
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-adiabatic quantum dynamics of the electronic quenching OH(A2Σ+) + Kr†
Pablo
Gamallo
 a,
Alexandre
Zanchet
bcd,
F. Javier
Aoiz
*b and
Carlo
Petrongolo
*e
a,
Alexandre
Zanchet
bcd,
F. Javier
Aoiz
*b and
Carlo
Petrongolo
*e
aDepartament de Ciència de Materials i Química Física & Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, c/Martí i Franquès 1-11, 08028 Barcelona, Spain
bDepartamento de Química Física, Facultad de Química, Universidad Complutense, 28040 Madrid, Spain. E-mail: aoiz@quim.ucm.es
cInstituto de Fisica Fundamental, CSIC, Serrano 123, 28006 Madrid, Spain
dDepartamento de Química Física, Universidad de Salamanca, 37008 Salamanca, Spain
eIstituto per i Processi Chimico Fisici, Consiglio Nazionale delle Ricerche, Via G. Moruzzi 1, 56124 Pisa, Italy. E-mail: carlopetrongolo41@gmail.com
First published on 18th June 2020
Abstract
We present the dynamics of the electronic quenching OH(A2Σ+) + Kr(1S) → OH(X2Π) + Kr(1S), with OH(A2Σ+) in the ground ro-vibrational state. This study relies on a new non-adiabatic quantum theory that uses three diabatic electronic states Σ+, Π′, and Π′′, coupled by one conical-intersection and nine Renner–Teller matrix elements, all of which are explicitly considered in the equation of the motion. The time-dependent mechanism and initial-state-resolved quenching probabilities, integral cross sections, thermal rate constants, and thermally-averaged cross sections are calculated via the real wavepacket method. The results point out a competition among three non-adiabatic pathways: Σ+ ↔ Π′, Σ+ ↔ Π′′, and Π′ ↔ Π′′. In particular, the conical-intersection effects Σ+–Π′ are more important than the Renner–Teller couplings Σ+–Π′, Σ+–Π′′, and Π′–Π′′. Therefore, Π′ is the preferred product channel. The quenching occurs via an indirect insertion mechanism, opening many collision complexes, and the probabilities thus present many oscillations. Some resonances are still observable in the cross sections, which are in good agreement with previous experimental and quasi-classical findings. We also discuss the validity of more approximate quantum methods.
1. Introduction
The role of the hydroxyl radical is paramount in atmospheric and combustion chemistry. It is also the product of many elementary reactions that in turn contribute to more complex processes involving the OH radical. Because its efficiency, its detection is usually carried out by laser induced fluorescence on the OH A2Σ+–X2Π electronic band,1,2 that can be used to measure the radical concentration very accurately.3 Hence, collisional electronic quenching of the A2Σ+ state is a process of considerable practical interest since it reduces radiative lifetimes and fluorescence quantum yield. It is thus not surprising that the quenching of OH(A2Σ+) has received a great deal of attention especially devoted to measure quenching cross sections with different collision partners4–7 and to investigate the mechanism associated with the transit through the conical intersection.8–12 Among other processes involving electronic quenching with OH(A2Σ+), collisions with rare gases (Rg) has received a special attention in the last few years as examples of processes in which collisional energy transfer and rotational depolarization may compete with electronic deactivation.13–23 In addition, Rg + OH(A2Σ+) collisions are amenable to rigorous electronic and dynamical calculations16–19,21–23 that can be compared with a considerable amount of experimental information, ranging from thermal rate coefficients7 and cross sections for selected spin-rotational initial states5,6,18,19,21,23 to rotational and lambda-doublet state resolved cross sections.21 Interestingly, whereas quenching cross sections for He, Ne, and Ar are almost negligible when compared with rotational energy transfer on the excited potential energy surface (PES), for Kr and Xe quenching cross sections are similar or larger than those with H2, O2, or N2.18,21,23 As such, adiabatic calculations carried out on the 2A′ excited PES for Kr and Xe cannot account for rotational initial state depopulation.21–23Previous calculations for the Kr + OH(A2Σ+) system using quasiclassical trajectories (QCT) and surface hopping (SH) on ab initio PESs21,22 demonstrated that the sole consideration of 2-PES transition (2A′–1A′) could not reproduced the magnitude of the quenching cross section dependence on the initial rotational state of OH(A2Σ+). It was necessary to include the participation of the 1A′′ and the roto-electronic couplings between 2A′ and 1A′′, and 1A′ and 1A′′, to recover the experimental values and the rotational state dependence. However, in that work the PESs and their couplings were calculated at a fixed O–H internuclear distance corresponding to its equilibrium value on the A2Σ+ electronic state. It remains to be seen how the approximate results on two-dimensional (2D) PESs compare with those obtained on 3D PESs. More importantly, the results reported in ref. 22 obtained using QCT-SH need to be validated with quantum dynamics calculations.
We here investigate the non-adiabatic quantum dynamics of the electronic quenching OH(A2Σ+) + Kr(1S) → OH(X2Π) + Kr(1S), extending experimental and quasi-classical previous works by Lehman et al.21 and by Perkins et al.22 Using reactant Jacobi coordinates r, R, and γ, where γ = 0 corresponds to the direction from O to H, we plot in Fig. 1R cuts of the three PESs required for this study for better understanding the present definitions some of which are different from those in ref. 22.
 | ||
| Fig. 1 R cuts of PESs for OH(A2Σ+) + Kr → OH(X2Π) + Kr. Adiabatic states 1A′, 2A′, and A′′. Diabatic states Σ+(1), Π′(2), and Π′′(3). Energy from this work, with respect to the reactants. Lower/higher energy in red/blue. The black dot labels the CI12 conical intersection. | ||
The doublet electronic states are labeled according to ref. 21 and 22, in both adiabatic and diabatic representations, and the non-adiabatic couplings between them are schematically shown.
Adiabatically, the reactants correlate with the excited 2A′ electronic species of OHKr and the products correlate with the nearly degenerate ground states 1A′ and 1A′′. 2A′ is considerably bound at γ = 180°, i.e. for a linear Kr–OH geometry, whereas 1A′ and A′′ are unbound and lie well below the reactant energy. At large R, the main configurations of 2A′ and 1A′ are Σ+ and Π′, respectively, and at small R these configurations exchange to Π′ and Σ+, respectively, owing to a conical intersection of the A′ states at R ∼ 3.8a0 and γ = 180°. Therefore, Fig. 1 shows that the electronic states can be also described in a diabatic representation as Σ+(1), Π′(2), and Π′′(3), which belong to the Cs irreducible representations A′, A′, and A′′, respectively. At linearity (z) these diabats are the eigenstates of the z component of the total and spinless electronic angular momentum ![[L with combining circumflex]](https://www.rsc.org/images/entities/b_i_char_004c_0302.gif) with eigenvalues 0 and ±1, respectively.
with eigenvalues 0 and ±1, respectively.
The electronic quenching is barrierless but closed in the Born–Oppenheimer approximation. Nevertheless, the electronic states interact via non-adiabatic effects that open the quenching channel: the linear conical intersection (CI12) between the A′ states [Σ+(1) and Π′(2)]21,22 and the -induced Renner–Teller24,25 couplings among all three species (i.e., RT12, RT13, and RT23).22 This is depicted in Fig. 1 which also shows that the 2A′ minimum and the CI12 point are very near, both in geometry and in energy, as we shall discuss in Section 3.
Extending the previous studies,21,22 the present work reports time-dependent wavepacket (WP) quantum dynamics of the electronic quenching OH(A2Σ+) + Kr(1S) → OH(X2Π) + Kr(1S), including three 3D PESs and all the non-adiabatic interactions (i.e., one CI and nine RT coupling surfaces) all of them at multi-reference configuration-interaction (MRCI) level. The paper is organized as follows. Section 2 presents theory and computational methods. Section 3 shows the results of the electronic calculations, the time-dependent quenching mechanism, initial-state-resolved reaction probabilities with OH(2Σ+) in the ground ro-vibrational state, some model probabilities, and initial-state-resolved cross sections, rate constants, and thermally-averaged cross sections. We finally report our main conclusions in Section 4 and other results in the ESI.†
2. Theory and computational methods
Unless otherwise specified, we employ atomic units (a.u.), Jacobi reactant coordinates, and a counter-clockwise body-fixed frame with the origin at the center of the nuclear masses, where OHKr lies in the z,x plane and the z axis is either along r (r-embedding) or R (R-embedding).We investigate the collision dynamics of the OH(A2Σ+) + Kr electronic quenching following our previous works22,26 on non-adiabatic effects in triatoms. The present theoretical treatment is somewhat similar to that of Zhou et al.,27 who considered three diabats coupled by both conical-intersection and Renner–Teller effects. Spin–orbit interactions are non negligible for this system, and accounting for them would permit to consider additional routes for the quenching, by coupling the 2A′(Σ+) to 1A′(Π′) and A′′(Π′′) states. In the vicinity of the conical intersection, spin–orbit couplings are of the order 200 cm−1 approximately, being slightly stronger between the two A′ states (230 versus 170 cm−1). Those values of ∼0.0009 a.u. are nevertheless three orders of magnitude smaller than other couplings here considered, as we shall see in Section 3, and including them is not expected to significantly change the results. Due to the huge additional cost required to include them in the treatment for a small expected change in the results, make us prefer to work within the non-relativistic approximation.
We therefore use a total spinless Ĥ containing all rovibronic couplings: (1) electronic, with diabatic electronic states coupled by the electronic Hamiltonian Ĥel; (2) vibrational-electronic, with adiabatic or diabatic states coupled by the vibrational Hamiltonian ![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) vib; and (3) roto-electronic, with adiabatic or diabatic species coupled by the total electronic angular momentum . In the last case, at least one electronic state is degenerate when the molecule is linear. We here employ strictly diabatic electronic states whose vibronic couplings are much smaller than other couplings, and are thus safely neglected. The electronic and roto-electronic couplings between these diabats are associated with conical-intersection (CI) and Renner–Teller (RT) effects, respectively. The former depend on just one operator, Ĥel, but the latters correspond to seven operators,
vib; and (3) roto-electronic, with adiabatic or diabatic species coupled by the total electronic angular momentum . In the last case, at least one electronic state is degenerate when the molecule is linear. We here employ strictly diabatic electronic states whose vibronic couplings are much smaller than other couplings, and are thus safely neglected. The electronic and roto-electronic couplings between these diabats are associated with conical-intersection (CI) and Renner–Teller (RT) effects, respectively. The former depend on just one operator, Ĥel, but the latters correspond to seven operators, ![[L with combining circumflex]](https://www.rsc.org/images/entities/i_char_004c_0302.gif) 2, 2z, xz, y∂/∂γ, y, z, and x, whose matrix elements are rarely computed on initio MRCI states.
2, 2z, xz, y∂/∂γ, y, z, and x, whose matrix elements are rarely computed on initio MRCI states.
2.1. Electronic structure calculations and computational details
Using the MOLPRO suite of ab initio codes,28 we have calculated twelve 3D adiabatic surfaces: three PESs 1A′, 2A′, and A′′, and nine r-embedding matrix elements of. Labeling r- or R-embedding with or without a bar, respectively, these are  ,
,  ,
,  ,
,  , 〈1A′|y|2A′〉,
, 〈1A′|y|2A′〉,  ,
,  , 〈1A′|2y|2A′〉, and
, 〈1A′|2y|2A′〉, and  , where the y and 2y terms are the same in both embeddings. Using again MOLPRO,28 we then obtain the 3D diabatic PESs Σ+, Π′, and Π′′, the 〈Σ+|Ĥel|Π′〉 diabatic coupling, and the surface of the adiabat-to-diabat transformation angle χ.
, where the y and 2y terms are the same in both embeddings. Using again MOLPRO,28 we then obtain the 3D diabatic PESs Σ+, Π′, and Π′′, the 〈Σ+|Ĥel|Π′〉 diabatic coupling, and the surface of the adiabat-to-diabat transformation angle χ.
As already pointed out, to treat the quenching of OH(Σ+ → Π), three electronic states have to be considered. The Π state is doubly degenerated, but this degeneracy is broken by the presence of the Kr atom when the three atoms are not aligned. This occurs because OH presents an unpaired electron, which can be located in a p orbital in the plane of the triatomic system (A′) or one perpendicular to the plane (A′′). Asymptotically, when Kr is too far to interact with OH, the two orthogonal p orbitals are equivalent and both states are degenerated. For symmetry reasons, when the three atoms are in a collinear configuration, the orbitals are also equivalent and both A′ and A′′ components are degenerated.
Previous calculations of the ground and first excited state PESs21,22 were performed at a fixed OH internuclear distance at its equilibrium value of the excited A2Σ+ state. Considering that this value is similar to that of the minimum of the ground X2Π state, and that collisional quenching was studied at thermal temperatures (0.025–0.039 eV), this was deemed as a reliable approximation. However, the effect of the OH vibration must be included for a more accurate and reliable description of the processes, especially if calculations are performed at higher collision energies. Therefore, to obtain the 3D analytical PESs and couplings, ab initio calculations were performed over a regular grid in Jacobi coordinates (R,r,γ) referred to the Kr + OH channel with γ = 180° corresponding to the HO–Kr linear configuration. The considered grid elements are R = [3.4, 3.6, 3.8, 4.0, 4.2, 4.4, 4.6, 4.8, 5.0, 5.2, 5.4, 5.6, 5.8, 6.0, 6.4, 6.8, 7.2, 7.6, 8.0, 8.5, 9.0, 9.5, 10, 15]a0, r = [1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.4]a0, and γ = [0, 15, 30, 45, 60, 75, 90, 105, 120, 135, 150, 165, 180] degrees for a total of 2808 points. Electronic structure calculations at each point of the grid were performed with a full electron Douglas–Kroll aug-cc-pVTZ basis set29 and using the Douglas–Kroll Hamiltonian to account for relativistic effects of Kr inner electrons. In a first step, the two A′ states and the A′′ state associated to the Σ+ and two components of the Π state of OH are calculated in a state-average CASSCF calculation30 using an active space of 5 electrons in 4 orbitals (16–18a′,6a′′). This active space ensures a good accuracy with an affordable computation time and, most importantly, keeps the continuity in the electronic wavefunctions in all the configuration space and for the three electronic states considered. The generated configurations and orbitals are then employed for an internally contracted MRCI treatment31 including Davidson correction.32 This scheme was employed within the quasi-diabatization procedure available in the MOLPRO program28 which allowed us to obtain directly the diabatic PESs of the states Σ+ and Π′, as well as the 〈Σ+|Ĥel|Π′〉 non-adiabatic coupling, and the surface of the mixing angle χ [see below, eqn (2.1)]. By construction, the diabatic Π′′ state corresponds to the adiabatic A′′ state. Finally, the nine r-embedding matrix elements of were computed at CASSCF level. Here we should remind that these elements are calculated in reference to the adiabatic states. When a change of character of the electronic wavefunction happens along a coordinate of the PES (after an avoided crossing for example), some of the r-embedding matrix elements are swapped. When this happens, they need to be rearranged properly in order to fulfill the conditions of continuity and derivability in the configuration space, if this requirement is not met, it would be impossible to obtain a reliable three dimensional fit.
The three diabatic PESs, the 〈Σ+|Ĥel|Π′〉 coupling surface, and the nine r-embedding surfaces in the adiabatic representation were then fitted independently using the Reproducing Kernel Hilbert Space (RKHS) method.33 This method is known to be well suited for fitting 3D PESs, and it is even better adapted to fit couplings surfaces. The advantage of this method stems from the use of its angle-like kernels which are convenient to fit angular variables such as Jacobi angle from which couplings between PESs are highly dependent. Angle-like kernels are also well adapted to obtain a good extrapolation for distance variables when surfaces converge to a fixed value in the asymptotic region, which is also the case of couplings (i.e. for Kr far away, one should recover the couplings of isolated OH).
The fitting procedure is extensively described in ref. 34 and 35. Here, only the most relevant details will be described. To fit the various PESs, the energy was decomposed in 1-, 2- and 3-body terms. 1- and 2-body terms were fitted implicitly together referring the zero of energy to the asymptotic value of the dissociated diatom. To account properly for the asymptotic limits of the three electronic states considered, five diatoms were considered: OH(X2Π), OH(A2Σ+), and three van der Waals diatomic clusters Kr–H, Kr–O(3P) and Kr–O(1D). Once the energy for each diatom is subtracted from the corresponding electronic states, the 3-body term, which also vanishes in asymptotic regions, are fitted independently. The global PESs over the whole configuration space are then obtained as the sum of the corresponding fitted 2- and 3-body terms. We should point out here that since all the fitted diatomics go to zero asymptotically; the ∼2 eV separation between O(3P) and O(1D) is added when the total PESs are reconstructed, a necessary step to keep the good relative energy differences between the three states. For the couplings, most of the terms were fitted directly (without decomposition) since most of them vanish in the asymptotic regions. Only the couplings elements depending on the OH bond length and which do not vanish asymptotically were decomposed in order to apply efficiently the 3D RKHS fitting. In all the cases, angle-like kernels were used for the 3D fitting. Three new angular variables (i.e. limited in the [0,1] interval) were thus defined from the Jacobi coordinates as follow: x = exp(−br), y = exp(−aR) and z = (1 + cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) γ)/2.
γ)/2.
2.2. Hamiltonian theory
 | (2.1) |
 | (2.2) |
 ,26 where σ is their parity and Λ their z eigenvalue at linearity. Note that they do not correspond to the complex species labelled in the same way in ref. 22.
,26 where σ is their parity and Λ their z eigenvalue at linearity. Note that they do not correspond to the complex species labelled in the same way in ref. 22.
The total Ĥ, the CI electronic operator Ĥel, the RT operators 2 and y, the vibrational operator vib, and the Jacobi coordinates do not depend on the embedding used but many operators of Ĥ become different in r- or R-embedding. Thus, for practical purposes we have labeled the operators with or without a bar depending on they are expressed in r- or R-embedding, respectively (the invariant operators are also unbarred). Like Ĥel, we define a RT roto-electronic operator  as the sum of all rotational terms of Ĥ that contain
as the sum of all rotational terms of Ĥ that contain  . Writing the full spinless rotational operator26 as
. Writing the full spinless rotational operator26 as  , where
, where  contains terms that depend only on the total angular momentum Ĵ, the r-embedding full RT operator is equal to
contains terms that depend only on the total angular momentum Ĵ, the r-embedding full RT operator is equal to
 | (2.3) |
 rotations and between both rotations and the angular coordinate γ. The most important operators are
rotations and between both rotations and the angular coordinate γ. The most important operators are  and
and 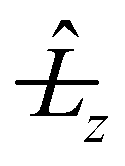 because they are multiplied by 1/sin2γ that diverges quadratically at the linearity. Nevertheless this degeneracy is analytically removed at linearity26 where Ĵz = z. The
because they are multiplied by 1/sin2γ that diverges quadratically at the linearity. Nevertheless this degeneracy is analytically removed at linearity26 where Ĵz = z. The  terms of
terms of  are called Coriolis operators.
are called Coriolis operators.
Real electronic species |e〉 fulfill
 | (2.4) |
 ,26 where
,26 where  is the r-embedding helicity, p is the total parity and σ1/2 = + or i for σ = + or −, respectively, the matrix elements of
is the r-embedding helicity, p is the total parity and σ1/2 = + or i for σ = + or −, respectively, the matrix elements of  are presented in eqn (2.5):
are presented in eqn (2.5): | (2.5) |
 . These matrix elements are real as the states, those of x, y, and z are purely imaginary, and the others are real. This equation defines both |σΛ〉-diagonal potential terms and off-diagonal coupling terms; it is also obtained by summing eqn (4.6) and (4.7) of ref. 26, changing appropriately the notation and omitting the unnecessary terms. It is also similar to eqn (44)–(46) of ref. 22, taking into account that the present diabatic states are real and that we call here “RT couplings” also Aij and Cij of eqn (44) and (46),22 following ref. 24 and 25.
need to be considered since they rule the quenching dynamics.37Eqn (2.5) then simplifies and specializes for the off-diagonal elements as
. These matrix elements are real as the states, those of x, y, and z are purely imaginary, and the others are real. This equation defines both |σΛ〉-diagonal potential terms and off-diagonal coupling terms; it is also obtained by summing eqn (4.6) and (4.7) of ref. 26, changing appropriately the notation and omitting the unnecessary terms. It is also similar to eqn (44)–(46) of ref. 22, taking into account that the present diabatic states are real and that we call here “RT couplings” also Aij and Cij of eqn (44) and (46),22 following ref. 24 and 25.
need to be considered since they rule the quenching dynamics.37Eqn (2.5) then simplifies and specializes for the off-diagonal elements as | (2.6) |
 | (2.7) |
J, the vibrational operator vib that does not work on strictly diabatic electronic states, and the normalized associated Legendre states |jK〉 with38 | (2.8) |
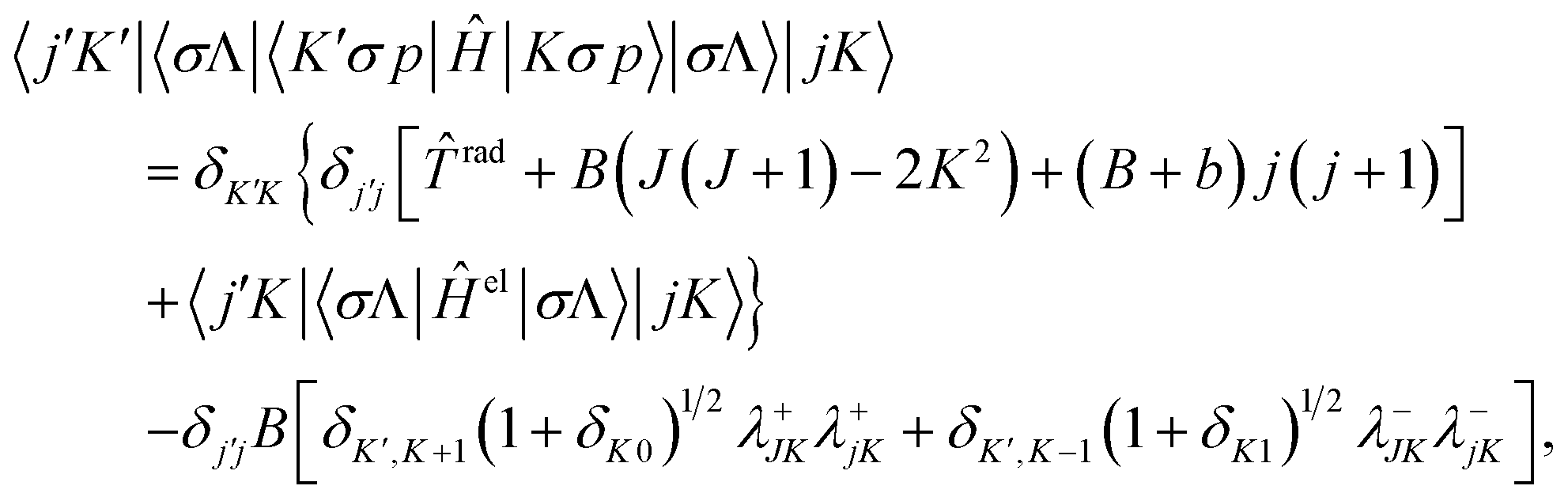 | (2.9) |
rad = −B∂2/∂R2 − b∂2/∂r2, and 〈σΛ|Ĥel|σΛ〉 are the diabatic potential energy surfaces.
The non-adiabatic CI and RT interactions between the diabats Σ+, Π′, and Π′′ are
 | (2.10) |
 | (2.11) |
 | (2.12) |
 | (2.13) |
 | (2.14) |
operators on the roto-electronic basis, the non-adiabatic couplings in Fig. 1 are equal to| CI12 = 〈Σ+|Ĥel|Π′〉, | (2.15) |
 | (2.16) |
 | (2.17) |
 | (2.18) |
| 〈j′K0| 〈Σ+|〈K0 + p|Ĥ|K0 + p〉|Π′〉|jK0〉 = 〈j′K0|CI12 + RT12|jK0〉, | (2.19) |
| 〈j′K0|〈Σ+|〈K0 + p|Ĥ|K0 − p〉|iΠ′′〉 |jK0〉 = 〈j′K0|RT13|jK0〉, | (2.20) |
| 〈j′K0|〈Π′|〈K0 + p|Ĥ|K0 − p〉|iΠ′′〉|jK0〉 = 〈j′K0|RT23|jK0〉, | (2.21) |
x, y, and z are purely imaginary and the others are real. In block-matrix form we have | (2.22) |
The coefficients of the components increase with J and depend on K0, those of 2z, xz, z, and x diverge at C∞v geometries, mainly for 2z and z, and their relative strength depends on the system under study and can be assessed only via numerical calculations. The centrifugal-sudden approximation is obtained with  . If J = 0, the RT12y term of eqn (2.16) vanishes and the Π′′ product channel in eqn (2.17) and (2.18) is closed. If K0 = 0, also the first lines of eqn (2.17) and (2.18) vanish. On the other hand, all RT couplings are present if K0 > 0.
. If J = 0, the RT12y term of eqn (2.16) vanishes and the Π′′ product channel in eqn (2.17) and (2.18) is closed. If K0 = 0, also the first lines of eqn (2.17) and (2.18) vanish. On the other hand, all RT couplings are present if K0 > 0.
We represent this roto-electronic angular Hamiltonian on a (R,r) 2D radial grid, where the rad matrix elements are obtained via a Fourier transform from the coordinate to the momentum representation, calculating the matrix elements in the latter space, and returning back to the former representation via an inverse Fourier transform. In conclusion, the full representation includes three diabats, two radial coordinates, Legendre states, and overall-rotational species, and it is used for computing the quenching dynamics as we briefly report in the next Section.
In closing this Section we summarize all our approximations.
(1) Spinless Hamiltonian: the spin is considered only in the calculation of the electronic states.
(2) OH(X2Π): the electronic angular momentum is omitted.
(3) Strictly diabatic electronic representation: ∂|e〉/∂Q ≡ 0, where |e〉 is an electronic state and Q is a Jacobi coordinate.
(4) Quenching approximation: only couplings 〈e′|Ô|e〉, e′ ≠ e.
(5) Diagonal approximation: 〈1A′| − − |1A′〉 = 〈2A′| − − |2A′〉 in the calculation of 〈Σ+| − − |Π′〉.
(6) K approximation: K′ = K ± 1 ≈ K = K0.
2.3. Wavepacket collision dynamics
Initial-state-resolved reaction probabilities are obtained through the time-dependent real wavepacket (WP) formalism of Gray and Balint-Kurti,39 essentially equal to the Chebyshev approach of Guo.40 To this end we scale and shift the Hamiltonian of Section 2.2.4, obtaining Ĥs, whose arccos mapping of the equation of the motion is solved recursively. We employ a complex initial WP| |ψ0〉 = |a0〉 + i|b0〉 = |Σ+〉|s0(R)〉|v0j0(r)〉|j0K0〉, | (2.23) |
 | (2.24) |
| |a1〉 = Ĥs|a0〉 − (1 − Ĥs2)1/2|b0〉, first complex propagation, | (2.25) |
| |an+2〉 = 2Ĥs|an+1〉 − |an〉, other real Chebyshev propagations. | (2.26) |
We have converged quenching probabilities  and
and 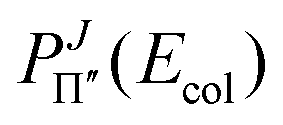 of OH(2Σ+), from the ground vibrational state and the ground rotational state j0 = K0 = 0 to the OH(2Π) final diabatic states Π′ and Π′′, propagating WPs at all J ≤ 140 with the numerical parameters of Table 1, that correspond to 7227394 basis states, and using 330 values of the collision energy Ecol, from 0.001 to 0.33 eV.
of OH(2Σ+), from the ground vibrational state and the ground rotational state j0 = K0 = 0 to the OH(2Π) final diabatic states Π′ and Π′′, propagating WPs at all J ≤ 140 with the numerical parameters of Table 1, that correspond to 7227394 basis states, and using 330 values of the collision energy Ecol, from 0.001 to 0.33 eV.
| Initial sinc s0(R): α, E0, R0, β | 17, 0.039 eV, 11, 0.01 |
| R range and number of grid points | 2.95–16 and 307 |
| r range and number of grid points | 1.45–7 and 79 |
| Number of Legendre states | 100 |
| R and r absorption start at | 12 and 5 |
| R and r absorption strengths | 0.05 and 0.01 |
| Asymptotic analysis at R∞ | 11 |
Finally, initial-state-resolved cross sections σ(Ecol) and thermal rate constants at the temperature T, k(T), are calculated via the usual expressions. Following ref. 21 and 42, we also obtain thermally averaged cross sections from the mean relative velocity, 〈v(T)〉 = (8kBT/πμR)1/2, as 〈σ(T)〉 = k(T)/〈v(T)〉. For analyzing some probabilities we also use thermally averaged partial cross sections 〈σJ(300)〉 resolved on the quantum numbers J and j0 = K0 = 0, at 300 K.
3. R-Embedding results and discussion
3.1. Electronic calculations
The present diabatic PESs and the respective couplings can be considered as 3D extensions of the previous 2D PESs and couplings presented in ref. 21, in which the OH bond distance r was fixed at its equilibrium value on the A2Σ+ state. The effect of varying r is best seen near the 2A′–1A′ CI seam at linearity, where the adiabats belong exactly to the Σ+ and Π′ irreps, respectively, and their Ĥel coupling vanishes by symmetry. As already pointed out in ref. 21 and 22, and also below Fig. 3, that seam plays a central role in the quenching and it is presented in Fig. 2 at γ = 180° (Kr–OH configuration). We clearly see that the initial Σ+ PES becomes fully repulsive at r > 2.5a0 and that the CI seam lies very near the PES minimum and then moves at larger R and r values and higher energies. We report further details and the implications of this result by discussing Fig. 3. | ||
| Fig. 2 γ = 180°. Σ+ PES (eV with respect to the reactants). The Σ+–Π′ CI seam is labeled by a green line. | ||
 | ||
| Fig. 3
r = 1.916a0. Σ+ PES (eV with respect to the reactants), conical-intersection coupling CI12 = 〈Σ+|Ĥel|Π′〉, Renner–Teller matrix element 〈Σ+|2|Π′〉. J = 70 and K0 = 0, total coupling CI12 + RT12 of eqn (2.15) + (2.16). Negative/zero/positive levels in red/black/blue. The Σ+–Π′ diabatic crossing is labeled by a green line. | ||
Nevertheless the main topographic characteristics of the three PESs are essentially the same and will be described mainly at the calculated equilibrium bond length of the OH asymptote in the Σ+ PES: r = 1.916a0. The top-left panel of Fig. 3 shows that the Σ+ PES (2A′ in Cs symmetry), which correlates with OH(A2Σ+), exhibits two pronounced van der Waals (vdW) wells in linear configurations. The global minimum corresponds to a collinear Kr–OH vdW complex at RvdW = 3.847a0, rvdW = 1.900a0, γvdW = 180°, and VvdW = −0.990 eV. This minimum is very near to a conical-intersection point with the Π′ species, at RCI = 3.827a0, rCI = 1.916a0, γCI = 180°, and VCI = −0.984 eV. It is the presence of this deep Kr–OH minimum that allows collisions at thermal energies to access the conical intersection, boosting the quenching cross sections to values comparable to those of collisions with H2 and somewhat higher than those with N2. This is in contrast to other Rg + OH(A) systems with lighter rare gases (He, Ne, Ar), whose Rg–OH(A) minima are much shallower and their conical intersection lies well above the respective asymptotes at a much shorter R distance, above the strong repulsive wall of the Rg–OH(X) PES. As a consequence, their respective quenching cross sections are two to three orders of magnitude smaller than those for Kr + OH(A). As can be expected, however, for Xe + OH(A) the situation is analogous to the Kr + OH(A), but the quenching cross section are almost a factor of two bigger for the former.5,23
The origin of the well between OH Σ+ state and the noble gas can be traced back to the electronic structure of the system. OH(Σ+) is characterized by the excitation of one electron from the σ bonding to the π orbital. In the same time, σ presents a favourable overlap with the collinear p orbital of Kr. This deficit of electron in the σ bonding orbital allows it to drag the electronic density of the p orbital providing a partial charge transfer of about −0.4 a.u., according to a Mulliken population analysis of the MRCI wavefunction, and therefore stabilizing the system. In the OH(Π) ground state, the σ bonding orbital is filled and cannot drag electronic density: in this case, Krypton remains neutral and the interaction with OH is purely repulsive. The OH Σ+ state can thus be considered as partial Lewis acid when it interacts with Krypton.
The secondary minimum, at a linear OH–Kr configuration with γ = 0, occurs at R ∼ 5.3a0, is shallower (V ∼ −0.28 eV), and is therefore less important for the quenching dynamics. In the adiabatic representation, both minima are the result of avoided crossings of the Σ+ and the repulsive wall of the Π′ ground state, which can cross in C∞v linear configuration where the A′ species take exactly their Σ+ and Π′ character and their electronic coupling vanishes. However, when the system departs from linearity, the A′ component of the Π state couples with the Σ+ state and crossing becomes avoided.
We also see that the Σ+ PES is strongly anisotropic: the linear configurations are indeed preferred whereas the Kr perpendicular approach to OH is inhibited. In contrast, the Π′ PES that correlates with OH(X2Π), shown in Fig. S1 (ESI†), is unbound, fairly isotropic, symmetric with respect to γ = 90°, and very similar to the Π′′ one that it is not shown. Our 3D results confirm essentially the previous 2D calculations21,22 at constant r, except that the present global minimum is deeper and takes place at somewhat smaller value of R than that of ref. 21, where RvdW = 4.16a0, r = 1.913a0, and VvdW = −0.754 eV.
Due to the topographic singularities at the conical intersections, the adiabatic representation is not ideally suited for dynamical calculations and the fit is much more problematic. The diabatic representation, where the Σ+ and Π′ character are preserved in the whole PES, becomes a much better option. In this representation, Σ+ and Π′ states can cross not only in linear conformation, but everywhere. However, the crossings only occur in the regions of the Σ+ wells. When Kr approaches perpendicularly to OH, the interaction is repulsive for both Σ+ and Π′ and they never cross. Since the couplings become effective only when the energy difference between the coupled states is small, the regions of the two wells will be more relevant for the efficiency of the quenching. In addition of the PESs, it is therefore interesting to discuss the evolution of the couplings depending on the geometry of the system, and in particular the couplings. We thus plot in Fig. 3 also some relevant couplings, at r = 1.916a0, and at J = 70 and K0 = 0 when they depend on these quantum numbers.
The CI12 term 〈Σ+|Ĥel|Π′〉 on the top-right panel of Fig. 3 vanishes by symmetry in C∞v and is nearly antisymmetric with respect to γ = 90°, with a maximum value equal to 0.14 a.u. at R = 3a0 and γ = 117° at the r value here considered. Nevertheless, this configuration is at a repulsive Σ+ energy, not important for the low-energy quenching that is favored by the Σ+ attractive region and by rather large coupling values near R ∼ 3.8a0 and γ ∼ 160°.
The eqn (2.14)–(2.21) use the R-embedding and the matrix elements of between diabatic states, which are calculated from the MOLPRO r-embedding and adiabatic species in two steps, through the properties of .26 (1) The R-embedding components are obtained from the r-ones by rotating counter-clockwise the body-fixed axes around y by γ. (2) The diabatic matrix elements are then calculated viaeqn (2.1) and (2.2), and the quenching approximation 〈1A′| − − |1A′〉 = 〈2A′| − − |2A′〉. For example
 | (3.1) |
| 〈Σ+|2z|Π′〉 = 〈1A′|2z|2A′〉(cos2χ − sin2χ). | (3.2) |
x(R) couplings in both embeddings and representations, and two  couplings in the r-embed and adiabatic representation, at r = 1.916a0 and γ = 175°. This figure also shows that our r-embed, adiab matrix elements
couplings in the r-embed and adiabatic representation, at r = 1.916a0 and γ = 175°. This figure also shows that our r-embed, adiab matrix elements  ,
,  , and
, and  are nearly equal to those of Fig. 2 of ref. 22, provided a A′′ change of sign, but that RAC is larger by ∼0.32a0 due to the present variation of r and to the sensitivity of the CI seam to the calculation accuracy.
are nearly equal to those of Fig. 2 of ref. 22, provided a A′′ change of sign, but that RAC is larger by ∼0.32a0 due to the present variation of r and to the sensitivity of the CI seam to the calculation accuracy.
Among the eight matrix elements of in eqn (2.16)–(2.18), the largest one is 〈Σ+|2|Π′〉 plotted in the bottom-left panel of Fig. 3. It is approximately antisymmetric with respect to γ = 90°, as the electronic coupling due to Ĥel, and varies sharply both in value and in sign near the linear conical intersections where it attains large absolute values. This result is due to sudden changes of χ in these regions where both numerator and denominator of eqn (2.2) almost vanish, as the above-left panel of Fig. S2 (ESI†) shows. Fig. S3 (ESI†) then reports the 2z, xz, and y matrix elements. The decidedly smaller 2z values with respect to 2 point out that the latter term is essentially due to 2x + 2y, and xz and y are nearly constant at R > 4a0.
According to eqn (2.16)–(2.18), all Renner–Teller couplings do not depend solely on the matrix elements but also on their coefficients, all of which are functions of the Jacobi coordinates and those of the components depend also on the overall-rotation quantum numbers J and K0. For example, the B coefficient of 2 in eqn (2.16) is 1/(2μRR2) ≤ 10−6 a.u. in the considered R range, and the coefficients of 2z, xz, z, and x are large near linear geometries if K0 > 0, especially that of z. We thus plot in the bottom-right panel of Fig. 3 the overall non-adiabatic Σ+(1)–Π′(2) coupling CI12 + RT12, eqn (2.15) + (2.16), at J = 70 and K0 = 0, showing that it is very similar to the Ĥel coupling. This fact implies that the Σ+–Π′ interaction is essentially due to the conical intersection, as we shall see also in Section 3.3.2.
After the non-adiabatic CI and RT matrix elements of operators belonging to the A′ irreducible representation of the Cs point group, we close this Session by presenting in Fig. 4 the total couplings Σ+(1)–Π′′(3) RT13, (2.17), and Π′(2)–Π′′(3) RT23, (2.18), due to z and x ∼ A′′.
 | ||
| Fig. 4 r = 1.916a0, J = 70 and K0 = 0. RT13, eqn (2.17), and RT23, eqn (2.18). Details as in Fig. 3. | ||
Even at J = 70 and K0 = 0, where λ070,0 is large and equal to 70.5 a.u., these Renner–Teller couplings RT13 and RT23 are smaller than the conical-intersection term CI12, confirming that the latter effects are more important than the former when OH(A2Σ+) is in the ground ro-vibrational state. In particular, both RT13 and RT23 are different from zero at large R values and RT23 is rather important at linearity, because z couples the nearly degenerate Π′ and Π′′ components of the OH(2Π) product via the cotγ factor of eqn (2.18). This is different from CI12 that is larger far from the linearity where it vanishes by symmetry. Since RT13 < RT23, these results show that the relative strength of the non-adiabatic couplings is CI12 ≫ RT23 > RT13 > RT12. Finally, other matrix operators are plotted in Fig. S4 (ESI†) and it is easy to check that all fulfill the C∞v selection rules at γ = 0 and 180°.
3.2. Time-dependent reaction mechanism
The time-dependent reaction mechanism on the three coupled diabats |e〉 is shown in Fig. 5 that presents normalized electronic norms and snapshots of the j-summed (R,r) density | (3.3) |
 | ||
| Fig. 5 J = 70 and j0 = K0 = 0. WP norms and γ-averaged (R,r) densities at 5 times. | ||
The initial Σ+ WP remains essentially on this PES up to ∼180 fs, moving towards the interaction region without being affected by the non-adiabatic couplings with the Π states. Contrasting this finding with other barrierless quenchings with smaller or equal exoergicity, as OH(X) + H+37 and OH(A) + H,44 we see that this collision is much slower than the others owing to the larger mass of the Kr atom. During this early stage of the reaction, only R decreases while both r and γ do not vary appreciably.
The attack of Kr to the O atom and the quenching process begins at ∼180 fs, when part of the WP enters into the strong interaction region at R < 5a0, where the WP jumps on the Π′ PES at R ∼ 3.8a0 and γ ∼ 160° owing to the Σ+–Π′ conical intersection. At 181 fs the hops from Σ+ to Π′ occur mainly at Rhop ∼ 3.5–4 and 4.5–5a0, where the WP forms KrOH metastable complexes near the CI seam. Nearly 20 fs later, that is at ∼200 fs, also the Π′′ surface begins to be populated at Rhop ∼ 3.5–4a0 and the collision complex reveals now its full non-adiabatic nature, spreading on all the three PESs.
At larger times we see both the non reactive WP on Σ+, due to recrossings of the CI seam, and the reactive densities on Π′ and Π′′, together with the formation of the quenching products resolved on these final states. Nevertheless, the major part of the WP is not reactive and is reflected back into the reactant channel.
By transforming the WP from the Legendre representation j to the angular one γ,45 Fig. S5 (ESI†) shows a complementary picture of the mechanism plotting r-summed (R,γ) densities. Because we are considering a rotationless OH(2Σ+) diatom with j0 = 0, the initial r-averaged density depends only on R, as the top panel of Fig. S5 (ESI†) shows. During the OH(A) + Kr collision many Legendre polynomials are occupied and the WP thus spreads on all the γ values, mainly at 180° near the Σ+ minimum and the most important CI point. Therefore, the Π metastable complexes begin at ∼180 fs with a Kr–OH linear geometry.
In conclusion, these results reflect nicely the non-adiabatic couplings and extend to three coupled electronic states and to (R,γ) densities the findings found with just two of them,37,44 showing the different role played by two final electronic states here investigated in two different conformational subspaces.
3.3. Initial-state-resolved quenching probabilities
 ,
,  , and
, and  , resolved and summed on the final Π states, at J = 0, 30, 70, 100, and 140 and at j0 = K0 = 0. Contrarily to the OH + H+ quenching,37 where only the 〈Π′|z|Π′′〉 matrix element was considered but it is closed at J = 0, the OH(A) + Kr quenching is open at any J value owing to other couplings. As Fig. 3 shows, this finding is mainly due to the 〈Σ+|Ĥel|Π′〉 conical intersection coupling, and the Π′′ channel is closed at J = 0 according to eqn (2.17) and (2.18).
, resolved and summed on the final Π states, at J = 0, 30, 70, 100, and 140 and at j0 = K0 = 0. Contrarily to the OH + H+ quenching,37 where only the 〈Π′|z|Π′′〉 matrix element was considered but it is closed at J = 0, the OH(A) + Kr quenching is open at any J value owing to other couplings. As Fig. 3 shows, this finding is mainly due to the 〈Σ+|Ĥel|Π′〉 conical intersection coupling, and the Π′′ channel is closed at J = 0 according to eqn (2.17) and (2.18).
 | ||
| Fig. 6 J = 0, 30, 70, 100, 140, and j0 = K0 = 0. Quenching probabilities, resolved and summed on the final Π diabats. | ||
The most striking feature of the probabilities is their resonance structure in all the energy range and at all J values, decreasing however as J increases, similar to that observed in other electronic quenchings.37 Many causes can contribute to this finding: (1) The strong exoergic character of the quenching, associated with an indirect Kr insertion, with the formation of many long-lived intermediate complexes on the Π surfaces as Fig. 5 and Fig. S5 (ESI†) show, and with an electronic-to-translational energy transfer. (2) The formation of OH(2Π) rotational states.21,22 (3) The presence of the van der Waals minimum on the Σ+ PES. (4) The K approximation used which enhances the oscillatory structure by omitting all K ≠ K0 terms that in general average the probability oscillations.
Because the quenching is barrierless, low-J probabilities do not present any threshold, which appears only around J = 30 and increases very slowly with the overall rotation. This J shifting is simply due to the centrifugal barrier BJ(J + 1) of the Born–Oppenheimer matrix elements, where B is small owing to the Kr large mass and to R ≥ 2.95a0, as we already found in comparing CI12 and CI12 + RT12 in Fig. 3. We also see that the Π′ electronic component is preferred with respect to Π′′, on account of the important Ĥel electronic coupling CI12, and that the Π′ and Π′′ populations decrease or increase with J, respectively. In fact, the former channel in essentially due to conical-intersection effects and the latter depends only on Renner–Teller couplings that increase with J.
 at J = 70 and j0 = K0 = 0. In fact, calculations of cross sections at j0 > 0 is beyond our computer facilities.
at J = 70 and j0 = K0 = 0. In fact, calculations of cross sections at j0 > 0 is beyond our computer facilities.
We plot in Fig. 7 the results of four models: (1) centrifugal sudden (CS) with λ070,0 = 0 in eqn (2.14)–(2.18); (2) x = y = 0 in eqn (2.16)–(2.18); (3) conical-intersection only (CI) with = 0; (4) Renner–Teller only (RT) with χ = 0 in eqn (2.1). The sharp and strong resonances make it difficult in some cases to compare quantitatively different probabilities. To this aim, we use the thermally averaged partial cross sections  of Table 2, resolved on the quantum numbers J = 70 and j0 = K0 = 0, and at T = 300 K.
of Table 2, resolved on the quantum numbers J = 70 and j0 = K0 = 0, and at T = 300 K.
 | ||
| Fig. 7 J = 70 and j0 = K0 = 0. Full and model total probabilities. | ||
| Calculation |
|
|---|---|
| CS | 0.11 |
|
x
= y = 0 |
0.14 |
| CI | 0.13 |
| RT | 0.01 |
| Full | 0.14 |

The left panel presents two widely used approximations. The CS one does not work well at this high J value, with the Π′′ channel closed at K0 = 0, according to eqn (2.17) and (2.18), and with larger resonances shifted to higher energies. Nevertheless, the Maxwell averaging on Ecol reduces strongly these differences with a CS  lower by ∼20%. On the other hand, the model probability obtained with x = y = 0 is practically equal to the full results and indistinguishable on the scale of Fig. 7. The right panel reports model probabilities when some non-adiabatic couplings are switched off. We see that both
lower by ∼20%. On the other hand, the model probability obtained with x = y = 0 is practically equal to the full results and indistinguishable on the scale of Fig. 7. The right panel reports model probabilities when some non-adiabatic couplings are switched off. We see that both  and
and  are large by considering only CI effects, and the opposite is true if only RT effects are taken into account. These two effects are not addictive and CI is by far the most important coupling when j0 = K0 = 0, because the geometries where Σ+ is attractive and the term 〈Σ+|Ĥel|Π′〉 is large are near, as we have seen in Fig. 3. The CI model slightly overestimates some resonances with respect to the full calculations. This finding implies that CI and RT effects can be in mutual competition because they are large at different geometries as we have seen in Section 3, and RT couplings can induce a few recrossings from Π′ and Π′′ to Σ+, which are small when K0 = 0 according to eqn (2.16) and (2.17). These findings are essentially confirmed by other model calculations at different values of J, j0, and K0.
are large by considering only CI effects, and the opposite is true if only RT effects are taken into account. These two effects are not addictive and CI is by far the most important coupling when j0 = K0 = 0, because the geometries where Σ+ is attractive and the term 〈Σ+|Ĥel|Π′〉 is large are near, as we have seen in Fig. 3. The CI model slightly overestimates some resonances with respect to the full calculations. This finding implies that CI and RT effects can be in mutual competition because they are large at different geometries as we have seen in Section 3, and RT couplings can induce a few recrossings from Π′ and Π′′ to Σ+, which are small when K0 = 0 according to eqn (2.16) and (2.17). These findings are essentially confirmed by other model calculations at different values of J, j0, and K0.
Therefore, the general trend of model probabilities, with respect to full calculations, is that the CS approximation cannot be used at large J, the x = y = 0 approximation works very well at low K0 values, and CI effects are larger than those RT which are enhanced when OH(A) is in an excited rotational state, as it is usually found.37 On the overall, the effects of model calculations are larger in quantum than in quasi-classical calculations22 where these effects are considered indirectly, via hopping probabilities from the couplings, and they are summed over J by the cross sections results.
3.4. Initial-state-resolved quenching cross sections and rate constants
Fig. 8 shows quenching cross sections σ at j0 = 0, from 0.001 to 0.33 eV, and Table 3 reports some representative numerical values. | ||
| Fig. 8 j 0 = 0. Quenching cross sections, resolved (black) and summed (red) on the final Π diabats. | ||
The Π′ channel is strongly preferred at low Ecol, in agreement with the reaction probabilities and with the bottom-left panel of Fig. 6 of ref. 22, and the collision energy enhances the Π′′ weight with a branching ratio σΠ′′/σΠ′ increasing from ∼0.1 at the threshold to ∼0.6 at 0.33 eV. We also see that at least nine resonances survive the partial wave sum of the probabilities, mainly for the quenching on the Π′ surface. The Π′ + Π′′ and Π′ cross sections increase sharply from the threshold up to maximum values equal to 14.8 Å2 at 0.045 eV and to 11.6 Å2 at 0.044 eV, respectively, that is at the third resonance. Their average values then decrease at larger energies because the quenching is barrierless and exoergic. This trend is less evident for σΠ′′ that presents smother variations with a maximum value equal to 4.2 Å2 at 0.083 eV, near the forth resonance. At 0.039 eV the total quantum cross section σΠ′+Π′′ is equal to 14.1 Å2, whereas quasi-classical22 value is 17.2 ± 0.4 Å2. We believe that this difference is mainly due to many and sharp oscillations of the quantum probabilities which are averaged by quasi-classical calculations.
We also report in Fig. 9 and Table 4 initial-state-resolved non-adiabatic thermal rate constants and thermally averaged cross sections at j0 = 0. Total and Π′-resolved k and 〈σ〉 increase sharply at low temperature and reach their asymptotic values at 900 K: 12.4 × 10−11 and 8.8 × 10−11 cm3 s−1 for k, and 10.7 and 7.6 Å2 for 〈σ〉. The rate constants increase monotonically whereas 〈σΠ′+Π′′〉 has a maximum at 450 K, equal to 11.9 Å2. At 300 K, 〈σΠ′+Π′′(300)〉 = 11.6 Å2 in good agreement with the experimental value21 of 10 ± 1 Å2.
 | ||
| Fig. 9 j 0 = 0. Thermal rate constants (left) and thermally averaged cross sections (right), resolved (black) and summed (red) on the final Π diabats. | ||
| T/K | k Π′+Π′′/10−11 cm3 s−1 | 〈σΠ′+Π′′〉/Å2 |
|---|---|---|
| 100 | 3.1 | 8.0 |
| 200 | 5.8 | 10.7 |
| 300 | 7.8 | 11.6 (10 ± 1)21 |
| 400 | 9.2 | 11.8 |
| 500 | 10.3 | 11.8 |
| 700 | 11.7 | 11.4 |
| 900 | 12.4 | 10.7 |
Quantum σΠ′+Π′′(0.039) and 〈σΠ′+Π′′(300)〉 differ by 2.5 Å2, owing to the averaging of the latter on its resonances, whereas quasi-classical22 observables are nearly equal since the oscillations are washed out in the quasi-classical opacities. We fit the total rate constant from 100 to 900 K to the extended Arrhenius expression kArr = A(T/K)xexp(−Ea/kBT), finding A = 3.02 × 10−11 cm3 s−1, x = 0.230, and Ea = 0.0094 eV. This is a small activation energy, because the Σ+ PES is barrierless, the centrifugal barrier is little important, the quenching is exoergic, and the WPs do not feel any repulsive potential on the Π′ and Π′′ PESs.
4. Summary and conclusions
Following a previous quasi-classical trajectory surface hopping study,22 this paper presents the non-adiabatic quantum dynamics of the electronic quenching OH(A2Σ+) + Kr(1S) → OH(X2Π) + Kr(1S), with OH(A2Σ+) in the ground ro-vibrational state. We use a new non-adiabatic quantum theory based on three diabatic electronic states, Σ+(1), Π′(2), and Π′′(3), coupled by nine conical-intersection (CI) and Renner–Teller (RT) matrix elements. Using a triatomic Hamiltonian Ĥ, all the matrix elements of the electronic Hamiltonian Ĥel and of the total electronic angular momentum are taken into account and ab initio calculated in three dimensions. The time-dependent mechanism, initial-state-resolved quenching probabilities, integral cross sections, thermal rate constants, and thermally averaged cross sections are calculated through the real wavepacket (WP) method and an asymptotic analysis.
Since the minimum structure of the initial Σ+ PES and its CI seam with the Π′ surface are very near, the quenching is opened by the Σ+–Π′ CI12 coupling at γ ∼ 160°, whereas RT couplings are more important near the linearity. When OH(A2Σ+) is in the ground ro-vibrational state, CI12 is the preferred non-adiabatic pathway, much more than those Σ+–Π′ RT12, Σ+–Π′′ RT13 and Π′–Π′′ RT23. In particular, the relative strength of the non-adiabatic couplings is CI12 ≫ RT23 > RT13 > RT12, with 〈Π′|z|Π′′〉 in RT23 more important than other matrix elements.
These electronic results are confirmed and strengthened by the analysis of the quantum dynamics, showing that the Π′ channel is preferred with respect to Π′′, and that conical-intersection and Renner–Teller effects are non-additive and in competition at some collision energies Ecol. When OH(A2Σ+) is in the ground ro-vibrational state, the quantum results also suggest that the conical-intersection coupling rules the dynamics but can overestimate the reactivity, which is reduced by the Renner–Teller effects that modulate the dynamics.
Time-dependent WP snapshots show that the quenching is rather slow and that the opening rates of the Π′ and Π′′ channels are ∼180 and 200 fs−1, respectively. Quenching probabilities P(Ecol) present many sharp oscillations that imply an insertion, complex-forming mechanism that is reflected on some smooth resonances of the cross sections σ(Ecol), as in other electronic quenching processes.37 We obtain total σ(Ecol = 0.039 eV) = 14.1 Å2 and thermally averaged 〈σ(300 K)〉 = 11.6 Å2. These quantum findings compare well with previous quasi-classical22 and experimental results,21 which are equal to 17.2 ± 0.4 and 10 ± 1 Å2, respectively. The total cross sections increase from small values at the threshold up to the maximum values σ(Ecol = 0.045 eV) = 14.8 Å2 and 〈σ(450 K)〉 = 11.9 Å2. On the other hand, the total rate constant increases monotonically in the temperature range here investigated, with a ∼T1/4 temperature dependence and a small activation energy.
In closing, we contrast the present quenching OH(A2Σ+) + Kr with other quenchings we investigated with similar quantum methods, namely O(1D) + N2,46 OH(A2Σ+) + H,44 and OH(X2Π) + H+ 37 with the reactant diatom also in the ground rotational state. When the quenching is due to non-adiabatic couplings of different nature, as in O(1D) + N2 with Spin–Orbit and Renner–Teller effects or in OH(A) + Kr, the RT couplings are less important, because they are significantly different from zero near the linearity whereas the others are important in larger geometry ranges. This is confirmed by the H2O photodissociation27 where CI and RT terms are in competition, like here. OH(A) + H has a smaller resonance structure and larger cross sections than those found here, probably because its reduced mass μR is 15 times smaller, giving both a faster collision with shorter-lived intermediate complexes and larger cross sections. On the other hand OH + H+ presents many resonances, like OH(A) + Kr, and larger cross sections owing to its smaller μR.
Clearly, this work should be extended to a coupled-channel formalism, to excited OH(A) rotational states, and to other cross sections investigated in ref. 21 and 22. Nevertheless, these quantum calculations are very expensive and much beyond our present computer resources. A final comment concerns the comparison of full 3D calculations with respect to those 2D21,22 with constant r. The real WP method of Gray and Balint-Kurti39 relies on the projection of the WP at R∞ onto the final ro-vibrational states of the products OH(Π′) and OH(Π′′) and it cannot used in reduced dimensionality. Nevertheless, we are planning to carry out surface-hopping QCT calculations on the new PESs with a fixed OH distance and with the full 3D PESs. In addition, not only the quenching cross section will be calculated but also the rotational energy transfer cross sections and the OH rotational distributions on the 1A′ and A′′ PESs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are very grateful to A. Bernini, P. Defazio, M. Olivucci, and to the Dipartimento di Biotecnologie, Chimica, e Farmacia, Università di Siena, Italy, for invaluable computing resources. This research is supported by the Spanish Ministry of Science, Innovation, and Universities through grants RTI2018-094757-B-I00, MCIU/AEI/FEDER, UE and MDM-2017-0767, and in part, by the Generalitat de Catalunya (2017SGR0013 and XRQTC). PG thanks Generalitat de Catalunya for his Serra Húnter Associate Professorship. Funding by the Spanish Ministry of Innovation and Universities (MCIU/FEDER-PGC2018-096444-B-100) is also acknowledged.References
- D. R. Crosley, Adv. Ser. Phys. Chem., 1995, 3, 256 Search PubMed.
- D. R. Crosley, J. Phys. Chem., 1989, 93, 6273 CrossRef.
- D. R. Crosley, J. Atmos. Sci., 1995, 52, 3299 CrossRef.
- D. E. Heard and D. A. Henderson, Phys. Chem. Chem. Phys., 2000, 2, 67 RSC.
- B. L. Hemming, D. R. Crosley, J. E. Harrington and V. Sick, J. Chem. Phys., 2001, 115, 3099 CrossRef.
- B. L. Hemming and D. R. Crosley, J. Phys. Chem. A, 2002, 106, 8992 CrossRef.
- P. H. Paul, J. L. Durant, Jr., J. A. Gray and M. R. Furlanetto, J. Chem. Phys., 1995, 102, 8378 CrossRef.
- B. C. Hoffman and D. R. Yarkony, J. Chem. Phys., 2000, 113, 10091 CrossRef.
- P. A. Cleary, L. P. Dempsey, C. Murray, M. I. Lester, J. Kłos and M. H. Alexander, J. Chem. Phys., 2007, 126, 204316 CrossRef.
- B. Fu, E. Kamarchik and J. M. Bowman, J. Chem. Phys., 2010, 133, 164306 CrossRef PubMed.
- J. H. Lehman, L. P. Dempsey, M. I. Lester, B. Fu, E. Kamarchik and J. M. Bowman, J. Chem. Phys., 2010, 133, 164307 CrossRef.
- J. H. Lehman and M. I. Lester, Annu. Rev. Phys. Chem., 2014, 65, 537 CrossRef PubMed.
- M. L. Costen, S. Marinakis and K. G. McKendrick, Chem. Soc. Rev., 2008, 37, 732 RSC.
- M. Brouard, A. Bryant, Y.-P. Chang, R. Cireasa, C. J. Eyles, A. M. Green, S. Marinakis, F. J. Aoiz and J. Kłos, J. Chem. Phys., 2009, 130, 044306 CrossRef CAS PubMed.
- M. L. Costen, R. Livingstone, K. G. McKendrick, G. Paterson, M. Brouard, H. Chadwick, Y.-P. Chang, C. J. Eyles, F. J. Aoiz and J. Kłos, J. Phys. Chem. A, 2009, 113, 15156 CrossRef CAS.
- J. Kłos, M. H. Alexander, M. Brouard, C. J. Eyles and F. J. Aoiz, J. Chem. Phys., 2008, 129, 054301 CrossRef PubMed.
- M. Brouard, H. Chadwick, Y.-P. Chang, C. J. Eyles, F. J. Aoiz and J. Kłos, J. Chem. Phys., 2011, 135, 084306 CrossRef CAS PubMed.
- H. Chadwick, M. Brouard, Y.-P. Chang, C. J. Eyles, T. Perkins, S. A. Seamons, J. Kłos, M. H. Alexander and F. J. Aoiz, J. Chem. Phys., 2012, 137, 154305 CrossRef.
- H. Chadwick, M. Brouard, Y.-P. Chang, C. J. Eyles, G. McCrudden, T. Perkins, S. A. Seamons, J. Kłos, M. H. Alexander, P. J. Dagdigian, D. Herráez-Aguilar and F. J. Aoiz, J. Chem. Phys., 2014, 140, 054306 CrossRef.
- H. Chadwick, M. Brouard, T. Perkins and F. J. Aoiz, Int. Rev. Phys. Chem., 2014, 33, 79 Search PubMed.
- J. H. Lehman, M. I. Lester, J. Kłos, M. H. Alexander, P. J. Dagdigian, D. Herráez-Aguilar, F. J. Aoiz, M. Brouard, H. Chadwick, T. Perkins and S. A. Seamons, J. Phys. Chem. A, 2013, 117, 13481 CrossRef PubMed.
- T. Perkins, D. Herráez-Aguilar, G. McCrudden, J. Kłos, F. J. Aoiz and M. Brouard, J. Chem. Phys., 2015, 142, 144307 CrossRef PubMed.
- J. Kłos, G. McCrudden, M. Brouard, T. Perkins, S. A. Seamons, D. Herráez-Aguilar and F. J. Aoiz, J. Chem. Phys., 2018, 149, 184301 CrossRef.
- P. R. Bunker and P. Jensen, Molecular Symmetry and Spectroscopy, NRC Research Press, Ottawa, Canada, 2nd edn, 1998, p. 370 Search PubMed.
- Ch. Jungen, J. Mol. Spectrosc., 2019, 363, 111172 CrossRef.
- C. Petrongolo, J. Chem. Phys., 1988, 89, 1297 CrossRef.
- L. Zhou, B. Jiang, D. Xie and H. Guo, J. Phys. Chem. A, 2013, 117, 6940 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. T. Manby, M. Schülz, et al., MOLPRO version 2012.1, a package of ab initio programs, 2012, http://www.molpro.net Search PubMed.
- V. Kellö and A. J. Sadlej, Theor. Chim. Acta, 1996, 94, 93 Search PubMed.
- H.-J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053 CrossRef.
- P. J. Knowles and H.-J. Werner, Chem. Phys. Lett., 1988, 145, 514 CrossRef.
- E. R. Davidson, J. Comput. Phys., 1975, 17, 87 CrossRef.
- T.-S. Ho and H. J. Rabitz, Chem. Phys., 1996, 104, 2584 Search PubMed.
- A. Zanchet, B. Bussery-Honvault and P. Honvault, J. Phys. Chem. A, 2006, 110, 12017 CrossRef.
- A. Zanchet, B. Bussery-Honvault, M. Jorfi and P. Honvault, Phys. Chem. Chem. Phys., 2009, 11, 6182 RSC.
- B. T. Sutcliffe and J. Tennyson, Mol. Phys., 1986, 58, 1053 CrossRef.
- P. Gamallo, S. Akpinar, P. Defazio and C. Petrongolo, Phys. Chem. Chem. Phys., 2017, 19, 4454 RSC and refs therein.
- E. U. Condon and G. H. Shortley, The Theory of Atomic Spectra, Cambridge University Press, Cambridge, UK, 1984, p. 52 Search PubMed.
- S. K. Gray and G. G. Balint-Kurti, J. Chem. Phys., 1998, 108, 950 CrossRef.
- H. Guo, Int. Rev. Phys. Chem., 2012, 31, 1 Search PubMed and refs therein.
- M. Hankel, G. G. Balint-Kurti and S. K. Gray, Int. J. Quantum Chem., 2003, 92, 205 CrossRef.
- H. Chadwick, M. Brouard, Y.-P. Chang, C. J. Eyles, T. Perkins, S. A. Seamons, J. Kłos, M. H. Alexander and F. J. Aoiz, J. Chem. Phys., 2012, 137, 154305 CrossRef.
- G. G. Balint-Kurti, A. I. Gonzalez, E. M. Goldfield and S. K. Gray, Faraday Discuss., 1998, 110, 169 RSC.
- P. Gamallo, S. Akpinar, P. Defazio and C. Petrongolo, J. Chem. Phys., 2013, 139, 094303 CrossRef.
- Z. Bacic and J. C. Light, J. Chem. Phys., 1986, 85, 4594 CrossRef.
- P. Gamallo, S. Akpinar, P. Defazio and C. Petrongolo, J. Chem. Phys., 2012, 136, 054308 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02512g |
| This journal is © the Owner Societies 2020 |