
Dissociation of dinitrogen on iron clusters: a detailed study of the Fe16 + N2 case†
 *b
Weiguo
Sun,
a
Xiaoyu
Kuang,
*a
Cheng
Lu,
*c
Lavrenty G.
Gutsev,
de
Sergey M.
Aldoshin
d
and
Bala R.
Ramachandran
e
*b
Weiguo
Sun,
a
Xiaoyu
Kuang,
*a
Cheng
Lu,
*c
Lavrenty G.
Gutsev,
de
Sergey M.
Aldoshin
d
and
Bala R.
Ramachandran
e
Abstract
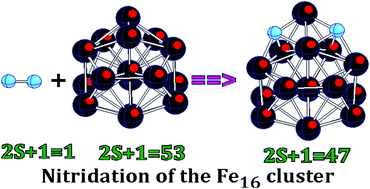
The coalescence of two Fe8N as well as the structure of the Fe16N2 cluster were studied using density functional theory with the generalized gradient approximation and a basis set of triple-zeta quality. It was found that the coalescence may proceed without an energy barrier and that the geometrical structures of the resulting clusters depend strongly on the mutual orientations of the initial moieties. The dissociation of N2 is energetically favorable on Fe16, and the nitrogen atoms share the same Fe atom in the lowest energy state of the Fe16N2 species. The attachment of two nitrogen atoms leads to a decrease in the total spin magnetic moment of the ground-state Fe16 host by 6 μB due to the peculiarities of chemical bonding in the magnetic clusters. In order to gain insight into the dependence of properties on charge and to estimate the bonding energies of both N atoms, we performed optimizations of Fe16N and the singly charged ions of both Fe16N2 and Fe16N. It was found that the electronic properties of the Fe16N2 cluster, such as electron affinity and ionization energy, do not appreciably depend on the attachment of nitrogen atoms but that the average binding energy per atom changes significantly. The lowering in total energy due to the attachment of two N atoms was found to be nearly independent of charge. The IR and Raman spectra were simulated for Fe16N2 and its ions, and it was found that the positions of the most intense peaks in the IR spectra strongly depend on charge and therefore present fingerprints of the charged states. The chemical bonding in the ground-state Fe16N20,±1 species was described in terms of the localized molecular orbitals.
 Please wait while we load your content...
Please wait while we load your content...

