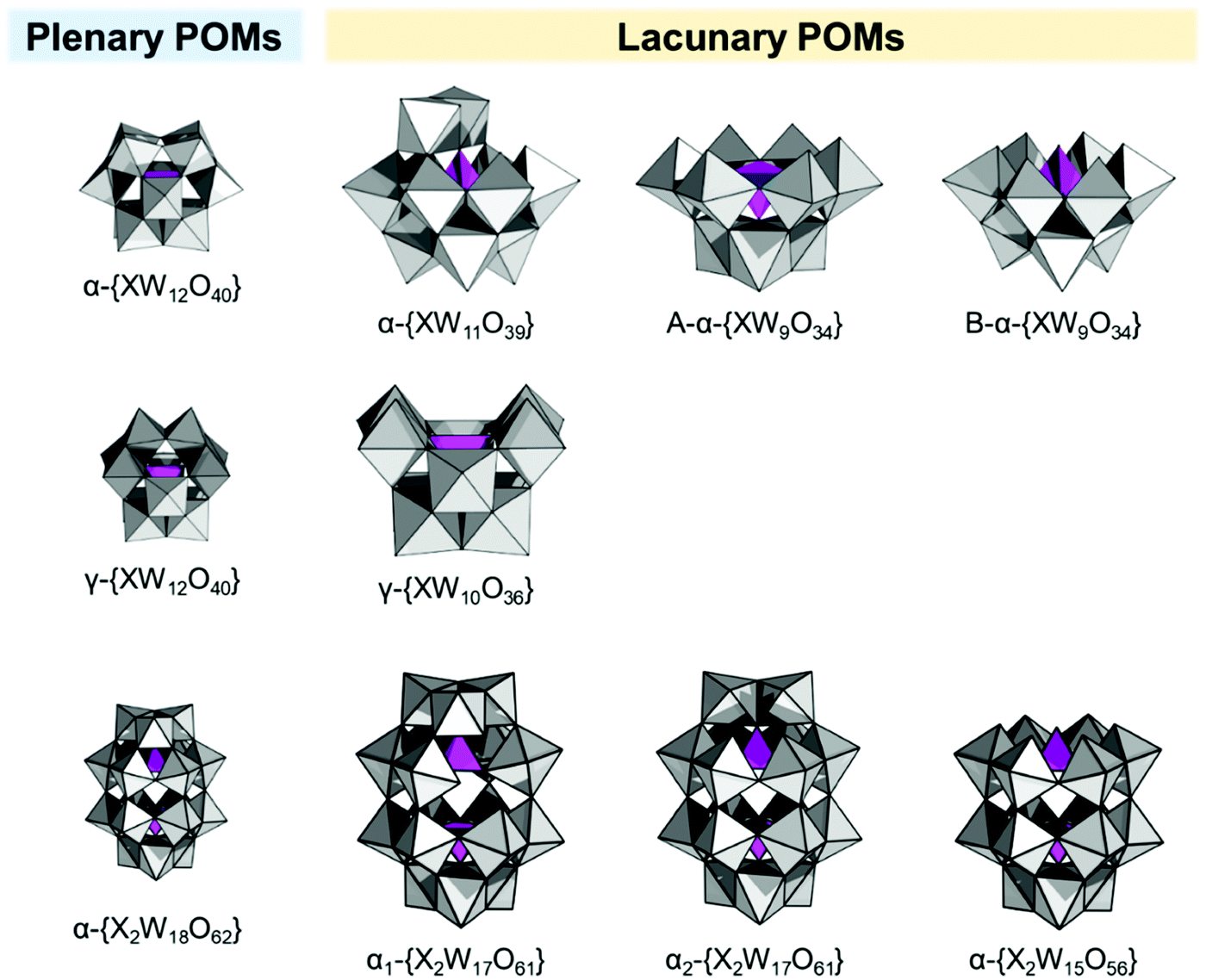 | ||
| Fig. 4 Polyhedral representation of the saturated POMs (α-/γ-Keggin structure and α-Wells–Dawson structure) and the related “unsaturated” POMs (lacunary POMs) relevant for the constitution of hybrid POM platforms. Colour code: {MO6} = grey octahedra, {XO4} = purple tetrahedra. | ||
Removing a triad fragment {M3O6} results in the so-called trilacunary POMs. In α-Keggin, there are two inequivalent triad positions which can be virtually removed leading to the formation of A-α and B-α trilacunary Keggin POMs. Depending on the encapsulated heterogroup, i.e. whether it is a tetrahedral {XO4} or trigonal pyramidal {XO3} group, two general formulae {XW9O34} and {XW9O33} are possible. The A-α isomer is the most commonly observed with the {XO4} heterogroups where X = PV, SiIV and GeIV, while the B-α isomer is common for the {XO3} groups with a lone pair of electrons such as X = AsIII, SbIII and BiIII which contain a lobe of electrons pointing towards the outer of the lacuna (Fig. 4).
All lacunary POMs contain terminal oxygen atoms that are very reactive towards nucleophiles. Nucleophiles can be different cationic or organometallic groups such as {SnR}. Some monolacunary but also dilacunary POMs can react with extended handle-like {R-X-O-X-R} groups where X = P, Si and Ge, affording double-linker sites which are suitable for post-functionalization (see more details in Section 2.4). The trilacunary POMs are also reactive, mainly towards p-block elements, but also to vanadates, allowing the tris-alkoxo functionalization. Recently, Mizuno and co-workers showed that tris-alkoxo functionalization of the trilacunary polytungstates is also possible.62
The Wells–Dawson [X2W18O62]n− structure exhibits a combination of geometric and configurational isomerism.58 Among the different geometry possibilities obtained by rotation of a triad or the guest heterogroup, the D3h structure (Fig. 4), referred to as the α-WD isomer, remains the most stable, and it shows two structurally inequivalent W centers. Substitution of α-WD structure with heterometals leads to two configurational isomers, namely α1-isomer if the substitution occurs at any of the twelve equivalent belt metal centers, or α2-isomer if the substitution occurs at any of the six addenda centres from the two caps. Loss of a triad leads to a single {P2W15O56} structure which can be isolated and further used as a polydentate ligand, thus resulting in the formation of {P2V3W15O62} species in the presence of a vanadium(V) source. The monolacunary α1-/α2-{P2W17O61} isomers and the vanadium-substituted WD polyanion {P2V3W15O62} remain the three main WD POM cores used in the preparation of the corresponding platforms.
2.2. Anderson–Evans (AE) structure
The Anderson–Evans structure can be described as a ring of edge-sharing polyhedra {M6O18} encapsulating a central {XO6} guest.18,63 Depending on the encapsulated guest unit and the overall charge, part of the sharing oxo ligands of the {MO6} guest unit can be formally replaced by tris-alkoxide ligands. Due to their more labile nature, this formal exchange is far more dominant among AE polyoxomolybdates than among polyoxotungstates. Careful control of the degree of protonation can lead to single- or double-sided functionalized hybrid POMs with the general formula δ-{XM6O21((OCH2)3CR)} and δ-{XM6O18((OCH2)3CR)2} respectively (Fig. 5a).63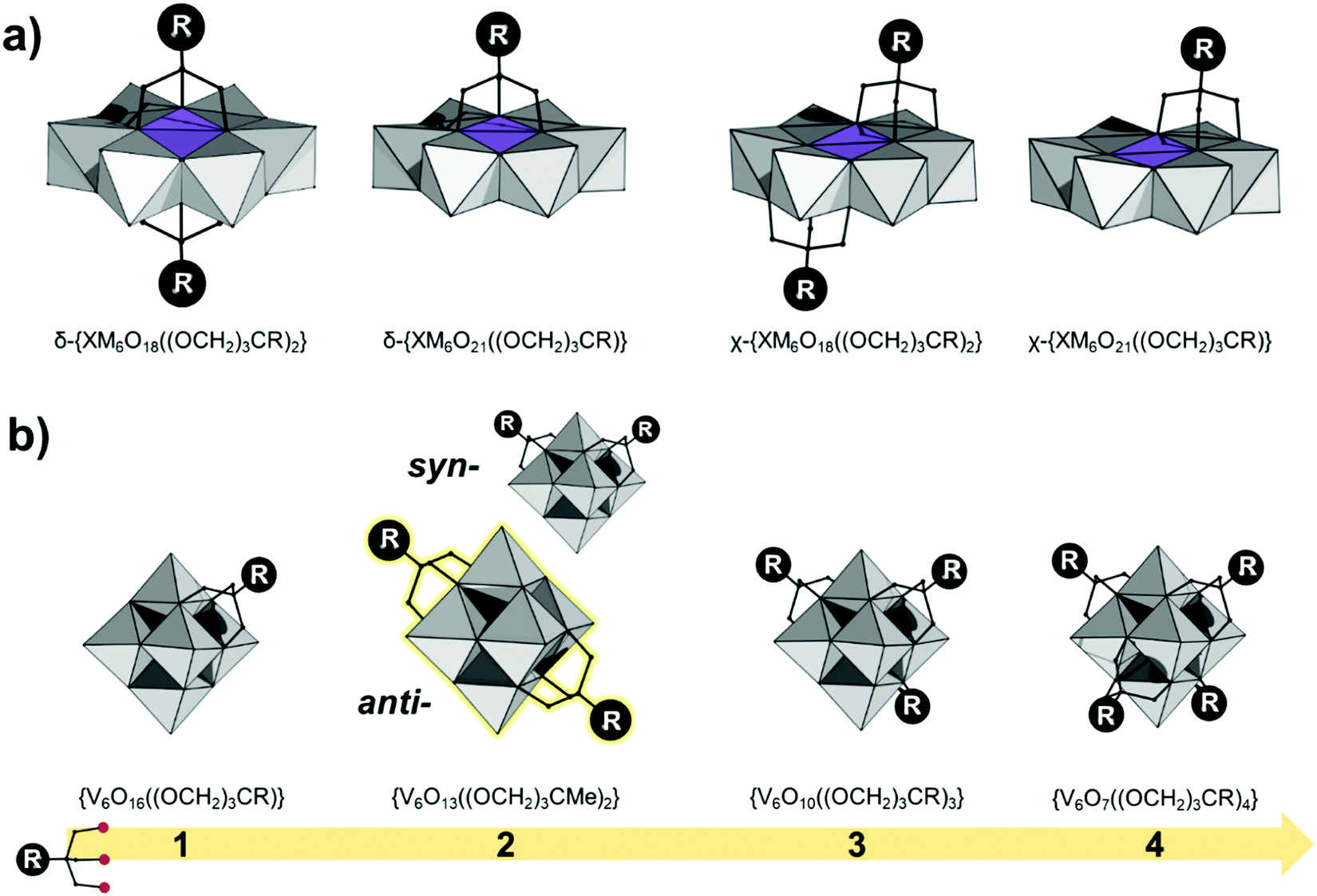 | ||
| Fig. 5 Combination of wireframe and polyhedral representation of (a) tris-alkoxo functionalized AE structures; (b) tris-alkoxo functionalized Lindqvist polyanions. Colour code: {MO6} = grey octahedra, {XO6} = purple octahedra, C = black frame. Hydrogen atoms are omitted for clarity. | ||
Occasionally tris-alkoxide substitution can bind over the triangular cavities of the plenary {XM6O24} structure, by replacing two μ3-oxo ligands from the {XO6} octahedron and a μ2-oxo from the {M6O18} ring. In this case χ-{XM6O18((OCH2)3CR)2} and χ-{XM6O18((OCH2)3CR)2} structures are derived (see Fig. 5a). Among the different hybrid AE platforms, compounds based on δ-{XM6O18((OCH2)3CR)2} are most commonly isolated and investigated (see Section 4).
2.3. Lindqvist (L) structure
The core Lindqvist topology is well-retained among the hexaniobates and hexatantalates; however the Lindqvist hexavanadate structure has not been reported yet.57 Comparing the Shannon–Prewitt ionic radii of VV to other VA and VIA group cations,64,65 Pope argued that the hypothetical [V6O19]8− polyoxoanion should be structurally unstable due to its high negative charge delocalized over the small molecular volume.21 Consequently, {V6O19} fragments are part of polyoxovanadates of larger nuclearity, such as [VV10O28]6− and [VV13O34]3−,19 which have been successfully isolated due to their lower molecular charge density.57However, owing to the stabilization of the tris-alkoxo units it has been possible to isolate the elusive {V6O19} topology.57 Depending on the vanadium precursor and the overall working conditions, different number of μ2-O ligands can be substituted by the terminal oxo groups of the tris-alkoxide, leading to {V6O19−3x−y(OH)y{(OCH2)3CR}x} species where x = 2, 3, and 4, which have been isolated and characterized (Fig. 5b).57 The bis-trisalkoxide stabilized hexavanadates exhibit two geometric isomers where the most frequently isolated isomer is capped in a transoid (anti-) fashion, although the cisoid (syn-) capped isomer has been seldom observed.57 The bis-functionalized anti-isomer is by far the most commonly isolated. Tri- and tetra-functionalized systems are often observed for highly reduced vanadates. Monofunctionalized trisalkoxide Lindqvist-type hexametalates of the type {V3Mo3O16{(OCH2)3CR}} with R = –Me, –CH2OH and –NH2 ligands have been only recently discovered by using microwave assisted synthesis.66 The latter polyanion exists in the form of configurational isomers, where trisalkoxide predominantly binds to the vanadium, while the overall Lindqvist structure is further stabilized by the less labile molybdenum–oxo component.
2.4. Degrees of modularity of the hybrid POM platforms
The modularity of the hybrid POM platforms can be defined by the degree of linker coordination, linker rigidity and linker post-functionalizability. The modularity determines the overall reactivity of the hybrid POM platforms and can influence the type of resulting materials. In this regard, herein we introduce a few additional descriptions which can help to describe the types of platforms that will be introduced over the next sections.Depending on the number of linker groups one can differentiate between “single-linker”, “double-linker”, “triple-linker” or “multiple-linker” modularity. The interlocking scenarios in Keggin and Wells–Dawson type hybrid POMs rely on tris-alkoxides or organo-tin and can be regarded as typical “single-linker” modularity (see Tables 1 and 2). The “double-linker” modularity can be further described based on the relative orientation between the two linkers, that is linear or bent, but also based on whether the same or different functional groups are present at the anchoring terminals (see Fig. 3a and Tables 3 and 4). Triple and multiple-linker functionalities are rare67 and usually relate to the Lindqvist hexavanadate where they can adopt idealized C3v and Td coordination symmetry (see Table 4).
Further on, the degree of the rigidity of the linkers, which is predominantly described by the coexistence of various conformers, can influence the type of interaction between the hybrid POM platforms and other type of materials. For instance, rigid and highly directing ligands may be necessary in the formation of reticular hybrid POM–metal coordinating materials,68 while flexible but strongly binding biotin linkers are suitable for integrating hybrid POMs to biotin binding proteins.69
In the Introduction we listed a number of reactions that can occur at the anchoring group sites and these can be generally grouped as condensation reactions, coupling reactions, click reactions, polymerization reactions and metal coordination reactions (Fig. 6b). It is also noteworthy to mention that depending on the post-functionalizing moiety, single or multiple-modifications introduced in a stepwise manner can be carried on.70
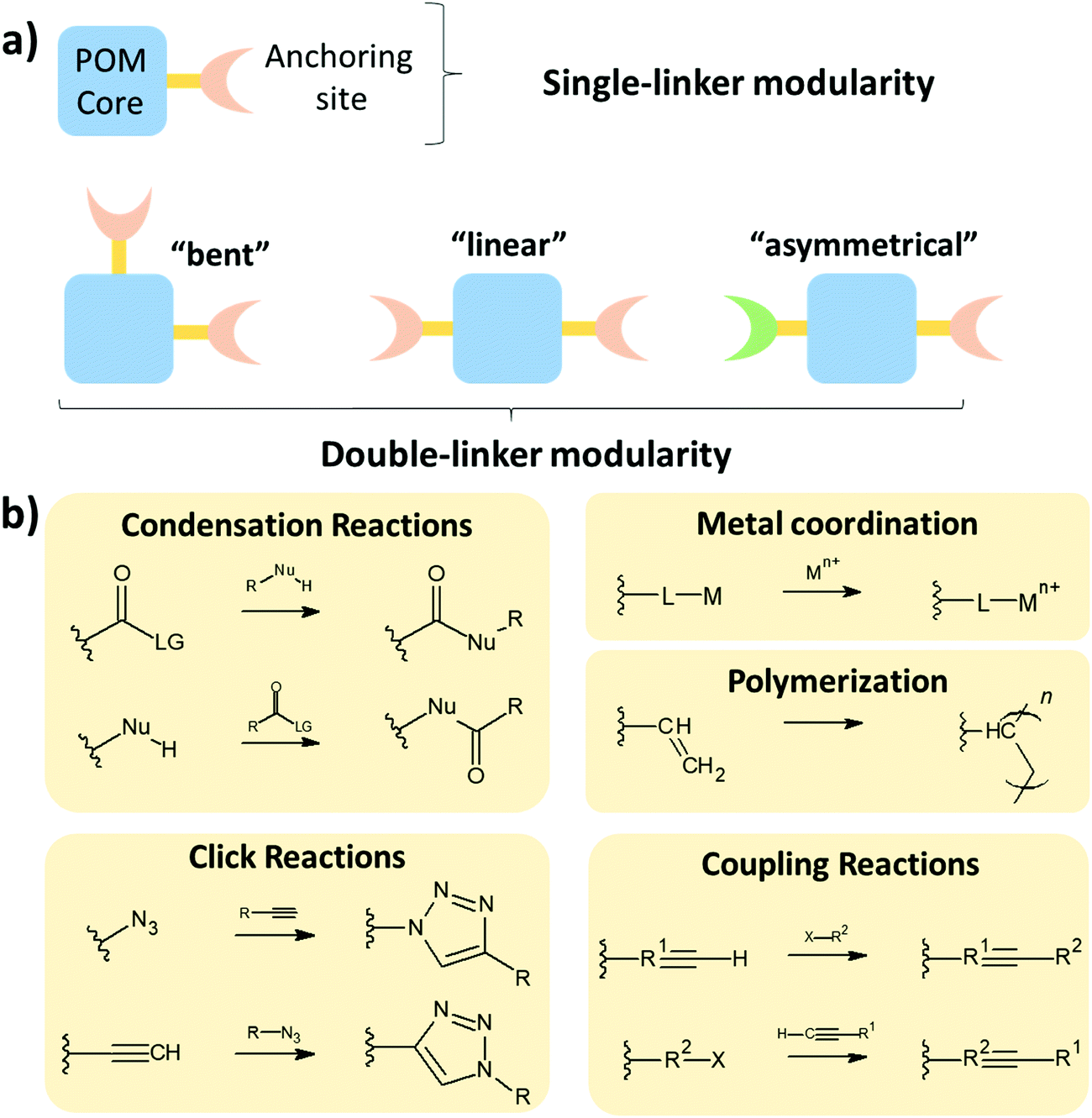 | ||
| Fig. 6 Schematic representation of (a) the most-common modularities of the hybrid POM platforms; (b) typical postfunctionalization reactions taking place at the anchoring terminals, where L = ligand and LG = leaving group. | ||
2.5. Atomistic fine-tuning of the electronic structure
As the hybrid POM platforms mainly consist of metal and oxo atoms, the type of the POM fragment and the bridging unit predominantly affect the overall electronic structure.71 One example is the hexavanadate [V6O13{(OCH2)3CCH2OCH2CCH}2]2− platform (Fig. 7a), which despite post-functionalization retains the highest basicity at the bridging and terminal oxo sites, as revealed by molecular electrostatic potential projections (Fig. 7b). The aforementioned platform exhibits the highest occupied molecular orbital (HOMO) predominantly delocalized over the {V6O19} core and resembling an “oxo-band” (Fig. 7c) and the lowest unoccupied molecular orbital (LUMO) resembling a “metal-band” (Fig. 7d).42 This implies that upon reduction most of the electrons will be (de)localized on the POM fragment rather than on the organic moiety.
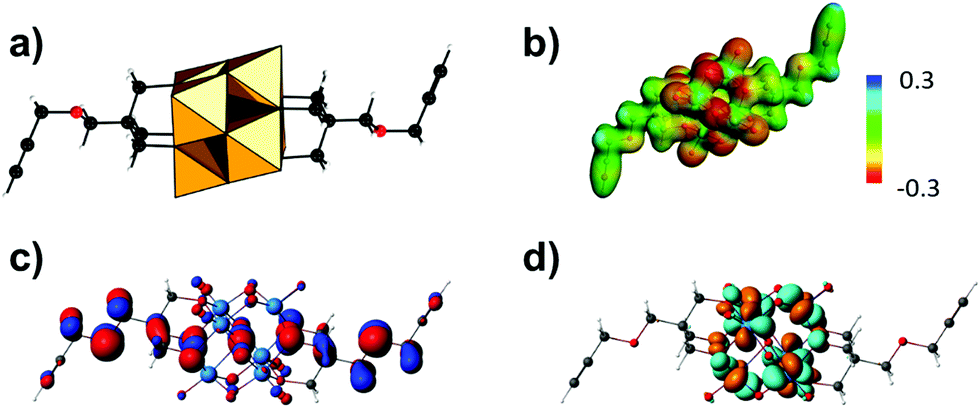 | ||
| Fig. 7 The hybrid hexavanadate platform [V6O13{(OCH2)3CCH2OCH2CCH}2]2−: (a) combination of polyhedral and ball-and-stick representation; (b) molecular electrostatic potential with most basic sites in red; (c) highest occupied molecular orbital (HOMO) resembling a hybridized “oxo-band”; (d) lowest unoccupied molecular orbital resembling a “metal-band”. Colour code: {MO6} = orange polyhedra, C = black, O = red, V = blue, H = white balls. | ||
Post-functionalization can dislocate the HOMO from the metal–oxo core to the (bio-)organic functionality, while the LUMO usually remains on the POM core fragment.42 This allows effective fine-tuning of the HOMO–LUMO gap and occasionally can promote electron transport from the organic to the POM core fragment, which is an important process for fluorescence and non-linear optical phenomena.56
3. Keggin and Wells Dawson-type hybrid platforms
The Keggin (K) and the Wells–Dawson (WD) polyanions show high similarities to one another as both represent shell shaped POMs encapsulating heterogroups. As comparable platforms have been developed over time it is convenient to cover the two POM topologies together, while keeping in mind the distinctions that exist between the Keggin and the Wells–Dawson hybrid platforms. The first significant difference is that the preparation of tris-alkoxo functionalized species is well-described in the case of the Wells–Dawson structure, as the mixed-addenda [P2W15V3O62]9− can be isolated and reacted with various tris-alkoxides.72–74 At the same time, only one {GeW9V3} Keggin POM conjugated with a tris-alkoxo ligand has been described.75 The second difference is in the reactivity of lacunary K and WD POMs. The lacunary Keggin heteropolytungstates such as trilacunary {EW9}, dilacunary {EW10}, and monolacunary {EW11} (E = P, Si, As, Ge) anions can react with (alkyl-O)3Si-R species. Such variety of starting lacunary polyoxometalates lead to diverse hybrid POM molecules containing the {R-SiOSi-R} fragment in mono- and dilacunary K POMs, and {(R-SiOH)3} or {(OSi-R)3(Si-R)} fragments in trilacunary POMs. However, in the case of WD, only the monolacunary [P2W17O61]10− anion was shown to interact with (alkyl-O)3Si-R, giving only one available type of the hybrid platform [P2W17O61{O(Si-R)2}]6−. The transformations of ligands coordinated to all-inorganic heterometal-substituted Keggin and Wells–Dawson POMs such as {XM11M′-L} or {P2W17M′-L} (where L is an inorganic ligand) have been described elsewhere76–87 and will not be the focus of this review. All reported platforms for Keggin and Wells–Dawson based POMs are summarized in Tables 1 and 2 respectively, where they are grouped based on the structure of the POM and the type of the terminal reactive group. Most platforms so far address connectivity between the POM and the functional organic molecules via amide, ester, thioester, sulfonamide and imide bond formation; however a few other types of organic couplings have been reported recently.3.1. Condensation reactions
This type of reactions lead to the formation of amide, ester, thioester, sulfonamide, imide, imine and other bonds, producing also water or other small molecules as a by-product. In the case of the Keggin species, a large series of carboxylic group-terminated Keggin POMs [XW11O39{M′(CH2)2COOH}]5− (M′ = Si, Ga, Ge) were reported by Sazani and Pope in 2004.219 The further post-functionalization of these hybrid POMs has been achieved by the formation of the glycinate derivative [SiW11O39{Ge(CH2)2CO-NHCH2COOCH3}]5− and ethyl ester [SiW11O39Ge(CH2)2CO-OCH2CH3]5−. Later, TBA4[α-PW11O39{Sn(CH2)2-COOH}] was conjugated step-by-step with a H2N–biotin derivative and streptavidin–agarose beads to produce a nanomolar non-competitive inhibitor of protein kinase CK2.146 [PW11O39{Ge(CH2)2COOH}]4− was synthesized by the same procedure and coupled with propargylamine in the presence of trimethylamine and isobutylchloroformate.91 Modification of tin-substituted Keggin with silsesquioxanes resulted in the formation of molecular Janus-like particles.128 In 2018 formation of nonmetric spheres based on the hybrid of phenylalanine and Sn–Ar-containing Keggin POM was observed.151The Keggin-type POM [PW11O39]7− has been also functionalized with two reactive amino groups through the condensation of (alkyl-O)3Si-Ph-4-NH2 into the lacuna of the POM.185 The resulting product [PW11O39{O(Si(CH2)3-NH2)2}]5− was successfully grafted on a modified Au(111) surface and used as a bridging nanostructured object for protein immobilization.119 The alkyl-containing [(SiW11O39)O{Si(CH2)3-NH2}2]4− POM hybridized with an azobenzene derivative opened a pathway to fabricate thermotropic liquid-crystalline nanomaterials134 (Fig. 8) that demonstrated the stabilization and optical switching of blue phases.220 Varying the length of the linker between POM and azobenzene provided an opportunity to tune the self-assembly by using different ratios of CH3CN/H2O solvents, which allowed the switching between 0D and 1D structures.135 The reaction of carbon nanotubes CNTs-COOH and [(SiW11O39)O{Si(CH2)3–NH2}2]4− resulted in a nanocomposite material of CNTs-{SiW11}, which remained stable after 100 cycles of charge–discharge, with high discharge capacity and good capacity retention.221 The sulfonamidation of an alternative NH2-terminated hybrid [γ-SiW10O36{O(Si(CH2)3-NH2)2}]4− POM was demonstrated by Carraro, Bonchio and co-workers by reaction with dansyl chloride222 and pyrene sulfonyl chloride110 in the presence of triethylamine.
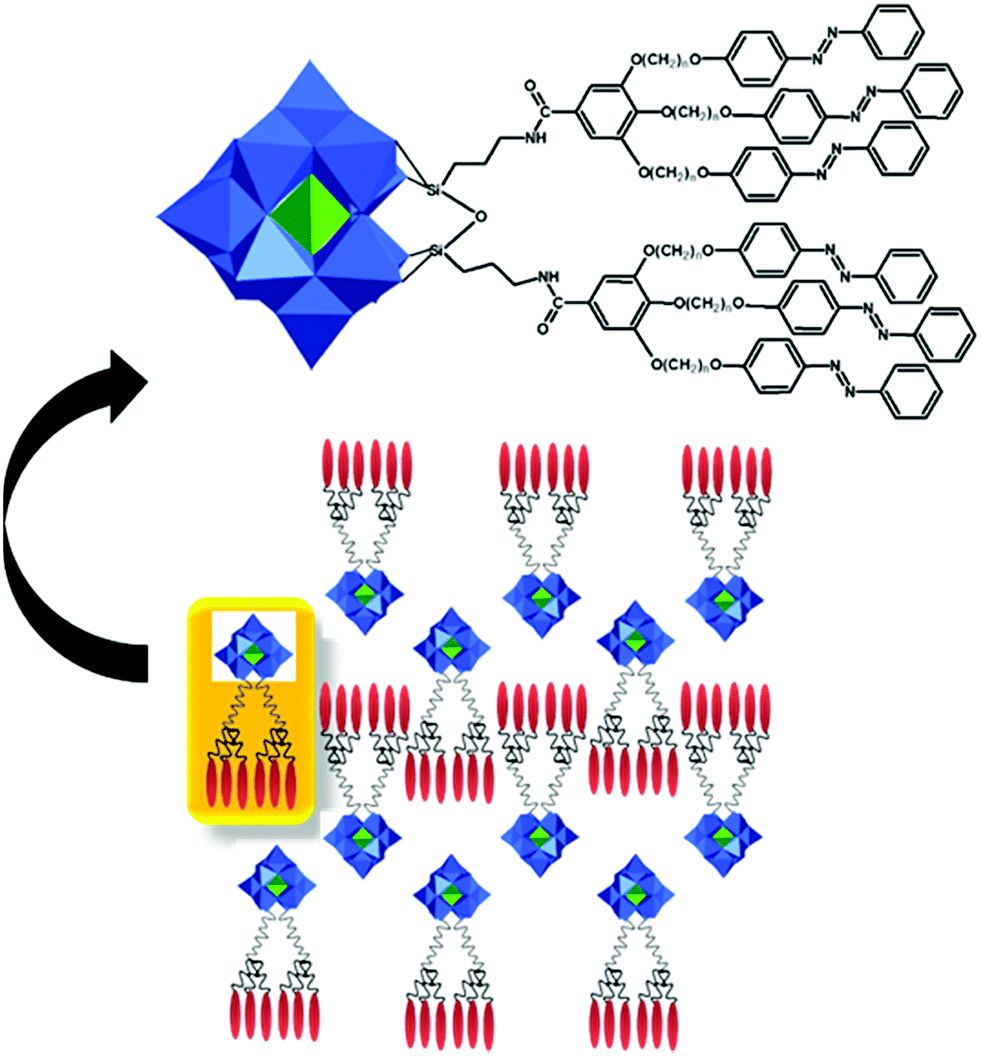 | ||
| Fig. 8 Schematic representation of azobenzene-modified Keggin POM hybrids. All structures are shown in ball-and-stick and polyhedral representation using the following colour code: C = black chains, {SiO4} = green octahedra and {WO6} = blue octahedra. | ||
In the case of the Wells–Dawson structure, in 2003 Bareyt et al. described the synthesis of two mono-organotin functionalized Wells–Dawson POMs, α1- and α2-[P2W17O61{Sn(CH2)2COOH}]7−, bearing a COOH group in the organic fragment.189 The coupling of these POMs with various amines and alcohols was achieved through activation of the carboxylic acid by either EEDQ or isobutylchloroformate in combination with tBuOK, as activating agents.184,189 Moreover, in the absence of any nucleophile an activated POM intermediate could be isolated where the organic chain was folded and inserted into the POM framework.194 Subsequently, this reactive intermediate was further interacted with amines and thiols to form the corresponding amide and thioester linked products.190–192,223 In a recent study these cycle-containing Wells–Dawson POMs were decorated with polyglycine chains which fold toward the POM resulting in zipper hydrogen bond networks depending on the length of the chain.198 A similar reactivity was demonstrated with [SiW11O39SnCH2CH2C(O)]4–223 POM, allowing a consecutive step-by-step functionalization with an amino–azide linker and ferrocene. The modified WD–DNA primers, which were synthesized by the reaction of an NH2-containing oligonucleotide and [P2W17O61SnCH2CH2C(O)]6−, demonstrated an opportunity for electrochemical detection of the PCR product.199 The 7-deaza-7-propargylamino purines were covalently attached to WD and K POMs (Fig. 9a) through PCR amplification, providing an easy electrochemical detection of the double-stranded DNA product (Fig. 9b).162 Using the same post-functionalization strategy, dideoxynucleotides were conjugated with WD and K Sn-substituted POMs through amide bond formation, and incorporated into DNA with relatively inexpensive Terminator® polymerase (Fig. 9b). Upon grafting on a gold electrode these hybrids were used for the detection of single nucleotide polymorphism during electrochemical primer extinction reaction.224
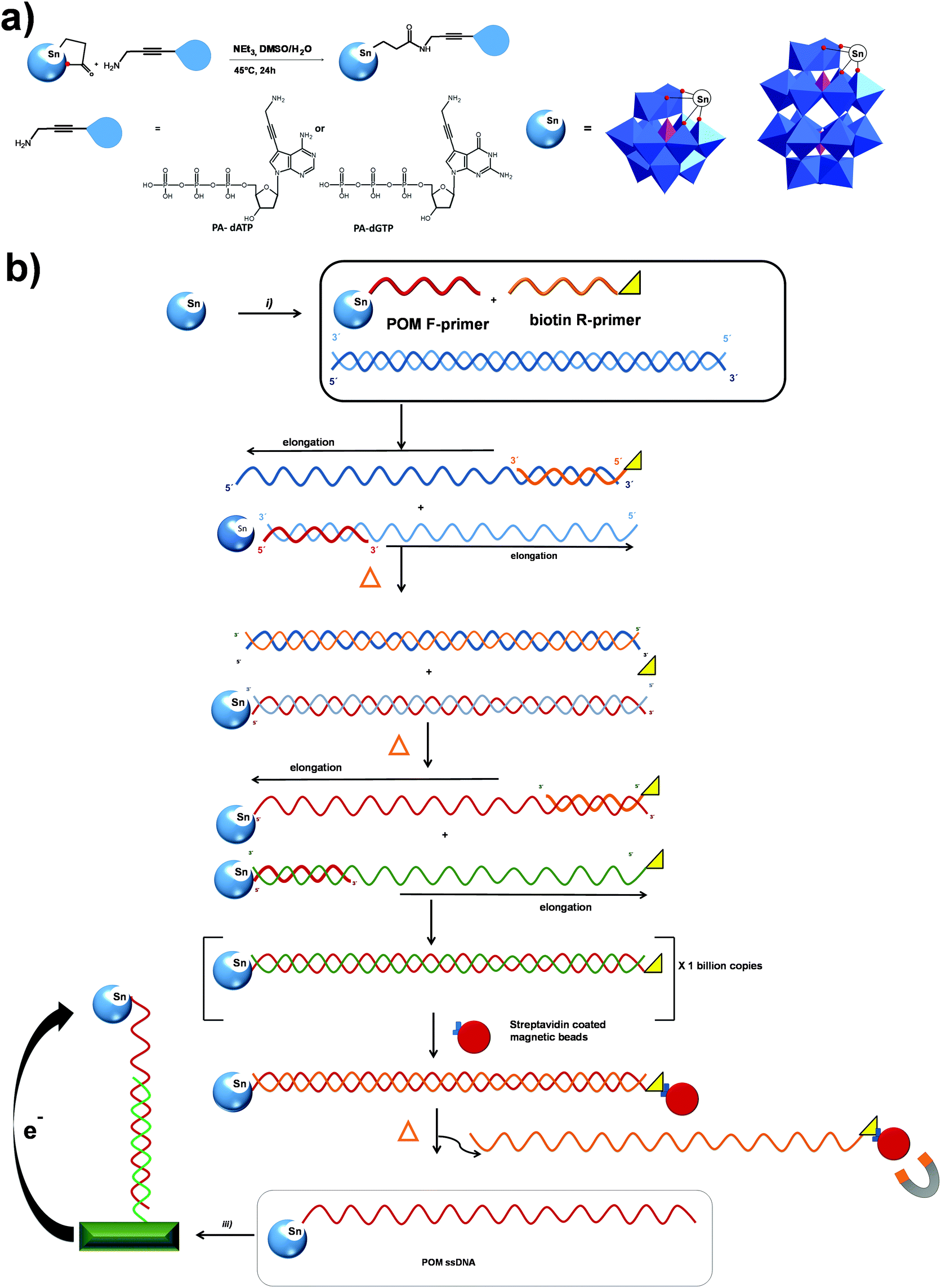 | ||
| Fig. 9 Schematic presentation of (a) the synthesis of POM-modified nucleotide triphosphates; (b) (i) covalent coupling of an oligonucleotide to a functionalized POM, (ii) PCR amplification with POM-forward primer and biotin-reverse primer, followed by streptavidin-coated magnetic-bead-mediated generation of single-stranded DNA (ssDNA). (iii) Hybridization of ssDNA to a surface-tethered probe and electrochemical detection. All polyoxometalate moieties are shown in polyhedral representation using the following colour code: {PO4} = pink octahedra and {WO6} = blue octahedra. | ||
The activated acyl-POMs [SiW11O39SnCH2CH2C(O)]4–223 and [P2W17O61SnCH2CH2C(O)]6− can react with amines,190194 offering a very convenient approach for the post-functionalization of polyoxometalates. The WD-POM-poly(N,N-diethylacrylamide) hybrid was obtained by the described method and showed thermoresponsive solubility/aggregation of micelles.195 Squaramide-containing organo-POMs were used as bifunctional catalytic systems for the activation of nitroalkenes via H-bonding.197 An imidazolidinone-POM achiral hybrid catalyst demonstrates a unique activity in asymmetric Diels–Alder reaction with good yields and high enantioselectivities, using POM as a source of chiral information (Fig. 10a).196 Alternatively, chlorine-functionalized tin-substituted82 [PW11O39{SnCl}]4− can react with various n-nucleophiles, including primary, secondary, and tertiary amines and tertiary phosphine, resulting in POM-Sn(Cl)-N+R′R′′R′′′ or POM-Sn-NR′R′′ derivatives of amines and amino acids.225 The unusual double functionalization was described for [α2-P2W17O61(Sn-Alk)]7− POMs that can react with isocyanates aryl-NCO producing regioselective double functionalized polyoxotungstates.183
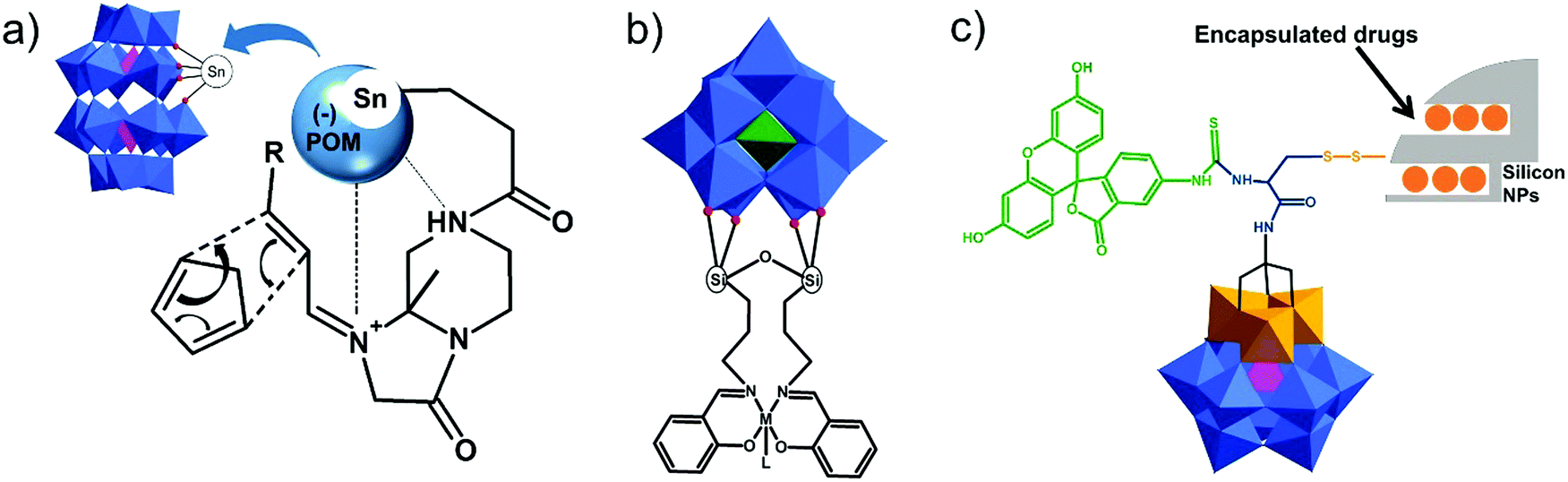 | ||
| Fig. 10 Schematic representation of (a) the mechanism of activation of the substrate by a chiral POM-based artificial Diels–Alderase; (b) the structure of catalytically active POM–salen–M compounds (M = Mn(II), Co(II), Ni(II), Pd(II), Cu(II), Fe(II), Mo(VI)); (c) the fluorescein–POM–NP hybrid molecule with the encapsulated drug. All polyoxometalate moieties are shown in polyhedral representation using the following colour code: O atoms = red sphere, {PO4} = pink octahedra, {SiO4} = green octahedra, {VO6} = orange octahedra and {WO6} = blue octahedra. | ||
A promising nanosystem for catalyst applications was prepared based on the reaction of [SiW10O36{O(Si(CH2)3-SH)2}]4− sulfide and gold nanoparticles, demonstrating the absence of the desorption of [POM(SH)2]4− from the particles.226 This POM platform can also stabilize the formation of palladium nanoparticles which are effective for the aerobic oxydehydrogenation of vinylcyclohexene and vinylcyclohexane to styrene.227 WD bearing the same sulfide-functionalized ligand was grafted on the benzyl chloride functionalized nanolatex cores.209 This nanocomposite material demonstrated colloidal stability in solution and due to the protected Wells–Dawson shell it also showed good contrast in TEM analysis.209
Only a few examples on the use of imine linkers between POMs and organic fragments can be found in the literature. In classic organic chemistry the reaction between amines and carbonyl functions to form imines is often followed by the reduction of the latter to create a hydrolytically stable amine bond. However, as POMs are extremely sensitive towards the addition of reducing agents, this reduction step is not suitable for POM post-functionalization. As a result, the application of these hybrids is restricted to non-aqueous conditions. The amino-terminated Keggin type hybrid platform [SiW11O39{O(Si(CH2)3-NH2)2}]4− has also been applied in post-functionalization by formation of imine bonds. The combination of noble metal complexes and POMs is one of the potential pathways towards novel catalytic systems with enhanced catalytic properties. The first example of a POM–salen hybrid with the imine link was synthesized by Neumann and co-workers,132 and later this and other M(salen)–POM complexes (Fig. 10b, M = Mn, Co, Ni, Pd, Mo) were tested as catalysts.132,228 Depending on the coordinated transition metal (Fig. 10b), the POM–salen–metal hybrid demonstrated activity in various reactions such as oxidation of alkanes and alkenes by hydrogen peroxide (M = Cu(II), Fe(II)),139 Suzuki coupling (M = Pd(II)),140 and alkene epoxidation (M = Mo(VI)).141,229
In contrast to monovacant and divacant POM derivatives, trivacant POM platforms are based on trisubstituted anions such as TBA3[PW9O34{tBuSiOH}3]88 that can react with various Cl3SiR88 or Cl3GeR91 to produce {PW9{(tBuSiO)3X-R}}, where R represents (CH2)n-CHCH2230 or COOH and X = Si or Ge. The amidation of the COOH function with amino acid derivatives leads to the chiral hybrid POM-based compounds, demonstrating the potential for creating novel stereoselective catalysts.231 Later {PW9{(tBuSiO)3Ge-R}} and {PW11{Ge-R}} decorated with the carboxylic group were coupled with amines including propargyl amine, resulting in an alkyne product which is suitable for click chemistry.91 A prospective functional Keggin-type platform was obtained by the reaction of [GeW9V3O40]7− with (HOCH2)3CNH2, followed by coupling to Boc-cysteine. This bifunctional Boc-protected construct [GeW9V3O37{(OCH2)3CNHC(O)CH(-NH-Boc)CH2-SH}]4− was further hybridized through the H2N-group with fluorescein-5(6)-isothiocyanate, and through the HS-group with doxorubicin-saturated thiolated mesoporous silica nanoparticles, leading to a multidrug system (Fig. 10c).232 The development of porous silica materials containing hybrid POMs was achieved through the reaction between lacunary POMs and tetraethylorthosilicate (TEOS).233 For example, a novel strategy to obtain covalently grafted silica-supported POMs was developed based on the interaction of the SiCl-modified SBA-15 mesoporous silica support and [SiW10O36{Si(Me)OH}2]4−.104 Trilacunary POMs also provide promising hybrid platforms for post-modification. For example, [AsW9O33{P(O)CH2CH2COOH}2]5− was covalently bound with benzylamine or with H2N-functionalized SBA-15,101 resulting in novel catalytically active materials.234 By the same approach, NH2-functionalized magnetic iron oxide nanoparticles were grafted with [AsW9O33{P(O)(CH2)2COOH}2]5− resulting in the increased stability of the combined material during processing.102 The covalent hybrid of SBA-COOH and [PW9O34{As(O)Ph-4-NH2}2]5−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 235 also demonstrated activity in the epoxidation of alkenes.234
235 also demonstrated activity in the epoxidation of alkenes.234
3.2. Coupling reactions
The Sonogashira reaction affords convenient coupling of terminal alkynes with aryl or vinyl halides and has motivated the design of various Keggin and Wells–Dawson hybrid platforms. In this context, Proust, Izzet and co-workers published a series of Keggin and Wells–Dawson platforms. Four different iodo-aryl functionalized POMs have been developed for this purpose: the mono-functionalized Keggin and Wells–Dawson POMs (Fig. 11a), [α-PW11O39(SnAr-I)]4− and [α2-P2W17O61(SnAr-I)]7−,147 [α-PW11O39(GeAr-I)]4−,153 [α-SiW11O39{GeAr-I}]5−,153 [α-PMo11O39{GeA-I}]4−,143 and di-functionalized Keggin and Wells–Dawson POMs (Fig. 11a), [α-PW11O39{O(SiAr-I)2}]3− and [α2-P2W17O61{O(SiAr-I)2}]6−,123,147,185 [α2-P2W17O61{P(O)ArCCH}]6−.205,213 These POM platforms have been mainly used for coordination to Ir(III)147,186 or Ru(II)/Ru(III)154,155,236 containing photosensitizers.123 In addition they have been applied for the production of photochromic compounds with grafted organic groups such as spiropyran124 or reacted with hydrophobic molecules to produce self-assemblies.213 The alkyne-containing [PW9O34{(tBuSiO)3Ge(CH2)2CONHCH2-CCH}]3− has been electrografted onto n-type Si(100) leading to electrochemically active films.91 In 2013 Rinfray et al. synthesized a Ge-substituted Keggin platform bearing NH2-, N3Et2-, and N2+-terminated derivatives for Sonogashira coupling.153 The diazonium-terminated hybrid was shown to be suitable for grafting on silica (Fig. 11b), leading to self-assembly of monolayers.152,153 Grafting on epitaxial graphene resulted in a hybrid material exhibiting POM-layer charge transfer.237 The same tungstophosphate153 was immobilized on gold or glassy carbon surfaces by electrografting (Fig. 11b), chemical grafting and peptidic coupling.156 The NH2-grafting derivative of the K-type POM was coupled with thioctic acid through amidation reaction. The resulting hybrid demonstrated a template effect for self-assembly of the 2D supramolecular honeycomb network built on hydrogen bonding of perylene-3,4,9,10-tetracarboxylic acid diimide and 1,3,5-triazine-2,4,6-triamine after grafting on the Au(111) surface.157 The sulphide-bearing {PW11Sn} POM was grafted on a Au electrode and the resulting conjugate was tested in electron transfer processes.149 Later, the [α-PMo11O39(SnAr-I)]4− platform143 (Fig. 11a) was developed using similar conditions and successfully coupled with alkyne ferrocene;143 the POM-linker-diazonium hybrid was obtained through a triazene derivative.238 Recent research has also demonstrated a rapid photoinduced intramolecular electron transfer in Keggin-boron-dipyrromethene hybrids (Fig. 11a).125
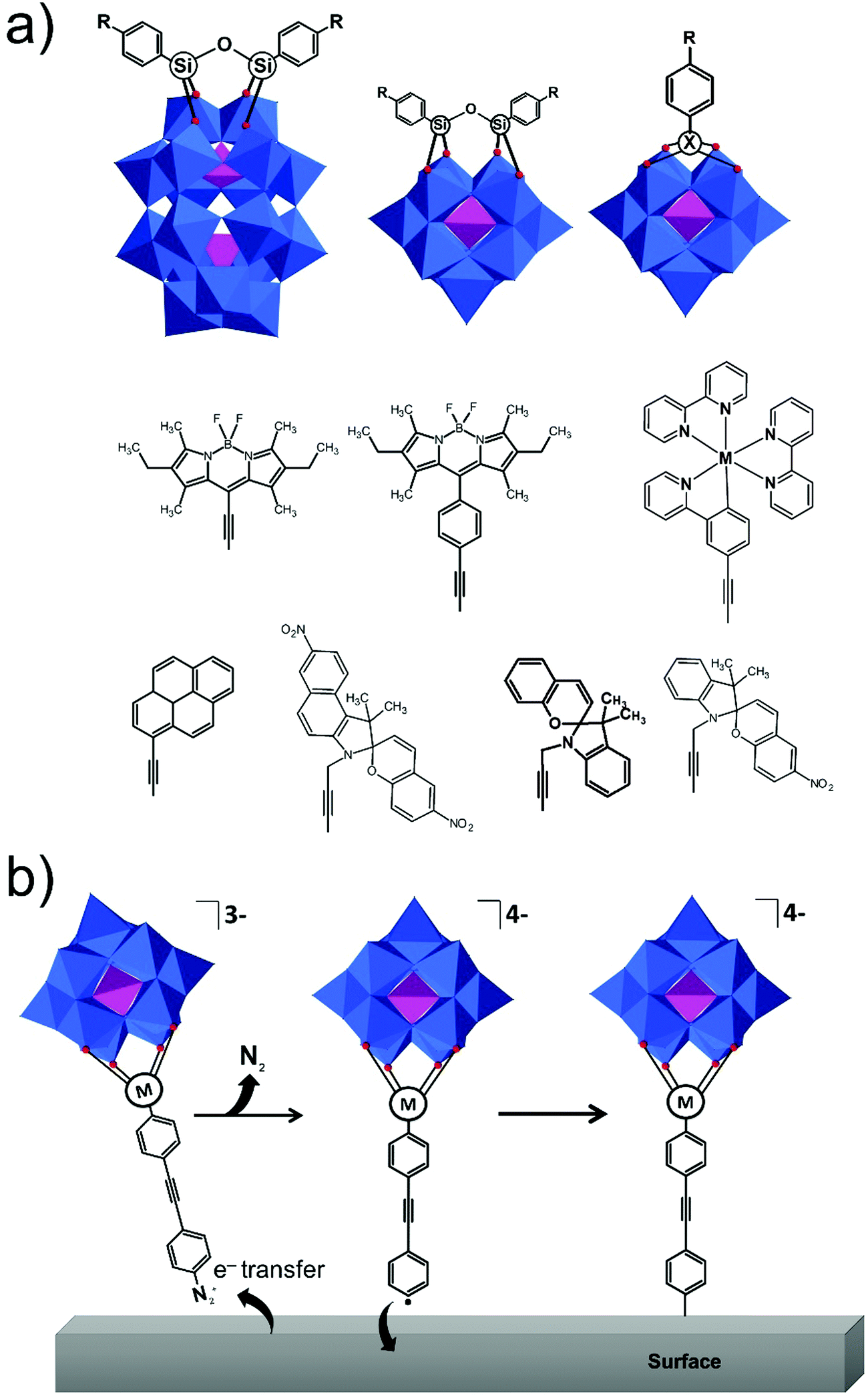 | ||
| Fig. 11 Schematic representation of (a) hybrid POMs suitable for the coupling reactions and various functional groups that have been employed so far; (b) the mechanism of covalent grafting of diazonium post-functionalized molybdenum and tungsten POMs on a surface S (S = Au(111), glassy carbon, silicon and graphene). All polyoxometalate moieties are shown in polyhedral representation using the following colour code: O atoms = red sphere, {PO4} = pink octahedra, {SiO4} = green octahedra, {MoO6} = orange octahedra and {WO6} = blue octahedra. | ||
3.3. Click chemistry
Over the past years, click reactions have been gaining increasing interest as they can be applied under relatively mild conditions allowing coupling of the POM hybrid platform with virtually any organic molecule with a suitable connecting functional group. The Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC) is a very popular reaction that has motivated the design of respective hybrid POM platforms. In 2007, Micoine et al. reported the synthesis of several N3− and HCC-terminated organotin Keggin and Wells–Dawson POMs and their subsequent use as a post-functionalization platform in a CuAAC reaction.159 The initial reactions in this study using typically low CuSO4/sodium ascorbate concentrations in a 1:2 acetonitrile/water solvent mixture only produced traces of the desired product. The lack of reactivity was ascribed to ion-pairing between the positively charged copper ions and the negatively charged surface of the POM, thereby decreasing the concentration of the catalyst near the reaction center. By switching the ratio of CuSO4/sodium ascorbate to 1:10 the reaction could be driven to completion. Due to the high copper concentration and ion-pairing, all copper ions had to be removed after the reaction by using ion-exchange in order to obtain the pure tetrabutylammonium salt of the hybrid POM. Following the former study several other works with minor modifications on the synthetic protocols have been reported.200,205,208,215,223,239 Hence WD-porphyrin and perylene (Fig. 12)205,239 hybrids were made and studied as intramolecular electron transfer systems and for artificial photosynthesis.215 The α2-WD bearing two N3 terminal groups also opens an easy way for covalent immobilization inside alkyne-grafted bezylamine-styrene-divinylbenzene resin, improving the sustainability of the catalytic system for oxidation of organic sulfides.207 The same POM was used to synthesize propargyl-terminated poly(3-caprolactone) hybrids as promising materials for use in medicine with precisely controlled molecular structure, controllable molecular weight, and narrow molecular weight distribution.208 A mild post-functionalization method based on copper(I) catalyzed cycloaddition was developed for less stable {P2W15V3} by functionalization of POM with an azide.169
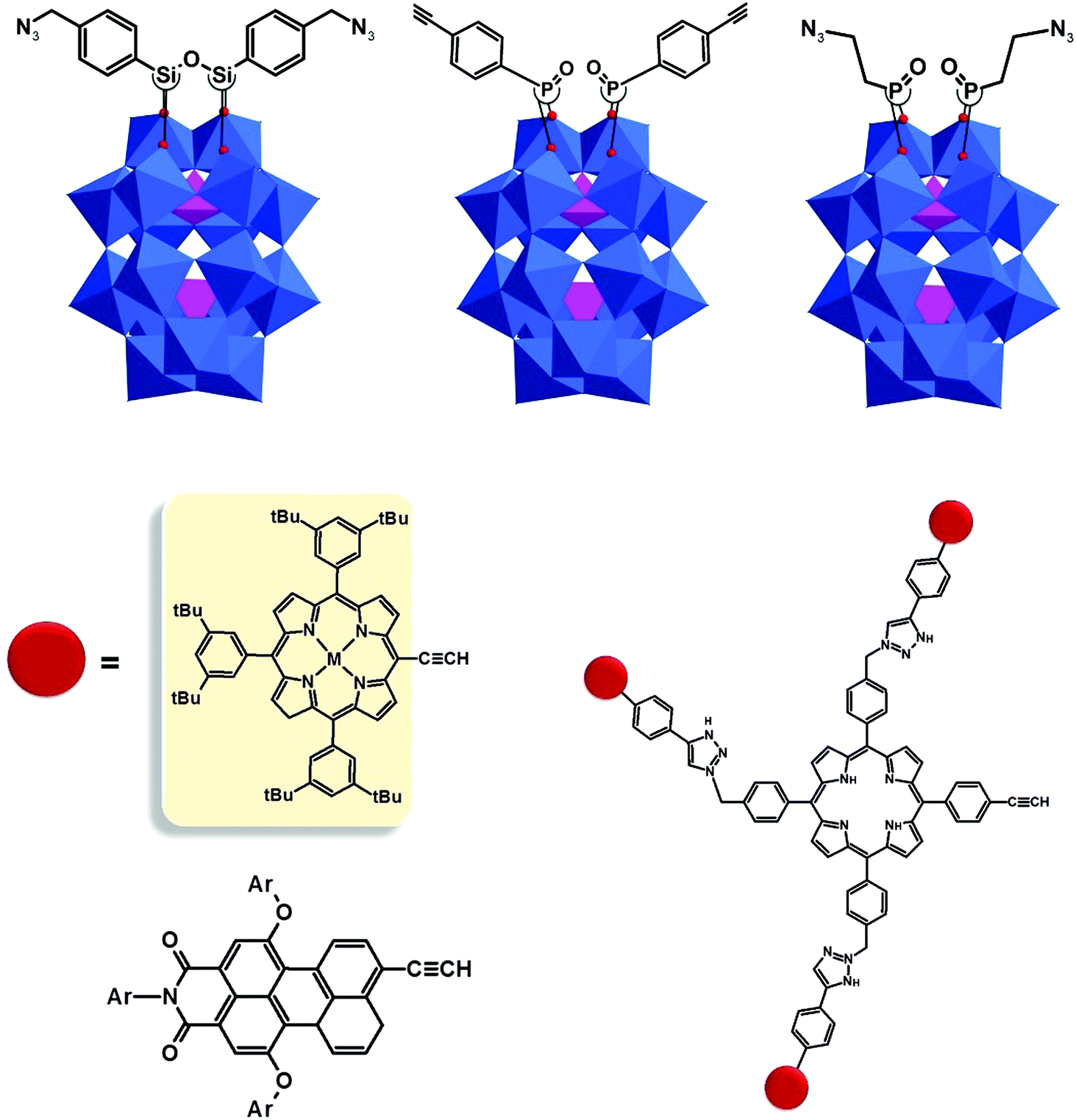 | ||
| Fig. 12 Schematic representation of WD POMs with alkyne and azide functions and clickable polyaromatic ligands suitable for artificial photosynthesis. All polyoxometalate moieties are shown in polyhedral representation using the following colour code: O atoms = red sphere, {PO4} = pink octahedra and {WO6} = blue octahedra. | ||
3.4. Polymerization reactions
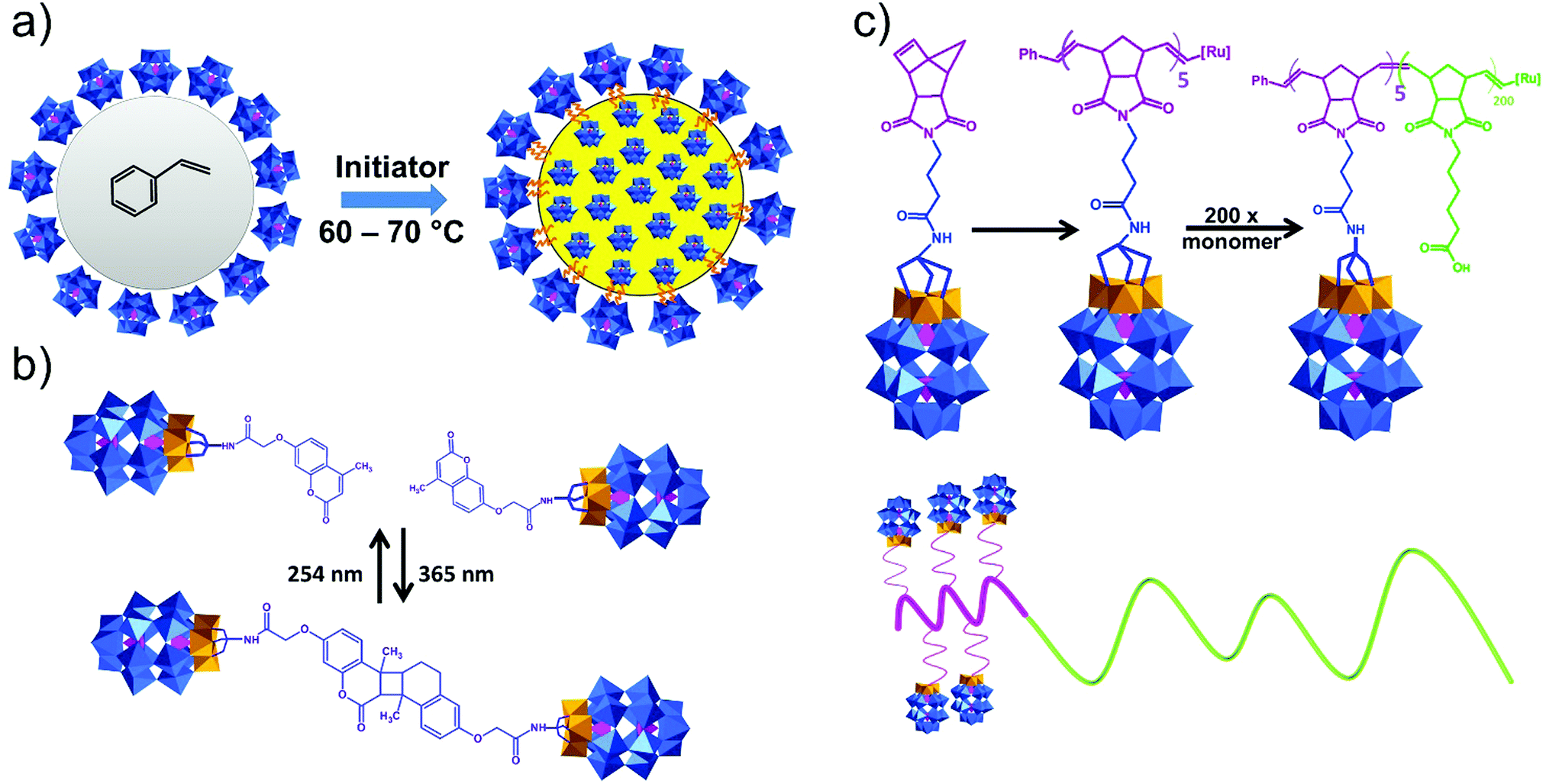
Post-functionalization of hybrid POMs by co-polymerization with various organic monomers provides a promising opportunity for combining the unique catalytic and redox properties of POMs with the flexibility, stability and plasticity of organic polymers. This combination allows the design of novel materials for application in catalysis and electronics. While monovacant Keggin and Wells–Dawson-type POMs provide a typical platform for difunctionalization, other multivacant Keggin-type POMs have shown more complex substitution patterns with organosilyl groups. These hybrid Keggin and Wells–Dawson based platforms can undergo polymerization reactions.240 In order to produce polymers, one approach is to devise polycondensation of difunctional precursors which link together POMs to form a polymer chain. Another approach is to post-functionalize already formed polymers or eventually to devise (co-)polymerization of hybrid POM monomers containing a polymerizable group such as vinyl (-CHCH2) or 3-(methacryloxy)propyl (-(CH2)3OC(O)C(Me)CH2) groups (Fig. 13).130,240,241 The first work on polymerization of functionalized TBA4[α-SiW11O39{O(Si-chain-CHCH2)2}] in the absence of a copolymerizing agent was reported by P. Judenstein in 1992 (chain = none, CH2, (CH2)3OC(O)Me, 4-Ph) (Fig. 13).130 Later it was found that a copolymer of H4[α-SiW11O39{O(Si-CHCH2)2}] and methyl acrylate coating resulted in high loadings of immobilized POM into films and provided high proton conduction.241 By carefully controlling the stoichiometry of the reagents, the group of Thouvenot was able to synthesize both the pure di- and tetrasubstituted derivatized divacant Keggin-type POMs with the general formula [γ-SiW10O36{O(SiR)2}]4− and [γ-SiW10O36(OSiR)4]4− (R = H, CHCH2, (CH2)3OCOC(Me)CH2).242 Both the di- and tetramethacrylate substituted compounds were further used as monomers in polymerization reactions to create hydrogels.91 A large array of copolymers based on the same POMs, butyl acrylate and hexanediol diacrylate have been characterized and examined as proton conductive membranes that demonstrated promising results.131 Several examples of covalently immobilized hybrid catalysts were reported, including several polymer composites obtained through copolymerization of hybrid POM monomers with acrylic monomers.106,243 A series of copolymers of Keggin type POMs, decorated with {Si(CH2)6-CHCH2}, and methyl acrylate were catalytically active in H2O2-based oxidation of sulfides and thiophenes.244 In the case of Keggin containing hybrid polymers, the copolymerization of [γ-SiW10O36{O(Si(CH2)3OC(O)-C(Me)CH2)2}]4− with methyl acrylate in acetonitrile at 70 °C in the presence of AIBN as the initiator is a typical example of polymerization reaction with POMs.115 Incorporation and copolymerization of the silane-NH2-modified {SiW11} POM into the polyimide matrix was shown as a convenient route for the synthesis of hybrid films with an ultra-low dielectric constant.112,133 Copolymerization of alkene-grafted [(SiW11O39)O{Si(CH2)3NHCO(CH2)2OOC-CHCH2}2]4− resulted in photopolymerizable POM-based nanoparticles, which formed a polymer network that exhibited a reduction of voltage during the switching of the blue phase material (Fig. 13).41
 | ||
| Fig. 13 Schematic representation of hybrid polyoxotungstate platforms bearing polymerizable organosilane pendants. Colour code: O atoms = red spheres, {PO4} = pink octahedra and {WO6} = blue octahedra. | ||
In the case of Wells–Dawson based hybrid platforms, Hasegawa et al. in 2007 reported the free radical copolymerization of the hybrid Wells–Dawson heteropolyacid H6[α2-P2W17O61{O(Si(CH2)3OC(O)-C(Me)CH2)2}] with methyl methacrylate.243 The acidity of the POMs was retained in the resulting polymer and hence created an interesting application as solid acidic catalysts.202 A different approach was used by Han et al., where the hybrid POM was not used as a monomer but as a macro-initiator for the polymerization of styrene, resulting in an amphiphilic compound containing a POM head group and a hydrophobic PS tail.170 For this purpose, the authors synthesized an alkyl bromide functionalized {P2W15V3} POM, after which the ATRP reaction with styrene was performed at 110 °C in DMF using a CuBr/PMDTA catalyst. A novel and convenient design was developed by Chen and colleagues to prepare covalently decorated polystyrene nanoparticles during the copolymerization reaction of styrene and the POM–methylmethacrylate surfactant agent (Fig. 14a).97 Copolymers of POM-based hybrid TBA5H[P2V3W15O59{(OCH2)3CNHCO-C(Me)CH2}] (Fig. 14b) with (methacryloyloxy)phenyldimethylsulfonium triflate and methylmethacrylate are shown to act as photoresists. Using electron beam/extreme ultraviolet lithographic techniques resulted in patterning up to 20 nm lines and the hybrid material exhibited enhanced sensitivity in comparison to the (methacryloyloxy)phenyldimethylsulfonium homopolymer.166 Norbornene-conjugated derivatives of {PW15V3} form well-defined linear poly(polyoxometalate)s with high molecular weights (Fig. 14b).167 A hybrid block copolymer (Fig. 14b) bearing poly(polyoxometalate) and poly(6-norbornene-hexanoic acid) reveals controllable molecular weight, low polydispersity, the formation of self-assembles in acetonitrile and a good oxidation catalytic activity.245 A hybrid POM-containing polymer typically shows a precisely controlled molecular weight and narrow molecular weight distribution.208 Thus, very homogeneous, colloidally stable silver nanoparticles were synthesized with the covalently grafted WD-poly(N,N-diethylacrylamide) polymer.200 These POMs lead to the formation of nano-size photoactive polystyrene-POM latexes and can assist spatially controlled nucleation of silver nanoparticles.193
 | ||
| Fig. 14 Schematic representation of (a) prepare covalently-decorated polystyrene nanoparticles during the copolymerization reaction of styrene and POM–methylmethacrylate surfactant agent; (b) emulsion polymerization process with hybrid WP POM–norbornene conjugate; (c) photo-induced dimerization process of WD-coumarone hybrid molecules. Colour code: {PO4} = pink octahedrons, {VO6} = orange octahedrons and {WO6} = blue octahedrons. | ||
Over the years four films of copolymers made by using pyridine and porphyrin terminated {P2W15V3} were reported.172 At the same time, soluble WD POMs covalently bound to porphyrin significantly enhanced nonlinear optical responses, including reverse saturation absorption of porphyrins.136 A reversible photo-dimerization of coumarin-modified POM has been achieved by the reaction of {P2W15V3} with a triol-substituted derivative of coumarin (Fig. 14c).171 Different linker designs provided electrostatic interaction between the ligand and the hybrid POM segment. For this purpose, diol ligands were combined with {P2W15V3} resulting in a new tripod ligand containing a carbonyl carbon atom of the organic fragment.171,177,178,181,246–248 Later the {P2W15V3} was functionalized with pyridine and copolymerized with Zn(II)-octaethylporphyrin, leading to redox active photosensitive films (Fig. 15).172,181
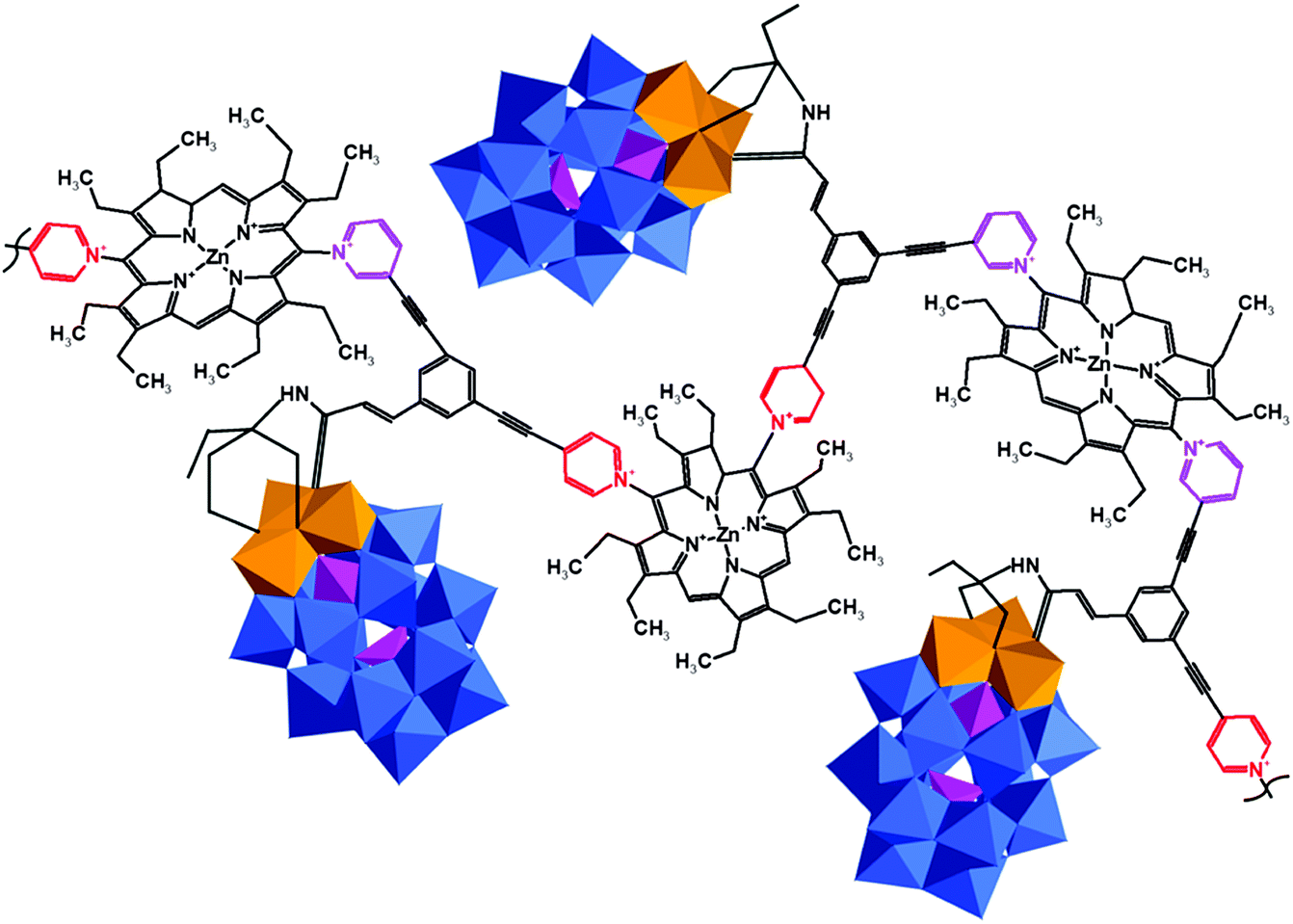 | ||
| Fig. 15 Schematic representation of the fragment of the polymeric photosensitive POM–(Zn)porphyrin film; the presence of 3-Py and 4-Py groups in the polymer leads to the different structure of polymers. Colour code: 4-Py = red, 3-Py = magenta, {PO4} = pink octahedra, {VO6} = orange octahedra and {WO6} = blue octahedra. | ||
3.5. Coordination to metal cations
The post-functionalization of hybrid POM platforms can be also extended to coordination to transition metal cations. The Wells–Dawson and the Keggin hybrids terminated with pyridine react with Pd(II) to afford self-assembly of charge-sensitive polygons.204 Following this, a terpyridine-modified ditopic POM-based building block was developed and its self-assembly behavior in the presence of Fe(II) ions was studied.203 Post-functionalized Keggin-type [PW11O39{O(Si(CH2)3NH2)2PtIV(NH3)2Cl2}]3− was encapsulated in DSPE-PEG2000 (Fig. 16a) and the resulting nanoparticles demonstrated highly efficient inhibition of cellular growth of human colorectal cancer cells in mice, and lower cytotoxicity against normal cells compared to cisplatin.122 Another platform, based on terpyridyl-linked {P2W15V3}, was designed and coordinated with Ru(III) to afford a photo-responsive molecular material.249 In a light-harvesting POM–bipyridine hybrid the electron transfer from the Re(I)-bipyridyl complex (Fig. 16b) to the POM was observed that led to complete quenching of the emission of the Re-bipy-carbonyl complex while enhancing the photosensitization of the hybrid POM in the visible region.174,175 Another promising property is “switching” between the “Dumb-bell” and “Triangular” shaped hybrid POM platforms176 that was followed by programmable assembly of hybrid POMs via Zn(II) ion coordination.177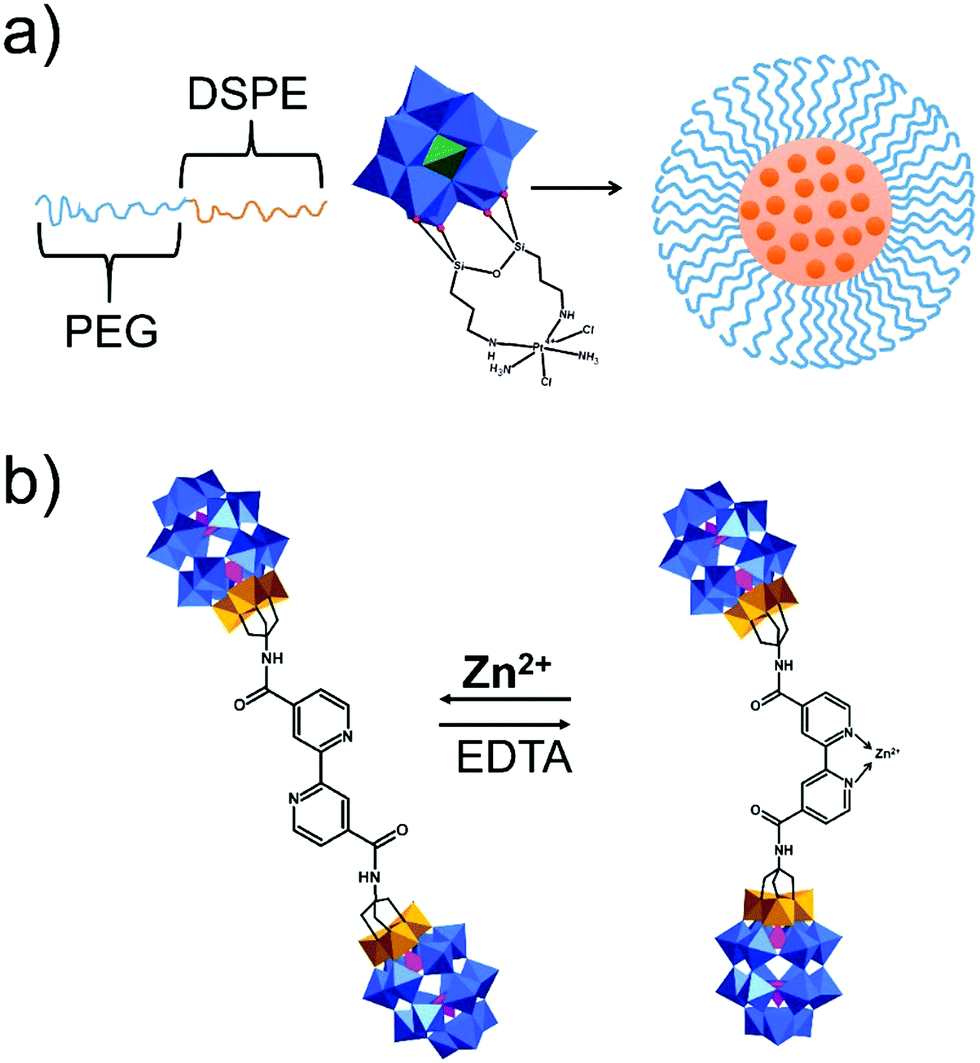 | ||
| Fig. 16 Schematic representation of (a) preparation of the {PW11O39PtIV} DSPE-PEG2000 nanoparticles; (b) the reversible conformational transformation of the WD-bipy POM hybrid molecule initiated by coordination of Zn2+ leading to the formation of the complexed dumbbell. Colour code: O atoms = red sphere, {PO4} = pink octahedra, {SiO4} = green octahedra, {VO6} = orange octahedra and {WO6} = blue octahedra. | ||
The addition of Pd(II) and its bridging coordination to the pyridyl-grafted K and WD hybrid POMs leads to the formation of trimer and tetramer species.126 Interestingly the Keggin derivatives can form both triangle and square coordination oligomers, while the Wells–Dawson POM forms only triangle assemblies (Fig. 17a) because of the higher charge of the WD anion (−6) in comparison with K (−3). An alternative way to obtain transition metal-functionalized POM hybrids was described by Lui et al. in 2015. Hagihara dehydrohalogenating coupling of TBA6[α2-P2W17O61{P(O)Ph-4-CCH}] with HgCl2212 or with trans-[Pt(PBu3)2Cl2]214 and CuI leads to metalorganic POM-CC-Hg and POM-CC-Pt respectively (Fig. 17b). The resulting oligomer products form films that demonstrated a photoelectric effect.
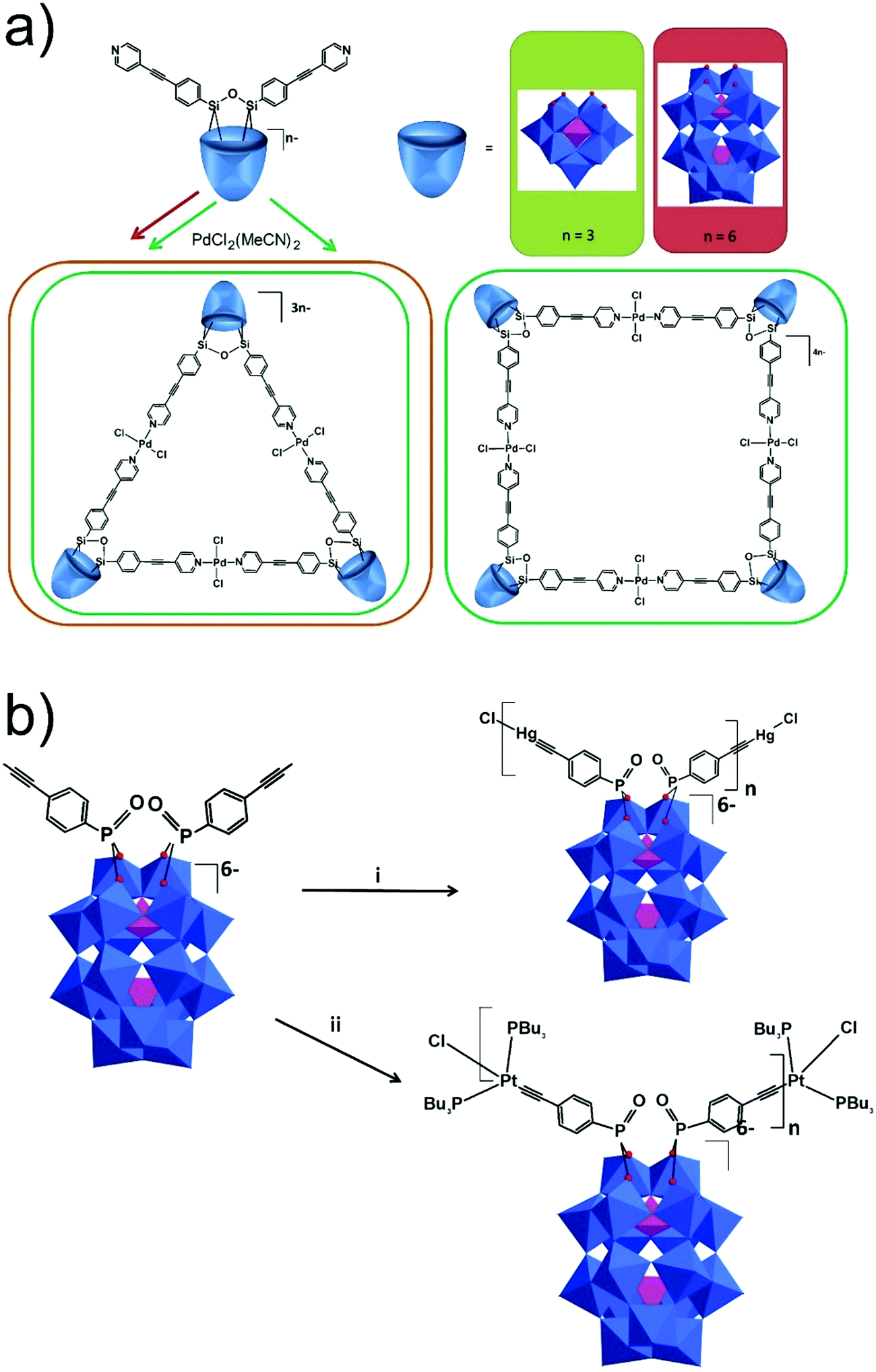 | ||
| Fig. 17 Schematic representation of (a) metal-driven formation of the molecular triangle and square from the POM hybrids; (b) synthetic routes to the organo-diphosphoryl POM-based oligomer hybrids, (i) HgCl2, CuI, Et3N, CH3CN, (ii) trans-[PtCl2(PBu3)2], CuI, Et3N, CH3CN. Colour code: O atoms = red sphere, {PO4} = pink octahedra, {SiO4} = green octahedra, {VO6} = orange octahedra and {WO6} = blue octahedra. | ||
The coupling of the {P2W15V3O62} mixed addenda Wells–Dawson with a series of diols leads to hybrid platforms with the general formula [P2W15V3O59{(OCH2)2C(Et)NHCOR}]5− (see WD polyoxometalates in Section 3.1, Fig. 15).246 In this design, a carbonyl carbon atom of the organic fragment inserts itself into the POM framework, thus serving as a third anchoring point. An important achievement made by this carbonyl insertion was the possibility to obtain electronic communication between the POM and the organic ligand.172,178,179,246–248 The formation of onion-like structures in bulk solutions of fullerene C60-Dawson POM hybrids revealed the strong intermolecular interactions, leading to rich magnetic, electrochemical and photodynamic properties.72 Upon the coordination of Pd(II) these hybrids were tested as dual site catalysts for simultaneous allylation of sulfonylimines and oxidation of mercaptanes and Mizoroki–Heck reaction.178 A 2:1 amphiphilic C60-Dawson POM hybrid was obtained by the same approach and was shown to form various structures from oligomers to vesicles, depending on the concentration and solution composition.165 The coordination of the Keggin-type POM–salen hybrid ligand to transition metal ions produced catalytically active POM–salen–M complexes (M = Mn(II),132,228 Co(II),132,228 Ni(II),132,228 Pd(II),131,140,228 Fe(II),139 Mo(VI)141,229) as mentioned above (see Section 3.1, Fig. 10).
Lacunary POMs {EW9} grafted with bisorganophosphonyl and bisorganoarselyl groups are able to coordinate with various transition metal ions in different ways, depending on the nature of the functional group in the organic ligand, and the type of d- or f-metal. The {PW9{P(O)tBu}2}250 forms sandwich-type dimers 2:2 with Ti(IV),235 La(III),235 and Zr(IV)100 in DMF with the coordination number of the metals being 6, 7, and 7 respectively, whereas the same reaction with Ca(II) produces a discrete 1:1 complex. La(III)- and Zr(IV)-containing complexes100 of {PW9{P(O)R}2} (R = Ph and Et)250 have the same structure as described for the P(O)tBu-grafted POM. Using the As-Ph-NH2-grafted POM instead of the PtBu-grafted POM does significantly affect the stoichiometry and structure of the complex, resulting in a 1:1 discrete complex.235 Interestingly, [AsW9O33{P(O)CH2CH2COOH}2]5−101 produces a discrete 2:2 complex with vanadyl, in which the coordination number of vanadium is 5, whereas the interaction of the same POM with La(III) leads to a coordination polymer in which only one carboxyl group of the ligand is bridging.235 Coordination of Ti(IV) to [XW9O34{Si(tBu)OH}3]3− (X = P(V), Sb(III)) gives monomeric [XW9O34{Si(tBu)O}3Ti(iPrO)]3− and dimeric [(SbW9O34{Si(tBu)O}3Ti)2O]6− species, which were studied as a model of the isolated titanium site on the surface of a catalyst.90 The results showed that inclusion of a silanol-functionalized trilacunary phosphotungstate anion significantly increases the effectiveness of the TiO2- or ZnO2-containing electron collecting interface and significantly improves the stability and efficiency of non-encapsulated cell elements.93 The dimerization of TBA4[γ-H2SiW10O36{Pd2(OAc)2}]113 by ligand exchange reaction with malonic, glutaric, and pimelic acids produced TBA8[(γ-H2SiW10O36)2{Pd2(OOC(CH2)nCOO)2}2] (n = 1, 3, 5), which was active in the catalytic hydration of nitriles.114
Minato and co-workers developed an alternative strategy to form mixed-sandwich POMs. Alkoxide-modified TBA4[A-α-SiW9O28{OMe}6] trivacant Keggin can form pentaerythritol derivative TBA4[A-α-SiW9O28{(OCH2)2C(CH2OH)2}3], which reacts with TBA4[A-α-XW9O28{OMe}6] giving dimeric TBA8[(α-A-SiW9O28){(OCH2)2C(CH2O)2}3(A-α-XW9O28)] (X = Si, Ge), which was isolated and studied by mass-spectrometry (Fig. 18).251 Transition metal ions interact with phosphonate-grafted WD-POM K6[P2W17O61(P(O)-Ph-4-COOH)2] leading to metal-functionalized hybrid polyoxometalates with different stoichiometry. If M = Zn(II) or Co(II) the M(POM)2 species were observed, while in the case of Mn(II) the M2(POM)2 hybrid was detected, in which manganese atoms form a Mn2(μ2-OH)2 bridging core between two POM subunits.218
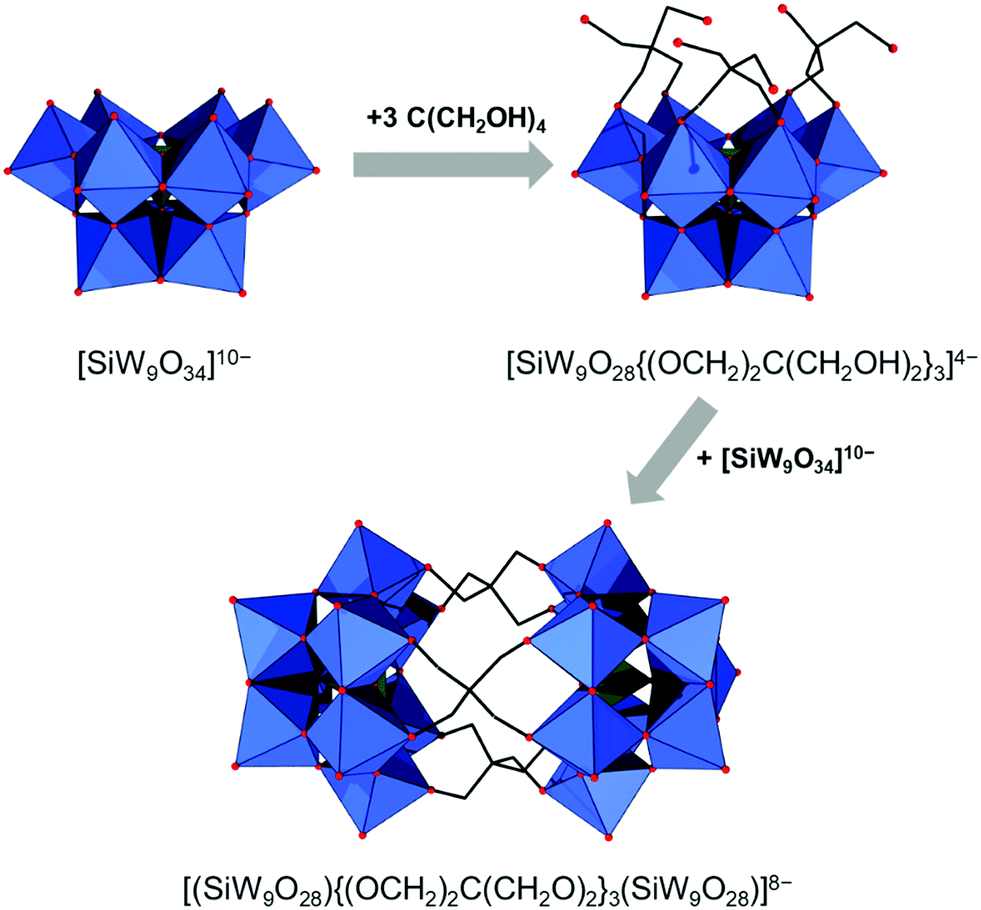 | ||
| Fig. 18 Schematic representation of the mixed-sandwich POM synthesis. Colour code: O atoms = red spheres, {WO6} = blue octahedra, {SiO4} = green tetrahedra; carbon–carbon bonds are represented as black sticks. | ||
4. Anderson–Evans type platforms
This section addresses the developments of the hybrid Anderson–Evans polyanions {MMo6O24} where M = Mn(III), Fe(III), Co(III), Cr(III) and Al(III). The encapsulated redox active centers make these polyanions typically suitable for one electron redox processes. All hybrid POM platforms in this section represent bis-trisalkoxo capped molecules with a variety of terminal functional groups. These platforms could be divided into mono-functionalized platforms such as [MnMo6O18{(OCH2)3CCH3}{(OCH2)3CNH2}]3−, where only one bis-trisalkoxo molecule is available for post-functionalization, and di-functionalized platforms such as [MnMo6O18{(OCH2)3CNH2}2]3−, where both bis-trisalkoxo molecules bear functional groups for further post-functionalization. The latter family of platforms usually are symmetric, i.e. they contain the same end functional groups in both linkers, but some of the di-functionalized platforms are asymmetric and contain 2 different functional groups, such as for example [MnMo6O18{(OCH2)3CNHC(O)Ph-4-N3}{(OCH2)3C-NH2}]3−. All previously reported AE-based platforms are grouped according to the type of the {MM′6O18} POM core and the terminal reactive group and are summarized in Table 3.4.1. Condensation reactions
To date Anderson–Evans hybrid platforms have been very commonly post-functionalized using amide, ester, thioester, sulfonamide and imide bond formations, in some cases resulting in “extended” platforms. In this regard, several amphiphilic hybrid POMs were obtained by the reaction of the double amine functionalized Anderson–Evans type POM [MnMo6O18{(OCH2)3CNH2}2]3− with long-chain acyl chlorides in the presence of a tertiary amine.252–254 Recently, different research groups focused on the post-functionalization of asymmetrical Anderson–Evans type POMs through amidation reaction. Several procedures were reported including the reaction between amine-functionalized POMs and acid anhydrides255–261 or acyl chlorides.262 Similarly, the reactions between amines and activated NHS–ester functionalized POMs263 or coupling in the presence of an activation agent such as EEDQ264–270 have been described (Fig. 19). Promising materials for lithium ion batteries have been developed by the reaction of the acyl chloride derivative of single-walled carbon nanotubes (SWNTs) and [MnMo6O18{(OCH2)3CNH2}2]3−, which showed significantly enhanced electrochemical performance, high discharge capacity and long-term cycling stability.262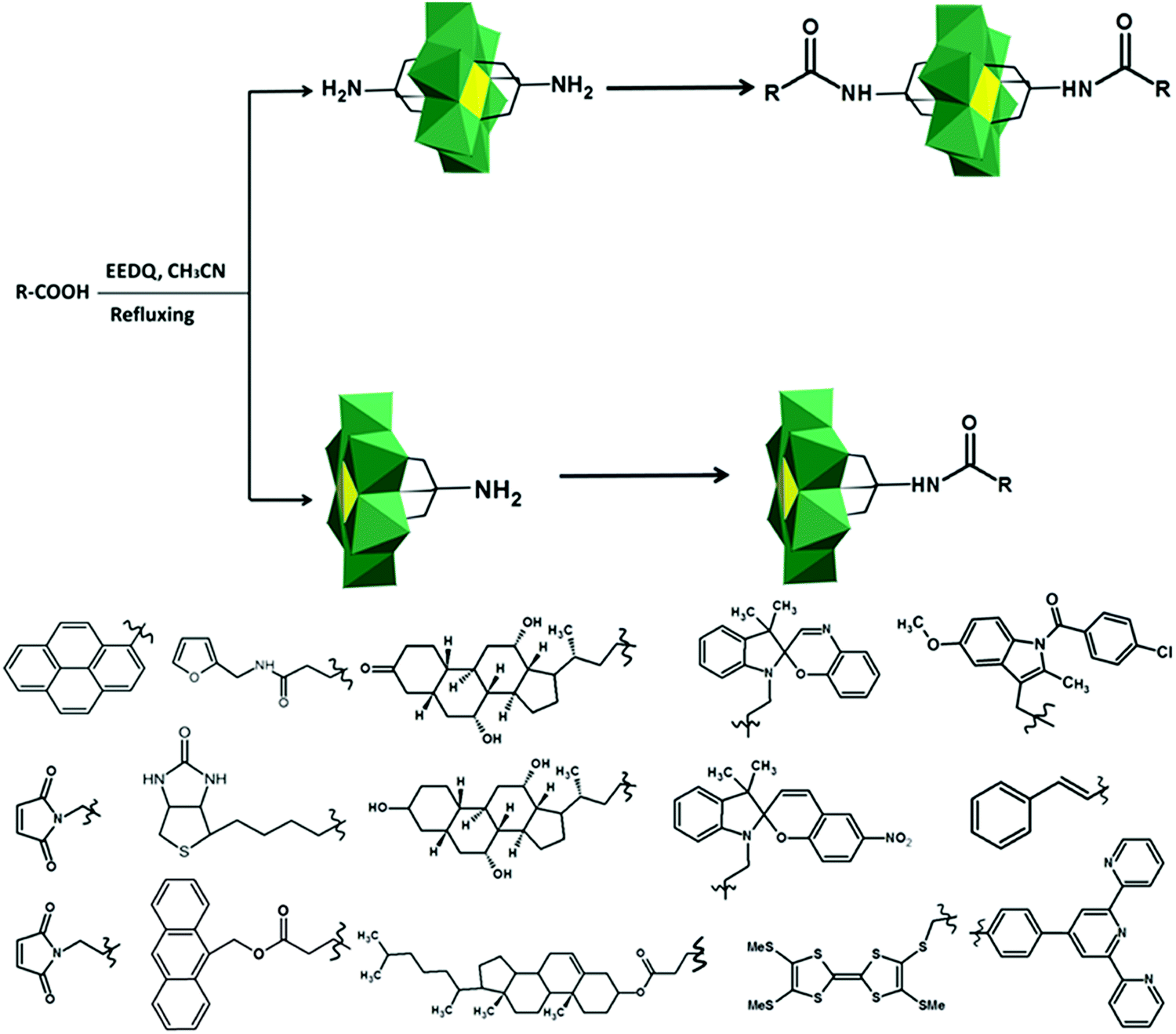 | ||
| Fig. 19 Scheme of amidation of AE-NH2 hybrid POMs in the presence of EEDQ as a coupling agent; the residues of carboxylic molecules are presented below. Colour code: {MO6} = green octahedra and {MO6} = yellow octahedra, where M = Al(III), Mn(III). | ||
By using the coupling agent methodology and FMOC protection/deprotection chemistry, Cronin and co-workers succeeded in developing an asymmetrical Anderson–Evans hybrid platform which was later used to insert peptide chains in solution and for grafting on the solid support (Fig. 20a).263 The peptide-functionalization of the Mn-AE POM was later successfully expanded by hybridization with the 15-amino acid bombesin-receptor antagonist peptide. In these hybrids the POM facilitated a specific partial α-helix peptide folding, causing the self-assembly of the hybrid POM–peptide molecules in water solution, and influencing the ability of the POM–peptide hybrid to interact with target receptors (Fig. 20a).259 Coupling of phenylcarboxyterpyridine with [MnMo6O18{(OCH2)3CNH2}2]3− resulted in a POM conjugate which exhibited complex self-assembly behavior depending on the solvent and the nature of the cation.271 The detailed investigation on using EEDQ as a coupling agent in the reaction of a diamino-derivative of Mn-AE and different carboxylic-bearing substrates, including biotin and cholesterol, has been reported (Fig. 19).272 The Al-centered hybrid platform [Al(OH)3Mo6O18(OCH2)3CNH2]3− was successfully conjugated with bioactive molecules such as indometacin or cinnamic acid, which could be interesting for the design of POM-based antibiotic compounds.270
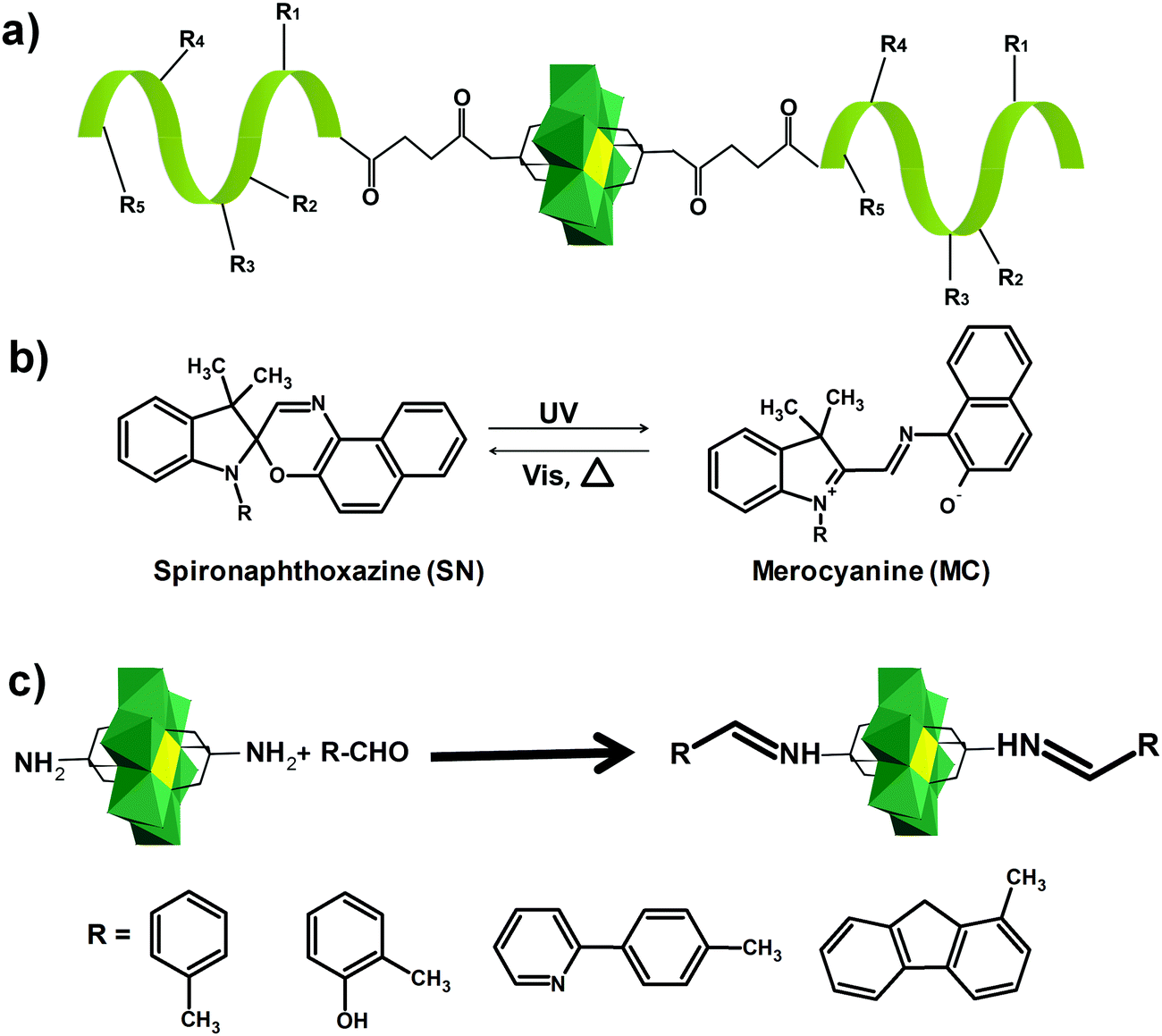 | ||
| Fig. 20 Schematic representation of (a) the POM–(oligopeptide)2 hybrid molecule where oligopeptide = Demobensin-1 or NH2-Ala-Asn-Thr-Leu-Ser-Ser-Thr-Ala-Ser-Thr-Leu-Glu-Ser-Tyr-Leu-OH; (b) photoinduced reversible transformation of spironaphthoxazine–merocyanine; (c) postfunctionalization of AE POMs with various aromatic aldehyde molecules via formation of imine bonds. Colour code: {MoO6} = green octahedra and {MO6} = yellow octahedra, where M = Co(III), Fe(III), Mn(III). | ||
The Fe(III) analogue of AE [FeMo6O18{(OCH2)3CNH2}2]3− has been also demonstrated to be a promising platform for postfunctionalization (Fig. 33c).273 The [MMo6O18{(OCH2)3CNH2}2]3− (M = Fe(III) and Mn(III)) Anderson–Evans hybrid platforms have been covalently linked with the Lindqvist hexamolybdate [Mo6O19]2− or octamolybdate274 [Mo8O26]4− using DCC as a coupling agent. The coupling resulted in two nanoscale rod-like chiral molecular triads273 that exhibit self-recognition during self-assembly and reversible aggregation in the presence of Zn2+ ions (Fig. 33c).274–276 The asymmetric umbrella-like azobenzene-conjugated Al(III)-centered POM forms a three-component system with α-cyclodextrins and cationic dye methylene blue through electrostatic and host–guest recognition interactions. Such a construct allows carrying out the consecutive transfer of chirality from α-cyclodextrin to the counter ion dye via the bridging POM hybrid molecule.265
The hybrid [MnMo6O18{(OCH2)3CNH2}2]3− platform has been also used to synthesize POM–biomolecule conjugates in the presence of coupling agents.277–279 The influence of molecular structure, solution components, and temperature on the self-assembly of POM–steroid277 and POM–cholesterol279 conjugates (Fig. 19) was also examined in order to create hybrid drugs for cancer therapeutics. The steroid–POM conjugate was investigated for its potential to act as an organic–inorganic hybrid material demonstrating tunable and temperature-dependent reversible gelation.280,281 The reaction of cholic acid derivatives and galactose with the Anderson–Evans platform (Fig. 19) has been also considered as an approach to create hybrid drugs for cancer therapeutics.278 Recently a simple nucleophilic substitution for the coupling of nucleophiles to Anderson–Evans POMs was developed as a versatile post-functionalization method by our group.169 Very recently, unusual one-side modification of the Mn-AE(NH2)2 POM was described by Zeng and co-workers by phenylthiourification reaction resulting in [MnMo6O18{(OCH2)3CNH-C(S)-NHPh}{(OCH2)3CNH2}]3−.282 Finally, a new platform [MnMo6O18{(OCH2)3CCH2OH}2]3− was developed for the consecutive synthesis of heteropolymers with ε-caprolactone as a potential antitumor agent.283
Non-linear optical (NLO) phenomena are interesting for the development of optical fibers and computers, data storage and transfer, photo switchable elements and other applications. The significant success in this area was brought by the development of step-by-step symmetric and asymmetric post-functionalization of the [MMo6O18{(OCH2)3CNH2}2]3− platform, opening a unique opportunity to combine two different building blocks within one hybrid POM resulting in a wide range of desired properties. Thus, the hybridization of porphyrins and AE POMs led to remarkable NLO response, depending on the length of the linker between the POM and the tetraphenylporphyrin ring.260 The first covalently conjugated POM–spiropyran dyads were shown to be electrochemically and optically active (Fig. 20b), demonstrating six states having different colours. These hybrids undergo reversible change without formation of aggregates and exhibit significant solid state photochromic properties.266 Mixed spiropyran–POM–spironaphthoxazine compounds form a unique system, containing a redox-active POM and two different chromophores, giving them remarkable electrochromic properties and highly effective room temperature solid-state photochromic properties (Fig. 20b).267 Formed by the self-assembly of an asymmetric spiropyran–POM–C16 hybrid, the hollow nanospheres showed a reversible assembly–disassembly light-driven process for the vesicles obtained in polar solvents. The reversed vesicles formed in non-polar medium are not sensitive to UV or visible light irradiation.268 The unusual second-order non-linear optical properties were demonstrated for the asymmetric tetrafulvalene–POM–spiropyran hybrid.269
In contrast to K and WD platforms a significant number of H2N-functionalized AE hybrid POMs have been functionalized by reaction with aldehydes forming imine bonds. As mentioned in Section 3.1 due to the hydrolytic instability of the imide bond and redox sensitivity of POMs, such post-functionalized products were mainly prepared and investigated in organic solvents. In 2003, Marcoux et al. reported the first use of imine post-functionalization with the reaction between an amine functionalized Anderson-type POM and 2-pyridinecarbaldehyde.284 The same coupling reaction was used by the group of Cronin for the asymmetrical post-functionalization of an Anderson-type POM containing a reactive amine on one side and an inert nitro-group on the other side of the planar POM.285 By employing the [MnMo6O18{(OCH2)3CNH2}2]3− hybrid Anderson–Evans platform, clusters with appended aromatic molecules were synthesized (Fig. 20c).286 Using polyoxometalates with photo-switchable azobenzene ligands it was demonstrated that ion-mobility mass spectrometry is a powerful tool to study the self-assembly of POMs based on their size and charge, and to separate POM hybrids having photoswitchable groups.287 The similar nano-sized azobenzene–POM–azobenzene hybrids resulted in self-assembly into two types of architectures which exhibited light-dependent switching: fiber-like layers in trans conformation and multilayer spheres in cis conformation.288 Boronic acid- and boronic ester-containing hybrid Anderson–Evans species have been also reported289 and demonstrated a relatively high degree of stability in solutions.
4.2. Coupling reactions
Wang and co-workers reported the AE-based platform [MnMo6O18{(OCH2)3CNH-Ph-4-I}2] for Sonogashira coupling reactions and probed its reactivity towards various alkynes.290 The Sonogashira–Hagihara cross-coupling of the Anderson–Evans tetrabromo-bifunctionalized POM and 1,3,5-triethynylbenzene allowed successful construction of microporous polymers, which showed remarkable improvements in the stability and catalytic performance of the incorporated POM (Fig. 21).264 These materials demonstrated high photocatalytic activity in the reductive photoactivation of molecular O2 under visible light which could be used for degradation of aromatic dyes under mild conditions.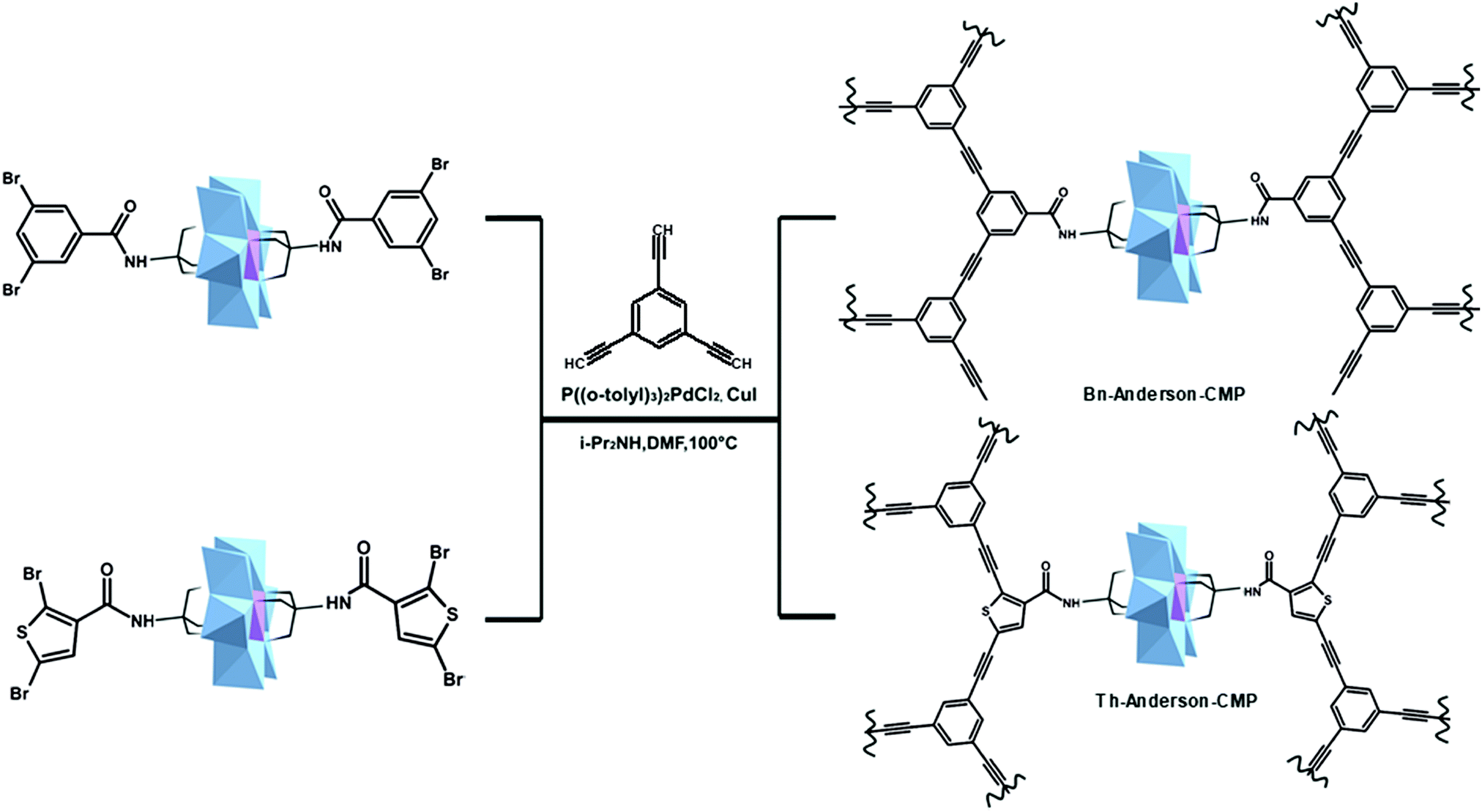 | ||
| Fig. 21 Scheme of the routes for the synthesis of two POM-based conjugated porous polymers. Colour code: {MoO6} = light blue octahedra and {MnO6} = magenta octahedra. | ||
4.3. Click reactions
In 2015, the CuAAC coupling of Anderson–Evans hybrid platforms has been applied for the controlled oligomerization of POM hybrids.291 In the same year, our group reported CuAAC using CuI as a catalyst instead of the classic CuSO4/sodium ascorbate approach.292 The same approach was used to reach the fluorescent spiropyran-polyoxometalate-BODIPY single-molecule that demonstrates high fatigue resistance and photoswitchable properties (Fig. 22a).293As an alternative to the CuAAC reaction, in 2013 Yang et al. described the post-functionalization of an AE-type POM using a metal-free Diels–Alder click reaction (Fig. 22b).278 For this purpose, the authors synthesized three different POM-platforms: a maleimide-(dienophile), a furan-(diene) and an anthracene-(diene) functionalized Anderson-type POM. Reacting these preformed POM-platforms with several complementary diene or dienophilic substrates in acetonitrile or DMF at 81 °C resulted in the desired Diels–Alder coupled products in high yields within reaction times ranging from a couple of hours to several days, depending on the nature of the substrate.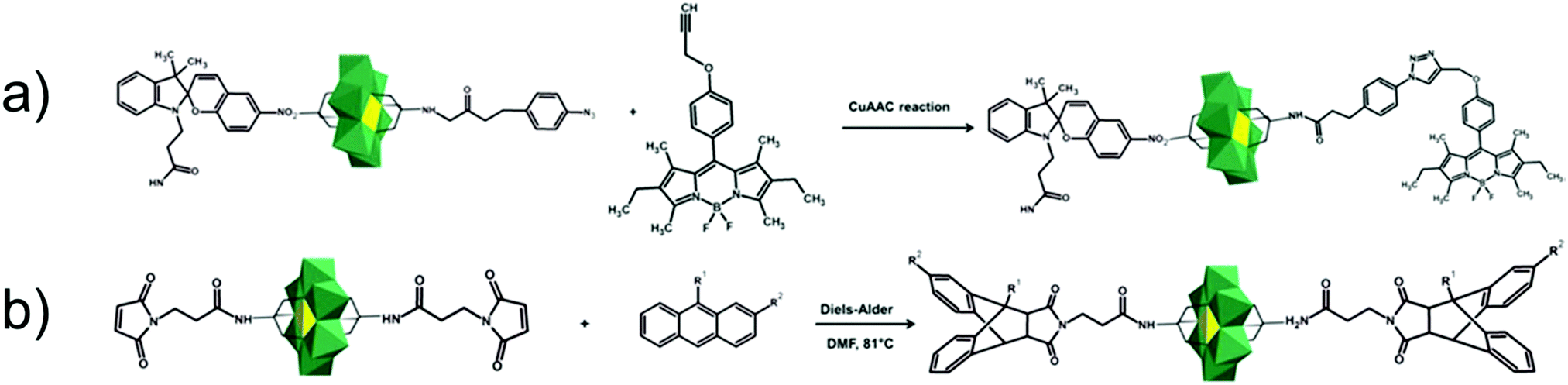 | ||
| Fig. 22 Schemes of reactions: (a) synthesis of the SP-POM-BODIPY hybrid. (b) metal-free Diels–Alder coupled products obtained in high-yield (>90%) under mild conditions. Colour code: {MoO6} = green octahedra and {MO6} = yellow octahedra, where M = Mn(III). | ||
4.4. Polymerization reactions
Polymerization reactions of Anderson–Evans hybrids have been of high interest in the design of POM-incorporating materials for NLO,261 development of metal-free Diels–Alder click reaction,278 self-organized and switchable materials294–297 and highly dense multilevel memory polymers with a switching mechanism.255,256 The POM–organic hybrid polymer was developed and employed as an active layer to achieve novel multi-redox materials for data storage devices with high-performance (Fig. 23a).256 Soon after, a multi-redox POM-based inorganic–organic hybrid polymer was designed having a ternary resistance switching memory (Fig. 23b).255 Following this, the Anderson–Evans-coumarin conjugate was able to undergo reversible photodimerization forming a polymer chain (Fig. 23c).294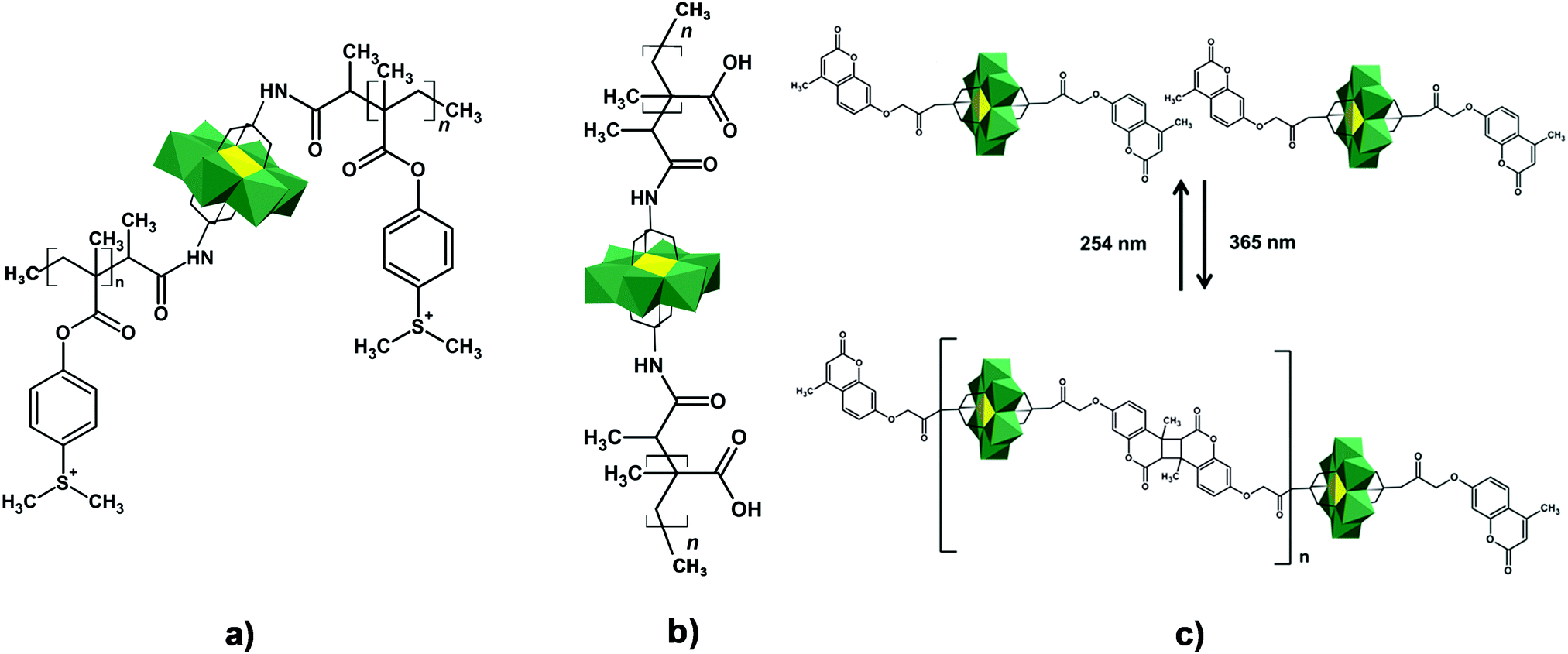 | ||
| Fig. 23 Schematic representation of (a) the structure of the AE POM–methyl methacrylate hybrid copolymer; (b) the structure of the AE POM–(methacryloyloxy)phenyldimethylsulfoniumtriflate hybrid copolymer; (c) the photopolymerization process of AE-coumarin hybrid molecules. Colour code: {MoO6} = green octahedra and {MnO6} = yellow octahedra. | ||
The one-step anodic electropolymerization of zinc-porphyrin with Mn-AE-{Py}2 resulted in co-polymeric films which were able to photocatalytically reduce Ag(I), leading to 1D and 2D metallic nanostructures (Fig. 24a).295 Co-polymerization of the spiropyran-AE-methacrylate unit with methyl methacrylate provides materials having a very low loading level of photochromic molecules within the polymer, but with good coloration and photoresponsive activity.261 In addition to covalent interactions, hydrogen bonding could also lead to POM-based polymeric structures. The interaction between adenine- and thymidine-AE hybrids forms base pair-matching chains, which can be transformed into a supramolecular polymer by the addition of tetrakis(decyl) ammonium which causes cross-bonding (Fig. 24b).296 The impact of the counter ion on the temperature-dependent behavior of the Mn-AE-adenine functionalized POM was investigated for TBA and DODA salts. While the TBA salt forms thin fiber structures at room temperature which transform into rod-like tubes upon heating, the DODA salt produces fibrous-like layers which are transformed into multilayer spheres by heating.297
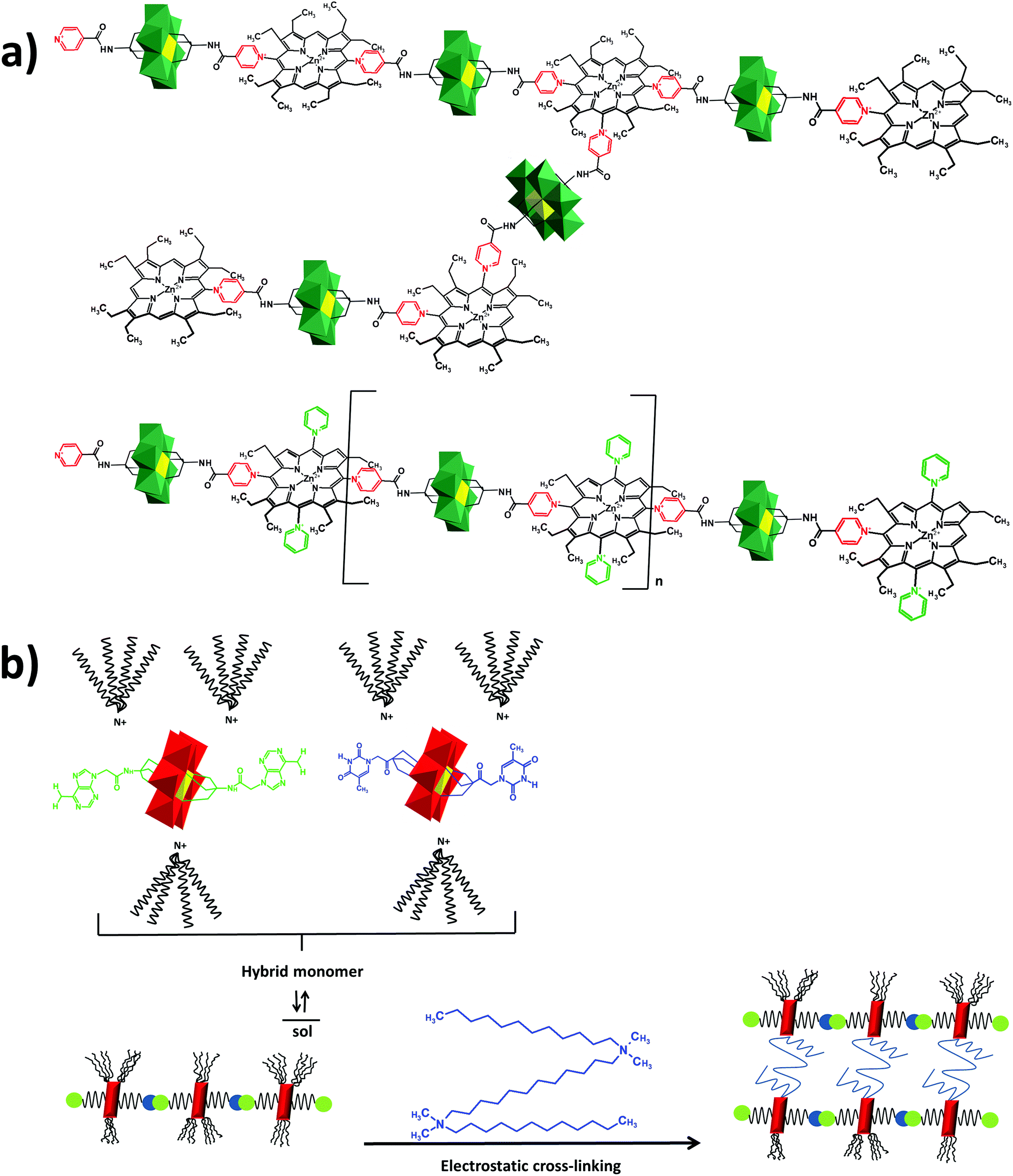 | ||
| Fig. 24 Schematic representation of (a) copolymer of POM AE-Py2 with ZnOEP and 5,15-ZnOEP(Py)22+ (colour code: {MoO6} = green octahedra and {MnO6} = yellow octahedra); (b) formation of a hybrid chain through the complementary hydrogen bonds between AE-(Adenine)2 and AE-(Thymidine)2 POM derivatives with consecutive aggregation into a gel through electrostatic cross linking (colour code: {MoO6} = red octahedra and {MnO6} = yellow octahedra). | ||
4.5. Coordination to metal cations
The Anderson–Evans hybrid platforms have been used in the coordination to metal cations including Zn(II), Cr(III), Mn(III), Fe(II), Cu(I) and Cu(II), Pd(II) and Ir(III). The reaction between pyridine-terminated [MnMo6O18{(OCH2)3CNH-4-Py}2]2− and Pd(II) affords the formation of an anisotropic gel,298 while the reaction with CuI68 or M(NO3)2 (M = Zn, Cu)299 leads to POM-(CuI)n (n = 2 or 4)68 and (POM)x-My299 frameworks (Fig. 25a). Similarly, the single pyridine terminated platform [CrMo6O18(OH)3{(OCH2)3CNH-CO-R}]4− produces a composite structure in the presence of CuI by stabilizing the Cu8I6 core (Fig. 25b).300 The bipy terminated platform [MMo6O18{(OCH2)3CNCH-4-bipy-4′-Me}2]3− (M = Mn(III), Fe(III), Co(III)) was coordinated to Cu(I) and the electrochemical properties of these complexes were investigated.301 The same hybrid POM platform has been used in coordination to Ir(III) cations resulting in effective light harvesting and charge separation (Fig. 26a).302,303 This has led to the successful application of these iridium coordinating POM hybrids as light-driven catalysts for hydrogen evolution. It is noteworthy that the POM–terpyridyl hybrid offers a modular strategy for post-functionalization by coordination to Pd(II), leading to discrete molecular complexes.249 Para-pyridyl-functionalized {MMo6} (M = Mn(III) and Fe(III)) POMs demonstrate axial coordination to the metal ion in Ru- and Zn-tetraphenylporphyrin giving redox active POM–porphyrin assemblies in solution (Fig. 26b).304 Depending on the stoichiometry of reagents, the direct reaction of {MnMo6}-(2,6-di(pyrazol-1-yl)-pyridine)2 with Fe(II) in solution produces 1D or 2D networks and polymers, which demonstrate a significant level of photoinduced spin-crossover in Fe(II) centers under irradiation with a green light (Fig. 26c).305
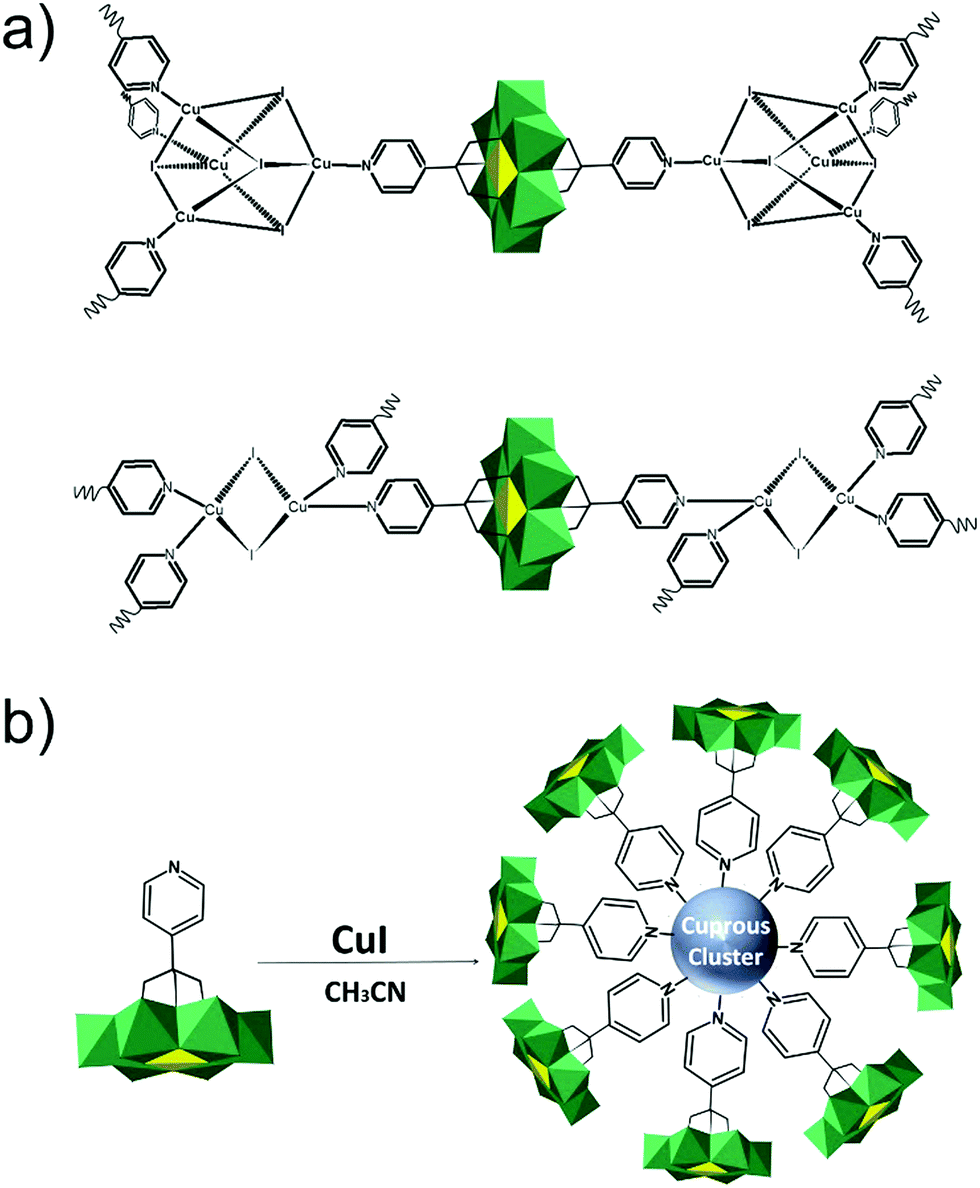 | ||
| Fig. 25 Schematic representation of AE-based hybrid POMs coordinated to CuI-clusters. Colour code: {MoO6} = green octahedra and {MnO6} = yellow octahedra. | ||
 | ||
| Fig. 26 Schematic representation of (a) the structure of [MMo6O18{(OCH2)3CNCH-4-bipy-4′-Me(Ir-py2)}2]3− (M = Mn(III), Fe(III), Co(III)) (colour code: {MoO6} = green octahedra and {MO6} = yellow octahedra); (b) the structure of [MnMo6O18{(OCH2)3CNHCO(4-C5H4N-Ru(CO)TTP)}2] (colour code: {MoO6} = green octahedra and {MnO6} = yellow octahedra); (c) the structure of the polymeric network of {MnMo6}-(2,6-di(pyrazol-1-yl)-pyridine)2–Fe(II) (colour code: {MoO6} = green octahedra and {MnO6} = yellow octahedra). | ||
5. Lindqvist type hybrid platforms
This section addresses Lindqvist type polyoxometalates comprised of vanadium and mixed vanadium–molybdenum (Section 5.1) and solely molybdenum centers (Section 5.2) as addenda atoms. The distinction in separate sections is primarily made based on the different reactivity of the oxo ligands which results in the formation of distinct hybrid platforms. Considering the strong bonding in the terminal vanadyl moieties {VO}, Lindqvist-type hexavanadates primarily undergo formal exchange of the bridging μ2-oxo ligands with trisalkoxides. In the case of hexamolybdates, exchange of the bridging μ2-oxo ligands remains rare, while the exchange of the terminal oxo with nitride or imido ligands is far more dominant. In both cases the overall charge can determine the number of exchanged ligands. Typically, highly reduced (all addenda metals in d1 electronic configuration) or super-reduced Lindqvist-type polyoxometalates (all addenda metals in d2 electronic configuration) undergo multiple formal substitutions of the terminal oxo or the bridging oxo ligands with other hetero-ligands with lower overall charge.47,57 In addition, modularity can be affected by formal substitution of the addenda atoms and the bridging oxo ligand centers with other heterometals or hetero-ligands respectively. All of these aspects are discussed in the following two sections.5.1. Hexavanadates and mixed vanadium–molybdenum hybrid platforms
Hybrid-type hexavanadates have been attracting research attention since the late 1980s when Klemperer and co-workers isolated the first organometallic derivatives.316 Motivated by the possibility to further expand the multielectron redox stable and optically active hexavanadate core, different types of organometallic and monoalkoxide substituted derivatives have been developed with relevance to homogeneous catalysis, energy storage, and materials science in general.57,317,318The anti-type bis-(trisalkoxo)hexavanadates are typically formed in the reactions of MVO3 (where M = NH4+ or Na+) or TBA[H3V10O28] and pentaerythritol as precursors.57 Fully reduced or super-reduced hexavanadates that form syn-type, tri- or four trisalkoxo stabilized hexavanadates are also reported;57 however their use as hybrid platforms suitable for post-functionalization has been limited so far. The mixed-valence trimodular Lindqvist-type hexavanadates suitable for post-functionalization have been recently reported and are part of this review (vide infra).
Lindqvist-type hybrid hexavanadate polyanions suitable for post-functionalization are listed in Table 4. These platforms can undergo amide and ester bond formation (5.1.1), nucleophilic substitution reactions (5.1.2), 1,3-alkyne–azide cycloadditions (5.1.3), imine bond formation (5.1.4) and coordination to metal cations (5.1.5).
 | ||
| Fig. 27 Combined ball-and-stick representation of (a) [VV6O13{(OCH2)3CNH2}2]2−; (b) [V6O13{(OCH2)3-C(NH(CO)CH2CH2CH2C16H9)}2]2−; (c) [V6O13{(OCH2)3CNH(COO)CH2C6H4CH2CH2(CF2)5CF3}2]2− next to a schematic representation of its hierarchical hollow and bilayer forming in the solvent mixture of acetonitrile/water; (d) [VV6O13{(OCH2)3CCH2OH}2]2−; (e) [VV6O13{(OCH2)3CCH2OC(O)(CH2)16CH3}2]2−. Colour code: C = black balls, N = blue balls, O = red balls, H = white balls, {VO6} = orange octahedra. | ||
The [VV6O13{(OCH2)3CNH2}2]2− platform typically undergoes simultaneous post-functionalization at both anchoring sites. An example of this is the reaction with anthracene-2,3-dicarboxylic acid anhydrides in DMA, which has been shown to lead to the formation of amide bond containing products.325 Under non-oxidative conditions, the formed products are redox responsive to mustard gas simulants and turn to a dark blue colour owing to the reduction of the {V6O19} core; however once exposed to air they easily regenerate.
The [VV6O13{(OCH2)3CNH2}2]2− platform has been applied for the formation of hybrid structures with long alkyl tails which are broadly regarded as polyoxoanionic surfactants. Symmetrical and asymmetric post-functionalization of [V6O13{(OCH2)3CNH2}2]2− with 1-pyrenebutyric acid N-hydroxysuccinimide ester has been achieved, leading to [V6O13{(OCH2)3CNHC(O)CH2CH2CH2C16H9}2]2− (Fig. 27b) and [V6O13{(OCH2)3CNH2}{(OCH2)3CNHC(O)CH2CH2CH2C16H9}]2− polyanions respectively.322 These polyanions have been isolated as TBA, TMA (=tetramethylammonium, Me4N+), TEA (=tetraethylammonium, Et4N+) salts and also as protonated species, and they have been observed to aggregate into spherical vesicular structures in aqueous DMSO solutions, while exhibiting pH-dependent fluorescence as a result of the pyrene fluorophores.322
The groups of Liu and Hill have further studied the application of the [VV6O13{(OCH2)3CNH2}2]2− platform in the preparation of hexavanadate-containing fluoro-surfactants and their self-assembly in solution.323 The formation of these types of surfactant hybrids was motivated by the highly hydrophobic nature of the fluorinated chains which have affinity to dissolve molecular oxygen, and the highly hydrophilic nature of the Lindqvist hexavanadate core. The reaction of this platform with N-hydroxysuccinimide-activated fluorocarbon chain carboxybenzyl compounds in anhydrous DMF has led to isolation of solutions containing dianionic products [V6O13{(OCH2)3CNH(COO)CH2C6H4CH2CH2(CF2)5CF3}2]2− (Fig. 27c) and [V6O13{(OCH2)3CNH(COO)CH2C6H4CH2CH2(CF2)7CF3}2]2−, which upon further treatment were isolated as orange coloured TBA salts. The negative overall charge of the fluorinated polyanions mainly resides at the hexavanadate core. The formed [V6O13{(OCH2)3CNH(COO)CH2C6H4CH2CH2(CF2)5CF3}2]2− is well-soluble in acetonitrile; however based on the static light scattering (SLS) measurements, it was revealed that upon addition of water (65–95 vol%) agglomeration and formation of micellar architectures occurred. Combination with dynamic light scattering (DLS), atomic force microscopy and transmission electron microscopy (TEM) revealed that the formed particles are spherical, bilayered and hollow vesicles that have a diameter of ca. 85 nm and a wall thickness of ca. 12 nm (Fig. 27c). It was argued that this thickness is comparable to the approximate sum of two POMs (ca. 0.7 nm each) and the zigzag length of the fluorocarbon chain (ca. 8.5 nm).
Another interesting example of asymmetrical functionalization is the single-tailed Lindqvist hexavanadate surfactant [VV6O13{(OCH2)3CNH2}-{(OCH2)3CNHCH2C6H4COOC16H33}]2− which has been isolated from the reaction of cetyl (4-chloromethyl)benzoate and [V6O13{(OCH2)3CNH2}2]2− in DMF in the presence of tetrabutyl ammonium hydroxide in methanol.324 Interestingly, the TBA countercations of the resulting product can be exchanged for H+ by using a biphasic ion exchange technology. In aqueous solutions, the proton-counterbalanced polyoxoanionic molecules were shown to undergo stepwise hierarchical self-assembly to from micelle structures. The further process includes spontaneous coagulation of these micelles into one-dimensional (1D) anisotropic nanobelt-like structures with a nanoscale thickness of ca. 20 nm. The 1D architecture is stabile in acetonitrile and exhibits heterogeneous catalytic activity for the oxidation of organic sulfides with hydrogen peroxide, yielding sulfide monoxides and sulfones.
In 2017, the Wei group reported the mixed-valent trimodular hexavanadate platform [VIV3VV3O10{(OCH2)3C-NH2}3]2− having three amine anchoring groups. This platform was obtained from the reactions of NH4VO3 and NH2C(CH2OH)3 under hydrothermal conditions in the presence of N2H4·H2O.67 The structure of the polyanion was confirmed based on X-ray diffraction. Reactions of [VIV3VV3O10{(OCH2)3C-NH2}3]2− with benzoic acid in DMA in the presence of EDCl and HOBt under a nitrogen atmosphere have led to the formation of the amidated [V6O10{(OCH2)3CNHCOC6H5}3]2− product. Full-oxidation of the six VIV to VV centers has been showcased on the basis of cyclic voltammogram.
The [VV6O13{(OCH2)3CCH2OH}2]2− platform is one of the first hexavanadate platforms reported by the Zubieta group already in 1992 (Fig. 27d).327 The solutions containing the platform were prepared by the reaction of pentaerythritol with decavanadate in hot acetonitrile. Following solvent evaporation and use of the etherate method for separation, solid air stable material can be obtained for further functionalization. The [VV6O13{(OCH2)3CCH2OH}2]2− platform can undergo a number of DMAP-catalyzed esterification reactions with acid anhydrides (RCO)2O (aliphatic R = –Me, –Et, –C5H11, and –C17H35) in acetonitrile and in the presence of Et3N to yield dianionic [VV6O13{(OCH2)3CCH2OC(O)R}2]2− polyanions.330 The isolated products exhibit good solubility in acetone, acetonitrile, chloroform, and DMF.
Esterification reactions of TBA2[V6O13{(OCH2)3CCH2OH}2] with stearic acid CH3(CH2)16COOH in acetonitrile,328 and in the presence of N,N-dicyclohexylcarbodiimide (DCC) and DMAP as Steglich reagents342 have successfully led to the preparation of another hybrid hexavanadate surfactant [VV6O13{(OCH2)3CCH2OC(O)(CH2)16CH3}2]2− (Fig. 27e), which in acetonitrile–water mixtures readily forms vesicles. Further on, the latter hybrid polyanion showed cation dependent blue luminescence emission bands upon excitation at 335 nm.
The Wei group has showcased that the [VV6O13{(OCH2)3CCH2OH}2]2− platform can further undergo oxidation and transformation into the carboxylate anchoring platform [V6O13{(OCH2)3C-COOH}2]2− that is suitable for post-functionalization with alcohols.336 In addition, other transformations of [VV6O13{(OCH2)3CCH2OH}2]2− can be achieved by two-step organic post-functionalization.70 In the first step this platform has been esterified with succinic anhydride in the presence of DMAP and the resulting carboxylic acid-terminated species [VV6O13{(OCH2)3CCH2OC(O)CH2CH2COOH}2]2− has been amidated with primary aliphatic or aromatic amines RNH2 (R = –C3H7, –C4H9, –CH2CH = CH2, –C6H13, and –(C6H4)Me-p) in the presence of 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline, leading to anti-[VV6O13{(OCH2)3CCH2OC(O)CH2CH2C(O)NHR}2]2− products (Fig. 28).
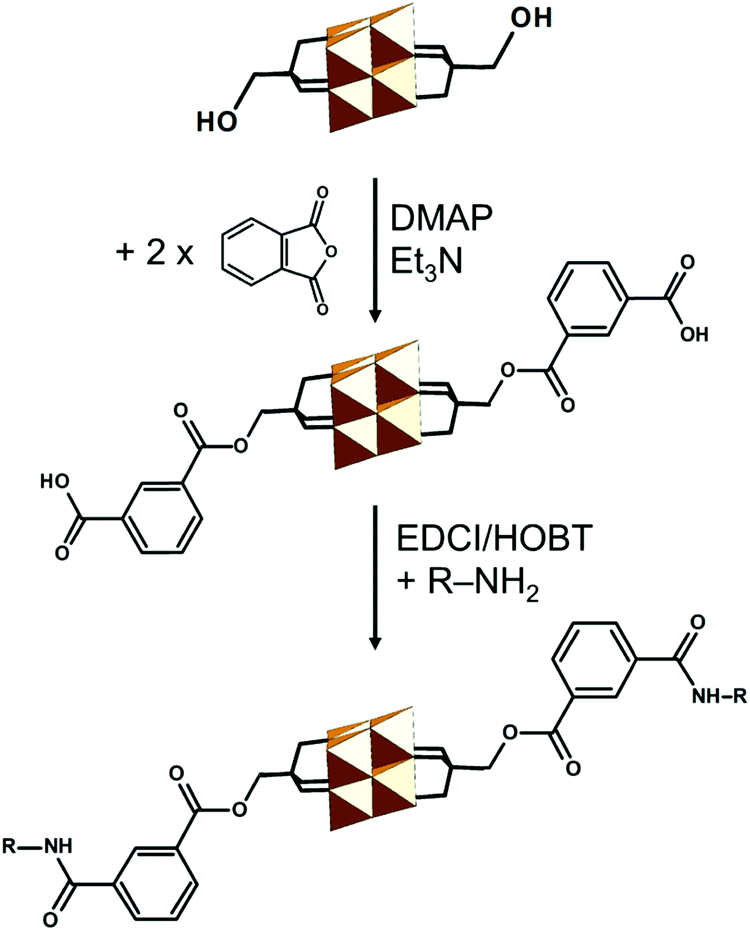 | ||
| Fig. 28 Schematic representation of the step-wise post-functionalization of the [VV6O13{(OCH2)3CCH2OH}2]2− platform with phthalic anhydride and the subsequent amidation reaction. All {VO6} are shown as orange octahedra. | ||
A non-classical approach aiming towards the development of monomodular mixed-addenda Lindqvist type platforms of the type [V3Mo3O16{(OCH2)3CCH2OH}]2− has been recently initiated by the Ritchie group.319 The former platform exhibits the {V3Mo3O19} POM core where the V and Mo centers may adopt different positions, leading to the coexistence of multiple positional or configurational isomers (Fig. 29), which is not uncommon among all-inorganic POMs.343–347 Following DFT calculations it was established that the two or three V centers were directly sharing oxo ligands with the tris-alkoxo bridging functionality.66 To form mixed addenda species, Ritchie and co-workers argued that microwave radiation was essential to obtain the hybrid platform product. The reaction of decavanadate, octamolybdate and pentaerythritol in acetonitrile in a sealed vial for 5 minutes at 110 °C, 4 bar led to the formation of the desired mixture of monomodular polyanions. In a subsequent reaction using acetic anhydride DMAP in acetonitrile heated to 50 °C overnight, an esterified product was obtained, demonstrating the application of [V3Mo3O16{(OCH2)3CCH2OH}]2− as an interesting platform.
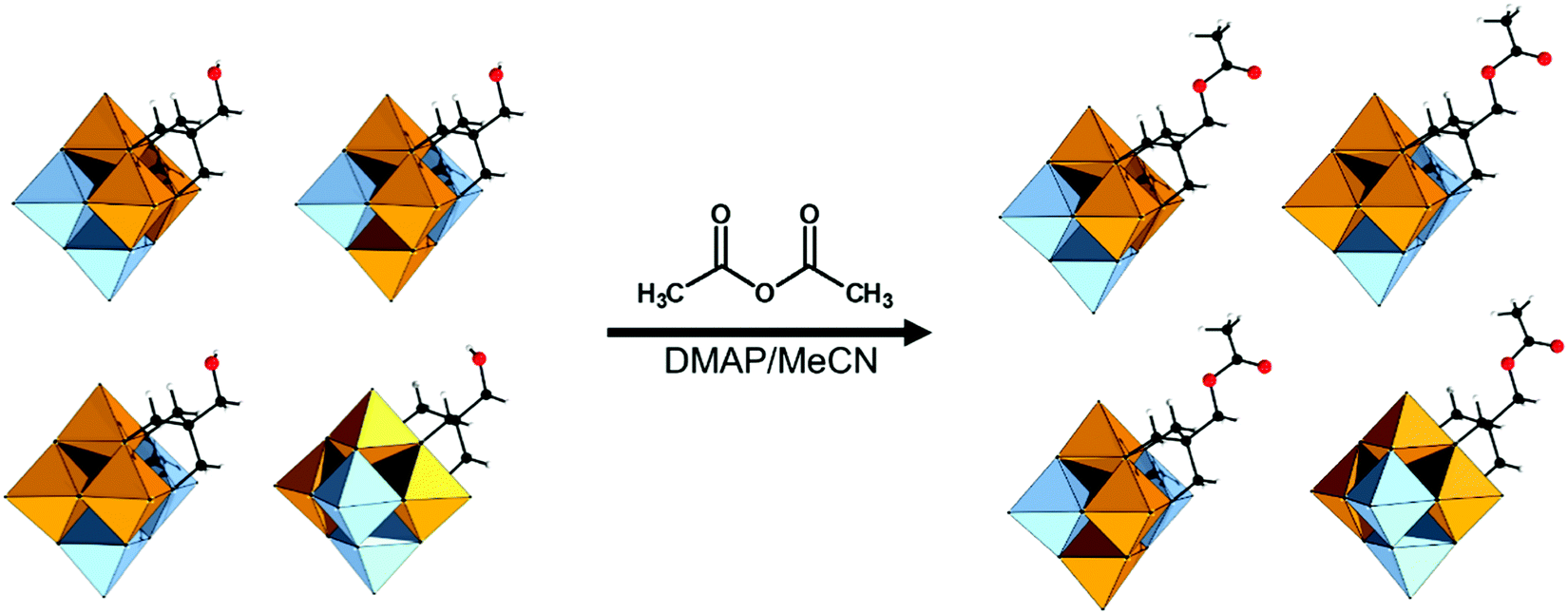 | ||
| Fig. 29 Schematic representation of the isomeric [V3Mo3O16{(OCH2)3CCH2OH}]2− polyanions (left) transforming to isomeric [V3Mo3O16{(OCH2)3CCH2OC(O)CH3}]2− (right). In the experiment the respective populations of different isomers may vary. All structures are shown in a ball-and-stick and polyhedral representation using the following colour code: C = black balls, O = red balls, H = white balls, {VO6} = orange octahedra and {MoO6} = blue octahedra. | ||
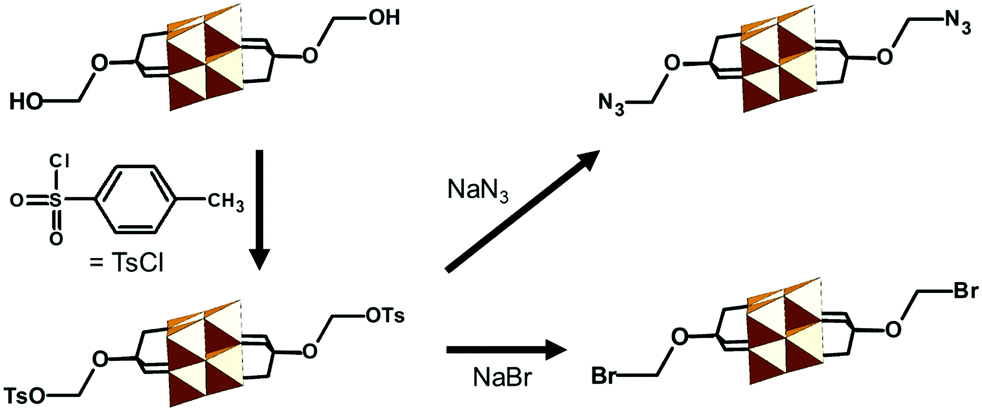 | ||
| Fig. 30 Schematic representation of the tosylation reaction of the [VV6O13{(OCH2)3CCH2OH}2]2− platform and its subsequent nucleophilic reaction with NaN3 and NaBr. All {VO6} are shown as orange octahedra. | ||
CH}2]2− platform whose electronic properties were discussed above (Section 2.5, Fig. 7a) can undergo facile copper(I)-catalysed 1,3-dipolar azide–alkyne cycloaddition with other azide-bearing organic ligands but also with bioactive molecules such as biotin (Fig. 31b). The platform was obtained in a reaction of 2,2-bis-hydroxymethyl-3-prop-2-ynyloxy-propan-1-ol with (NBu4)3[H3V10O28] at 80 °C in DMF solution in the absence of reducing agents.42 Following this, reactions of [V6O13{(OCH2)3CCH2OCH2CCH}2]2− with organoazide complexes in acetonitrile under a N2 atmosphere and in the presence of CuI and DIPEA have led to a large variety of post-functionalized hybrid POMs that were produced in 65–95% yields after 3 hours of stirring. The structural features of the platform and its post-functionalized products have been assessed on the basis of X-ray diffraction. Further characterization has revealed that the formed compounds exhibit good solution stability and solubility in various solvents depending on the properties of the post-functionalizing moiety. Further on, all compounds have exhibited reversible 1 electron reduction and good thermal stability until ca. 200 °C. On the other hand, Wei's azide-anchoring [V6O13{(OCH2)3CCH2-N3}2]2− platform can react with different terminal alkynes in DMF in the presence of CuI and DIPEA; however the reported reactions have been conducted at 70 °C for 3 days, leading to a number of post-functionalized products.334
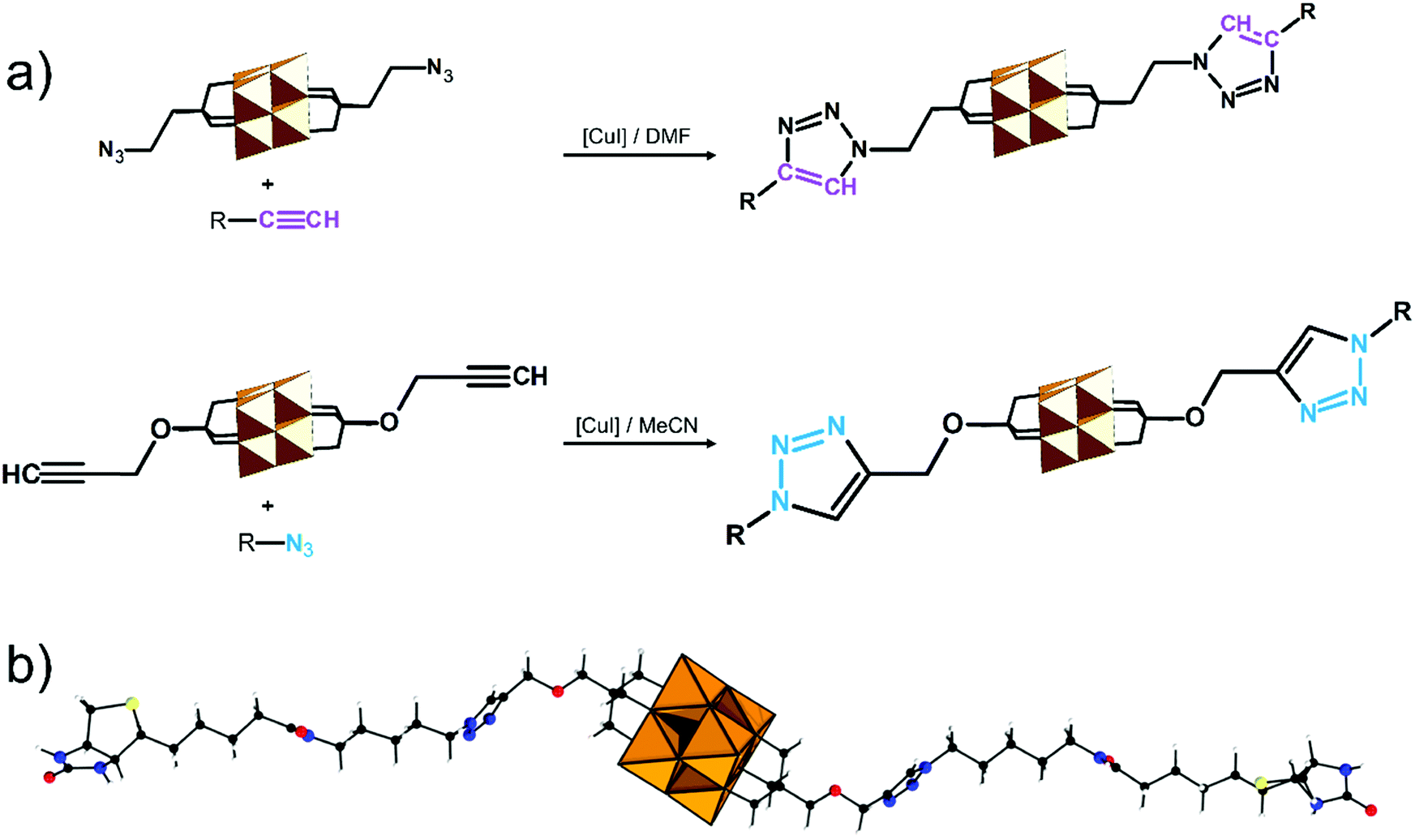 | ||
| Fig. 31 (a) Schematic representation of the two different Lindqvist-type hybrid hexavanadate platforms suitable for the CuI-catalyzed 1,3-alkyne–azide cycloaddition; (b) combined ball-and-stick and polyhedral representation of the biotinylated product deriving from the [V6O13{(OCH2)3CCH2OCH2CCH}2]2− platform. Colour code: C = black balls, N = blue balls, O = red balls, H = white balls, {VO6} = orange octahedra. | ||
In 2018, Nachtigall and Spandl have reported the reduced monomodal platform [V6O7(OCH3)9(OCH2)3CCH2-N3], in which nine μ2-oxo ligands are formally exchanged with μ2-methoxo ligands (Fig. 32a).320 The large number of methoxy ligands hinders the functionalization with a second tris-alkoxo bridging unit. The reactive platform suitable for 1,3-dipolar azide–alkyne cycloaddition is formed by reacting VO(OtBu)3 and (HOCH2)3C-CH2N3 in hot methanol under an argon atmosphere in a sealed vessel for one day. After filtering and evaporating the obtained green solution, a green crude product was obtained which was cleaned in DCM and then crystallized in n-hexane. The platform was then reacted with R–CCH molecules (where R stands for different derivatives of phenylacetylene) in the presence of copper catalyst and potassium carbonate in acetonitrile under an argon atmosphere at room temperature for 1 day. Based on cyclic voltammetry, few distinct {VVxVIV6−x} states were obtained, showing the excellent redox activity of the platform and of the obtained derivatives.
 | ||
| Fig. 32 (a) Schematic representation of typical CuI-catalyzed 1,3-alkyne–azide cycloaddition of the [V6O7(OCH3)9(OCH2)3CCH2-N3] platform (left) leading to the respective 4-bromophenyl post-functionalized derivative (right); (b) reaction of the [V6O7(OCH3)9(OCH2)3C-NH2] platform (left) with tris(4-formylphenyl)amine leading to the formation of trimeric macromolecules (right). Colour code: C = black balls, N = blue balls, O = red balls, H = white balls, {VO6} = orange octahedra. | ||
CH-Ph]3N macromolecule that has good solubility in organic solvents. Cyclic voltammetry of [V6O7(OMe)9(OCH2)3C-NCH-Ph]3N in DCM revealed the absence of notable electronic interaction between the three hexavanadate multielectron reservoirs, indicating that the redox behaviour of the platform building units can be well retained in oligomers and hybrid polymers.
Following the development of the rod-shaped macroions based on Lindqvist and Anderson–Evans structures,275 the groups of Wei and Liu reported a new rod-type architecture comprised of two peripheral Lindqvist hexamolybdates and a central tris-alkoxo stabilized Lindqvist hexavanadate. The polyanion was produced in a rational synthesis following the reaction of two equivalents of hexamolybdate with the [VV6O13{(OCH2)3CNH2}2]2− platform in the presence of DCC.326 The reaction undergoes typical exchange of terminal oxo ligands from the molybdate POM core with the nitride ligand originating from the anchoring group of the primary platform (Fig. 33). The formed structure exhibits photochromic behavior under sunlight illumination, and one-electron reversible reduction of the vanadium and molybdenum Lindqvist cores simultaneously.
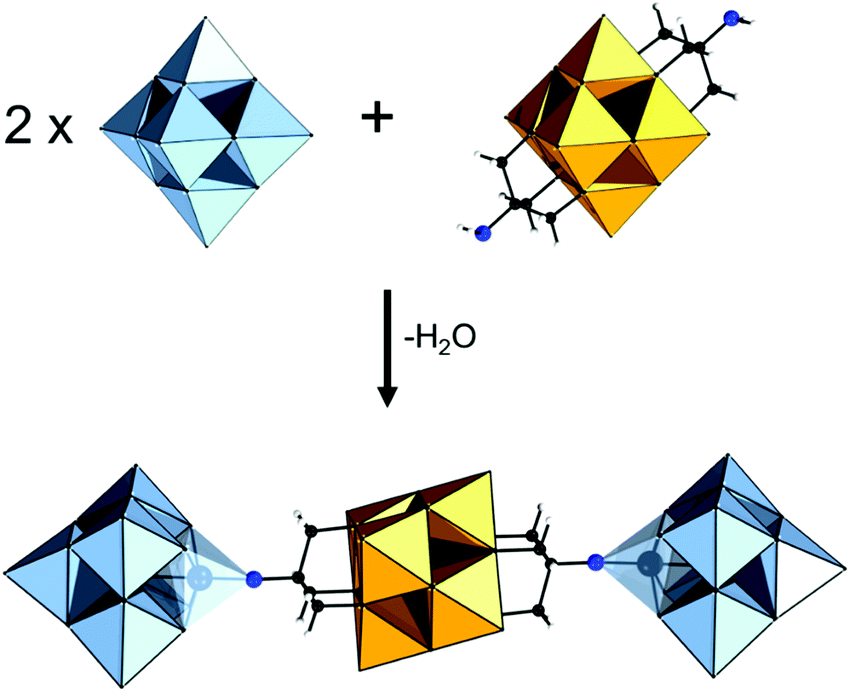 | ||
| Fig. 33 Combined ball-and-stick and polyhedral representation of the formation of the rod-shaped [VV6O13{(OCH2)3CNMo6O18}2]6− macroanion (bottom) following a condensation reaction between two [MoVI6O19]2− units and the [VV6O13{(OCH2)3CNH2}2]2− platform in an anhydrous acetonitrile/pyridine mixed solution. Color code: H = white balls; C = black balls; N = blue balls; O = red balls; Mo = large black balls, {VO6} = orange octahedra and {MoO6} = blue octahedra. | ||
Hybrid hexavanadates with pyridyl anchoring groups such as the [VV6O13{(OCH2)3CCH2OC(O)-4-Py}2]2− polyanion have been employed by Hasenknopf, Ruhlmann and coworkers for coordination to [RuII(CO)TPP] (TPP = meso-tetraphenylporphyrin).304 It has been proposed that the N-centers of each of the two pyridyl functionalities axially coordinate to the RuII centers of the metalloporphyrin unit (Fig. 34a). The formed moiety has been characterized on the basis of elemental analysis, IR, and 1H and 13C NMR spectroscopy. Based on the electrochemical studies it has been shown that the formed molecules exhibit redox irreversible behaviour. Another type of integration of metalloporphyrin with hybrid hexavanadate is by the electrosynthetic copolymerization of anti-[VV6O13{(OCH2)3CNHC(O)-4-Py}2]2− with zinc-β-octaethylporphyrin (Fig. 34b).348 Unlike in the previous example, here the pyridyl termini are covalently bonded to the opposite meso positions of porphyrin. The obtained materials are characterized by UV-Vis, XPS, AFM and CV.
 | ||
| Fig. 34 Schematic representation of: (a) the dumbbell-shaped macrostructure comprised of two [Ru(CO)TPP]-units anchored on the [VV6O13{(OCH2)3CCH2OC(O)-4-Py}2]2− platform; (b) a segment of the copolymeric film made of the interlocked [VV6O13{(OCH2)3CNHC(O)-4-Py}2]2− platform with zinc-β-octaethylporphyrin. All {VO6} units are shown as orange octahedra. | ||
When the platform is furnished with 3-pyridyl instead of 4-pyridyl anchoring terminals, the topology construction takes a different path due to the difference in internal binding angles. Hanan, Hasenknopf and coworkers have reported that the reaction of the [V6O13{(OCH2)3CNHC(O)-3-Py}2]2− platform with [PdCl2(MeCN)2] in DMA leads to trimeric complexes interlocked by {PdCl2} moieties (Fig. 35a).340 Although no single-crystal X-ray structure of the compound has been obtained, its composition and constitution in solution have been determined based on the combination of elemental analysis, high-resolution electrospray ionization (HRESI) mass spectrometry and NMR spectroscopy. On the other hand, by employing the [VV6O13{(OCH2)3CNHC(O)-4-Py}2]2− platform, Hill and coworkers have isolated 1D coordination polymers of the type [M(DMF)2(H2O)2(V6O13{(OCH2)3CNHC(O)-4-Py}2)]n where M = Mn2+, Co2+, Ni2+, or Zn2+ (Fig. 35b).338 Each M2+ ion adopts quasi-octahedral coordination and binds to two DMF, two aqua and 4-pyridyl ligands from the hybrid platform. The copolymers have been isolated in the form of crystals, and have been further characterized on the basis of elemental analysis, 1H and 51V NMR, IR and UV-vis and TGA. Single crystal X-ray diffraction has revealed that in the solid state there was a close packing of the copolymers as a result of hydrogen bonding between CH moieties and the 4-pyridyl rings. The 1-D copolymers have low solubility in polar aprotic solvents (DMSO, DMF and MeCN); however in CCl4 they undergo reversible redox processes without degradation by phenylhydrazine, NaBH4, O2, Br2, CeIV and S2O82−.
 | ||
| Fig. 35 Schematic representation of (a) the PdII-directed trimerization of the [VV6O13{(OCH2)3CNHC(O)-3-Py}2]2− hexavanadate platform; (b) the step-wise formation of the [Co(DMF)2(H2O)2(V6O13{(OCH2)3CNHC(O)-4-Py}2)]n coordination polymer using the [V6O13{(OCH2)3CNHCH2C6H4-4-COOH}2]2− platform; (c) reacting components (left) leading to the formation of TbIII-connecting hexavanadate based hybrid networks (right). The details of the connectivity along single chains are provided in a yellow box. Colour code: C = black balls, N = blue balls, O = red balls, H = white balls, Co = cyan balls, Tb = purple balls, {VO6} = orange octahedra. | ||
By reacting the carboxylate terminated platform TBA2[V6O13{(OCH2)3CNHCH2C6H4-4-COOH}2] with Co(NO3)2·6H2O and 4,4′-bipyridine in a mixed solvent of DMSO/DMF, Hill and co-workers observed the formation of chain-like assemblies which are interlocked by 4,4′-bipyridine moieties at the CoII centers. These copolymers have been characterized on the basis of elemental analysis, IR, and single crystal XRD, and have been shown to catalyze air-based oxidations of thiols (R-SH) to disulfides (R-SS-R, R = Prn).341
The carboxylate anchoring platform [V6O13{(OCH2)3CNHCH2C6H4-4-COOH}2]2− has been further used by the Hill group to coordinate with lanthanide cations such as TbIII.339 Reactions with 4,4′-bis(pyridine-N-dioxide) (= bpdo) and Tb(NO3)3 in DFM/ethylene glycol have led to the formation of a 3-D superstructure elucidated on the basis of single crystal X-ray crystallography. This structure can be described as a 2-D coordinative network made of the primary hybrid platform and the TbIII centers, which is interconnected by the bpdo ligands at the TbIII sites (Fig. 35c). The overall network is porous and exhibits pore diameters between 5.2 Å and 8.0 Å, allowing solvent molecules to reside inside. The network has been further characterized on the basis of elemental analysis, TGA and IR and was found to exhibit catalytic activity for aerobic oxidation of R-SH to R-SS-R (R = Prn) under mild conditions. Following these efforts, the group of Wei also reported that the [V6O13{(OCH2)3C-COOH}2]2− platform can be employed in a similar design of crystalline hybrid networks.337
5.2. Hexamolybdate hybrid platforms
The preparation of hybrid inorganic–organic hexamolybdates can be traced back to the late 1980s and early 1990s when the groups of Zubieta,349 Maatta,350–354 Proust,355 and Errington356 were exploring the reaction of [Mo6O19]2− with phosphinimines, isocyanates, and aromatic amines. In the early 2000s the Peng group discovered that besides the reaction time, addition of N,N′-dicyclohexylcarbodiimide (DCC) in certain concentrations and ratios can allow facile preparation of hybrid hexamolybdate platforms under mild conditions. The method resulted in rapid preparation of hybrid platforms (Table 5) for N-alkylation reactions (Section 5.2.1), esterification reactions (Section 5.2.2), Sonogashira coupling (Section 5.2.3), Heck reaction (Section 5.2.4), addition polymerization (Section 5.2.5) and formation of zwitterions (Section 5.2.6).Typically, these platforms can exhibit single or double anchoring groups, where the anchoring groups are placed in cis rather than in the trans position. The reaction with aromatic amines is also influenced by the bulkiness of the aromatic units, so to achieve successful Mo–N bond formation one should consider repulsion between the bridging unit and the POM core (Fig. 3). A number of platforms have been prepared so far, and the significant research interest in the study of Lindqvist-type hybrid hexamolybdate platforms is due to their unique photoelectronic and non-linear optical properties.357–359
NCH2CH2CH2Cl}]2− platform is obtained in a reaction of octamolybdate with 3-chloropropylamine hydrochloride in hot anhydrous acetonitrile and in the presence of DCC.379 After filtering and washing, red crystals of the TBA salt were isolated and their structure was elucidated on the basis of single crystal XRD (Fig. 36). Subsequent substitution of the anchoring Cl atom with iodine or nitrate has been achieved by reaction of the platform with AgNO3 or NaI in warm acetonitrile under a N2 atmosphere, and single crystal XRD characterization of crystalline solids has validated the post-functionalization.
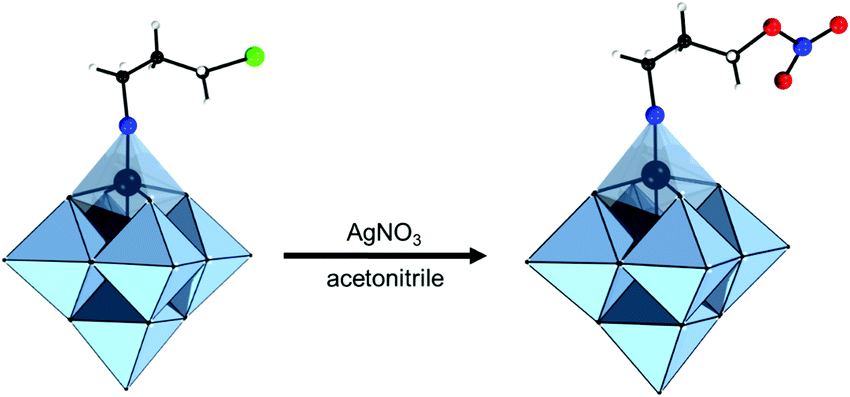 | ||
| Fig. 36 Combined ball-and-stick representation of the [Mo6O18{NCH2CH2CH2Cl}]2− platform (left) and its transformation to the respective nitrated derivative (right). Colour code: C = black balls, N = blue balls, O = red balls, H = white balls, Mo = large black balls, and {MO6} = blue octahedra. | ||
N-3-Py}]2− platforms that are suitable for alkylation. These platforms are formed in reactions of [Mo6O19]2− 3-aminopyridine in hot anhydrous acetonitrile and in the presence of DCC.375 In a similar fashion the Wei group could prepare 4-aminopyridine and 3-methyl-2-aminopyridine derivatives of the same platform; however, it was shown that they either form in very low yields or cause steric hindrance that prevents facile post-functionalization, respectively. Subsequent reactions of [Mo6O18{N-3-Py}]2− with iodomethylane or iodoethylane in acetonitrile without stirring at 50 °C for one week led to N-alkylated products that were isolated in a crystalline form. The group of Wei also reported the [Mo6O18{N-Ph(2,6-Me)-4-CH2-N(C2H4)2O}]2− platform which was prepared under analogous conditions as [Mo6O18{N-3-Py}]2−; however the platform preparation reaction was carried out in the presence of 2,6-dimethyl aniline-para-CH2-morpholine instead of 3-aminopyridine (Fig. 37).375 Wei and co-workers have been also successful in isolating crystals of this platform and characterizing them by single crystal XRD and with other techniques. The platform has been showcased to react with benzyl iodide or allyl iodide in acetonitrile solution at 50 °C for one week, leading to crystalline product formation.
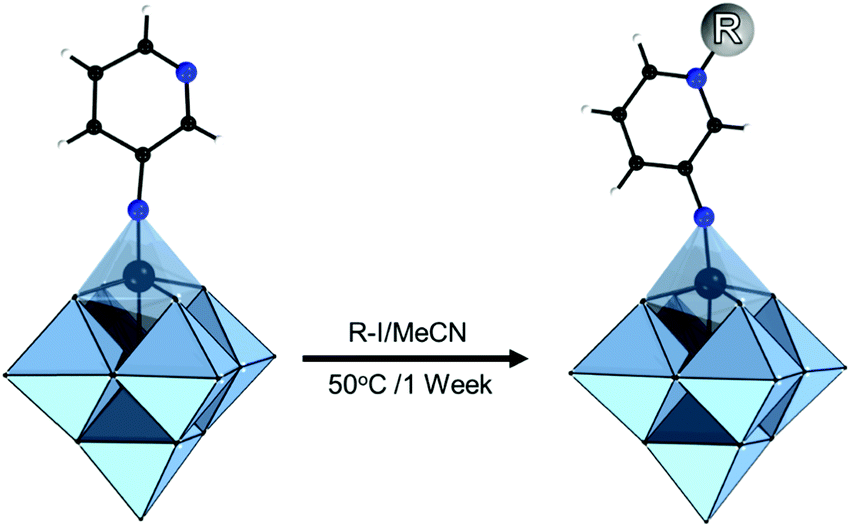 | ||
| Fig. 37 Combined ball-and-stick representation of the [Mo6O18{N-3-Py}]2− platform (left) and its transformation to the respective N-alkylation derivative (right). Color code: C = black balls, N = blue balls, O = red balls, H = white balls, Mo = large black balls, R = grey balls, and {MO6} = blue octahedra. | ||
N-Ar-OH}]2− that can undergo a facile esterification reaction with carboxylic acids in the presence of dicyclohexylcarbodiimide (DCC) to form esters. The platform itself is produced from the reactions of [α-Mo8O26]4− and hydrochloride salt of 4-amino-m-cresol in acetone, under a N2 atmosphere and in the presence of DCC.372 After precipitation, re-dissolving and filtration steps, black-brown crystals were isolated from acetone/ethanol solution. The platform has been reacted with a number of small carboxylate-bearing molecules such as acetic acid, p-methylbenzoic acid, p-chlorobenzoic acid, propanoic acid or acrylic acid, leading to post-functionalized molecules, as confirmed on the basis of single crystal XRD analysis (Fig. 38a).372,373
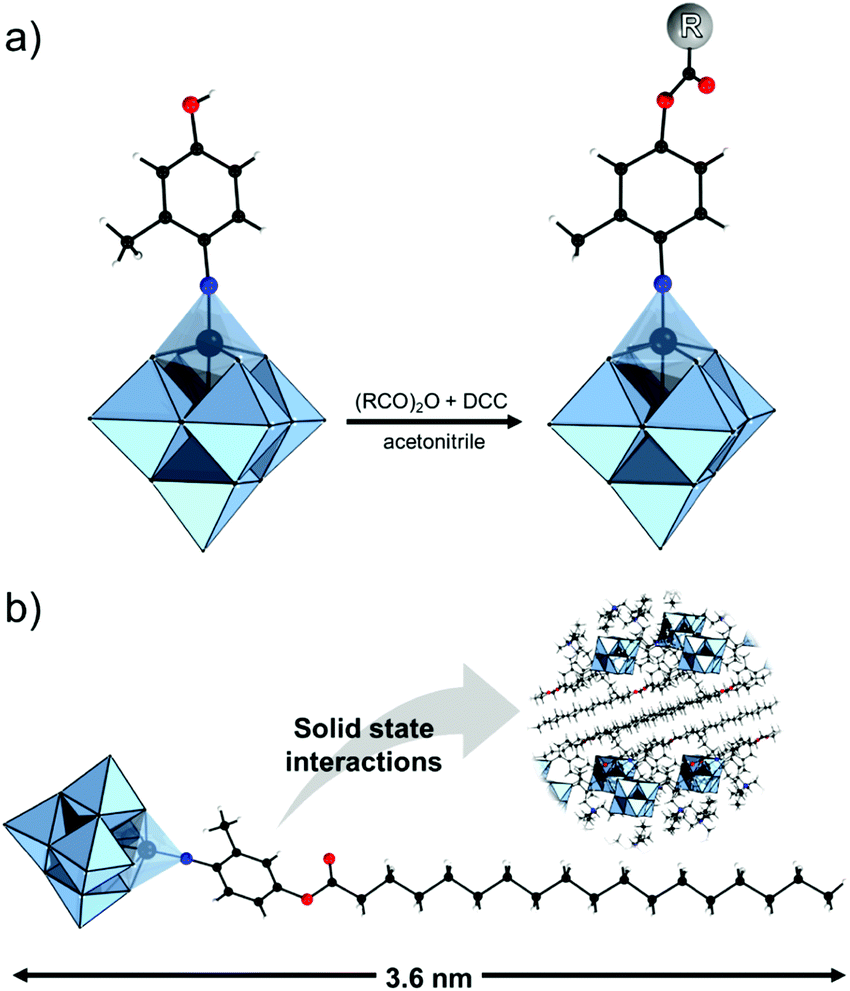 | ||
| Fig. 38 (a) Synthetic representation of the esterification reaction of the [Mo6O18{N-Ar-OH}]2− platform with various RCOOH functionalities. (b) Combined ball-and-stick representation of the layer-by-layer packing of the hybrid [Mo6O18{N-Ar-OOCC17H35}]2− surfactants. Color code: C = black balls; O = red balls, N = blue balls, H = white balls, Mo = large black balls, and {MoO6} = blue octahedra. | ||
In a subsequent work, Wei, Yin and co-workers have reacted the [Mo6O18{N-Ar-OH}]2− platform with alkyl chains with the formula CnH2n+1COOH where n = 5, 9, 13, and 17, leading to the formation of large surfactant molecules which were successfully crystallized and further characterized on the basis of single crystal XRD.374 Interestingly, in the crystal structure of the stearic ester [Mo6O18{N-Ar-OOCC17H35}]2− layer-by-layer packing was observed, where the alkyl chains interact with each other and form domains of close-packed alkyl chains which are sandwiched by two domains of the hexamolybdate POM cores (Fig. 38b). The formed layers have a thickness of 18.28 Å and further ionically interact with TBA countercations from both sides. Differential scanning calorimetry (DSC) analysis revealed melting of the alkyl domain at relatively low temperatures (i.e. 45.84 °C and 80.86 °C) while the interactions of the POM core with the countercations have remained intact, thus exhibiting a typical liquid crystal behaviour.
N-Ph(2,6-Me)-4-CCH}]2− (Fig. 39a) and [Mo6O18{N-Ph(2,6-Me)-4-I}]2− (Fig. 39b),360 which were later showcased to be excellent platforms for Sonogashira coupling reactions.362 These platforms are produced by the reactions of [Mo6O19]2− with aromatic amines in refluxing acetonitrile and in the presence of 1 equivalent of DCC. Following this, by using [Mo8O26]4− as a precursor and doubling the ratio of the 2,6-diisopropyl-4-ethynylaniline ligands and respectively the DCC, it was found that the double anchoring platforms [Mo6O17{N-Ph(2,6-iPr)-4-CCH}2]2− (Fig. 39c) and [Mo6O17{N-Ph(2,6-iPr)-4-I}2]2− (Fig. 39d) could be formed in high yields.367,380 All these platforms have been effectively employed in combination with other molecules, resulting for example in the creation of molecular dumbbells.363 They have been also connected with ferrocene molecules,364 and used to produce cation binding platforms.361 Polymers with unique solution assembly properties361,366 and suitable hybrid molecules for post-functionalization of Si surfaces365 have been also produced based on these platforms.
 | ||
| Fig. 39 Combined ball-and-stick and polyhedral representation of different Lindqvist-type hexamolybdate platforms suitable for Sonogashira coupling: (a) [Mo6O18{N-Ph(2,6-Me)-4-CCH}]2−; (b) [Mo6O18{N-Ph(2,6-Me)-4-I}]2−; (c) [Mo6O17{N-Ph(2,6-iPr)-4-I}2]2−; (d) [Mo6O17{N-Ph(2,6-iPr)-4-CCH}2]2−. Colour code: C = black balls; O = red balls, N = blue balls, H = white balls, Mo = large black balls, I = pink balls, and {MoO6} = {MoO6} = blue octahedra. | ||
Both iodo- or ethynyl-phenylimido anchoring platforms can undergo Pd-catalyzed Sonogashira coupling with compatible molecules in acetonitrile at room temperature under a N2 atmosphere (see for instance the post-functionalized product in Fig. 40a).362 The products are typically formed in about twenty minutes in high yields (>80%), as confirmed on the basis of thin-layer chromatography. Longer reaction times and excess of base were found to lower the amount of the formed products, as they seem to break the Mo–N bond from the POM core.
 | ||
| Fig. 40 Combined ball-and-stick and polyhedral representation of different Lindqvist-type hybrid hexamolybdate platforms obtained following Sonogashira coupling of the respective platforms: (a) [Mo6O18{N-Ph(2,6-Me)-4-CC-1-Ph(4-Me)}]2−; (b) [Mo6O18{N-Ph(2,6-Me)-4-CCH-4-Ph(2,6-Me)-N-}Mo6O18]4− [Mo6O18{N-Ph(2,6-Me)-4-I}]2−; (c) [Mo6O18{N-Ph(2,6-Me)-4-CC-(C5H4)Fe(C5H5)}]2−. Color code: C = black balls; O = red balls, N = blue balls, H = white balls, Mo = large black balls, Fe = brown balls, and {MoO6} = blue octahedra. | ||
Early studies by the Peng and Powell groups have shown the possibility to form dumbbell-like assemblies, simply by coupling both [Mo6O18{N-Ph(2,6-Me)-4-CCH}]2− and [Mo6O18{N-Ph(2,6-Me)-4-I}]2− platforms (Fig. 40b).362 Coupling of two [Mo6O18{N-Ph(2,6-Me)-4-I}]2− platforms with a bridging agent 2,5-bis(2,2-dimethylpropoxy)-1,4-diethynylbenzene is also possible leading to extended dumbbell-like assemblies.362,363 The formation of the dumbbells from platforms has been followed on the basis of UV-Vis spectroscopy, which showed a significant bathochromic shift of the two major absorptions 362 and 368 nm to 426 and 438 nm, respectively. The bathochromic shift can be justified on the basis of the changes of the π bond delocalization around the conjugated π system. In contrast to the phenylene, the dumbbell macroions formed from the ethynylene based platform do not exhibit fluorescence under excitation from 200 to 500 nm, which implies that the conjugating bridge is an efficient fluorescence quencher. More recently, the double anchoring [Mo6O17{N-Ph(2,6-iPr)-4-I}2]2− platform was reported that can undergo reactions with the bridging agent 2,5-bis(2,2-dimethylpropoxy)-1,4-diethynylbenzene to form conjugated polymers.366 In the presence of DMF these conjugated polymers appear in a stretched or extended conformation while in the presence of DMF/water they form cylindrical hollow self-assemblies. The self-assembly process was found to be driven by the presence of counterions, which induce ionic interactions that are similar to those of assembling small inorganic macroions.
The double anchoring [Mo6O17{N-Ph(2,6-iPr)-4-I}2]2− platform has been also successfully coupled with ferrocenyl groups in yields of 65%.364 The attachment was realized by a π-conjugated bridge, allowing communication between the ferrocenyl moiety and the POM core (Fig. 40c). Based on UV-Vis spectroscopic analysis it was revealed that the resulting species exhibit a broad band beyond 550 nm, indicating low frontier orbital gap energy and charge-transfer transition from the ferrocenyl donor to the POM core, even though the components of the formal “donor–acceptor” system were separated by 1.13 nm. Cyclic voltammetry of the hybrid with one ferrocenyl unit has revealed a single-electron oxidation wave at 0.328 V and a single electron reduction wave at −0.831 V.
Coupling of [Mo6O18{N-Ph(2,6-Me)-4-I}]2− with 4-ethynylphenyldiethyltriazene forms [Mo6O18{NPh(2,6-Me)-4-CC-Ph-N3Et2}]2− which was subsequently treated with a strong acid, HBF4, to remove the ethyl-moieties. This led to the formation of [Mo6O18{NPh(2,6-Me)-4-CC-Ph-N2}]− which has a positively charged {N2+} anchoring group, suitable for covalent binding to the Si(111) surface.365 Such Si-post-functionalized surfaces were reported by the Tour group, which demonstrated that depending on the interaction time and the concentration of the applied platform, monolayers and multilayers can be formed. Based on X-ray photoelectron spectroscopy two signals having a 1:5 ratio in the Mo3d region were observed, which were assigned to the Mo-atoms bound to N/O and O centres exclusively, indicating that the hybrid POM remains intact during the surface functionalization. Electrochemical analyses have revealed two reversible redox waves in the range of −1.5 to 0 V, which were different compared to the platform alone in solution and could be explained by surface confinement.
The [Mo6O18{N-Ph(2,6-Me)-4-I}]2− platform can also undergo coupling with suitable 4′-ethynyl-2,2′,6′,2′′-terpyridine moieties, thus generating a derivative platform that can undergo binding to cationic moieties (vide infra).361
N-Ph(2,6-R′)-4-X}]2−, R′ = Me, Et, X = Br, I, which were formed following the procedure developed by the Peng group,368 were further employed to undergo post-functionalization following the Pd-catalyzed Heck coupling reaction. The platform was reacted with the bromomagnesium form of aromatic amines and 1 mol% of [Pd2(dba)3] (dba = dibenzylideneacetone) in hot THF under a nitrogen atmosphere (Fig. 41). Validations of the post-functionalization were obtained on the basis of single crystal analysis, and the procedure has been also confirmed for the post-functionalization of the platform with styrene, 4-bromostyrene, ethyl acrylate and acrylonitrile.
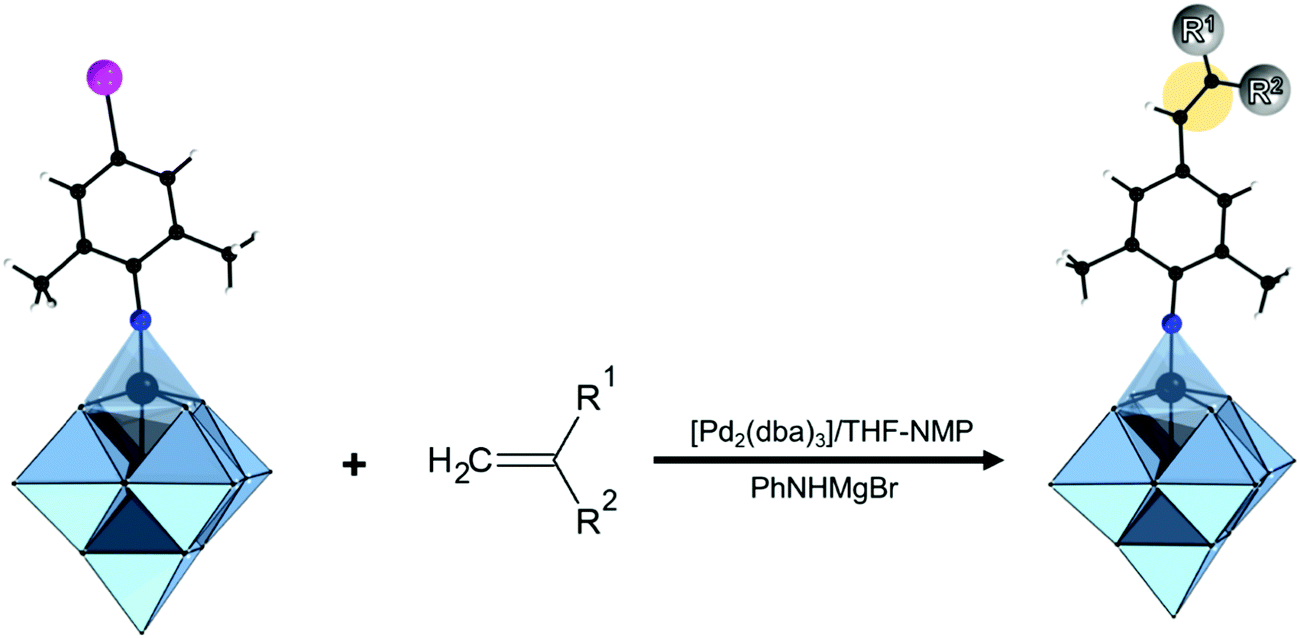 | ||
| Fig. 41 Schematic representation of the Heck reaction transformation of [Mo6O18{N-Ph(2,6-Me)-4-I}]2− towards its corresponding hybrid derivatives. Colour code: C = black balls, N = blue balls, O = red balls, H = white balls, Mo = large black balls, and {MO6} = blue octahedra. | ||
N-Ph-4-COOH}]2− has been obtained in a reaction of [Mo8O26]4− with p-aminobenzoic acid hydrochloride in hot acetonitrile and in the presence of DCC.377 In acetonitrile the carboxylic anchoring groups can coordinate to two copper(II) cations to form paddle-wheel-like tetrapolyoxometalate macroions (Fig. 42a). Both compounds were characterized on the basis of single crystal XRD analysis. DLS analysis of the tetrapolyoxometalate microions in an acetone/toluene mixture has revealed the formation of large, vesicular and stable blackberry-type structures of ca. 100 nm. The size of the blackberry-structures can be effectively tuned by relative proportions of acetone/toluene. These aspects indicate hierarchical POM platform coordination and organization to macroions and further to nanoscopic supramolecular architectures.
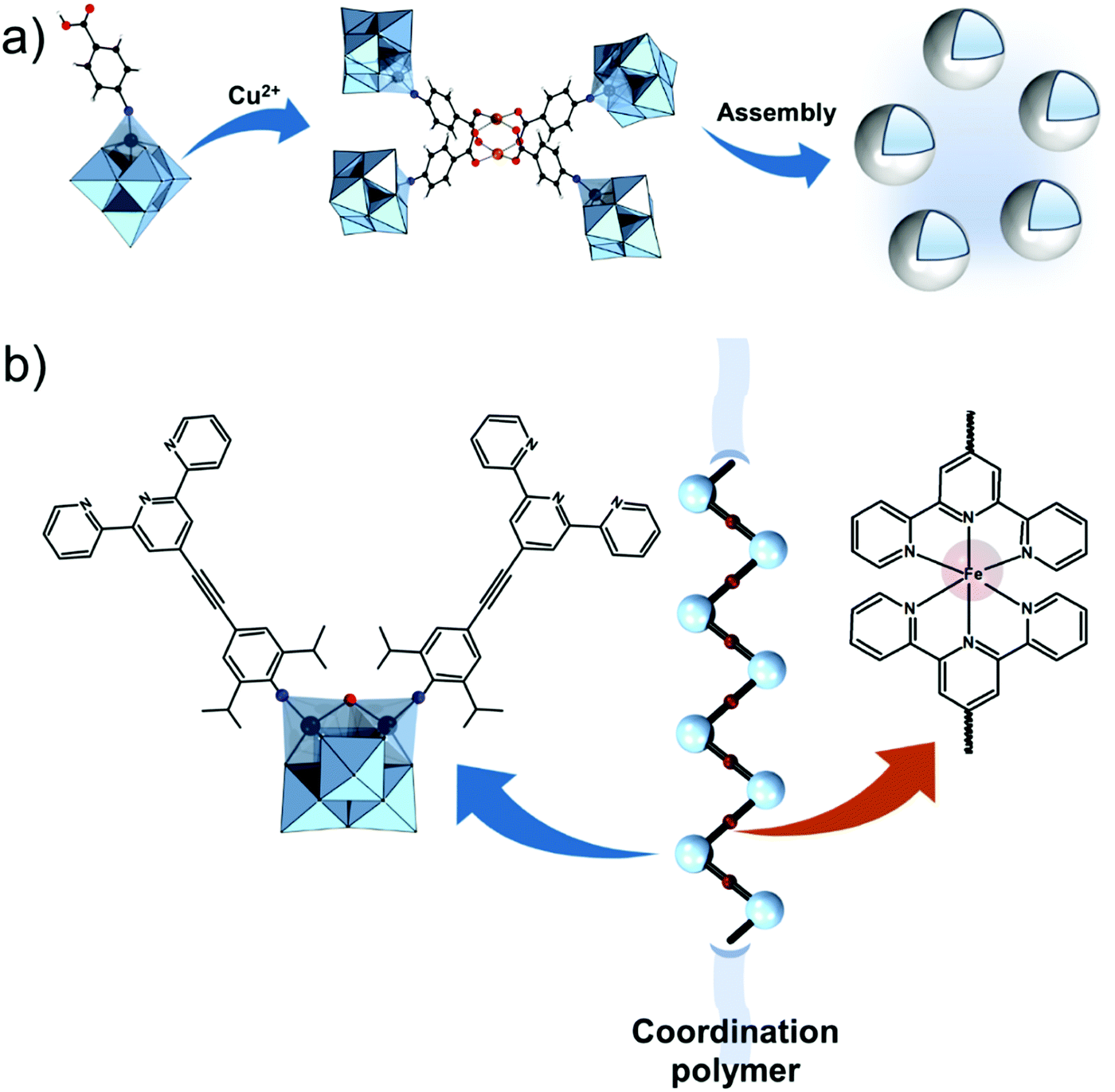 | ||
| Fig. 42 Schematic representation of (a) the hierarchical assembly of [Mo6O18{N-Ph-4-COOH}]2− to tetramers by coordination to Cu2+ cations and subsequent organization of the tetramers to vesicular structures in acetone/toluene mixtures; (b) the constituents of the metallopolymer made of a bimodular [Mo6O17{N-Ph(2,6-iPr)-4-CC-terpyridyl}2]2− platform and Fe2+ cations. Colour code: C = black balls, N = blue balls, O = red balls, H = white balls, Mo = large black balls, and {MO6} = blue octahedra. | ||
The [Mo6O18{NPh(2,6-Me)-4-CC-terpyridyl}]2− platform was synthesized by a reaction of [Mo6O19]2− with the terpyridine ligand in hot acetonitrile and in the presence of DCC, and its structure was confirmed on the basis of single crystal XRD.378 The platform has been subsequently used for coordination with Zn2+ and Ru2+ cations in DMSO, while the overall process has been observed using UV-Vis absorption spectroscopy. The UV-Vis analysis in both cases exhibits bathochromic shift which can be rationalized based on the decrease in the energy of the frontier orbitals. In a follow-up study, the interactions of Fe2+ with the [Mo6O18{NPh(2,6-Me)-4-CC-terpyridyl}]2− platform have been further investigated. The isolated material has shown that two platform units chelate a single Fe2+ center, leading to a linear macroanion. The linear structure of the formed species has been confirmed on the basis of 1H NMR spectroscopy, and ESI-MS showed a peak with m/z: 1266.0 that can be assigned to [Mo12O36N8C50H36Fe]2− species. The double modular platform [Mo6O17{N-Ph(2,6-iPr)-4-CC-terpyridyl}2]2− has similar affinity to interact with Fe2+ cations in acetonitrile or DMSO at room temperature; however it forms coordination polymers that have low solubility in DMSO (Fig. 42b).361
Finally, the platform [Mo6O18{NPh-4-CH2-N3C2H2}]2−, which was formed in the reactions of octamolybdate with 4-(1,2,4-triazolylmethyl)phenylamine hydrochloride in hot acetonitrile and in the presence of DCC,376 has been isolated as a crystalline material but in low yields (ca. 10%). The platform has been successfully reacted with fac-[Re(phen)(CO)3(H2O)], leading to the formation of the [Mo6O18NC6H4-CH2-N3C2H2-Re-phen(CO)3]− complex (Fig. 43). DFT calculations have shown that the HOMO of the platform is distributed over the organic functionality and exhibits electronic conjugation similar to the π-orbital. Binding of ReI increases the energy of the HOMO and dispositions it partially over the ReI center, while the LUMO remains on the hexamolybdate core. The reduction of the frontier energy gap energy was regarded as the dominant effect leading to the observed UV-Vis bathochromic shift, and was also confirmed on the basis of time-dependent density functional theory calculations.
 | ||
| Fig. 43 Schematic representation of the binding of the [Mo6O18{NPh-4-CH2-N3C2H2}]2− platform to Ru(tPy)Cl3. All {MO6} are represented as blue octahedra. | ||
N-Ph-4-CHCH2}]2− can undergo facile addition polymerization with 4-methylstyrene in 1,2-dichloroethane.371 The free radical copolymerization is induced by AIBN (2,2′-azobis(2-methylpropionitrile)) which is also added in the reaction mixture. The polymer formation was confirmed on the basis of IR spectroscopy, which showed that the band corresponding to the CC bond stretch was diminished, while the spectrum contained stretching and bending vibrations associated with the POM core. Based on 1H NMR intensities of the protons deriving from the counter-cations and the styrenic –CH3 group, it was proposed that ca. three 4-methyl styrene units are bound per p-styrenylimido hexamolybdate (Fig. 44). This implies that the reactivity of the [Mo6O18{N-Ph-4-CHCH2}]2− platform is comparable to that of the styrene and can be further utilized in the preparation of POM containing inorganic–organic hybrid polymers.
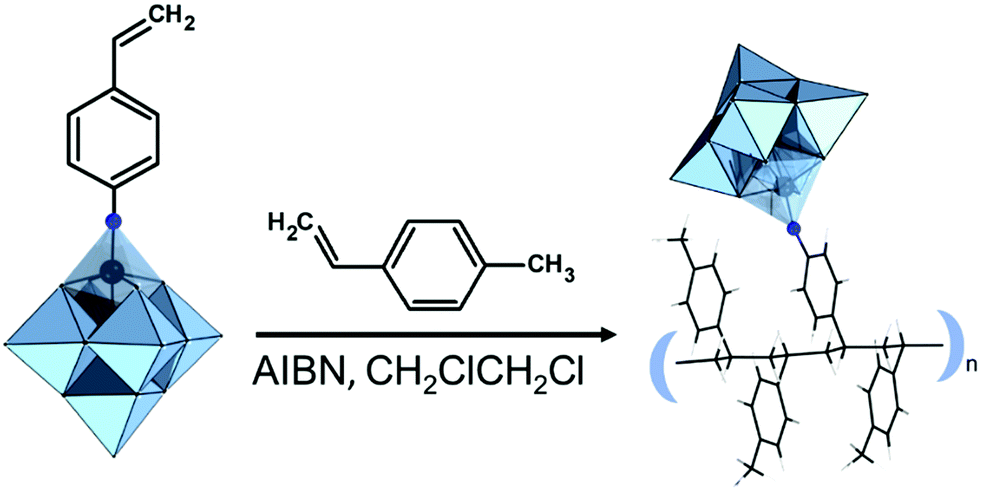 | ||
| Fig. 44 Schematic representation of free radical-induced copolymerization between the [Mo6O18{N-Ph-4-CHCH2}]2− platform and 4-methylstyrene. All {MO6} are represented as blue octahedra. | ||
6. Summary and outlook
Over the past two decades we have witnessed a vibrant development in the area of hybrid polyoxometalates that are suitable for facile post-functionalization with other organic or inorganic molecules, leading to an advanced material design. In this review we have surveyed the currently available hybrid POM platforms suitable for post-functionalization, and classified them on the basis of topology, elemental content, modularity, type of organic bridging and spacing units, and available post-functionalization approaches. Through this review it was revealed how the post-functionalization approaches can generate POMs with novel and improved physicochemical properties relevant to the development of novel (photo)catalysts, photosensitizers, surfactants, energy materials, biocompatible materials, POM-functionalized surfaces and optoelectronic materials, among others. It was also apparent that in the course of designing novel hybrid POMs, the post-functionalization strategy needs to be carefully chosen and well-implemented, as otherwise the improper choice of reaction conditions may prove to be detrimental to the POM unit or to the bridging moiety that connects the POM with the reacting anchoring functional group.In the coming years we expect that prospective advances in the post-functionalization approaches of hybrid POM platforms will further increase the importance of these methods in POM applications. Computer-aided design and rational synthesis of novel POM hybrids that match POM units with respective (bio)organic or inorganic counterparts are having an increasingly important role in the advanced POM engineering, and can lead to materials with superior properties that are relevant to specific domains. The ability to perform multiple modifications in a regioselective, stereoselective and chemoselective manner will be crucial for producing materials with functionalities that are positioned in desirable configurations suitable for tandem catalysis, photosensing, molecular recognition and surface immobilization. Finally, the development of novel platforms which are based on mixed-addenda or late-transition metal addenda may surpass the realms of complexity exerted by classical POMs, leading to full realization of the POM potential in a variety of technological domains.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TPV thanks Research Foundation – Flanders (FWO) and KU Leuven for funding. AA (on leave from Nikolaev Institute of Inorganic Chemistry SB RAS, Novosibirsk) thanks KU Leuven for funding. AK thanks FWO for post-doctoral fellowship (166497/12Y9218N LV 5457). The authors thank Ms. Lara Delevic for help with composing some of the figures.References
- A. Kondinski and T. N. Parac-Vogt, Front. Chem., 2018, 6, 346 CrossRef PubMed.
- S. L. Sass, The Substance of Civilization: Materials and Human History from the Stone Age to the Age of Silicon, Skyhorse Publishing, 2011 Search PubMed.
- J. L. Provis, Front. Mater., 2015, 2, 31 CrossRef.
- O. Hayden, Front. Mater., 2014, 1, 5 CrossRef.
- T. Heine, Front. Mater., 2014, 1, 7 CrossRef.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- H. Zhang, G. Chen and D. W. Bahnemann, J. Mater. Chem., 2009, 19, 5089–5121 RSC.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- C. Yuan, H. Bin Wu, Y. Xie and X. W. (David) Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS.
- S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. (David) Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- A. Seliverstov, J. Forster, J. Tucher, K. Kastner and C. Streb, Discovering the Future of Molecular Sciences, 2014 Search PubMed.
- H. N. Miras, Discovering the Future of Molecular Sciences, 2014 Search PubMed.
- A. Kondinski and K. Y. Monakhov, Chem. – Eur. J., 2017, 23, 7841–7852 CrossRef CAS PubMed.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, 1983, vol. 8 Search PubMed.
- A. V. Anyushin, A. I. Smolentsev, D. A. Mainichev, C. Vicent, A. L. Gushchin, M. N. Sokolov and V. P. Fedin, Chem. Commun., 2014, 50, 9083–9085 RSC.
- G. Hervé, A. Tézé and R. Contant, in Polyoxometalate Molecular Science, ed. J. J. Borrás-Almenar, E. Coronado, A. Müller and M. Pope, Springer, Netherlands, Dordrecht, 2003, pp. 33–54 Search PubMed.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 112 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464–7478 RSC.
- A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239–272 CrossRef PubMed.
- D. Gatteschi, L. Pardi, A. L. Barra and A. Müller, Mol. Eng., 1993, 3, 157–169 CrossRef CAS.
- G. Absillis, E. Cartuyvels, R. Van Deun and T. N. Parac-Vogt, J. Am. Chem. Soc., 2008, 130, 17400–17408 CrossRef CAS PubMed.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed.
- A. Bijelic and A. Rompel, Coord. Chem. Rev., 2015, 299, 22–38 CrossRef CAS PubMed.
- G. Kickelbick, Introduction to Hybrid Materials, in Hybrid Materials, ed. G. Kickelbick, 2007, pp. 1–48, DOI:10.1002/9783527610495.ch1.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- B. Kowalewski, J. Poppe, U. Demmer, E. Warkentin, T. Dierks, U. Ermler and K. Schneider, J. Am. Chem. Soc., 2012, 134, 9768–9774 CrossRef CAS PubMed.
- A. Müller and D. Rehder, Molecular Metal Oxides in Protein Cages/Cavities, in Coordination Chemistry in Protein Cages, ed. T. Ueno and Y. Watanabe, 2013, pp. 25–42, DOI:10.1002/9781118571811.ch2.
- D. Janell, A. Tocilj, I. Kölln, F. Schlünzen, M. Glühmann, H. A. S. Hansen, J. Harms, A. Bashan, I. Agmon, H. Bartels, M. Kessler, S. Weinstein, F. Franceshi and A. Yonath, Ribosomal Crystallography and Heteropolytungstates, in Polyoxometalate Chemistry From Topology via Self-Assembly to Applications, ed. M. Pope and A. Müller, Springer, Netherlands, Dordrecht, 2001, pp. 391–415 Search PubMed.
- Y.-F. Song, D.-L. Long, C. Ritchie and L. Cronin, Chem. Rec., 2011, 11, 158–171 CrossRef CAS PubMed.
- S. Radhakrishnan, J. Thomas and M. Yoon, Polyoxometalate-based molecular/nano composites: Advances in environmental remediation by photocatalysis and biomimetic approaches to solar energy conversion, 2012, vol. 13 Search PubMed.
- S. Herrmann, C. Ritchie and C. Streb, Dalton Trans., 2015, 44, 7092–7104 RSC.
- J. Wang, C. G. Lin, J. Li, J. Wei, Y. F. Song and J. Guo, Mol. Cryst. Liq. Cryst., 2016, 634, 12–23 CrossRef CAS.
- O. Linnenberg, A. Kondinski, C. Stöcker and K. Y. Monakhov, Dalton Trans., 2017, 46, 15636–15640 RSC.
- A. Miolati and R. Pizzighelli, J. Prakt. Chem., 2004, 77, 417–456 CrossRef.
- A. Rosenheim and R. Bilecki, Ber. Dtsch. Chem. Ges., 1913, 46, 539–557 CrossRef CAS.
- K. M. Barkigia, L. M. Rajkovic, M. T. Pope and C. O. Quicksall, J. Am. Chem. Soc., 1975, 97, 4146–4147 CrossRef CAS.
- K. Y. Monakhov, W. Bensch and P. Kögerler, Chem. Soc. Rev., 2015, 44, 8443–8483 RSC.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC.
- W. Kwak, L. M. Rajkovic, M. T. Pope, C. O. Quicksall, K. Y. Matsumoto and Y. Sasaki, J. Am. Chem. Soc., 1977, 99, 6463–6464 CrossRef CAS.
- J. Yan, X. Zheng, J. Yao, P. Xu, Z. Miao, J. Li, Z. Lv, Q. Zhang and Y. Yan, J. Organomet. Chem., 2019, 884, 1–16 CrossRef CAS.
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef CAS.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- R. Pütt, A. Kondinski and K. Monakhov, Chem. Unserer Zeit, 2018, 52, 384–389 CrossRef.
- J. R. Winkler and H. B. Gray, Electronic Structures of Oxo-Metal Ions, in Molecular Electronic Structures of Transition Metal Complexes I, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 17–28 Search PubMed.
- X. López, J. A. Fernández and J. M. Poblet, Dalton Trans., 2006, 1162–1167 RSC.
- X. López, J. M. Maestre, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2001, 123, 9571–9576 CrossRef PubMed.
- X. López, J. J. Carbó, C. Bo and J. M. Poblet, Chem. Soc. Rev., 2012, 41, 7537–7571 RSC.
- O. Linnenberg, A. Kondinski and K. Y. Monakhov, The Lindqvist hexavanadate: A platform for coordination-directed assembly, in Supramolecular Systems: Chemistry, Types and Applications, ed. C. Pena, Nova Science Publishers, 2017, pp. 39–66 Search PubMed.
- L. C. W. Baker and J. S. Figgis, J. Am. Chem. Soc., 1970, 92, 3794–3797 CrossRef CAS.
- H. Chermette and F. Lefebvre, C. R. Chim., 2012, 15, 143–151 CrossRef CAS.
- N. H. Nsouli, B. S. Bassil, M. H. Dickman, U. Kortz, B. Keita and L. Nadjo, Inorg. Chem., 2006, 45, 3858–3860 CrossRef CAS PubMed.
- J. Canny, A. Teze, R. Thouvenot and G. Herve, Inorg. Chem., 1986, 25, 2114–2119 CrossRef CAS.
- T. Minato, K. Suzuki, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2017, 23, 14213–14220 CrossRef CAS PubMed.
- A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64 CrossRef CAS.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925–946 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. Spillane, R. Sharma, A. Zavras, R. Mulder, C. A. Ohlin, L. Goerigk, R. A. J. O’Hair and C. Ritchie, Angew. Chem., Int. Ed., 2017, 56, 8568–8572 CrossRef CAS PubMed.
- X. Hu, Z. Xiao, B. Huang, X. Hu, M. Cheng, X. Lin, P. Wu and Y. Wei, Dalton Trans., 2017, 46, 8505–8513 RSC.
- L. Xin-Xiong, W. Yang-Xin, W. Rui-Hu, C. Cai-Yan, T. Chong-Bin and Y. Guo-Yu, Angew. Chem., Int. Ed., 2016, 55, 6462–6466 CrossRef PubMed.
- V. A. Zamolo, G. Modugno, E. Lubian, A. Cazzolaro, F. Mancin, L. Giotta, D. Mastrogiacomo, L. Valli, A. Saccani, S. Krol, M. Bonchio and M. Carraro, Front. Chem., 2018, 6, 278 CrossRef PubMed.
- Z. Xiao, K. Chen, B. Wu, W. Li, P. Wu and Y. Wei, Eur. J. Inorg. Chem., 2016, 808–811 CrossRef CAS.
- A. J. Kibler and G. N. Newton, Polyhedron, 2018, 154, 1–20 CrossRef CAS.
- S. Zhou, Y. Feng, M. Chen, Q. Li, B. Liu, J. Cao, X. Sun, H. Li and J. Hao, Chem. Commun., 2016, 52, 12171–12174 RSC.
- Y. Hou and C. L. Hill, J. Am. Chem. Soc., 1993, 115, 11823–11830 CrossRef CAS.
- C. P. Pradeep, D. L. Long, G. N. Newton, Y. F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388–4391 CrossRef CAS PubMed.
- D. Karimian, B. Yadollahi and V. Mirkhani, Microporous Mesoporous Mater., 2017, 247, 23–30 CrossRef CAS.
- P. Putaj and F. Lefebvre, Coord. Chem. Rev., 2011, 255, 1642–1685 CrossRef CAS.
- N. V. Izarova, M. T. Pope and U. Kortz, Angew. Chem., Int. Ed., 2012, 51, 9492–9510 CrossRef CAS PubMed.
- M. Nyman and L. Fullmer, Small Angle X-ray Scattering of Group V Polyoxometalates, in Trends in Polyoxometalates Research, ed. L. Ruhlmann and D. Schaming, Nova Science Publishers, Inc., 2015, pp. 151–170, ISBN: 978-1-63482-656-3 Search PubMed.
- C. Vicent, S. A. Adonin, A. V. Anyushin, D. A. Mainichev and M. N. Sokolov, Eur. J. Inorg. Chem., 2014, 5618–5624 CrossRef CAS.
- C. Dablemont, C. G. Hamaker, R. Thouvenot, Z. Sojka, M. Che, E. A. Maatta and A. Proust, Chem. – Eur. J., 2006, 12, 9150–9160 CrossRef CAS PubMed.
- V. Artero, D. Laurencin, R. Villanneau, R. Thouvenot, P. Herson, P. Gouzerh and A. Proust, Inorg. Chem., 2005, 44, 2826–2835 CrossRef CAS PubMed.
- W. H. Knoth, P. J. Domaille and D. C. Roe, Inorg. Chem., 1983, 22, 198–201 CrossRef CAS.
- B. Liu, J. Yan, Y.-F. Wang and X.-Y. Yi, Dalton Trans., 2015, 44, 16882–16887 RSC.
- X. Wei, R. E. Bachman and M. T. Pope, J. Am. Chem. Soc., 1998, 120, 10248–10253 CrossRef CAS.
- J. Karcher, K. Mijares, E. Maatta, V. Lahootun, F. Launay, C. Courillon and A. Proust, Eur. J. Inorg. Chem., 2008, 4899–4905 Search PubMed.
- M. N. Sokolov, S. A. Adonin, D. A. Mainichev, C. Vicent, N. F. Zakharchuk, A. M. Danilenko and V. P. Fedin, Chem. Commun., 2011, 47, 7833–7835 RSC.
- M. Sadakane, S. Moroi, Y. Iimuro, N. Izarova, U. Kortz, S. Hayakawa, K. Kato, S. Ogo, Y. Ide, W. Ueda and T. Sano, Chem. – Asian J., 2012, 7, 1331–1339 CrossRef CAS PubMed.
- A. Mazeaud, N. Ammari, F. Robert and R. Thouvenot, Angew. Chem., Int. Ed. Engl., 1996, 35, 1961–1964 CrossRef CAS.
- D. Agustin, C. Coelho, A. Mazeaud, P. Herson, A. Proust and R. Thouvenot, Z. Anorg. Allg. Chem., 2004, 630, 2049–2053 CrossRef CAS.
- T. Zhang, L. Mazaud, L.-M. Chamoreau, C. Paris, A. Proust and G. Guillemot, ACS Catal., 2018, 8, 2330–2342 CrossRef CAS.
- N. Joo, S. Renaudineau, G. Delapierre, G. Bidan, L. M. Chamoreau, R. Thouvenot, P. Gouzerh and A. Proust, Chem. – Eur. J., 2010, 16, 5043–5051 CrossRef CAS PubMed.
- G. Guillemot, E. Matricardi, L. M. Chamoreau, R. Thouvenot and A. Proust, ACS Catal., 2015, 5, 7415–7423 CrossRef CAS.
- M. Tountas, Y. Topal, A. Verykios, A. Soultati, A. Kaltzoglou, T. A. Papadopoulos, F. Auras, K. Seintis, M. Fakis, L. C. Palilis, D. Tsikritzis, S. Kennou, A. Fakharuddin, L. Schmidt-Mende, S. Gardelis, M. Kus, P. Falaras, D. Davazoglou, P. Argitis and M. Vasilopoulou, J. Mater. Chem. C, 2018, 6, 1459–1469 RSC.
- S. Aoki, T. Kurashina, Y. Kasahara and T. Nishijima, Dalton Trans., 2011, 01, 2–7 Search PubMed.
- M. Carraro, G. Modugno, G. Fiorani, C. MacCato, A. Sartorel and M. Bonchio, Eur. J. Org. Chem., 2012, 281–289 CrossRef CAS.
- J. Niu, M. Li and J. Wang, J. Organomet. Chem., 2003, 675, 84–90 CrossRef CAS.
- X. Chen, H. Li, P. Yin and T. Liu, Chem. Commun., 2015, 51, 6104–6107 RSC.
- C. R. Mayer and R. Thouvenot, J. Chem. Soc., Dalton Trans., 2002, 7–14 Search PubMed.
- R. Villanneau, A. Ben Djamâa, L. M. Chamoreau, G. Gontard and A. Proust, Eur. J. Inorg. Chem., 2013, 1815–1820 CrossRef CAS.
- R. Villanneau, D. Racimor, E. Messner-Henning, H. Rousselière, S. Picart, R. Thouvenot and A. Proust, Inorg. Chem., 2011, 50, 1164–1166 CrossRef CAS PubMed.
- R. Villanneau, A. Marzouk, Y. Wang, A. Ben Djamaa, G. Laugel, A. Proust and F. Launay, Inorg. Chem., 2013, 52, 2958–2965 CrossRef CAS PubMed.
- O. Makrygenni, E. Secret, A. Michel, D. Brouri, V. Dupuis, A. Proust, J. M. Siaugue and R. Villanneau, J. Colloid Interface Sci., 2018, 514, 49–58 CrossRef CAS PubMed.
- M. Carraro, G. Modugno, A. Sartorel, G. Scorrano and M. Bonchio, Eur. J. Inorg. Chem., 2009, 5164–5174 CrossRef CAS.
- E. Grinenval, J. Basset and F. Lefebvre, J. Inorg. Chem., 2013, 2013, 1–8 CrossRef.
- C. R. Mayer, I. Fournier and R. Thouvenot, Chem. – Eur. J., 2000, 6, 105–110 CrossRef CAS.
- C. R. Mayer, R. Thouvenot and T. Lalot, Chem. Mater., 2000, 12, 257–260 CrossRef CAS.
- C. R. Mayer, S. Neveu and V. Cabuil, Angew. Chem., Int. Ed., 2002, 41, 501–503 CrossRef CAS PubMed.
- M. Carraro, L. Sandei, A. Sartorel, G. Scorrano and M. Bonchio, Org. Lett., 2006, 8, 3671–3674 CrossRef CAS PubMed.
- M. Carraro, G. Fiorani, L. Mognon, F. Caneva, M. Gardan, C. MacCato and M. Bonchio, Chem. – Eur. J., 2012, 18, 13195–13202 CrossRef CAS PubMed.
- G. Modugno, Z. Syrgiannis, A. Bonasera, M. Carraro, G. Giancane, L. Valli, M. Bonchio and M. Prato, Chem. Commun., 2014, 50, 4881–4883 RSC.
- M. Carraro, G. Bergamini, M. Di Lauro, G. Modugno, M. Baroncini, P. Ceroni and M. Bonchio, Eur. J. Inorg. Chem., 2016, 3405–3410 CrossRef CAS.
- H. Chen, L. Xie, H. Lu and Y. Yang, J. Mater. Chem., 2007, 17, 1258–1261 RSC.
- T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425–6433 CrossRef CAS PubMed.
- T. Hirano, K. Uehara, S. Uchida, M. Hibino, K. Kamata and N. Mizuno, Inorg. Chem., 2013, 52, 2662–2670 CrossRef CAS PubMed.
- C. Mayer, R. Thouvenot and T. Lalot, Macromolecules, 2000, 33, 4433–4437 CrossRef CAS.
- C. R. Mayer, V. Cabuil, T. Lalot and R. Thouvenot, Angew. Chem., Int. Ed., 1999, 38, 3672–3675 CrossRef CAS PubMed.
- A. Mazeaud, Y. Dromzee and R. Thouvenot, Inorg. Chem., 2000, 39, 4735–4740 CrossRef CAS PubMed.
- B. Matt, C. Coudret, C. Viala, D. Jouvenot, F. Loiseau, G. Izzet and A. Proust, Inorg. Chem., 2011, 50, 7761–7768 CrossRef CAS PubMed.
- D. Mercier, S. Boujday, C. Annabi, R. Villanneau, C.-M. Pradier and A. Proust, J. Phys. Chem. C, 2012, 116, 13217–13224 CrossRef CAS.
- D. Agustin, J. Dallery, C. Coelho, A. Proust and R. Thouvenot, J. Organomet. Chem., 2006, 692, 746–754 CrossRef.
- J. L. Horan, A. Genupur, H. Ren, B. J. Sikora, M.-C. Kuo, F. Meng, S. F. Dec, G. M. Haugen, M. A. Yandrasits, S. J. Hamrock, M. H. Frey and A. M. Herring, ChemSusChem, 2009, 2, 226–229 CrossRef CAS PubMed.
- T. Sun, W. Cui, M. Yan, G. Qin, W. Guo, H. Gu, S. Liu and Q. Wu, Adv. Mater., 2016, 28, 7397–7404 CrossRef CAS PubMed.
- V. Duffort, R. Thouvenot, C. Afonso, G. Izzet and A. Proust, Chem. Commun., 2009, 6062–6064 RSC.
- A. Parrot, G. Izzet, L.-M. Chamoreau, A. Proust, O. Oms, A. Dolbecq, K. Hakouk, H. El Bekkachi, P. Deniard, R. Dessapt and P. Mialane, Inorg. Chem., 2013, 52, 11156–11163 CrossRef CAS PubMed.
- F. A. Black, A. Jacquart, G. Toupalas, S. Alves, A. Proust, I. P. Clark, E. A. Gibson and G. Izzet, Chem. Sci., 2018, 9, 5578–5584 RSC.
- M. Piot, S. Hupin, H. Lavanant, C. Afonso, L. Bouteiller, A. Proust and G. Izzet, Inorg. Chem., 2017, 56, 8490–8496 CrossRef CAS PubMed.
- S. Aoki, T. Kurashina, Y. Kasahara, T. Nishijima and K. Nomiya, Dalton Trans., 2011, 40, 1243–1253 RSC.
- H. Liu, J. Luo, W. Shan, D. Guo, J. Wang, C. H. Hsu, M. Huang, W. Zhang, B. Lotz, W. Bin Zhang, T. Liu, K. Yue and S. Z. D. Cheng, ACS Nano, 2016, 10, 6585–6596 CrossRef CAS PubMed.
- R. Zhang and C. Yang, J. Mater. Chem., 2008, 18, 2691–2703 RSC.
- P. Judeinstein, Chem. Mater., 1992, 4, 4–7 CrossRef CAS.
- J. L. Horan, A. Lingutla, H. Ren, M.-C. Kuo, S. Sachdeva, Y. Yang, S. Seifert, L. F. Greenlee, M. A. Yandrasits, S. J. Hamrock, M. H. Frey and A. M. Herring, J. Phys. Chem. C, 2014, 118, 135–144 CrossRef CAS.
- I. Bar-Nahum, H. Cohen and R. Neumann, Inorg. Chem., 2003, 42, 3677–3684 CrossRef CAS PubMed.
- L. Tan, S. Liu, F. Zeng, Z. Ling and J. Zhao, Polym. Adv. Technol., 2010, 21, 435–441 CAS.
- C.-G. Lin, W. Chen, S. Omwoma and Y.-F. Song, J. Mater. Chem. C, 2015, 3, 15–18 RSC.
- W. Chen, D. Ma, J. Yan, T. Boyd, L. Cronin, D.-L. Long and Y.-F. Song, ChemPlusChem, 2013, 78, 1226–1229 CrossRef CAS.
- S. ul Hassan, Y. Zhou, L. Zhang, Z. Shi, D. Yang, H. M. Asif and N. Qu, J. Phys. Chem. C, 2016, 120, 7757–7766 CrossRef.
- W. Chen, L. Huang, J. Hu, T. Li, F. Jia and Y. F. Song, Phys. Chem. Chem. Phys., 2014, 16, 19668–19673 RSC.
- M. De Bruyn and R. Neumann, Adv. Synth. Catal., 2007, 349, 1624–1628 CrossRef CAS.
- V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork and N. Rasouli, Catal. Commun., 2008, 9, 2411–2416 CrossRef CAS.
- J. Tong, H. Wang, X. Cai, Q. Zhang, H. Ma and Z. Lei, Appl. Organomet. Chem., 2014, 28, 95–100 CrossRef CAS.
- M. Moghadam, V. Mirkhani, S. Tangestaninejad, I. Mohammadpoor-Baltork and M. M. Javadi, Polyhedron, 2010, 29, 648–654 CrossRef CAS.
- M. Moghadam, V. Mirkhani, S. Tangestaninejad, I. Mohammadpoor-Baltork and M. M. Javadi, Inorg. Chem. Commun., 2010, 13, 244–249 CrossRef CAS.
- C. Rinfray, S. Renaudineau, G. Izzet and A. Proust, Chem. Commun., 2014, 50, 8575–8577 RSC.
- W. H. Knoth, P. J. Domaille and D. C. Roe, Inorg. Chem., 2005, 22, 198–201 CrossRef.
- F. F. Jian, X. Wang, J. Wang, H. L. Xiao and R. R. Zhuang, Polyhedron, 2010, 29, 886–896 CrossRef CAS.
- R. Prudent, V. Moucadel, B. Laudet, C. Barette, L. Lafanechère, B. Hasenknopf, J. Li, S. Bareyt, E. Lacôte, S. Thorimbert, M. Malacria, P. Gouzerh and C. Cochet, Chem. Biol., 2008, 15, 683–692 CrossRef CAS PubMed.
- B. Matt, J. Moussa, L. M. Chamoreau, C. Afonso, A. Proust, H. Amouri and G. Izzet, Organometallics, 2012, 31, 35–38 CrossRef CAS.
- F. A. Black, A. Jacquart, G. Toupalas, S. Alves, A. Proust, I. P. Clark, E. A. Gibson and G. Izzet, Chem. Sci., 2018, 9, 5578–5584 RSC.
- M. Yaqub, J. J. Walsh, T. E. Keyes, A. Proust, C. Rinfray, G. Izzet, T. McCormac and R. J. Forster, Langmuir, 2014, 30, 4509–4516 CrossRef CAS PubMed.
- M. M. Lorion, B. Matt, S. Alves, A. Proust, G. Poli, J. Oble and G. Izzet, Chem. – Eur. J., 2013, 19, 12607–12612 CrossRef CAS PubMed.
- A. G. G. Coutsolelos, E. Nikoloudakis, K. Karikis, M. Laurans, C. Kokotidou, A. Solé-Daura, J. J. J. Carbó, A. Charisiadis, G. Charalampidis, G. Izzet, A. Mitraki, A. M. Douvas, J. M. Poblet and A. Proust, Dalton Trans., 2018, 47, 6304–6313 RSC.
- F. Volatron, J. M. Noël, C. Rinfray, P. Decorse, C. Combellas, F. Kanoufi and A. Proust, J. Mater. Chem. C, 2015, 3, 6266–6275 RSC.
- R. Corentin, I. Guillaume, P. Jean, G. D. Sarra, G. Jean-Jacques, C. Catherine, K. Frédéric and P. Anna, Chem. – Eur. J., 2013, 19, 13838–13846 CrossRef PubMed.
- K. Barthelmes, M. Sittig, A. Winter and U. S. Schubert, Eur. J. Inorg. Chem., 2017, 3698–3706 CrossRef CAS.
- Y. Luo, M. Wächtler, K. Barthelmes, A. Winter, U. S. Schubert and B. Dietzek, Chem. Commun., 2018, 54, 2970–2973 RSC.
- S. G. Derouich, C. Rinfray, G. Izzet, J. Pinson, J.-J. Gallet, F. Kanoufi, A. Proust and C. Combellas, Langmuir, 2014, 30, 2287–2296 CrossRef PubMed.
- A. Lombana, C. Rinfray, F. Volatron, G. Izzet, N. Battaglini, S. Alves, P. Decorse, P. Lang and A. Proust, J. Phys. Chem. C, 2016, 120, 2837–2845 CrossRef CAS.
- S. Piligkos, B. Hasenknopf, P. Gouzerh, S. Thorimbert, M. Malacria and M. Curie, J. Am. Chem. Soc., 2005, 127, 6788–6794 CrossRef PubMed.
- K. Micoine, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Org. Lett., 2007, 9, 3981–3984 CrossRef CAS PubMed.
- A. M. Debela, M. Ortiz, C. K. Ósullivan, S. Thorimbert and B. Hasenknopf, Polyhedron, 2014, 68, 131–137 CrossRef CAS.
- S. Thorimbert, C. K. O’Sullivan, R. B. Cole, D. Lesage, V. Beni, M. Ortiz, B. Hasenknopf and A. M. Debela, Chem. – Eur. J., 2015, 21, 17721–17727 CrossRef PubMed.
- M. Ortiz, A. M. Debela, M. Svobodova, S. Thorimbert, D. Lesage, R. B. Cole, B. Hasenknopf and C. K. O’Sullivan, Chem. – Eur. J., 2017, 23, 10597–10603 CrossRef CAS PubMed.
- N. Chahin, L. A. Uribe, A. M. Debela, S. Thorimbert, B. Hasenknopf, M. Ortiz, I. Katakis and C. K. O’Sullivan, Biosens. Bioelectron., 2018, 117, 201–206 CrossRef CAS PubMed.
- G. Sazani and M. T. Pope, Dalton Trans., 2004, 1989 RSC.
- S. Zhou, L. Wang, Z. Yuan, M. Chen, G. Zhang and H. Li, Eur. J. Inorg. Chem., 2018, 4255–4264 CrossRef CAS.
- V. Singh, V. Kalyani, K. E. Gonsalves, S. K. Sharma, S. Ghosh, V. S. V. Satyanarayana and C. P. Pradeep, Chem. – Eur. J., 2015, 21, 2250–2258 CrossRef.
- Y. Xiao, C.-H. Wang, W.-K. Miao, L.-X. Ren, Y.-K. Yan, P. Zheng, L.-J. Ren, X.-L. Wang and W. Wang, ACS Macro Lett., 2014, 3, 211–215 CrossRef.
- W. K. Miao, A. Yi, Y. K. Yan, L. J. Ren, D. Chen, C. H. Wang and W. Wang, Polym. Chem., 2015, 6, 7418–7426 RSC.
- S. Vanhaecht, T. Quanten and T. Parac-Vogt, Dalton Trans., 2017, 46, 10215–10219 RSC.
- Y. Han, Y. Xiao, Z. Zhang, B. Liu, P. Zheng, S. He and W. Wang, Macromolecules, 2009, 42, 6543–6548 CrossRef CAS.
- W. Chen, U. Tong, T. Zeng, C. Streb and Y.-F. Song, J. Mater. Chem. C, 2015, 3, 4388–4393 RSC.
- I. Azcarate, I. Ahmed, R. Farha, M. Goldmann, X. Wang, H. Xu, B. Hasenknopf, E. Lacôte and L. Ruhlmann, Dalton Trans., 2013, 42, 12688–12698 RSC.
- M. P. Santoni, A. K. Pal, G. S. Hanan, A. Proust and B. Hasenknopf, Inorg. Chem., 2011, 50, 6737–6745 CrossRef CAS PubMed.
- T. Auvray, M.-P. Santoni, B. Hasenknopf and G. S. Hanan, Dalton Trans., 2017, 46, 10029–10036 RSC.
- M. P. Santoni, A. K. Pal, G. S. Hanan, M. C. Tang, A. Furtos and B. Hasenknopf, Dalton Trans., 2014, 43, 6990–6993 RSC.
- H. N. Miras, C. P. Pradeep, L. Cronin, F.-Y. Li, L. Xu, D.-L. Long and C. Lydon, Chem. – Eur. J., 2011, 17, 7472–7479 CrossRef PubMed.
- X. Zuo, L. Cronin, T. Li, T. Liu, C. Lydon, B. Lee, P. Yin, Z. N. Zheng, R. S. Forgan and D. Long, J. Am. Chem. Soc., 2013, 135, 13425–13432 CrossRef PubMed.
- B. Riflade, J. Oble, L. Chenneberg, E. Derat, B. Hasenknopf, E. Lacôte and S. Thorimbert, Tetrahedron, 2013, 69, 5772–5779 CrossRef CAS.
- B. Riflade, D. Lachkar, J. Oble, J. Li, S. Thoribert, B. Hasenknopf and E. Lacôte, Org. Lett., 2014, 16, 3860–3863 CrossRef CAS PubMed.
- L. David, V. Debora, D. Elise, L. Moreno and L. Emmanuel, Angew. Chem., Int. Ed., 2016, 55, 5961–5965 CrossRef PubMed.
- Z. Huo, I. Azcarate, R. Farha, M. Goldmann, H. Xu, B. Hasenknopf, E. Lacôte and L. Ruhlmann, J. Solid State Electrochem., 2015, 19, 2611–2621 CrossRef CAS.
- K. Micoine, M. Malacria, E. Lacôte, S. Thorimbert and B. Hasenknopf, Eur. J. Inorg. Chem., 2013, 1737–1741 CrossRef CAS.
- K. Micoine, M. Malacria, E. Lacôte, S. Thorimbert and B. Hasenknopf, Eur. J. Inorg. Chem., 2013, 1737–1741 CrossRef CAS.
- S. Bareyt, S. Piligkos, B. Hasenknopf, P. Gouzerh, E. Lacôte, S. Thorimbert and M. Malacria, J. Am. Chem. Soc., 2005, 127, 6788–6794 CrossRef CAS PubMed.
- B. Matt, S. Renaudineau, L.-M. Chamoreau, C. Afonso, G. Izzet and A. Proust, J. Org. Chem., 2011, 76, 3107–3112 CrossRef CAS.
- B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504–1508 RSC.
- M. M. Lorion, B. Matt, S. Alves, A. Proust, G. Poli, J. Oble and G. Izzet, Chem. – Eur. J., 2013, 19, 12607–12612 CrossRef CAS PubMed.
- C. Bosch-Navarro, B. Matt, G. Izzet, C. Romero-Nieto, K. Dirian, A. Raya, S. I. Molina, A. Proust, D. M. Guldi, C. Martí-Gastaldo and E. Coronado, Chem. Sci., 2014, 5, 4346–4354 RSC.
- B. Sébastian, P. Stergios, H. Bernold, G. Pierre, L. Emmanuel, T. Serge and M. Max, Angew. Chem., Int. Ed., 2003, 42, 3404–3406 CrossRef.
- M. Kévin, H. Bernold, T. Serge, L. Emmanuel and M. Max, Angew. Chem., Int. Ed., 2009, 48, 3466–3468 CrossRef PubMed.
- D. Nathalie, B. Christian, F. Louis, M. Max, T. Serge, H. Bernold and L. Emmanuel, Chem. – Eur. J., 2012, 18, 12962–12965 CrossRef PubMed.
- B. Christian, D. Nathalie, M. Max, H. Bernold, L. Emmanuel and T. Serge, Chem. – Eur. J., 2014, 20, 16074–16077 CrossRef PubMed.
- E. Lacôte, J. Lesage de La Haye, J. Rieger, J.-M. Guigner, B. Hasenknopf, E. Marceau and L. Ruhlmann, Chem. – Eur. J., 2015, 21, 2948–2953 CrossRef PubMed.
- C. Boglio, K. Micoine, É. Derat, R. Thouvenot, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, J. Am. Chem. Soc., 2008, 130, 4553–4561 CrossRef CAS PubMed.
- J. Rieger, T. Antoun, S. H. Lee, M. Chenal, G. Pembouong, J. Lesage De La Haye, I. Azcarate, B. Hasenknopf and E. Lacôte, Chem. – Eur. J., 2012, 18, 3355–3361 CrossRef CAS PubMed.
- W. J. Xuan, C. Botuha, B. Hasenknopf and S. Thorimbert, Chem. – Eur. J., 2015, 21, 16512–16516 CrossRef CAS PubMed.
- D. Lachkar and E. Lacôte, C. R. Chim., 2016, 19, 113–116 CrossRef CAS.
- D. Vilona, D. Lachkar, E. Dumont, M. Lelli and E. Lacôte, Chem. – Eur. J., 2017, 23, 13323–13327 CrossRef CAS PubMed.
- A. M. Debela, O. Mayreli, B. Valerio, T. Serge, L. Denis, R. B. Cole, C. K. O’Sullivan and H. Bernold, Chem. – Eur. J., 2015, 21, 17721–17727 CrossRef CAS PubMed.
- L. de la Haye Jennifer, B. Patricia, R. Laurent, H. Bernold, L. Emmanuel and R. Jutta, ChemPlusChem, 2014, 79, 250–256 CrossRef.
- T. Hasegawa, H. Murakami, K. Shimizu, Y. Kasahara, S. Yoshida, T. Kurashina, H. Seki and K. Nomiya, Inorg. Chim. Acta, 2008, 361, 1385–1394 CrossRef CAS.
- T. Hasegawa, K. Shimizu, H. Seki, H. Murakami, S. Yoshida, K. Yoza and K. Nomiya, Inorg. Chem. Commun., 2007, 10, 1140–1144 CrossRef CAS.
- G. Izzet, B. Abécassis, D. Brouri, M. Piot, B. Matt, S. A. Serapian, C. Bo and A. Proust, J. Am. Chem. Soc., 2016, 138, 5093–5099 CrossRef CAS PubMed.
- I. Guillaume, M. Andrew, R. Corentin, P. Madeleine, R. Séverine, D. Etienne, A. Benjamin, A. Carlos and P. Anna, Chem. – Eur. J., 2015, 21, 19010–19015 CrossRef.
- A. Harriman, K. J. Elliott, M. A. H. Alamiry, L. Le Pieux, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer and F. Odobel, J. Phys. Chem. C, 2009, 113, 5834–5842 CrossRef CAS.
- F. Odobel, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer, K. J. Elliott and A. Harriman, Chem. – Eur. J., 2009, 15, 3130–3138 CrossRef CAS PubMed.
- Y. Xiao, D. Chen, N. Ma, Z. Hou, M. Hu, C. Wang and W. Wang, RSC Adv., 2013, 3, 21544–21551 RSC.
- M. B. Hu, N. Xia, W. Yu, C. Ma, J. Tang, Z. Y. Hou, P. Zheng and W. Wang, Polym. Chem., 2012, 3, 617–620 RSC.
- C. Cannizzo, C. R. Mayer, F. Sécheresse and C. Larpent, Adv. Mater., 2005, 17, 2888–2892 CrossRef CAS.
- C. N. Kato, Y. Kasahara, K. Hayashi, A. Yamaguchi, T. Hasegawa and K. Nomiya, Eur. J. Inorg. Chem., 2006, 4834–4842 CrossRef CAS.
- C. Cannizzo, C. R. Mayer, F. Sécheresse and C. Larpent, Adv. Mater., 2005, 17, 2888–2892 CrossRef CAS.
- F.-B. Li, D.-H. Yao, L. Li, Z.-L. Du, S.-Z. Liu, L. Liu, Q. Liu, L. Peng, W.-Y. Wong and L. Hu, J. Organomet. Chem., 2015, 792, 102–108 CrossRef.
- J. Ni, Z. Zhou, L. Liu, S. Z. Liu, F. B. Li, G. H. Li and Z. L. Du, Aust. J. Chem., 2015, 68, 106–112 CrossRef CAS.
- L. Liu, L. Hu, Q. Liu, Z. L. Du, F. B. Li, G. H. Li, X. J. Zhu, W. Y. Wong, L. Wang and H. Li, Dalton Trans., 2015, 44, 306–315 RSC.
- K. J. Elliott, A. Harriman, L. Le Pleux, Y. Pellegrin, E. Blart, C. R. Mayer and F. Odobel, Phys. Chem. Chem. Phys., 2009, 11, 8767–8773 RSC.
- S. Fujimoto, J. M. Cameron, R. J. Wei, K. Kastner, D. Robinson, V. Sans, G. N. Newton and H. Oshio, Inorg. Chem., 2017, 56, 12169–12177 CrossRef CAS PubMed.
- J. M. Cameron, S. Fujimoto, K. Kastner, R. J. Wei, D. Robinson, V. Sans, G. N. Newton and H. H. Oshio, Chem. – Eur. J., 2017, 23, 47–50 CrossRef CAS PubMed.
- J. M. Cameron, S. Fujimoto, R. J. Wei, G. N. Newton and H. Oshio, Dalton Trans., 2018, 47, 10590–10594 RSC.
- G. Sazani and M. T. Pope, Dalton Trans., 2004, 1989–1994 RSC.
- J. Wang, C.-G. Lin, J. Zhang, J. Wei, Y.-F. Song and J. Guo, J. Mater. Chem. C, 2015, 3, 4179–4187 RSC.
- W. Chen, L. Huang, J. Hu, T. Li, F. Jia and Y. F. Song, Phys. Chem. Chem. Phys., 2014, 16, 19668–19673 RSC.
- M. Carraro, G. Modugno, G. Fiorani, C. Maccato, A. Sartorel and M. Bonchio, Eur. J. Org. Chem., 2012, 281–289 CrossRef CAS.
- A. M. Debela, M. Ortiz, C. K. ÓSullivan, S. Thorimbert and B. Hasenknopf, Polyhedron, 2014, 68, 131–137 CrossRef CAS.
- N. Chahin, L. A. Uribe, A. M. Debela, S. Thorimbert, B. Hasenknopf, M. Ortiz, I. Katakis and C. K. O’Sullivan, Biosens. Bioelectron., 2018, 117, 201–206 CrossRef CAS PubMed.
- I. Bar-Nahum, J. Ettedgui, L. Konstantinovski, V. Kogan and R. Neumann, Inorg. Chem., 2007, 46, 5798–5804 CrossRef CAS PubMed.
- C. R. Mayer, S. Neveu and V. Cabuil, Angew. Chem., Int. Ed., 2002, 41, 501–503 CrossRef CAS PubMed.
- M. De Bruyn and R. Neumann, Adv. Synth. Catal., 2007, 349, 1624–1628 CrossRef CAS.
- V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork and N. Rasouli, Inorg. Chem. Commun., 2007, 10, 1537–1540 CrossRef CAS.
- M. Moghadam, V. Mirkhani, S. Tangestaninejad, I. Mohammadpoor-Baltork and M. M. Javadi, Inorg. Chem. Commun., 2010, 13, 244–249 CrossRef CAS.
- D. Agustin, C. Coelho, A. Mazeaud, P. Herson, A. Proust and R. Thouvenot, Z. Anorg. Allg. Chem., 2004, 630, 2049–2053 CrossRef CAS.
- M. Carraro, G. Modugno, A. Sartorel, G. Scorrano and M. Bonchio, Eur. J. Inorg. Chem., 2009, 5164–5174 CrossRef CAS.
- D. Karimian, B. Yadollahi and V. Mirkhani, Microporous Mesoporous Mater., 2017, 247, 23–30 CrossRef CAS.
- R. C. Schroden, C. F. Blanford, B. J. Melde, B. J. S. Johnson and A. Stein, Chem. Mater., 2001, 13, 1074–1081 CrossRef CAS.
- F. Bentaleb, O. Makrygenni, D. Brouri, C. Coelho Diogo, A. Mehdi, A. Proust, F. Launay and R. Villanneau, Inorg. Chem., 2015, 54, 7607–7616 CrossRef CAS PubMed.
- R. Villanneau, A. Ben Djamâa, L. M. Chamoreau, G. Gontard and A. Proust, Eur. J. Inorg. Chem., 2013, 1815–1820 CrossRef CAS.
- Y. Luo, M. Wächtler, K. Barthelmes, A. Winter, U. S. Schubert and B. Dietzek, Phys. Chem. Chem. Phys., 2018, 20, 11740–11748 RSC.
- L. Huder, C. Rinfray, D. Rouchon, A. Benayad, M. Baraket, G. Izzet, F. Lipp-Bregolin, G. Lapertot, L. Dubois, A. Proust, L. Jansen and F. Duclairoir, Langmuir, 2016, 32, 4774–4783 CrossRef CAS PubMed.
- C. Rinfray, V. Brasiliense, G. Izzet, F. Volatron, S. Alves, C. Combellas, F. Kanoufi and A. Proust, Inorg. Chem., 2016, 55, 6929–6937 CrossRef CAS PubMed.
- F. Odobel, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer, K. J. Elliott and A. Harriman, Chem. – Eur. J., 2009, 15, 3130–3138 CrossRef CAS PubMed.
- M. Carraro, S. Gross, M. Carraro and S. Gross, Materials, 2014, 7, 3956–3989 CrossRef CAS PubMed.
- J. L. Horan, A. Genupur, H. Ren, B. J. Sikora, M.-C. Kuo, F. Meng, S. F. Dec, G. M. Haugen, M. A. Yandrasits, S. J. Hamrock, M. H. Frey and A. M. Herring, ChemSusChem, 2009, 2, 193 CrossRef.
- C. R. Mayer, I. Fournier and R. Thouvenot, Chem. – Eur. J., 2002, 6, 105–110 CrossRef.
- T. Hasegawa, H. Murakami, K. Shimizu, Y. Kasahara, S. Yoshida, T. Kurashina, H. Seki and K. Nomiya, Inorg. Chim. Acta, 2008, 361, 1385–1394 CrossRef CAS.
- M. Carraro, G. Fiorani, L. Mognon, F. Caneva, M. Gardan, C. MacCato and M. Bonchio, Chem. – Eur. J., 2012, 18, 13195–13202 CrossRef CAS PubMed.
- W. K. Miao, A. Yi, Y. K. Yan, L. J. Ren, D. Chen, C. H. Wang and W. Wang, Polym. Chem., 2015, 6, 7418–7426 RSC.
- J. Li, I. Huth, L. M. Chamoreau, B. Hasenknopf, E. Lacôte, S. Thorimbert and M. Malacria, Angew. Chem., Int. Ed., 2009, 48, 2035–2038 CrossRef CAS PubMed.
- A. Iban, H. Zhaohui, F. Rana, G. Michel, X. Hualong, H. Bernold, L. Emmanuel and R. Laurent, Chem. – Eur. J., 2015, 21, 8271–8280 CrossRef.
- J. Oble, B. Riflade, A. Noël, M. Malacria, S. Thorimbert, B. Hasenknopf and E. Lacôte, Org. Lett., 2011, 13, 5990–5993 CrossRef CAS.
- M.-P. P. Santoni, A. K. Pal, G. S. Hanan, A. Proust, B. Hasenknopf, G. S. Hanan, M.-P. P. Santoni, B. Hasenknopf and A. K. Pal, Inorg. Chem., 2011, 50, 6737–6745 CrossRef CAS PubMed.
- C. R. Mayer and R. Thouvenot, J. Chem. Soc., Dalton Trans., 1998, 7–13 RSC.
- T. Minato, K. Suzuki, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2017, 23, 14213–14220 CrossRef CAS PubMed.
- J. Zhang, Y.-F. Song, L. Cronin and T. Liu, J. Am. Chem. Soc., 2008, 130, 14408–14409 CrossRef CAS PubMed.
- S. Encapsulation, Y. Song, N. Mcmillan, D. Long, J. Thiel and Y. Ding, Chem. – Eur. J., 2008, 14, 2349–2354 CrossRef PubMed.
- J. Zhang, Y. F. Song, L. Cronin and T. Liu, Chem. – Eur. J., 2010, 16, 11320–11324 CrossRef CAS.
- B. Hu, C. Wang, J. Wang, J. Gao, K. Wang, J. Wu, G. Zhang, W. Cheng, B. Venkateswarlu, M. Wang, P. S. Lee and Q. Zhang, Chem. Sci., 2014, 5, 3404–3408 RSC.
- V. Kalyani, V. S. V. Satyanarayana, A. S. Sarkar, A. Kumar, S. K. Pal, S. Ghosh, K. E. Gonsalves and C. P. Pradeep, RSC Adv., 2015, 5, 36727–36731 RSC.
- M. Hutin, C. Yvon, J. Yan, A. Macdonell, D.-L. Long and L. Cronin, CrystEngComm, 2013, 15, 4422–4430 RSC.
- C. Yvon, A. Macdonell, S. Buchwald, A. J. Surman, N. Follet, J. Alex, D.-L. Long and L. Cronin, Chem. Sci., 2013, 4, 3810–3817 RSC.
- D. Ventura, A. Calderan, C. Honisch, S. Krol, S. Serratì, M. Bonchio, M. Carraro and P. Ruzza, Pept. Sci., 2018, 110, e24047 CrossRef.
- S. ul Hassan, H. M. Asif, Y. Zhou, L. Zhang, N. Qu, J. Li and Z. Shi, J. Phys. Chem. C, 2016, 120, 27587–27599 CrossRef.
- I. Bazzan, P. Bolle, O. Oms, H. Salmi, N. Aubry-Barroca, A. Dolbecq, H. Serier-Brault, R. Dessapt, P. Roger and P. Mialane, J. Mater. Chem. C, 2017, 5, 6343–6351 RSC.
- J. Yuanchun, H. Jun, H. Lujiang, C. Wei, S. Carsten and S. Yu-Fei, Chem. – Eur. J., 2015, 21, 6469–6474 CrossRef PubMed.
- Y. Carine, A. J. Surman, H. Marie, A. Jennifer, B. O. Smith, L. De-Liang and C. Leroy, Angew. Chem., Int. Ed., 2014, 53, 3336–3341 CrossRef PubMed.
- Y. Li, M. Liu and L. Chen, J. Mater. Chem. A, 2017, 5, 13757–13762 RSC.
- B. Zhang, L. Yue, Y. Wang, Y. Yang and L. Wu, Chem. Commun., 2014, 50, 10823–10826 RSC.
- O. Oms, K. Hakouk, R. Dessapt, P. Deniard, S. Jobic, A. Dolbecq, T. Palacin, L. Nadjo, B. Keita, J. Marrot and P. Mialane, Chem. Commun., 2012, 48, 12103–12105 RSC.
- A. Saad, O. Oms, J. Marrot, A. Dolbecq, K. Hakouk, H. El Bekkachi, S. Jobic, P. Deniard, R. Dessapt, D. Garrot, K. Boukheddaden, R. Liu, G. Zhang, B. Keita and P. Mialane, J. Mater. Chem. C, 2014, 2, 4748–4758 RSC.
- Y. Chu, A. Saad, P. Yin, J. Wu, O. Oms, A. Dolbecq, P. Mialane and T. Liu, Chem. – Eur. J., 2016, 22, 11756–11762 CrossRef CAS.
- A. Boulmier, A. Vacher, D. Zang, S. Yang, A. Saad, J. Marrot, O. Oms, P. Mialane, I. Ledoux, L. Ruhlmann, D. Lorcy and A. Dolbecq, Inorg. Chem., 2018, 57, 3742–3752 CrossRef CAS PubMed.
- N. I. Gumerova, A. Blazevic, T. Caldera Fraile, A. Roller, G. Giester and A. Rompel, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 1378–1383 CrossRef CAS PubMed.
- X. Zhuang, W. Wang and J. Hao, J. Colloid Interface Sci., 2018, 519, 81–87 CrossRef CAS.
- H.-K. Yang, H. Zhao, X.-M. Wang and Y.-K. Li, Colloids Surf., A, 2018, 538, 711–719 CrossRef CAS.
- J. Zhang, J. Hao, Y. Wei, F. Xiao, P. Yin and L. Wang, J. Am. Chem. Soc., 2010, 132, 14–15 CrossRef CAS PubMed.
- J. Zhang, P. Yin, J. Hao, F. Xiao, L. Chen and Y. Wei, Chem. – Eur. J., 2012, 18, 13596–13599 CrossRef CAS.
- P. Yin, J. Zhang, T. Li, X. Zuo, J. Hao, A. M. Warner, S. Chattopadhyay, T. Shibata, Y. Wei and T. Liu, J. Am. Chem. Soc., 2013, 135, 4529–4536 CrossRef CAS.
- L. Jiancheng, C. Kun, Y. Panchao, L. Tao, W. Gang, Z. Jin, Y. Songtao, B. Xiaoman, P. Yi, W. Yongge and L. Tianbo, Angew. Chem., Int. Ed., 2018, 57, 4067–4072 CrossRef PubMed.
- H.-K. Yang, RSC Adv., 2016, 6, 66431–66437 RSC.
- Y. Hai-Kuan, S. Ming-Ming, R. Li-Jun, T. Jing, Y. Yu-Kun, M. Wen-Ke, Z. Ping and W. Wei, Eur. J. Inorg. Chem., 2013, 1381–1389 Search PubMed.
- H.-K. Yang, L.-J. Ren, H. Wu and W. Wang, New J. Chem., 2016, 40, 954–961 RSC.
- H.-K. Yang, L.-L. Liu, X. Yuan and S.-M. Wu, J. Colloid Interface Sci., 2017, 496, 150–157 CrossRef CAS PubMed.
- H.-K. Yang, M.-M. Su, L.-J. Ren, P. Zheng and W. Wang, RSC Adv., 2014, 4, 1138–1145 RSC.
- X. Zeng, C. Gong, H. Guo, H. Xu, J. Zhang and J. Xie, Polyhedron, 2018, 151, 37–42 CrossRef CAS.
- N. Xia, W. Yu, Y. Wang, Y. Han, P. Zheng, W. Wang, G. Sakaguchi, K. Matsuda, K. Saijo, M. Takenaka and H. Hasegawa, Polymer, 2011, 52, 1772–1780 CrossRef CAS.
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2406–2412 CrossRef CAS.
- Y. F. Song, D. L. Long, S. E. Kelly and L. Cronin, Inorg. Chem., 2008, 47, 9137–9139 CrossRef CAS PubMed.
- Y.-F. Song, D.-L. Long and L. Cronin, CrystEngComm, 2010, 12, 109–115 RSC.
- J. Thiel, D. Yang, M. H. Rosnes, X. Liu, C. Yvon, S. E. Kelly, Y.-F. Song, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2011, 50, 8871–8875 CrossRef CAS PubMed.
- Y. Yan, H. Wang, B. Li, G. Hou, Z. Yin, L. Wu and V. W. W. Yam, Angew. Chem., Int. Ed., 2010, 49, 9233–9236 CrossRef CAS.
- H. Karoui and C. Ritchie, Dalton Trans., 2016, 45, 18838–18841 RSC.
- L. S. Wang, Y. Lu, G. M. Ó. Máille, S. P. Anthony, D. Nolan and S. M. Draper, Inorg. Chem., 2016, 55, 9497–9500 CrossRef CAS PubMed.
- A. Macdonell, N. A. B. Johnson, A. J. Surman and L. Cronin, J. Am. Chem. Soc., 2015, 137, 5662–5665 CrossRef CAS PubMed.
- S. Vanhaecht, J. Jacobs, L. Van Meervelt and T. Parac-Vogt, Dalton Trans., 2015, 44, 19059–19062 RSC.
- A. Saad, O. Oms, A. Dolbecq, C. Menet, R. Dessapt, H. Serier-Brault, E. Allard, K. Baczko and P. Mialane, Chem. Commun., 2015, 51, 16088–16091 RSC.
- T. UnSong, C. Wei, R. Chris, W. Xiaoting and S. Yu-Fei, Chem. – Eur. J., 2014, 20, 1500–1504 CrossRef.
- D. Schaming, C. Allain, R. Farha, M. Goldmann, S. Lobstein, A. Giraudeau, B. Hasenknopf and L. Ruhlmann, Langmuir, 2010, 26, 5101–5109 CrossRef CAS PubMed.
- Z. He, B. Li, H. Ai, H. Li and L. Wu, Chem. Commun., 2013, 49, 8039–8041 RSC.
- Z. He, Y. Yan, B. Li, H. Ai, H. Wang, H. Li and L. Wu, Dalton Trans., 2012, 41, 10043–10051 RSC.
- S. Favette, B. Hasenknopf, J. Vaissermann, P. Gouzerh and C. Roux, Chem. Commun., 2003, 2664–2665 RSC.
- F.-J. Yazigi, C. Wilson, D.-L. Long and R. S. Forgan, Cryst. Growth Des., 2017, 17, 4739–4748 CrossRef CAS.
- X.-X. Li, X. Ma, W.-X. Zheng, Y.-J. Qi, S.-T. Zheng and G.-Y. Yang, Inorg. Chem., 2016, 55, 8257–8259 CrossRef CAS PubMed.
- S. Schönweiz, D. Sorsche, B. Schwarz, S. Rau and C. Streb, Dalton Trans., 2017, 46, 9760–9764 RSC.
- S. Stefanie, S. A. Rommel, K. Joachim, M. Mathias, D. Benjamin, R. Sven and S. Carsten, Chem. – Eur. J., 2016, 22, 12002–12005 CrossRef PubMed.
- S. Schönweiz, M. Heiland, M. Anjass, T. Jacob, S. Rau and C. Streb, Chem. – Eur. J., 2017, 23, 15370–15376 CrossRef.
- C. Allain, S. Favette, L.-M. Chamoreau, J. Vaissermann, L. Ruhlmann and B. Hasenknopf, Eur. J. Inorg. Chem., 2008, 3433–3441 CrossRef CAS.
- A. Abhervé, M. Palacios-Corella, J. M. Clemente-Juan, R. Marx, P. Neugebauer, J. van Slageren, M. Clemente-León and E. Coronado, J. Mater. Chem. C, 2015, 3, 7936–7945 RSC.
- S. Stefanie, H. Magdalena, A. Montaha, J. Timo, R. Sven and S. Carsten, Chem. – Eur. J., 2017, 23, 15370–15376 CrossRef.
- W. Yongliang, W. Xiaole, Z. Xinjun, X. Nan, L. Bo, Y. Jie, Y. Wei, H. Minbiao, Y. Miao and W. Wei, Chem. – Eur. J., 2010, 16, 12545–12548 CrossRef PubMed.
- B. Liu, J. Yang, M. Yang, Y. Wang, N. Xia, Z. Zhang, P. Zheng, W. Wang, I. Lieberwirth and C. Kubel, Soft Matter, 2011, 7, 2317–2320 RSC.
- H.-K. Yang, Y.-X. Cheng, M.-M. Su, Y. Xiao, M.-B. Hu, W. Wang and Q. Wang, Bioorg. Med. Chem. Lett., 2013, 23, 1462–1466 CrossRef CAS PubMed.
- Y.-F. Song, N. McMillan, D.-L. Long, S. Kane, J. Malm, M. O. Riehle, C. P. Pradeep, N. Gadegaard and L. Cronin, J. Am. Chem. Soc., 2009, 131, 1340–1341 CrossRef CAS PubMed.
- S. Vanhaecht, T. Quanten and T. N. Parac-Vogt, Inorg. Chem., 2017, 56, 3095–3101 CrossRef CAS PubMed.
- Z. He, H. Wang, Y. Wang, Y. Wu, H. Li, L. Bi and L. Wu, Soft Matter, 2012, 8, 3315–3321 RSC.
- S. Schönweiz, D. Sorsche, B. Schwarz, S. Rau and C. Streb, Dalton Trans., 2017, 46, 9760–9764 RSC.
- B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Eur. J. Inorg. Chem., 2002, 1081–1087 CrossRef CAS.
- H. Ai, Y. Wang, B. Li and L. Wu, Eur. J. Inorg. Chem., 2014, 2766–2772 CrossRef CAS.
- H. K. Chae, W. G. Klemperer and V. W. Day, Inorg. Chem., 1989, 28, 1423–1424 CrossRef CAS.
- J.-K. Li, X.-Q. Huang, S. Yang, H.-W. Ma, Y.-N. Chi and C.-W. Hu, Inorg. Chem., 2015, 54, 1454–1461 CrossRef CAS PubMed.
- L. E. VanGelder and E. M. Matson, J. Mater. Chem. A, 2018, 6, 13874–13882 RSC.
- H. Karoui and C. Ritchie, New J. Chem., 2018, 42, 25–28 RSC.
- O. Nachtigall and J. Spandl, Chem. – Eur. J., 2018, 24, 2785–2789 CrossRef CAS.
- A. Bayaguud, K. Chen and Y. Wei, CrystEngComm, 2016, 18, 4042–4045 RSC.
- D. Li, J. Song, P. Yin, S. Simotwo, A. J. Bassler, Y. Aung, J. E. Roberts, K. I. Hardcastle, C. L. Hill and T. Liu, J. Am. Chem. Soc., 2011, 133, 14010–14016 CrossRef CAS PubMed.
- B. Zhang, J. Song, D. Li, L. Hu, C. L. Hill and T. Liu, Langmuir, 2016, 32, 12856–12861 CrossRef CAS PubMed.
- Y. Panchao, B. Aruuhan, C. Peng, H. Fadi, H. Lang, W. Joy, V. Dmitri, R. E. Winans, H. Jian, L. Tao, W. Yongge and L. Tianbo, Chem. – Eur. J., 2014, 20, 9589–9595 CrossRef PubMed.
- J. Song, D. A. Hillesheim, J. W. Han, R. Cao, Y. V. Geletii and C. L. Hill, Organic Esters of {V6O19}, Synthons that Detect and Catalytically Decontaminate Using Air, https://apps.dtic.mil/dtic/tr/fulltext/u2/a591189.pdf.
- A. Bayaguud, J. Zhang, R. N. N. Khan, J. Hao and Y. Wei, Chem. Commun., 2014, 50, 13150–13152 RSC.
- Q. Chen, D. P. Goshorn, C. P. Scholes, L. T. Xiao and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667–4681 CrossRef CAS.
- P. Yin, P. Wu, Z. Xiao, D. Li, E. Bitterlich, J. Zhang, P. Cheng, D. V. Vezenov, T. Liu and Y. Wei, Angew. Chem., Int. Ed., 2011, 50, 2521–2525 CrossRef CAS PubMed.
- B. Huang, Z. Xiao, B. Wu, X. Hu, X. Hu, P. Wu and Y. Wei, Inorg. Chem. Front., 2017, 4, 165–170 RSC.
- P. Wu, Z. Xiao, J. Zhang, J. Hao, J. Chen, P. Yin and Y. Wei, Chem. Commun., 2011, 47, 5557–5559 RSC.
- B. Huang, M. Cheng, J. Cai, B. Wu, W. Xiong, X. Hu, Z. Xiao and P. Wu, J. Chem. Crystallogr., 2017, 47, 95–100 CrossRef CAS.
- P. Yin, J. Wang, Z. Xiao, P. Wu, Y. Wei and T. Liu, Chem. – Eur. J., 2012, 18, 9174–9178 CrossRef CAS PubMed.
- H. Jia, Q. Li, A. Bayaguud, S. She, Y. Huang, K. Chen and Y. Wei, Sci. Rep., 2017, 7, 12523 CrossRef PubMed.
- H. Jia, Q. Li, A. Bayaguud, Y. Huang, S. She, K. Chen and Y. Wei, Dalton Trans., 2018, 47, 577–584 RSC.
- B. Huang, N. Wang, K. Yang, D. Ke, Y. Fang, X. Hu, B. Wu, Z. Xiao, P. Wu and Y. Wei, New J. Chem., 2018, 42, 5853–5858 RSC.
- A. Bayaguud, K. Chen and Y. Wei, Nano Res., 2016, 9, 3858–3867 CrossRef CAS.
- A. Bayaguud, J. Li, S. She and Y. Wei, Dalton Trans., 2017, 46, 4602–4608 RSC.
- J. W. Han, K. I. Hardcastle and C. L. Hill, Eur. J. Inorg. Chem., 2006, 2598–2603 CrossRef CAS.
- W. H. Jong and C. L. Hill, J. Am. Chem. Soc., 2007, 129, 15094–15095 CrossRef PubMed.
- M.-P. Santoni, A. K. Pal, G. S. Hanan, M.-C. Tang, K. Venne, A. Furtos, P. Ménard-Tremblay, C. Malveau and B. Hasenknopf, Chem. Commun., 2012, 48, 200–202 RSC.
- C. L. Hill, T. M. Anderson, J. W. Han, D. A. Hillesheim, Y. V. Geletii, N. M. Okun, R. Cao, B. Botar, D. G. Musaev and K. Morokuma, J. Mol. Catal. A: Chem., 2006, 251, 234–238 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., 1978, 90, 556–557 CrossRef CAS.
- M. T. Pope and T. F. Scully, Inorg. Chem., 1975, 14, 953–954 CrossRef CAS.
- D. P. Smith and M. T. Pope, Inorg. Chem., 1973, 12, 331–336 CrossRef CAS.
- M. T. Pope, S. E. O’Donnel and R. A. Prados, in Inorganic Compounds with Unusual Properties, ed. R. B. King, American Chemical Society, 1976, vol. 150, pp. 8–85 Search PubMed.
- N. V. Izarova, A. Kondinski, N. Vankova, T. Heine, P. Jäger, F. Schinle, O. Hampe and U. Kortz, Chem. – Eur. J., 2014, 20, 8556–8560 CrossRef CAS PubMed.
- L. K. Mahnke, A. Kondinski, U. Warzok, C. Näther, J. van Leusen, C. A. Schalley, K. Y. Monakhov, P. Kögerler and W. Bensch, Angew. Chem., Int. Ed., 2018, 57, 2972–2975 CrossRef CAS PubMed.
- Z. Huo, D. Zang, S. Yang, R. Farha, M. Goldmann, B. Hasenknopf, H. Xu and L. Ruhlmann, Electrochim. Acta, 2015, 179, 326–335 CrossRef CAS.
- H. Kang and J. Zubieta, J. Chem. Soc., Chem. Commun., 1988, 1192–1193 RSC.
- Y. Du, A. L. Rheingold and E. A. Maatta, J. Am. Chem. Soc., 1992, 114, 345–346 CrossRef CAS.
- J. L. Stark, A. L. Rheingold and E. A. Maatta, J. Chem. Soc., Chem. Commun., 1995, 1165–1166 RSC.
- J. L. Stark, V. G. Young Jr. and E. A. Maatta, Angew. Chem., Int. Ed. Engl., 1995, 34, 2547–2548 CrossRef CAS.
- J. B. Strong, B. S. Haggerty, A. L. Rheingold and E. A. Maatta, Chem. Commun., 1997, 1137–1138 RSC.
- J. B. Strong, G. P. A. Yap, R. Ostrander, L. M. Liable-Sands, A. L. Rheingold, R. Thouvenot, P. Gouzerh and E. A. Maatta, J. Am. Chem. Soc., 2000, 122, 639–649 CrossRef CAS.
- A. Proust, R. Thouvenot, M. Chaussade, F. Robert and P. Gouzerh, Inorg. Chim. Acta, 1994, 224, 81–95 CrossRef CAS.
- W. Clegg, R. J. Errington, K. A. Fraser, S. A. Holmes and A. Schäfer, J. Chem. Soc., Chem. Commun., 1995, 455–456 RSC.
- A. Al-Yasari, N. Van Steerteghem, H. Kearns, H. El Moll, K. Faulds, J. A. Wright, B. S. Brunschwig, K. Clays and J. Fielden, Inorg. Chem., 2017, 56, 10181–10194 CrossRef CAS PubMed.
- R. N. N. Khan, C. Lv, J. Zhang, J. Hao and Y. Wei, Dalton Trans., 2015, 44, 4568–4575 RSC.
- Y. Li, K. Shetye, K. Baral, L. Jin, J. D. Oster, D. M. Zhu and Z. Peng, RSC Adv., 2016, 6, 29909–29919 RSC.
- Y. Wei, B. Xu, C. L. Barnes and Z. Peng, J. Am. Chem. Soc., 2001, 123, 4083–4084 CrossRef CAS PubMed.
- J. Kang, B. Xu, Z. Peng, X. Zhu, Y. Wei and D. R. Powell, Angew. Chem., Int. Ed., 2005, 44, 6902–6905 CrossRef CAS PubMed.
- B. Xu, Y. Wei, C. L. Barnes and Z. Peng, Angew. Chem., Int. Ed., 2001, 40, 2290–2292 CrossRef CAS PubMed.
- M. Lu, Y. Wei, B. Xu, C. F. C. Cheung, Z. Peng and D. R. Powell, Angew. Chem., Int. Ed., 2002, 41, 1566–1568 CrossRef CAS PubMed.
- J. Kang, J. A. Nelson, M. Lu, B. Xie, Z. Peng and D. R. Powell, Inorg. Chem., 2004, 43, 6408–6413 CrossRef CAS PubMed.
- M. Lu, W. M. Nolte, T. He, D. A. Corley and J. M. Tour, Chem. Mater., 2009, 21, 442–446 CrossRef CAS.
- F. Haso, R. Wang, J. He, J. Luo, S. A. Eghtesadi, Z. Peng and T. Liu, New J. Chem., 2016, 40, 910–913 RSC.
- M. Lu, B. Xie, J. Kang, F. C. Chen, Y. Yang and Z. Peng, Chem. Mater., 2005, 17, 402–408 CrossRef CAS.
- Y. Zhu, L. Wang, J. Hao, P. Yin, J. Zhang, Q. Li, L. Zhu and Y. Wei, Chem. – Eur. J., 2009, 15, 3076–3080 CrossRef CAS PubMed.
- A. Al-Yasari, N. Van Steerteghem, H. El Moll, K. Clays and J. Fielden, Dalton Trans., 2016, 45, 2818–2822 RSC.
- B. Xu, M. Lu, J. Kang, D. Wang, J. Brown and Z. Peng, Chem. Mater., 2005, 17, 2841–2851 CrossRef CAS.
- A. R. Moore, H. Kwen, A. M. Beatty and E. A. Maatta, Chem. Commun., 2000, 1793–1794 RSC.
- L. Zhu, Y. Zhu, X. Meng, J. Hao, Q. Li, Y. Wei and Y. Lin, Chem. – Eur. J., 2008, 14, 10923–10927 CrossRef CAS PubMed.
- L. Zhu, Y. F. Zhong and X. Y. Cao, Z. Kristallogr. – New Cryst. Struct., 2012, 227, 547–549 CrossRef CAS.
- L. Zhu, K. Chen, J. Hao, Z. Wei, H. Zhang, P. Yin and Y. Wei, Inorg. Chem., 2015, 54, 6075–6077 CrossRef CAS PubMed.
- C. Lv, J. Hu, R. N. N. Khan, J. Zhang and Y. Wei, Dalton Trans., 2015, 44, 16698–16702 RSC.
- P. Hermosilla-Ibáñez, K. Wrighton-Araneda, G. Prado, V. Paredes-García, N. Pizarro, A. Vega and D. Venegas-Yazigi, Dalton Trans., 2017, 46, 8611–8620 RSC.
- Y. Zhu, P. Yin, F. Xiao, D. Li, E. Bitterlich, Z. Xiao, J. Zhang, J. Hao, T. Liu, Y. Wang and Y. Wei, J. Am. Chem. Soc., 2013, 135, 17155–17160 CrossRef CAS PubMed.
- B. Xu, Z. Peng, Y. Wei and D. R. Powell, Chem. Commun., 2003, 2562 RSC.
- Q. Li, J. Zhang, L. Wang, J. Hao, P. Yin and Y. Wei, New J. Chem., 2016, 40, 906–909 RSC.
- L. Xu, M. Lu, B. Xu, Y. Wei, Z. Peng and D. R. Powell, Angew. Chem., Int. Ed., 2002, 41, 4129–4132 CrossRef CAS PubMed.
- R. Wang, Y. Li, K. Shetye, T. Dutta, L. Jin, S. Li and Z. Peng, Eur. J. Inorg. Chem., 2015, 656–663 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |