
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvidence of a new phase in gypsum–anhydrite transformations under microwave heating by in situ dielectric analysis and Raman spectroscopy†
Angel M.
López-Buendía
 *a,
Beatriz
García-Baños
*b,
M. Mar
Urquiola
a,
José M.
Catalá-Civera
b and
Felipe L.
Peñaranda-Foix
b
*a,
Beatriz
García-Baños
*b,
M. Mar
Urquiola
a,
José M.
Catalá-Civera
b and
Felipe L.
Peñaranda-Foix
b
aCEINNMAT (INNCEINNMAT, SL), PCUV, C/Catedrático Agustín Escardino, 9, 46980, Paterna, Valencia, Spain. E-mail: angel.lopez@ceinnmat.com
bInstituto ITACA, Universitat Politècnica de València, Camino de Vera, s/n, 46022, Valencia, Spain. E-mail: beagarba@upvnet.upv.es
First published on 26th November 2020
Abstract
Mineral transformations of the gypsum–anhydrite system under microwave heating have been studied using in situ dielectric thermal analysis (MW-DETA) and Raman spectroscopy simultaneously. The dielectric properties of samples that were measured under microwave heating provided thorough information about the dynamics of the gypsum–anhydrite system transformations and its significance from the mineralogical point of view. In particular, the MW-DETA technique revealed a new intermediate phase with a γ-anhydrite structure. This phase corresponds to the soluble stage of γ-anhydrite, and it is characterized by a high ionic charge inside the crystal channels. The complete sequence is gypsum → 0.625-subhydrate → bassanite → hydro γ-anhydrite → anhydrous γ-anhydrite → β-anhydrite. The transformations were also assessed using DSC, TG, DTA and dielectric measurements at room temperature, as well as other techniques including X-ray powder diffraction (XRPD) and high-temperature XRD (HT-XRD). Correlations between the dielectric properties with temperature and the rest of the techniques elucidated the heating mechanisms of this material under microwave energy during the different stages. The in situ combination of the MW-DETA and the Raman analysis appears to be a powerful technique, providing new insights about the mechanisms which govern the volumetric heating of this and other materials.
Introduction
Microwave treatment of materials is a well-established technique that is increasingly employed in industry1,2 with applications in many fields, such as the food, pharmaceutical, and chemical industries,3 as well as for agriculture, medicine, and industrial mineral processing,4–10 showing clear advantages in terms of efficiency, processing time, and product quality when compared to the more conventional heating technologies.Although numerous theories attempt to explain the origin, nature, or magnitude of the mechanisms behind the advantages shown by microwave processing, these processes are still a matter of study.11–13 Since the interaction of materials with microwave energy is determined by their dielectric properties, many studies in the last decades have focused on measuring these properties as a function of frequency, time, or temperature.14 Some previous attempts measuring dielectric measurements as a function of temperature have shown certain limitations (a review can be found in ref. 15). For example, samples were heated in conventional furnaces and then moved to a microwave measurement set-up, and therefore, the dielectric properties measured under conventional heating may not have been representative of the specific microwave heating process.15,16
Microwave analytical methods, whose objective is to gain a thorough and deeper understanding of microwave-matter interactions, have recently been developed, such as the in situ determination of dielectric properties under microwave heating (MW-DETA).13,17 These advanced methods are able to provide real-time and accurate dielectric measurements during the reactions or transformations which occur under an applied microwave field, thereby yielding more comprehensive and realistic insights into the mechanisms that can offer greater benefits than those achieved by conventional heating.
Gypsum–anhydrite is a relevant mineral system of high interest for many areas of science and industry, such as geology,18–20 engineering materials for construction,21,22 heritage conservation,23 industrial processing,24,25 geotechnical engineering,26–28 and also for artistic, painting, medical, or pharmaceutical applications. Gypsum is a basic mineral commodity whose worldwide production from geological resources was estimated at 150 Tg (million tonnes) in 2018,29 with an additional production of nearly 150–200 Tg including synthetic gypsum (own data), mainly produced as flue gas desulfurization gypsum (FGD), which is a by-product of coal power plants.
Application of microwave energy was studied for gypsum treatment in industrial applications like gypsum plaster drying30 or gypsum plaster production.31 In spite of the obtained results, the authors acknowledged there was a lack of information about the effect of microwave fields on the gypsum–anhydrite system at different temperatures.
In order to contribute to a deeper understanding of how microwave fields affect mineral transformations, in this study, advanced microwave equipment17 has been applied to this mineral system composed of dipolar H2O molecules and non-polar domain molecules of CaSO4. As a result, new data emerged on the transitional vibration stages, where microwaves partially polarize water molecules, thus affecting the measured dielectric properties. The findings obtained by this technique unveiled the microwave heating mechanisms in each phase, providing useful information for potential industrial applications. Correlation with other analysis techniques, such as simultaneous Raman spectroscopy during microwave heating and X-ray powder diffraction, verified the new technique's information and supported the conclusions about the novelties in the observed transformations.
The gypsum–anhydrite system
Gypsum is a calcium sulfate mineral based on non-polar CaSO4 molecules and 2 dipolar molecules of H2O (CaSO4·2H2O) with a very crystalline, stable and monoclinic structure, which some authors assign to the C12/c1 space group.32 The water of crystallization presents hydrogen (H) in two positions at different distances of the SO4 tetrahedron. Its structure shows two sheets that comprise SO4 tetrahedra with Ca–O polyhedra, sharing six O atoms between them, and where two O atoms are shared with H2O molecules.33According to the literature,34–43 when heated over 95 °C, water is partially sublimated, and gypsum is transformed through several sub-hemihydrates into a more stable mineral, which is bassanite. The latter, in turn, is transformed into anhydrite, which has three known structures (Table 1). The thermal sequence of the minerals, which shows increasing stability, is: Gypsum → Bassanite → γ-anhydrite → β-anhydrite → α-anhydrite.
| Mineral | Chemical composition | Other terms | Density g cm−3 | Temp./pressure of transitions | Lattice | Space group |
|---|---|---|---|---|---|---|
| Gypsum | CaSO4·H2O | Gypsum | 2.31–2.3351 | <80 °C | Monoclinic | I2/a52 |
| C2/c53 | ||||||
| 0.625-subhydrate | CaSO4·0.625H2O | 2.6351 | 65–180 °C | Trigonal | C24,5 | |
| Bassanite | CaSO4·0.5H2O | α-Hemihydrate | 2.75751 | 95–235 °C | Monoclinic | I24–8 |
| β-Hemihydrate | 2.63732 | I26–8 | ||||
| P319 | ||||||
| γ-Anhydrite | ‘γ-CaSO4’ | Anhydrite III46 (soluble anhydrite) | 2.5851 | 160–380 °C | Hexagonal | C22235 |
| P62222,9,10 | ||||||
| β-Anhydrite | ‘β-CaSO4’ | Anhydrite II46 (insoluble anhydrite) | 2.93–2.9751 | 260 to >1228 °C | Orthorhombic | (Bmmb54,55) |
| (Cmcm(63)56) | ||||||
| Amma 9,30,31,45,47,57 | ||||||
| α-Anhydrite46 | ‘α-CaSO4’ | Anhydrite I50 | 960 to >1450 °C | Cubic41 | ||
| Trigonal50 | ||||||
| Barite-type | CaSO4 | >8 GPa | Orthorombic | Pnma 49 | ||
| Monazite type | CaSO4 | >5 GPa | Monoclinic | P21/n48,49,58 |
Each stage has its own industrial relevance for different applications, and this leads to the involvement of different disciplines in their study. The terminology related to these materials in the literature uses mineral, chemical, and technical nomenclature,44–58 whose equivalences are summarized in Table 1.
Dielectric properties
Microwave processing applications are restricted to ISM (Industrial, Medical and Scientific) standardized frequencies in the 2.45 GHz and 915 MHz bands. At these frequencies, the capacity of materials to absorb microwave energy has been reported to be dominated by the dipolar effect (orientation of dipoles at the molecular level) together with the ionic effect (orientation of free charges, as electrons or ions).1The parameter that defines the behavior of materials exposed to a microwave electric field is the complex permittivity (ε*):
| ε* = ε′ − jε″ | (1) |
| tan(δ) = ε″/ε′ | (2) |
It is a well-known fact that the water molecule is a dipolar molecule with water dimer resonances in the microwave frequency band59 which is easily polarized by the electric field and transforms the electromagnetic energy into heat when it is in a liquid state.60,61
The water of crystallization in the gypsum crystal structure is, however, very stable, with compensated charges, and shows a very low capacity to be polarized by the electromagnetic fields.
Sulfate is a unipolar molecule showing Ca–SO4–Ca with highly compensated charges, as demonstrated by the difficulty in obtaining anhydrite crystal growth in CaSO4 solutions.62
Dielectric properties allow the direct measurement of the polarizability of the materials and, consequently, of their interaction with microwave fields. In this work, given that transformations will occur in the calcium sulfate system, important changes are expected to be seen in their capacity to convert microwave energy into heat. In addition, dramatic and abrupt changes are often observed in the materials’ dielectric properties when temperature changes,15 and thus analyzing them during the heating process is convenient.
Experimental section
Materials
A sample of high purity gypsum of collection quality was selected for the study. This sample is natural polycrystalline gypsum of specular habit from Soneja (Spain) of secondary formation associated with geological Keuper facies.Experimental techniques
Samples were examined through mineralogical (XRD, HT-XRD), micro-morphological (SEM), thermo-analytical (DSC, TG, DTA), and dielectric thermal analyses during microwave heating (MW-DETA).In order to carry out a comparative study of the molecular influence, a review of the crystallography of gypsum–anhydrite series was also conducted using the most recent crystallographic data available, which was introduced and treated with CrystalMaker® V9.2.8 software (CrystalMaker Software Ltd).
High-temperature X-ray diffractions (HT-XRD) were analyzed with a high-temperature chamber model, Anton Paar HTK-1600, adapted to the Bruker D8 X-ray diffractometer, operating in air at atmospheric pressure, with temperatures ranging from 30 °C up to 1100 °C (heating rate of 1 °C min−1), and measured at regular intervals of 10 °C. Samples were spread evenly covering the Pt strip sampler. At the end of the test, an additional diffractogram was recorded at 30 °C to calibrate the peak displacement exclusively due to the change in temperature. The same measurement conditions were applied in the HT-XRD and XRPD experiments.
The thermo-gravimetric measurements to obtain TG and the DTA analyses were carried out with a Mettler Toledo TGA/SDTA851e equipped with a horizontal furnace, which is able to heat up to 1600 °C under air conditions.
Additionally, a PerkinElmer Pyris Diamond thermal analyzer running up to 1100 °C was employed for the differential thermal analyses (TG-DTA).
A second VNA is installed for the measuring mode, and also, a cross-coupling filter is used to avoid interferences with the heating signal. At each temperature, the dielectric properties (ε′ and ε″) of the sample are determined by a cavity perturbation method based on the measured values of the cavity resonance frequency and the quality factor. With this technique, accuracy is estimated to be 3% for the dielectric constant and 10% for the loss factor. More details about this method can be found in ref. 17.
In the experiments, fine powdered gypsum (<50 µm) was placed in a standard fused quartz sample holder with a diameter of 10 mm, filling it up to a height of 15 mm (sample volume = 1.13 ± 0.08 cm3). A compressed sample (350 MPa) of powdered gypsum with a diameter of 9.95 ± 0.03 mm and 15 mm height was also measured to evaluate the effect of porosity.
For comparison purposes with the rest of the techniques, the microwave heating rate was set at 5 °C min−1. During the microwave heating, the sample temperature was measured continuously with a pyrometer through a cut-off cylindrical window of 7 mm diameter in the cavity wall. The bulk sample temperature was calculated from the quartz holder surface measurement by following a calibration curve obtained in a thorough procedure described in ref. 63.
Additionally, visual observation of samples was made through another cut-off inspection hole in the cavity wall and recorded with a video camera for subsequent analysis.
Dielectric measurements were based on the resonance frequency shift and the quality factor of the microwave cavity with the sample inside. The complex permittivity of the sample was calculated based upon the structure's electromagnetic model and a numerical procedure controlled by Labview software.64
For the experiments, milled gypsum (<50 µm grain size) was prepared, and subsamples were heated in a conventional electric furnace at selected temperatures, as follows: 140 °C (30 min) to obtain bassanite, 250 °C (30 min) for γ-anhydrite, 550 °C (30 min) for β-anhydrite, and 1200 °C (30 min) to obtain α-anhydrite. Cooled samples were stored in a closed dry environment until they were placed in a standard 1 mL glass vial for the dielectric measurements.
Results and discussion
Samples of natural polycrystalline gypsum were prepared and characterized according to the procedures described in the previous section. The main results are discussed below.X-ray diffraction analysis (XRPD and HT-XRD)
Fig. 1 presents the X-ray diffractograms of the gypsum powder (XRPD), showing the absence of impurities at the resolution level of the technique.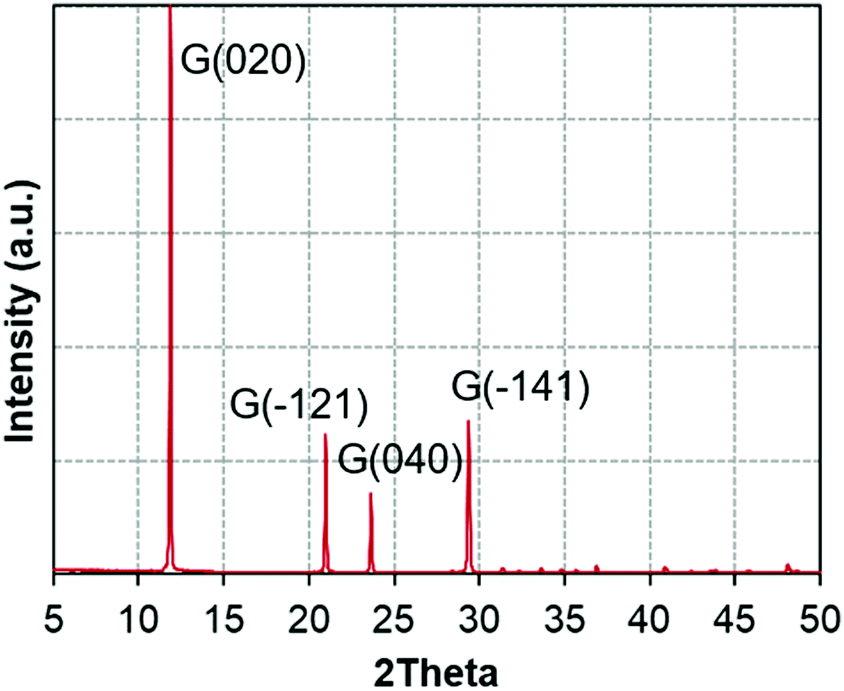 | ||
| Fig. 1 X-ray diffractograms of the gypsum powder (XRPD). | ||
Fig. 2a and b depict the described transformations of the gypsum sample using HT-XRD and their temperatures, following this sequence: gypsum (up to 80 °C), subhydrates (65–180 °C), bassanite (95–235 °C), γ-anhydrite (160–400 °C), β-anhydrite (260–1100 °C), and α-anhydrite (960–1100 °C). This sequence agrees with previous findings in the literature,34–43 with intermediate phases overlapping the different stages, due to the progressive increase of temperature.
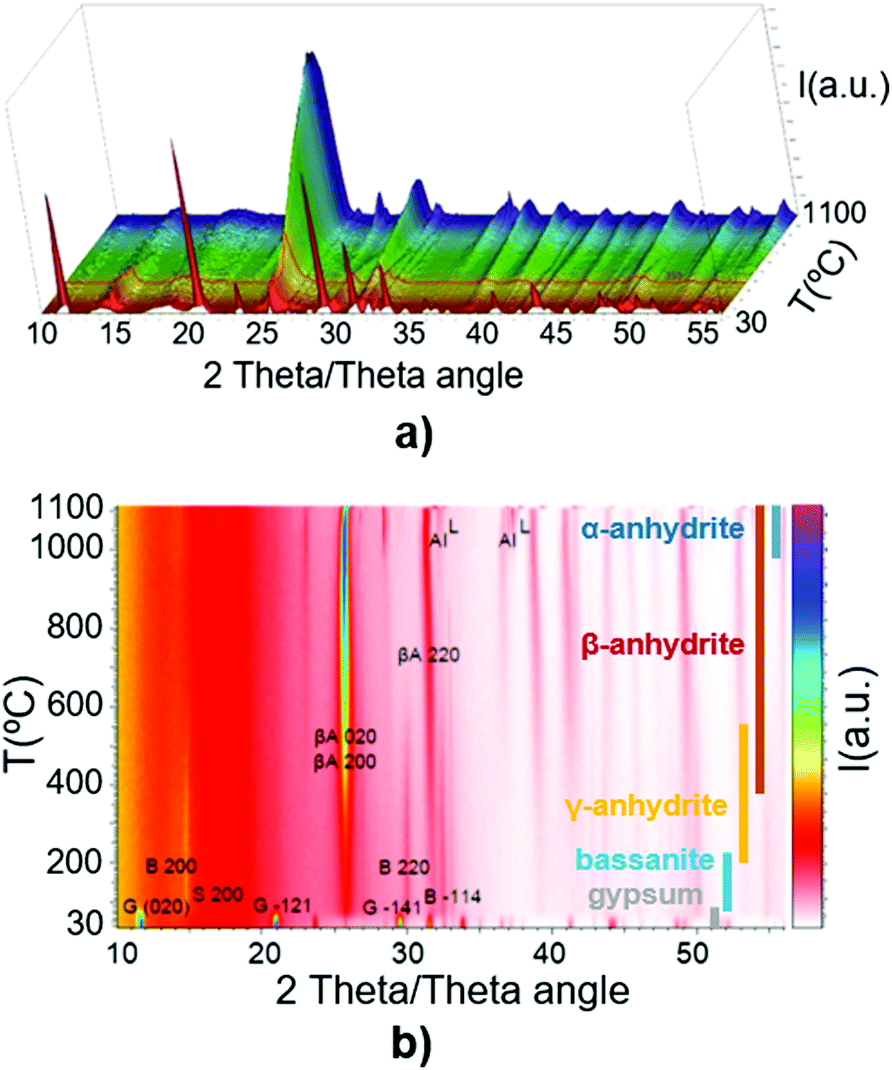 | ||
| Fig. 2 (a) HT-XRD of gypsum in the temperature range of 30–1100 °C (3D view); (b) HT-XRD of gypsum in the range of 30–1100 °C in the different phases. | ||
A more detailed study of the temperatures at which the transitions occur can be carried out by examining parameters such as the intensity or full width at half maximum values in the HT-XRD diagram. Fig. 3 plots the areal intensity (IA) of the reflection (121) of gypsum (d-space of 2.87 Å), which decreases until fully disappearing at 100 °C. It also shows the reflection (102) of anhydrite at 2.853 Å, which appears approximately at 160 °C.
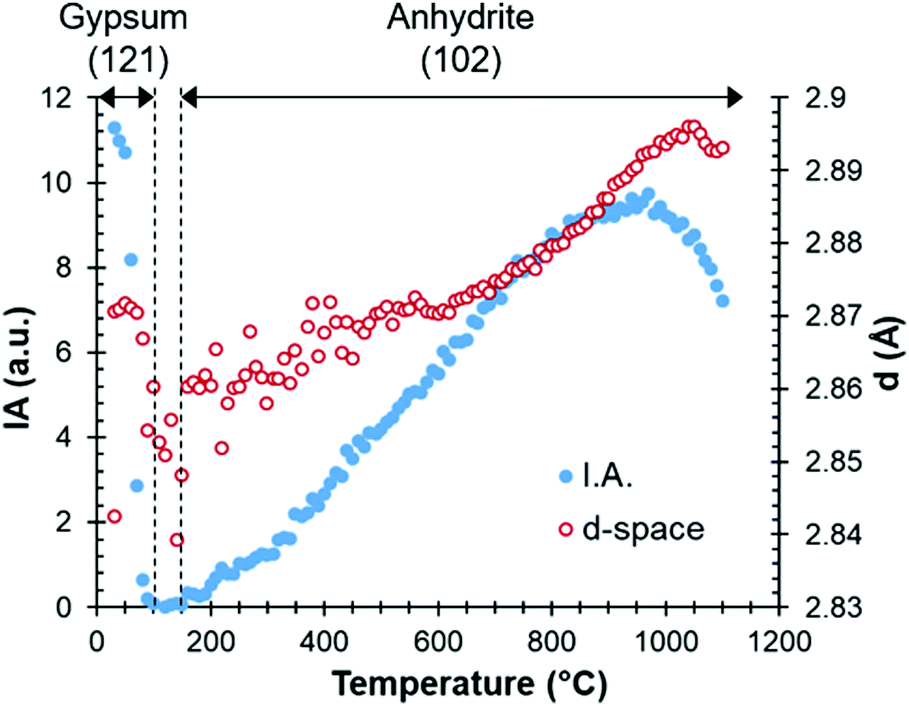 | ||
| Fig. 3 Areal intensity (IA) and d-spacing as a function of temperature, with the gypsum (up to 100 °C) and anhydrite (from 160 °C) phases. | ||
Fig. 4 represents the intensity and full width at half maximum (FWHM) of the anhydrite (200) reflection located at 3.5 Å, which appears at low temperatures of between 60 and 80 °C. At 190–330 °C, this reflection presents its minimum crystal order or maximum width, as shown by the FWHM values, due to the transition from bassanite to anhydrite.
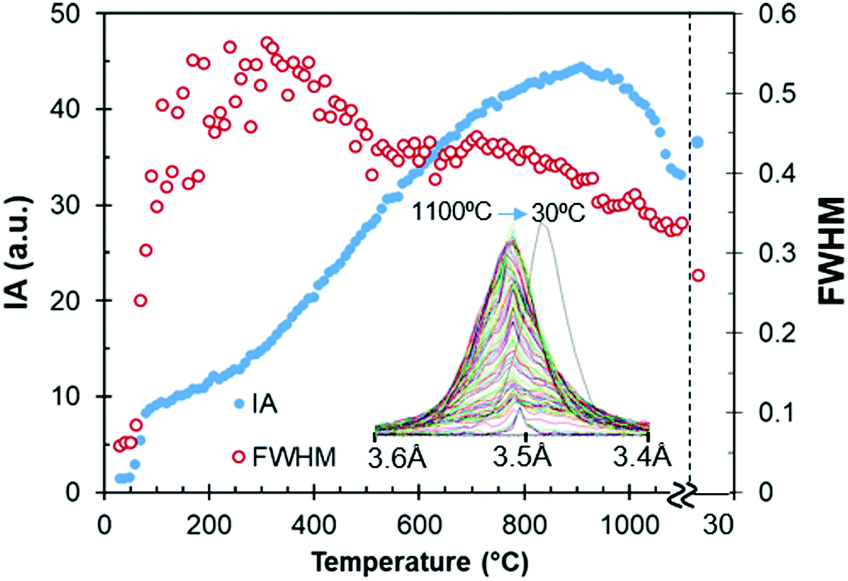 | ||
| Fig. 4 Intensity (IA) and FWHM of the anhydrite (200) reflection at 3.5 Å during the stage of heating up until 1100 °C. XRD after cooling at 30 °C is also included for comparison. | ||
Fig. 2–4 illustrate how the HT-XRD analytical technique can identify the crystalline transitions in the CaSO4–H2O system. However, the coexistence of phases highlights the complexity of these transformations, and the difficulty in distinguishing these phases is aggravated by using conventional heating methods which rely on a slow heat transfer within the sample. This in fact hinders the precise identification of the stages and their corresponding temperatures.
Thermal analysis techniques
Fig. 5a and b represent the measurements from DTA-TG and DSC analyses of gypsum samples. The endothermic area in the range of 100–210 °C is the most relevant characteristic shown by the DTA and DSC curves, which is associated with a significant weight loss in the TG (20.73%). This is attributed to water liberation during the bassanite formation with an intermediate subhydrate phase. The minimum value at 163 °C corresponds to the formation of bassanite, and water loss continues up to 190 °C when γ-anhydrite is definitively formed.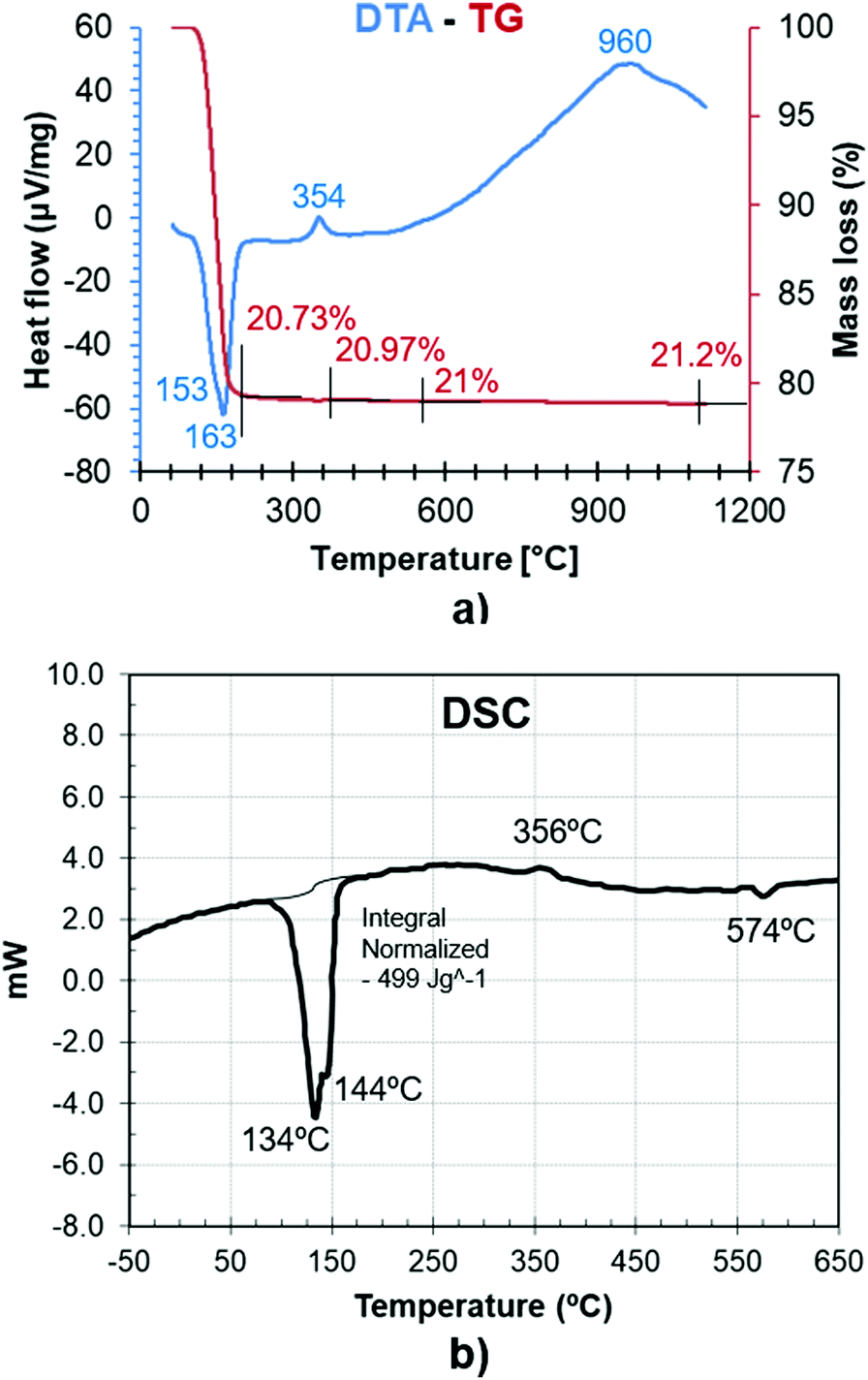 | ||
| Fig. 5 (a) DTA and TG of a gypsum sample. (b) DSC of a gypsum sample. | ||
Additionally, an exothermic heat flow peak around 354 °C is related to the structural reorganization in the transition from β-anhydrite to γ-anhydrite without any significant mass variation in the TG.
Dielectric analysis at room temperature
The dielectric properties of samples heated at different temperatures in a conventional furnace and measured at room temperature (22 °C) are presented in Table 2.| Processing temperature [°C] | Main phase | Mass loss (%) | ε′ (at 22 °C) | tan(δ) (at 22 °C) |
|---|---|---|---|---|
| 22 | Gypsum | — | 2.99 ± 0.01 | <1 × 10−5 |
| 140 | Bassanite | 16.6 ± 0.2 | 2.62 ± 0.13 | 7.09 × 10−3 ± 4.81 × 10−3 |
| 250 | γ-anhydrite | 20.6 ± 0.15 | 2.45 ± 0.52 | 1.18 × 10−2 ± 1.03 × 10−2 |
| 550 | β-anhydrite | 21.1 ± 0.05 | 2.70 ± 0.02 | 6.44 × 10−3 ± 1.78 × 10−3 |
| 1200 | α,β-anhydrite | 22.1 ± 0.3 | 2.70 ± 0.01 | <1 × 10−5 |
| Reference of compressed gypsum | ||||
| 22 | Gypsum | — | 4.11 ± 0.01 | 4.13 × 10−3 ± 1.22 × 10−4 |
The dielectric constant is very similar for all treated samples, but differences are observed in the loss tangent, which can be attributed to the different phases’ microstructure. Gypsum, with nonpolar CaSO4 molecules, and given the polar structure of the water molecules, shows a low dielectric constant (ε′ = 2.99 ± 0.01) and a very low loss tangent, indicating that there is a lack of interaction with microwave energy. The same loss tangent is observed for the sample treated at 1200 °C, with α- and β anhydrite as the main phases, which indicates that the crystal structure is very stable with a low capacity to be polarized. The rest of the samples show higher values of loss tangent, particularly in the case of the sample treated at 250 °C, evidencing the presence of phases (γ-anhydrite) which have a higher interaction with microwave energy. The fact that the dispersion of dielectric measurements is higher in this sample informs us of the irregular presence of intermediate phases and of an uneven distribution of different transformation stages within the sample.
A compressed sample (350 MPa) of gypsum was also measured to assess the effect of apparent density on the dielectric properties. As expected, the measured dielectric values are higher because since the air volume in the sample is insignificant, it has a minor effect. However, the difference with respect to the non-compacted samples indicates that the apparent density has a small influence on the dielectric results.
Raman spectroscopy
As opposed to the analysis techniques described in previous sections, which were based on the conventional heating of samples, the results presented hereafter relied on microwave heating.Fig. 6 shows some examples of Raman spectra at different temperatures during microwave heating. The temperatures have been selected to be representative of the main different phases. Changes occurring in the gypsum–anhydrite system can be followed by inspecting the Raman modes, in particular, the 3 vibration modes65 identified in the frequency mode of SO4: (i) ν1, the symmetric stretch vibration mode, around a Raman frequency of 1009 cm−1; (ii) ν2, symmetric bending, around a frequency of 417 cm−1; and (iii) ν3, asymmetric stretch vibration, around a frequency of 1166 cm−1.
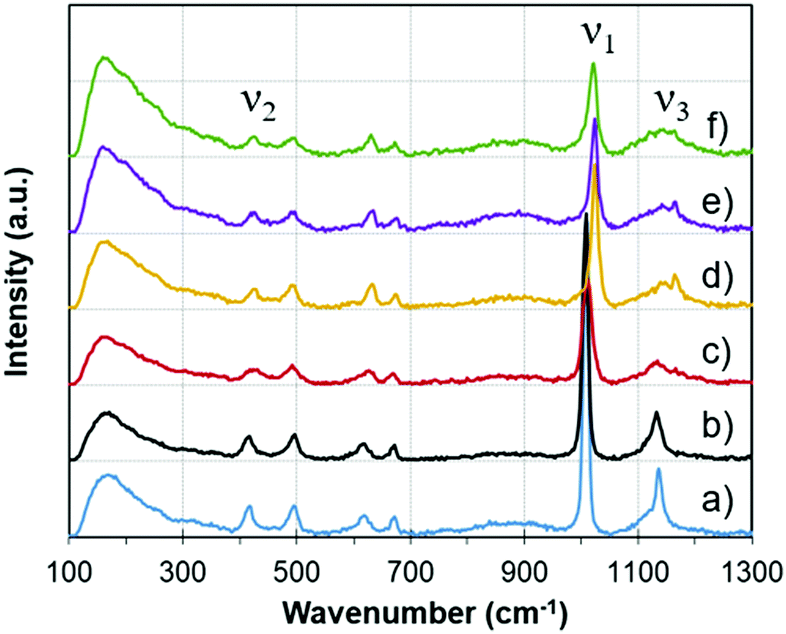 | ||
| Fig. 6 Raman spectra at different temperatures during microwave heating, representative of the different phases: (a) gypsum (30 °C), (b) subhydrate (145 °C), (c) bassanite (180 °C), (d) γ-anhydrite (225 °C), (e) γ-anhydrite (296 °C), (f) β-anhydrite (365 °C); with the three main SO4 vibration modes (ν1, ν2 and ν3). | ||
Fig. 7 illustrates the variations in the ν3 SO4 vibration mode as a function of temperature. New emissions appear during heating, suggesting the complexity of multiple transitional structural phases. In particular, new vibration modes appear from approximately 300 °C, which demonstrates the formation of new structures from γ-anhydrite.
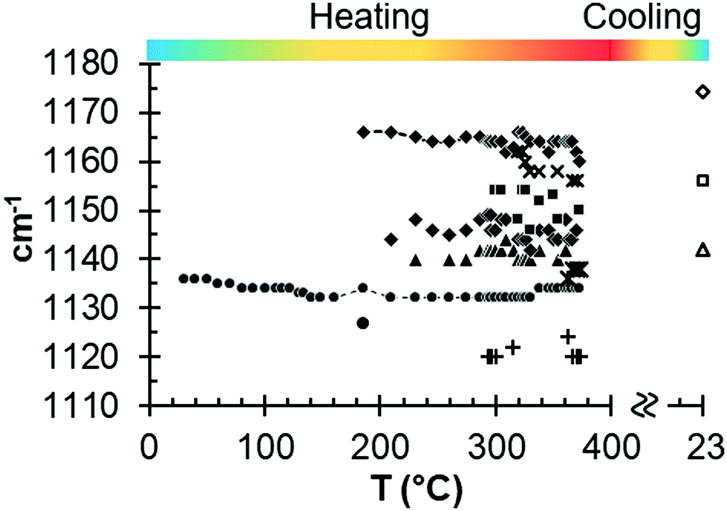 | ||
| Fig. 7 Results from in situ Raman measurements for the ν3 SO4 vibration mode as a function of temperature. | ||
In the case of the ν2 SO4 vibration mode (see Fig. S2, ESI†), only minor variations occur, indicating that they are mainly caused by relaxation due to thermal dilatation.
Nevertheless, it is clear that the principal peak in intensity corresponds to the ν1 SO4 mode, with a frequency of 1008 cm−1. This mode will be discussed in detail in the next section, together with the MW-DETA analysis, since data correlation between both techniques helps to interpret the different phases.
Dielectric thermal analysis (MW-DETA)
Fig. 8 depicts the loss tangent of the gypsum sample measured under microwave heating (MW-DETA). The Raman spectroscopy (ν1 SO4 mode) and DTA measurement of gypsum samples are also included in the figure for comparison purposes. Fig. S3, ESI† plots the specific dielectric constant and loss factor variations as a function of temperature, both for the microwave heating and cooling cycles.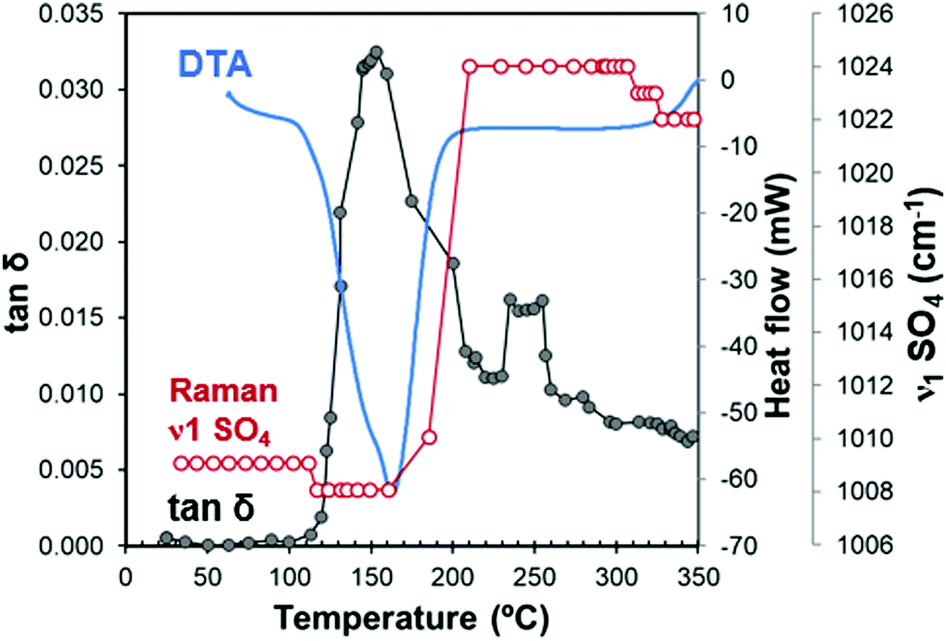 | ||
| Fig. 8 Combined diagram with in situ simultaneous MW-DETA (tan(δ)) and Raman (ν1 of S–O bond) measurements during microwave heating. For illustrative purposes, the DTA measurement of gypsum aliquot is also included. | ||
Fig. 8 shows different stages, which can be clearly distinguished from the loss tangent variations. For example, in the temperature range from 22 °C to 110 °C, the loss tangent of approximately 10−3 reflects the low capacity of the sample to absorb microwave energy, which is in agreement with the dielectric properties at room temperature shown in Table 2. The Raman frequency of the ν1 SO4 mode remains constant because no changes in the crystal structure occur in this range.
According to the dielectric characterization, the water of crystallization in gypsum does not exhibit any dipolar effect.
This very low polarization is consistent with the fact that water of crystallization (H–Ow–H) shows a stable chemical bond of Ca (Ow–Ca) and CaSO4 (see Fig. 9a), with a short distance (2.38 Å) between both. This hinders dipolar molecular movements and leads to the formation of a layered structure.66 The surface between the two water slices facing each other forms weak planes along the (010) layer with an electrically neutral resultant, and thus, a macroscopic dipole moment cannot be generated.67
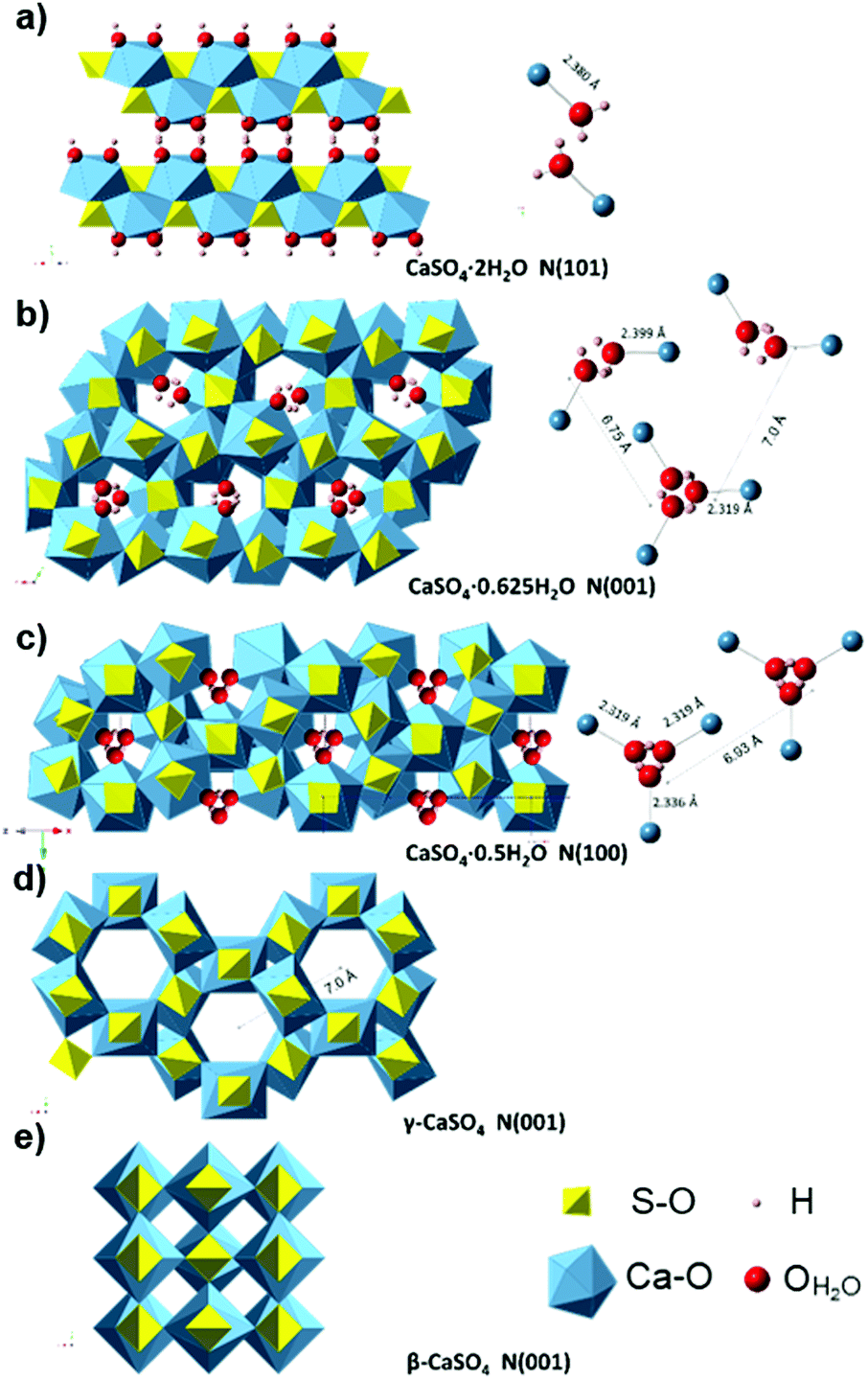 | ||
| Fig. 9 Crystal structure of the gypsum–anhydrite series structure showing H2O molecules (ball and stick representation) in polyhedral projection. (a) Gypsum (CaSO4·2H2O) structure perpendicular to (101), H atoms are forming the plane (010). (b) CaSO4·0.625H2O structure (C2) showing the two types of water channels in projection normal to (001) plane (data from ref. 34). (c) Bassanite (CaSO4·0.5H2O) structure (I2) in a projection normal to (100) (data from ref. 34). (d) γ-anhydrite lattice P6222 (data from ref. 35) perpendicular to (001). (e) Anhydrite Amma in a projection normal to (001), data based on ref. 68. | ||
In the range of 110–160 °C, the loss tangent (Fig. 8) exhibits a steep increase of one order of magnitude along with the endothermic area in the DTA, and a slight decrease in the ν1 SO4 mode frequency. These changes can be attributed to the crystal structure transformation during water loss in the gypsum–anhydrite system (Fig. 9b). The movement of water molecules and the subsequent liberation process lead to a notable increase in the dielectric properties. The well-compensated layers of water in gypsum are transformed into water channels in the 0.625-subhydrate. Two types of water channels are formed, namely the 2W and 3W channels with 2 or 3 water molecules, respectively, these having uncompensated bonds with an excess of negative charge.34 Detailed information about the structure of the water channels is provided in the ESI† (Fig. S4).
Fig. 8 shows a sudden decrease in the loss tangent in the temperature range of 160 °C to 220 °C. Around 200 °C, the ν1 SO4 vibration mode presents a steep increase of frequency of up to 1024 cm−1. These changes indicate that the formation of bassanite involves the transformation of all the channels into the 3W type, with closer Ow–Ca bonds (with a distance of 2.336 Å) and with a slightly higher distance in the Ow–Ow bonds (4.458 Å) (see Fig. 9c), which contributes to the decrease in dielectric properties in this stage.
In the range of 220 °C to 347 °C, there is a sharper increase in the loss tangent between 235 °C and 255 °C, whereas the Raman response does not show any change in the ν1 SO4 vibration mode frequency up to 309 °C. In this range, γ-anhydrite water molecules are removed from the 3W channels maintaining a similar structure (see Fig. 9d). As the channels are still present, γ-anhydrite shows its typical high capacity to absorb water in this stage.
It should be noted here that evidence of this intermediate phase was not well detected by the XRD method (Fig. 2a and b), nor by the TG analysis, which showed a water release but only of 0.065% between 225 °C and 265 °C (Fig. 5a). In addition, the DSC analysis did not reveal any heat changes (Fig. 5b), and no thermal changes were observed in the DTA curve (Fig. 8). However, the dielectric changes clearly suggest a reorganization of the ionic charges in the channels and that the remaining water molecules disappear entirely. The observed impact on the MW-DETA curve of this water liberation from the γ-anhydrite structure justifies the existence of a new intermediate phase (hydro γ-anhydrite), combined with the fact that the Raman response shows minor variations in the ν3 modes (Fig. 7), reflecting minor structural variations at these temperatures. The identification of this hydro γ-anhydrite stage is coherent with the structure mentioned by Bezou35 and studied by Khasanov69 using electron spin resonance (ESR) techniques.
Under microwave fields, and once the binding energy has been achieved, water molecules behave as isolated dipoles, absorbing microwave energy with high efficiency, which produces a sudden and fast liberation of water through the structure channels. This leads to an abrupt change in the microwave response between phases, which helps us identify the phase limits. In contrast, when using conventional heating, water is liberated by thermal conductivity from the external heating source, causing the water loss and re-structuration to occur from the surface to the inner parts. This difference can be appreciated in the resulting particle morphology shown in Fig. 10 and 11.
 | ||
| Fig. 10 SEM images of gypsum crystal at 250 °C during conventional heating, where the particles’ morphology evidences the re-structuration and water loss from the surface to the inner parts. | ||
 | ||
| Fig. 11 SEM images where water channels can be appreciated in the structure (microwave heating at 350 °C). During microwave heating, water is liberated along water channels in the whole crystal volume. | ||
From 255 °C, a gradual decrease is observed in the loss tangent (Fig. 8). This trend indicates a decreasing capacity of the sample to absorb microwave energy, preventing the material from being heated beyond 347 °C. In the Raman ν1 SO4 mode, a small decrease occurs in this range, which then remains stable up to the end of the test. This decrease in the MW-DETA and Raman curves evidences the continuous re-structuration of anhydrite from a hexagonal to a more stable orthorhombic structure of β-anhydrite (Fig. 9e) from approximately 300 °C. This finding is in agreement with the strong exothermic energy reaction observed in the DSC analysis at 354 °C (Fig. 5b). Similar observations were reported by Smykatz70 or Carvalho,71 who determined the transformation taking place at 380 °C.
Based upon the new evidence shown by the combined techniques (Fig. 8), the transformation sequence identified during microwave heating, including the newly identified phase, is compared to conventional heating analyses (HT-XRD, DTA) and summarized in Table 3.
| T [°C] | Conventional heating (HT-XRD, DTA) | Microwave heating (DETA-MW, RAMAN) | |||
|---|---|---|---|---|---|
| Appear | Domain | Present | Identified | Description | |
| G = gypsum, Ba = bassanite, Sh = subhydrate, hγ-A = hydro γ-anhydrite, γ-A = γ-anhydrite, β-A = β-anhydrite, α-A = α-anhydrite. | |||||
| 23 | G | G | A very small amount of water is lost, with hemihydrates at temperatures above 60 °C | ||
| 65 | Sh | Sh | G | ||
| 80 | |||||
| 96 | Ba | ||||
| 110 | |||||
| 115 | Sh | Water is liberated from gypsum by sublimation and the peak in the dielectric properties matches perfectly with the maximum loss of water molecules. Bassanite formation starts with the restructuring of the 2W water channels towards 3W channels, with minor structural changes | |||
| 140 | B | Sh | |||
| 160 | γ-A | ||||
| 170 | B | Bassanite stage with reduction of water loss and decrease of dielectric properties | |||
| 210 | B | hγ-A | Hydro γ-anhydrite stage with channels containing residual water | ||
| 235 | hγ-A + γ-A | γ-Anhydrite dehydration with residual water sublimation from the channels and a local increase in dielectric properties | |||
| 260 | β-A | γ-A | γ-A | γ-Anhydrite fully dehydrated | |
| 309 | |||||
| 315 | |||||
| 320 | γ → β-A | Transition to β-anhydrite with a decrease in the loss factor and dielectric constant. | |||
| 330 | β-A | β-Anhydrite domain | |||
| 350 | γ-A | β-A | |||
| 380 | β-A | γ-A | |||
| 470 | β-A | — | |||
| 960 | α-A | β-A | |||
Identified mechanisms of microwave absorption
The variations in the dielectric properties during heating are a consequence of the changes in the material that affect its interaction with microwave fields. In general, dielectric properties are influenced by impurities, porosity, grain size, grain boundaries, or sample density.72,73 At the crystal lattice level, some factors affecting the polarizability of the material have been previously identified: electrons traveling through the material, adjacent dipoles, lattice distortion,74 as well as bulk ionic heating.74In the gypsum–anhydrite system the contribution of dipolar and ionic effects changes as the different phases evolve. In gypsum, most charges of the water molecules are compensated in the double water layers except in the surface of the grains, where water molecules show an ionic charge. This mechanism is also observed during the subhydrate stage because the water molecule charges in the 2W channels are not fully compensated. Orientation of the E-field causes the polarization of these bounded water molecules (Ca2–H2O), which enhances the precession movement (Fig. 12) that would contribute to the local heating. It should be noted here that this movement appears in response of the applied alternating electric field and thus, the polarisation effect is maintained as long as the microwave field is applied.
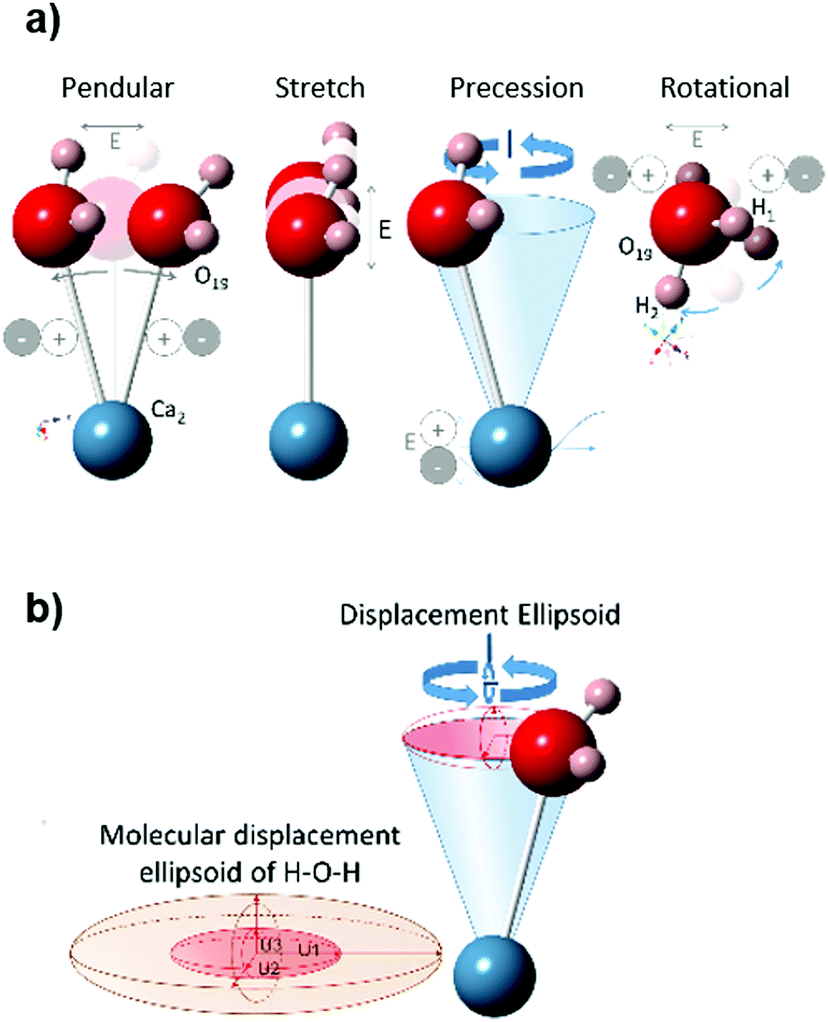 | ||
| Fig. 12 (a) Main displacements of molecule components due to material polarization when exposed to the electrical field (E) in the frequency of microwaves, which causes a precession movement (resultant displacement ellipsoid) shown in (b). Example of a water molecule (H1–O19–H2) placed in a 2W water channel of 0.625-hemihydrate, at room temperature. Translational movement (H1–O19–H2) rotate fixed Ca2 of the O16–Ca2 bond in 2W, and rotational along the O16–Ca2 bond axis. | ||
Also, in the dehydrated γ-anhydrite stage, the free electric charges along the structure's channels are suitable for ionic polarizability. The presence of SO3− radical ion molecules, particularly in γ-anhydrite, was detected by ESR,69 these molecules contributing to the ionic heating. After the structural recrystallization to β-anhydrite, with a more balanced distribution of the charges, this ionic mechanism is reduced.
On the other hand, a dipolar effect is observed as the water is liberated from the crystal structure, mainly in the subhydrate and hydro γ-anhydrite stages.
Thus, the main mechanisms governing microwave heating in the gypsum–anhydrite system can be summarized as follows:
– An ionic effect, related to free ions and the non-compensated charges of the sulfates, mainly: (i) crystal structure with free ions (SO3−); (ii) vibration of the water of crystallization with non-compensated charges, in the water of the gypsum grain surfaces, 2W channels of subhydrates and 3W channels of bassanite.
– A dipolar effect when water is liberated from the structure and where sublimation from the sources containing bounded water takes place: (i) gypsum > 100 °C; (ii) subhydrates > 140–155 °C; (iii) bassanite > 150 °C (171 °C maximum); and (iv) hydro γ-anhydrite (235–255 °C).
Conclusions
A gypsum–anhydrite system has been analyzed by dielectric measurements under microwave heating (MW-DETA), simultaneously to Raman spectroscopy, and correlated with conventional thermal analytical techniques (DTA, TG, DSC).MW-DETA has proven to be a powerful analytical tool for mineral and material characterization which can assess mineral transformations, including crystal structure changes, showing a very high sensitivity to water loss. With this technique, volumetric and selective heating at the crystal level has detected phase transformations with clear transitions as opposed to that observed with conventional heating methods, which rely on a heat transfer from the surface to the inner parts of the structure and lead to gradual transformations and the coexistence of different phases.
As a result, this technique has allowed the identification of a new intermediate phase (hydro γ-anhydrite) in the temperature range between 230 °C and 260 °C, characterized by uncompensated ionic charges inside the structural channels of the γ-anhydrite phase, which also supports the well-known solubility of this phase.
The dielectric properties of the mineral phases of thermally treated samples have also been quantified, providing information about the capacity of the material to absorb microwave energy in each phase. The results have demonstrated that microwave absorption by gypsum and β-anhydrite is particularly low compared to that in the transitional phases.
The information obtained by this technique has unveiled the mechanisms behind the microwave heating of the gypsum–anhydrite system and provides evidence of the influence of crystal water and of water channels in the crystal structure as a function of temperature.
Moreover, the capacity of the simultaneous application of optical (i.e., Raman spectroscopy) and other in situ techniques to detect microwave-matter interactions in this and other dielectric materials offers unique opportunities for further in-depth and thorough analytical studies of these interactions.
Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 26 Nov 2020, which contained errors where “anhydrite” had been changed to “anhydride” throughout during editing.References
- A. C. Metaxas and R. J. Meredith, Industrial microwave heating, Peregrinus, London, 1983 Search PubMed.
- R. R. Mishra and A. K. Sharma, Compos. Part. A Appl. Sci. Manuf., 2016, 81, 78 CrossRef CAS.
- J. D. Gutiérrez, J. M. Catalá-Civera, J. Bows and F. L. Peñaranda-Foix, J. Food Eng., 2017, 202, 1–8 CrossRef.
- S. W. Kingman and N. A. Rowson, Miner. Eng., 1998, 11, 1081 CrossRef CAS.
- S. W. Kingman, Int. Mater. Rev., 2006, 51, 1 CrossRef CAS.
- M. Lovás, M. Kováčová, G. Dimitrakis, S. Čuvanová, I. Znamenáčková and Š. Jakabský, Int. J. Heat Mass Transf., 2010, 53, 3387 CrossRef.
- S. M. J. Koleini and K. Barani in The development and application of microwave heating, ed. Wenbin C., IntechOpen, London, 2012, ch. 4, p. 79 Search PubMed.
- A. M. López-Buendía, B. García-Baños, J. Bastida, G. Llorens-Vallés, M. M. Urquiola and J. M. Catalá-Civera, presented at 3GCMEA, Cartagena, Spain, July 2016.
- J. J. Reinosa, B. García-Baños, J. M. Catalá-Civera, Á. M. López-Buendía, L. Guaita and J. F. Fernández, Int. J. Appl. Glass. Sci., 2019, 10, 208–219 CAS.
- J. J. Reinosa, B. García-Baños, J. M. Catalá-Civera and J. F. Fernández, Appl. Clay Sci., 2019, 168, 237–243 CrossRef CAS.
- H. J. Kitchen, S. R. Vallance, J. R. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114, 1170–1206 CrossRef CAS.
- J. Sun, W. Wang and Q. Yue, Materials, 2016, 9, 231 CrossRef.
- L. Wu, Y. Zhang, F. Wang, W. Ma, T. Xie and K. Huang, An On-Line System for High, Materials, 2019, 12(4), 665 CrossRef CAS.
- R. N. Clarke, A. P. Gregory, D. Cannell, M. Patrick, S. Wylie, I. Youngs and G. Hill, Technical Report, Institute of Measurement and Control/National Physical Laboratory, 2003.
- B. Garcia-Baños, J. M. Catalá-Civera, F. L. Peñaranda-Foix, P. Plaza-González and G. Llorens-Vallés, Materials, 2016, 9(5), 349 CrossRef.
- B. García-Baños, J. M. Catalá-Civera, J. R. Sánchez, L. Navarrete, A. M. López-Buendía and L. Schmidt, Metals, 2020, 10, 693 CrossRef.
- J. M. Catalá-Civera, A. J. Canós, P. Plaza-González, J. D. Gutierrez, B. Garcia-Baños and F. L. Peñaranda-Foix, IEEE Trans. Microwave Theory Tech., 2015, 63, 2905 Search PubMed.
- R. C. Hildyard, S. Llana-Funez, J. Wheeler, D. R. Faulkner and D. J. Prior, Electron Backscatter Diffraction (EBSD), J. Petrol., 2011, 52, 839 CrossRef CAS.
- A. Kasprzyk, J. Sed. Res., 1995, 65, 348 Search PubMed.
- S. Azam, Bull. Eng. Geol. Environ., 2007, 66, 177 CrossRef CAS.
- P. Tesárek, J. Drchalová, J. Kolísko, P. Rovnaníková and R. Černý, Constr. Build. Mater., 2007, 21, 1500 CrossRef.
- M. Singh and M. Garg, Cem. Concr. Res., 2000, 30, 571 CrossRef CAS.
- A. Charola, J. Pühringer and M. Steiger, Environ. Geol., 2007, 52, 339 CrossRef CAS.
- K. K. Kelley, C. T. Anderson and J. C. Southartd, USD Interior Tech, Paper 625.
- P. S. Valimbe and V. M. Malhotra, Fuel, 2002, 81, 1297 CrossRef CAS.
- G. Brune, AEG, 1965, 2, 26 Search PubMed.
- A. N. James and A. R. R. Lupton, Geotechnique, 1978, 28, 249 CrossRef CAS.
- S. Ko, D. L. Olgaard and U. Briegel, Geophys. Res. Lett., 1995, 22, 1009 CrossRef CAS.
- Anonymous Mineral Commodity Summaries 2019, Reston, VA, 2019.
- W. Chun, S. Kim and K. Lee, ‘EuroDrying2011’, 2011.
- D. Kasparaitė, L. Lukošenkinaitė, Z. Valančius, N. Kybartienė and V. Leškevičienė, Chem. Technol., 2013, 63, 1 Search PubMed.
- J. C. A. Boeyens and V. V. H. Ichharam, Z. Kristallogr. – New Cryst. Struct., 2002, 217, 9 CAS.
- P. F. Schofield, K. S. Knight and I. C. Stretton, Am. Mineral., 1996, 81, 847 CAS.
- H. Schmidt, I. Paschke, D. Freyer and W. Voigt, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 467 CrossRef CAS.
- C. Bezou, A. Nonat, J. Mutin, A. N. Christensen and M. S. Lehmann, J. Solid State Chem., 1995, 117, 165 CrossRef CAS.
- R. J. Hand, Br. Ceram. Trans., 1997, 96, 116 CAS.
- A. N. Christensen, M. Olesen, Y. Cerenius and T. R. Jensen, Chem. Mater., 2008, 20, 2124 CrossRef CAS.
- S. Follner, A. Wolter, A. Preusser, S. Indris, C. Silber and H. Follner, Cryst. Res. Technol., 2002, 37, 1075 CrossRef CAS.
- R. J. Morris, Nature, 1963, 198, 1298 CrossRef CAS.
- C. Vellmer, B. Middendorf and N. Singh, J. Therm. Anal. Calorim., 2006, 86, 721 CrossRef CAS.
- N. Prieto-Taboada, A. Larrañaga, O. Gómez-Laserna, I. Martínez-Arkarazo, M. A. Olazabal and J. M. Madariaga, Microchem. J., 2015, 122, 102 CrossRef CAS.
- E. Sipple, P. Bracconi, P. Dufour and J. Mutin, Solid State Ionics, 2001, 141, 447 CrossRef.
- M. Carbone, P. Ballirano and R. Caminiti, Eur. J. Mineral., 2008, 20, 621 CrossRef CAS.
- A. M. López-Buendía, C. Suesta, J. M. Cuevas, M. M. Urquiola and J. Bastida, presented at 12th EMABM, 2009, 443.
- Z. M. Zhao, C. J. Wang, C. Q. Li and X. M. Zhang, Appl. Mech. Mater., 2013, 368-370, 780 Search PubMed.
- O. W. Flörke, Neues. Jb. Miner. Abh., 1952, 4, 180 Search PubMed.
- A. Kirfel and G. Wil, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, B36, 2881 CrossRef CAS.
- L. Gracia, A. Beltrán, D. Errandonea and J. Andrés, Inorg. Chem., 2012, 51, 1751 CrossRef CAS.
- W. A. Crichton, J. B. Parise, S. M. Antao and A. Grzechnik, Am. Mineral., 2005, 90, 22 CrossRef CAS.
- F. Hanic, L. Galikova, J. Havlica, I. Kapralik and V. Ambruz, Trans. J. Br. Ceram. Soc., 1985, 84 Search PubMed.
- Anonymous Calcium sulfate MAK Value Documentation, 1991. The MAK-Collection for Occupational Health and Safety 2012.
- B. F. Pedersen and D. Semmingsen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1074 CrossRef.
- Á. G. De la Torre, M. López-Olmo, C. Álvarez-Rua, S. García-Granda and M. A. G. Aranda, Powder Diffr., 2004, 19, 240 CrossRef.
- P. Hartmann, Eur. J. Mineral., 1989, 1, 721 CrossRef.
- H. Morikawa, I. Minato, T. Tomita and S. Iwai, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2164 CrossRef.
- N. Prieto-Taboada, O. Gómez-Laserna, I. Martínez-Arkarazo, M. Á. Olazabal and J. M. Madariaga, Anal. Chem., 2014, 86, 10131 CrossRef CAS.
- S. M. Antao, Powder Diffr., 2011, 26, 326 CrossRef CAS.
- I. Y. Borg and D. K. Smith, Contrib. Mineral. Petrol., 1975, 50, 127 CrossRef CAS.
- A. Mukhopadhyay, W. T. S. Cole and R. J. Saykally, Chem. Phys. Lett., 2015, 633, 13e CrossRef.
- U. Kaatze, J. Phys. Chem. Ref. Data, 1989, 34, 371 CrossRef CAS.
- W. J. Ellison, J. Phys. Chem. Ref. Data, 2007, 36, 1 CrossRef CAS.
- L. A. Hardie, Am. Mineral., 1967, 52, 171 CAS.
- B. García-Baños, J. J. Reinosa, F. L. Peñaranda-Foix, J. F. Fernández and J. M. Catalá-Civera, Sci. Rep., 2019, 9, 10809 CrossRef.
- J. D. Gutiérrez-Cano, P. Plaza-González, A. J. Canós, B. García-Baños, J. M. Catalá-Civera and F. L. Peñaranda-Foix, IEEE Trans. Instrum. Meas., 2020, 69(6), 3595–3605 Search PubMed.
- B. J. Berenblut, P. Dawson and G. R. Wilxinson, Spectrochim. Acta, 1973, 29A, 29–36 CrossRef.
- B. Simon and M. Bienfait, Acta Crystallogr., 1965, 19, 750 CrossRef CAS.
- F. R. Massaro, M. Rubbo and D. Aquilano, Cryst. Growth Des., 2010, 10, 2870 CrossRef CAS.
- F. C. Hawthorne and R. B. Ferguson, Can. Mineral., 1975, 13, 289 Search PubMed.
- R. A. Khasanov, N. M. Nizamutdinov, N. M. Khasanova, A. T. Gubaĭdullin and V. M. Vinokurov, Crystallogr. Rep., 2008, 53, 806 CrossRef CAS.
- W. Smykatz-Kloss, K. Heide and W. Klinke in Handbook of Thermal Analysis and Calorimetry, ed. M. E. Brown and P. K. Gallagher, Elsevier, 2003, ch. 11, vol. 2, p. 451 Search PubMed.
- M. Carvalho, M. Leles and R. Tubino, J. Therm. Anal. Calorim., 2008, 91, 621 CrossRef CAS.
- B. Gacía-Baños, A. M. López-Buendía, C. Suesta, J. M. Catalá-Civera., J. Jiménez Reinosa and J. F. Fernández, 15th International Conference on Microwave and High Frequency Heating, 2015, Krakow (Poland), p. 107.
- N. M. Alford, J. Breeze, X. Wang, S. J. Penn, S. Dalla, S. J. Webb, N. Ljepojevic and X. Aupi, J. Eur. Ceram. Soc., 2001, 21, 2605 CrossRef CAS.
- J. Baker-Jarvis and S. Kim, J. Res. Natl. Inst. Stand. Technol., 2012, 117, 1 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04926c |
| This journal is © the Owner Societies 2020 |