
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVibronic optical spectroscopy of cryogenic flavin ions: the O2+ and N1 tautomers of protonated lumiflavin†
David
Müller
and
Otto
Dopfer
 *
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, D-10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de; Fax: +49 30 314 23018
First published on 4th August 2020
Abstract
Flavins are key compounds in many photochemical and photophysical processes used by nature, because their optical properties strongly depend on the (de-)protonation site and solvation. Herein, we present the vibronic optical spectrum of protonated lumiflavin (H+LF), the parent molecule of the flavin family, obtained by visible photodissociation (VISPD) spectroscopy in a cryogenic ion trap. By comparison to time-dependent density functional theory (TD-DFT) calculations at the PBE0/cc-pVDZ level coupled to multidimensional Franck–Condon simulations, the spectrum recorded in the 420–500 nm range is assigned to vibronic bands of the optically bright S1 ← S0(ππ*) transition of the two most stable H+LF tautomers protonated at the O2+ and N1 position. While the most stable O2+ protomer has been identified previously by infrared spectroscopy, the N1 protomer is identified here for the first time. The S1 band origins of H+LF(O2+) and H+LF(N1) at 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 128 and 23202 cm−1 are shifted by 1617 and 1691 cm−1 to the blue of that of bare LF measured in He droplets, indicating that the proton affinity of both tautomers is slightly reduced upon S1 excitation. This view is consistent with the molecular orbitals involved in the assigned ππ* transition. The spectrum of both protomers is rich in vibrational structure indicating substantial geometry changes by ππ* excitation. Interestingly, while the O2+ protomer is planar in both electronic states, the N1 protomer is slightly nonplanar giving rise to large vibrational activity of low-frequency out-of-plane modes. Comparison with protonated lumichrome and metalated lumiflavin reveals the impact of functional groups and the type of the attached cation (proton or alkali ion) on the geometric and electronic structure of flavins.
128 and 23202 cm−1 are shifted by 1617 and 1691 cm−1 to the blue of that of bare LF measured in He droplets, indicating that the proton affinity of both tautomers is slightly reduced upon S1 excitation. This view is consistent with the molecular orbitals involved in the assigned ππ* transition. The spectrum of both protomers is rich in vibrational structure indicating substantial geometry changes by ππ* excitation. Interestingly, while the O2+ protomer is planar in both electronic states, the N1 protomer is slightly nonplanar giving rise to large vibrational activity of low-frequency out-of-plane modes. Comparison with protonated lumichrome and metalated lumiflavin reveals the impact of functional groups and the type of the attached cation (proton or alkali ion) on the geometric and electronic structure of flavins.
1. Introduction
Flavins are yellow dye molecules and represent a fundamental class of photochemically active biomolecules. They are derived from the tricyclic heteroaromatic 7,8-dimethyl-10-alkyl-isoalloxazine chromophore, which is responsible for their photophysical properties. The various flavins differ by their substituent R at the N10 position. Important members of the flavin family include iso-lumichrome (iso-LC, R = H), lumiflavin (LF, R = CH3, Fig. 1), riboflavin (RF, vitamin B2, R = ribityl), flavin mononucleotide (FMN, R = ribophosphate), and flavin adenine dinucleotide (FAD, R = ribophosphate + adenine). Because iso-LC is less stable than LC (R = H at N1 instead of N10) and thus a metastable species, LF is often considered as the parent molecule of the flavin family. Flavins absorb light in a broad spectral range ranging from the visible to the ultraviolet, and their photophysical properties are determined by the interplay between the optically bright ππ* excitations of the aromatic π electrons and the optically dark nπ* excitations originating from the lone pair electrons of the heterocyclic N atoms and the oxygen atoms of the CO groups in the tricyclic ring. This interplay can readily be modulated by the flavin environment, such as the substitution of functional groups, (de-)protonation, the oxidation state, metalation, and solvation. As a result, nature utilizes flavins for a plethora of photochemical processes, including light harvesting, DNA repair, and enzyme catalysis.1–7 | ||
| Fig. 1 Top and side views of the structures of the two most stable protomers of H+LF (O2+ and N1) in the S0 state obtained at the PBE0/cc-pVDZ level. The O2+ protomer has a planar ring system (Cs), whereas the N1 protomer is slightly bent along the N5-N10 axis (C1). Atoms are labelled according to IUPAC nomenclature. The structures of further protomers (O2−, O4±, N5, OH+±) are shown in Fig. S3 in the ESI.† | ||
Flavins have extensively been investigated by spectroscopy in the condensed phase and quantum chemical calculations.8–18 Concerning LF, the first excited singlet state (S1) observed near 445 nm is attributed to the first optically allowed ππ* state. The large geometry changes upon S1 excitation result in a substantial difference between the vertical and adiabatic excitation energies and thus a large Stokes shift of 90 nm in fluorescence.9 While the maximum of the S1 band of LF occurs in a narrow spectral window (441–447 nm) and thus is relatively independent of the solvent, the second absorption band assigned to the next ππ* state is highly sensitive to solvation (332–367 nm), because of its sensitivity to hydrogen bonding with the solvent.9 Protonation of LF has a drastic impact on its absorption spectrum and shifts its first absorption band in aqueous solution from 441 nm (neutral, pH 6–8) to 394 nm (protonated, pH −1.1).8,11 Hydrogen bonding with the solvent leads to a redshift of the optically bright S3 state und hence reduces the gap between S1 and S3, resulting in the single broad absorption band at 394 nm observed experimentally.15 In the spectroscopic study,11 protonation at N1 of LF was assumed although there was no evidence for the protonation site from the experimental data. Computational studies on the photophysics of H+LF consider only protonation at N1 and N5 in both gas phase and aqueous solution,15 although O2+ was computed to be the most stable protomer at the HF level.17 The former computation15 predicts the N1 protomer to be more stable than the N5 protomer in the S0 state, while the energy order is reversed in the S1(ππ*) state. Significantly, all optical spectra of flavins recorded in the condensed phase at room temperature are broad and vibrationally unresolved. Consequently, details of individual environmental effects on the optical properties cannot be extracted. To this end, it is essential to cool the molecules down to cryogenic temperatures.19,20 Indeed, the recent optical laser-induced fluorescence (LIF) excitation spectrum of neutral LF doped into superfluid He nanodroplets (T = 0.4 K) was assigned to vibronic structure of the S1 ← S0(ππ*) transition, with an adiabatic S1 origin at 21511 cm−1 (464.87 nm).20 It was argued that the He environment has typically only a minor impact on the transition energy (<1%, ∼250 cm−1). The low temperature in the He droplet allows to resolve the rich vibronic structure in the S1 excited state of LF, which illustrates the large geometry change upon electronic ππ* excitation.
To reveal the intrinsic optical properties of isolated flavins completely free from external perturbations, spectroscopic studies in the gas phase are required. However, such studies are scarce because of the difficulties involved in transferring such biomolecules in large abundance into the gas phase. This obstacle can be circumvented by electrospray ionization (ESI) techniques, which can be used to efficiently transfer flavin ions into the gas phase for interrogation with action spectroscopy. In recent years, this approach has been applied to a few cationic and anionic flavins of various complexity to study their photophysical properties.21–26 However, all these experiments were conducted at room temperature, leading to broad and unresolved electronic spectra of similar appearance as in the condensed phase. As a result, the clear discrimination between environmental effects and intrinsic properties is challenging if not impossible. Concerning flavins, it is not even possible to reliably determine the (de-)protonation sites. To this end, cryogenic cooling is required to obtain high-resolution electronic spectra at the level of vibrational resolution, which then provides precise details about the isomeric structure of the flavin ion and the nature of the electronic states responsible for the absorption process.
To characterize the structure and intrinsic optical properties of flavins isolated in the gas phase, we have initiated a few years ago a research program to systematically investigate the effects of protonation, metalation, and solvation on flavins with increasing complexity, ranging from LC to FMN. In the first step of the research strategy, the geometric structure of protonated and metalated flavins is determined in the ground electronic state (S0) by infrared multiple photon dissociation (IRMPD) of mass-selected ions generated by ESI. Although these experiments are conducted at room temperature, the IRMPD spectra recorded in the fingerprint range exhibit sufficient resolution to determine the preferred protonation and metalation sites by probing the highly structure-sensitive CO stretch vibrations.27–29 With the aid of density functional theory (DFT) calculations at the B3LYP/cc-pVDZ level, the IRMPD spectrum of protonated LF (H+LF) has been assigned to the most stable O2+ protomer, while significant contributions of higher energy isomers have been excluded.27 Significantly, these IRMPD studies present the first spectroscopic data of any flavin molecule in the gas phase. In a second step, we have extended our initial spectroscopic characterization of flavins in the ground electronic state to electronically excited states. For this purpose, we employ a recently commissioned cryogenic ion trap tandem mass spectrometer (BerlinTrap)30 to record visible photodissociation (VISPD) spectra of mass-selected flavin ions generated by ESI and cooled to cryogenic temperatures (T < 20 K). Significantly, cooling of the ions enables us for the first time to obtain vibrationally resolved electronic spectra of isolated flavins. Analysis of these vibronic VISPD spectra with time-dependent DFT (TD-DFT) calculations coupled to multidimensional Franck–Condon (FC) simulations provides reliable and precise information about the protonation and metalation sites as well as the nature and adiabatic energy of the involved electronic excitation along with the resulting structural changes. The initial application to the S1 ← S0(ππ*) transition of H+LC confirms N5 as the preferred protonation site already inferred from its IRMPD spectrum.27,31 N5 protonation of LC causes a remarkably large S1 redshift of ∼6000 cm−1, which can be rationalized by the molecular orbitals involved in this ππ* excitation. Subsequent studies characterize the effects of metalation of LC and LF with alkali metal cations (M = Li–Cs) on their optical properties, illustrating that the M+⋯LF/LC interaction strength and thus the S1 excitation energy shifts depend strongly on the metalation site.32,33 Herein, we extend these VISPD studies to H+LF. The major questions to be addressed are the observed protonation sites, the effects of protonation on the optical properties of LF, and the differences between H+LF and H+LC/M+LF studied previously by the same spectroscopic and computational approach.
2. Experimental and computational details
Vibronic VISPD spectra of H+LF are measured in a cryogenic ion trap coupled to a tandem mass spectrometer (BerlinTrap) and an ESI source.30 Briefly, ions are generated in the ESI source by spraying at a flow rate of 2 ml h−1 a solution of 2–4 mg LF (Sigma Aldrich, >99%) dissolved in 19 ml methanol, 1 ml water, and 2 ml formic acid to enhance protonation. The produced ions are skimmed, accumulated, and thermalized for 90 ms in a short quadrupole located directly behind the skimmer. After passing through a hexapole ion guide, the H+LF ions are mass-selected by a quadrupole, deflected by a quadrupole bender, and guided through an octopole into a temperature-controlled cryogenic 22-pole ion trap (T = 4–300 K) held at 6 K. In the trap, the ions are stored for around 90 ms and cooled down via He buffer gas cooling to vibrational temperatures below 20 K.30 The cold ions extracted from the 22-pole are transferred via a series of electrostatic lenses toward the extraction region of an orthogonal reflectron time-of-flight mass spectrometer, where they are irradiated by a laser pulse around 40 μs before pulsed ion extraction. The generated photofragment ions are detected simultaneously with the remaining parent ions using a dual microchannel plate detector. Photons in the visible range are generated by an optical parametric oscillator (OPO, GWU, VersaScan, bandwidth of 4 cm−1) pumped by the third harmonic of a nanosecond Q-switched Nd:YAG laser (Innolas, Spitlight 1000, 355 nm, 180 mJ per pulse) operating at a repetition rate of 10 Hz to be synchronized with the cycle of the tandem mass spectrometer setup. A selected part of the VISPD spectrum is also recorded at higher resolution using a dye laser (Radiant Dyes, NarrowScan, Coumarin 120 and Stilbene 3 dyes dissolved in ethanol, bandwidth of 0.014 cm−1) pumped by the same type of Nd:YAG laser (355 nm, 100 mJ per pulse). The wavelength of the OPO and dye laser outputs is calibrated by a wavemeter. VISPD spectra measured between 23050 and 23750 cm−1 (421–434 nm) are obtained by normalising the integrated signal of all fragment ions by the parent ions and the laser intensity measured at the exit of the instrument by a pyroelectric detector. The mass spectra of the ions extracted from the trap do not show any peaks arising from tagging of H+LF with He or N2. At a trap temperature of 6 K, the N2 impurity gas in the He buffer gas line freezes out at the cold walls of the trap. Tagging with He is not observed because the binding energy is too low for He attachment to H+LF at an effective vibrational ion temperature of T = 15–20 K.
The vibronic VISPD spectra are assigned by comparison to quantum chemical (TD-)DFT calculations at the PBE0/cc-pVDZ level using the GAUSSIAN16 package.34–36 To this end, the structures of all low-energy protomers are optimized in the ground electronic state (S0). All reported proton affinities (here taken as protonation energies at T = 0 K) and relative energies (E0) are corrected for harmonic vibrational zero-point energy. Vertical energies and oscillator strengths for electronic excitations are computed for both singlet (up to S4) and triplet states (up to T4). Adiabatic energies are obtained (up to S2 and T1) by optimizing the excited states using the converged ground state geometry as starting point. Cartesian coordinates and energies of all relevant structures are provided in the ESI.† Multidimensional Franck–Condon simulations at T = 0 K are carried out to generate vibronic excitation spectra.37 In previous studies on related flavins (H+LC, M+LC/LF with M = Li–Cs),31–33 the employed PBE0/cc-pVDZ level was shown to be a computationally efficient and reliable DFT approach for calculating vibronic spectra. Orbitals contributing to the observed transitions are determined using the natural transition orbital (NTO) approach.38 Energy barriers at transition states (TS) between different protomers are calculated using the QST2 method and are not corrected for zero-point energy. Harmonic frequency analysis is employed to determine the nature of stationary points as minima or transition states. The atomic charge distribution of the ground and excited states is evaluated using the natural bond orbital analysis (NBO).
3. Results and discussion
Initial overview VISPD spectra of H+LF are recorded at a larger step size of 0.5 nm starting from 500 nm (20000 cm−1) and scanning to the blue to search for structured resonant absorption. This spectral range is suggested by the adiabatic S1 origin prediction at 443.75 nm (22535 cm−1) for the most stable O2+ protomer, which was identified as the single H+LF isomer present in the ESI source by our previous IRMPD experiments.27 The first pronounced transition is indeed detected at 432.38 nm (23128 cm−1), and thus the VISPD spectrum is investigated in the vicinity of this peak at higher resolution using a smaller step size (22700–23800 cm−1). Resonant VISPD of the H+LF parent ion (m/z 257) at 23128 cm−1 results in a variety of fragment ions with m/z 145, 156, 159, 171, 186, 199, 214, and 242 appearing with different branching ratios (Fig. S1 and Table S1 in the ESI†). The H+LF ions stored in the ion trap are cold and thus no fragmentation is observed without laser excitation. In particular, there are no fragments resulting from metastable decay and collision-induced dissociation (CID) upon extraction of the cold ions from the trap. Previous IRMPD spectra of H+LF reveal the photodissociation products m/z 145, 159, 171, 186, and 214 in the ground electronic state.27 Apart from m/z 145, these can be explained by formal loss of OCNH (isocyanic acid) and combinations of OCNH with CO. New mass peaks observed exclusively upon VISPD of H+LF include m/z 242 and 199, which are rationalized by loss of CH3 and CH3 + OCNH. Hence, electronic excitation of H+LF opens a new fragmentation channel not operating upon IRMPD occurring in the S0 state. This new channel involves loss of CH3, probably the methyl group attached to N10. The VISPD fragmentation pattern of H+LF is much more complex than that observed for S1(ππ*) excitation of H+LC, for which only two fragment channels are observed for both VISPD and CID, namely loss of CO + NH3 (or HCN + H2O) and loss of OCNH + CO. Consequently, the ring opening reaction upon VISPD is different for H+LF and H+LC, confirming that the photochemistry of H+LC differs strongly from that of H+LF.
The obtained vibronic action spectra of H+LF generated for each individual fragment channel (Fig. S2 in the ESI†) are very similar with respect to the positions, widths, and relative intensities of all transitions observed. This result is in contrast to the corresponding IRMPD spectra of H+LF, which are complicated by further resonant IR absorption of the some of the initial fragment ions.27 The maximum fragmentation efficiency for VISPD of H+LF is of the order of a few percent (∼2.5% for the most intense fragment channel), and the total VISPD spectrum generated by adding all action spectra of the individual fragments shown in Fig. 2 is used for further analysis. The total VISPD yield amounts to around 10% suggesting reasonable overlap between the ion and laser beams. Interestingly, although the VISPD spectrum observed for the m/z 242 ion (loss of CH3) and the related m/z 199 ion (loss of CH3 + OCNH) also matches the other action spectra well in the range from 23130 to 23750 cm−1, we detect substantial unstructured signal in these particular channels down to at least 20000 cm−1 (500 nm) indicating that the process leading to these fragments is different from the others (as already indicated by their different behaviour in IRMPD and VISPD mass spectra). Similarly, also for the other mass channels there is slowly decaying and structureless background fragmentation signal to the red of the first absorption peak. In general, cooling of the ions to temperatures below 20 K is sufficient to completely suppress hot band transitions and to measure well-resolved vibronic VISPD spectra for such biomolecular ions. The VISPD spectrum of H+LF exhibits rich vibrational structure indicating substantial geometry changes upon electronic excitation.
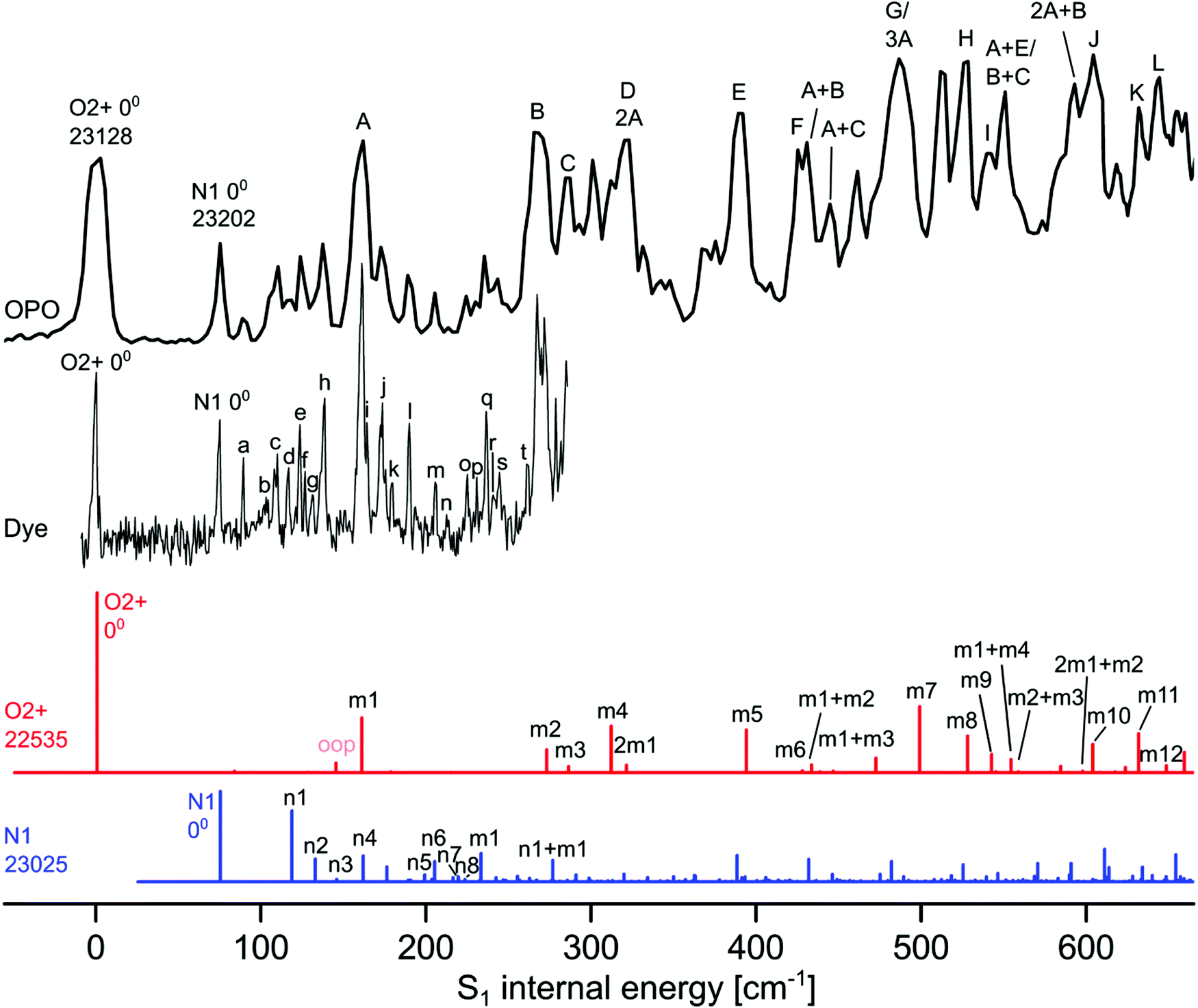 | ||
| Fig. 2 Experimental VISPD spectrum of H+LF recorded with the OPO and dye laser compared to Franck–Condon (FC) simulations of the two most stable protomers O2+ (red) and N1 (blue). All spectra are referenced to the experimental S1 internal energy of the O2+ protomer. The absolute values of the origins are given in cm−1. The calculated FC spectra are scaled vertically to match the associated intensity of the experimental S1 origins. The calculated FC intensity of the O2+ origin is one order of magnitude higher than that of the N1 protomer. Peak positions and suggested vibrational and isomer assignments are given in Tables 4 and 5. | ||
To assign the electronic VISPD spectrum by TD-DFT calculations, we first optimize various low-energy protomers in their ground electronic state (S0) and compute their vertical and adiabatic singlet excitations and their corresponding vibronic spectra. The proton affinities (PA), computed here as protonation energies for T = 0 K, and the relative energies (E0) in the S0 ground state obtained for the most stable H+LF protomers at the PBE0/cc-pVDZ level are listed in Table 1. The PBE0 values agree well with previously reported B3LYP data using the same basis set.27 In the low-energy protomers shown in Fig. 1 and Fig. S3 in the ESI,† the proton is attached to one of the nucleophilic lone pairs of the N atoms (N1 and N5) or carbonyl O atoms (O2 and O4). For the O protonated tautomers, the ± sign indicates the orientation of the OH proton with respect to the N3H group (+ away, − toward).27 The most stable protomer is O2+ with PA = 972 kJ mol−1, and the previous IRMPD spectrum was exclusively assigned to this H+LF protomer.27 The next protomer close in energy is N1 at E0 = 15 kJ mol−1, while all further protomers obtained by direct protonation of LF (O2−, N5, O4+, O4−) or protonation coupled to lactam–lactim tautomerization (OH+−, OH++), in which the N3H proton migrates to a CO group, are significantly higher in energy (>22 kJ mol−1). In the following, we focus mostly on the most stable O2+ and N1 tautomers, while corresponding results for higher energy protomers are given in the ESI.†
| Protomer | PBE0/cc-pVDZ | B3LYP/cc-pVDZ |
|---|---|---|
| PA (E0) | PA (E0) | |
| O2+ | 971.7 (0) | 974.0 (0) |
| N1 | 956.7 (15.0) | 961.0 (13.0) |
| O2− | 945.6 (26.1) | 948.6 (25.4) |
| O4+ | 936.1 (35.6) | 939.5 (34.5) |
| N5 | 919.8 (51.9) | 924.6 (49.4) |
| OH++ | 944.6 (27.1) | 945.4 (28.6) |
| OH+− | 949.3 (22.4) | 950.5 (23.5) |
| O4− | 904.6 (67.1) | 909.4 (64.6) |
As a next step, we compute vertical excitation energies (Ev) and oscillator strengths (f) for the S1–S4 states of the various H+LF protomers (Table 2 and Fig. S4 in the ESI†). These excitations include optically bright ππ* and optically dark nπ* excitations of bonding aromatic π electrons and nonbonding lone pair electrons (n) of N and O into empty nonbonding π* orbitals. Among the S1–S4 states, there are two ππ* and two nπ* states. The oscillator strengths of the bright ππ* states are typically several orders of magnitude larger than those of the dark nπ* states. Concerning the vertical transitions, the S1 state is bright (ππ*), while the S2 state is dark (nπ*) for all considered protomers, while the order of the second ππ* and nπ* states is switched for some of the tautomers. For O2+, the oscillator strength for S1(ππ*), f = 0.063, is two orders of magnitude higher than for S2 (f = 0.0007), which is located 1951 cm−1 above S1. The second optically bright S3(ππ*) state lying 2850 cm−1 above S1 has a substantially higher oscillator strength (f = 0.34) than S1, while S4(nπ*) at 4417 cm−1 above S1 is again dark (f = 0.0001). The N1 tautomer has a similar electronic coarse structure as O2+. The bright S1(ππ*) state (f = 0.025) lies 435 cm−1 below the dark S2(nπ*) state (f = 0.004). The S3(ππ*) state is 2606 cm−1 above S1 and again much brighter (f = 0.39). These results are in good accordance with quantum chemical calculations performed by other groups for related systems.9,13,15,39
| S1(ππ*) | S2(nπ*) | S3(nπ*/ππ*) | S4(nπ*/ππ*) | |||||
|---|---|---|---|---|---|---|---|---|
| E v (Ea) | f (× 103) | E v (Ea) | f (× 103) | E v | f (× 103) | E v | f (× 103) | |
| LF | 25236 (22448) |
212.6 | 26121 (21620) |
0.4 | 27585 |
0.4 | 32250 |
136.3 |
| O2+ | 25553 (22535) |
63.2 | 27504 (23703) |
0.7 | 28403 |
339.2 | 29970 |
0.1 |
| N1 | 26388 (23025) |
25.4 | 26823 (22488) |
3.6 | 28994 |
385.3 | 30227 |
1.8 |
| O2− | 25295 (22341) |
67.7 | 27453 (23568) |
0.8 | 28322 |
334.7 | 30491 |
0.0 |
| O4+ | 20229 (17682) |
95.4 | 26568 (22280) |
0.2 | 26724 |
239.3 | 28613 |
0.0 |
| N5 | 18207 (15622) |
122.0 | 20342 (14731) |
0.0 | 25583 |
0.1 | 25808 |
186.4 |
| OH++ | 23595 (20762) |
62.1 | 24751 (20828) |
0.0 | 27928 (26389) |
377.3 | 33085 |
20.1 |
| OH+− | 23987 (21074) |
51.4 | 25125 (21734) |
0.2 | 28305 (26919) |
404.2 | 34001 |
1.0 |
| O4− | 20683 (18046) |
87.5 | 25701 (20994) |
0.4 | 26948 |
263.0 | 29828 |
0.0 |
Direct comparison with the experimental vibronic spectra requires the computation of adiabatic excitation energies (Ea) and vibrational normal modes as input for the FC simulations. To this end, we optimized the S1 and S2 states for all considered protomers (Table 2 and Fig. S5 in the ESI†). The resulting S1 origins are listed in Table 3, along with the protonation-induced shifts from S1 of bare LF. The S1(ππ*) origin computed for LF as 22448 cm−1 agrees well with the value measured in He droplets (21511 cm−1), which should be close to the origin of the bare molecule with an estimated error of less than 250 cm−1.20 The small difference between the experimental and computed S1 origins of ∼1000 cm−1 (∼0.1 eV) confirms that the chosen DFT level describes the electronic structure of this type of flavin well.31–33 In general, the protonation-induced ΔS1 shifts depend strongly on the site of protonation and span a large range from −6826 to +578 cm−1. Interestingly, for the N1 protomer the order of the excited S1 and S2 states changes, when going from vertical to adiabatic energies. Thus, for this protomer, the first dark nπ* state is adiabatically located slightly lower than the first bright ππ* state by 537 cm−1 (nonetheless, we keep herein the notation of the order of the states obtained from the vertical transitions). Significantly, only the S1 origins of O2+ and N1 exhibit S1 blueshifts upon protonation.
The total VISPD spectrum of H+LF presented in Fig. 2 is referenced to the first peak at 23128 cm−1 (432.38 nm) observed in the range above 20000 cm−1 (below 500 nm), which is assigned to the origin band (00) of the observed electronic transition. This spectrum is recorded using the OPO laser with a spectral resolution of 4 cm−1 and maximal available intensity (3–4 mJ per pulse at a beam diameter of 5 mm) at a step size of 2.5 cm−1. The spectral range covers up to ∼600 cm−1 of S1 internal energy. A higher-resolution spectrum covering the first ∼300 cm−1 is also recorded using the dye laser with a spectral resolution of 0.014 cm−1 and similar intensity (3 mJ per pulse at a beam diameter of 5 mm) at a step size of ∼0.5 cm−1 (Fig. 2). The peak positions and suggested vibrational and protomer assignments are given in Tables 4 and 5. The OPO spectrum shows a series of broader transitions (∼10 cm−1) labelled with capital letters A–L built on the origin at 0 cm−1 internal energy, along with a series of significantly narrower transitions (∼5 cm−1) starting from an origin at 75 cm−1 and labelled with lowercase letters (a–t). These two series are assigned to the S1 ← S0(ππ*) electronic transitions of the two most stable O2+ and N1 protomers of H+LF, respectively. Their derived S1 origins of 23128 and 23202 cm−1 agree well with the predicted values of 22535 and 23025 cm−1, respectively. The experimental blueshifts with respect to LF of ΔS1 = 1617 and 1691 cm−1 are in accord with the computed shifts of ΔS1 = 87 and 578 cm−1, when taking into account the error in both experiment (effect of the He droplet on the S1 origin of LF)20 and computation. The previous VISPD study on the S1 ← S0 transitions of the related O4+ protomers of M+LF (M = Li–Cs) using the same computational approach yields differences of less than 650 cm−1 between measured and computed adiabatic S1 origins.33 Additionally, deviations in the same order of magnitude have been reported for the S1 transitions of H+LC(N5) (809 cm−1) and M+LC(O4+) (up to 1154 cm−1).31,32 Clearly, the S1 origins predicted for the other H+LF protomers fit less well. In particular, the computed S1 origins of the N5 and O4± tautomers exhibit large redshifts of ∼4000–7000 cm−1, similar to those observed for O4+ of M+LF.33 The OH+± tautomers also have substantial computed S1 redshifts of ∼1500 cm−1. The only other protomer with a predicted nearby S1 origin is O2− with ΔS1 = −107 cm−1. As expected, the Sn energies of the O2± tautomers are similar (as are those of O4±) because their electronic structure is not much affected by the different orientation of the excess proton. However, we may exclude O2− at this stage from an assignment to any of the two identified S1 origins, because of its significantly higher energy relative to O2+ (ΔE0 = 26 kJ mol−1). For the N1 and O2± isomers, the second optically bright S3(ππ*) state is predicted to be substantially higher in energy than S1 (by ΔEv = 2606–3027 cm−1) so that we can also safely exclude this option. On the other hand, the S3 states of the N5 and O4± are calculated to be nearby considering their vertical energies (Table 2). Unfortunately, all efforts to optimize these higher excited states have failed (because of running into conical intersections) so that we cannot predict the adiabatic transition energies. Thus, we may at this stage exclude this option just by their rather low stability (E0 > 35 kJ mol−1). In case of the other high-energy OH+± protomers, the bright S3 states have predicted adiabatic origins far away from the observed transitions (>3000 cm−1) so that an assignment of these isomers may safely been rejected. An assignment to a triplet state appears unlikely (Fig. S4 in the ESI†). In summary, by considering both the predicted optically bright adiabatic and vertical S1 and S3/4 energies and the relative stabilization energies, the only assignment of the band origins observed at 23128 and 23202 cm−1 is to S1 of the two most stable O2+ and N1 protomers and we adapt this scenario for the remaining analysis.
| Band | ν (exp.) | ν (calc.) | Assignment | LF (exp.)a | LF (calc.) |
|---|---|---|---|---|---|
| a Data measured in He droplets (ref. 20). | |||||
| 00 | 23128 |
22535 |
00 | 21511 |
22448 |
| A | 160 | 160 | m1 | 164 | 165 |
| B | 268 | 272 | m2 | 274 | 276 |
| C | 285 | 286 | m3 | 294 | |
| D | 317 | 312 | m4 | 322 | |
| E | 389 | 393 | m5 | 403 | 409 |
| F | 425 | 427 | m6 | 440 | 444 |
| G | 486 | 498 | m7 | 489 | |
| H | 526 | 527 | m8 | 513 | 521 |
| I | 539 | 542 | m9 | 545 | |
| J | 603 | 603 | m10 | 593 | 603 |
| K | 633 | 631 | m11 | 635 | |
| L | 645 | 648 | m12 | 669 | |
| Band | ν (exp.) | ν (calc.) | Assignment |
|---|---|---|---|
| 00 | 23202 |
23025 |
00 |
| a | 15 | ||
| b | 29 | ||
| c | 36 | 43 | n1 |
| d | 42 | ||
| e | 49 | 58 | n2 |
| f | 52 | ||
| g | 57 | ||
| h | 63 | 70 | n3 |
| i | 90 | ||
| j | 98 | 87 | n4 |
| k | 105 | ||
| l | 115 | 124 | n5 |
| m | 131 | 132 | n6 |
| n | 139 | ||
| o | 150 | 144 | n7 |
| p | 156 | 148 | n8 |
| q | 162 | 158 | m1 |
| r | 166 | ||
| s | 170 | ||
| t | 187 |
The magnitude and direction of the S1 shifts upon protonation for the different H+LF protomers can be rationalized by the molecular orbitals involved in the first ππ* excitation and the resulting charge reorganisation. To this end, we consider in Fig. 3 the natural transition orbitals (HOMO and LUMO) for LF and the O2+ and N1 protomers of H+LF and in Fig. S6 and Table S2 in the ESI† the NBO charge distribution in both electronic states of LF. S1 excitation of LF decreases the negative charge on both N1 (by 0.152 e from −0.660 to −0.508 e) and O2 (by 0.028 e from −0.598 to −0.570 e). Thus, S1 excitation reduces the attraction of the proton at these binding sites, leading to a reduced proton affinity in the S1 state, which directly translates into the S1 blueshifts for the two protomers N1 and O2+ (and also the small redshift for O2−). On the other hand, S1 excitation increases the negative partial charge on N5 (by 0.078 e from −0.376 to −0.454 e) and O4 (by 0.015 e from −0.577 to −0.592 e), which increases the proton affinity and thus produces S1 redshifts for N5 and O4+ (and also O4−). This view is confirmed by the molecular orbital wavefunctions in Fig. 3, which show large changes in the amplitudes near O4 and N5 but only small ones near O2 and N1, explaining the large S1 redshifts for N5 and O4± and the small blueshifts (redshift) for N1 and O2+ (O2−) upon protonation.
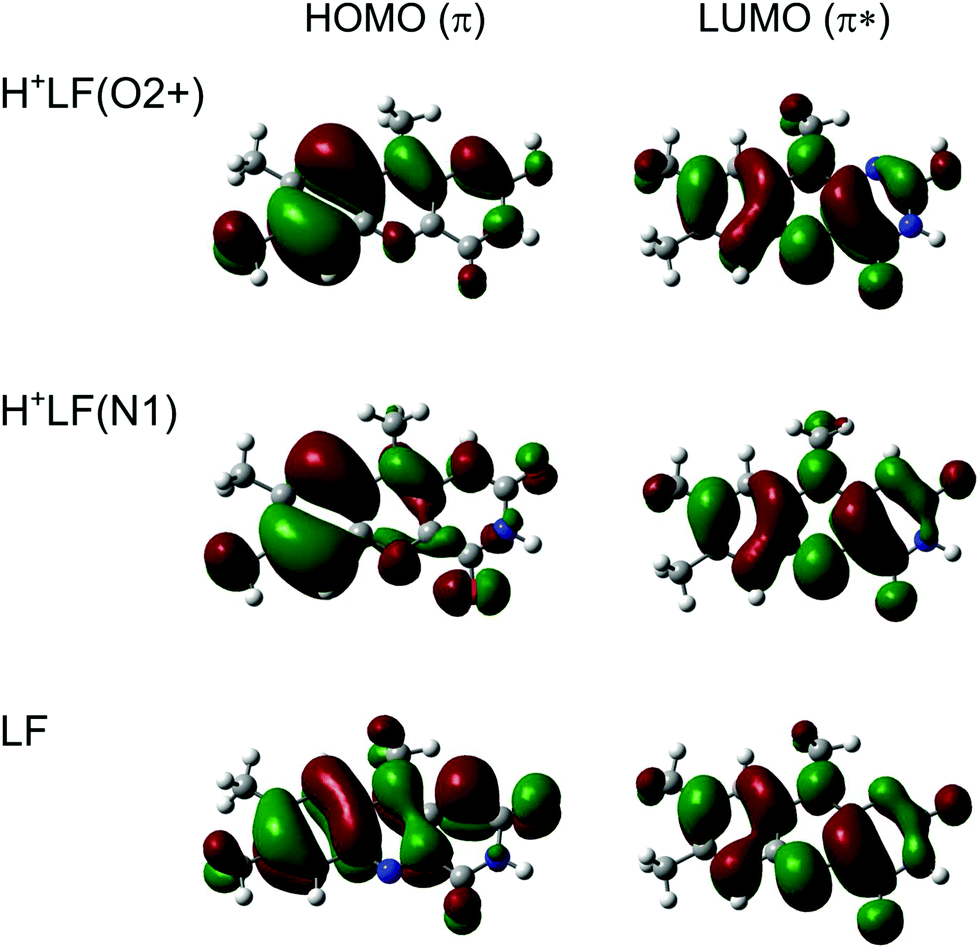 | ||
| Fig. 3 Natural transition orbitals involved in the electronic S1 ← S0(ππ*) transition of LF, H+LF(O2+), and H+LF(N1) calculated at the PBE0/cc-pVDZ level. | ||
In the next step, we assign the vibrational structure in the S1 state of the O2+ protomer by comparison to FC simulations (Fig. 2). As already mentioned, the broader peaks in the OPO spectrum are attributed to this most stable protomer, and the FC simulations provide convincing evidence for this assignment (Table 4). Apart from the three CH3 groups, H+LF(O2+) is planar in both the S0 and S1 states (Cs), so that only modes with a′ symmetry are FC active. This includes all in-plane fundamentals, their combination bands and their overtones. Moreover, even quanta of out-of-plane modes (a′′) are also FC allowed. Indeed, the O2+ protomer has 12 a′ fundamentals below 650 cm−1 and all are observed in the VISPD spectrum (A–L, m1–m12, Table 4) with various intensities. A full set of calculated vibrational frequencies sorted by symmetry for the S0 and S1 states for both protomers (O2+ and N1) is available in the ESI† (Tables S3 and S4). The experimental frequencies of O2+ agree well with the computed values, with maximum and average deviations of 12 and 3 cm−1, respectively. In addition, the relative intensities of fundamentals and overtone/combination bands predicted by the FC simulations reproduce satisfactorily the measured pattern. Some deviations in relative intensities may result from power broadening and saturation effects described below. The overall convincing agreement confirms the given isomer assignment and illustrates again the suitability of the computational approach. The normal modes denoted m1–m12 were described in some detail in our previous study on M+LF clusters and are shown in Fig. S7 and S8 in the ESI† for LF and H+LF(O2+).33 The three modes associated with the excess proton are the in-plane OH stretch and bend modes, which are outside the investigated spectral range (σOH = 3779 cm−1, βOH = 1208 cm−1), and the out-of-plane OH torsion, which is FC forbidden (τOH = 537 cm−1). The observed m1–m12 modes are essentially in-plane ring deformation modes of the LF moiety and only little affected by the excess proton at O2. Thus, their frequencies deviate only little from those of bare LF, with maximum and average differences of 21 and 9 cm−1, respectively. Indeed, the vibrational structure of the LIF excitation spectrum of the S1 state of LF in He droplets20 is very similar to the VISPD spectrum of the H+LF(O2+) protomer with respect to both the band positions and intensities (Fig. S9 in the ESI†), indicating that the same electronic ππ* transition is observed in both molecules. This view is further confirmed by the molecular orbitals involved in this S1 ← S0 transition, which have essentially no amplitude at the excess proton. For example, the LIF spectrum of LF is dominated by progressions of mode m1 = 164 cm−1 with several other fundamentals,20 and the corresponding progression for H+LF(O2+) has a frequency of m1 = 160 cm−1. The m1 mode is an in-plane bending motion of the outer aromatic rings I and III. Mode m2 describes a rocking motion of the methyl group at C8 coupled to a shear deformation of ring II. Mode m3 is similar to mode m2 but contains mainly a rocking motion of the methyl group at C7. For details of all detected in-plane normal modes m1–m12 the reader is referred to Fig. S8 in the ESI.† The FC simulations of the O2+ protomer predict a few low-intensity combination bands and overtones of out-of-plane (oop) modes with a′′ symmetry, for example at 84 and 144 cm−1 (2 × 42 and 2 × 72 cm−1). As expected for an electronic transition between two states with Cs symmetry, their FC activity is rather weak.
In addition to the broad transitions in the OPO spectrum labelled A–L, which are readily assigned to the O2+ protomer by the FC simulations, there are many sharper transitions starting from the band at 75 cm−1 internal energy. They are better resolved in the spectrum recorded with the dye laser and some 20 transitions are detected below 300 cm−1 (Fig. 2). These rather intense transitions are not predicted by the FC simulations for the O2+ protomer. Most of them could, in principle, be explained by FC forbidden a′′ modes of the O2+ protomer (and combinations thereof) and such an assignment is presented in Table S5 in the ESI.† The only explanation for such pronounced transitions is vibronic coupling of the S1(ππ*) state with a dark nπ* state. For example, the S2(nπ*) state of O2+ is calculated to be only 1168 cm−1 above the S1(ππ*) state. Although, we cannot exclude such a scenario, it is not favoured here because it is difficult to rationalize such high-intensity FC forbidden transitions by vibronic coupling, while the FC approximation works well for the in-plane modes. Moreover, the rather different widths of the transitions point toward two different species or two different electronic states. Initially, the VISPD spectrum has been measured at the highest laser intensities available to detect as many transitions as possible. In the OPO spectrum measured with 4 mJ per pulse, the widths of the band origins at 0 and 75 cm−1 are 13.7 and 4.3 cm−1, respectively. The latter value is close to the bandwidth of the laser (∼4 cm−1). Other factors contributing to the large width of the origin at 0 cm−1 include lifetime broadening, unresolved rotational substructure, and power broadening. The widths of the two transitions in the spectrum taken with the dye laser decrease to 2.6 and 1.4 cm−1 at reduced laser power, indicating that most of the large width of the O2+ transitions in the OPO spectrum arises from power broadening rather than from a short lifetime. However, at any laser power tried (until the signal disappears at the achieved signal-to-noise ratio), the transition at 0 cm−1 is broader than that at 75 cm−1.
Our mass spectra do not show any evidence for cluster formation of mass-selected H+LF ions stored in the cold trap during He buffer gas cooling. Thus, we can exclude any H+LF(O2+)–Ln clusters (e.g., L = He, N2, or H2O) as carriers for the sharp bands in the OPO spectrum. Thus, as an alternative and here favoured interpretation, these transitions are attributed to another protomer of H+LF, namely the second most stable N1 protomer, which is rather low in energy (E0 = 15 kJ mol−1). Interestingly, the N1 protomer is slightly nonplanar in both the S0 and S1 excited state, leading to a reduction in symmetry from Cs to C1 upon protonation.15 The N1 protomer is slightly bent along the N5–N10 axis leading to a butterfly-type deformation, which is somewhat more pronounced in the S1 state than in the S0 state (7° versus 3°). Moreover, the CH3 group at N10 rotates out of the plane (by ∼30°), probably because of steric hindrance between the H atom that is in the aromatic plane of neutral LF and the excess proton added at N1. The barrier to planarity is rather small, and amounts to Vb = 1.6/0.4 kJ mol−1 at the transition state with Cs symmetry in the S0/S1 state with an imaginary frequency of i113/i137 cm−1 for the concerted motion of CH3 rotation and bending of the aromatic ring. The close proximity of the excess proton in O2+ and N1 leads to a double minimum potential for proton transfer. The barrier for this tautomerization process is very high in both the S0 and S1 states (Vb = 156 and 157 kJ mol−1 for O2+ → N1 in S0 and S1) so that these isomers can be treated as two distinct protomers cooling down in their own deep potential wells.
The vibronic S1 ← S0 spectrum predicted for the N1 protomer by the FC simulations is compared in Fig. 2 to the VISPD spectrum of H+LF. We reiterate that the order between the lowest bright ππ* and dark nπ* excited states changes for this protomer when going from vertical to adiabatic excitations, and that the bright ππ* state is in fact the S2 state. The origin of the computed spectrum is aligned with the peak at 75 cm−1 internal energy, requiring a redshift of merely 177 cm−1 for the origin calculated at 23025 cm−1. The calculated FC spectra are scaled vertically to match the associated intensity of the experimental S1 origins. The calculated FC intensity of the O2+ origin is one order of magnitude higher than that of the N1 protomer, while the oscillator strengths differ by a factor of only 2.5 (0.063 versus 0.025). This difference may indicate a somewhat higher abundance of the more stable O2+ protomer, although it is rather difficult to estimate the abundance ratio of both protomers in a more quantitative fashion because of different FC pattern and possibly different photodissociation cross sections. Due to the loss of symmetry, all modes of the N1 protomer become FC allowed, and thus the density of predicted transitions is much greater than for the O2+ protomer. This is particularly noticeable in the vicinity of the S1 origins, because the low-frequency out-of-plane fundamentals forbidden for O2+ become allowed and quite active for N1. The FC simulations for N1 clearly do not match the experimental spectrum with the same quality as the corresponding simulations for O2+ with respect to both frequency and relative intensities. A tentative assignment for the transitions a–t is suggested in Table 5. The first four modes (n1–n4) above the S1 origin are predicted at 43, 58, 70, and 87 cm−1, with corresponding experimental frequencies of 36 (c), 49 (e), 63 (h), and 98 (j) cm−1, respectively. Mode n1 is a butterfly motion describing the motion leading to nonplanarity of the molecular ion, while mode n2 describes shearing or twisting of the tricyclic ring around its long axis. Mode n3 corresponds to a hindered internal rotation of the CH3 group at C7, while mode n4 is a rocking motion of the whole CH3 group at N10 (see Fig. S10 in the ESI† for a graphical representation of the a′′ normal modes). There are more peaks in the experimental spectrum than in the FC simulation, such as the low-frequency transition at 15 cm−1 (a). We find indeed several pronounced transitions with a spacing of 14–16 cm−1, which may indicate a very low-frequency out-of-plane mode not properly predicted by the harmonic calculations. Several reasons may cause the failure of the simple harmonic FC calculations of these low-frequency a′′ modes. First, the barrier of the bent N1 structure to linearity is rather low in both S0 and S1 (<2 kJ mol−1), which may cause substantial errors in the harmonic force field and/or large anharmonic corrections for the butterfly motion (mode n1) and resulting FC intensities. Second, several low-frequency modes involve hindered internal methyl rotations,40 which may not be described well by the harmonic approach. Third, out-of-plane modes may be affected by vibronic coupling to the dark nπ* state lying just 537 cm−1 below the bright ππ* state. In fact, the near in-plane modes are described well by the FC simulation, as shown by the lowest-frequency m1 mode (158 cm−1) assigned to the intense transition q at 162 cm−1. A FC simulation of the planar transition state of the N1 protomer with that of the slightly bent minimum shown in Fig. S11 in the ESI† clearly reveals that the slight bending angle causes a huge change in the appearance of the vibronic spectrum due to strong activation of the low-frequency a′′ fundamentals. A more sophisticated analysis of the vibronic structure of the bent N1 protomer beyond the harmonic FC simulations presented herein is rather challenging and beyond the scope of this work. FC simulations for the S1 transitions of the other planar protomers are shown in Fig. S12 in the ESI† and do not differ too much in the spectral range below 600 cm−1 because the binding site of the excess proton has not a drastic effect on the skeletal normal modes of the tricyclic (H+)LF ring.
The extensive FC activity in the VISPD spectra of the two assigned H+LF protomers O2+ and N1 is indicative of a large geometry change upon electronic ππ* excitation. To this end, we consider in Fig. 4 and 5 the computed structures of both protomers in their S0 and S1 states. As expected from the similar molecular orbitals involved in S1 excitation, the geometry changes are similar for both protomers, in particular because the group carrying the excess proton is only little affected by excitation. The most pronounced geometry changes occur in ring I and II. We find a strong elongation of the C8–C5a axis of 8.9 (O2+) and of 9.8 pm (N1), combined with a contraction of the C6–C9 axis of −6.0 and −5.8 pm. Ring II elongates along the N5–N10 axis by 8.9 (O2+) and 6.8 pm (N1). Ring III, where the excess proton is located in both protomers, experiences a smaller deformation. We find as a maximum change a contraction of the N3–C10a distance of 2.2 pm (O2+) and 3.1 pm (N1). The two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds elongate by 0.4–0.5 pm in O2+ and N1. As expected, the changes in the O–H and N–H bond lengths of the excess proton are negligible (≤0.1 pm) for both protomers. According to the FC principle, these structural changes upon electronic excitation translate directly into the excitation of the vibronic transitions. For example, the geometry changes of O2+ reflect closely the amplitudes of normal mode m1, giving rise to the progression observed in the electronic spectrum. Concerning the N1 protomer, the angles between the planes of ring I and III with a folding axis along N5–N10 decreases from 177° in S0 to 173° in S1, thus inducing excitation of the butterfly mode c.
O bonds elongate by 0.4–0.5 pm in O2+ and N1. As expected, the changes in the O–H and N–H bond lengths of the excess proton are negligible (≤0.1 pm) for both protomers. According to the FC principle, these structural changes upon electronic excitation translate directly into the excitation of the vibronic transitions. For example, the geometry changes of O2+ reflect closely the amplitudes of normal mode m1, giving rise to the progression observed in the electronic spectrum. Concerning the N1 protomer, the angles between the planes of ring I and III with a folding axis along N5–N10 decreases from 177° in S0 to 173° in S1, thus inducing excitation of the butterfly mode c.
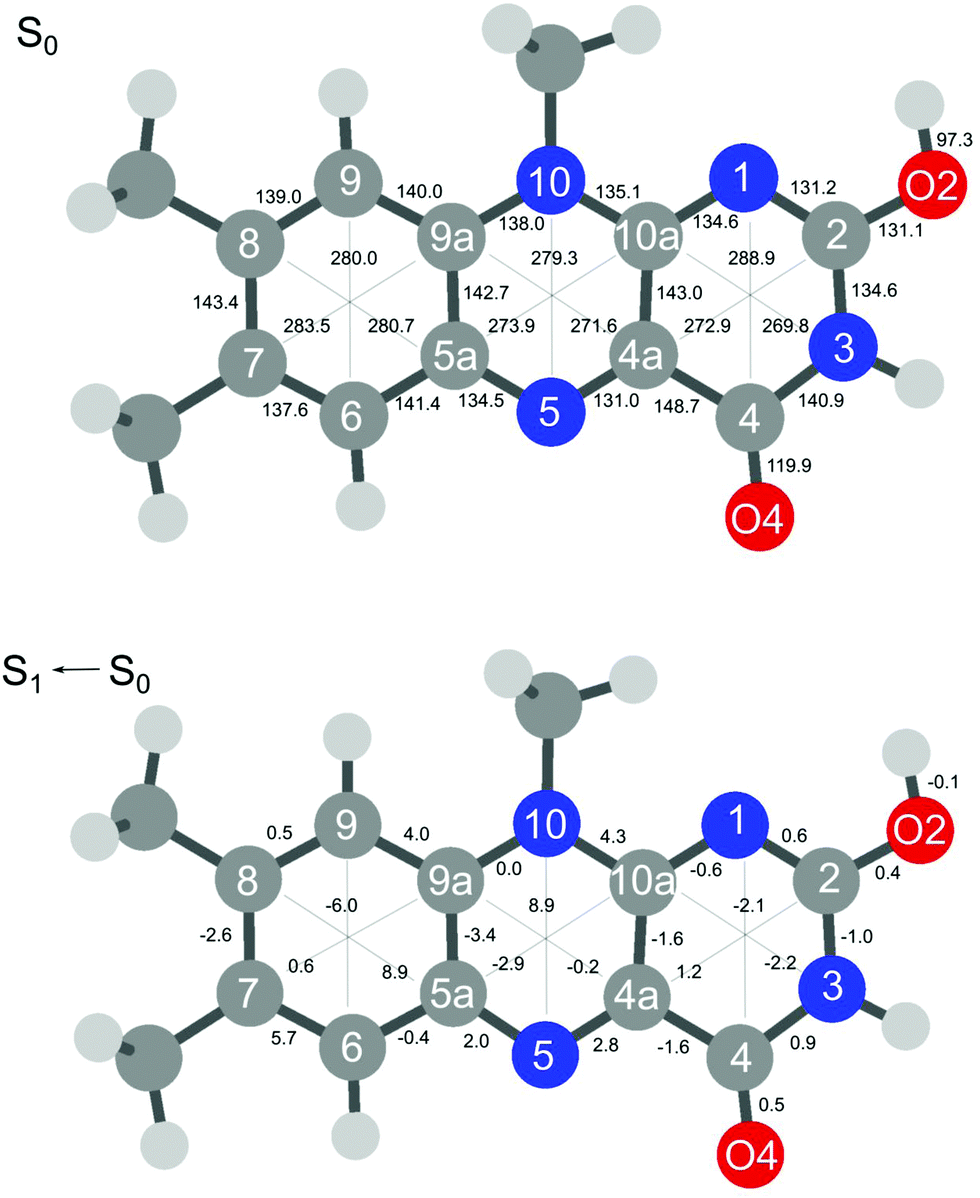 | ||
| Fig. 4 Absolute bond distances of H+LF(O2+) in the ground state S0 (top) and geometry changes upon S1 ← S0(ππ*) electronic excitation (all values in pm). | ||
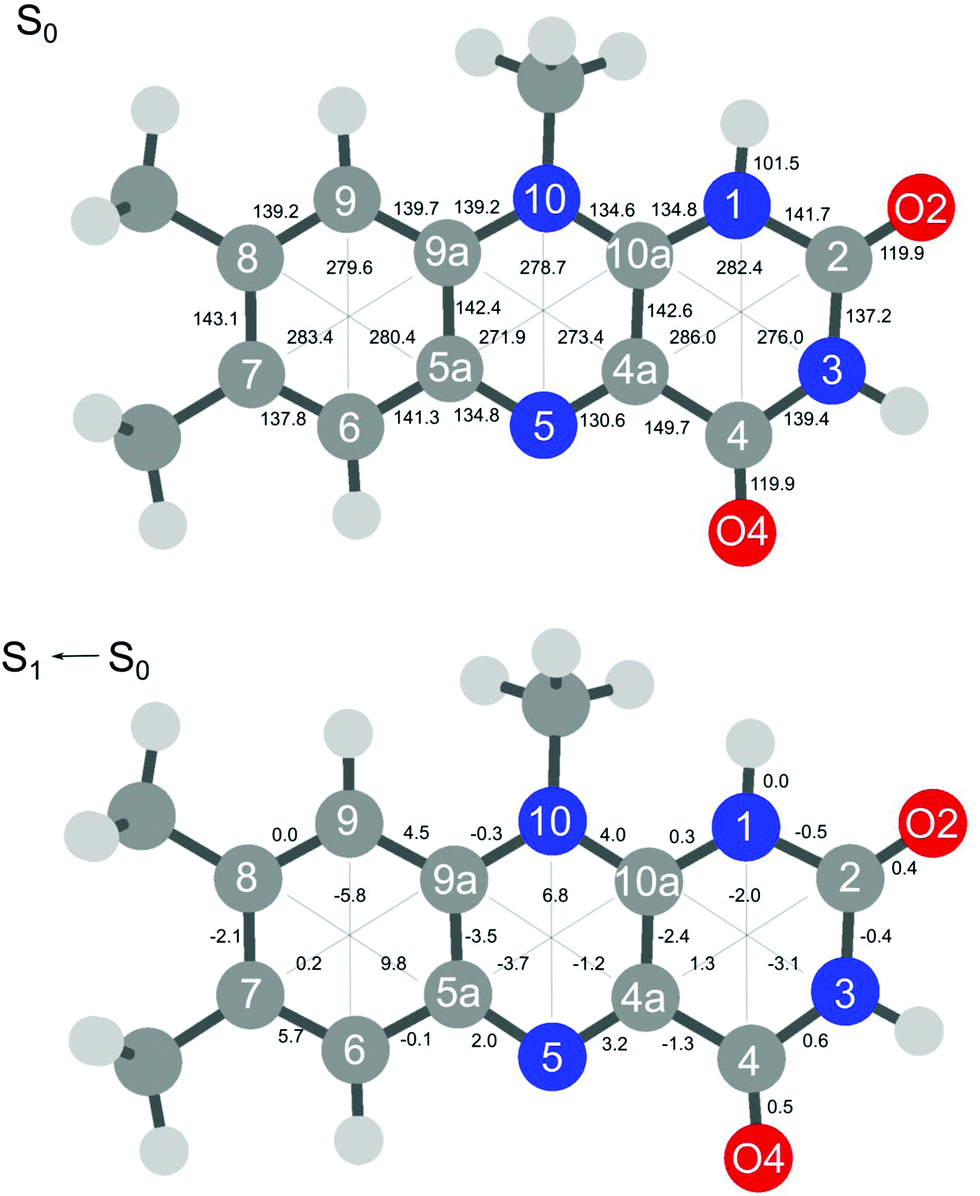 | ||
| Fig. 5 Absolute bond distances of H+LF(N1) in the ground state S0 (top) and geometry changes upon S1 ← S0(ππ*) electronic excitation (all values in pm). | ||
For completeness, the geometry changes upon O2+/N1 protonation are reported in Fig. S13 in the ESI.† Briefly, protonation at N1 and O2+ at ring III leaves the remote ring I nearly unaffected, with the largest deformation being an elongation along the C9–C6 axis of 2.1 and 1.7 pm for O2+ and N1, respectively. Within ring II, the maximum change is a contraction of the C5a–C10a axis of −2.9 and −4.9 for O2+ and N1. In contrast, ring III is highly affected by protonation because protonation occurs there. For example, O2+ protonation strongly elongates the C2–O2 bond (by 10 pm) because of the change from a double bond to a single bond (CO → C–OH), while the second C4–O4 bond slightly contracts (by 1.2 pm). On the other hand, N1 protonation hardly affects the C–O bond lengths, with a small contraction of 1.2 pm. As expected prototonation at both N1 and O2+ has a large impact on the skeleton of ring III due to changes in the conjugation. Protonation at O2+ substantially contracts the C4a–C2 axis (by 9.5 pm) and to a smaller extent the N1–C4 axis (by 1.4 pm), while the N3–C10a axis slightly elongates (by 2.0 pm). Protonation at N1 deforms ring III in a different way. The N1–C4 distance contracts by 7.9 pm and the N3–C10a axis is stretched by 8.2 pm, while the third axis C4a–C2 is less affected (by +3.6 pm). In the earlier IRMPD study of H+LF, the CO stretching frequencies are used as a sensitive indicator for the proton binding sites.27
The substitution of functional groups, protonation, and metalation has a large impact on the photophysical properties of flavins. To this end, it is instructive to compare the H+LF results obtained herein with those previously obtained for H+LC and M+LF.31,33 LC differs from LF such that LC has no functional group at N10 but a H atom at N1, while LF has a CH3 group at N10 and no group at N1. Consequently, the N1 position is not available for protonation of LC. Instead, IRMPD and VISPD spectra of H+LC reveal preferred protonation at N5, in line with quantum chemical calculations, and only the most stable H+LC(N5) protomer has been detected so far. Thus, when going from LC to LF, the preferred protonation site changes from N5 to O2+/N1. This switch in protonation site has a drastic effect on the optical spectrum, and herein we consider specifically the behaviour of the lowest ππ* state. While N5 protonation of LC induces a drastic redshift in the S1 excitation energy (by around 6000 cm−1 from ∼25000 to 19962 cm−1 or by 51 nm from ∼400 to 501 nm), protonation of LF at O2+ and N1 causes a smaller blueshifts (of the order of ∼1500 cm−1 from ∼21500 to ∼23000 cm−1 or by ∼30 nm from ∼465 to ∼435 nm). Actually, the molecular orbitals involved in S1 excitation of LC and LF do not differ much,33 and thus the magnitude and direction of the shifts upon protonation at the same protonation site are not so different for LC and LF (Table S6 in the ESI†). For example, large ΔS1 redshifts are also predicted herein for N5 and O4+ protonation of LF (6826 and 4766 cm−1, Table 2), with predicted S1 origins at 15622 and 17682 cm−1 (640 and 565 nm), a spectral range not yet investigated. However, the H+LF(N5) and H+LF(O4+) protomers are relatively high in energy (E0 = 52 and 36 kJ mol−1) and were thus not been detected in the previous IRMPD study.27 Similarly, the calculated S1 energies of LC/LF(O2+) are quite similar (22422/22535 cm−1) but again the H+LC(O2+) protomer has not been identified in its IRMPD spectrum because of its elevated relative energy (E0 = 70 kJ mol−1).27,31 As a result of the different protonation sites and position of the functional H/CH3 groups, the fragmentation processes of H+LC(N5) and H+LF(O2+/N1) upon VISPD are rather different. While for H+LC only two main fragments are observed, namely loss of CO + NH3 (or HCN + H2O) and loss of OCNH + CO, the photodissociation mass spectrum of H+LF(O2+/N1) is much richer, with many more fragmentation channels, indicating that the ring opening process upon VISPD is quite different for both protonated flavins.
In recent IRMPD experiments for M+LF with M = Li–Cs, the O4+ and O2(+) isomers have been identified by their characteristic CO stretch frequencies.28 In these metalated ions, which are isovalent to H+LF, the smaller alkali cations Li–K are large enough to simultaneously benefit from interacting with the nucleophilic lone pairs of the neighboring N and carbonyl O atoms by forming chelates of the type O4–M–N5 and O2–M–N1. Thus, there is only a single potential minimum between O2 and N1 and between O4 and N5. The ionic radius of the larger alkali ions Rb–Cs causes Pauli repulsion with the CH3 group at N10, so that no O2–M–N1 chelate can be formed, and the resulting O2 isomers feature a linear C2–O2–M bond. In contrast, the proton is much smaller than all alkali ions, and thus double minimum potentials are developed for H+LF, with two deep minima separated by high barriers for the N1 and O2+ as well as the N5 and O4+ protomers. In general, the O4+ and O2(+) isomers of M+LF have quite similar stabilities to within ΔE0 ≤ 16 kJ mol−1. The VISPD spectra of M+LF(O4+) exhibit large metalation-induced S1 redshifts of 2480–3866 cm−1 for M = Cs–Li from the S1 origin of bare LF, which increase in magnitude for decreasing ionic radii of M.33 In line with this trend, the protonation-induced redshifts predicted for H+LF(O4+/N5) are even larger (4766/6826 cm−1). For the O2(+) isomers of M+LF, much smaller ΔS1 blueshifts of 710–1048 cm−1 are calculated,33 and preliminary unpublished VISPD spectra confirm this prediction. The calculated corresponding protonation-induced blueshifts are of similar magnitude (87 and 578 cm−1 for O2+ and N1), in line with the experimentally estimated values (1617 and 1691 cm−1). The fragmentation processes upon VISPD of the isovalent M+LF and H+LF are quite different. While M+LF fragments exclusively into M+ and LF by simply breaking the mostly electrostatic M+⋯LF bond (with binding energies of D0 = 150–300 kJ mol−1), the proton affinities of the O2+ and N1 protomers of H+LF are much larger (PA = 957–972 kJ mol−1), leading to a more complex fragmentation pattern involving ring opening processes. The detailed mechanism for VISPD of the H+LF protomers remains unclear. Measurements of the VISPD yield as a function of laser power indicate a linear dependence rather than a quadratic one (Fig. S14 in the ESI†). This result may be taken as evidence for single-photon dissociation, which for example may involve S1 excitation followed by internal conversion to the S0 state and statistical dissociation on this ground state. However, the VISPD mass spectra differ from those of the CID and IRMPD process, suggesting dissociation upon visible excitation occurs on an excited state potential. This may occur by electronic predissociation from S1 to the continuum of S0 (one-photon process) or by resonant two-photon or multiphoton excitation via S1 to a higher excited Sn state followed by dissociation. While a quadratic or higher order dependence would be indicative of the latter process, the observed linear dependence is consistent with both scenarios.
In general, the absorption spectra of flavins and their derivatives recorded in the condensed and gas phase at room temperature are broad and without any vibrational structure. Thus, they do not provide reliable and precise information about the nature of the observed electronic states and the impact of the environment on their photophysical properties. In contrast, early work in cryogenic matrices demonstrated that cryogenic cooling to low temperature is required to achieve vibronic spectra of the excited states.19 The only spectroscopic work on cryogenic flavins includes the LIF spectrum of LF recorded in He droplets20 and our VISPD spectra on protonated and metalated flavin cations recorded in cryogenic ion traps.30–33 To illustrate the effect of isolating and cooling the flavin ion, the cryogenic VISPD spectrum of isolated H+LF measured herein is compared in Fig. S15 in the ESI† to the absorption spectrum of H+LF in aqueous solution at room temperature, in which the protonated flavin is generated at low pH.11 Significantly, while the VISPD spectrum reveals numerous vibronic transitions in the spectral range 420–440 nm with the S1 origins of the O2+ and N1 protomers at 432.38 and 431.00 nm, the solution phase spectrum exhibits a single broad transition peaking at 394 nm with a width of ∼60 nm and no information about the protonation site (although the authors assume N1 protonation).11 Our vertical transition energies computed for the O2+ and N1 protomers (Ev = 391 and 379 nm) are within 3 and 15 nm of the solution-phase spectrum, suggesting that hydration effects are probably not very substantial. It is however difficult to extract any reliable quantitative information about the solvation effects on the S1 transition energy. To this end, future spectroscopic measurements of microhydrated H+LF–(H2O)n clusters under controlled solvation conditions are required.
4. Concluding remarks
Herein, we present the first optical spectrum of isolated protonated lumiflavin (H+LF), the simplest member of the photochemically important flavin family. The vibronic spectrum, measured by electronic photodissociation (VISPD) of cold ions in a tandem mass spectrometer coupled to an electrospray source and a cryogenic ion trap, exhibits rich vibrational structure arising from large geometry changes upon electronic excitation. The analysis of the measured VISPD spectrum by TD-DFT calculations coupled to multidimensional FC simulations suggests an assignment of the spectral features near 430 nm to the optically bright S1 ← S0(ππ*) transition of the two most stable H+LF tautomers protonated at either the O2+ or the N1 position (E0 = 0 and 15 kJ mol−1), with band origins at 23128 and 23202 cm−1, respectively. While the O2+ global minimum has been identified in previous IRMPD experiments, the less stable N1 local minimum separated by a large barrier of ∼150 kJ mol−1 is assigned herein for the first time. Although this discrepancy may be due to different ESI conditions in the two studies (IRMPD versus VISPD), we ascribe the detection of the less stable isomer by the much higher spectral resolution and sensitivity of cryogenic electronic spectroscopy compared to the room-temperature IRMPD approach. While the planar O2+ protomer has Cs symmetry with a regular FC pattern in totally symmetric in-plane modes, the N1 isomer is slightly bent giving rise to a much denser vibronic structure produced by low-frequency out-of-plane modes. The observed modest blueshifts in the S1 origins upon protonation are in line with the molecular orbitals involved in this ππ* transition and the related atomic charge distributions of both electronic states, which are indicative of a small reduction of the proton affinity in the excited electronic state of both protomers. In that sense, the O2+ and N1 tautomers of H+LF show a rather similar photochemical response. Their spectral behaviour is, however, very different from that of the related H+LC ion, which prefers protonation at N5 and thus has a drastically different optical spectrum, confirming that substitution of flavins with functional groups can indeed drastically change their photochemical properties. Future efforts of extending this work include (i) the confirmation of the detection of the two different H+LF tautomers by double resonance techniques (IR–VIS or VIS–VIS), (ii) the study of larger flavin ions with higher biochemical relevance such as FMN and FAD, and (iii) the investigation of microsolvated flavin ions with particular focus on hydration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Deutsche Forschungsgemeinschaft (DFG, project DO 729/6-2). We thank Alkwin Slenczka for providing a digital version of the spectrum of LF.References
- V. Massey, Biochem. Soc. Trans., 2000, 28, 283 CrossRef CAS.
- P. F. Heelis, Chem. Soc. Rev., 1982, 11, 15 RSC.
- A. Losi, Photochem. Photobiol., 2007, 83, 1283 CrossRef CAS PubMed.
- A. Losi and W. Gärtner, Photochem. Photobiol., 2011, 87, 491 CrossRef CAS PubMed.
- A. Sancar, Chem. Rev., 2003, 103, 2203 CrossRef CAS PubMed.
- W. Buckel and R. K. Thauer, Chem. Rev., 2018, 118, 3862 CrossRef CAS PubMed.
- E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Chem. Rev., 2018, 118, 1742 CrossRef CAS PubMed.
- K. H. Dudley, A. Ehrenberg, P. Hemmerich and F. Müller, Helv. Chim. Acta, 1964, 47, 1354 CrossRef CAS.
- E. Sikorska, I. V. Khmelinskii, W. Prukła, S. L. Williams, M. Patel, D. R. Worrall, J. L. Bourdelande, J. Koput and M. Sikorski, J. Phys. Chem. A, 2004, 108, 1501 CrossRef CAS.
- W. Holzer, J. Shirdel, P. Zirak, A. Penzkofer, P. Hegemann, R. Deutzmann and E. Hochmuth, Chem. Phys., 2005, 308, 69 CrossRef CAS.
- A. Tyagi and A. Penzkofer, J. Photochem. Photobiol., A, 2010, 215, 108 CrossRef CAS.
- A. Tyagi and A. Penzkofer, Photochem. Photobiol., 2011, 87, 524 CrossRef CAS PubMed.
- C. Neiss, P. Saalfrank, M. Parac and S. Grimme, J. Phys. Chem. A, 2003, 107, 140 CrossRef CAS.
- J. Hasegawa, S. Bureekaew and H. Nakatsuji, J. Photochem. Photobiol., A, 2007, 189, 205 CrossRef CAS.
- S. Salzmann and C. M. Marian, Chem. Phys. Lett., 2008, 463, 400 CrossRef CAS.
- S. Salzmann, J. Tatchen and C. M. Marian, J. Photochem. Photobiol., A, 2008, 198, 221 CrossRef CAS.
- M. Meyer, H. Hartwig and D. Schomburg, J. Mol. Struct.: THEOCHEM, 1996, 364, 139 CrossRef CAS.
- E. Sikorska, I. V. Khmelinskii, J. Koput, J. L. Bourdelande and M. Sikorski, J. Mol. Struct., 2004, 697, 137 CrossRef CAS.
- R. J. Platenkamp, H. D. van Osnabrugge and A. J. W. G. Visser, Chem. Phys. Lett., 1980, 72, 104 CrossRef CAS.
- A. Vdovin, A. Slenczka and B. Dick, Chem. Phys., 2013, 422, 195 CrossRef CAS.
- M. H. Stockett, Phys. Chem. Chem. Phys., 2017, 19, 25829 RSC.
- E. Matthews, R. Cercola and C. E. H. Dessent, Molecules, 2018, 23, 2036 CrossRef PubMed.
- K. Lincke, J. Langeland, A. Ø. Madsen, H. V. Kiefer, L. Skov, E. Gruber, K. V. Mikkelsen, L. H. Andersen and M. B. Nielsen, Phys. Chem. Chem. Phys., 2018, 20, 28678 RSC.
- J. N. Bull, E. Carrascosa, L. Giacomozzi, E. J. Bieske and M. H. Stockett, Phys. Chem. Chem. Phys., 2018, 20, 19672 RSC.
- E. Matthews and C. E. H. Dessent, J. Phys. Chem. Lett., 2018, 9, 6124 CrossRef CAS PubMed.
- L. Giacomozzi, C. Kjær, J. L. Knudsen, L. H. Andersen, S. B. Nielsen and M. H. Stockett, J. Chem. Phys., 2018, 148, 214309 CrossRef CAS PubMed.
- J. Langer, A. Günther, S. Seidenbecher, G. Berden, J. Oomens and O. Dopfer, ChemPhysChem, 2014, 15, 2550 CrossRef CAS PubMed.
- P. Nieto, A. Günther, G. Berden, J. Oomens and O. Dopfer, J. Phys. Chem. A, 2016, 120, 8297 CrossRef CAS PubMed.
- A. Günther, P. Nieto, G. Berden, J. Oomens and O. Dopfer, Phys. Chem. Chem. Phys., 2014, 16, 14161 RSC.
- A. Günther, P. Nieto, D. Müller, A. Sheldrick, D. Gerlich and O. Dopfer, J. Mol. Spectrosc., 2017, 332, 8 CrossRef.
- A. Sheldrick, D. Müller, A. Günther, P. Nieto and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 7407 RSC.
- P. Nieto, D. Müller, A. Sheldrick, A. Günther, M. Miyazaki and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 22148 RSC.
- D. Müller, P. Nieto, M. Miyazaki and O. Dopfer, Faraday Discuss., 2019, 217, 256 RSC.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- V. Barone, J. Bloino, M. Biczysko and F. Santoro, J. Chem. Theory Comput., 2009, 5, 540 CrossRef CAS PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- S. Salzmann, V. Martinez-Junza, B. Zorn, S. E. Braslavsky, M. Mansurova, C. M. Marian and W. Gärtner, J. Phys. Chem. A, 2009, 113, 9365 CrossRef CAS PubMed.
- P. J. Breen, J. A. Warren, E. R. Bernstein and J. I. Seeman, Acc. Chem. Res., 1987, 87, 1917 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp03650a |
| This journal is © the Owner Societies 2020 |