
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anisotropy of Pt nanoparticles on carbon- and oxide-support and their structural response to electrochemical oxidation probed by in situ techniques†
Henrike
Schmies
 a,
Arno
Bergmann‡
a,
Elisabeth
Hornberger
a,
Jakub
Drnec
b,
Guanxiong
Wang
c,
Fabio
Dionigi
a,
Stefanie
Kühl
a,
Daniel J. S.
Sandbeck
d,
Karl J. J.
Mayrhofer
d,
Vijay
Ramani
c,
Serhiy
Cherevko
d and
Peter
Strasser
*a
a,
Arno
Bergmann‡
a,
Elisabeth
Hornberger
a,
Jakub
Drnec
b,
Guanxiong
Wang
c,
Fabio
Dionigi
a,
Stefanie
Kühl
a,
Daniel J. S.
Sandbeck
d,
Karl J. J.
Mayrhofer
d,
Vijay
Ramani
c,
Serhiy
Cherevko
d and
Peter
Strasser
*a
aDepartment of Chemistry, Chemical Engineering Division, Technical University of Berlin, Berlin, Germany. E-mail: pstrasser@tu-berlin.de
bEuropean Synchrotron Radiation Facility (ESRF), Grenoble, France
cSchool of Engineering & Applied Science, Washington University in St. Louis, St. Louis, USA
dHelmholtz-Institute Erlangen-Nürnberg for Renewable Energy (IEK-11), Forschungszentrum Jülich, Erlangen, Germany
First published on 8th September 2020
Abstract
Identifying the structural response of nanoparticle–support ensembles to the reaction conditions is essential to determine their structure in the catalytically active state as well as to unravel the possible degradation pathways. In this work, we investigate the (electronic) structure of carbon- and oxide-supported Pt nanoparticles during electrochemical oxidation by in situ X-ray diffraction, absorption spectroscopy as well as the Pt dissolution rate by in situ mass spectrometry. We prepared ellipsoidal Pt nanoparticles by impregnation of the carbon and titanium-based oxide support as well as spherical Pt nanoparticles on an indium-based oxide support by a surfactant-assisted synthesis route. During electrochemical oxidation, we show that the oxide-supported Pt nanoparticles resist (bulk) oxide formation and Pt dissolution. The lattice of smaller Pt nanoparticles exhibits a size-induced lattice contraction in the as-prepared state with respect to bulk Pt but it expands reversibly during electrochemical oxidation. This expansion is suppressed for the Pt nanoparticles with a bulk-like relaxed lattice. We could correlate the formation of d-band vacancies in the metallic Pt with Pt lattice expansion. PtOx formation is strongest for platelet-like nanoparticles and we explain this with a higher fraction of exposed Pt(100) facets. Of all investigated nanoparticle–support ensembles, the structural response of RuO2/TiO2-supported Pt nanoparticles is the most promising with respect to their morphological and structural integrity under electrochemical reaction conditions.
1. Introduction
Since the use of fossil fuel combustion devices in the automotive sector has to be largely reduced, the development of clean energy conversion devices such as polymer electrolyte membrane fuel cells (PEMFCs) has gained increasing interest. Catalyst systems at the cathode side of these fuel cells have been studied intensively with respect to the used materials and their properties.1,2 Supported Pt3–5 or Pt alloy nanoparticles6–12 are commonly used as an electrocatalyst for the oxygen reduction reaction (ORR). Degradation processes such as Ostwald ripening/agglomeration,13–15 carbon corrosion16,17 and dissolution18–23 of the active component decrease the overall performance and lifetime of the fuel cell. Surface processes such as Pt oxidation occurring at potentials higher than 1 V have an extensive influence on catalyst stability. Degradation phenomena due to dissolution are directly linked to the formation of PtOx-species and the consecutive reductive dissolution of Pt. Therefore, unraveling the origin of these surface processes is of great interest to understand the degradation behavior at a fundamental level.On Pt single crystal surfaces it was found that Pt oxidation occurs in the form of place exchange of Pt and O atoms on the Pt(111) surface.24,25 By in situ surface X-ray diffraction (XRD) it was shown that with increasing potential the oxide structure consists of a Pt-rich inner and an O-rich outer layer which transforms into a more homogeneous oxide at above 1.1 V. By cycling back to more negative potentials, the authors could prove that up to a critical oxide coverage the process of place exchange is quasi-reversible because place-exchanged atoms are located on top of their original positions. In another study by Feliu et al. no changes in the voltammograms were observed by consecutive cycling up to 1.15 V evidencing reversible ordering of Pt(111).26 Pt oxidation in the presence of oxygen was studied by coulometric and gravimetric measurements on a smooth polycrystalline Pt surface. Place exchange was assumed to occur in the presence of O2 at lower potentials (0.75 V).27 The structural sensitivity of intermediate stages of Pt oxidation was investigated by surface-enhanced Raman spectroscopy on Pt(111) and Pt(100). Therein, it was shown that place-exchange and the formation of amorphous 3D α-PtO2 proceed sequentially on Pt(111) whereas on Pt(100) both processes take place simultaneously above 1.1 V.28 Investigation of the dissolution trends of Pt single crystals using the scanning flow cell setup by Sandbeck et al. revealed a correlation between dissolution behavior and surface energies of the studied single crystals. It was found that low coordinated Pt surface sites such as Pt(110) exhibit the lowest dissolution rates.29 Recently, Fuchs et al. explicitly demonstrated that such surface-dependent dissolution is correlated to different place exchange mechanisms on Pt(111) and Pt(100) single crystals, using in situ high-energy surface X-ray diffraction, online inductively coupled plasma mass spectrometry and density functional theory calculations.30 Studies on a monolayer of Pt deposited on a Rh(111) single crystal showed the evolution of a Pt oxide structure by electrochemical oxidation up to high potentials of 1.6 V while at potentials below 1 V chemisorbed oxygen-containing species at the Pt surface were proposed. The onset of Pt oxide formation was found to be dependent on the kinetic stability of chemisorbed species resulting from different surfaces (single crystal vs. nanoparticle). By a combination of computational and experimental methods Pt3O4 was in best agreement with both methods.31
High-energy-resolution X-ray absorption spectroscopy showed that for size-selected Pt nanoparticles of ∼1 nm different adsorbed species on the Pt surface (hydrogen, O/OH and Pt oxide) can be distinguished in certain potential regions.32 Formation of Pt oxides by broadening of the white line was found to begin at 0.96 V, which is lower than seen on single crystals and was explained with a stronger bond of Pt nanoparticle surface to oxygen. In another study by Adzic et al., a wide potential range up to 2.6 V was investigated by in situ X-ray methods.33 According to the authors, O and OH adsorption at around 0.7 V and place exchange onset at 0.9 V were observed resulting in the formation of α-PtO2 which was dissolved at high potentials above 1.9 V. Additionally, Pt oxidation only occurs at the outer Pt layers and the inner, bulk part remains metallic. Imai et al. by time-resolved in situ X-ray diffraction and absorption experiments on Pt nanoparticles also proposed the formation of α-PtO2 by analyzing the Pt–O bond length.34
A study on potential dependent structural transformation of Pt on different carbon supports showed that, after aging, Pt nanoparticle surfaces were oxidized to Pt(2+)O on reaching potentials above 1.4 V. Differences were observed for the onset of oxide formation and the reduction to the metallic state for different supports with behaviors varying from full reversibility to irreversibility.35
Pt oxidation was also studied on nanoparticles supported on a metal oxide. For a ceria support it was shown by XPS and TEM that the oxide support has an impact on the formation of Pt oxide as a composite of Pt oxide and Ce was formed, but it was reversibly reconverted to metallic Pt.36 The authors interpreted this phenomenon as evidence for strong interaction between CeO2 and Pt. Formation of Pt–O–Ce bonds stabilizes Pt from sintering under an oxidative atmosphere. Another study on a model catalyst of Pt/CeO2(111) showed the possibility to quantify the charge transfer between small Pt particles on a well-defined ceria support by combining STM and XPS techniques. The electronic metal–support interactions in the form of transferred electrons per Pt atom were found to be dependent on Pt nanoparticle size.37 Even though these studies were conducted at different temperatures and gas atmospheres and not in realistic electrochemical environments, one could draw first conclusions of Pt oxidation/reduction in oxide-supported Pt catalysts from them. Brummel et al. reported a stabilizing effect from the support of Pt/CeO2 with low Pt loadings and Pt particles up to 30 atoms during methanol oxidation.38 In another study, the authors further explored the stabilizing interaction between metal and oxide support by analyzing a Pt/Co3O4(111) model system. They found an interface Pt oxide which is stable between 0.5 and 1.4 V and believed this to be the reason for the catalyst's high stability towards particle sintering.39 Studies on model electrodes made from doped-SnO2 thin films supporting Pt nanoparticles showed that electronic metal–support interactions are the cause for superior specific ORR activity when compared to Pt on carbon.40,41
Recently, we showed that the density of oxidizable Pt sites is an important property governing catalytic activity for ORR as we investigated the degradation pathways of ITO-supported Pt nanoparticles under simulated fuel cell conditions using combined in situ X-ray diffraction and scattering techniques.42
In this work, we studied the structural response of Pt nanoparticles on different supports to the electrochemical oxidation using different in situ X-ray techniques. We tracked the Pt dissolution rate as well as the voltammetric profiles of Pt nanoparticles and monitored the crystalline properties and the electronic structure during oxidation.
2. Experimental section
2.1. Synthesis of oxide supports
Indium Tin Oxide (ITO) was synthesized as reported in earlier works.42,43 Propylene epoxide was added to a mixture of In(Cl)3 and Sn(Cl)4 in water and ethanol. After aging and solvent exchange with acetone the resulting gel was dried by supercritical CO2 extraction and calcined at 820 °C in air.In a typical synthesis of Ruthenium Titanium Oxide (RTO),44,45 the ruthenium precursor was mixed with TiO2 and the pH was adjusted to 7 by adding KOH. The resulting powder was calcined in two steps at 120 and 450 °C under an air atmosphere.
2.2. Pt deposition on oxide supports
Pt was deposited on ITO as reported in a previous work.42 In short, a solvothermal synthesis route was applied in which the support and the Pt(acac)2 precursor were mixed prior to the heating protocol to ensure sufficient dispersion of Pt nanoparticles on ITO. Long-chain surfactants were used to control Pt particle size and a reducing agent was used to yield metallic Pt from the Pt precursor. Surfactants were removed by centrifugation and washing with ethanol. No further heat treatment was applied.For the RTO support, Pt was deposited via a wet chemical approach as reported elsewhere.44,45 In a typical synthesis, a suspension of support, hexachloroplatinic acid precursor and reducing agent was heated to 80 °C for 2 h and washed with water afterwards.
A commercially available carbon-supported Pt catalyst (20 wt% Pt on Vulcan, BASF) was used for comparison.
2.3. Inductively coupled plasma optical emission spectroscopy (ICP-OES)
In order to determine the Pt weight loading on the supports, compositional analysis using ICP-OES (Varian 715-ES) was performed. 5–10 mg of catalyst powder was digested in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3-mixture of HNO3/H2SO4/HCl using a microwave treatment for 20 min at 180 °C and 18 bar. Elemental standards with concentrations 1, 5 and 10 mg L−1 and Pt emission lines at 203.646, 204.939, 212.863, 214.424, 217.468, and 224.552 nm were used for quantitative analysis.
:1:3-mixture of HNO3/H2SO4/HCl using a microwave treatment for 20 min at 180 °C and 18 bar. Elemental standards with concentrations 1, 5 and 10 mg L−1 and Pt emission lines at 203.646, 204.939, 212.863, 214.424, 217.468, and 224.552 nm were used for quantitative analysis.
2.4. Transmission electron microscopy (TEM)
TEM images were obtained using a FEI Tecnai G2 20 S-TWIN with a LaB6 cathode operating with 200 kV acceleration voltage; the resolution limit was 0.24 nm. For sample preparation, a small amount of catalyst powder was dispersed in absolute ethanol using an ultrasonic horn and afterwards drop cast on a Cu grid and dried in air at 60 °C for 7 min.2.5. Electrochemical characterization
For ink preparation a defined mass of catalyst powder was added to 1.99 mL of water, 500 μL of iPrOH (≥99.95%, Sigma Aldrich) and 10 μL of Nafion solution (5 wt%, Sigma Aldrich). The mixture was sonicated using an ultrasonic horn for 30 min. 10 μL of the ink was drop cast on a 5 mm diameter glassy carbon (GC) electrode and dried in air at 60 °C in an oven for 7 min resulting in a geometric Pt loading of 25 μgPt cm−2 for all RDE measurements. Two polishing-steps were applied to the GC electrode using Buehler alumina polishing solution with diameter 1.0 and 0.05 μm.For rotating disc electrode (RDE) measurements a commercial three electrode cell was used equipped with a mercury/mercury sulfate (MMS) reference electrode (Hg/Hg2SO4, Ametek, potential −0.722 V vs. reversible hydrogen electrode (RHE)) and a Pt mesh (furled Pt 5 × 5 cm2) as a counter electrode. 0.1 M HClO4 electrolyte (diluted with Milli-Q from 70% HClO4, 99.999% trace metal bases, Sigma Aldrich) was used for all measurements. To control potential and rotation a SP-150 or SP-200 Potentiostat (BioLogic Instruments) and a commercial AFMSRCE rotator (Pine Research) were used.
Cyclic voltammograms (CVs) to determine the electrochemical surface area based on underpotential deposition of H (Hupd-ECSA) were recorded between 0.05 and 1.0 V with a scan rate of 100 mV s−1 for 100 cycles in a nitrogen-saturated electrolyte. For Pt electrooxidation, three CVs each from 0.05 to 0.5 V to an increasing upper potential limit of 1.4 V in steps of 0.1 V were measured under a nitrogen-saturated atmosphere. For CO stripping experiments, a nitrogen-saturated electrolyte was generated. Then the electrode was immersed in the electrolyte under potential control of 0.05 V and a rotation speed of 400 rpm and CO was bubbled through the electrolyte for 15 min. To remove excess CO from the electrolyte, the gas was changed to nitrogen for another 15 min. CO oxidation was performed by cycling between 0.05 and 1.00 V for 5 cycles with a scan rate of 50 mV s−1.
Determination of Hupd-ECSA was done by integration in the Hupd region between 0.05 and 0.40 V while subtracting capacitive currents and assuming a theoretical value for a one electron transfer (QtheoH = 210 μC cm−2) for the adsorption/desorption of hydrogen. For CO-ECSA, the charge QCO was estimated by subtracting the first cycle from the second cycle of the stripping protocol and normalizing this to the theoretical value for a two-electron transfer (QtheoCO = 420 μC cm−2).
For all in situ electrochemical X-ray measurements, a homemade three electrode cell allowing measurements in transmission geometry was used.11 In this setup, the working electrode consists of a carbon paper sheet on which the catalyst ink (10 × 10 μL) was deposited and dried. A Pt wire as the counter electrode and an Ag/AgCl (3 M) reference electrode (freshly calibrated against a Pt/H2 electrode) were used. A SP-200 potentiostat (BioLogic Instruments) was used to control the potential and to apply electrochemical protocols. Prior to in situ X-ray characterization, the electrode potential was applied for at least 10 min for equilibration.
All potentials shown are referred to the RHE scale and are iR-corrected by the high frequency resistance RHF determined by potentiostatic electrochemical impedance spectroscopy (PEIS) at 0.5 V.
2.6. In situ high energy X-ray diffraction (HE-XRD)
In situ HE-XRD measurements were conducted at the European Synchrotron Radiation Facility (ESRF) in Grenoble at beamline ID 31. X-ray diffractograms were obtained using a focused, monochromatized X-ray beam with an energy of 77 keV. A large area Pilatus 3X CdTe 2 M detector was used to record the images. The obtained diffraction patterns were corrected by the background and a CeO2 standard (NIST SRM 674b) was used for calibration of the working distance.HE-XRD patterns were integrated and the intensity plots fitted using TOPAS®. Therein, the unit cell parameter was fitted and the occupancy of the tetrahedral sites with O atoms in the Pt lattice refined. The size- and strain-induced broadening of the Pt diffraction peaks was fitted. The size-induced broadening was fitted using the anisotropy approach developed by Ectors et al.46 In the case of the HE-XRD pattern of Pt nanoparticles on ITO, spherical nanoparticles with all three main axes of the ellipsoids identical (Rx = Ry = Rz) give the best fit results. In the case of Pt nanoparticles on RTO and on carbon, the size-induced broadening was fitted with three independently fitted principal axes of the ellipsoids (Rx ≠ Ry ≠ Rz). The polydispersity of the nanoparticles representing the particle size distribution was also taken into account.
The crystal properties of each catalyst were individually fitted in each investigated state but a joint fit approach was applied to determine zero offset and sample displacement.
The ECSA-normalized changes of the volume of Pt crystallites and the ECSA-normalized changes of the Pt scale factor as shown in Fig. 3 were obtained by normalization with respect to Hupd-ECSA.
2.7. In situ Pt L3-edge X-ray absorption near-edge spectroscopy
In situ X-ray absorption spectroscopy was performed at the μSpot beamline of Bessy II operated by the Helmholtz-Zentrum Berlin für Materialien und Energie GmbH. A Si(111) double crystal monochromator was used and the X-ray energy was aligned for each spectrum by shifting the first derivative of the simultaneously measured X-ray absorption spectra of Pt foil reference to 11654 eV. In situ X-ray absorption spectra were recorded in fluorescence yield using a scintillation counter as the detector and Al foil applied to reduce the background radiation.
The fit of the white line area and the resonances of the Pt L3 XANES spectra has been performed inspired by Merte et al.32 Three different resonances were used to fit with Res1 at ∼11566 eV, Res2 at ∼11570 eV (according to ref. 32) and an additional Res3 at ∼11580 eV. The ECSA-normalized white line areas as shown in Fig. 4 were obtained by normalization with respect to Hupd-ECSA.
2.8. In situ scanning flow cell (SFC) inductively coupled plasma mass spectrometry (ICP-MS)
For in situ SFC-ICP-MS measurements, inks of Pt/RTO, Pt/ITO and Pt/C were prepared from a solution of 25 μL of 5% Nafion (Sigma-Aldrich) and 4.975 mL of ultrapure water (18.2 MΩ cm, PureLab Plus System, Elga). The ink was sonicated and subsequently 0.3 μL drops were deposited onto the glassy carbon sheet (SIGRADUR® G, HTW) which was polished with a 1 μm Al2O3 suspension and cleaned with ultrapure water earlier. The spots of catalyst ink had a size of ca. 0.01 cm2 and a final loading of approximately 1.6 μgPt cm−2 for Pt/ITO and Pt/C and 8.33 μgPt cm−2 for Pt/RTO. In situ dissolution measurements were conducted using the previously described SFC-ICP-MS setup (NexION 300X, PerkinElmer).47,48 The contact area between the working electrode and the cell was 0.35 cm2. A freshly calibrated saturated Ag/AgCl electrode and graphite rod were used as reference and counter electrodes, respectively. Argon-saturated 0.1 M HClO4 prepared from ultrapure water and Suprapur 70% perchloric acid (Merck) was used as an electrolyte. The flow rate of the electrolyte in the SFC was approximately 170 μL min−1. 10 μg L−1 of Re (187Re was monitored) was used as an internal standard for Pt. The ECSA-normalized dissolution profiles as shown in Fig. 5 and Fig. S8, S10a (ESI†) were obtained by normalization with respect to Hupd-ECSA.3. Results and discussion
3.1. Structure and morphology
In order to investigate the differences in the electrochemical oxidation of Pt nanoparticles on various kinds of support, a commercial carbon supported Pt electrocatalyst was compared to two oxide-supported materials, in particular Indium Tin Oxide (ITO) and RuO2–TiO2 (RTO). Compositional analysis by ICP-OES revealed a Pt weight loading of 46.1 wt% for Pt/RTO, 29.9 wt% for Pt/ITO and 20 wt% for Pt/C (Table S1, ESI†). Fig. 1 shows the diffraction pattern and TEM images of the three catalysts in the as-prepared state.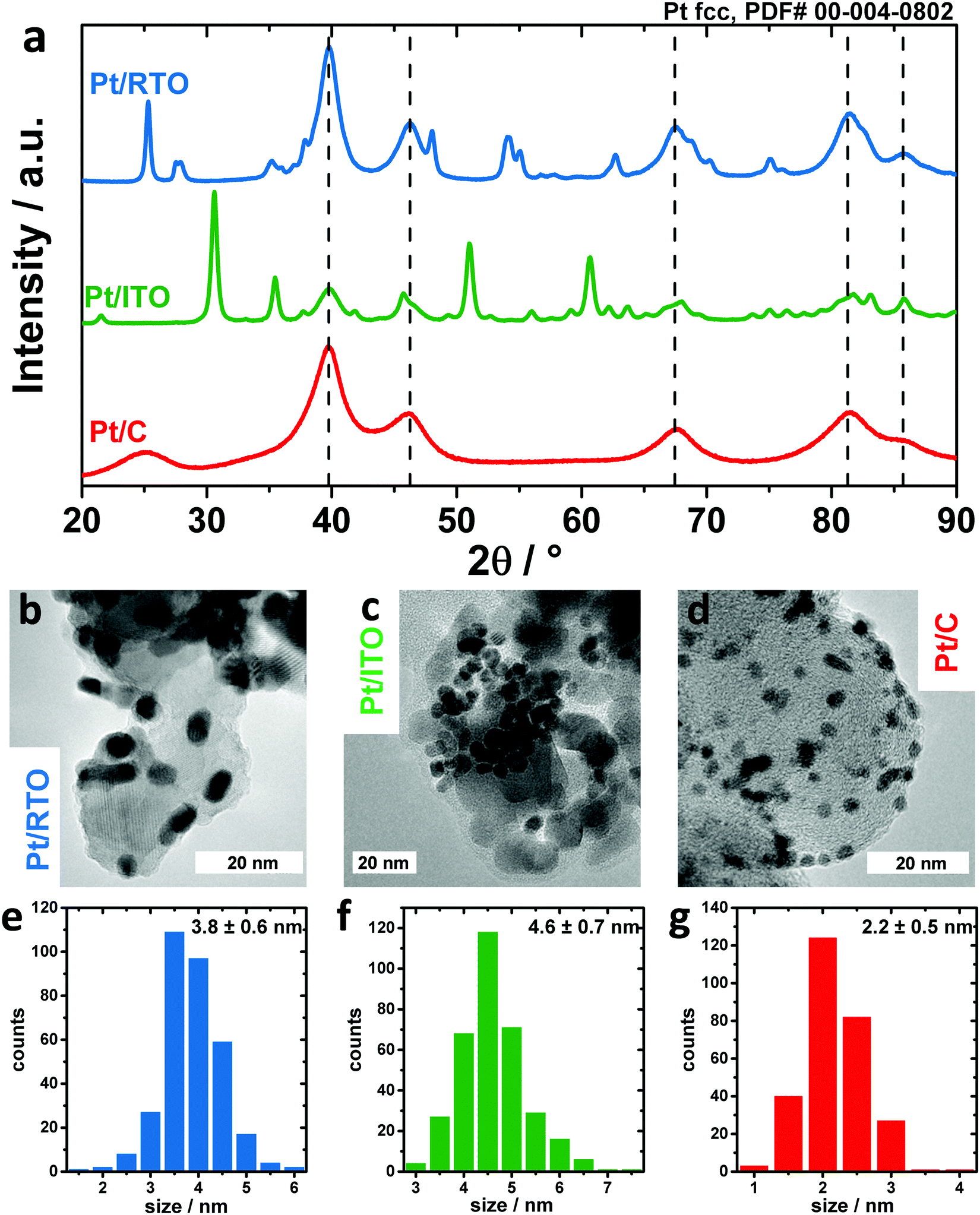 | ||
| Fig. 1 Structure and morphology of Pt nanoparticles supported on RTO (blue), ITO (green) and carbon (red) as determined by the powder X-ray diffraction pattern (a) and transmission electron microscopy images (b–d). The Pt particle size as depicted in the histograms (e–g) was determined from analyzing >200 Pt particles along their shortest principal axes. | ||
All three diffractograms in Fig. 1a show broad reflexes corresponding to a face-centered cubic Pt phase. In the case of the ITO support, a second phase is present that can be clearly assigned to In2O3 (see also Fig. S1, ESI† for more detailed information on XRD) and no SnOx phase was identified, meaning that Sn was likely incorporated into the indium oxide lattice. In the case of the RTO support, three crystalline phases can be identified in addition to the Pt phase: an anatase TiO2 as well as a rutile-type TiO2 and RuO2 as depicted in Fig. S1b (ESI†). In the case of the carbon support, broad reflexes of carbon are observed.
The morphology of the catalysts is depicted in Fig. 1b–d in TEM images. For all three supports, the Pt nanoparticles are homogeneously distributed on the supports (see also Fig. S2, ESI†). From the analysis of the Pt particle size along the shortest particle axes (Fig. 1e–g), ITO-supported nanoparticles exhibit the largest average size with 4.6 ± 0.7 nm, RTO-supported nanoparticles 3.8 ± 0.6 nm and carbon-supported nanoparticles the smallest size with 2.2 ± 0.5 nm. However, the shape of the Pt nanoparticles on C and RTO supports deviates from a sphere. Their anisotropy was investigated by determining their particle size along the longest particle axis. For Pt/RTO and Pt/C the particle size estimated in this way is around 1.5 and 1.1 nm larger, proving the anisotropic ellipsoidal shape of these nanoparticles (see also Fig. S3, ESI†). In the case of Pt/ITO, the nanoparticle size determined along the shortest and longest particle axes from the TEM images is 4.6 ± 0.7 nm and 5.0 ± 0.7 nm, respectively, and these values are in good agreement with each other indicating the spherical shape of the Pt NPs. The anisotropy of the Pt nanoparticles was additionally investigated in a TEM tilting study. Fig. S4 (ESI†) shows a series of TEM micrographs for Pt/RTO and Pt/C recorded under tilting angles between 0 and 30°. The tilting of the sample plane with respect to the incident electron beam changes the apparent shape of individual Pt nanoparticles showing their non-spherical shape.
Furthermore, Rietveld refinement of the HE-XRD pattern in Fig. S5 (ESI†) confirmed that the morphology of the nanoparticles is correlated to the applied synthesis routes as shown by the particle diameters D that were extracted from the principal axes of the ellipsoids. For Pt/C Dx and Dy are ∼2.4 nm and Dz is ∼1.8 nm whereas for Pt/RTO Dx, Dy, and Dz are ∼3.3 nm, 4.5 nm and 2.4 nm, respectively (see also Table S2, ESI†). Thus, impregnation of the support as applied for RTO and C supports leads to more ellipsoidal and platelet-like nanoparticles, respectively. In contrast, the surfactant-assisted route yields spherical Pt nanoparticles with a structural coherence length of ∼4 nm on the ITO. The Pt lattice parameter as determined by Rietveld refinement (Table S3, ESI†) decreases from Pt/ITO with a = 3.930 Å to Pt/RTO with a = 3.920 Å to Pt/C with a = 3.914 Å. The lattice parameter of bulk-like Pt is typically 3.923 Å (ICSD #243678). The contraction of the Pt lattice is correlated and can be explained by the decreasing structural coherence length.49 The most expanded Pt lattice on ITO might suggest minor Sn incorporation (<5%). The intensity ratio between the (111) and (200) diffraction peaks indicates a structural deviation from bulk Pt, which could not be explained by an anisotropy. The deviation can be interpreted by interstitial atoms like carbon or oxygen as residues from the surfactant-assisted synthesis route, which were not removed by calcination. E.g. Rietveld refinement suggests interstitial oxygen on the tetrahedral sites (OTd) in the Pt lattice with an occupancy of 0.15 ± 0.02. In contrast, the occupancy of OTd for the Pt nanoparticles on RTO and C was zero within the fit error (see also Table S3, ESI†). Furthermore, any kind of metal incorporation in the Pt nanoparticles on the oxide supports was not verifiable via Rietveld refinement but cannot be ruled out completely.
Thus, we conclude that the impregnation route of RTO and C supports leads to smaller ellipsoidal and platelet-like Pt nanoparticles exhibiting a contracted crystal lattice whereas the surfactant-assisted synthesis route leads to spherical Pt nanoparticles with a rather bulk-like Pt lattice wherein the presence of interstitial atoms like oxygen or carbon on the tetrahedral sites is likely.
Regarding the crystalline properties of the support, the interaction of oxides (especially of reducible oxides) with metallic nanostructures has extensively been studied with a special focus on the influence of surface oxygen vacancies.50 Oxygen vacancies at the metal support interface are known to cause charge transfer from the oxide to the metal facilitating the binding of the metal to the oxide. However, it has also been discussed that the electronic interaction between an oxide support and a metal nanoparticle has only a limited range and declines within several atomic layers.37
3.2. Electrochemical characterization
To identify the differences in the electrochemical behavior towards Pt oxidation of the three catalysts, a series of cyclic voltammograms (CV) in perchloric acid were recorded. Fig. 2a–c shows CVs from 0.05 V with a stepwise increasing upper potential limit from 0.5 to 1.4 V after an initial activation step for each catalyst of 100 CVs between 0.05 and 1.0 V. The typical Pt–Hupd and Pt–O/OH features are clearly visible for all three materials between 0.05 and 0.4 V as well as above 0.6 V, respectively.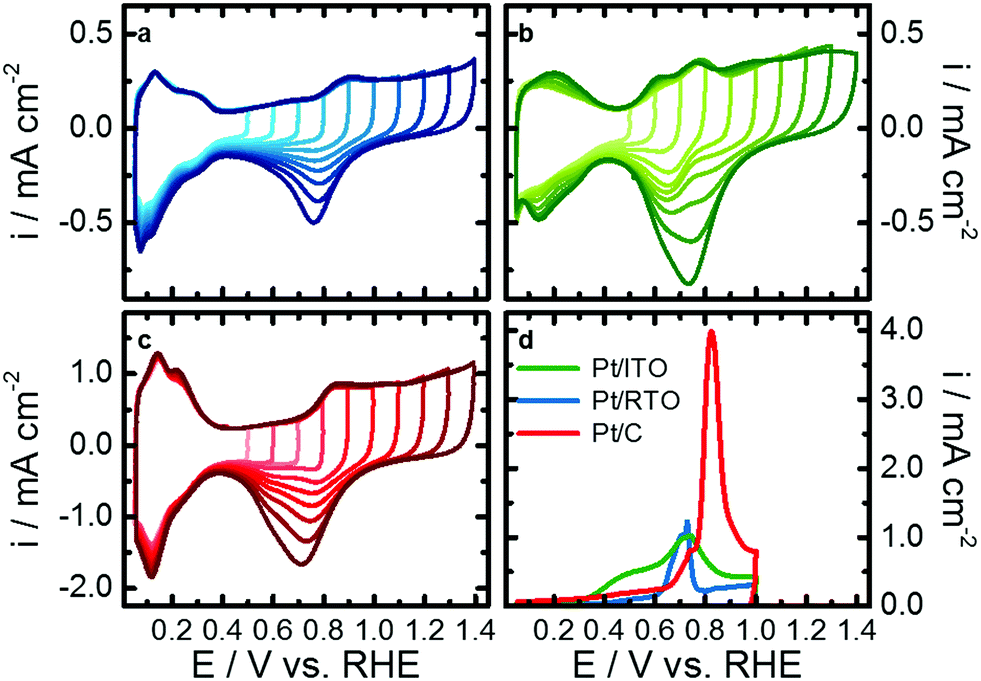 | ||
| Fig. 2 Electrochemical characterization of Pt nanoparticles on the RTO (a, blue), ITO (b, green) and carbon (c, red) support by the evolution of cyclic voltammogram with increasing upper potential limit in N2-saturated 0.1 M HClO4 from 0.5 to 1.4 V and from CO stripping experiments (d). | ||
By increasing the upper potential limit, the oxidative currents in the H-desorption region between 0.05 and 0.4 V stay rather stable compared to the H-adsorption region of the reductive scan. The H-desorption region of Pt/ITO shows the strongest growth with increasing upper potential limit. Additionally, Pt-oxidation peak(s) arising from around 0.6 V are stable, while the corresponding reduction peak(s) steadily increase. These findings indicate an increasing degree of Pt surface oxidation/reduction with higher upper potential limit. The larger currents in the Hupd region can be explained with an increasing roughening or more likely a cleaning of the nanoparticle surface e.g. from Sn with prolonged reduction of the Pt oxides under these dynamic conditions of the cyclic voltammograms.
Furthermore, the position of the Pt–O/OH reduction peak differs between the catalysts and shifts cathodic with upper potential limit for Pt/C whereas it shifts anodic for Pt/ITO. In the case of Pt/RTO it stays more or less constant. Two individual reduction features convolute the reduction peak of Pt/C and Pt/RTO but its binary character is less pronounced for RTO-supported Pt nanoparticles. The shape of the reduction peak does not strongly vary with increasing upper potential limit. In the case of Pt/ITO, the Pt–O/OH reduction peak consists of a single reduction at low upper potential limits (<0.9 V) but a binary shape developed with increasing upper potential limit similar to the reductive behavior determined for Pt on the other two supports. These support-dependent variations in peak positions indicate that not only the degree of Pt surface oxidation changes but also Pt oxide with different stability forms. Furthermore, the surface of the Pt nanoparticles, especially on the ITO, itself might alter with increasing upper potential.
To further characterize the Pt surface on the different supports, the electrochemical active surface area was determined from the Hupd region and from CO-stripping experiments. Fig. 2d shows the CO-oxidation profiles, and the carbon-support catalyst shows the largest oxidation peak corresponding to a CO-ECSA of 67.5 m2 gPt−1. For Pt/ITO the CO-stripping profile is rather broad and the overall current is lower, resulting in a CO-ECSA of 25.6 m2 gPt−1. CO electrooxidation on Pt/RTO caused a smaller and narrower peak at lower potentials compared to Pt/C, and Pt/RTO exhibits the lowest CO-ECSA of 16.7 m2 gPt−1 (see Table S4, ESI†). Correlations can be drawn to a study by Chattot et al. where structural defects originating from microstrain resulted in different CO oxidation profiles and peak positions.51
3.3. In situ electrochemical Pt oxidation
To identify the structural response of the Pt nanoparticles on the different supports to electrochemical oxidation, in situ high-energy X-ray diffraction patterns were recorded during step-wise oxidation from 0.6 to 1.4 V (see Fig. S6, ESI†). Again, all three catalysts were first electrochemically activated as described above. Fig. 3 shows the changes of the Pt lattice parameter and of the Pt scale factor as determined by Rietveld refinement, the latter normalized to the respective Hupd-ECSA. We choose to normalize using Hupd-ECSA as it probes comparable to the X-ray beam all the accessible Pt NPs.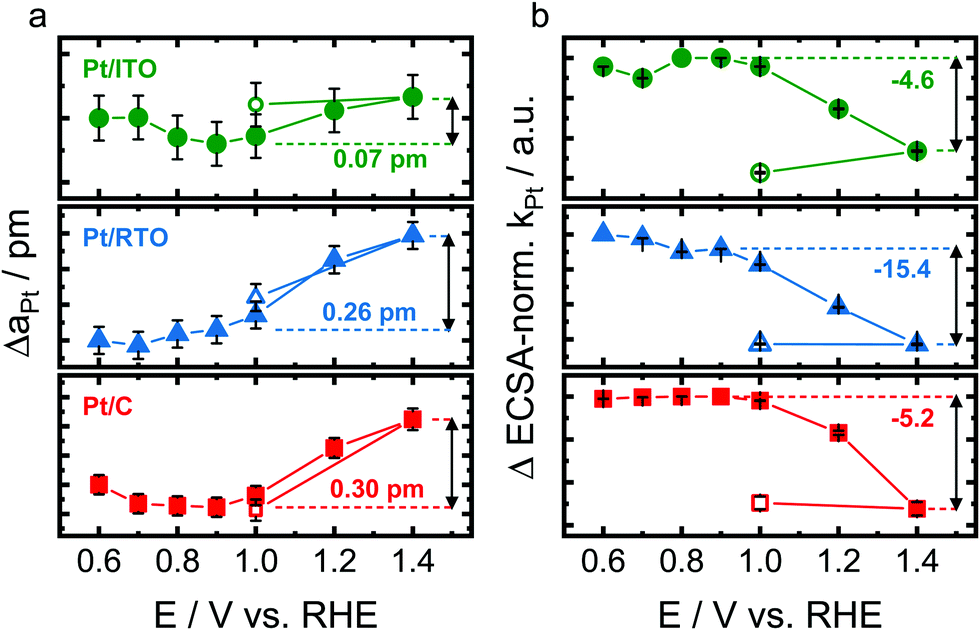 | ||
| Fig. 3 Structural response of crystalline Pt nanoparticles supported on ITO (green), RTO (blue) and carbon (red) to electrochemical oxidation with respect to the change of the Pt lattice parameter (a) and Hupd-ECSA-normalized change of the Pt scale factor (b) as determined by Rietveld refinement of the in situ high-energy X-ray diffraction pattern. The unfilled data points reflect the measurement at 1.0 V after the step-wise Pt oxidation. The dashed horizontal line represents the reference state at the onset of Pt oxidation. Error bars represent the standard error provided by Rietveld refinement. | ||
The metallic Pt lattice parameter (aPt) as shown in Fig. 3a increases almost linearly and reversibly with electrode potential above 0.9–1.0 V for the carbon- and RTO-supported Pt as a response to the electrochemical Pt oxidation. The metallic Pt lattice expands to a similar degree for Pt/RTO and Pt/C with ΔaPt ≅ 0.25–0.3 pm at 1.4 V. The lattice of the ITO-supported Pt nanoparticles however expands significantly less strongly with ΔaPt ≅ 0.07 pm at 1.4 V. In contrast, the Pt lattice contracts between 0.6 and 0.7 V in the case of the carbon support.
The scale factor (kPt) as extracted from Rietveld refinement is directly correlated to the absolute Pt metal diffraction intensity and can thus be treated as a measure of the absolute amount of metallic Pt probed by the X-ray beam. Fig. 3b shows the evolution of the Hupd-ECSA-normalized change in the Pt scale factor (ΔkPt), and the Pt scale factor decreases irreversibly above 1.0 V for all three catalysts. The Hupd-ECSA-normalized changes are similar for the ITO- and carbon-supported nanoparticles and strongest for Pt/RTO. For all three catalysts, no variation with electrode potential was identified for the occupancy with interstitial atoms on the tetrahedral sites.
Thus, the oxidizing electrochemical conditions induce a structural response of the metallic Pt domains of the nanoparticles with respect to their Pt lattice and amount of the metallic Pt domain as shown in Fig. 3. The structural transformation of the near-surface of the Pt nanoparticles starts at ∼0.9–1.0 V as shown by the evolution of the scale factor of the metallic Pt domains (Fig. 3b) and is irreversible between 1.0 and 1.4 V. Furthermore, the electrochemically oxidizing conditions cause a reversible lattice expansion of the metallic bulk of Pt nanoparticles on the different supports (Fig. 3a).52 This phenomenon contrasts the correlation between Pt domain size and lattice parameter because the formation of an oxide shell on the smaller metallic Pt core (probed by the scale factor kPt) should let the Pt lattice contract (probed by the lattice parameter aPt) due to a particle size effect.53
Therefore other processes have to lead to an expansion of the Pt lattice as observed in this study. On the one hand, the formation of a surface oxide and the place-exchange mechanism expel the surface atomic layers24,25 and thus, the bulk of the Pt nanoparticles responds to this expansive force with an expansion of the lattice. On the other hand, the anodic charging of a metal surface can induce expansive strains due to surface relaxations and charge reorganization.54,55 The identified reversibility of the lattice expansion between 1.0 and 1.4 V (Fig. 3a) rather supports a structural response to charging possibly initiated by Pt–O formation at ∼0.9 V. We propose that the formation of a surface Pt oxide as the cause for lattice expansion is unlikely because PtOx is reduced below 1.0 V (see electrochemical data, Fig. 2) and thus, we expect PtOx (shell) yet to be present on the non-expanded metallic Pt core.
Interestingly, the expansion of the Pt lattice is significantly lower for the largest Pt nanoparticles on ITO (see Fig. 3a) and might be suppressed by the non-contracted (bulk-like) Pt lattice and/or by the presence of interstitial atoms.
In a next step, in situ X-ray absorption spectra were recorded at selected electrode potentials to study the influence of electrochemical oxidation on the (electronic) structure of the Pt nanoparticles in more detail. Fig. S7 (ESI†) shows the normalized Pt L3-edge absorption spectra for Pt on the three supports in potentials steps between 0.6 and 1.4 V. The so-called white line (WL) between ∼11564 and 11572 eV originates from 2p3/2 to 5d orbital transitions and thus, its spectral weight is proportional to the total density of unoccupied 5d states in Pt. A higher WL area therefore indicates a higher density of d-band vacancies associated with oxidized Pt.
Fig. 4a shows the integrated WL area for Pt on ITO, RTO, and carbon. The WL area of all three catalysts increases almost linearly with an onset potential between 0.8 and 1.0 V indicating electrochemical oxidation of Pt and formation of additional d-band vacancies. Three different resonances were used to fit the Pt L3 XANES spectra with Res1 at ∼11566 eV and Res2 at ∼11570 eV (Fig. 4b) describing the WL following Merte et al.32 as well as Res3 at ∼11580 eV (Fig. 4c) corresponding to the first local maximum above the white line. We attribute the individual resonances to the spectral weight of d-band vacancies of metallic Pt (Res1), of Ptn+ (Res2) as well as of the metallic Pt phase (Res3). Below 0.9 V the (electronic) structure of the metallic Pt does not strongly vary for all three catalysts because the spectral weight of Res1 and Res3 does not change. For Pt/C and Pt/RTO even the degree of oxidized Pt remained constant as seen in Res2. Above ∼0.9 V the spectral weight of Res1 increases for all three catalysts and in the case of the oxide-supported Pt nanoparticles even linearly up to 1.4 V with a similar slope. Thus, the number of d-band vacancies in the metallic Pt increases with potential above ∼0.9 V due to Pt oxidation similar to the identified expansion of the Pt lattice. In the case of Pt/C, the spectral weight of Res2 increases above 1.1 V accompanied by a decreasing contribution of Res1. Thus, the carbon-supported metallic Pt nanoparticles partially restructure above 1.1 V and form Ptn+. The increasing spectral weight of Res2 can be explained with a growing PtOx shell on the metallic Pt core. In the case of the oxide supported Pt nanoparticle, the spectral weight of Res2 does not increase showing the absence of a similar PtOx shell formation. The spectral weight of Res3 decreases above 1.1 V for all three catalysts and even saturates for Pt/C. This depletion of Res3 suggests a conversion of metallic Pt for all three catalysts as extracted from in situ HE-XRD analysis but a limited growth of the oxide shell.
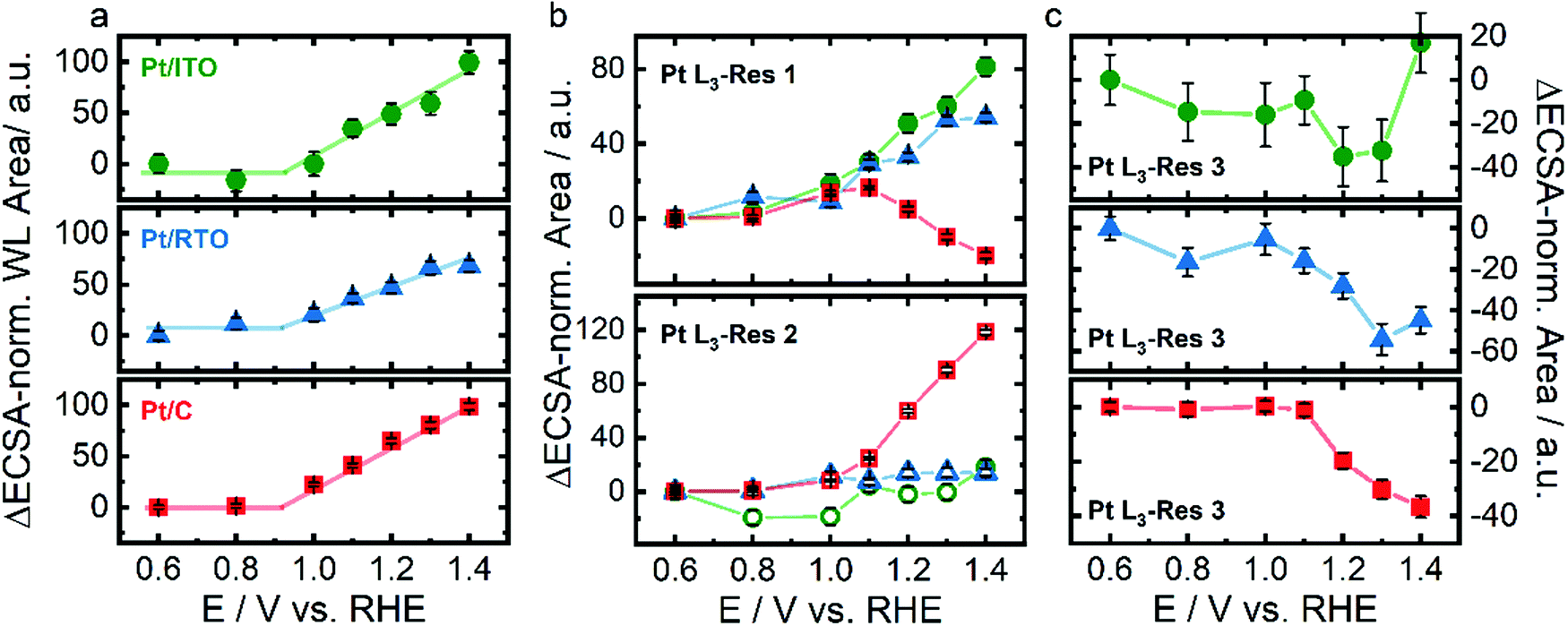 | ||
| Fig. 4 Structural response of Pt nanoparticles supported on ITO (green), RTO (blue), and carbon (red) to electrochemical oxidation with respect to the Hupd-ECSA-normalized white line area (a) and the Hupd-ECSA-normalized integrated area as the resonances 1 (Pt L3-Res 1) at ∼11566 eV and Pt L3-Res 2 at ∼11570 eV (b) and Pt L3-Res 3 at ∼11580 eV (c) as determined by peak fitting of in situ Pt L3-edge X-ray near-edge absorption spectra shown in Fig. S7 (ESI†). Error bars represent the standard error provided by the fitting of the WL. | ||
The differences in the structural response to the electrochemical oxidation can be partially explained with the morphology of the Pt nanoparticles. The rather platelet-like Pt nanoparticles on the carbon support may potentially exhibit a higher projected area of (100) facets which are known to oxidize differently than the (111) facets. For Pt(111), the place-exchange and the formation of amorphous 3D α-PtO2 proceed sequentially at 1.1 V and above 1.3 V, respectively, whereas on Pt(100) both processes occur simultaneously above 1.1 V,28 which is also accompanied by varying atomistic reaction mechanisms.30 The rather spherical/ellipsoidal Pt nanoparticles on the oxide support potentially exhibit more Pt sites similar to the Pt(111) or high-index surfaces limiting the formation of an amorphous 3D α-PtO2 adaption layer.
In addition to the formation of PtOx adaption layers, the Pt NPs are prone to exhibit Pt dissolution at those high potentials. To be able to differentiate these restructuring and dissolution processes, which would yield the same response of coherence length, the potential-dependent dissolution of Pt on the different supports was followed by in situ SFC-ICP-MS. Fig. 5 shows the Hupd-ECSA-normalized Pt dissolution of the initial anodic potential scan to 1.5 V mimicking the electrochemical oxidation tracked by the in situ X-ray experiments after conducting CVs between 0.05 and 1.5 V after electrochemical activation of 100 CVs (Fig. S8, ESI†). The anodic Pt dissolution has an onset potential at 1.1–1.2 V and continuously increases up to 1.5 V. The Hupd-ECSA-normalized Pt dissolution is strongest for Pt/C, lowest for Pt/RTO and Pt/ITO shows an intermediate Pt dissolution rate. The onset of Pt dissolution corresponds to the decrease in the Pt scale factor as well as to the loss of spectral weight for Res1 and Res3. Therefore, the detected Pt dissolution rate might be caused by dissolution of Ptn+ species during place-exchange between surface-O and Pt and PtOx formation. Thus, a fraction of the electrochemically formed Ptn+ species is dissolved into the electrolyte. On the other hand, a constant anodic dissolution of Ptn+ could limit the growth of an oxide shell around the nanoparticle or lead to the formation of a “stable” PtOx on the surface. Additionally, one has to consider the difference between the dynamic in situ SFC-ICP-MS measurements and the quasi-stationary in situ X-ray experiments.
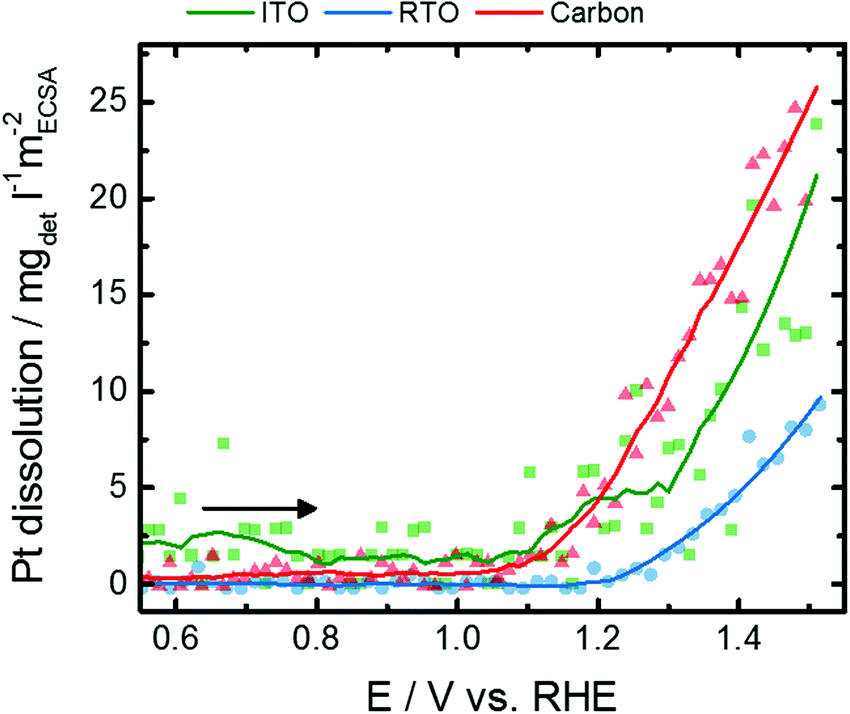 | ||
| Fig. 5 Evolution of the Hupd-ECSA-normalized Pt dissolution rate for Pt nanoparticles supported on ITO (green), RTO (blue), and carbon (red) during the initial anodic potential sweep from 0.05 to 1.5 V with a scan rate of 10 mV s−1 as determined by SFC-ICP-MS measurements. The complete dissolution profiles and the corresponding cathodic dissolution profiles are shown in the ESI,† Fig. S8–S10. | ||
Noteworthily, strong Pt dissolution peaks arise in the reductive scan below 1.0 V (Fig. S10, ESI†) with similar dissolution rates for Pt on C and ITO and the lowest dissolution for Pt/RTO. Compared to anodic Pt dissolution, electrochemical reduction of the formed PtOx causes Pt dissolution which is one order of magnitude higher and thus represents an important degradation pathway of Pt-based electrocatalysts. Note that cathodic dissolution is known to be more prominent than anodic dissolution, while anodic dissolution has a much stronger dependence on Pt loading.18,56–59 The onset of the cathodic Pt dissolution rate is below 1.0 V and might exhibit a binary profile especially for Pt/C. It is known that Pt in the PtO2 phase is reduced at lower electrode potentials than in PtOx compounds with lower average Pt oxidation state.60,61 These differences furthermore suggest that PtO2 formation is suppressed for the oxide-supported nanoparticles but stronger for the carbon-supported nanoparticles.
The Pt dissolution rates (and total quantities) are the lowest for the RTO supported material, and our previous studies have demonstrated the RTO support to be much more stable towards dissolution than ITO. This suggests RTO to be an interesting and promising support material for further investigations.62 However, the degradation and dissolution of fuel cell catalysts is highly complex, and firm conclusions would be enhanced by perfectly defined materials with identical particle size, shape, loading and interparticle distance which are all known to impact Pt dissolution.56–59 Furthermore, continued technique development that improves resolution could deconvolute closely spaced dissolution peaks. Therefore, the close collaboration of researchers with varied expertise will be required for continued progress in the future.
4. Conclusion
In this study, we investigated the structural response of Pt nanoparticles on different supports to electrochemical oxidation in order to establish a more comprehensive understanding of stability determining properties of fuel cell catalysts.We found that the applied synthesis route determines the morphology of the Pt nanoparticles. A surfactant assisted synthesis as used for the ITO support yields ∼4 nm spherical nanoparticles potentially with interstitial atoms on tetrahedral sites of the Pt lattice and a bulk-like Pt lattice. Impregnation of the support on the other hand leads to ellipsoidal/platelet-like nanoparticles in smaller sizes without interstitial atoms on the tetrahedral sites and a size-induced contraction of the Pt lattice. The anisotropic shape of Pt on carbon (more platelet-like) and RTO (more ellipsoidal) was confirmed from TEM micrographs.
Our in situ HE-XRD investigations revealed that the Pt lattice parameter of the nanoparticles on carbon and RTO increases reversibly and linearly with potential above 1.0 V while only slight changes are observed for Pt/ITO. We attribute this expansion to charge induced lattice strain facilitated by surface oxide formation. From in situ X-ray absorption spectroscopy we show that the formation of Ptn+ species is much more pronounced for Pt/C but follows (to a certain extent) Hupd-ECSA and particle size. Pt nanoparticles on the oxide support show suppressed Pt oxide formation which can be primarily attributed to their shape. Furthermore, in situ dissolution experiments show anodic Pt dissolution that follows the trend as observed from the X-ray investigations.
Most importantly, Pt on RTO shows the lowest Pt dissolution representing an improved total electrochemical stability of an RTO-supported Pt electrocatalyst which we have previously shown.62
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received financial support from German Research Foundation (DFG) through grant STR 596/4-1 (“Pt-Stability”) and grant STR 596/5-2 (“In situ TEM KAT”), the German Federal Ministry of Education and Research (BMBF) through grant 03SF0531B (“HT-linked”) and grant 03XP0251 (“KorrZellKat”) and the Alexander von Humboldt foundation, Bonn, Germany. We thank ESRF and BESSY II for allocation of synchrotron radiation beamtime and I. Zizak and T. Merzdorf for their help during beamtimes. ZELMI of Technical University Berlin is acknowledged for their support with TEM measurements.References
- Y. Shao, G. Yin, Z. Wang and Y. Gao, J. Power Sources, 2007, 167, 235–242 CrossRef CAS.
- J. C. Meier, C. Galeano, I. Katsounaros, J. Witte, H. J. Bongard, A. A. Topalov, C. Baldizzone, S. Mezzavilla, F. Schuth and K. J. J. Mayrhofer, Beilstein J. Nanotechnol., 2014, 5, 44–67 CrossRef.
- J. Speder, I. Spanos, A. Zana, J. J. K. Kirkensgaard, K. Mortensen, L. Altmann, M. Bäumer and M. Arenz, Surf. Sci., 2015, 631, 278–284 CrossRef CAS.
- N. V. Long, C. M. Thi, M. Nogami and M. Ohtaki, New J. Chem., 2012, 36, 1320 RSC.
- J. Speder, L. Altmann, M. Roefzaad, M. Baumer, J. J. K. Kirkensgaard, K. Mortensen and M. Arenz, Phys. Chem. Chem. Phys., 2013, 15, 3602–3608 RSC.
- I. Spanos, J. J. K. Kirkensgaard, K. Mortensen and M. Arenz, J. Power Sources, 2014, 245, 908–914 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315(5811), 493–497 CrossRef CAS.
- R. Chattot, T. Asset, P. Bordet, J. Drnec, L. Dubau and F. Maillard, ACS Catal., 2017, 7, 398–408 CrossRef CAS.
- F. Hasche, M. Oezaslan and P. Strasser, J. Electrochem. Soc., 2012, 159, B25–B34 CAS.
- V. Beermann, M. Gocyla, E. Willinger, S. Rudi, M. Heggen, R. E. Dunin-Borkowski, M.-G. Willinger and P. Strasser, Nano Lett., 2016, 16, 1719–1725 CrossRef CAS.
- X. Tuaev, S. Rudi, V. Petkov, A. Hoell and P. Strasser, ACS Nano, 2013, 7, 5666–5674 CrossRef CAS.
- F. Dionigi, C. C. Weber, M. Primbs, M. Gocyla, A. M. Bonastre, C. Spori, H. Schmies, E. Hornberger, S. Kuhl, J. Drnec, M. Heggen, J. Sharman, R. E. Dunin-Borkowski and P. Strasser, Nano Lett., 2019, 19, 6876–6885 CrossRef CAS.
- F. Hasche, M. Oezaslan and P. Strasser, Phys. Chem. Chem. Phys., 2010, 12, 15251–15258 RSC.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285–305 CrossRef CAS.
- H. Yano, M. Watanabe, A. Iiyama and H. Uchida, Nano Energy, 2016, 29, 323–333, DOI:10.1016/j.nanoen.2016.02.016.
- J. Speder, A. Zana, I. Spanos, J. J. K. Kirkensgaard, K. Mortensen, M. Hanzlik and M. Arenz, J. Power Sources, 2014, 261, 14–22 CrossRef CAS.
- A. Riese, D. Banham, S. Ye and X. Sun, J. Electrochem. Soc., 2015, 162, F783–F788 CrossRef CAS.
- G. P. Keeley, S. Cherevko and K. J. J. Mayrhofer, ChemElectroChem, 2016, 3, 51–54 CrossRef CAS.
- C. A. Rice, P. Urchaga, A. O. Pistono, B. W. McFerrin, B. T. McComb and J. Hu, J. Electrochem. Soc., 2015, 162, F1175–F1180 CrossRef CAS.
- A. A. Topalov, I. Katsounaros, M. Auinger, S. Cherevko, J. C. Meier, S. O. Klemm and K. J. Mayrhofer, Angew. Chem., Int. Ed., 2012, 51, 12613–12615 CrossRef CAS.
- L. Y. Xing, M. Hossain, M. Tian, D. Beauchemin, K. Adjemian and G. Jerkiewicz, Electrocatalysis, 2014, 5, 96–112 CrossRef CAS.
- M. Matsumoto, T. Miyazaki and H. Imai, J. Phys. Chem. C, 2011, 115, 11163–11169 CrossRef CAS.
- S. Cherevko, N. Kulyk and K. J. J. Mayrhofer, Nano Energy, 2016, 29, 275–298 CrossRef CAS.
- M. Ruge, J. Drnec, B. Rahn, F. Reikowski, D. A. Harrington, F. Carla, R. Felici, J. Stettner and O. M. Magnussen, J. Electrochem. Soc., 2017, 164, H608–H614 CrossRef CAS.
- J. Drnec, M. Ruge, F. Reikowski, B. Rahn, F. Carlà, R. Felici, J. Stettner, O. M. Magnussen and D. A. Harrington, Electrochim. Acta, 2017, 224, 220–227 CrossRef CAS.
- A. M. Gómez-Marín and J. M. Feliu, Catal. Today, 2015, 244, 172–176 CrossRef.
- A. Kongkanand and J. M. Ziegelbauer, J. Phys. Chem. C, 2012, 116, 3684–3693 CrossRef CAS.
- Y.-F. Huang, P. J. Kooyman and M. T. M. Koper, Nat. Commun., 2016, 7, 12440 CrossRef CAS.
- D. J. S. Sandbeck, O. Brummel, K. J. J. Mayrhofer, J. Libuda, I. Katsounaros and S. Cherevko, ChemPhysChem, 2019, 20, 2997–3003 CrossRef CAS.
- T. Fuchs, J. Drnec, F. Calle-Vallejo, N. Stubb, D. J. S. Sandbeck, M. Ruge, S. Cherevko, D. A. Harrington and O. M. Magnussen, Nat. Catal., 2020, 3, 754–761, DOI:10.1038/s41929-020-0497-y.
- D. Friebel, D. J. Miller, C. P. O'Grady, T. Anniyev, J. Bargar, U. Bergmann, H. Ogasawara, K. T. Wikfeldt, L. G. Pettersson and A. Nilsson, Phys. Chem. Chem. Phys., 2011, 13, 262–266 RSC.
- L. R. Merte, F. Behafarid, D. J. Miller, D. Friebel, S. Cho, F. Mbuga, D. Sokaras, R. Alonso-Mori, T.-C. Weng, D. Nordlund, A. Nilsson and B. Roldan Cuenya, ACS Catal., 2012, 2, 2371–2376 CrossRef CAS.
- K. Sasaki, N. Marinkovic, H. S. Isaacs and R. R. Adzic, ACS Catal., 2016, 6, 69–76 CrossRef CAS.
- H. Imai, K. Izumi, M. Matsumoto, Y. Kubo, K. Kato and Y. Imai, J. Am. Chem. Soc., 2009, 131, 6293–6300 CrossRef CAS.
- K. Nagasawa, S. Takao, K. Higashi, S. Nagamatsu, G. Samjeske, Y. Imaizumi, O. Sekizawa, T. Yamamoto, T. Uruga and Y. Iwasawa, Phys. Chem. Chem. Phys., 2014, 16, 10075–10087 RSC.
- M. Hatanaka, N. Takahashi, N. Takahashi, T. Tanabe, Y. Nagai, A. Suda and H. Shinjoh, J. Catal., 2009, 266, 182–190 CrossRef CAS.
- Y. Lykhach, S. M. Kozlov, T. Skala, A. Tovt, V. Stetsovych, N. Tsud, F. Dvorak, V. Johanek, A. Neitzel, J. Myslivecek, S. Fabris, V. Matolin, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284–288 CrossRef CAS.
- O. Brummel, F. Waidhas, F. Faisal, R. Fiala, M. Vorokhta, I. Khalakhan, M. Dubau, A. Figueroba, G. Kovács, H. A. Aleksandrov, G. N. Vayssilov, S. M. Kozlov, K. M. Neyman, V. Matolín and J. Libuda, J. Phys. Chem. C, 2016, 120, 19723–19736 CrossRef CAS.
- O. Brummel, Y. Lykhach, M. Vorokhta, B. Šmíd, C. Stumm, F. Faisal, T. Skála, N. Tsud, A. Neitzel, K. Beranová, K. C. Prince, V. Matolín and J. Libuda, J. Phys. Chem. C, 2019, 123, 8746–8758 CrossRef CAS.
- E. Fabbri, A. Patru, A. Rabis, R. Kotz and T. J. Schmidt, Chimia, 2014, 68, 217–220 CrossRef CAS.
- E. Fabbri, A. Rabis, Y. Chino, M. Uchida and T. J. Schmidt, Electrochem. Commun., 2017, 83, 90–95 CrossRef CAS.
- H. Schmies, A. Bergmann, J. Drnec, G. Wang, D. Teschner, S. Kühl, D. J. S. Sandbeck, S. Cherevko, M. Gocyla, M. Shviro, M. Heggen, V. Ramani, R. E. Dunin-Borkowski, K. J. J. Mayrhofer and P. Strasser, Adv. Energy Mater., 2018, 8, 1701663 CrossRef.
- G. Wang, K. Bhattacharyya, J. Parrondo and V. Ramani, Chem. Eng. Sci., 2016, 154, 81–89 CrossRef CAS.
- C.-P. Lo, G. Wang, A. Kumar and V. Ramani, Appl. Catal., B, 2013, 140–141, 133–140 CrossRef CAS.
- J. Parrondo, T. Han, E. Niangar, C. Wang, N. Dale, K. Adjemian and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 45–50 CrossRef CAS.
- D. Ectors, F. Goetz-Neunhoeffer and J. Neubauer, J. Appl. Crystallogr., 2015, 48, 1998–2001 CrossRef CAS.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, G. P. Keeley and K. J. J. Mayrhofer, J. Electrochem. Soc., 2014, 161, H501–H507 CrossRef CAS.
- A. K. Schuppert, A. A. Topalov, I. Katsounaros, S. O. Klemm and K. J. J. Mayrhofer, J. Electrochem. Soc., 2012, 159, F670–F675 CrossRef CAS.
- P. M. Diehm, P. Agoston and K. Albe, ChemPhysChem, 2012, 13, 2443–2454 CrossRef CAS.
- G. Pacchioni, Phys. Chem. Chem. Phys., 2013, 15, 1737–1757 RSC.
- L. Dubau, J. Nelayah, S. Moldovan, O. Ersen, P. Bordet, J. Drnec, T. Asset, R. Chattot and F. Maillard, ACS Catal., 2016, 6, 4673–4684 CrossRef CAS.
- I. Martens, R. Chattot, M. Rasola, M. V. Blanco, V. Honkimäki, D. Bizzotto, D. P. Wilkinson and J. Drnec, ACS Appl. Energy Mater., 2019, 2, 7772–7780 CrossRef CAS.
- I. N. Leontyev, A. B. Kuriganova, N. G. Leontyev, L. Hennet, A. Rakhmatullin, N. V. Smirnova and V. Dmitriev, RSC Adv., 2014, 4, 35959–35965 RSC.
- Y. Umeno, C. Elsässer, B. Meyer, P. Gumbsch, M. Nothacker, J. Weissmüller and F. Evers, Europhys. Lett., 2007, 78, 13001 CrossRef.
- J. Weissmuller, R. N. Viswanath, D. Kramer, P. Zimmer, R. Wurschum and H. Gleiter, Science, 2003, 300, 312–315 CrossRef CAS.
- D. J. S. Sandbeck, PhD thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2020.
- D. J. S. Sandbeck, N. M. Secher, F. D. Speck, J. E. Sørensen, J. Kibsgaard, I. Chorkendorff and S. Cherevko, ACS Catal., 2020, 10, 6281–6290 CrossRef CAS.
- D. J. S. Sandbeck, M. Inaba, J. Quinson, J. Bucher, A. Zana, M. Arenz and S. Cherevko, ACS Appl. Mater. Interfaces, 2020, 12, 25718–25727 CrossRef CAS.
- D. J. S. Sandbeck, N. M. Secher, M. Inaba, J. Quinson, J. E. Sørensen, J. Kibsgaard, A. Zana, F. Bizzotto, F. D. Speck, M. T. Y. Paul, A. Dworzak, C. Dosche, M. Oezaslan and I. Cho, The Dissolution Dilemma for low Pt Loading Polymer Electrolyte Membrane Fuel Cell Catalysts, 2020 Search PubMed.
- H. Angerstein-Kozlowska, B. E. Conway and W. B. A. Sharp, J. Electroanal. Chem. Interfacial Electrochem., 1973, 43, 9–36 CrossRef CAS.
- B. E. Conway, Prog. Surf. Sci., 1995, 49, 331–452 CrossRef CAS.
- E. Hornberger, A. Bergmann, H. Schmies, S. Kühl, G. Wang, J. Drnec, D. J. S. Sandbeck, V. Ramani, S. Cherevko, K. J. J. Mayrhofer and P. Strasser, ACS Catal., 2018, 8, 9675–9683 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional structural and morphological characterization and supporting electrochemical characterization. See DOI: 10.1039/d0cp03233f |
| ‡ Present address: Fritz-Haber-Institute of the Max-Planck-Society, Department of Interface Science, Faradayweg 4-6, 14195 Berlin, Germany. |
| This journal is © the Owner Societies 2020 |