
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Following the unusual breathing behaviour of 17O-enriched mixed-metal (Al,Ga)-MIL-53 using NMR crystallography†
Cameron M.
Rice‡
a,
Zachary H.
Davis‡
a,
David
McKay
 a,
Giulia P. M.
Bignami
a,
Ruxandra G.
Chitac
a,
Daniel M.
Dawson
a,
Russell E.
Morris
*ab and
Sharon E.
Ashbrook
*a
a,
Giulia P. M.
Bignami
a,
Ruxandra G.
Chitac
a,
Daniel M.
Dawson
a,
Russell E.
Morris
*ab and
Sharon E.
Ashbrook
*a
aSchool of Chemistry, EaStCHEM and Centre of Magnetic Resonance, University of St Andrews, St. Andrews, KY16 9ST, UK. E-mail: rem1@st-andrews.ac.uk; sema@st-andrews.ac.uk
bDepartment of Physical and Macromolecular Chemistry, Faculty of Sciences, Charles University, Hlavova 8, 128 43 Prague 2, Czech Republic
First published on 16th June 2020
Abstract
The breathing behaviour of 17O-enriched (Al,Ga)-MIL-53, a terephthalate-based metal–organic framework, has been investigated using a combination of solid-state nuclear magnetic resonance (NMR) spectroscopy, powder X-ray diffraction (PXRD) and first-principles calculations. These reveal that the behaviour observed for as-made, calcined, hydrated and subsequently dehydrated mixed-metal MIL-53 materials differs with composition, but cannot be described as the compositionally weighted average of the breathing behaviour seen for the two end members. Although the form of MIL-53 adopted by the as-made material is independent of metal composition, upon calcination, materials with higher levels of Al adopt an open pore (OP) form, as found for the Al end member, but substitution of Ga results in mixed pore materials, with OP and narrow pore (NP) forms co-existing. Although the Ga end member is prone to decomposition under the calcination conditions used, a low level of Al in the starting synthesis (5%) leads to an OP mixed-metal MOF that is stable to calcination. Upon hydration, all materials almost exclusively adopt a closed pore (CP) structure, with strong hydrogen bonding interactions with water leading to two distinct resonances from the carboxylate oxygens in 17O NMR spectra. When dehydrated, different framework structures are found for the two end members, OP for Al-MIL-53 and NP for Ga-MIL-53, with the proportion of NP MOF seen to increase systematically with the Ga content in mixed-metal materials, in contrast to the forms seen upon initial calcination. 17O NMR spectra of mixed-metal MIL-53 materials show an increased preference for clustering of like cations as the Ga content increases. This is not a result of the small-scale dry gel conversion reactions used for enrichment, as a similar cation distribution and clustering is also observed for (Al0.5,Ga0.5)-MIL-53 synthesised hydrothermally and enriched with 17O via post-synthetic steaming.
Introduction
Metal–organic frameworks (MOFs) are a class of microporous materials comprising metal cations (nodes) connected through organic molecules (linkers) to form three-dimensional, voided structures.1 Since their first report by Hoskins and Robson in 1989,2 the range of MOF materials produced has expanded at a phenomenal rate, with over 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 frameworks published in the Cambridge Crystallographic Data Centre by 2016.3,4 Driving this growth is the potential diversity of the framework components; different cations may be combined with a large number of varying polytopic organic linkers to produce functionalised MOFs with pre-determined properties.1,5 Consequently, MOFs have many potential applications in the fields of gas storage, biomedicine, catalysis and energy materials.1,6–9
000 frameworks published in the Cambridge Crystallographic Data Centre by 2016.3,4 Driving this growth is the potential diversity of the framework components; different cations may be combined with a large number of varying polytopic organic linkers to produce functionalised MOFs with pre-determined properties.1,5 Consequently, MOFs have many potential applications in the fields of gas storage, biomedicine, catalysis and energy materials.1,6–9
One sub-class of MOFs of particular interest are the terephthalate frameworks, whose rod-like linkers and structural ‘pivot-points’ can facilitate breathing behaviour within the solid.10–12 The most documented breathing terephthalate MOF is MIL-53 (MIL = Materiaux Insitut Lavoisier). First synthesised in 2002 by the Férey group as a CrIII MOF,13,14 (and subsequently extended to Al,15 Fe,16 In,17 Ga18 and Sc19) this material exhibits significant structural changes (causing the pore volume to vary by up to 40%) as a consequence of changes in external temperature, humidity or guest loading.13,14,20 Furthermore, the exact breathing behaviour (i.e., the size and shape of the pores formed at specified conditions) exhibited by a MIL-53 material is known to depend on its constituent cations.21–24
The range of MOFs that can be produced can be further extended by combining one or more metal nodes and/or one or more organic linkers.22,25–30 A number of examples exist in the literature that show improved activity in catalysis, in gas storage and release, or an enhanced framework stability, by combining or doping a MOF with a second cation.22,28,31–38 In this work, we explore the effect on the breathing properties of MIL-53 materials when two different metal cations are incorporated into the framework.22,27,28 The monometallic Al- and Ga-MIL-53 MOFs exhibit different breathing behaviours, as shown in Fig. 1.15,21,22 The breathing enabled by the ‘wine-rack’ like structure of MIL-53 hinges around the M3+ cation centres, which are joined through carboxylate linkages to the 1,4-benzenedicarboxylate (BDC) linkers in two dimensions, and through bridging μ2(OH) (i.e., (M–O(H)–M) hydroxyl) linkages in the third dimension (see ESI†).13 This suggests that the properties exhibited by mixed-metal materials could vary with the composition of the MOF and, importantly, with the distribution or order/disorder of the cations.
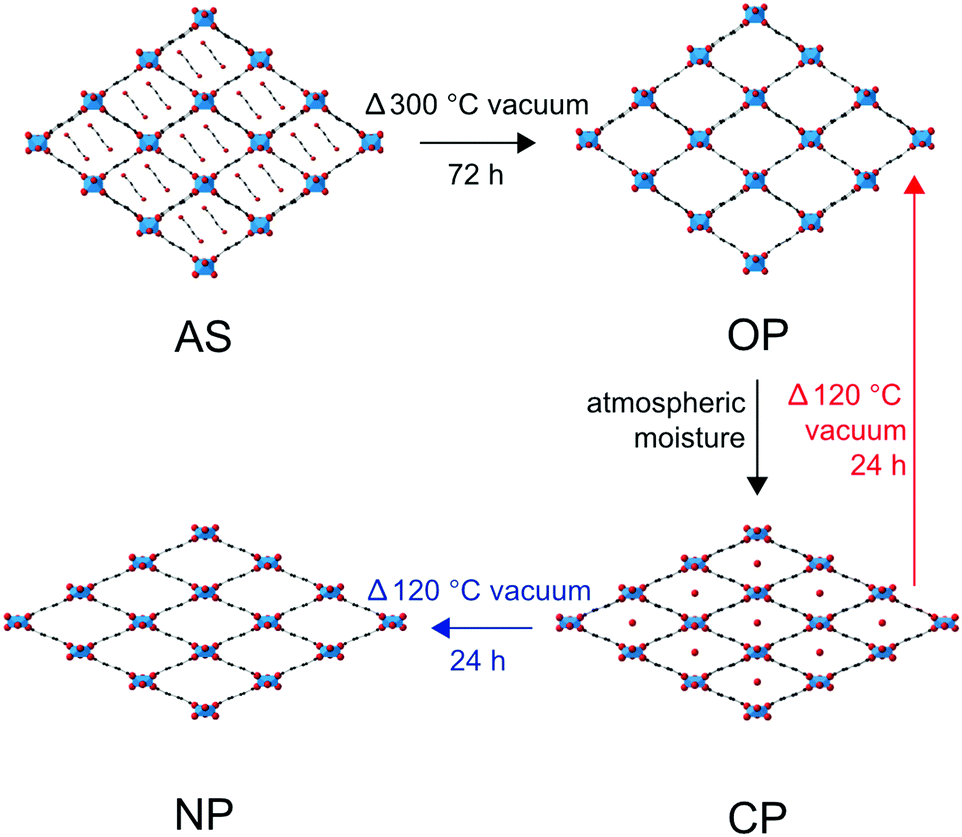 | ||
| Fig. 1 Breathing behaviour exhibited by Al-MIL-53 and Ga-MIL-53 when calcined, hydrated and subsequently dehydrated. Transformations in black are observed for both Al- and Ga-MIL-53 end members, that in red by Al-MIL-53 only and that in blue by Ga-MIL-53 only. | ||
The framework disorder present in mixed-metal MOFs (and possible consequent structural disorder) can pose a significant challenge for structural characterisation, with commonly employed diffraction methods providing information only on the average structure. However, the combination of diffraction with solid-state NMR spectroscopy offers an ideal approach for understanding structure, and structure–property relationships, in MOFs, particularly those with disordered frameworks. NMR spectroscopy provides an element-specific measurement that is sensitive to the local, atomic-scale environment (without the need for any long-range order), is able to probe dynamic processes that take place over a very wide range of timescales,39–41 and has been widely applied for the study of MOFs.42–44 Many studies have exploited 13C, 15N or 1H (I = 1/2) NMR spectroscopy, to provide information on the number, type and additional functionalisation of the linkers and guest molecules present.40–42 Many of the metal centres present in MOFs have high spin quantum number (I > 1/2), leading to lines broadened by the quadrupolar interaction,39–41 increasing the challenge of acquiring and interpreting experimental NMR spectra. One element of particular interest in NMR spectroscopy of MOFs, and of MIL-53 in particular, is oxygen, with connections between metal centres and the organic linkers, and between adjacent metal centres, both occurring via O atoms. Although 17O, the only NMR-active isotope of O, is also quadrupolar (I = 5/2), it has one of the smallest quadrupole moments of all nuclides, usually enabling spectra to be acquired at moderate magnetic field strengths.45,46 However, routine study is hindered by its extremely low natural abundance (0.037%), and isotopic enrichment is typically employed to enable spectra to be acquired on a reasonable timescale. The high cost of isotopically enriched reagents (∼£3000 mL−1 for 90% H2O(l)) requires cost effective, atom-efficient approaches for enrichment, such as the dry gel conversion (DGC) reactions and steaming approaches exploited in this work.22,47
Here, multinuclear NMR spectroscopy is combined with powder XRD (PXRD) measurements and energy dispersive X-ray (EDX) spectroscopy to investigate cation disorder in 17O-enriched mixed-metal (Al,Ga)-MIL-53 materials, and to explore any subsequent effect on their breathing behaviour. Experimental results are supported by first-principles calculations, in an NMR crystallographic approach,48–51 facilitating interpretation and assignment of the complex spectral resonances observed for these disordered materials, and providing additional insight into the relative energies of the different materials that can be formed. We show that the mixed-metal MOFs have different compositions from those expected from the starting synthesis, and demonstrate that the cation distribution in the final product is independent of the route used for synthesis and 17O enrichment. The mixed-metal materials also exhibit different breathing behaviour from that observed for the two end members in calcined, hydrated and dehydrated forms, with ‘mixed pore’ materials being observed in many cases. We also show that the incorporation of even a very small amount of Al (∼5% within the starting synthesis) into the Ga-MIL-53 framework is sufficient to overcome the previously observed instability of this material to high-temperature calcination. The combination of NMR spectroscopy, with its sensitivity to local structural changes, with information on the long-range positional order from diffraction provides a unique insight into the structure and properties of these interesting materials.
Methods
Synthetic procedures
(Al1−xGax)-MIL-53 materials were synthesised either using a previously reported DGC route or a cation-corrected variation of the original hydrothermal literature procedure.13,14,22,47 For DGC reactions, Al(NO3)3·9H2O, Ga(NO3)3·nH2O and terephthalic acid (H2BDC) were combined in a Teflon cup in the ratios given in Table 1. The cup was placed inside a Teflon autoclave liner containing 130 μL of H217O(l) (90%, Cortecnet), before sealing and heating to 220 °C for 72 h. For standard hydrothermal syntheses, the quantities of reagents in Table 1 were again used; however, these were combined with 5.0 mL of distilled water directly within the Teflon liner of the autoclave, which was sealed and heated under the same conditions. Upon cooling, solids were washed with the minimum volume of distilled water and left to dry in air. For 17O enrichment, the as-made hydrothermally produced materials were then placed in a Teflon cup inside a Teflon autoclave liner containing 130 μL of H217O(l) (90%, Cortecnet), before sealing and heating to 150 °C for 72 h.22 For both enrichment methods 17O levels were estimated to be ∼15% (by comparison to materials previously studied using secondary ion mass spectrometry22). As-made materials were calcined under vacuum at 300 °C for 72 h at pressures approaching 10−4 Torr and then stored in an argon atmosphere in flame-sealed glass vials. Vials were stored on the benchtop for a maximum of 72 hours before PXRD or NMR characterisation. Materials were hydrated by exposure to air for at least 24 h, and their subsequent dehydration was performed in the same manner as calcination, but with heating at 120 °C for 24 h. The calcination procedure used for Ga-MIL-53 (i.e., x = 1) was adapted to prevent the occurrence of the reported collapse of the framework under the conditions above.18,21,22 Ga-MIL-53 samples were heated under vacuum at 260 °C for 96 h (with most samples decomposing when subjected to heating at temperatures above this). As described in later sections the conditions used for calcination can affect the forms (i.e., OP or NP) of the material produced.| x | Al(NO3)3·9H2O/mmol | Ga(NO3)3·nH2O/mmol | H2BDC/mmol |
|---|---|---|---|
| 0.0 | 1.40 | 0.00 | 1.80 |
| 0.2 | 1.12 | 0.28 | 1.80 |
| 0.5 | 0.70 | 0.70 | 1.80 |
| 0.8 | 0.28 | 1.12 | 1.80 |
| 0.9 | 0.14 | 1.26 | 1.80 |
| 0.95 | 0.07 | 1.33 | 1.80 |
| 1.0 | 0.00 | 1.40 | 1.80 |
Powder X-ray diffraction
PXRD data were collected on a STOE STADIP diffractometer (Cu Kα1), monochromated with a curved Ge(111) crystal in transmission Debye–Scherrer mode. For capillary PXRD of air-sensitive (i.e., calcined and dehydrated) materials, the MOF was packed into 0.5 mm capillary tubes before dehydration/calcination using a Schlenk apparatus. The capillaries were then removed from the dehydration rig under a flow of argon, flame sealed and capped with hydrocarbon wax. Structureless Le Bail full-pattern fits, with refinement of lattice parameters, were performed between 5° and 50° 2θ for as-made MIL-53 materials, using GSAS, implemented within the EXPGUI interface. The published structure of Al-MIL-53 (Pnma) was used as a starting model.14Scanning electron microscopy and energy dispersive X-ray spectroscopy
Scanning electron microscopy (SEM) and EDX measurements were performed using a conventional Jeol JSM-5600 (3.5 nm resolution) scanning electron microscope (tungsten filament), with integrated Oxford Inca X-ray source for EDX spectroscopy. For SEM, a working distance of 10 mm and acceleration voltage of 5 kV was used, with values of 20 mm and 20 kV for EDX analysis. EDX analyses were performed on several batches of calcined, hydrated MOFs for each composition with a minimum of 21 crystallites scanned in each instance.Solid-state NMR spectroscopy
Solid-state NMR spectra were acquired using Bruker Avance III spectrometers equipped with 14.1 or 20.0 T wide-bore magnets. Samples were packed into 3.2 mm ZrO2 rotors and magic angle spinning (MAS) spectra were acquired at spinning speeds of 12.5 kHz (1H and 13C) and 20 kHz (17O) using a conventional 3.2 mm HX probe. Spectra were acquired at Larmor frequencies (at 14.1 T) of 600.13 MHz and 150.87 MHz, for 1H and 13C, respectively, and for 17O at 81.34 MHz (14.1 T) and 115.3 MHz (20.0 T). Radiofrequency (rf) field strengths were calibrated to be 100 kHz (1H) and 55–77 kHz (17O). Spectra are referenced to Si(CH3)4 for 1H and 13C (using a secondary reference of L-alanine (δ(N![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3) = 8.5 ppm and δ(
3) = 8.5 ppm and δ(![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3) = 20.5 ppm)) and H2O (17O) at room temperature. For 13C MAS NMR experiments, transverse magnetisation was obtained by cross-polarisation52 (CP) from 1H using a 5 ms contact pulse (ramped for 1H) and TPPM-15531H decoupling (100 kHz) in acquisition. 17O and 1H MAS NMR spectra were acquired either using a rotor-synchronised spin echo pulse sequence to avoid baseline distortions. At 14.1 and 20.0 T, 17O multiple-quantum MAS (MQMAS)54 experiments were carried out using a triple-quantum z-filtered (0 → ±3 → 0 → 1) pulse sequence.55 These are shown after a shearing transformation to enable projection of the isotropic spectrum directly onto δ1, and are shown referenced using the convention outlined in ref. 56.
H3) = 20.5 ppm)) and H2O (17O) at room temperature. For 13C MAS NMR experiments, transverse magnetisation was obtained by cross-polarisation52 (CP) from 1H using a 5 ms contact pulse (ramped for 1H) and TPPM-15531H decoupling (100 kHz) in acquisition. 17O and 1H MAS NMR spectra were acquired either using a rotor-synchronised spin echo pulse sequence to avoid baseline distortions. At 14.1 and 20.0 T, 17O multiple-quantum MAS (MQMAS)54 experiments were carried out using a triple-quantum z-filtered (0 → ±3 → 0 → 1) pulse sequence.55 These are shown after a shearing transformation to enable projection of the isotropic spectrum directly onto δ1, and are shown referenced using the convention outlined in ref. 56.
Calculations
Density functional theory (DFT) calculations were carried out using the gauge-including projector augmented wave (GIPAW)57 approach as implemented in the CASTEP58 code (version 18.1), using the PBE59 exchange correlation functional (with the semi-empirical dispersion correction scheme of Tkatchenko and Scheffler60 applied – see also ESI†). Ultrasoft pseudopotentials61 were generated with ZORA relativistic effects,62 and a planewave energy cut-off of 60 Ry was applied. Sampling of the first Brillouin zone was performed on a Monkhorst–Pack63 grid with k-point spacing of 0.04 2π Å−1. Structural models were generated by substituting cations into the relevant structural models (i.e., OP or NP) for Al-MIL-53, as described in more detail in the ESI.† Optimisation of the geometry was carried out for all models, with atomic coordinates and unit cell parameters allowed to vary. The calculation of mixing energies, Emix, is described in the ESI.† Calculations generate the absolute shielding tensor (σ) in the crystal frame, and diagonalisation yields the three principal components, σXX, σYY and σZZ, and the isotropic shielding, σiso = (1/3)Tr{σ}. The isotropic chemical shift, δiso, is given by −(σiso − σref), where σref is a reference shielding, determined as described in the ESI.† The quadrupolar coupling constant, CQ = eQVZZ/h and asymmetry parameter, ηQ = (VXX − VYY)/VZZ are obtained from the principal components of the electric field gradient (EFG) tensor, V, where Q is the nuclear quadrupole moment (for which values of −25.58, 146.6 and 107 mb were used for 17O, 27Al and 71Ga respectively).64
Results and discussion
Mixed-metal (Al,Ga)-MIL-53 materials were prepared using DGC as described above. PXRD measurements confirmed MIL-53 was produced in all cases, and all materials are consistent with the Pnma space group exhibited by the end members (see ESI†). EDX analysis (of calcined, hydrated MOFs – see later), as shown in Table 2, confirms that although both Al and Ga are incorporated into all the crystallites analysed, the level of Ga found in the final product is much lower than the stoichiometry of the starting reagents, or ‘nominal’ composition, in agreement with a preliminary observation in previous work.22 The range of compositions for different crystallites within each sample (as determined by EDX) is shown in the ESI,† and confirms a small variation is present, with a typical standard deviation of 10–12%. Calcination of the as-made materials removes the excess terephthalic acid (and any water) occluding the pores. For Al-MIL-53, this produces the expected OP form of the MOF (see Fig. 1), with the PXRD pattern and position of the carboxyl resonance at δ = 171 ppm in the 13C CP MAS NMR spectrum characteristic of this form (Fig. 2).22 However, for the Ga end member, the calcination conditions used (300 °C for 72 h) resulted in partial decomposition of the MOF, as seen in previous work.22 To investigate this further, Ga-MIL-53 was calcined under a range of different conditions, as shown in the ESI.† Although it appears possible to prevent the collapse of the MOF using lower calcination temperatures (e.g., 260 °C), this produces either the NP form (evidenced by the 13C carboxyl resonance at δ = 175 ppm) or, more usually, a mixture of OP and NP forms. The relative proportion of the two forms obtained varies when the calcination is repeated, and with the time between calcination and spectral acquisition (see ESI†). The PXRD pattern of Ga-MIL-53 shown in Fig. 2 (calcined at 260 °C) shows reflections similar to those found for the as-made form at 8.9° and 10.1°. However, following hydration, in which the closed pore (CP) form is obtained, and subsequent dehydration, in which the NP form is seen, these reflections disappear (see ESI†). It was not possible to find calcination conditions that enabled only the OP form of Ga-MIL-53 to be produced without significant decomposition. However, it is interesting to note that the incorporation of only small amounts of Al into Ga-MIL-53 (e.g., a nominal composition of x = 0.95) prevents decomposition even at the higher calcination temperature of 300 °C (as shown in Fig. 2).| x | Nominal Al:Ga |
EDX Al:Ga |
Difference (%) |
|---|---|---|---|
| 0.0 | 100:0 |
100:0 |
0 |
| 0.2 | 80:20 |
84:16 |
4 |
| 0.5 | 50:50 |
74:26 |
24 |
| 0.8 | 20:80 |
42:58 |
22 |
| 0.9 | 10:90 |
35:65 |
25 |
| 0.95 | 5:95 |
15:85 |
10 |
| 1.0 | 0:100 |
0:100 |
0 |
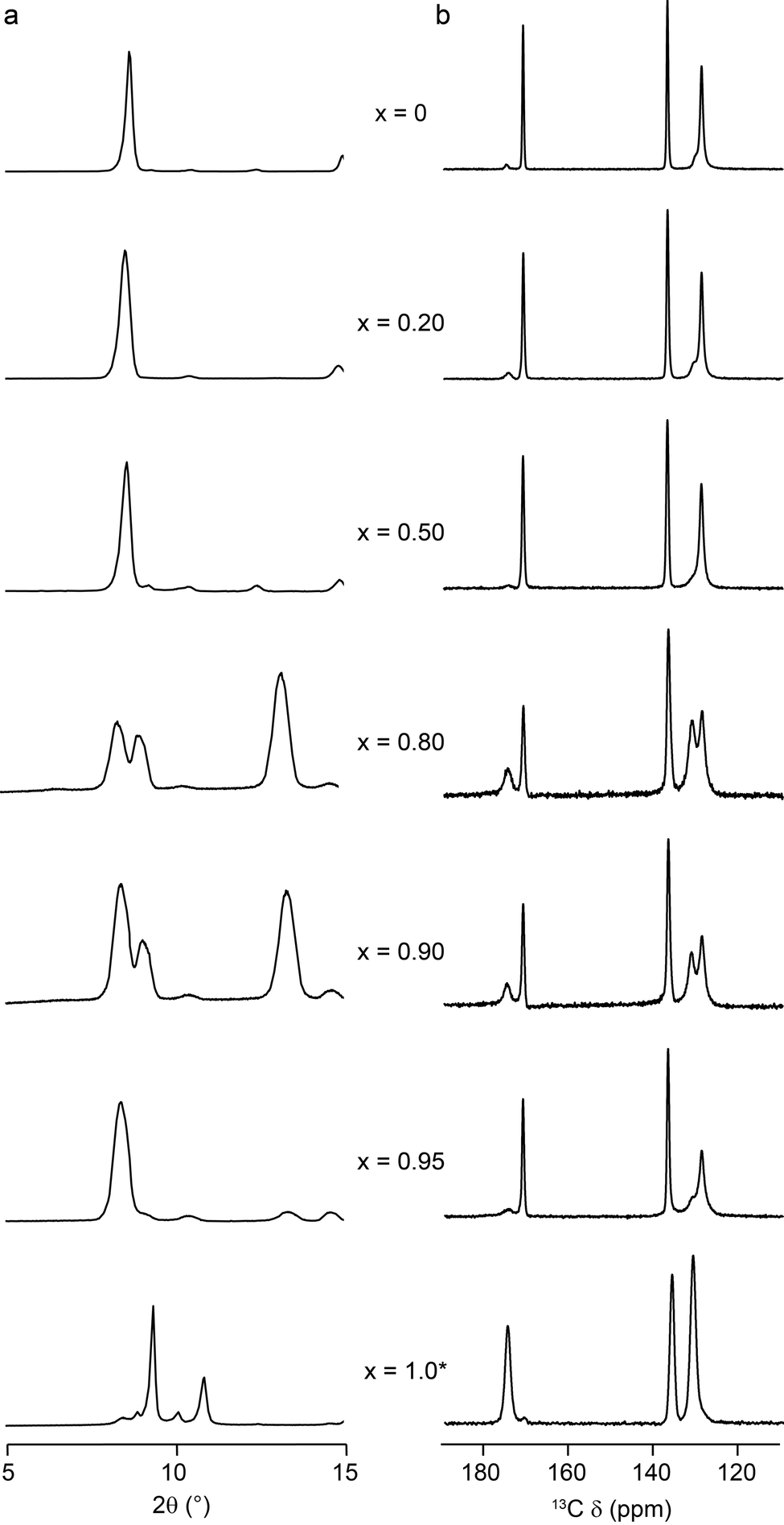 | ||
| Fig. 2 (a) Partial PXRD patterns and (b) 13C (14.1 T, 12.5 kHz) CP MAS NMR spectra of calcined (Al1−xGax)-MIL-53 for nominal compositions of x = 0, 0.2, 0.5, 0.8, 0.9, 0.95 and 1. *Note that Ga-MIL-53 was calcined at a lower temperature (260 °C) than other materials (300 °C) to prevent framework degradation. | ||
As shown in Fig. 2, for (Al0.8Ga0.2)-MIL-53 (i.e., x = 0.2), calcination at 300 °C for 72 h produces almost exclusively the OP form of MIL-53, but an increasing amount of NP form is observed as x increases to 0.90. A further increase in Ga content to x = 0.95 results in a lower proportion of the NP form, with almost exclusively OP present. (It should also be noted that although Ga-MIL-53 begins to decompose when calcined under these conditions, the parts of the MOF that remain intact adopt the OP form). It is challenging to quantify the absolute proportion of the two forms of MIL-53 present in each case, owing to the non-quantitative nature of 13C CP MAS spectra. However, the relative intensities of the resonances attributed to the carboxylate peaks in the NP and OP forms (at δ = 175 ppm and δ = 171 ppm, respectively) do not vary with the contact time in the CP MAS experiment, and so can be used to investigate the relative changes in the proportions of each form with composition, as shown in the ESI.† (Note that although changes are also observed in the aromatic region, the overlap of signals from different aromatic carbons (see also later DFT calculations) mean these signals cannot easily be used to follow changes in the proportions of each pore form.) 1H MAS NMR spectra (ESI†) confirm that the samples have not been exposed to H2O (which would be expected to result in a closing of the pores owing to hydrogen bonding to water – see below).
17O MAS NMR spectra of calcined (Al,Ga)-MIL-53, shown in Fig. 3a, exhibit two distinct areas of signal: between −80 and 20 ppm (resulting from the bridging hydroxyl species) and between 100 and 250 ppm (resulting from the carboxylate oxygens). There is a clear change in both sets of signals with composition. Resolution is improved in triple-quantum MAS NMR spectra, as seen in Fig. 3b and c, which show the regions corresponding to the carboxylate oxygens (acquired at 14.1 T) and the hydroxyl oxygens (acquired at 20.0 T), respectively. For Al-MIL-53, a single carboxylate resonance is observed (δ1 = 146 ppm), in agreement with previous work, and as would be expected given the symmetry (Imma)14 of the OP form. However, the MQMAS spectrum Ga-MIL-53 where only the NP form is present in Fig. 3 contains two distinct carboxylate resonances (δ1 = 146 and 155 ppm). The change in symmetry for this pore form results in a difference between the two carboxylate 17O species (although only one distinct carboxylate group and, therefore, one distinct carboxylate 13C signal, is present), with one oxygen having a much shorter distance to a bridging hydroxyl group across the pore than the second (see ESI†).14 The mixed-metal materials show the single carboxyl resonance associated with the OP form of MIL-53 for x = 0.2 and 0.5, but two resonances for x = 0.8 and 0.9. Although this confirms the NP form is present, the apparent overlap of the carboxylate resonance from the OP with the signal at lower δ1 in the NP form makes a detailed and quantitative analysis challenging. For x = 0.95, an intense carboxylate signal is seen for the OP form, with signal from the very small amount of NP form present considerably weaker, and not apparent at the contour levels shown.
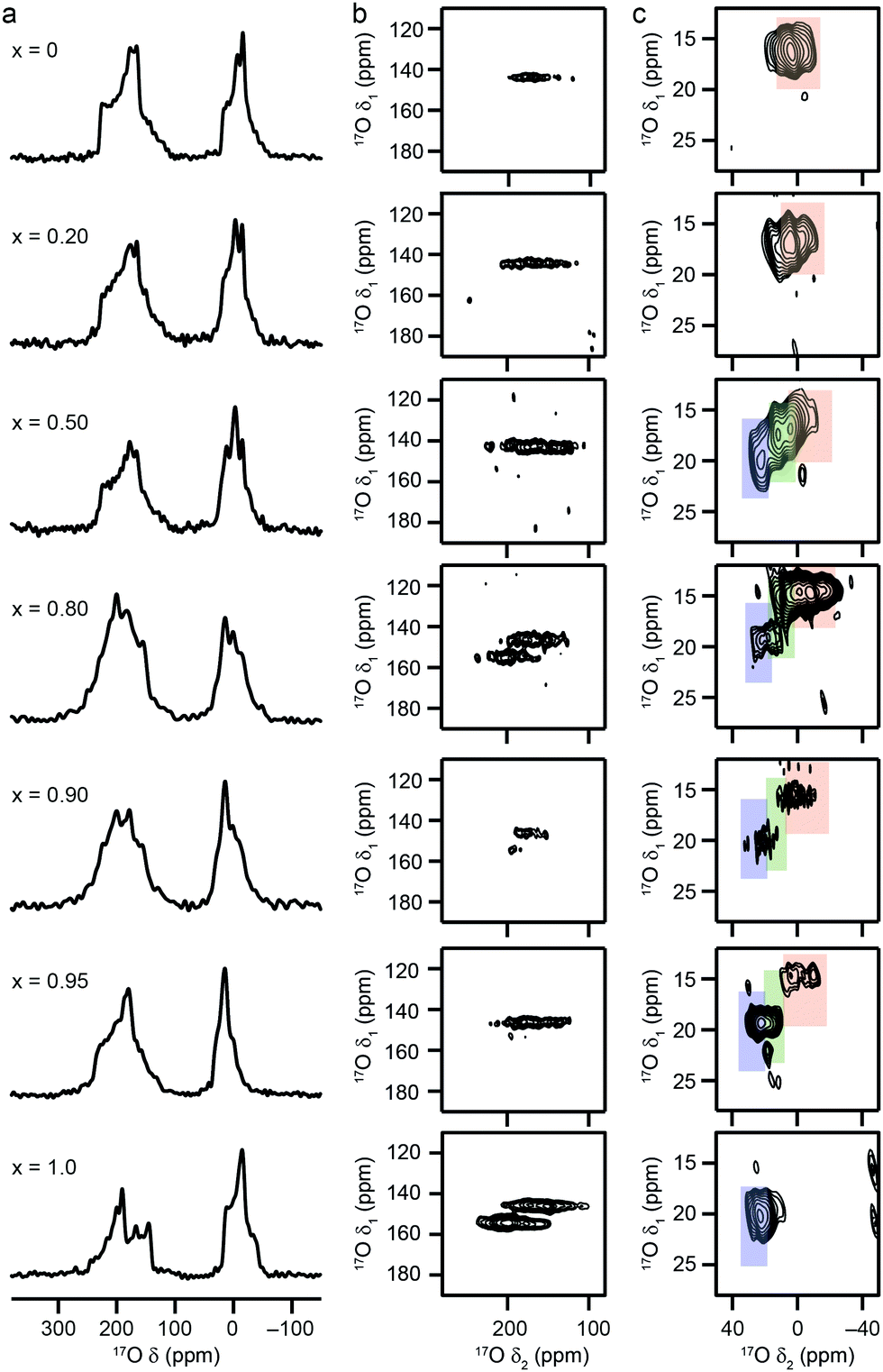 | ||
| Fig. 3 17O (a) MAS and (b and c) MQMAS NMR spectra of calcined (Al1−xGax)-MIL-53 for nominal compositions of x = 0, 0.2, 0.5, 0.8, 0.9, 0.95 and 1. Spectra have been acquired at (a and b) 14.1 T and (c) 20.0 T, with an MAS rate of 20 kHz. (b and c) Show expansions of the regions corresponding to carboxyl and hydroxyl oxygens, respectively. Signal corresponding to Al–OH–Al, Al–OH–Ga and Ga–OH–Ga is highlighted in red, green and blue, respectively. See also ref. 22. | ||
As the hydroxyl groups bridge between two adjacent metal centres in MIL-53, it is likely that the 17O NMR parameters will be sensitive to the nature of the two bound cations. However, these signals cannot be resolved in MQMAS NMR spectra at 14.1 T, and higher field measurements are required. Fig. 3c shows 17O MQMAS spectra of calcined (Al,Ga)-MIL-53, acquired at 20.0 T. Signals from Al–OH–Al and Ga–OH–Ga species are seen for the two end members (highlighted in red and blue), respectively, with an additional signal (resulting from Al–OH–Ga, highlighted in green) present in the mixed metal materials with low x. Previous work showed that for x = 0.5 the relative proportions of the signals from the three different linkages could be determined from a quantitative (i.e., short flip angle) 17O MAS NMR spectrum.22 This confirmed the composition of the MOF as 74:26 (Al:Ga), but also suggested a preference for some ordering/clustering of like cations, with the proportion of Al–OH–Al and Ga–OH–Ga linkages higher than expected for a truly random cation distribution. Given the materials have been prepared using DGC, where the solvent has been significantly reduced to facilitate cost effective isotopic enrichment in 17O, it could be that this might affect both the solubility of the metal salts (and therefore the overall composition of the MOF) and diffusion, perhaps promoting clustering rather than disorder.
To investigate the effect the synthesis route has on the composition and cation distribution in a mixed-metal MOF, the x = 0.5 material was remade using a standard hydrothermal synthesis (see above) and enriched in 17O post synthetically, using the steaming approach introduced in previous work.22 As shown in Fig. 4a, the composition of the MOF produced in this way, determined via EDX spectroscopy, is similar to that found in the material prepared using DGC, with Al:Ga = 75:25, suggesting that the differences between real and nominal compositions do not result from any relative differences in solubility in DGC reactions. The 13C CP MAS NMR spectrum, shown in Fig. 4b, shows essentially only the OP form of MIL-53 is present. The full 17O MAS NMR spectrum (after enrichment by steaming with 130 μL of 90% H217O(l)) is shown the ESI,† with an expansion of the hydroxylregion shown in Fig. 4c. The fitting of this lineshape confirms the presence of the three bridging hydroxyl environments, resulting from Al–OH–Al, Al–OH–Ga and Ga–OH–Ga species. The NMR parameters and relative intensities for these three components are in very good agreement with those seen for the same material synthesised using DGC,22 as shown in the ESI† and in Table 3, confirming a slight preference for ordering/clustering of like cations. This confirms a similar cation distribution is present in MIL-53 synthesised by both hydrothermal and DGC approaches, and suggests the preference for clustering of like cations is not due to limited diffusion in the latter.
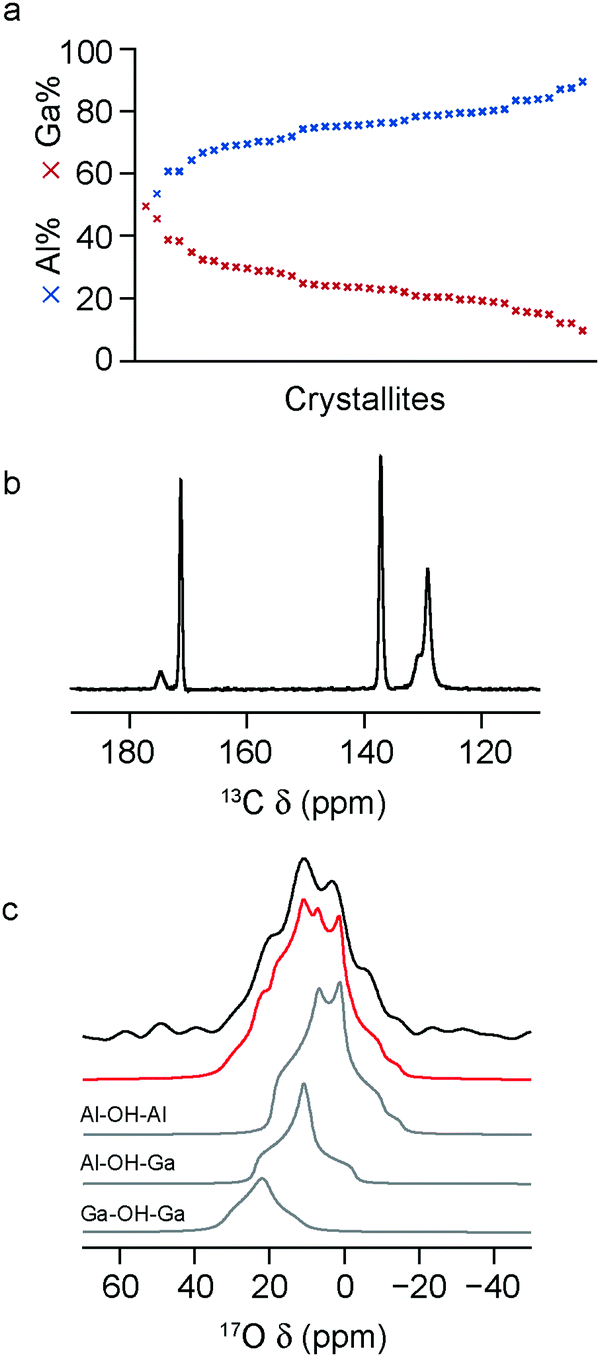 | ||
| Fig. 4 (a) Plot showing the compositions determined by EDX for a range of crystallites of calcined (Al0.5Ga0.5)-MIL-53 prepared hydrothermally and subsequently steamed with 17O-enriched water (90%). (b) Corresponding 13C (14.1 T, 12.5 kHz) CP MAS NMR spectrum. (c) Corresponding 17O (20.0 T, 20 kHz) MAS NMR spectrum (hydroxyl region only) and fit, showing the decomposition into signals resulting from Al–OH–Al, Al–OH–Ga and Ga–OH–Ga species. | ||
| Hydroxyl species | Relative intensity (%) | δ iso (ppm) | C Q/MHz | η Q |
|---|---|---|---|---|
| DGC synthesis (ref. 22) | ||||
| Al–OH–Al | 60(2) | 20(3) | 5.4(2) | 0.7(1) |
| Al–OH–Ga | 23(2) | 24(3) | 4.5(2) | 1.0(2) |
| Ga–OH–Ga | 17(2) | 32(3) | 3.9(2) | 1.0(2) |
| Hydrothermal synthesis (Fig. 4c) | ||||
| Al–OH–Al | 58(1) | 23(3) | 5.4(2) | 0.7(1) |
| Al–OH–Ga | 26(1) | 25(3) | 4.5(2) | 1.0(1) |
| Ga–OH–Ga | 16(1) | 31(3) | 3.9(2) | 1.0(1) |
Interestingly, as x increases from 0.5 in the mixed-metal materials, there is a decreasing amount of the signal attributed to Al–OH–Ga (relative to Ga–OH–Ga and Al–OH–Al), suggesting an increased preference for clustering of like cations as the Ga content rises. However, It should be noted that the presence of both NP and OP forms of MIL-53 in many of the samples hinders detailed analysis of the cation distribution (with the variation in composition of the individual crystallites possibly affecting the form of the MOF adopted and the spectral lineshapes observed – see later discussion). However for x = 0.95, where the vast majority of crystallites adopt the OP form, only significant signals from Al–OH–Al and Ga–OH–Ga appear to be present (despite predicted intensities of 2.25%, 25.5% and 72.5% for Al–OH–Al, Al–OH–Ga and Ga–OH–Ga if a random distribution of cations were present, and assuming an actual composition of 15:85 (see EDX in ESI†)). When combined with the observation from EDX that all crystallites studied contain a mix of Al and Ga (rather than phase separation), this suggests that increased Ga content in MIL-53 promotes the local ordering/clustering of like cations.
Upon hydration, most MIL-53 materials adopt a closed pore form, as shown in Fig. 1, as a result of hydrogen bonding interactions with the occluded water molecules.13,14,18,22,23,28Ref. 65 discusses the differences in water adsorption between Al-MIL-53 and Ga-MIL-53 (where an additional hydrated intermediate phase is seen for the latter upon initial hydration). However, PXRD and 13C CP MAS NMR spectra confirm that under the conditions used here Ga-MIL-53 is fully hydrated and adopts the same CP form as the Al end member. The mixed-metal materials studied here also all adopt a CP framework on hydration, although very small amounts of an OP form remain for some samples, as shown in the 13C CP MAS NMR spectra (see ESI† and Fig. 5 for x = 0.9), perhaps suggesting a very small fraction of the material is not fully hydrated. It should be noted for these materials that PXRD patterns do not contain any reflections relating to the OP form. The 17O MAS NMR spectra show small changes with composition, particularly in the hydroxyl region. 17O MQMAS spectra of the carboxylate region (ESI† and Fig. 5 for x = 0.9) differ very little with composition, with two distinct carboxyl resonances seen for all materials. Previous work suggested that these resulted from carboxylate O species with (δ1 = 142 ppm) and without (δ1 = 158 ppm) significant hydrogen bonding interactions with water – also see later.22
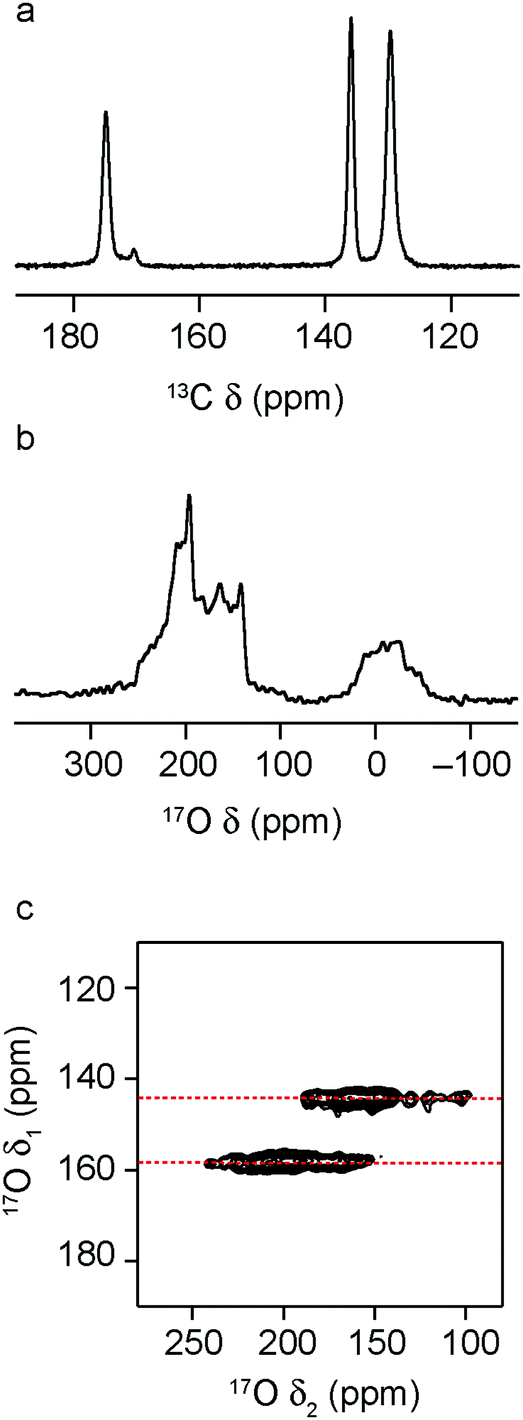 | ||
| Fig. 5 (a) 13C (14.1 T, 12.5 kHz) CP MAS, (b) 17O (14.1 T, 12.5 kHz) MAS and (c) 17O (14.1 T, 12.5 kHz) MQMAS NMR spectra of hydrated (Al0.1Ga0.9)-MIL-53. In (c), only the region of the spectrum containing the carboxyl species is shown. | ||
Fig. 6a shows 13C CP MAS NMR spectra of (Al,Ga)-MIL-53 materials that have been calcined, hydrated and subsequently dehydrated (at 120 °C). For Al-MIL-53, the MOF adopts predominantly the OP form (as shown by 13C NMR spectra and PXRD patterns), as observed for the calcined material. However, for Ga-MIL-53, the NP form is seen almost exclusively. This is in good agreement with previous work, which showed two forms of dehydrated Ga-MIL-53, a NP form said to be stable at lower temperatures, and the OP form produced when dehydration was carried out at higher temperatures (i.e., closer to those used for the calcination).65,66 The mixed-metal materials have both OP and NP forms present, with the relative amount of the latter increasing fairly systematically with Ga content (see ESI†). 1H MAS NMR spectra (ESI†) show that the presence of two different pore sizes does not result from inefficient dehydration, with no signals from H2O observed for these samples. 17O MQMAS spectra in Fig. 6b (expanded to show the carboxyl region) also confirm this observation, showing small but significant shifts of the two carboxylate resonances from the two shifts seen for the hydrated CP form (shown by the dashed red lines). Although mixed pore materials are found for both calcined and (calcined, rehydrated and subsequently) dehydrated mixed-metal MOFs, the relative proportions of the two forms seen, and the variation with composition, are different. For calcination, the higher temperature used results in the presence of NP MOF only for the more substituted (and presumably most disordered) mixed-metal materials, with lower levels of substitution favouring OP MOFs. However, upon dehydration of the mixed-metal calcined, hydrated materials (where the temperature used is lower) increasing amounts of NP form are seen with increasing Ga content. This is in good agreement with the observation of a NP form for dehydrated Ga-MIL-53 in previous work.65,66 Furthermore, as noted above, the calcination of Ga-MIL-53 at 300 °C produces only the OP form (as with the Al end member), but results in some framework degradation. Lowering the temperature prevents this decomposition but produces an increasing amount of NP MOF.
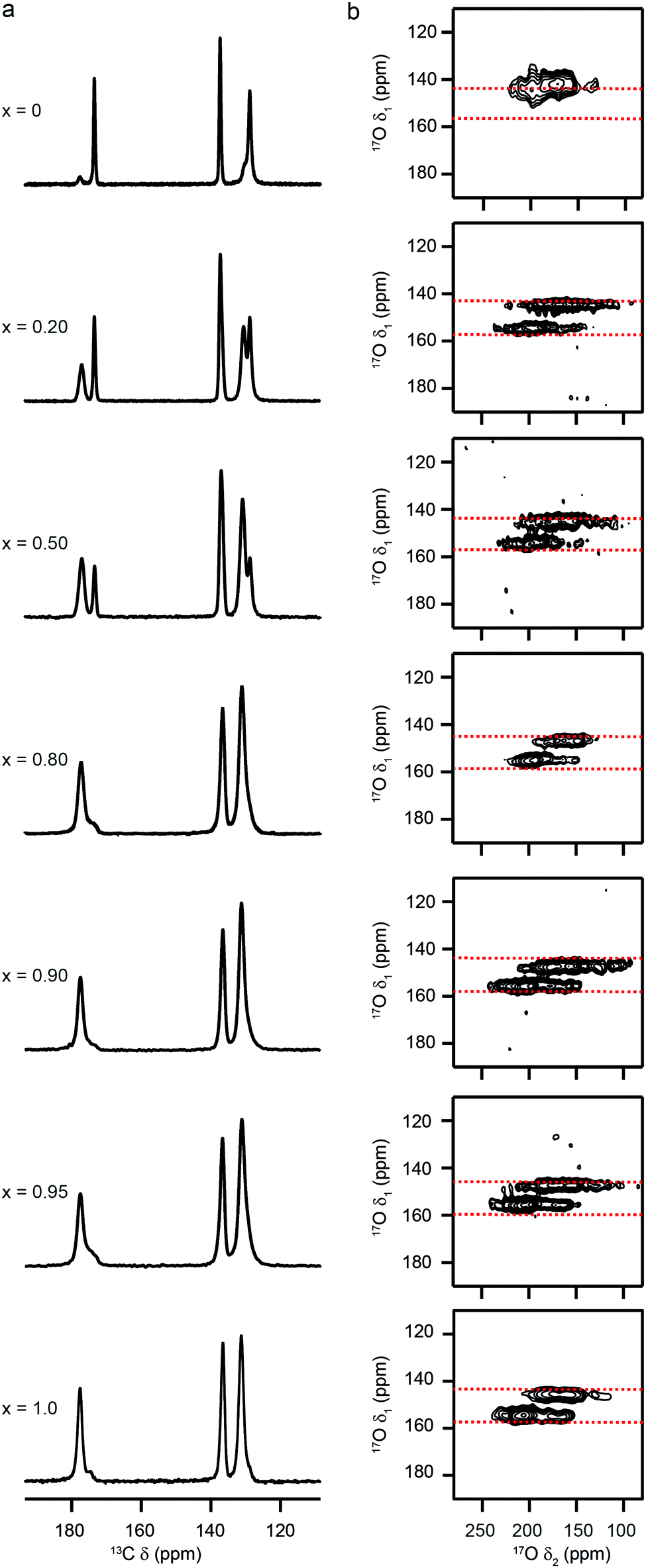 | ||
| Fig. 6 (a) 13C (14.1 T, 12.5 kHz) CP MAS and (b) 17O (14.1 T, 12.5 kHz) MQMAS NMR spectra of calcined, hydrated and subsequently dehydrated (Al1−xGax)-MIL-53 for nominal compositions of x = 0, 0.2, 0.5, 0.8, 0.9, 0.95 and 1. In (b), only the region of the spectrum containing the carboxyl species is shown. | ||
In order to probe the relative energies of the different forms of MIL-53 as the composition varies, and to investigate if there is any energetic preference for cation ordering, first-principles DFT calculations were carried out for a set of model structures (in both OP and NP forms), as described in the ESI.†Fig. 7a plots the difference in energy between the OP and NP forms of MIL-53 as a function of composition. This predicts that the NP form is favoured across the compositional range, although the relative energy difference between the two forms is lower with increasing Al content.
 | ||
| Fig. 7 Calculated (a) energy difference between the OP and NP forms and (b) mixing energy for the NP form of mixed-metal (Al1−xGax)-MIL-53, plotted as a function of composition, and separated by the type of ordering/mixing present (see ESI† for descriptions). | ||
However, it should be noted that this energy difference is very different if dispersion is not accounted for in the calculations. Previous work has demonstrated the importance of including a dispersion correction when determining the geometry (and energy) of flexible and microporous materials.44,67 As shown in the ESI† (Fig. S2.2), if a dispersion correction is not applied at all in the calculations, the OP form is predicted to be more stable for all compositions. This change reflects the greater importance of dispersion interactions in the NP form, where the linkers are in much closer proximity across the narrower pore width. As shown in the ESI† (Fig. S2.3), there is a significant difference in the absolute energy of the NP form of MIL-53 (a decrease of ∼11 eV) upon the inclusion of dispersion interactions (although this energy does not vary appreciably with composition). In contrast, there is a much smaller decrease in energy of the OP form (of ∼2 eV) when dispersion interactions are included. This change, however, does vary significantly with composition, with an increase in Ga content leading to less stabilisation from dispersion. This reflects the greater increase in volume observed for the OP form with increased Ga content. In contrast, almost no change in volume is observed for the NP form with a variation in composition (see ESI,† Fig. S2.4). Although the neglect of dispersion interactions does not provide a good description of the real system, the differences seen between Fig. 7a and Fig. S2.2 (ESI†) reflect the importance of including accurate dispersion corrections, and the possible changes (both in the absolute values and in the resulting conclusions) that would be observed for any under or overestimation of these in any chosen methodology. Although the DFT calculations predict that the NP form of MIL-53 is more stable across all Al/Ga mixed-metal compositions (at least at 0 K, as no effects of temperature are included in the calculations reported here), the relative increase in stability of the OP form at higher Al content goes some way to explaining the experimental observation of the OP form of Al-MIL-53 upon calcination and dehydration, and the higher levels of the OP form seen for mixed-metal materials with higher Al content. The differences in the relative levels of OP and NP forms seen for calcined and for calcined, hydrated and subsequently dehydrated mixed-metal materials suggests that kinetics, rather than simply thermodynamics, plays a role in the forms that are present experimentally. The higher temperatures used for calcination, and the excess BDC in the pore of the as-made materials, which leads to an initial framework with larger pores, result in more of the OP form of MIL-53. In contrast, the lower temperatures used for dehydration (and the strong hydrogen bonding that results in the CP form of MIL-53 for the hydrated MOF) appear insufficient to open the framework when the Ga content is higher (and the OP form is relative higher in energy).
The DFT calculations can also be used to confirm the assignment and interpretation of the signals seen in the experimental NMR spectra. Fig. 8 shows calculated 13C δiso for OP and NP forms of mixed-metal MIL-53 materials as a function of composition. There is a clear difference for the carboxyl resonance between the two different forms, with a lower δ in the OP form, and, interestingly, the range of δ predicted for the NP form is significantly greater, both in good agreement with the experimental results. The resonance associated with the ipso C on the aromatic ring is found at higher δ than those for the C–H. There is very little change in δ with composition for all species. Fig. 9 shows similar plots for 17O δiso. Signals from the different types of hydroxyl groups (i.e., Al–O–Al, Ga–O–Ga and Al–O–Ga) have different δiso, but there is more overlap in values of δiso for the different types of carboxyl O. Small downfield shifts are seen for each type of OH on going from NP to OP, but these changes are less significant than those seen for the variation in composition. Two relatively distinct groups of shifts are seen for the NP carboxyls – resulting from a rotation of the linker moving one carboxyl 17O species, at the higher δiso of ∼255 ppm, away from the pore and the other towards the pore at lower δiso of ∼230 ppm. While the latter is moved closer to the hydroxyl species across the pore by this rotation, the resulting O⋯H distance of ∼3.5 Å (depending on composition), and its similar 17O chemical shift to those of OP carboxyls, suggests the absence of a hydrogen-bonding interaction across the pore. Although this is in good qualitative agreement with experiment, with only one resonance seen in the carboxyl region of the MQMAS spectrum for OP MOFs and two distinct resonances for NP materials, the position of a signal in an MQMAS spectrum depends also on the magnitude of the quadrupolar interaction. As shown in Fig. 10, all OH species have similar CQ values in the NP form, while for the OP form, OH groups bound to at least one Al have slightly lower CQ than those bound to two Ga cations. Small differences in CQ are seen for different types of carboxyl groups, but the separation in the MQMAS experiment arises largely from the differences in δiso. This is confirmed in the ESI† (Fig. S2.6), where the positions of the centre-of-gravity of resonances in a (sheared) MQMAS spectrum (i.e., values of δ1 and δ2) are plotted for B0 = 14.1 T and 20.0 T. This predicts that the resonances from hydroxyls bridging different metal cations can be resolved at high field for both OP and NP forms, as seen in experiment, while for the carboxyl there is little separation of O bound to different cations at either field (and particularly so for the NP form), but more significant changes are seen between the two pore forms of MIL-53. (For completeness, information on the calculated 1H and 27Al NMR parameters for mixed-metal MIL-53 are shown in the ESI.†) It is clear from Fig. 7b that different spatial arrangements of Al and Ga in NP mixed-metal MIL-53 have different relative energies. In general, this figure shows that lower energy arrangements have less cation ordering, although layers of similar cations are generally preferred to chains of like cations (see ESI† for details of the models used). For all compositions, there are arrangements that are favourable (i.e., have a negative mixing energy, Emix) relative to phase separation, although it should be noted that (i) no entropic contribution is accounted for here and (ii) the cation arrangement obtained in the synthetic samples should reflect the relative energies of the as-made, rather than the calcined (or dehydrated) forms, of MIL-53. Although it is not easy to calculate these (because of the static and dynamic disorder of the occluded BDC and water molecules), it should be noted that the plot of the mixing energy for OP mixed-metal MIL-53 shown in the ESI† is similar, with more random arrangements lower in energy, and layers preferred over chains of similar cations. This perhaps suggests this is a more general result for different forms of MIL-53 and might well be applicable to as-made materials. However, it should be noted that only a single unit cell was considered in the calculations performed in this work (because of restrictions on computational cost and time), limiting the possible cation arrangements that were studied. In particular, the unit cell contains only two cation sites in the chains found along the x axis, meaning that substitution of the two cations here leads to the formation of infinite chains of like cations in the structural models. Fig. 7b shows that such chains are relatively less favoured than more random cation distributions, or layers of like cations. This perhaps appears in poor agreement with the experimental 17O MQMAS spectra, which suggest that mixed metal (i.e., Al–O–Ga) linkages are less prevalent (at least at higher Ga content) relative to OH groups bridging between like cations. However, the necessity for infinite chains (in the x direction) of Al–O–Al and Ga–O–Ga linkages upon substitution into a single unit cell, and that in such a model layers and chains are mutually exclusive rather than potentially coexisting as in experimental samples, means that it is difficult to make unambiguous conclusions about the preference (or otherwise) for clustering or ordering of like cations – but does suggest that regions containing long chains of like cations are not likely to be favourable, and that clustering must be fairly local. As the cation distribution observed experimentally should reflect the relative energies of the as-made materials, a potentially interesting consideration for the pore form of the MOF observed upon calcination is the relative difference in the energies of the OP and NP forms for a particular cation arrangement. However, Fig. 7a shows that there is relatively little correlation between energy difference between OP and NP forms, and whether substituted cations are found in layers, chains or are more randomly distributed.
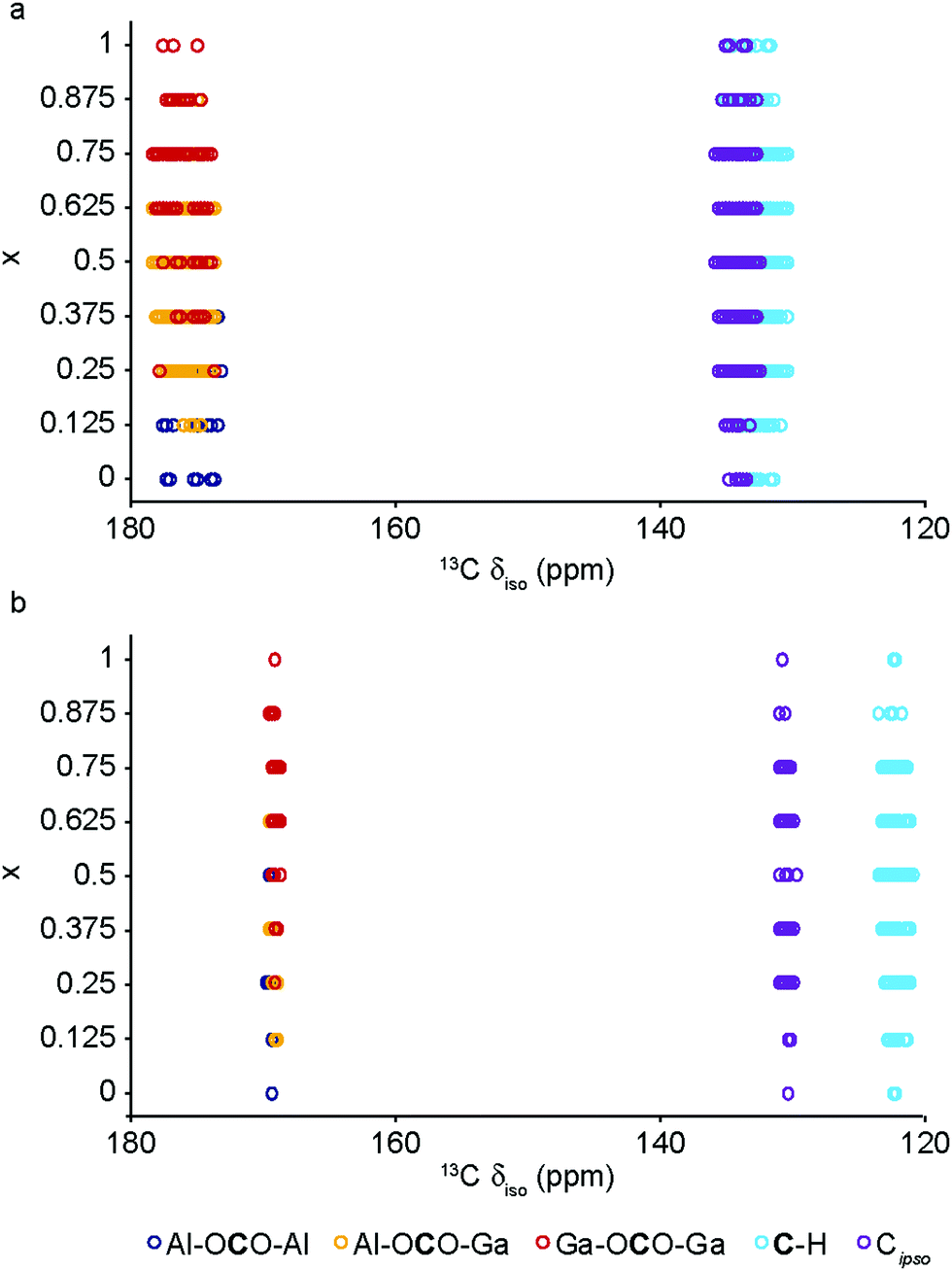 | ||
| Fig. 8 Calculated 13C δiso for (a) NP and (b) OP forms of mixed-metal (Al1−xGax)-MIL-53. | ||
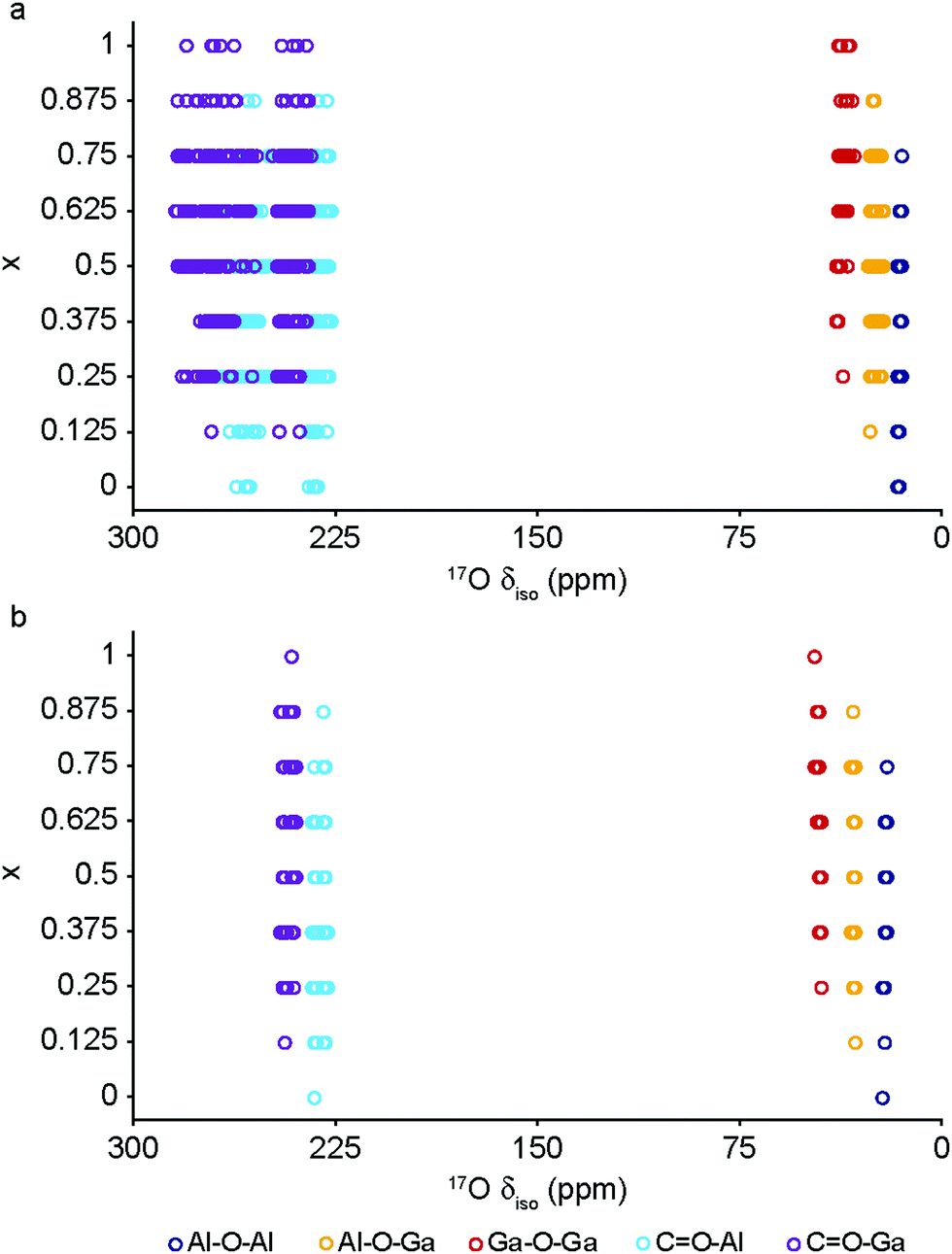 | ||
| Fig. 9 Calculated 17O δiso for (a) NP and (b) OP forms of mixed-metal (Al1−xGax)-MIL-53. | ||
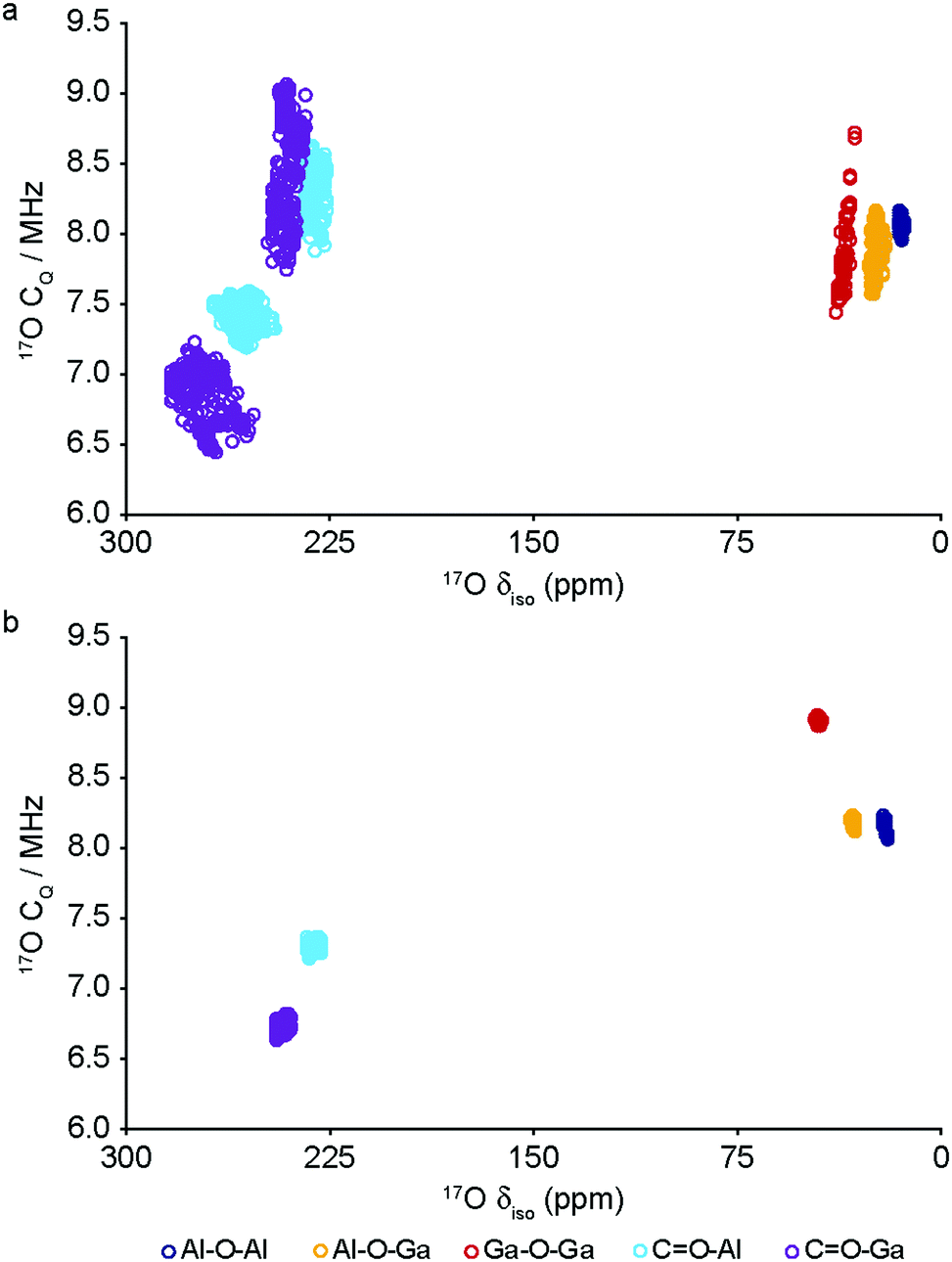 | ||
| Fig. 10 Calculated 17O NMR parameters (δiso and CQ) for (a) NP and (b) OP forms of mixed-metal (Al1−xGax)-MIL-53. | ||
The EDX analysis shown in the ESI† (and discussed above) reveals that while the level of Al/Ga in each crystallite is relatively similar, some variation (typically 7–12%) is observed between crystallites. Given the variation in the relative energy of the OP and NP forms with composition predicted using DFT, it is possible that the presence of mixed pore forms of MIL-53 for mixed-metal compositions reflects the different levels of substitution in each crystallite (i.e., crystallites with lower levels of Al adopt one form and those with higher level a different form). However, the relative proportions of OP and NP forms seen for calcined MOFs do not correlate linearly with the distribution of compositions of the crystallites seen from EDX. This is perhaps not too surprising given the implication that both kinetics and thermodynamics play a role in the forms seen (i.e., the proportions are different depending on the conditions used to produce the empty framework), the observation from the computational work that different distributions of cations affects the relative energies of the OP and CP forms and previous work which has shown that the form seen can also be affected by crystallite size.18,68 However, the fairly systematic change from OP to NP forms seen in Fig. 6 for calcined, hydrated and subsequently dehydrated (Al,Ga)-MIL-53 suggests a general trend that crystallites with higher average levels of Al are more likely to adopt the OP form, while those with lower levels are more likely to adopt the NP form for lower temperature dehydration.
Conclusions
The use of the atom-efficient DGC reactions have enabled the cost-effective synthesis of 17O-enriched mixed-metal (Al,Ga)-MIL-53 materials, whose chemical structure and breathing behaviour were investigated using an NMR crystallographic approach. Although EDX analysis confirms all of the crystallites studied contain a mix of metal cations, the average level of Ga in the final product (determined from both EDX and 17O NMR spectroscopy) is significantly lower than expected from the stoichiometry of the starting reagents. Upon calcination, we show that materials with higher levels of Al adopt an OP form of the MOF, as is found for the Al end member, but substitution of Ga results in mixed pore materials, with OP and NP forms co-existing. For further increases in Ga substitution, the OP form is once again favoured. It was only possible to obtain calcined Ga-MIL-53 without any decomposition by reducing the calcination temperature, resulting in the formation of a NP form of the MOF. 13C CP MAS NMR spectra were shown to be very sensitive to the pore structure(s) that a framework adopts, with first-principles calculations predicting a significant shift to higher δ and a broadening of the resonances resulting from the carboxylate carbon as the pores close, in good agreement with experiment. Signals resulting from carboxyl and hydroxyl O species were easily resolved in 17O MAS NMR spectra (confirming efficient isotopic enrichment of both types of O during the DGC reaction); however, the second-order quadrupolar broadening limits the resolution of different types of carboxyl or hydroxyl O species. MQMAS spectra showed clear differences in the carboxylate region for OP and NP MOFs, with one carboxylate resonance observed for the former, but two for the latter. The overlap of the carboxylate signal at lowest δ1 with that from the OP form hinders a detailed quantitative analysis when mixed pore forms are present. Although relatively little change in the 13C or 17O spectra was seen with composition, high-field (20.0 T) MQMAS spectra were able to resolve chemically different bridging hydroxyl species, and the relative ratios of Al–O(H)–Al, Al–O(H)–Ga and Ga–O(H)–Ga species seen supported the compositions determined using EDX and demonstrated an increased preference for clustering of like cations as the Ga content increases (i.e., a reduction in the Al–O(H)–Ga linkages over those expected assuming a random cation distribution). It has been confirmed that this preferential clustering is not a result of the small-scale DGC synthesis method, as a similar cation distribution and clustering effect is observed for (Al0.5,Ga0.5)-MIL-53 synthesised hydrothermally and enriched with 17O via post-synthetic steaming, as shown by EDX and 17O NMR spectroscopy.For empty frameworks, the pore structure adopted by a material depends both on the composition and on the thermal history. For calcined MOFs, the amount of the NP form seen is greatest for the most disordered materials (i.e., those closer to a 50:50 composition). Interestingly, although the Ga end member is prone to decomposition under the calcination conditions used (300 °C for 72 h), a low level of Al in the starting synthesis (5%) leads to mixed-metal MOF (with ∼15% Ga substitution) that is stable to calcination and adopts a predominantly OP framework. Although the decomposition of Ga-MIL-53 can be prevented by using lower temperatures, this leads to a NP calcined framework, and it was not possible to find calcination conditions that resulted in a purely OP material. Upon hydration all materials almost exclusively adopted a CP structure, with the strong hydrogen bonding interactions with water leading to a significant difference between the two distinct resonances from the carboxylate oxygens seen in this form. When dehydrated at 120 °C different framework structures are found for the two end members, with OP and NP forms adopted by Al- and Gal-MIL-53, respectively. For mixed metal materials, evidence for the presence of both OP and NP framework is provided by PXRD, and 13C and 17O NMR spectra, with the proportion of NP MOF seen to increase fairly systematically with the Ga content, in contrast to the materials seen upon initial calcination of as-made MOFs. This suggests that materials with higher Ga content favour the adoption of a NP structure when empty, but the higher temperatures used in the initial calcination are more likely to force more crystallites to adopt an OP structure.
DFT calculations on calcined MIL-53 materials predict that the NP form is favoured across the compositional range, although the relative energy difference is lower with increasing Al content, perhaps suggesting why higher levels of the OP form are seen for mixed-metal materials with high Al content upon calcination and dehydration. However, it should be noted that the results are significantly affected by the inclusion of a dispersion correction scheme in the calculations (with the OP more stable if this is not included). While it is clear such a scheme is required to take into account the van der Waals interactions not modeled in DFT, this significant change does highlight the importance of including accurate dispersion corrections, and the possible changes (in the absolute energies and resulting structures) that could be observed for any significant under or overestimation within the chosen computational methodology. Calculations also showed that spatial arrangements of Al and Ga in mixed-metal MIL-53 lead to different energies for OP and NP material, and different relative energy differences between them. While this will undoubtedly affect the propensity of a material to adopt an OP or NP framework on calcination or dehydration, it is difficult to compare the values obtained directly to experimental observations, as the cation distribution seen should reflect the energies of the as-made forms of the MOFs produced in the initial synthesis.
Although a challenging system to study, we have shown that the sensitivity of NMR spectroscopy to changes in pore size and shape and to compositional variation suggests that this will be a useful tool for the characterisation of structure and disorder in mixed-metal MOFs, particularly when combined with PXRD and DFT calculations in the NMR crystallographic approach employed here. Detailed characterisation of local and long-range structure, and an understanding of the effect this has on properties, will be crucial for the future design of functional MOFs and their industrial application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the ERC (EU FP7 Consolidator Grant 614290 EXONMR and Advanced Grant 787073 ADOR), and EPSRC (EP/N509759/1) for a studentship for CMR. SEA would like to thank the Royal Society and Wolfson Foundation for a merit award. We acknowledge support from the Collaborative Computational Project on NMR Crystallography (CCP-NC) funded by EPSRC (EP/M022501/1) and the UKCP consortium funded by EPSRC (EP/K013564/1). For computational resources we are grateful to the UK Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/P020194/1) and the UK HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202). The research data (and/or materials) supporting this publication can be accessed at DOI: http://10.17630/31529c8b-f197-484c-929b-ef993a5bea68.69References
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- L. Li, S. Wang, T. Chen, Z. Sun, J. Luo and M. Hong, Cryst. Growth Des., 2012, 12, 4109–4115 CrossRef CAS.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- G. Xu, P. Nie, H. Dou, B. Ding, L. Li and X. Zhang, Mater. Today, 2017, 20, 191–209 CrossRef CAS.
- L. N. McHugh, M. J. McPherson, L. J. McCormick, S. A. Morris, P. S. Wheatley, S. J. Teat, D. McKay, D. M. Dawson, C. E. F. Sansome, S. E. Ashbrook, C. A. Stone, M. W. Smith and R. E. Morris, Nat. Chem., 2018, 10, 1096–1102 CrossRef CAS PubMed.
- A. Schoedel, M. Li, D. Li, M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS.
- M. O'Keeffe, M. Eddaoudi, H. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3–20 CrossRef.
- M. Alhamami, H. Doan and C.-H. Cheng, Materials, 2014, 7, 3198–3250 CrossRef PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- T. R. Whitfield, X. Wang, L. Liu and A. J. Jacobson, Solid State Sci., 2005, 7, 1096–1103 CrossRef CAS.
- E. V. Anokhina, M. Vougo-Zanda, X. Wang and A. J. Jacobson, J. Am. Chem. Soc., 2005, 127, 15000–15001 CrossRef CAS PubMed.
- M. Vougo-Zanda, J. Huang, E. Anokhina, X. Wang and A. J. Jacobson, Inorg. Chem., 2008, 47, 11535–11542 CrossRef CAS.
- J. P. S. Mowat, S. R. Miller, A. M. Z. Slawin, V. R. Seymour, S. E. Ashbrook and P. A. Wright, Microporous Mesoporous Mater., 2011, 142, 322–333 CrossRef CAS.
- Y. Zhang, B. E. G. Lucier, V. V. Terskikh, R. Zheng and Y. Huang, Solid State Nucl. Magn. Reson., 2017, 84, 118–131 CrossRef CAS PubMed.
- F. Millange, C. Serre and G. Férey, Chem. Commun., 2002, 822–823 RSC.
- C. Volkringer, T. Loiseau, N. Guillou, G. Férey, E. Elkaïm and A. Vimont, Dalton Trans., 2009, 2241–2249 RSC.
- G. P. M. Bignami, Z. H. Davis, D. M. Dawson, S. A. Morris, S. E. Russell, D. McKay, R. E. Parke, D. Iuga, R. E. Morris and S. E. Ashbrook, Chem. Sci., 2018, 9, 850–859 RSC.
- F. Millange and R. I. Walton, Isr. J. Chem., 2018, 58, 1019–1035 CrossRef CAS.
- F.-X. Coudert, A. Boutin and A. H. Fuchs, Mol. Phys., 2014, 112, 1257–1261 CrossRef CAS.
- L. J. Wang, H. Deng, H. Furukawa, F. Gańdara, K. E. Cordova, D. Peri and O. M. Yaghi, Inorg. Chem., 2014, 53, 5881–5883 CrossRef CAS PubMed.
- R. Q. Liu, H. Cong and H. Deng, J. Am. Chem. Soc., 2016, 138, 13822–13825 CrossRef PubMed.
- O. Kozachuk, M. Meilikhov, K. Yusenko, A. Schneemann, B. Jee, A. V. Kuttatheyil, M. Bertmer, C. Sternemann, A. Pöppl and R. A. Fischer, Eur. J. Inorg. Chem., 2013, 4546–4557 CrossRef CAS.
- F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J.-M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyn and C. Serre, Chem. Commun., 2015, 48, 10237–10239 RSC.
- J.-S. Qin, S. Yuan, Q. Wang, A. Alsalme and H.-C. Zhou, J. Mater. Chem. A, 2017, 5, 4280–4291 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Catal. Sci. Technol., 2016, 6, 5238–5261 RSC.
- S. Horike and S. Kitagawa in Metal-Organic Frameworks: Application from Catalysis to Gas Storage, ed. D. Farrusseng, Wiley-VCH, Weinheim, 1st edn, 2011, ch. 1, vol. 1, pp. 1–22 Search PubMed.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, C. J. Kelsall, M. K. Doherty, P. D. Whitfield, I. L. Megson and R. E. Morris, RSC Adv., 2016, 6, 14059–14067 RSC.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- J. Kahr, R. E. Morris and P. A. Wright, CrystEngComm, 2013, 15, 9779–9786 RSC.
- J. D. Howe, C. R. Morelock, Y. Jiao, K. W. Chapman, K. S. Walton and D. S. Sholl, J. Phys. Chem. C, 2017, 121, 627–635 CrossRef CAS.
- R. M. Marti, J. D. Howe, C. R. Morelock, M. S. Conradi, K. S. Walton, D. S. Sholl and S. E. Hayes, J. Phys. Chem. C, 2017, 121, 25778–25787 CrossRef CAS.
- D. Ge, G. Qu, X. Li, K. Geng, X. Cao and H. Gu, New J. Chem., 2016, 40, 5531–5536 RSC.
- S. H. Kim, Y. J. Lee, D. H. Kim and Y. J. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 660–667 CrossRef CAS PubMed.
- S. E. Ashbrook, D. M. Dawson and J. M. Griffin in Local Structure Characterisation, ed. D. W. Bruce, D. O’Hare and R. I. Walton, John Wiley & Sons, Oxford, 1st edn, 2014, ch. 1, vol. 1, pp. 1–88 Search PubMed.
- R. F. Moran, D. M. Dawson and S. E. Ashbrook, Int. Rev. Phys. Chem., 2017, 36, 39–115 Search PubMed.
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid State NMR Basic Principles and Practice, Momentum Press, New York, 2012 Search PubMed.
- A. Sutrisno and Y. Huang, Solid State Nucl. Magn. Reson., 2013, 49–50, 1–11 CrossRef CAS PubMed.
- S. E. Ashbrook, D. M. Dawson and V. R. Seymour, Phys. Chem. Chem. Phys., 2014, 16, 8223–8242 RSC.
- H. C. Hoffmann, M. Debowski, P. Mueller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537–2572 CrossRef CAS.
- S. E. Ashbrook and M. E. Smith, Chem. Soc. Rev., 2006, 35, 718–735 RSC.
- S. E. Ashbrook and M. E. Smith, 17O NMR of Inorganic Materials, eMagRes, 2011 DOI:10.1002/9780470034590.emrstm1213.
- P. He, J. Xu, V. V. Terskikh, A. Sutrisno, H.-Y. Nie and Y. Huang, J. Phys. Chem. C, 2013, 117, 16953–16960 CrossRef CAS.
- NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, John Wiley & Sons, Hoboken, NJ, 2009 Search PubMed.
- S. E. Ashbrook and D. McKay, Chem. Commun., 2016, 52, 7186–7204 RSC.
- C. Martineau, J. Senker and F. Taulelle, NMR Crystallography, Annu. Rep. NMR Spectrosc., 2014, 82, 1–57 CrossRef.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- A. Pines, M. G. Gibby and J. S. Waugh, J. Chem. Phys., 1972, 56, 1776–1777 CrossRef CAS.
- A. E. Bennett, C. M. Reinstra, M. Auger, K. V. Lakshmi and R. G. Griffin, J. Chem. Phys., 1995, 103, 6951–6958 CrossRef CAS.
- L. Frydman and J. S. Harwood, J. Am. Chem. Soc., 1995, 117, 5367–5368 CrossRef CAS.
- J.-P. Amoureux, C. Fernandez and S. Steuernagel, J. Magn. Reson., Ser. A, 1996, 123, 116–118 CrossRef CAS PubMed.
- K. J. Pike, R. P. Malde, S. E. Ashbrook, J. McManus and S. Wimperis, Solid State Nucl. Magn. Reson., 2000, 16, 203–215 CrossRef CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 1865–1868 CrossRef PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- J. R. Yates, C. J. Pickard, M. C. Payne and F. Mauri, J. Chem. Phys., 2003, 118, 5746–5753 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. Pyykko, Mol. Phys., 2018, 116, 1328–1338 CrossRef CAS.
- F.-X. Coudert, A. U. Ortiz, V. Haigis, D. Bousquet, A. H. Fuchs, A. Ballandras, G. Weber, I. Bezverkhyy, N. Geoffroy, J.-P. Bellat, G. Ortiz, G. Chaplais, J. Patarin and A. Boutin, J. Phys. Chem. C, 2014, 118, 5397–5405 CrossRef CAS.
- A. Boutin, D. Bousquet, A. U. Ortiz, F.-X. Coudert, A. H. Fuchs, A. Ballandras, G. Weber, I. Bezverkhyy, J. P. Bellat, G. Ortiz, G. Chaplias, J.-L. Paillaud, C. Marichal, H. Nouali and J. Patarin, J. Phys. Chem. C, 2013, 117, 8180–8188 CrossRef CAS.
- S. Sneddon, D. M. Dawson, C. J. Pickard and S. E. Ashbrook, Phys. Chem. Chem. Phys., 2014, 16, 2660–2673 RSC.
- T. Kundu, M. Wahiduzzaman, B. B. Shah, G. Maurin and D. Zhao, Angew. Chem., 2019, 28, 8073–8077 CrossRef PubMed.
- C. M. Rice, Z. H. Davis, D. McKay, G. P. M. Bignami, R. G. Chitac, D. M. Dawson, R. E. Morris and S. E. Ashbrook, Following the Unusual Breathing Behaviour of 17O-Enriched Mixed-Metal (Al,Ga)-MIL-53 using NMR Crystallography, Dataset, University of St Andrews Research Portal, 2020, DOI:10.17630/31529c8b-f197-484c-929b-ef993a5bea68.
Footnotes |
| † Electronic supplementary information (ESI) available: Further information on the MIL-53 structure, the calcination of Ga-MIL-53, enrichment of a sample prepared using hydrothermal synthesis, the results of DFT calculations and structural models used, EDX, PXRD and NMR measurements. See DOI: 10.1039/d0cp02731f |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2020 |