
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational and experimental characterization of novel ultraviolet filters†
Jack M.
Woolley‡
 a,
Raúl
Losantos‡
b,
Diego
Sampedro
*b and
Vasilios G.
Stavros
*a
a,
Raúl
Losantos‡
b,
Diego
Sampedro
*b and
Vasilios G.
Stavros
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: V.Stavros@warwick.ac.uk
bDepartamento de Química, Centro de Investigación en Síntesis Química (CISQ), Universidad de La Rioja, Madre de Dios, 53, 26006 Logroño, La Rioja, Spain. E-mail: diego.sampedro@unirioja.es
First published on 21st October 2020
Abstract
Many current ultraviolet filters present potential hazards both to humans and to the natural environment. As such there is a new impetus to develop, through intimate characterisation, ultraviolet filters for use in cosmeceuticals. Here we report a new class of organic molecules which have a strong absorption band across the ultraviolet-A and -B regions of the electromagnetic spectrum and high photostability. We have performed ultrafast transient electronic absorption spectroscopy and steady-state spectroscopies, alongside computational studies to track and manipulate photoprotection mechanisms. Our results present a potentially new generation of ultraviolet filters for use in commercial formulations.
Introduction
The use of chemical products to afford photoprotection against harmful ultraviolet (UV) radiation is an increasingly popular area for research and development for many cosmeceutical industries.1,2 Many of the available products often employ mixtures of UV filters from an approved list to protect across both the UVA (315–400 nm) and UVB (280–315 nm) regions of the electromagnetic spectrum.2,3 Recently it has been shown that many currently approved filters, particularly those providing UVA protection, are responsible for photoallergic reactions as well as coral bleaching in tropical climates.4–6 Alongside these environmental concerns, the relaxation mechanisms employed by these UV filters to dissipate the absorbed energy often lead to unfavourable photoproducts; radical formation and photodegradation of the initial starting molecule being prime candidates.7,8 In an effort to expand the repertoire of available UV filters available for use in commercial formulations, research has moved towards the use of natural UV absorbers such as mycosporine-like amino acids (MAAs); their large absorption cross-section across the UVA region and impressive photostability over prolonged irradiation being prime, motivating factors towards their study.9–12 While these avenues of research show promising results, the chemical synthesis of MAAs is a complex process, involving many steps with low product yields.13,14 Recently there has been commercial products made available, namely Helioguard 365, which contains the MAA porphyra-334.15,16 Furthermore, there has been recent publications exploring their photochemical properties.17–19 The present study takes these ideas and proposes a novel set of chemically synthesised, but nature inspired, UV filters which provide the same levels of UVA protection as their natural counterparts but without the negative drawbacks.To bypass these aforementioned problems, we have performed a relatively simple synthesis of two MAA-inspired compounds termed 1 and 2 henceforth, see Fig. 1 inset for structures, and performed steady-state irradiation and femtosecond (fs) transient electronic absorption spectroscopy (TEAS) studies to monitor both the long term photostability and ultrafast dynamics following UV absorption and how this is linked to such photostability. To aid our interpretation, our proof-of-concept studies presented herein are in dioxane. This choice of solvent was specifically chosen given that it is weakly perturbing, enabling us to compare our TEAS data with our complementary density functional theory (DFT) and time-dependent DFT (TD-DFT) results. This combined experimental and theoretical approach allows us to assign the underlying photodynamics of this new class of molecules. Having grasped a thorough understanding of these photodynamics, we can then begin to increase the perturbation to the system through the addition of a more strongly interacting solvent or the addition of alternative substituents that are either electron donating, withdrawing or both (through multiple substitutions).
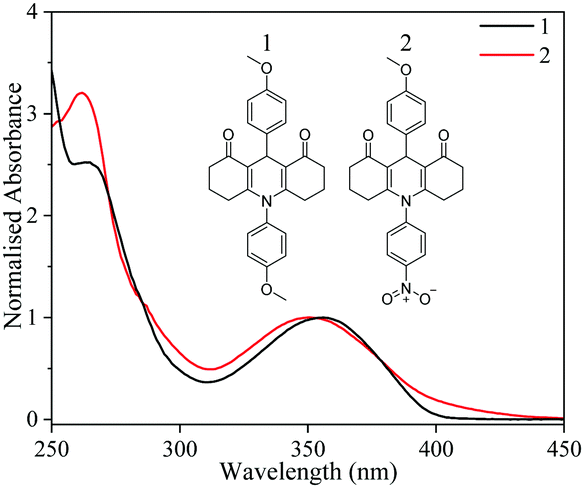 | ||
| Fig. 1 Absorption spectra of 1 and 2 (inset) normalised to the UVA maximum solvated in dioxane. | ||
Experimental
Synthesis
The compounds were prepared by neat condensation of reagents heating up to 160 °C, which is a modification of previously reported procedures in solution.20 These are summarised by Scheme 1 and detailed below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, was poured into a capped flask with a magnetic stirrer. It was heated to 160 °C and when the reagents melted, the reaction was allowed to proceed for 5 minutes. After this time, the flask was cooled to room temperature and the inner solid was triturated with AcOEt to afford the desired compound 1 as a pale grey solid (yield: 90%) (Scheme 1).
:1:1, was poured into a capped flask with a magnetic stirrer. This mixture was heated to 160 °C and when the reagents melted, the reaction proceeds for 12 minutes. After this time, the flask was cooled to room temperature and the inner solid was triturated with AcOEt to afford the desired compound 2 as a yellow solid (yield: 80%) (Scheme 1).
:1:1, was poured into a capped flask with a magnetic stirrer. It was heated to 160 °C and when the reagents melted, the reaction was allowed to proceed for 5 minutes. After this time, the flask was cooled to room temperature and the inner solid was triturated with AcOEt to afford the desired compound 1 as a pale grey solid (yield: 90%) (Scheme 1).
:1:1, was poured into a capped flask with a magnetic stirrer. This mixture was heated to 160 °C and when the reagents melted, the reaction proceeds for 12 minutes. After this time, the flask was cooled to room temperature and the inner solid was triturated with AcOEt to afford the desired compound 2 as a yellow solid (yield: 80%) (Scheme 1).
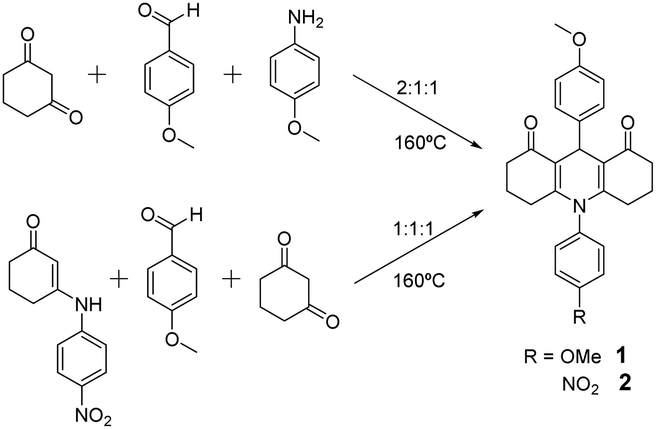 | ||
| Scheme 1 Synthetic procedure for 1 and 2. | ||
Transient electronic absorption spectroscopy
TEAS measurements were recorded at the Warwick Centre for Ultrafast Spectroscopy, (http://go.warwick.ac.uk/WCUS) the capabilities of which have been previously detailed and thus only summarised here.12 800 nm pulses (12 W, 1 kHz, 40 fs) were generated by a commercially available Ti-sapphire regenerative amplified laser system (Spectra-Physics, Dual Ascend Pumped Spitfire Ace) seeded by a Mai Tai (Spectra-Physics). The beam was split into four fractions, each having an independent recompression grating. One of these beams (3.5 W) was further split through the use of a 75/25 beam splitter with each beam used to produce the pump and probe pulses respectively. The pump beam (approximately 2.5 W) seeds an optical parametric amplifier (TOPAS-Prime with UV extension, Light Conversion) allowing variability in pump wavelength. The wavelengths of excitation were: 355 nm for compound 1 and 350 nm compound 2. The pump beam was focussed beyond the sample holder, to give a beam diameter of 500 μm at the sample. 5% of the remaining 1 W of fundamental 800 nm was further attenuated and irised (<0.05 W), before being focussed into a vertically translated 2 mm thick CaF2 window to generate a white light supercontinuum from 320 to 720 nm. The relative polarisation between the pump and probe pulses was held at magic angle (54.7°) to negate dynamics from ensemble molecular reorientation. Pump probe time delays (Δt) were achieved out to the maximum temporal delay of 3 ns using a motorized optical delay line in the probe beam (pre-supercontinuum generation). Changes in optical density (ΔOD) were measured through the change in probe intensities using a fibre-coupled spectrometer (Avantes, AvaSpec-ULS1650F). Collated spectra were subsequently chirp corrected using the KOALA package.21 Samples were prepared in 1 mM concentrations and delivered to the interaction region through a demountable liquid cell (Harrick Scientific Products Inc.) with a sample thickness of 100 μm placed between two CaF2 windows 1 mm and 2 mm respectively. The sample was flowed through the cell by a diaphragm pump (SIMDOS 02) at a rate to ensure that fresh sample was interrogated with each laser pulse.Steady-state spectroscopy
Steady-state UV/vis absorption measurements were taken on a Cary-60 spectrometer. Irradiation of the samples was performed through a solar simulator (Oriel Instruments, 91191-1000) using a neutral density filter to achieve an irradiation power equivalent to one sun (∼1000 W m−2). A quartz cuvette with a 1 cm path length was used for UV/vis and steady-state irradiation studies.Critical wavelength calculations
The critical wavelength for a sunscreen is defined as the wavelength at which the integrated area underneath the spectral absorbance curve reaches 90% of the total area between 290 and 400 nm.4 To attain the critical wavelengths of 1 and 2, UV-visible spectra of each compound were taken in dioxane using a Cary 60 instrument (Agilent). The area under each absorption curve between 290 and 400 nm was determined using the cumulative trapezoidal method function in MATLAB, which is defined mathematically as follows: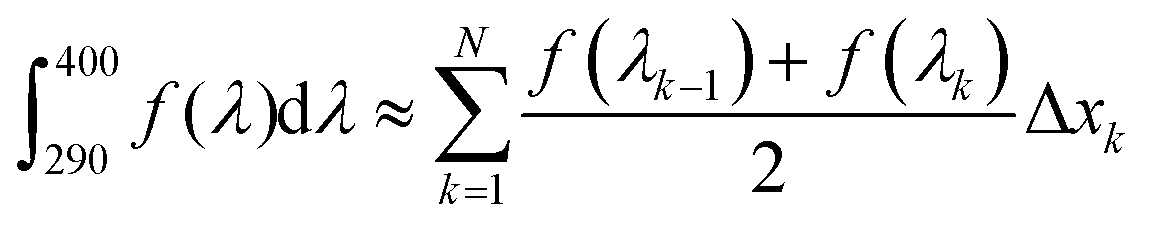 | (1) |
Computational details
All calculations were carried out using the B3LYP functional included in the Gaussian16 program package.22,23 We used the standard basis set 6-31+G**.24 TD-DFT calculations were performed considering the 10 first triplets and singlets. Minimum energy crossing points (MECP) were computed using the easymecp software,25 which is a simplified Python wrapper around the original MECP Fortran code from J. Harvey (2003) developed by J. Rodríguez-Guerra and I. Funes-Ardóiz. Linear interpolation on internal coordinates (LIIC) were carried out between the Franck–Condon and MECP (for 1) or the minimum in S1 and the MECP (for 2) obtaining 10 intermediate geometries. All calculations were computed in solution with implicit solvent applying the polarization continuum model (PCM) solvation model with 1,4-dioxane as solvent (ε = 2.2099).26Results
Compounds 1 and 2 are straightforwardly prepared by neat condensation in a fast, versatile and efficient one-pot process.This is extremely important for any potential application, as the large-scale preparation is usually a hindrance in the development of new commercial products. Both compounds are slightly coloured, a pale grey and pale yellow for 1 and 2 respectively, which is an ideal feature for their use in sunscreen formulations. The inclusion of electron-withdrawing (NO2) or electron-donating (OMe) substituents allows for various environments to be explored both in the synthesis and photophysical features.
We begin by discussing our results from our steady-state measurements. As shown in Fig. 1, 1 and 2 have peak UVA absorptions centred at 355 and 350 nm respectively. Indeed, their spectral absorptions span the entire region of the UVA, with critical wavelengths of 376 nm and 379 nm calculated for 1 and 2 respectively. Critical wavelengths provide a measure of capacity to absorb UVA, with a value of >370 nm being described as a broad spectrum absorber, a key requirement for new UV filters.4 Furthermore, both show strong absorption coefficients at the UVA absorption maximum of 11100 M−1 cm−1 and 10700 M−1 cm−1 for 1 and 2 respectively. Both critical wavelength and absorption strength are crucial performance criteria for emerging UVA filters which 1 and 2 satisfy.3,4
TD-DFT calculations performed at the B3LYP/6-31+G** level of theory provide absorption maxima (368 and 364 nm) in good agreement with the experimental absorption maximum for both 1 and 2. For both species, the excitation is initially located on the central ring π system; with similar transition orbitals (see ESI† for details). The results show that the strongest absorption feature in the UVA for 1 involves photoexcitation to the S1 (1ππ*) state, whilst for 2, this corresponds to the S7 (1ππ*) state.
Our next step was to determine the long-term photostability of 1 and 2. Steady-state irradiation was conducted using a solar simulator; measuring the drop in UV absorption post-irradiation provides a good reporter of the long term photostability of the molecule. As shown in Fig. 2, the absorption across the UVA region is maintained for 2 while the absorption of 1 is depleted over a two-hour period. This period of time is the maximum time of exposure, after which reapplication of commercial formulations is then recommended.27 For 1 and 2 the calculated degradation at the peak UVA absorption was found to be 30% and 13% drop for 1 and 2 respectively. Both of these results compare favourably with the current UV filter, avobenzone which loses 60% of its absorbance in 2 hours, when solvated in DMSO, a solvent of similar polarity to dioxane.7 These results highlight that the addition of the nitrophenyl group alters the electronic structure, providing more efficient relaxation back to the electronic ground state.
 | ||
| Fig. 2 Steady-state UV-vis of both (a) 1 and (b) 2 following irradiation by a solar simulator. Legend shows the time delay since beginning the irradiation. | ||
We now move to present results from our time-resolved studies. Transient absorption spectra (TAS) resulting from the photoexcitation of 1 at 355 nm in dioxane are displayed in Fig. 3a as a false colour heat map of all pump probe time delays (Δt), and Fig. 3c at selected Δt. The TAS display two excited state absorption (ESA) features, the first centred at 450 nm which blue-shifts as time evolves and secondly, a more intense ESA centred at 625 nm which forms as Δt increases and persists to the maximum Δt of the experiment (3 ns). Alongside these features, a ground state bleach (GSB) centred at 355 nm (photoexcitation wavelength) is present Δt = 3 ns. The TAS of 2 are also displayed as a false colour heat map, and shown in Fig. 3b for all Δt, and Fig. 3d at selected Δt. Following photoexcitation in dioxane at 350 nm, there are three ESA features evident; one centred at 340 nm, the second centred on 400 nm and is spectrally narrow and finally a third broad absorption centred on 550 nm which blue-shifts as time evolves. All three of these ESA features decay as Δt increases. Lastly, a final ESA is (mildly) visible around 600 nm at large Δt, and is more clearly illustrated in Fig. 3d.
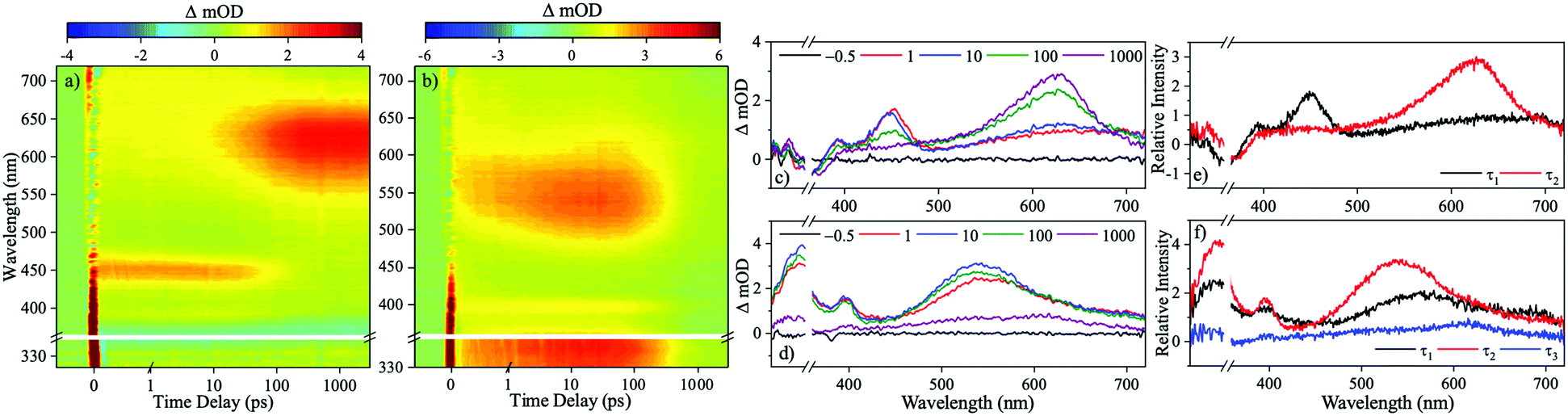 | ||
| Fig. 3 TAS displayed as false colour heat maps for (a) 1 following photoexcitation at 355 nm and (b) 2 following photoexcitation at 350 nm. Time delay is plotted linearly up to 1 ps and then logarithmically up to the maximum time delay of 3 ns. (c and d) Show selected transient absorption spectra of 1 and 2 following photoexcitation at the respective wavelengths. EADS (e) 1 following photoexcitation at 355 nm and (f) 2 following photoexcitation at 350 nm. Breaks are across the photoexcitation wavelength due to imperfect pump subtraction. | ||
Discussion
To gain quantitative insight into the ultrafast dynamics of both 1 and 2, a global sequential fitting method across all the probe wavelengths (320–720 nm) using the TIMP package in R interfaced through Glotaran was employed.28,29 Each TAS was sequentially fit, , with a multi-step kinetic model which incorporated a convolution with a Gaussian to model our instrument response (FWHM ≈ 100 fs). The fit returned time-constants for each step, along with associated errors to 2σ. The extracted time-constants are displayed in Table 1 and associated evolutionary associated difference spectra (EADS) are displayed in Fig. 3e and f for 1 and 2 respectively. The quality of the fit was evaluated through the residuals which are displayed in the ESI.†
, with a multi-step kinetic model which incorporated a convolution with a Gaussian to model our instrument response (FWHM ≈ 100 fs). The fit returned time-constants for each step, along with associated errors to 2σ. The extracted time-constants are displayed in Table 1 and associated evolutionary associated difference spectra (EADS) are displayed in Fig. 3e and f for 1 and 2 respectively. The quality of the fit was evaluated through the residuals which are displayed in the ESI.†
| Sample | τ 1 | τ 2 | τ 3 |
|---|---|---|---|
| 1 | 71.41 ± 3.36 ps | ≫3 ns | — |
| 2 | 2.90 ± 0.06 ps | 386.47 ± 7.30 ps | ≫3 ns |
We now link the extracted time-constants shown in Table 1 to dynamical processes, aided by our computation of critical points across the potential energy landscape, illustrated by Fig. 4. The TAS of 1 was fit with two time-constants with associated EADS shown in Fig. 3e. A 1ππ* transition initially populates the S1 electronic excited state, described by the first EADS. From the initially populated Franck–Condon region, the excited state population traverses the S1 state towards the crossing point between the S1 and T4 states. The population then evolves onto the T4 surface via an S1/T4 crossing. This is described by the first extracted time-constant, τ1 = 71.41 ± 3.36 ps. This manifests in the TAS as a decay of the ESA centred on 450 nm and the growth of the ESA centred on 625 nm assigned to the triplet absorption (vide infra); the absorption at 625 nm persists to the maximum temporal delay of the experiment, described by the second extracted time-constant τ2 ≫ 3 ns. This final state is shown by the second EADS. Throughout the experiment, there is a GSB centred of the photoexcitation wavelength (355 nm) indicating that there is little recovery of the initial ground electronic state population within Δt = 3 ns. The computational results (discussed in greater detail infra), shown in Fig. 4, agree well with our assignment of the overall dynamics.
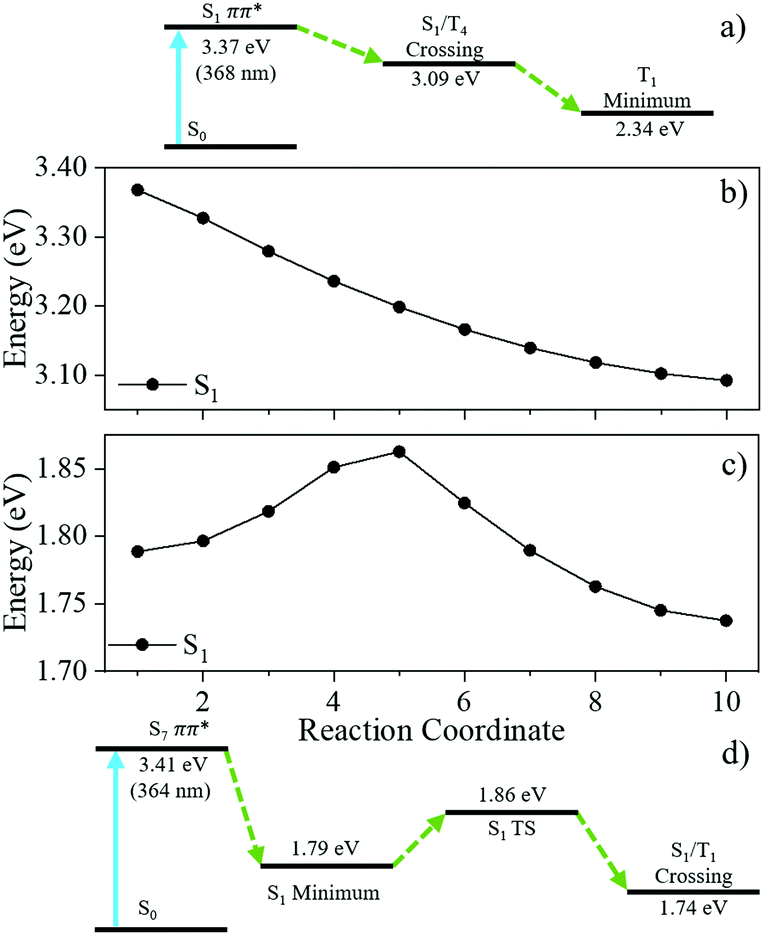 | ||
| Fig. 4 (a) Schematic of the relaxation pathway for 1 and (b) LIIC between the S1 Franck–Condon region and the minimum energy crossing point between the S1 and T4 for 1. (c) LIIC between the S1 minimum energy geometry and the minimum energy crossing point between the S1 and T1 for 2. (d) Schematic of the relaxation pathway for 2. All energies are relative to the S0 for each respective molecule. | ||
The TAS of 2 was fitted with three time-constants as shown in Table 1 with associated EADS displayed in Fig. 3f. We draw reference to the EADS here to explain the dynamics. The first EADS displays three absorptions centred at 340 nm, 410 nm and 540 nm. All three of these features increase in intensity with a time-constant of τ1 = 2.90 ± 0.06 ps. These changes in intensity, and lack of spectral shifts with associated τ1 leads us to propose that following photoexcitation to the S7 state, the excited state population evolves from the Franck–Condon region, internally converting to the S1 electronic state, within the instrument response (≈100 fs), either sequentially through the series of Sn electronic states, or through a more direct conversion from S7 to S1. Once on the S1 electronic state this population undergoes a geometry and solvent rearrangement with the time-constant of τ1. Following this geometry rearrangement on the S1 electronic surface, the excited state population moves towards an S1/T1 crossing, ultimately leading to population of the T1 state with a time-constant of τ2 = 386.47 ± 7.30 ps; this correlates to the decay of all three ESA features. The triplet state then persists for the maximum Δt of our experiment, described by the third extracted time-constant τ3 ≫ 3 ns. We draw attention to the weak ESA associated with τ3 centred at 600 nm.
Drawing comparisons between the observed dynamics for 1 and 2, they both appear to undergo intersystem crossing (ISC) to the triplet state (T4 and T1, respectively). For 1, this triplet state is observed as a strong ESA at large Δt, while for 2 it is observed as a weak absorption, again extended Δt. The changes in the intensity of these signals are further evidenced by EADS for τ2 (Fig. 3e) and τ3 (Fig. 3f), implying that less excited state population is transferred to Tn for 2 compared to 1.
To reconcile the change in population-transfer facilitated by S1/Tn in both 1 and 2, we once again turn to theory. LIIC, Fig. 4 from the initial Franck–Condon region to the minimum energy crossing point S1/T4 shows that for 1 there is no energy barrier along the reaction towards S1/T4. In this case, upon photoexcitation, the molecule evolves in the S1 PES by central ring stretching until a MECP with a spin–orbit coupling of 5.5 cm−1 is encountered. Here, T4 is populated and then a rapid conversion to T1 may take place. The molecule will be trapped in the minimum in the T1 PES for some time. From here, triplet absorption could take place (computed absorption 599 nm, see ESI† for details; cf. experimental absorption 625 nm). In contrast for 2, LIIC reveals the presence of an energy barrier between the minimum in S1 and the MECP for S1/T1 (Fig. 4d). Thus, the molecule will spend some time in the minimum in S1 before surmounting the barrier (1.8 kcal mol−1, 0.08 eV) and eventually reaching the MECP. These differences in the potential energy landscape between 1 and 2 could resolve both the increased time-constant for ISC for 2 compared to 1 (386 ps compared to 71 ps) and the reduced population of the triplet state, as the excited state population will have to surmount the barrier overpassing the minimum in S1 to populate the triplet state.
Our calculations also highlight the different characteristics of the triplet state. While vertical excitations for both 1 and 2 are located on the central ring, both having very similar optical properties, the calculated (T1) minima feature different electronic structures (see ESI†). For 1 the excitation mainly remains in the central ring with some contribution from the ketone and one of the methoxyphenyl moieties. However, for 2 there is a clear contribution from the nitrophenyl substituent. It is apparent that the addition of the stronger electron withdrawing nitrophenyl group alters the photochemical properties and the details of the photoprotection mechanism without compromising the optical properties nor photostability. This opens the door for the generalisation of the structures of this family of absorbers. Both of the species we have studied here display long lived electronic excited states, in contrast to many of the reported relaxation mechanisms of current UV filters.3 The photostability for species 2 shown in Fig. 2 indicates that the excited state population will return back to S0 ready to absorb another photon. While we can only speculate on the exact relaxation mechanism from T1 back to S0 (either radiative or nonradiative) this evidence does suggest it occurs with high efficiency given the steady-state irradiation studies. The steady-state results also agree with the excited state dynamics as the barrier to the triplet state prevents ISC allowing for recovery to the ground electronic state.
Furthermore, the effects of different substituent groups on the electronic excited states evidently alters the ability of the molecule to transfer effectively from one state to another; viz. the decrease in triplet absorption between 1 and 2. Importantly, the nitrophenyl group in 2 increases the available relaxation pathways owing to the initially populated S7 state (cf. S1 for 1), meaning there are more pathways for 2 to return to S0.
Conclusions
To conclude, the photodynamics of two potential new UVA filters have been explored and their electronic excited state dynamics characterised through transient electronic absorption spectroscopy alongside complementary steady-state spectroscopy and computational methods. Our results show that following photoexcitation, the excited state population evolves onto a triplet state. The extent of this transfer is influenced by the nature of substitution on the chromophore. The net result, in our present studies, is that transfer to the triplet state can be significantly thwarted through the addition of a highly electron withdrawing nitrophenyl group, allowing for internal conversion and/or luminescence back to the electronic ground state. These results highlight the promising nature of these UVA filters and presents new avenues of further research. For instance, exploration into the effects of a more strongly perturbing solvent and/or the role of different substituent groups on the dynamics. These future studies will undoubtedly illuminate on how one is able to tune the photochemistry and photophysics of this class of UVA filters for potential use in sunscreen formulations.With regards to photoprotection, while 1 and 2 exhibit long lived nanosecond dynamics, the UVA absorbance of the systems is maintained throughout prolonged irradiation, highlighting a potentially large quantum yield of ground state recovery. While these dynamics are contrary to those expected of UV filters (particularly the formation of triplet states which can produce reactive oxygen species),30 the electronic excited state lifetimes are comparable to the currently used UV filters homosalate and menthyl anthranilate. Both of these filters exhibit long lived excited state dynamics (greater than 3 ns).31,32 This study highlights the importance of maintaining a strong UV absorption spectrum throughout prolonged irradiation and the effects of electron withdrawing group on the photodynamics both of these are key steps in constructing the next generation of UV filters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. M. W. is grateful to EPSRC and Newport Spectra-Physics Ltd for a joint studentship. R. L. is grateful to Universidad de La Rioja for his predoctoral fellowship. D. S. and R. L. thank the Spanish Ministerio de Economía y Competitividad (MINECO)/Fondos Europeos para el Desarrollo Regional (FEDER) (CTQ2017-87372-P). V. G. S. thanks the HO2020 FET-OPEN Grant BoostCrop for financial support.References
- M. D. Horbury, E. L. Holt, L. M. M. Mouterde, P. Balaguer, J. Cebrián, L. Blasco, F. Allais and V. G. Stavros, Nat. Commun., 2019, 10, 1–8 CrossRef CAS.
- S. Forestier, J. Am. Acad. Dermatol., 2008, 58, 133–138 CrossRef.
- E. L. Holt and V. G. Stavros, Int. Rev. Phys. Chem., 2019, 38, 243–285 Search PubMed.
- B. L. Diffey, P. R. Tanner, P. J. Matts and J. F. Nash, J. Am. Acad. Dermatol., 2000, 43, 1024–1035 CrossRef CAS.
- C. A. Downs, E. Kramarsky-Winter, R. Segal, J. Fauth, S. Knutson, O. Bronstein, F. R. Ciner, R. Jeger, Y. Lichtenfeld, C. M. Woodley, P. Pennington, K. Cadenas, A. Kushmaro and Y. Loya, Arch. Environ. Contam. Toxicol., 2016, 70, 265–288 CrossRef CAS.
- J. F. Nash and P. R. Tanner, Photodermatol., Photoimmunol. Photomed., 2014, 30, 88–95 CrossRef CAS.
- S. Afonso, K. Horita, J. P. Sousa e Silva, I. F. Almeida, M. H. Amaral, P. A. Lobão, P. C. Costa, M. S. Miranda, J. C. G. Esteves Da Silva and J. M. Sousa Lobo, J. Photochem. Photobiol., B, 2014, 140, 36–40 CrossRef CAS.
- M. E. Burnett and S. Q. Wang, Photodermatol., Photoimmunol. Photomed., 2011, 27, 58–67 CrossRef CAS.
- J. M. Woolley and V. G. Stavros, Sci. Prog., 2019, 102, 1–17 CrossRef.
- R. Losantos, I. Lamas Frejo, R. Montero, A. Longarte and D. Sampedro, Phys. Chem. Chem. Phys., 2019, 21, 11376–11384 RSC.
- R. Losantos, I. Funes-Ardoiz, J. Aguilera, E. Herrera-Ceballos, C. García-Iriepa, P. J. Campos and D. Sampedro, Angew. Chem., Int. Ed., 2017, 56, 2632–2635 CrossRef CAS.
- J. M. Woolley, M. Staniforth, M. D. Horbury, G. W. Richings, M. Wills and V. G. Stavros, J. Phys. Chem. Lett., 2018, 9, 3043–3048 CrossRef CAS.
- J. D. White, J. H. Cammack and K. Sakuma, J. Am. Chem. Soc., 1989, 111, 8970–8972 CrossRef CAS.
- J. D. White, J. H. Cammack, K. Sakuma, G. W. Rewcastle and R. K. Widener, J. Org. Chem., 1995, 60, 3600–3611 CrossRef CAS.
- D. Schmid, S. Cornelia and Z. Fred, Cosmet. Toilet. Manuf. Worldw., 2004, 139–143 Search PubMed.
- D. Schmid, C. Schürch and F. Zülli, Pers. Care, 2004, 29–31 Search PubMed.
- F. Lalegerie, S. Lajili, G. Bedoux, L. Taupin, V. Stiger-Pouvreau and S. Connan, Mar. Environ. Res., 2019, 147, 37–48 CrossRef CAS.
- T. T. Abiola, A. L. Whittock and V. G. Stavros, Molecules, 2020, 25, 1–36 CrossRef.
- M. Hatakeyama, K. Koizumi, M. Boero, K. Nobusada, H. Hori, T. Misonou, T. Kobayashi and S. Nakamura, J. Phys. Chem. B, 2019, 123, 7649–7656 CrossRef CAS.
- P. Sarkar and C. Mukhopadhyay, Green Chem., 2016, 18, 6556–6563 RSC.
- M. P. Grubb, A. J. Orr-Ewing and M. N. R. Ashfold, Rev. Sci. Instrum., 2014, 85, 064104 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci and H. P. Hratchian, Gaussian, Inc., Wallingford, CT, 2016.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- https://github.com/jaimergp/, easymecp repository, publication in preparation.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS.
- B. L. Diffey, J. Am. Acad. Dermatol., 2001, 45, 882–885 CrossRef CAS.
- K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2007, 18, 1–46 CrossRef.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- J. Kockler, M. Oelgemöller, S. Robertson and B. D. Glass, J. Photochem. Photobiol., C, 2012, 13, 91–110 CrossRef CAS.
- N. D. N. Rodrigues, N. C. Cole-Filipiak, M. D. Horbury, M. Staniforth, T. N. V. Karsili, Y. Peperstraete and V. G. Stavros, J. Photochem. Photobiol., A, 2018, 353, 376–384 CrossRef CAS.
- E. L. Holt, K. M. Krokidi, M. A. P. Turner, P. Mishra, T. S. Zwier, N. D. N. Rodrigues and V. G. Stavros, Phys. Chem. Chem. Phys., 2020, 22, 15509–15519 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectroscopic and computational data, fitting residuals and power dependency measurements. See DOI: 10.1039/d0cp04940a |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2020 |