
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing the weights in the molecular work-out of cis- and trans-formic acid: extension of the vibrational database via deuteration†
Arman
Nejad
 ,
Martin A.
Suhm
and
Katharina A. E.
Meyer
*
,
Martin A.
Suhm
and
Katharina A. E.
Meyer
*
Institute of Physical Chemistry, University of Göttingen, Tammannstr. 6, 37077 Göttingen, Germany. E-mail: katharina.meyer@chemie.uni-goettingen.de
First published on 10th November 2020
Abstract
The higher-energy cis- as well as the global minimum trans-rotamers of the four H/D isotopologues of the formic acid monomer have been examined with Raman jet spectroscopy extending the vibrational gas phase reference database by eleven new cis-band positions for HCOOD, DCOOH, and DCOOD. With these new additions, all O–H/D, C–H/D, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching as well as the O–D in-plane bending vibrations of these higher-energy rotamers are known in addition to the previously determined C–O stretch and OH torsion of cis-HCOOH. Further, a comparison of the vibrational spectra of all four H/D isotopologues of the globally stable trans-rotamer of formic acid is shown to be very helpful in revealing similarities and differences in these systems, particularly with regard to Fermi resonances. Amongst the most prominent ones is the ν5/2ν9 resonance doublet of trans-HCOOH, for which we provide more insight into a recently suggested label switch of the resonance partners via the comparison of infrared and Raman jet spectra.
O stretching as well as the O–D in-plane bending vibrations of these higher-energy rotamers are known in addition to the previously determined C–O stretch and OH torsion of cis-HCOOH. Further, a comparison of the vibrational spectra of all four H/D isotopologues of the globally stable trans-rotamer of formic acid is shown to be very helpful in revealing similarities and differences in these systems, particularly with regard to Fermi resonances. Amongst the most prominent ones is the ν5/2ν9 resonance doublet of trans-HCOOH, for which we provide more insight into a recently suggested label switch of the resonance partners via the comparison of infrared and Raman jet spectra.
1 Introduction
As an important contributor to atmospheric chemistry,1,2 the formic acid monomer, particularly its global minimum trans-conformation, has received much attention over the years both from the experimental3–10 as well as the theoretical11–13 side. Despite the low abundance of merely 0.1% at room temperature, the first gas phase detection of the higher-energy cis-conformer was achieved with microwave spectroscopy in 1976 by Hocking,14 enabled by the threefold larger dipole moment of cis- (3.79 D) compared to trans-HCOOH (1.42 D).15 The first vibrational characterisation of cis-formic acid was accomplished in 1997 for HCOOH in an argon matrix via OH overtone excitation from the global minimum trans-rotamer16 and was subsequently extended to DCOOH17 and HCOOD.18,19 For a direct comparison to theory,20 however, these values are not so practical, as the host–guest interaction needs to be captured on the theoretical side which can be challenging, as recently shown by Ito for the formic acid dimer.21 The first and until 2018 only cis-formic acid fundamental measured in the gas phase was the OH torsion connecting both minima22 – the vibrational mode with the largest (predicted) spectral separation between both conformers. Four new band positions of cis-HCOOH have been determined in 2018 and 2019 with Raman jet spectroscopy, utilising thermal excitation to increase the cis-formic acid abundance by an order of magnitude23,24 prior to the expansion. Here, we present the first perturbation-free band positions of partially and fully deuterated cis-formic acid, extending the available cis-database by more than 200% from five to sixteen fundamentals. In 2019, cis-formic acid has also been detected as a distinct species in solution,25 where it has a much higher abundance than in the gas phase. The O–D and CO stretches were shown to be higher in frequency than those in the trans-form, as observed here under vacuum isolation, but only in weakly hydrogen-bonded solvents. In water, the sequence inverts for the O–D stretch, underscoring the importance of reliable reference values in the gas phase for theory.
Despite the completion of the vibrational gas phase database for the trans-rotamers of the H/D isotopologues of formic acid (with the exception of ν8 of HCOOD), there is still ambiguity in the assignment of Fermi resonance pairs. One prominent example is the ν5/2ν9 Fermi resonance of trans-HCOOH. This resonance is part of a larger resonance polyad involving half a dozen of states26 and problems with the ν5 assignment were noted early on. In 2019, Hull et al. presented convincing experimental evidence that the overtone 2ν9 is – contrary to previous beliefs – lower in energy than the fundamental ν5,27 in agreement with recent high-level calculations.11,12
As we will highlight in this contribution, one of the most crucial and insightful aspects that improves our understanding of near-degeneracies in these systems is the comparison of vibrational spectra of all four H/D isotopologues. The work of Redington, who has analysed 24 isotopologues of the formic acid monomer in a neon matrix,28 is an impressive example of such rigorous comparison. Further, we showcase the indispensability of Raman spectroscopy for a thorough vibrational characterisation of the formic acid monomer, which is so far underrepresented4,5,23,24,29–31 in comparison to a wealth of infrared studies (see for example ref. 3, 6–10, 32–41 and references therein). We extend and update the significant Raman gas phase work on hydrogenated and deuterated formic acid by Bertie et al.,4,5 which was focussed on the characterisation of the dimer.
From a computational point of view, the small size of only five atoms and two conformational isomers (energy difference of 16.3(4) kJ mol−1 (ref. 14)) makes the formic acid monomer particularly suitable for benchmarking quantum chemical models. The availability of benchmarking data in higher-energy regimes of the potential energy hypersurface (PES), e.g., local minima, is especially important, as it enables the assessment of the globality of the PES description. The need for higher-energy reference data is illustrated by two recent high-level variational anharmonic calculations, namely vibrational configuration interaction (VCI)11 and multi-configuration time-dependent Hartree (MCTDH),12 where the mean absolute deviation (MAD) between both models is 4 cm−1 for the global minimum trans-, but nearly three times as large (12 cm−1) for the cis-conformer. For trans-HCOOH, all nine fundamentals were considered for this analysis, but for the higher-energy cis-rotamer only eight, as ν1 was not reported in ref. 11. Another excellent test to reveal weaknesses in theoretical models are near-degeneracies, as recently showcased for the glycolic acid monomer,42 and extended in this work to the trans-rotamer of the formic acid monomer. A full characterisation of the rotational and vibrational states of cis- and trans-formic acid, which contribute to their partition function, together with an independent experimental value of the equilibrium constant between the two species could provide a more accurate experimental value for the energy difference between the two species,43 which so far relies on a single microwave analysis.14
2 Experimental and computational methods
The Raman jet set-up used to record all spectra has been described in detail before.24,44,45 Briefly, formic acid was seeded into helium and expanded at different temperatures through a vertical slit nozzle at 0.5 bar into an evacuated jet chamber (background pressures of 1–2 mbar during the expansion). Before the expansion, the acid-in-helium mixture was further diluted with helium yielding acid concentrations of <0.2–0.4% (Table S1 in the ESI†). Both the nozzle and its feed-line are heatable, which can be exploited to enhance the relative population of higher-energy conformers before the expansion,24 while cluster formation is suppressed. To ensure stable conditions during the long exposures needed for the low spontaneous Raman scattering photon flux, the gas expansion is continuous (see ref. 45 for further details). A 25 W continuous-wave Spectra Physics Millennia eV 532 nm laser was used to probe the expansion. For the HCOOD measurements, a slightly lower laser power of 24 W was employed. The distance between the slit nozzle and the laser beam was set to 1 mm. The scattered light was collected perpendicular to the laser and to the nozzle flow via a camera lens and focussed onto a 1 m monochromator (McPherson) which disperses the photons onto 1340 pixel columns of a 1340 × 400 liquid nitrogen-cooled CCD-camera (Princeton Instruments, PyLoN 400B), that was operated in vertical binning mode (400 pixels). The combination of laser and monochromator results in a resolution of about 1.5–2.0 cm−1, depending on the spectral range. Therefore, we generously assign band centre errors of ±2 cm−1. Exposure times of 200–300 s were used per scan and typically 5–9 scans were co-added for each spectral window. To calibrate the raw spectra in the wavenumber domain, neon lines were measured and compared to the known vacuum transitions in the NIST database. Spikes due to cosmic rays were eliminated by comparing multiple exposures for the same pixel column.The FTIR jet spectra were recorded with a Bruker IFS 66v spectrometer equipped with a globar, a potassium bromide beam splitter, and potassium bromide optics. The modulated IR beam is gently focussed on the pulsed jet expansion from a 600 × 0.2 mm2 slit nozzle. Behind it, the beam is focussed onto a mercury cadmium telluride (MCT) detector. A comparison of the expansion conditions of the FTIR and Raman set-up can be found in ref. 46 and further details on the FTIR set-up in ref. 47.
Geometry optimisations and the calculation of harmonic vibrational frequencies, IR intensities, and Raman activities have been performed with Gaussian 09 Rev. E.01.48 Keyword specifications for all calculations are summarised in the ESI† (Table S2). From the computed Raman activity Ai, the Raman scattering cross-section σi was calculated as
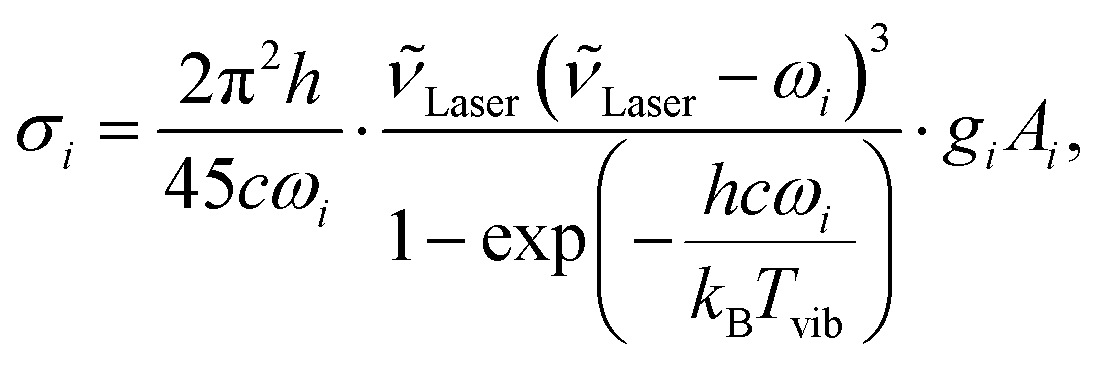 | (1) |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) Laser = (532 nm)−1 is the laser wavenumber, Tvib the vibrational temperature, ωi the harmonic wavenumber, gi the degeneracy of the vibration, and c the speed of light in vacuum. To roughly match the vibrational temperature in our jet, a temperature of 100 K was assumed in all our calculations.
Laser = (532 nm)−1 is the laser wavenumber, Tvib the vibrational temperature, ωi the harmonic wavenumber, gi the degeneracy of the vibration, and c the speed of light in vacuum. To roughly match the vibrational temperature in our jet, a temperature of 100 K was assumed in all our calculations.
For spectral assignments, the B3LYP functional49,50 was employed using two-body dispersion corrections (D3),51 Becke–Johnson damping,52 and the aug-cc-pVTZ basis set,53 hereafter denoted as aVTZ. All harmonic vibrational frequencies were scaled to the respective trans-formic acid band in each spectral window and in case of Fermi resonances, to the resonance centre ascertained from the overall scattering intensity. A list of the calculated harmonic vibrational frequencies, IR intensities, and Raman scattering cross-sections can be found in Table S3 in the ESI.†
In order to analyse harmonic mode mixing (see Section 3.3), additional harmonic vibrational frequency calculations were carried out at the PBE0-D3(BJ),54 B2PLYP-D3(BJ),55 HF, MP256 (all Gaussian 09 Rev. E.0148), and CCSD(T)57 levels (CFOUR version 158,59).
3 Results and discussion
3.1 cis-Formic acid fundamentals
To detect the cis-conformers of DCOOH, HCOOD, and DCOOD, Raman jet spectroscopy has been exploited in combination with thermal excitation between 100 and 190 °C utilising a heatable nozzle and feed-line, followed by a supersonic expansion to rapidly freeze out the enhanced cis-population24 of up to 1–2%. This technique is a variant of earlier trapping methods60 with the advantage that the trapped species are generated in a state which is easily accessible to quantum chemical modelling. By intensity-scaling the spectra of a temperature series to the respective trans-fundamental in each spectral region, cis-formic acid bands and non-isomeric hot bands, i.e., transitions from thermally populated states localised in trans-formic acid, can be easily distinguished from cold monomer and cluster bands, as they increase in intensity with temperature, whereas the cluster bands decrease. An illustrative example of this can be seen in the ν2 spectra of HCOOH in Fig. S3 in the ESI.†There are two measures to identify the molecular origin of a hot band – the band position difference to the trans-fundamental and the intensity. In the perturbational picture,61 the spectral shift between a fundamental νi and the hot band νi + νj − νj amounts to the anharmonic matrix element xij (2xii for 2νi − νi), which mediates binary coupling between two vibrational modes i and j (diagonal anharmonicity along mode i). The intensity of a non-isomeric hot band can be estimated from the expected Boltzmann population of that low-lying energy level j assuming similar Raman scattering cross-sections (see ref. 24 for further details). In case of an isomeric hot band, the expected population can be estimated from the energy difference between both conformers (1–2% at 190 °C24) and the difference in band position corresponds to the cis–trans-shift.
Fig. 1 shows the normalised relative Raman scattering cross-sections for all four H/D isotopologues and both rotamers of formic acid, indicating which vibrations are accessible with our experimental approach. For reasons of simplification and unification, we employ the Herzberg nomenclature of HCOOH for all isotopologues and rotamers (see Table S4 in the ESI† for comparison).
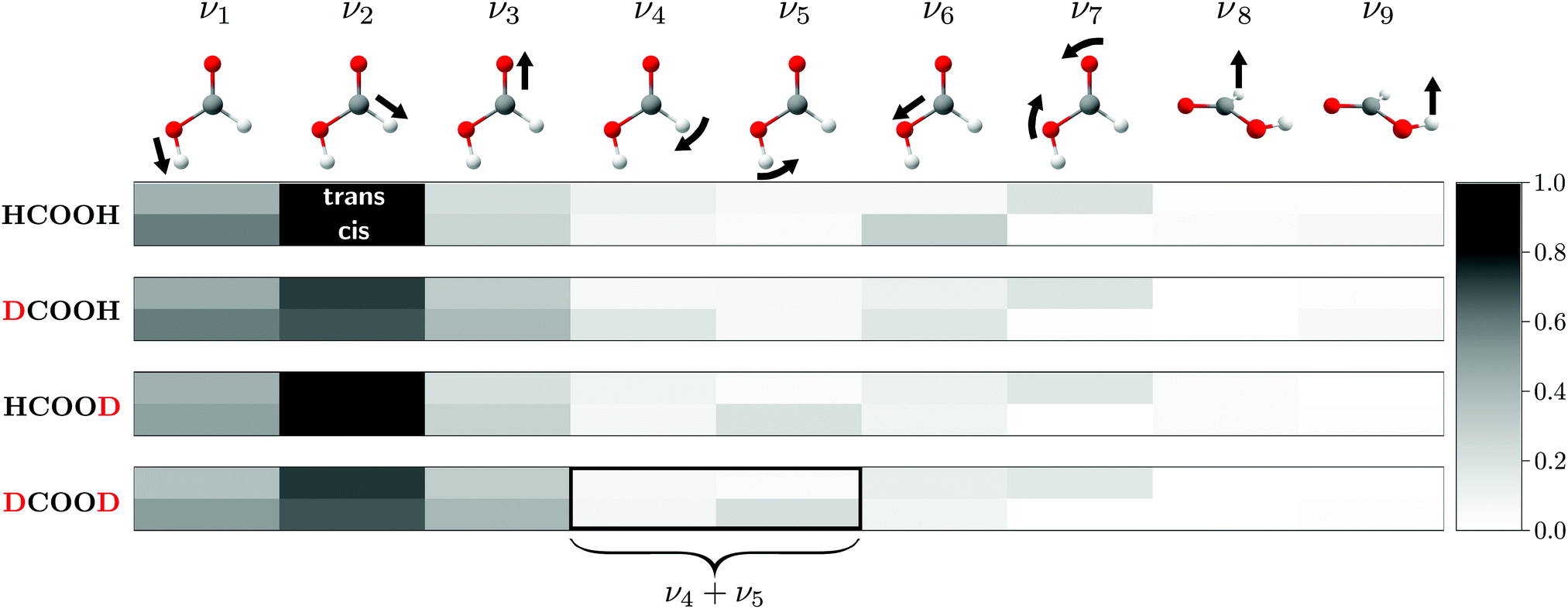 | ||
| Fig. 1 Harmonic (B3LYP-D3(BJ)/aVTZ) Raman scattering cross-sections of the formic acid monomer for both rotamers of all four H/D isotopologues, normalised to ν2 of HCOOD (scaling factor 175.9 × 10−36 m2 sr−1). A darker grey tone for the cis-rotamer indicates higher visibility than for the trans-rotamer, but the lower cis-abundance in the experiment must be kept in mind. The normal modes of cis-formic acid are visualised by arrows and labelled according to the Herzberg nomenclature for HCOOH, which is adopted for the deuterated isotopologues. For ν4 and ν5 of DCOOD, harmonic mode mixing into a symmetric and antisymmetric combination is predicted. See text for further details. | ||
As for HCOOH,24 the most Raman active modes of the deuterated isotopologues are the O–H/D, C–H/D, and CO stretches (ν1–ν3), for which all missing cis-fundamentals were determined in this work. We note that the assignment of ν2 of cis-DCOOH is somewhat tentative due to the prominent rovibrational and hot band structure of the respective trans-band (cf. Fig. S3 in the ESI†). The same applies to ν6 of cis-HCOOH.62 Due to the abundance disadvantage of cis-formic acid, most of the remaining cis-fundamentals are more difficult to access with our experimental approach. The notable exception is the O–D in-plane bending vibration ν5 of HCOOD and DCOOD. For other cis-fundamentals with seemingly high intensity such as ν4 and ν6 of DCOOH (cf.Fig. 1), spectral congestion due to an excessive hot band structure currently limits further conclusions. Guidance from theory would be particularly helpful for these spectral regions. ν8 and ν9 are close to the detection limit for the trans-rotamer, and below it for the cis-rotamer impurity.
A list of all available hydrogenated and newly determined deuterated (perturbation-free) cis-fundamentals of the formic acid monomer can be found in Table 1 alongside the corresponding trans-bands and high resolution literature values wherever available. A detailed view of the cis-formic acid spectra can be found in the ESI† (Fig. S2–S5). The agreement between the newly determined Raman jet and literature band positions for trans-formic acid is generally within the experimental uncertainty of our set-up (±2 cm−1, cf. Section 2). A preliminary Raman jet study from our laboratory31 generally agrees with the trans-formic acid results reported here, within the previous, somewhat larger calibration error and apart from a few assignments. For cis-formic acid, the vibrational reference database has been extended by eleven new band positions. With one gas phase band position22 and four Raman jet values from previous studies23,24 for cis-HCOOH, the total number of cis-formic acid fundamentals now amounts to sixteen.
| HCOOH | DCOOH | HCOOD | DCOOD | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ra. jet | Lit. | Ra. jet | Lit. | Ra. jet | Lit. | Ra. jet | Lit. | |||||
| a Ref. 23 and 24. b Ref. 22. c Ref. 6; the band listed for ν5 was originally assigned to 2ν9. d Ref. 40 and 63. e Ref. 10. f Ref. 9; the band was originally assigned to the fundamental ν5. g Ref. 64. h Ref. 8 and 39. i Ref. 5; the second band of the ν2 resonance doublet is reported at 2941.8 cm−1, but corresponds to an impurity of HCOOH in the spectra and is therefore not listed here. j Ref. 37. k Ref. 65. l Ref. 66. m Ref. 67. n Ref. 68. o Ref. 69. p Ref. 70, see also ref. 71 and 72. q Ref. 73. r Ref. 74. s Ref. 36. t Ref. 75. u Ref. 4. v Ref. 7. w Ref. 76. x Ref. 33. | ||||||||||||
| cis | ||||||||||||
| ν 1 | 3637a | 3635 | 2685 | 2685 | ||||||||
| ν 2 | 2873a | 2167 | 2871 | 2145 | ||||||||
| ν 3 | 1818a | 1790 | 1819 | 1789 | ||||||||
| ν 5 | 904 | 883 | ||||||||||
| ν 6 | 1093 | |||||||||||
| ν 9 | 493.42 | |||||||||||
| trans | ||||||||||||
| ν 1 |
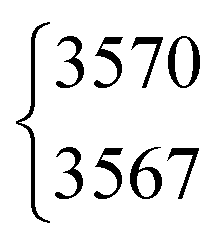
|
3570.5 | 3569 | 3566 | 2631 | 2631.64 | 2632 | 2631.87 | ||||
| ν 2 | 2942 | 2942.06 | 2219 | 2219.69 |
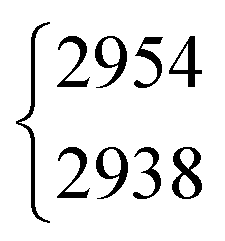
|
2938.2 |
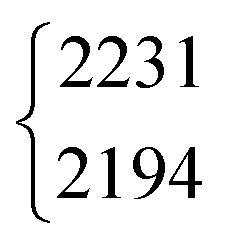
|
2231.8 | ||||
| 2195.1 | ||||||||||||
| ν 3 | 1776 | 1776.83 |
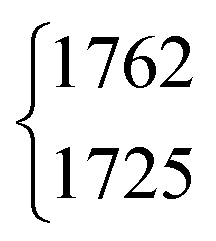
|
1762.9 | 1772 | 1772.12 |
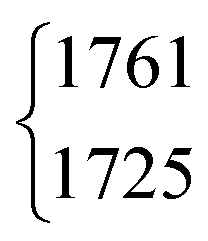
|
1760.0 | ||||
| 1725.87 | 1725.12 | |||||||||||
| ν 4 | 1379 | 1379.05 | 971 | 970.89 | 1365 | 1366.48 | 1039 | 1042 | ||||
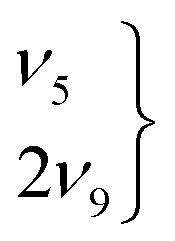
|
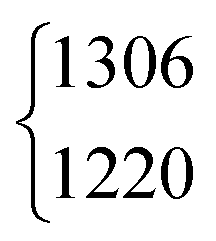
|
1306.2 |
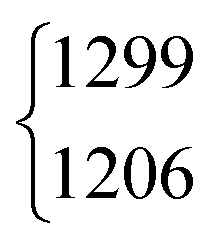
|
1297 |
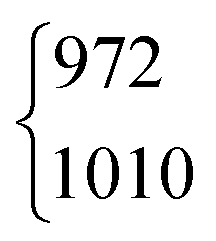
|
972.85 | 945 | 945.0 | ||||
| 1220.83 | 1011.68 | |||||||||||
| ν 6 | 1104 | 1104.85 | 1142 | 1142.31 | 1176 | 1177.09 | 1170 | 1170.80 | ||||
| ν 7 | 626 | 626.17 | 620 | 620.57 | 558 | 558.27 | 554 | 554.44 | ||||
| ν 8 | 1033.47 | 873.39 | 873.2 | |||||||||
| ν 9 | 640.73 | 631.54 | 508.13 | 492.23 | ||||||||
A recent example of a high-level variational anharmonic ab initio study on the formic acid monomer is an MCTDH study by Aerts et al. from 202013 who have characterised the cis- and trans-conformers of all three deuterated isotopologues. Due to the lack of environment-free experimental data on the higher-energy structure, the accuracy of their description could solely be evaluated for the global minimum trans-form. The new cis-formic acid band positions reported in this work also facilitate a performance evaluation for the higher-energy rotamer. For most modes, the deviations are below 2–3 cm−1. The largest band position discrepancy is observed for ν1 of cis-DCOOH and amounts to 10 cm−1 followed by 9, 4, and 7 cm−1 for ν3 of cis-DCOOH, cis-HCOOD and cis-DCOOD, respectively.
3.2 Comparison of the Raman spectra of all four H/D isotopologues
An overview of the Raman spectra of the four H/D (trans-)formic acid isotopologues is shown in Fig. 2 for a nozzle temperature of 160 °C – a compromise between the absence of cluster signals and signal intensity. Fermi resonance doublets of the trans-rotamers are indicated by brackets. Hot bands are marked by ‘h’ and impurities due to H/D exchange reactions and/or the manufacturing process by a double dagger. The maximum of such impurities observed in the spectra amounts to 6(2)% (see Table S1 in the ESI† for further details). To visualise and systematically analyse the spectral similarities and differences observed for the four H/D isotopologues, the cis–trans-shift is plotted against the absolute cis-band position for the five vibrations characterised with Raman jet spectroscopy and the sixth vibrational mode obtained from high resolution gas phase studies by Baskakov et al.8,22 in Fig. 3. In case of Fermi resonances of the respective trans-band, the band position of both resonance partners is shown and the symbol size reflects their relative intensity. For cis-formic acid, no signs of resonances were observed in our spectra, but this might be attributed to the relatively low intensity governed by the low abundance in the expansion.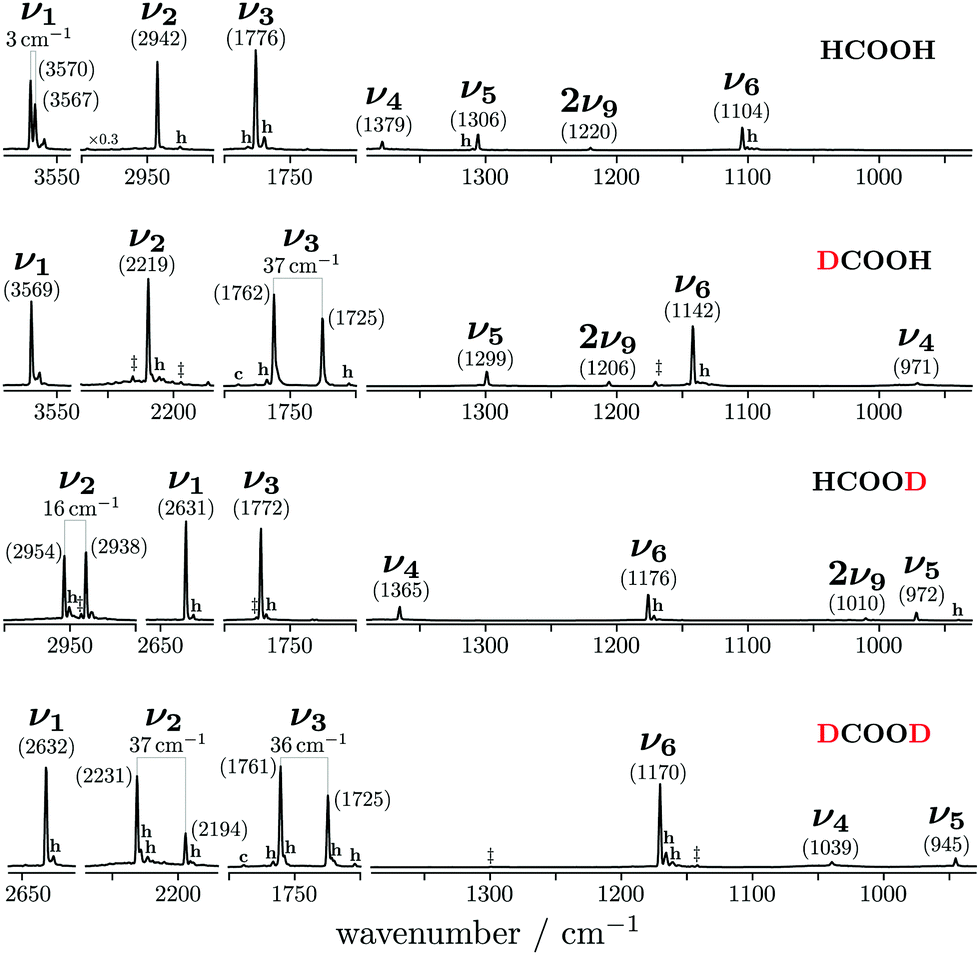 | ||
| Fig. 2 Overview of the Raman jet spectra of the O–H/D, C–H/D, CO, C–O stretching as well as C–H/D and O–H/D in-plane bending vibrations of HCOOH, DCOOH, HCOOD, and DCOOD recorded at a nozzle temperature of 160 °C. Bands of pronounced resonance doublets between fundamentals νi and combination or overtone bands are indicated by brackets and H, D impurities by double daggers. Non-isomeric hot and cis-rotamer bands are marked ‘h’ and ‘c’, respectively. | ||
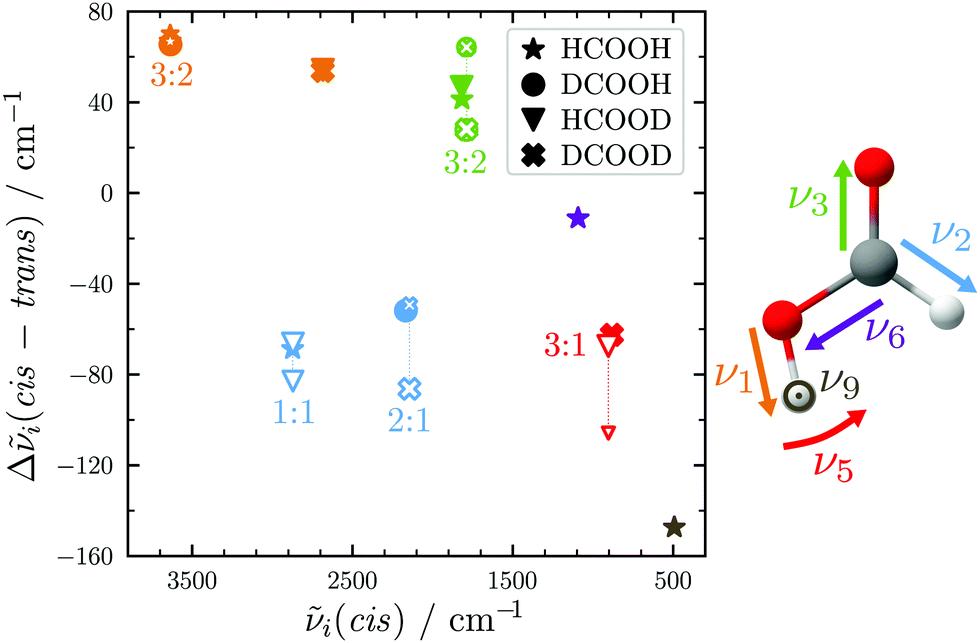 | ||
| Fig. 3 cis–trans vibrational band position shifts plotted against the cis-band positions for all H/D isotopologues of the formic acid monomer. The colour illustrates the type of vibration. For Fermi resonances, unfilled symbols are used and the band positions of both resonance partners are connected by dotted lines with the symbol size representing their relative intensity. | ||
The absolute band positions as well as cis–trans-shifts of the O–H and O–D stretching vibrations are insensitive to C–H isotope exchange (Fig. 3). However, next to the OH stretching band of trans-HCOOH (3570 cm−1), one fairly strong band with an intensity ratio of one third of ν1 can be seen at 3567 cm−1 in addition to a smaller third band at 3559 cm−1 (Fig. 2). Since the band position difference between the fundamental and the second band is very small, this does not affect the cis–trans-shift significantly (cf.Fig. 3). A similar ν1-triad of trans-HCOOH has also been observed in helium nanodroplets,38 which the authors attributed to Fermi and Coriolis resonances. For a full understanding of the OH stretching dynamics in formic acid, a detailed characterisation of skeletal modes and their associated coupling pathways is required.77
The CO stretch ν3 of trans-DCOOH and -DCOOD has a pronounced resonance (intensity ratio 3(0.5)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) with the C–D out-of-plane bending vibration 2ν8 (Fig. 2).65 The cis–trans-shift is again very similar for all isotopologues if one compares it against the resonance centre (Fig. 3).
:2) with the C–D out-of-plane bending vibration 2ν8 (Fig. 2).65 The cis–trans-shift is again very similar for all isotopologues if one compares it against the resonance centre (Fig. 3).
The situation becomes different for the C–H/D stretching vibration ν2. For trans-HCOOD, Bertie et al. reported a resonance with the (ν3 + ν6) combination band, yet assigned it to an impurity of HCOOH (2941.8 cm−1) in their spectra.5 We do, however, observe a resonance doublet with an intensity ratio close to 1:1 and an experimental splitting of 16 cm−1. The second band of the resonance doublet at 2954 cm−1 (Table 1) is likely overlayed by a dimer band in the spectra of Bertie et al. which they report at 2951.4 cm−1. The clear distinction between monomeric and dimeric contributions in the spectra via the temperature series is one of the advantages of the new Raman spectra reported in this work. Comparing against the resonance centre, the C–H cis–trans-shift is insensitive to O–H deuteration (Fig. 3). The same applies to the respective absolute cis-band position. The larger difference between the cis–trans-shifts of the C–D stretching vibrations (Fig. 3) can at least partially be ascribed to the Fermi resonance between ν2 and the (ν4 + ν6) combination band of trans-DCOOD.4 Interestingly, the isotope effect on the cis-C–D stretch is with 22 cm−1 much larger than for the C–H, O–D/H stretches, where these differences only amount to ≤2 cm−1 (cf.Table 1). This anomaly could be a result of an anharmonic perturbation that only occurs in one O–H/D isotopologue of the cis-rotamer, though this remains speculative due to the low intensity of the cis-contributions in our spectra. Another reason for this larger difference could be a misassignment of one of the two cis-ν2 bands, most likely that of cis-DCOOH due to the spectral congestion governed by the rovibrational structure of the trans-band (cf. Fig. S3 in the ESI†). However, the good agreement of all cis-ν2 bands with the high-level prediction of Aerts et al.13 (deviations below 3 cm−1) does not support this conjecture. Besides, close to the cis-band position of cis-DCOOD (2145 cm−1), there are no other hot bands in the DCOOH spectrum and vice versa (Fig. S3 in the ESI†).
Similar observations with regard to the absolute cis-band position as well as Fermi resonance governed difference in cis–trans-shift (cf.Table 1 and Fig. 3) apply to the O–D in-plane bending vibration ν5. However, there is another factor that sets the trans- (and cis-)DCOOD bending vibration apart from that of the other H/D isotopologues, which can be understood by taking a closer look at the ν5/2ν9 Fermi resonance across all H/D isotopologues.
3.3 The ν5/2ν9 Fermi resonance of trans-formic acid
The ν5/2ν9 Fermi resonance is amongst the most prominent ones found for trans-HCOOH and involves the O–H in-plane bend ν5 and the overtone of the large-amplitude O–H torsion 2ν9 (Fig. 1). In a high resolution study of the respective hot bands in 2019, Hull et al. showed that the labels of the two resonance partners at 1306 cm−1 and at 1220 cm−1 need to be switched, as the overtone was shown to be lower in energy than the fundamental.27 This Fermi resonance is also present in trans-DCOOH as well as in trans-HCOOD but seemingly absent in trans-DCOOD. For the cis-conformers, this Fermi resonance is not predicted,11–13 which makes a combination of both species valuable for vibrational benchmarking.A more detailed view of the Raman spectra of this resonance for all four isotopologues can be found in Fig. 4, which facilitates new insight into the strength of the resonance as well as into the suggested label switch for trans-HCOOH. Interestingly, the higher-energy band of trans-HCOOH at 1306 cm−1 is about seven times more intense than the band at 1220 cm−1, though the latter was previously assigned to the fundamental. The infrared spectra of HCOOH shown alongside the corresponding Raman spectra in Fig. 5 indicate an inverse situation where the lower energy band at 1220 cm−1 is more intense and the 1306 cm−1 band is barely visible at the employed conditions. This explains why based on solely the infrared spectra, the more intense band at 1220 cm−1 was previously assigned to the fundamental transition. A differing intensity ratio of a Fermi resonance in infrared and Raman spectra is rather unexpected, as usually the overtone (or combination band) is ‘dark’, meaning that it obtains intensity primarily via the anharmonic resonance with the ‘bright’ fundamental. As such, this infrared/Raman intensity difference implies that the overall comparably low-intense fundamental ν5 (cf.Fig. 1 for relative Raman scattering cross-sections and Table S3 in the ESI† for the predicted IR intensities) ‘steals’ intensity from the brighter overtone in one of the spectra and the dark overtone ‘steals’ intensity from the brighter fundamental in the other. Considering that Raman scattering cross-sections of overtones (or combination bands) are typically about two orders of magnitude lower than those of fundamentals,78 whereas this difference typically amounts to about one order of magnitude in the infrared, it is more plausible that the infrared ν9 overtone is brighter than the ν5 fundamental. This is in line with the suggested label switch of the resonance partners by Hull et al.,27 which was also proposed in the VCI and MCTDH studies from 201611 and 2018.12
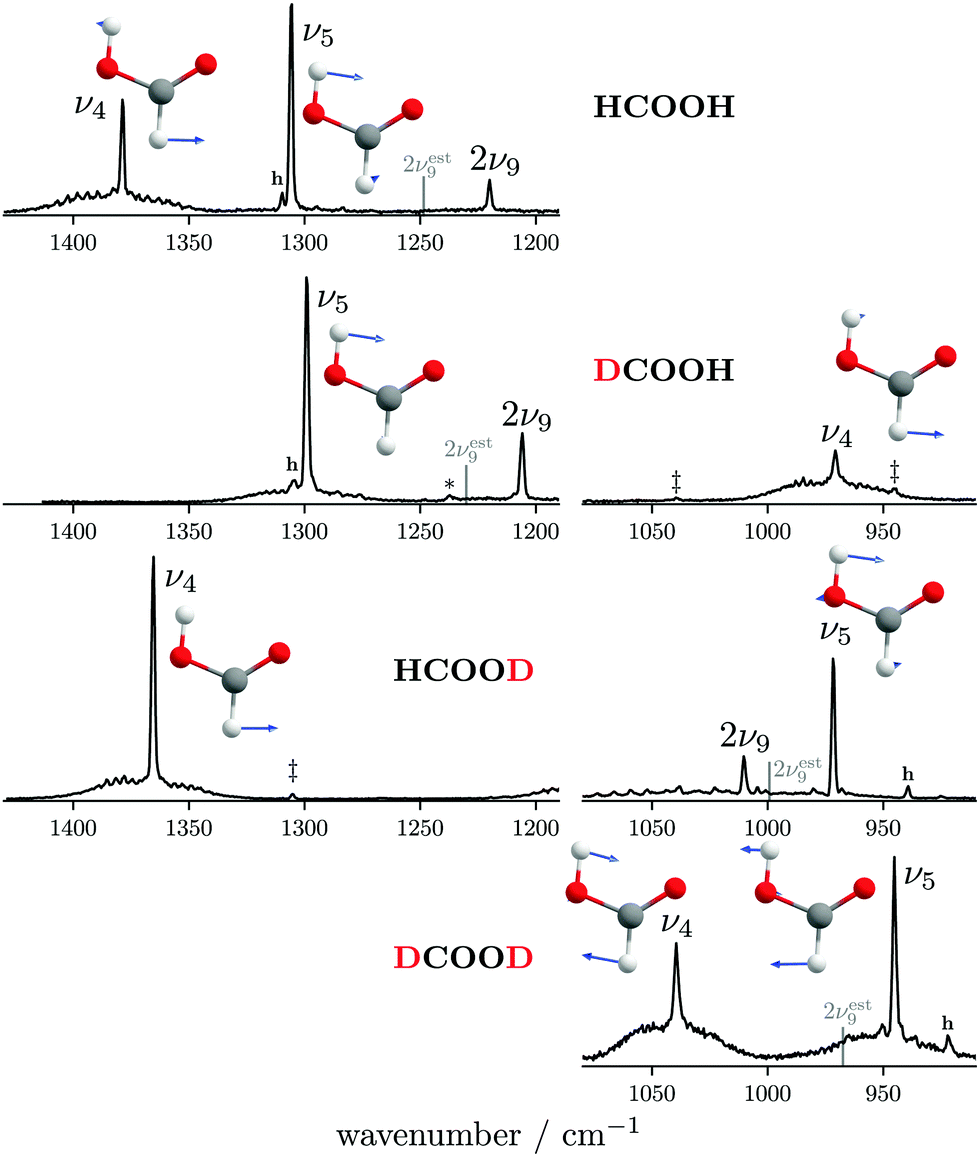 | ||
| Fig. 4 Raman jet spectra of the C–H/D in-plane bending vibration ν4 and the O–H/D ν5/2ν9 Fermi resonance of all four H/D isotopologues of the formic acid monomer recorded at a nozzle temperature of 160 °C. Cluster bands are marked with an asterisk, hot bands with ‘h’, and H, D impurities by double daggers. The band position of the overtone 2ν9 has been estimated from twice the experimental band position of ν9 (see Table 1 for band positions) by subtracting twice the diagonal anharmonicity matrix element reported in ref. 27 and is shown by a grey line. The normal modes of ν5 and ν4 are shown as an inset. Additional experimental details can be found in Section 2 and in the ESI.† | ||
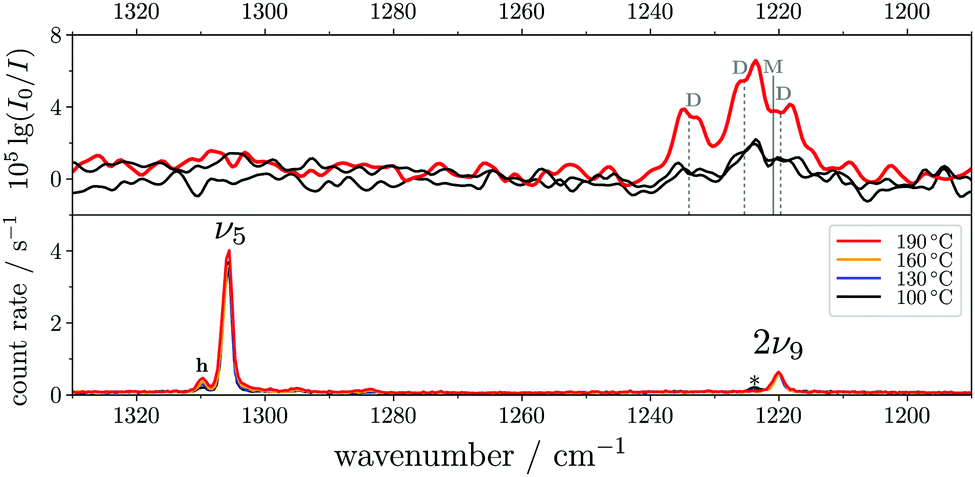 | ||
| Fig. 5 FTIR (top) and Raman (bottom) jet spectra of trans-HCOOH in the O–H in-plane bending region (ν5). The FTIR spectra have been recorded at increasing concentrations of <0.01–0.05% in helium at a reservoir pressure of 560 mbar with 1750–2130 co-added scans. The Raman spectra (<0.2% in helium, reservoir pressure 500 mbar, recording time 6 × 300 s) have been intensity-scaled to the ν6 band (not shown) of trans-HCOOH with the lowest intensity amongst the four nozzle temperatures (100–190 °C). Assignments of monomer (M) and dimer (D) contributions to the band between 1240–1210 cm−1 in the FTIR spectra were taken from ref. 9. Clusters in the Raman spectrum are marked with an asterisk. | ||
As aforementioned, the ν5/2ν9 Fermi resonance is also present in DCOOH with a slightly larger splitting (93 cm−1versus 86 cm−1 for HCOOH) and a similar intensity ratio (7(2):1). In case of DCOOH, Bertie et al.5 correctly assigned the band at 1299 cm−1 (1297 cm−1 in ref. 5) to the O–H in-plane bending vibration ν5, whereas the corresponding band of HCOOH at 1306 cm−1 (1307 cm−1 in ref. 4) was assigned to 2ν9 in their publication.4 For the O-deuterated isotopologues, the ν5/2ν9 Fermi resonance is only observed for HCOOD, where the ν9 overtone is higher in energy (1011.68 cm−1) than the ν5 fundamental (972.85 cm−1).73 The intensity ratio and splitting between both bands is distinctly smaller (1(1):3 versus 7(2):1 and 38 instead of 86/93 cm−1), which is consistent with a weakening of the resonance for the smaller OD amplitudes.
The absence or at least pronounced weakness of the ν5/2ν9 resonance in trans-DCOOD (cf. expected band position of 2ν9 in Fig. 4 which has been estimated from 2 × (ν9) assuming an anharmonic correction of twice the anharmonicity matrix element x99 as reported in ref. 27) can be understood in the comprehensive analysis of all four isotopologues. Fig. 2 shows that the C–D bend of DCOOH and O–D bend of HCOOD, which are estimates for the expected band positions of ν4 and ν5 in trans-DCOOD, are nearly isoenergetic at 971/972 cm−1. In the DCOOD spectrum, they are shifted up (1039 cm−1) and down (945 cm−1) in energy, indicating harmonic mixing due to near-degeneracies. This mixing is further supported by the unusually low ratio of the Q branch with respect to the rotational contour found for ν5 of trans-DCOOD (a feature of ν4, cf.Fig. 4), which might result from substantial mixing with ν4. Pointing in that same direction are different harmonic frequency calculations that unanimously predict a mixing into a symmetric and an antisymmetric combination for cis- and trans-DCOOD (HF, MP2, CCSD(T), and DFT, all with an aVTZ basis set, cf. inset in Fig. 4 for normal modes of the trans-rotamers).
For a closer scrutiny of this mixing across the four H/D isotopologues, harmonic frequencies of trans-formic acid were scanned for C–H proton masses between 1 and 2m(1H). The harmonic wavenumbers of ν4, ν5, ν6, 2ν7, and 2ν9 (all A′ symmetry) for both possible scans (trans-HCOOH → trans-DCOOH, trans-HCOOD → trans-DCOOD) are plotted for B3LYP-D3(BJ)/aVTZ in Fig. 6. Strong mixing between ν4, ν5, and ν6 is observed in these mass-scans with avoided crossings on the order of 60–110 cm−1. Accidentally, however, an avoided crossing of ν4 and ν534 coincides with an integer (even) mass in one case, i.e., DCOOD. Additional scans at other levels of theory (Fig. S1 in the ESI†) show the same qualitative behaviour. As such, the potential absence of the ν5/2ν9 Fermi resonance in DCOOD is in part a coincidence of an avoided crossing that detunes two otherwise moderately resonant states. This alone, however, does not explain the absence of the overtone 2ν9 in the DCOOD spectra entirely, as 2ν9 could gain in intensity via coupling to ν4 which exhibits ν5 character. The 2ν9 band of DCOOD might not gain sufficient intensity in this coupling triad to be observed under the employed experimental conditions. A comparison of measurements with perpendicular and parallel laser polarisation78 can be used to reduce the rotational contour via subtraction. This depolarised spectrum of DCOOD (cf. Fig. S5 in the ESI†) shows that no distinct 2ν9 band is hidden under the rotational contour of ν5 of DCOOD in our spectra, providing additional affirmation of its weakness.
 | ||
| Fig. 6 Harmonic wavenumbers (in cm−1, at the B3LYP-D3(BJ)/aVTZ level) of A′ symmetric fundamentals and overtones in the spectral windows between 900 and 1500 cm−1 as a function of the relative C–H proton mass of trans-formic acid (left O–H, right O–D). Grey bars indicate fractional masses which equal the mass of hydrogen or deuterium. Harmonically avoided crossing due to mode-mixing is indicated by a red disk in the interaction region, in which twice the coupling constant 2W is printed in bold letters. Green disks highlight the Fermi resonance-coupled states ν5 and 2ν9 in trans-HCOOH, -DCOOH, and -HCOOD. | ||
Overall, this harmonic mode mixing could be another reason for the differences observed for the cis–trans-shifts as well as cis-band positions (mixing is also predicted for cis-DCOOD) of HCOOD and DCOOD (Fig. 3). Subtle mass changes such as 13C isotopic substitution28 might allow for more insight, though this is experimentally too elaborate without further theoretical support.
3.4 Fermi resonance analysis for the H/D isotopologues of trans-formic acid
For vibrational benchmarking of near-degeneracies, not only the energetic order of the two interacting states needs to be predicted correctly by a quantum chemical model, but also their energy difference (spectral splitting |Δ|) as well as the spectral intensity ratio R (here Ihigh/Ilow). From these two quantities, the Fermi resonance coupling constant |WexpFermi| can be calculated from eqn (2) under the assumption that the dark state has a negligible intensity,79 which allows for an estimate of the strength of the resonance.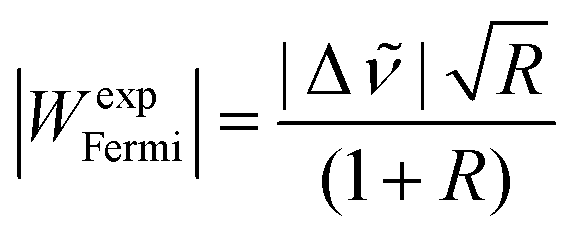 | (2) |
These effective values |WexpFermi| for all trans-formic acid Fermi resonances observed in this work are listed in Table 2 alongside model values |WcalcFermi| calculated from a quartic force field based on the PES of Tew and Mizukami11 (cf. ESI† for further details). For ν2 of HCOOD, only the Q branches of the vibrational bands were integrated to determine |WexpFermi| (cf. Fig. S3 in the ESI†). The neglect of the overlapping rotational contours for ν2/(ν3 + ν6) of HCOOD should not impact |WexpFermi| much, as the two resonance partners are nearly equal in intensity (cf.Fig. 2 and Table 2), so that their contributions to the rotational contour should also be similar. For ν5/2ν9 of HCOOD, there seems to be a broad rotational substructure which cannot be easily disentangled. Therefore, additional depolarisation measurements were performed (Fig. S5 in the ESI†) and |WexpFermi| was determined from these spectra.
| System | ν i |
high
|
low
|
I high | I low | |WFermi| | |
|---|---|---|---|---|---|---|---|
| Exp | Calc | ||||||
| a Only Q branches (without additional substructure) were integrated to determine |WexpFermi|. b Determined from a depolarised spectrum (see bottom of Fig. S5 in the ESI). | |||||||
| HCOOH | ν 1 | 3570 | 3567 | 3(0.5) | 2 | 2(2) | 0.3 |
| HCOOD | ν 2 | 2954 | 2938 | 1(0.5) | 1 | 8(2)a | 8.7 |
| DCOOD | ν 2 | 2231 | 2194 | 2(1) | 1 | 17(2) | 18.9 |
| DCOOH | ν 3 | 1762 | 1725 | 3(0.5) | 2 | 18(2) | 18.3 |
| DCOOD | ν 3 | 1761 | 1725 | 3(0.5) | 2 | 18(2) | 18.1 |
| HCOOH | ν 5 | 1306 | 1220 | 7(2) | 1 | 28(4) | 39.6 |
| DCOOH | ν 5 | 1299 | 1206 | 7(2) | 1 | 31(4) | 42.7 |
| HCOOD | ν 5 | 1010 | 972 | 1(1) | 3 | 17(2)b | 16.8 |
| DCOOD | ν 5 | 945 | 3.3 | ||||
As expected from the small spectral separation Δ = 3 cm−1, the Fermi resonance of ν1 of HCOOH is the weakest observed in this work with a coupling matrix element |WexpFermi| of solely 2(2) cm−1. It agrees with the very small predicted value |WcalcFermi| between ν1 and the (ν2 + ν7) combination band, although the third band in the Raman spectra and the resonance triad observed in helium nanodroplets38 indicate a more complex interaction than a simple two level resonance. Also rather weak is the ν2 Fermi resonance of HCOOD with 8(2) cm−1. Again, |WcalcFermi| and |WexpFermi| match rather well, which supports the assignment by Bertie et al.5 who ascribed the resonance partner to the (ν3 + ν6) combination band. The strength of the ν3/2ν8 Fermi resonance observed for the C-deuterated isotopologues is near-identical and accidentally very similar to that of ν2 of DCOOD (cf.Table 2). For all three resonances, the experimental and predicted Fermi coupling matrix elements agree within the experimental error bars.
For the ν5/2ν9 resonance doublet, a distinct strength variation is observed across the four isotopologues – it is largest for DCOOH (31(4) cm−1), decreases from HCOOH (28(4) cm−1) to HCOOD (17(2) cm−1), and remains undetected for DCOOD (cf.Fig. 4). As for most other resonance doublets, |WcalcFermi| of HCOOD matches the experimental value within the error bars. The calculated coupling constants |WcalcFermi| of HCOOH and DCOOH are 12 cm−1 larger than the experimental values. These findings seem to contradict the assumption of a completely dark state in case of 2ν9 of the two O–H isotopologues (cf.eqn (2)). Further investigation of this might be worthwhile. Overall, this is just one of many examples where anharmonic Raman intensities could prove to be very helpful.
4 Conclusions
After over eighty years of experimental research since the first publication on the formic acid monomer (and dimer) by Bonner and Hofstadter in 1938,32 the vibrational spectra of the simplest carboxylic acid are still not yet fully understood. The most notable missing vibrational information concerns its higher-energy cis-conformation, particularly that of the deuterated isotopologues of formic acid, where no perturbation-free band positions were known prior to this work. Recently, Raman jet spectroscopy in combination with thermal excitation was used to increase the number of cis-HCOOH fundamentals from one to five.24 In this work, we have extended this technique to cis-HCOOD, -DCOOH, and -DCOOD and were able to assign the bands of all O–H/D, C–H/D, CO stretching as well as those of the two O–D in-plane bending vibrations.
For the trans-formic acid monomer, although all but one fundamental vibration of the H/D isotopologues have already been determined, there is still ambiguity concerning assignments of overtones, combination bands, and Fermi resonance partners.11,12 A comparison of the latter amongst all four isotopologues has proven to be very insightful, particularly for the ν5/2ν9 resonance. This resonance is found to be weakest for HCOOD and appears to be absent in DCOOD. For HCOOH and DCOOH, similar coupling strengths are predicted, though from our Raman spectra we obtain smaller experimental coupling constants for both. This discrepancy between theory and experiment hints at a more complicated resonance mixing than that of a ‘bright’ fundamental interacting with a ‘dark’ overtone. The reason for the absence of this Fermi resonance in DCOOD might be harmonic mode mixing between ν4 and ν5 that detunes the resonance, yet for exact conclusions, further theoretical investigation is necessary.
Non-isomeric hot bands (see for example Fig. S2–S5 in the ESI†) are another valuable benchmarking target in addition to the cis-fundamentals and the Fermi resonance coupling matrix elements |WFermi|, as these facilitate an analysis of weaker anharmonicity signatures xij.24,44 For a thorough analysis of the large number of these hot bands observed in our spectra, a close collaboration between theory and experiment is vital. This represents one of the future directions of this work. The wealth of isomerically hot, yet rotationally cold formic acid monomer data provided in this contribution significantly advances its standing as a benchmarking reference system and hopefully triggers further theoretical investigation on this system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455 and 405832858. The authors thank M. Schwarzer for his help with early DCOOD measurements and Z. Xue for a preliminary Raman jet study of the trans-formic acid spectra.31 A. Nejad thanks the Fonds der Chemischen Industrie (FCI) for an attractive scholarship.Notes and references
- R. Zander, P. Duchatelet, E. Mahieu, P. Demoulin, G. Roland, C. Servais, J. V. Auwera, A. Perrin, C. P. Rinsland and P. J. Crutzen, Atmos. Chem. Phys., 2010, 10, 10047–10065 CrossRef CAS.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef CAS.
- I. C. Hisatsune and J. Heicklen, Can. J. Spectrosc., 1973, 18, 135–142 CAS.
- J. E. Bertie and K. H. Michaelian, J. Chem. Phys., 1982, 76, 886–894 CrossRef CAS.
- J. E. Bertie, K. H. Michaelian, H. H. Eysel and D. Hager, J. Chem. Phys., 1986, 85, 4779–4789 CrossRef CAS.
- M. Freytes, D. Hurtmans, S. Kassi, J. Liévin, J. Vander Auwera, A. Campargue and M. Herman, Chem. Phys., 2002, 283, 47–61 CrossRef CAS.
- F. Madeja, A. Hecker, S. Ebbinghaus and M. Havenith, Spectrochim. Acta, Part A, 2003, 59, 1773–1782 CrossRef.
- O. I. Baskakov, E. A. Alekseev, R. A. Motiyenko, J. Lohilahti, V.-M. Horneman, S. Alanko, B. P. Winnewisser, I. R. Medvedev and F. C. De Lucia, J. Mol. Spectrosc., 2006, 240, 188–201 CrossRef CAS.
- K. G. Goroya, Y. Zhu, P. Sun and C. Duan, J. Chem. Phys., 2014, 140, 164311 CrossRef.
- W. Luo, Y. Zhang, W. Li and C. Duan, J. Mol. Spectrosc., 2017, 334, 22–25 CrossRef CAS.
- D. P. Tew and W. Mizukami, J. Phys. Chem. A, 2016, 120, 9815–9828 CrossRef CAS.
- F. Richter and P. Carbonnière, J. Chem. Phys., 2018, 148, 064303 CrossRef.
- A. Aerts, P. Carbonnière, F. Richter and A. Brown, J. Chem. Phys., 2020, 152, 024305 CrossRef CAS.
- W. H. Hocking, Z. Naturforsch., A, 1976, 31, 1113–1121 Search PubMed.
- M. Winnewisser, B. P. Winnewisser, M. Stein, M. Birk, G. Wagner, G. Winnewisser, K. M. T. Yamada, S. P. Belov and O. I. Baskakov, J. Mol. Spectrosc., 2002, 216, 259–265 CrossRef CAS.
- M. Pettersson, J. Lundell, L. Khriachtchev and M. Räsänen, J. Am. Chem. Soc., 1997, 119, 11715–11716 CrossRef CAS.
- E. M. S. Maçôas, J. Lundell, M. Pettersson, L. Khriachtchev, R. Fausto and M. Räsänen, J. Mol. Spectrosc., 2003, 219, 70–80 CrossRef.
- A. Domanskaya, K. Marushkevich, L. Khriachtchev and M. Räsänen, J. Chem. Phys., 2009, 130, 154509 CrossRef.
- L. O. Paulson, D. T. Anderson, J. Lundell, K. Marushkevich, M. Melavuori and L. Khriachtchev, J. Phys. Chem. A, 2011, 115, 13346–13355 CrossRef CAS.
- E. L. Sibert III, J. Chem. Phys., 2019, 150, 090901 CrossRef.
- F. Ito, Comput. Theor. Chem., 2019, 1161, 18–25 CrossRef CAS.
- O. I. Baskakov, V.-M. Horneman, J. Lohilahti and S. Alanko, J. Mol. Struct., 2006, 795, 49–53 CrossRef CAS.
- K. A. E. Meyer and M. A. Suhm, J. Chem. Phys., 2018, 149, 104307 CrossRef.
- K. A. E. Meyer and M. A. Suhm, Chem. Sci., 2019, 10, 6285–6294 RSC.
- G. Giubertoni, O. O. Sofronov and H. J. Bakker, J. Phys. Chem. Lett., 2019, 10, 3217–3222 CrossRef CAS.
- J. Demaison, M. Herman and J. Liévin, J. Chem. Phys., 2007, 126, 164305 CrossRef CAS.
- K. Hull, T. Wells, B. E. Billinghurst, H. Bunn and P. L. Raston, AIP Adv., 2019, 9, 015021 CrossRef.
- R. L. Redington, J. Mol. Spectrosc., 1977, 65, 171–189 CrossRef CAS.
- L. G. Bonner and J. S. Kirby-Smith, Phys. Rev., 1940, 57, 1078 CrossRef CAS.
- A. Olbert-Majkut, J. Ahokas, J. Lundell and M. Pettersson, Chem. Phys. Lett., 2009, 468, 176–183 CrossRef CAS.
- Z. Xue, Raman spectroscopy of carboxylic acid and water aggregates, PhD thesis, Logos Berlin, Berlin, 2011 Search PubMed.
- L. G. Bonner and R. Hofstadter, J. Chem. Phys., 1938, 6, 531–534 CrossRef CAS.
- V. Z. Williams, J. Chem. Phys., 1947, 15, 243–251 CrossRef CAS.
- J. K. Wilmshurst, J. Chem. Phys., 1956, 25, 478–480 CrossRef CAS.
- R. C. Millikan and K. S. Pitzer, J. Chem. Phys., 1957, 27, 1305–1308 CrossRef CAS.
- O. I. Baskakov, H. Bürger and W. Jerzembeck, J. Mol. Spectrosc., 1999, 193, 33–45 CrossRef CAS.
- T. L. Tan, K. L. Goh, P. P. Ong and H. H. Teo, J. Mol. Spectrosc., 1999, 198, 387–392 CrossRef CAS.
- F. Madeja, P. Markwick, M. Havenith, K. Nauta and R. E. Miller, J. Chem. Phys., 2002, 116, 2870–2878 CrossRef CAS.
- A. Perrin, J.-M. Flaud, B. Bakri, J. Demaison, O. Baskakov, S. Sirota, M. Herman and J. Auwera, J. Mol. Spectrosc., 2002, 216, 203–213 CrossRef CAS.
- A. Perrin, J. Vander Auwera and Z. Zelinger, J. Quant. Spectrosc. Radiat. Transfer, 2009, 110, 743–755 CrossRef CAS.
- P. Das, C. J. Knapp and W. Jäger, J. Mol. Spectrosc., 2017, 341, 17–22 CrossRef CAS.
- A. Nejad, E. Meyer and M. A. Suhm, J. Phys. Chem. Lett., 2020, 11, 5228–5233 CrossRef CAS.
- F. Kollipost, R. Wugt Larsen, A. V. Domanskaya, M. Nörenberg and M. A. Suhm, J. Chem. Phys., 2012, 136, 151101 CrossRef CAS.
- T. Forsting and M. Suhm, Curry-Jet SETUP, DOI: 10.6084/m9.figshare.6395840.v1.
- M. Gawrilow and M. A. Suhm, Phys. Chem. Chem. Phys., 2020, 22, 15303–15311 RSC.
- K. A. E. Meyer and M. A. Suhm, J. Chem. Phys., 2017, 147, 144305 CrossRef.
- M. A. Suhm and F. Kollipost, Phys. Chem. Chem. Phys., 2013, 15, 10702–10721 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E.01, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- J. Gauss and J. F. Stanton, Chem. Phys. Lett., 1997, 276, 70–77 CrossRef CAS.
- J. F. Stanton, J. Gauss, L. Cheng, M. E. Harding, D. A. Matthews and P. G. Szalay, CFOUR, Coupled-Cluster techniques for Computational Chemistry, a quantum-chemical program package, With contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, Y. J. Bomble, O. Christiansen, F. Engel, R. Faber, M. Heckert, O. Heun, M. Hilgenberg, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, T. Kirsch, K. Klein, W. J. Lauderdale, F. Lipparini, T. Metzroth, L. A. Mück, D. P. O'Neill, D. R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, C. Simmons, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J. D. Watts and the integral packages MOLECULE (J. Almlöf and P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. A. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen, for the current version, see http://www.cfour.de.
- D. A. Matthews, L. Cheng, M. E. Harding, F. Lipparini, S. Stopkowicz, T.-C. Jagau, P. G. Szalay, J. Gauss and J. F. Stanton, J. Chem. Phys., 2020, 152, 214108 CrossRef CAS.
- P. Huber-Wälchli and H. H. Günthard, Spectrochim. Acta, Part A, 1981, 37, 285–304 CrossRef.
- H. H. Nielsen, Rev. Mod. Phys., 1951, 23, 90–136 CrossRef CAS.
- We note that the hot band (2ν6–ν6) of trans-HCOOH in Chem. Sci., 2019, 10, 6285–6294 (ref. 24) was incorrectly assumed to be shifted by x66 rather than 2x66 from the ν6-fundamental. By considering the missing factor of two, the predicted shift of this non-isomeric hot band is similar to the cis–trans-shift of ν6, i.e., it could contribute to the band assigned to ν6 of cis-HCOOH. The expected intensity of the (2ν6–ν6) hot band, however, is distinctly too small for it to be the sole contribution to the band at 1093 cm−1. As such, the cis-HCOOH assignment remains largely unaffected.
- W. H. Weber, P. D. Maker, J. Johns and E. Weinberger, J. Mol. Spectrosc., 1987, 121, 243–260 CrossRef CAS.
- O. I. Baskakov and J. Demaison, J. Mol. Spectrosc., 2002, 211, 262–272 CrossRef CAS.
- K. L. Goh, P. P. Ong and T. L. Tan, Spectrochim. Acta, Part A, 1999, 55, 2601–2614 CrossRef.
- O. I. Baskakov, S. Alanko and M. Koivusaari, J. Mol. Spectrosc., 1999, 198, 40–42 CrossRef CAS.
- K. L. Goh, P. P. Ong, T. L. Tan, W. F. Wang and H. H. Teo, J. Mol. Spectrosc., 1998, 190, 125–129 CrossRef CAS.
- O. I. Baskakov, J. Lohilahti and V.-M. Horneman, J. Mol. Spectrosc., 2003, 219, 191–199 CrossRef CAS.
- R. A'dawiah, T. L. Tan and L. L. Ng, J. Mol. Spectrosc., 2018, 349, 43–48 CrossRef.
- O. I. Baskakov, J. Mol. Spectrosc., 2002, 213, 1–7 CrossRef CAS.
- L. Nemes, A. R. W. McKellar and J. W. C. Johns, J. Opt. Soc. Am. B, 1987, 4, 1165–1172 CrossRef CAS.
- K. L. Goh, P. P. Ong, H. H. Teo and T. L. Tan, J. Mol. Spectrosc., 1999, 197, 322–323 CrossRef CAS.
- T. L. Tan, K. L. Goh, P. P. Ong and H. H. Teo, J. Mol. Spectrosc., 1999, 198, 110–114 CrossRef CAS.
- O. I. Baskakov, J. Mol. Spectrosc., 2001, 208, 194–196 CrossRef CAS.
- K. L. Goh, P. P. Ong, H. H. Teo and T. L. Tan, Spectrochim. Acta, Part A, 2000, 56, 991–1001 CrossRef CAS.
- T. L. Tan, K. L. Goh, P. P. Ong and H. H. Teo, J. Mol. Spectrosc., 1999, 195, 324–327 CrossRef CAS.
- D. Luckhaus, M. Quack and M. Willeke, Z. Phys. Chem., 2000, 214, 889 Search PubMed.
- D. A. Long, The Raman effect: A unified treatment of the theory of Raman scattering by molecules, Wiley, Chichester and New York, 2002 Search PubMed.
- S. J. Daunt and H. F. Shurvell, J. Mol. Spectrosc., 1976, 62, 373–395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: cis-Formic acid spectra, comparison of formic acid vibrational nomenclature, harmonic avoided crossing calculated at other levels than B3LYP-D3, as well as computational raw data and further experimental details. See DOI: 10.1039/d0cp04451b |
| This journal is © the Owner Societies 2020 |