
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characteristics of oligomers formed by pyroglutamate-modified amyloid β peptides studied by solid-state NMR†
Holger A.
Scheidt
 *a,
Anirban
Das
b,
Alexander
Korn
a,
Martin
Krueger
c,
Sudipta
Maiti
b and
Daniel
Huster
ab
*a,
Anirban
Das
b,
Alexander
Korn
a,
Martin
Krueger
c,
Sudipta
Maiti
b and
Daniel
Huster
ab
aInstitute for Medical Physics and Biophysics, Leipzig University Härtelstr. 16-18, D-04107 Leipzig, Germany. E-mail: holger.scheidt@medizin.uni-leipzig.de
bDepartment of Chemical Sciences, Tata Institute of Fundamental Research, Homi Bhabha Road, Colaba, Mumbai 400 005, India
cInstitute of Anatomy, Leipzig University, Liebigstraße 13, 04103 Leipzig, Germany
First published on 9th July 2020
Abstract
Neuronal plaques of amyloid β (Aβ) peptides of varying length carrying different posttranslational modifications represent a molecular hallmark of Alzheimer's disease. It is believed that transient oligomeric Aβ assemblies associating in early fibrillation events represent particularly cytotoxic peptide aggregates. Also, N-terminally truncated (in position 3 or 11) and pyroglutamate modified peptides exhibited an increased toxicity compared to the wildtype. In the current study, the molecular structure of oligomeric species of pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) was investigated using solid-state NMR spectroscopy. On the secondary structure level, for both modified peptides a large similarity between oligomers and mature fibrils of the modified peptides was found mainly based on 13C NMR chemical shift data. Some smaller structural differences were detected in the vicinity of the respective modification site. Also, the crucial early folding molecular contact between residues Phe19 and Leu34 could be observed for the oligomers of both modified peptide species. Therefore, it has to be concluded that the major secondary structure elements of Aβ are already present in oligomers of pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40). These posttranslationally modified peptides arrange in a similar fashion as observed for wild type Aβ(1–40).
Introduction
In the development of Alzheimer's disease, especially the early aggregates in the fibrillation process of amyloid β (Aβ) peptides of varying length and posttranslational modifications are considered the most cytotoxic species, which initiate the development of the disease on the molecular level.1–5 While the molecular structure of the final mature fibrils of Aβ has been very well studied,6–14 it still remains a significant challenge to catch these more disease-relevant intermediate oligomeric assemblies for structural investigations. Along with the application of special (and sometimes non-physiological) fibrillation conditions,15,16 spectral filtering in NMR measurements17 or the use of antibodies that specifically bind to folding intermediates,18,19 also freeze-drying of oligomeric assemblies in liquid nitrogen20 has been used for such challenging endeavors. Most of these studies on oligomers suggest that the secondary structure elements – the two β-strands which are connected by a short loop – of Aβ are formed very early in the fibrillation process16,18,20–22 even if the individual β-strands differ in their length.19 More differences between early species in the fibrillation process and mature fibrils were observed on the tertiary structure level: while in the mature fibrils the hydrogen bonds are intermolecular between the monomeric units leading to the formation of parallel β-sheets, in early Aβ oligomers antiparallel β-sheets forming intramolecular hydrogen bonds were observed.18,22–25It is now well described that during processing of the amyloid precursor protein by γ-secretase, amyloid peptides of varying lengths are released that exhibit altered cytotoxic potential.26 In addition, different posttranslational modifications as well as chemical reactions after processing further modify these Aβ fragments.27–31 These different variants of the Aβ peptides also include N-terminally truncated peptides, where the most N-terminal glutamate residue in position 3 or 11 is converted into a cyclic pyroglutamate.29,32 For both peptide species produced in this pathway (pGlu3-Aβ and pGlu11-Aβ) this posttranslational modification affects the fibrillation properties leading not only to increased oligomerization and fibrillation33–38 but also to higher cytotoxicity.34,39,40 Both modifications are abundant in the brain of AD and also Down syndrome patients41,42 and were found in the core of senile plaques of Alzheimer's patients.37,43 Consequently, it is assumed that the pyroglutamate modified peptides play an essential role in the (early) development of AD.29,32,39 It has been shown in mice that the inhibition of the pGlu lactam ring producing enzyme reduced amyloid plaque deposition and retarded memory decline.35
Interestingly, the structural features of the mature fibrils formed by these pyroglutamate-modified peptides have been found to be very similar compared to WT-Aβ.44,45 In fact, most modified and/or mutated Aβ peptides that exhibit a much altered cytotoxic characteristics46 show relatively mild structural alterations in the mature fibrillary state compared to the WT.47–50 Although the exact mechanism how oligomeric Aβ peptides exert their toxic potential to neurons remains speculative, it is clear that the structural features of these aggregates must differ from those of mature fibrils.51 Here, we set out to characterize structural features of oligomers formed by pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) peptide oligomers using the freeze trapping method that has been introduced by Sarkar et al.20
Materials and methods
Oligomer preparation
The differently isotope-labelled Aβ peptides were synthesized using standard Fmoc protocols. Uniformly 13C/15N-labeled amino acids were included as follows: pGlu3-Aβ(3–40) three differently labeled peptides: Peptide I: Ser8, Val12 Phe19, Leu34; Peptide II: Phe4, Glu11, Gly29, Val36; and Peptide III: Asp7, Gly9, Glu22, Ile31. For pGlu11-Aβ(11–40) four different labeled peptides: Peptide IV: Val12 Phe19, Ala21, Leu34 Gly37; Peptide V: Leu17, Glu22, Gly25, Ile31; Peptide VI: Asp23, Lys28, Gly33, Val39 and Peptide VII: pGlu11, Gly29. For a graphical depiction of the labelling scheme, see also ESI,† Scheme S1. The labelling scheme was chosen to avoid spectral overlap in the NMR spectra. Labelled amino acids were concentrated close to the peptide modifications. Additionally, amino acids which are known to be involved in tertiary molecular contacts in other Aβ preparations were labelled in the same peptide (i.e., Phe19-Leu34, Glu22-Ile31).The oligomer preparation followed the procedure described in the literature,20 where also a comprehensive biophysical characterization of these oligomers has been conducted.52,53 In brief, the peptides were dissolved as stock in 1 to 2 ml ammonia solution at pH 11 to start from a nearly monomeric state. This solution was diluted into 175 mM ammonium acetate buffer (pH 7.4) to 25 μM Aβ solution. After 30 min of incubation, the oligomeric solution was flash-frozen by dropwise addition into liquid nitrogen and afterwards lyophilized to remove the water as well as the NH4OAc. Finally, about 1.5 to 3.5 mg of dry powder was obtained and transferred into 3.2 mm MAS rotors for NMR measurements.
Fluorescence measurements for oligomer characterization
Peptide stocks were prepared at pH 11 in a similar way as mentioned above. They were stored at −80 °C after flash-freezing in liquid nitrogen. Before starting the experiments, they were thawed quickly and used. For TPE-TPP (bis(triphenylphosphonium) tetraphenylethene), 1 mg of the dye was dissolved in 1 mL of DMSO to prepare a stock solution of 1 mM. The aggregation of each of the Aβ peptides were started from an initial concentration of 25 μM in 175 mM NH4OAc buffer (pH 7.5), incubated with 10 μM TPE-TPP.Fluorescence emission spectra of TPE-TPP were recorded at different time-points (t = 0 to 2 hours, at 30 minutes interval) using a quartz cuvette (1 cm × 1 cm path length) on a FluoroMax-3 (Jobin Yvon, Horiba) spectrofluorimeter. TPE-TPP was excited at 330 nm and the emission was collected from 390 to 620 nm. Excitation and emission slits both were kept at 5 nm, and the integration time was fixed at 0.1 s. Five consecutive spectra were recorded at each of the time-points and were averaged later on. Samples were thoroughly mixed before each scan.
Electron microscopy
The fibril morphology was evaluated and analyzed by electron microscopy (EM). Droplets of the prepared fibril solutions (1 μl each) were placed on formvar- coated copper grids, allowed to dry for about 1 h and subsequently stained with 1% uranyl acetate in pure water. Electron microscopy was performed using a Zeiss SIGMA electron microscope, (Zeiss NTS, Oberkochen, Germany) equipped with a STEM detector and Atlas Software.Solid-state MAS NMR spectroscopy
The MAS NMR experiments were conducted on a Bruker Avance III 600 MHz or a Bruker Avance Neo 700 MHz spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) using triple channel 3.2 mm MAS probes. Typical pulse lengths were 4 μs for 1H and 13C and 5 μs for 15N. 1H–13C and 1H–15N CP contact time were 1 ms at a spin lock field of ∼50 kHz. During acquisition, 1H dipolar decoupling with a radio frequency field strength of 65 kHz was applied using Spinal64. The MAS frequency was 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 777 Hz. 13C chemical shifts were referenced externally relative to TMS. The relaxation delay in all experiments was 2.5 s. All NMR experiments were carried out at a temperature of 30 °C.
777 Hz. 13C chemical shifts were referenced externally relative to TMS. The relaxation delay in all experiments was 2.5 s. All NMR experiments were carried out at a temperature of 30 °C.
For all samples, 13C–13C DARR NMR spectra were acquired. If the 15N signal intensity allowed additional the detection 15N–13Cα correlation spectra, they were acquired at the same time using dual-acquisition.54 In one experiment, a two dimensional 13C–13C DARR NMR spectrum with 128 data points and four identical 15N–13Cα correlation spectra with 32 data points in the indirect dimensions were measured. The DARR mixing time was 500 ms, in some cases, additional DARR spectra with a mixing time of 50 ms were acquired to exclude cross peaks from long range interactions.
Results and discussion
A very comprehensive characterizations of the biophysical properties of early Aβ oligomers prepared by the procedure of Sarkar et al.20 or very similar protocols can be found in the literature.52,53 In addition, we checked the morphology of the oligomer preparations by recording electron micrographs of the respective samples (Fig. 1). For both pyroglutamated peptides, no fibrillar structures were observable. Instead a number of small spherical aggregates of varying size which sometimes cluster together are visible. Comparable structures were observed in the electron micrographs of different oligomer preparations.16,17,22,24,25,34,52,55 | ||
| Fig. 1 Scanning transmission electron micrograph of the oligomer preparation of pGlu3-Aβ(3–40) (left) and pGlu11-Aβ(11–40) (right). The scale bar represents 1.0 μm. | ||
For further characterization of these oligomers, we investigated the state of aggregation of the modified Aβ peptide by monitoring the fluorescence emission of the dye bis(triphenylphosphonium) tetraphenylethene (TPE-TPP), which shows a clear spectral change with maturation of the WT Aβ(1–40) fibrils, and it can identify even the early small oligomeric species.52 In the comparison of WT and both modified Aβ peptides it is expected that if the similar spectral characteristics report similar structural characteristics.
Fig. S1 (ESI†) shows the fluorescence emission of TPE-TPP as a function of time incubated with WT-Aβ(1–40). A control solution (10 μM TPE-TPP in 175 mM NH4OAc buffer) containing only TPE-TPP at the same concentration showed very low or negligible intensity (data not shown), as observed previously.52 The observed emission maximum at 462 nm (till 1 to 1.5 hours), show that the solution contains mostly the small oligomeric aggregates (n < 10).52 For pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) very similar emission maxima of 461 nm (till about 1.5 hours) and 463 nm (till about 1 hour of incubation) are observed (Fig. 2). We would like to note here that the solution conditions in the current experiment (175 mM NH4OAC pH), is different from that used earlier (20 mM phosphate and 150 mM NaCl).52 However, the emission spectra for the WT-Aβ40 appear to be very similar in both the buffers, so the results should be comparable. We therefore conclude that the species present after 30 minutes of incubation (as used in NMR experiments) in both pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) solutions are almost exclusively the small oligomers.
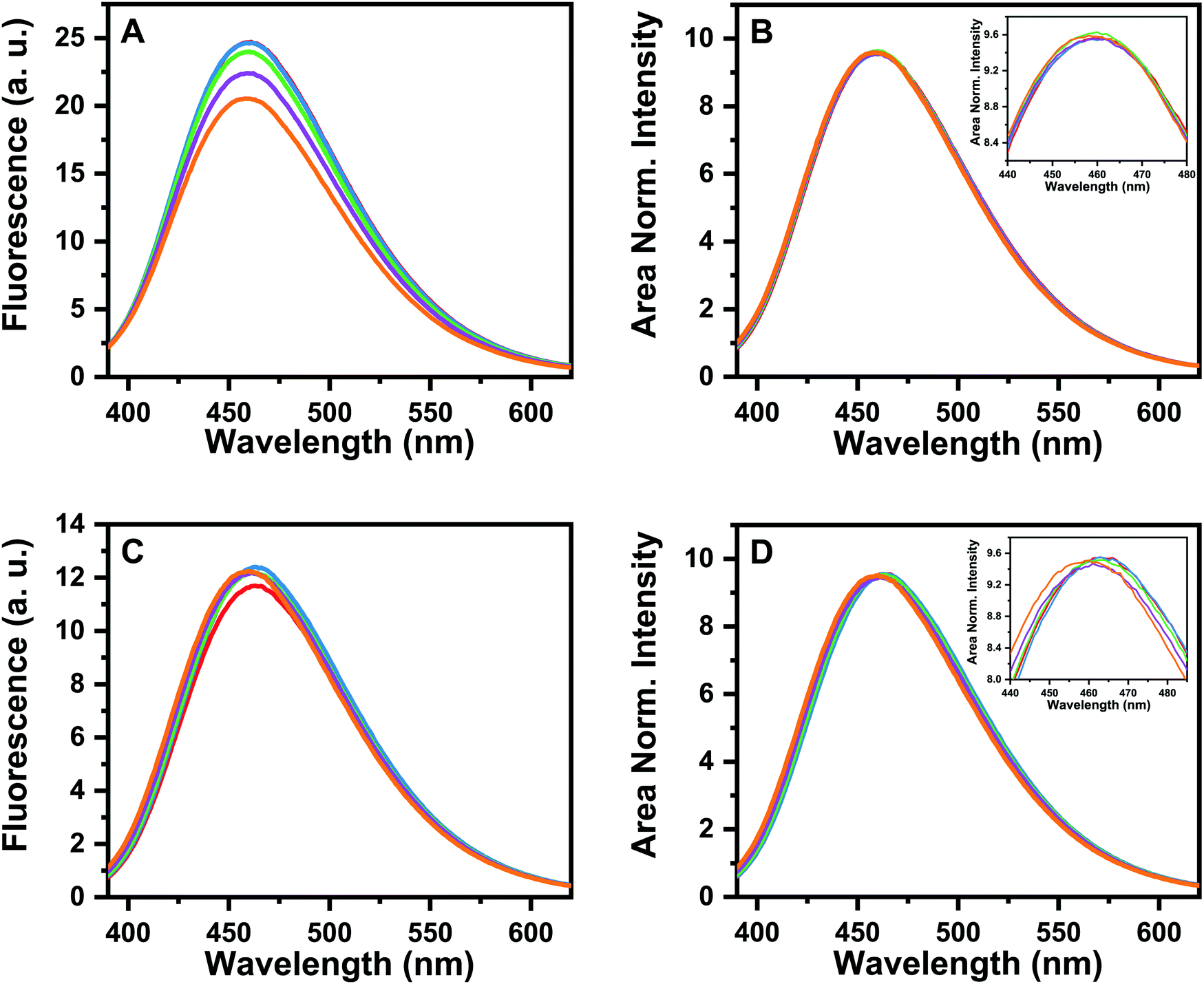 | ||
| Fig. 2 Fluorescence emission of TPE-TPP as a function of time, when incubated with (A) pGlu3-Aβ(3–40) and (C) pGlu11-Aβ(11–40). Emission spectra were recorded at 5 different time points, at an interval of 30 min [t = 0 (red), 30 min (blue), 1 h (green), 1.5 h (purple) and 2 hours (orange)]. (B and D) Same data as in panel A and C respectively, but area normalized. The respective insets show the magnified views of the peak regions of the spectra presented in (B and D). | ||
For more atomistic insights into the secondary structure of the respective peptides in the oligomeric aggregates, solid-state-NMR measurements were performed. The 13C/15N-labeled amino acids were concentrated in the regions close to the pyroglutamyl modification site of the respective peptide. For the oligomers of both modified peptides, the 1D 13C CP MAS NMR spectra exhibit an increased line width compared to the spectra of the respective mature fibrils (see ESI,† Fig. S2 and S3). This is probably caused by structural polymorphism in the samples, which is known for WT-Aβ peptides in different preparations6,19,20,56,57 but also a result of the current sample preparation since different oligomeric species will be present in the investigated samples.
For the assignment of the 13C and in part also the 15N NMR signals, 13C–13C DARR (if necessary with short and long mixing times) and in cases where sufficient peptide quantities for good NMR sensitivity were achieved also 15N–13Cα correlation spectra were acquired. Typical 13C–13C DARR spectra with a mixing time of 500 ms are shown in ESI,† Fig. S4. For some residues, more than one NMR signal was observed, which indicates structural polymorphism. The obtained chemical shift values are summarized in ESI,† Tables S1 and S2. Since the 13Cα and 13Cβ chemical shift values are sensitive for secondary structure, they are shown in Fig. 3 as differences to the random coil values (taken from literature58) for pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) oligomers. For comparison, also the chemical shift data for the mature fibrils of the respective peptides44,45 are plotted into the same diagrams. According to the chemical shift values, the differences between oligomers and mature fibrils with regard to their secondary structure are rather small for both pyroglutamate modified peptides. The two β-strands in the region of Glu11–Glu22 and Gly29–Gly38, which are typical for Aβ(1–40), are already formed in the oligomeric state. Some small deviations between the oligomers and the mature fibrils can be found in the vicinity of the pyroglutamate modification: for pGlu11-Aβ(11–40) oligomers only for the direct neighbor in the amino acid sequence Val12, for pGlu3-Aβ(3–40) oligomers in addition to Phe4 also for Asp7. Differences in the chemical shift values can also be observed for Phe19. Here, some unusual values were reported for the mature fibrils of pGlu3-Aβ(3–40) (two signals)44 and pGlu11-Aβ(11–40),45 therefore, here the values for the oligomers of these peptides match with those for wild type Aβ(1–40) fibrils.59
 | ||
| Fig. 3 Comparison of the secondary 13C MAS NMR chemical shifts of the oligomers and the respective mature fibrils of (A) pGlu3-Aβ(3–40) and (B) pGlu11-Aβ(11–40) for 13Cα (squares) and 13Cβ (circles). Data are plotted as the difference of a measured chemical shifts to random coil chemical shifts taken from the literature.58 For the pGlu residues the random coil values of Glu were used as no reference values for pGlu are available. The values for the mature fibrils were taken from ref. 44 or 45 respectively. The arrows above the plots indicate β-strand secondary structure, the gray area in the plots highlight the 13C labeled amino acids. | ||
For a further comparison between oligomers of this study and the respective mature fibrils Fig. 5(A and B) exhibit the correlation plots for the Cα–Cβ chemical shift difference (which is very sensitive to changes in the secondary structure and independent of chemical shift referencing) between pyroglutamate modified oligomers and mature fibrils. For both variants, a very good correlation is obtained.
To compare the oligomers of the pyroglutamyl-modified peptides with oligomers of WT-Aβ(1–40), Fig. 4 shows the 13Cα and 13Cβ chemical shifts values of pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) oligomers together with literature data from WT-Aβ(1–40) oligomers,20 which were prepared according the same procedure used in this study, in a similar way as above. A comparison is somewhat hampered by the fact that the labeling pattern of the studied peptides investigated is different. Nevertheless, overall, the pattern for all three oligomers is quite similar and the secondary structure elements are present for all three samples. Small differences in these values are only observed for amino acids close to the pyroglutamate modification at the N-terminus. Only for Phe19, some additional differences in the chemical shift values for Cβ are observed. For additional comparison, Fig. 5C and D exhibits the correlation plots for the Cα–Cβ chemical shift difference between the here investigated oligomers of pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) and the oligomers of WT-Aβ(1–40).20 Again, at least for the amino acid labeled in both studies a very good correlation is archived.
 | ||
| Fig. 4 Comparison of the secondary 13C MAS NMR chemical shifts for Cα (top) and Cβ (bottom) of the oligomers of pGlu3-Aβ(3–40) (red) and pGlu11-Aβ(11–40) (blue) and WT-Aβ(1–40) (black, data taken from literature20). Data are plotted as the difference of a measured chemical shifts to random coil chemical shifts for each residue.58 For the pGlu residues the random coil values of Glu were used as no reference values for pGlu are available. The arrows above the plots indicate β-strand secondary structure, the gray area in the plots indicate the 13C labeled amino acids in a least one of the studies. | ||
 | ||
| Fig. 5 Correlation plots of the Cα–Cβ chemical shift differences from the data shown in Fig. 3 and 4 for (A) pGlu3-Aβ(3–40) oligomers vs. mature fibrils,44 (B) pGlu11-Aβ(11–40) oligomers vs. mature fibrils,45 (C) oligomers of pGlu3-Aβ(3–40) vs. WT-Aβ(1–40)20 and (D) oligomers of pGlu11-Aβ(11–40) vs. WT-Aβ(1–40).20 Residue 11 was excluded from plot D, since pGlu should not be compared to Glu. Note that the glycine residues are excluded from all plots since Cα–Cβ chemical shift differences are shown. The Pearson correlation coefficient is given on each plot. | ||
To obtain insights into the three-dimensional structural arrangement of the two β-strands of Aβ(1–40), the well-studied hydrophobic tertiary contact between the side chains of Phe19 and Leu34,7,8,47,60 which is already formed during early stages of the fibrillation,16,19,20,24,53,61,62 was addressed. Using NMR REDOR measurements, the 15N–13CO distance between these two residues was determined to about 4 Å for oligomers and about 7 Å for mature fibrils.53 Also, in other structural models for oligomers and mature fibrils, similar distances between the side chains of these residues can be found.7,9,18,60
The formation of this molecular contact was investigated using 13C–13C DARR experiments with a long mixing time of 500 ms. The respective NMR spectra for oligomers for both modified peptides are shown in ESI,† Fig. S4. In both cases, a clear cross peak, indicating magnetization exchange between the aromatic ring of Phe19 and the side chain of Leu34, is observed. Therefore, also in the oligomers of the pyroglutamated variants of Aβ(1–40) both residues are close in space and the contact between these amino acids seems already established, which means that the two β-strands are already in an arrangement close to each other. This again indicates a structural similarity between the oligomers of pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) and the known structures of WT-Aβ(1–40).
One interesting molecular detail which was found for Aβ oligomers and also protofibrils on the one hand and mature fibrils on the other is the structural arrangement of the edges of the two β-strands. Previous work has shown that alterations in this region results in structural models, where the respective hydrogen bonds between β-strands are intramolecular for oligomers instead of the known intermolecular organization for mature fibrils.24,51 From these findings, a rearrangement of the hydrogen bond pattern during the fibrillation process was suggested. This structurally interesting region can be addressed by studying the molecular contact between the residues Glu22 and Ile31, since the side chains of these residues point towards each other in antiparallel β-sheets connected by intramolecular hydrogen bonds for oligomers and away from each other in the parallel β-sheets with intermolecular hydrogen bonds of mature fibrils. Consequently, this molecular contact was found in structural studies of Aβ oligomers and protofibrils18,63 but not in mature fibrils of WT-Aβ(1–40)63 or both variants of pyroglutamated Aβ.44,45 For the oligomers of both pyroglutamated variants of Aβ(1–40) of the current study, the cross peak between Glu22 and Ile31 could not be observed in the respective 13C–13C DARR NMR spectra. Also, the contact between Gly25 and Ile31, which was only observed for mature fibrils of pGlu11-Aβ(11–40)45 could not be observed. So far one can only speculate about the reason for this: too weak signal intensities of the respective cross peaks due to the small amount of sample after the oligomer preparation in this study is probable. Also possible is an influence of the affibodies/antibodies, which were used to stabilize the intermediate states during Aβ fibrillation in the studies were the contact Glu22 and Ile31 was observed.18,63
Conclusions
The procedure for Aβ oligomer preparation used here leads to an ensemble of small oligomeric structures, which is characterized by structural polymorphism in the samples as well as alterations in the molecular dynamics inferred from the observation of increased NMR line widths. The interpretation of such complex samples in terms of a simple molecular model of Aβ oligomers is quite complex. Nevertheless, overall one has to conclude that oligomers of pGlu3-Aβ(3–40) and pGlu11-Aβ(11–40) exhibit a close similarity to mature fibrils of the same peptides with regard to secondary structure. Also, known tertiary contacts between the two β-strands were observed to be similar. The structural elements (unstructured N-terminus, two β-strands connected by a short loop) of Aβ(1–40) in fibrils seems already well formed in these oligomers. At least for the investigated residues, a high similarity to oligomeric species of WT-Aβ(1–40) has been found.15,20 The small differences in the N-terminal region can be related to the pyroglutamate modification, but may also point to a possible role of the N-terminus during the fibrillation process. Also for WT-Aβ(1–40) oligomers, differences in the NMR chemical shift values compared to mature fibrils were observed in this region of the molecule,20 and additional differences in the dynamics of N-terminal residues in different fibrillation stages of WT-Aβ(1–40) argue in this direction.59,64 Furthermore, also for a phosphorylation at Ser8 structural changes in the N-terminus that influence the maturation of the fibrils were reported.31 Overall, modifications in the flexible N-terminal region seem to have only minimal effects on the secondary structure of the Aβ oligomers, especially the parts in β-strand conformation are not influenced. However, the properties of the disordered N-terminal are crucial in determining the toxicity. It was recently shown that an interaction between the N-terminus and an intracellular component is important for toxicity.65 This manuscript reinforces that notion.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Yuning Hong (La Trobe University) for her generous gift of the TPE-TPP dye. SM acknowledges support of the Department of Atomic Energy, Government of India, provided under project no. RTI4003. The study was supported by the Deutsche Forschungsgemeinschaft, DFG, project number 189853844-SFB TRR 102 (A06).References
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed.
- I. Benilova, E. Karran and S. B. De, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- F. Bemporad and F. Chiti, Chem. Biol., 2012, 19, 315–327 CrossRef CAS PubMed.
- M. Diociaiuti, G. Macchia, S. Paradisi, C. Frank, S. Camerini, P. Chistolini, M. C. Gaudiano, T. C. Petrucci and F. Malchiodi-Albedi, Biochim. Biophys. Acta, 2014, 1842, 1622–1629 CrossRef CAS PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez Del Amo, B. A. Gruning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fandrich, R. F. Frank, B. Reif, S. Gunther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- A. K. Paravastu, R. D. Leapman, W. M. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef CAS PubMed.
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, J. Am. Chem. Soc., 2011, 133, 16013–16022 CrossRef CAS PubMed.
- J. X. Lu, W. Qiang, W. M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Cell, 2013, 154, 1257–1268 CrossRef CAS PubMed.
- M. A. Walti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Bockmann, P. Guntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef CAS PubMed.
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef CAS.
- L. Gremer, D. Scholzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schroder, Science, 2017, 358, 116–119 CrossRef CAS PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fandrich, Nat. Commun., 2019, 10, 4760 CrossRef PubMed.
- S. Chimon and Y. Ishii, J. Am. Chem. Soc., 2005, 127, 13472–13473 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS PubMed.
- S. A. Kotler, J. R. Brender, S. Vivekanandan, Y. Suzuki, K. Yamamoto, M. Monette, J. Krishnamoorthy, P. Walsh, M. Cauble, M. M. Holl, E. N. Marsh and A. Ramamoorthy, Sci. Rep., 2015, 5, 11811 CrossRef PubMed.
- W. Hoyer, C. Gronwall, A. Jonsson, S. Stahl and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5099–5104 CrossRef CAS PubMed.
- H. A. Scheidt, I. Morgado, S. Rothemund, D. Huster and M. Fändrich, Angew. Chem., Int. Ed., 2011, 50, 2837–2840 CrossRef CAS PubMed.
- B. Sarkar, V. S. Mithu, B. Chandra, A. Mandal, M. Chandrakesan, D. Bhowmik, P. K. Madhu and S. Maiti, Angew. Chem., Int. Ed., 2014, 53, 6888–6892 CrossRef CAS PubMed.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CrossRef CAS PubMed.
- J. C. Stroud, C. Liu, P. K. Teng and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7717–7722 CrossRef CAS PubMed.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrene, V. Narayanaswami, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 CrossRef CAS PubMed.
- A. Sandberg, L. M. Luheshi, S. Sollvander, B. T. Pereira de, B. Macao, T. P. Knowles, H. Biverstal, C. Lendel, F. Ekholm-Petterson, A. Dubnovitsky, L. Lannfelt, C. M. Dobson and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15595–15600 CrossRef CAS PubMed.
- Y. Gao, C. Guo, J. O. Watzlawik, P. S. Randolph, E. J. Lee, D. Huang, S. M. Stagg, H. X. Zhou, T. L. Rosenberry and A. K. Paravastu, J. Mol. Biol., 2020, 2836, 30354–30355 Search PubMed.
- H. Steiner, A. Fukumori, S. Tagami and M. Okochi, Cell Stress., 2018, 2, 292–310 CrossRef PubMed.
- M. P. Kummer and M. T. Heneka, Alzheimer's Res. Ther., 2014, 6, 28 CrossRef PubMed.
- E. Cabrera, P. Mathews, E. Mezhericher, T. G. Beach, J. Deng, T. A. Neubert, A. Rostagno and J. Ghiso, Biochim. Biophys. Acta, 2018, 1684, 208–225 CrossRef PubMed.
- S. Jawhar, O. Wirths and T. A. Bayer, J. Biol. Chem., 2011, 286, 38825–38832 CrossRef CAS PubMed.
- Y. K. Al-Hilaly, T. L. Williams, M. Stewart-Parker, L. Ford, E. Skaria, M. Cole, W. G. Bucher, K. L. Morris, A. A. Sada, J. R. Thorpe and L. C. Serpell, Acta Neuropathol. Commun., 2013, 1, 83 CrossRef PubMed.
- N. Rezaei-Ghaleh, S. Kumar, J. Walter and M. Zweckstetter, J. Biol. Chem., 2016, 291, 16059–16067 CrossRef CAS PubMed.
- A. P. Gunn, C. L. Masters and R. A. Cherny, Int. J. Biochem. Cell Biol., 2010, 42, 1915–1918 CrossRef CAS PubMed.
- A. P. Gunn, B. X. Wong, T. Johanssen, J. C. Griffith, C. L. Masters, A. I. Bush, K. J. Barnham, J. A. Duce and R. A. Cherny, J. Biol. Chem., 2016, 291, 6134–6145 CrossRef CAS PubMed.
- D. Schlenzig, R. Ronicke, H. Cynis, H. H. Ludwig, E. Scheel, K. Reymann, T. Saido, G. Hause, S. Schilling and H. U. Demuth, J. Neurochem., 2012, 121, 774–784 CrossRef CAS PubMed.
- S. Schilling, U. Zeitschel, T. Hoffmann, U. Heiser, M. Francke, A. Kehlen, M. Holzer, B. Hutter-Paier, M. Prokesch, M. Windisch, W. Jagla, D. Schlenzig, C. Lindner, T. Rudolph, G. Reuter, H. Cynis, D. Montag, H. U. Demuth and S. Rossner, Nat. Med., 2008, 14, 1106–1111 CrossRef CAS PubMed.
- M. Wulff, M. Baumann, A. Thummler, J. K. Yadav, L. Heinrich, U. Knupfer, D. Schlenzig, A. Schierhorn, J. U. Rahfeld, U. Horn, J. Balbach, H. U. Demuth and M. Fandrich, Angew. Chem., Int. Ed., 2016, 55, 5081–5084 CrossRef CAS PubMed.
- W. He and C. J. Barrow, Biochemistry, 1999, 38, 10871–10877 CrossRef CAS PubMed.
- C. Dammers, L. Gremer, K. Reiss, A. N. Klein, P. Neudecker, R. Hartmann, N. Sun, H. U. Demuth, M. Schwarten and D. Willbold, PLoS One, 2015, 10, e0143647 CrossRef PubMed.
- M. Morawski, S. Schilling, M. Kreuzberger, A. Waniek, C. Jager, B. Koch, H. Cynis, A. Kehlen, T. Arendt, M. Hartlage-Rubsamen, H. U. Demuth and S. Rossner, J. Alzheimer's. Dis., 2014, 39, 385–400 CAS.
- J. M. Nussbaum, S. Schilling, H. Cynis, A. Silva, E. Swanson, T. Wangsanut, K. Tayler, B. Wiltgen, A. Hatami, R. Ronicke, K. Reymann, B. Hutter-Paier, A. Alexandru, W. Jagla, S. Graubner, C. G. Glabe, H. U. Demuth and G. S. Bloom, Nature, 2012, 485, 651–655 CrossRef CAS PubMed.
- T. C. Saido, W. Yamao-Harigaya, T. Iwatsubo and S. Kawashima, Neurosci. Lett., 1996, 215, 173–176 CrossRef CAS.
- C. Russo, T. C. Saido, L. M. DeBusk, M. Tabaton, P. Gambetti and J. K. Teller, FEBS Lett., 1997, 409, 411–416 CrossRef CAS.
- C. P. Sullivan, E. A. Berg, R. Elliott-Bryant, J. B. Fishman, A. C. McKee, P. J. Morin, M. A. Shia and R. E. Fine, Neurosci. Lett., 2011, 505, 109–112 CrossRef CAS PubMed.
- H. A. Scheidt, J. Adler, M. Krueger and D. Huster, Sci. Rep., 2016, 6, 33531 CrossRef CAS PubMed.
- H. A. Scheidt, J. Adler, U. Zeitschel, C. Hofling, A. Korn, M. Krueger, S. Rossner and D. Huster, Chemistry, 2017, 23, 15834–15838 CrossRef CAS PubMed.
- A. K. Das, A. Rawat, D. Bhowmik, R. Pandit, D. Huster and S. Maiti, ACS Chem. Neurosci., 2015, 6, 1290–1295 CrossRef CAS PubMed.
- J. Adler, H. A. Scheidt, M. Kruger, L. Thomas and D. Huster, Phys. Chem. Chem. Phys., 2014, 16, 7461–7471 RSC.
- F. Hoffmann, J. Adler, B. Chandra, K. R. Mote, G. Bekcioglu-Neff, D. Sebastiani and D. Huster, J. Phys. Chem. Lett., 2017, 8, 4740–4745 CrossRef CAS PubMed.
- A. Korn, S. McLennan, J. Adler, M. Krueger, D. Surendran, S. Maiti and D. Huster, ACS Chem. Neurosci., 2018, 9, 790–799 CrossRef CAS.
- A. K. Schutz, T. Vagt, M. Huber, O. Y. Ovchinnikova, R. Cadalbert, J. Wall, P. Guntert, A. Bockmann, R. Glockshuber and B. H. Meier, Angew. Chem., Int. Ed., 2015, 54, 331–335 CrossRef PubMed.
- T. Härd, FEBS J., 2011, 278, 3884–3892 CrossRef.
- A. Das, A. Gupta, Y. Hong, J. A. Carver and S. Maiti, Biochemistry, 2020, 59, 1813–1822 CrossRef CAS PubMed.
- B. Chandra, D. Bhowmik, B. K. Maity, K. R. Mote, D. Dhara, R. Venkatramani, S. Maiti and P. K. Madhu, Biophys. J., 2017, 113, 805–816 CrossRef CAS PubMed.
- T. Gopinath and G. Veglia, Angew. Chem., Int. Ed., 2012, 51, 2731–2735 CrossRef CAS PubMed.
- S. T. Kumar, J. Leppert, P. Bellstedt, C. Wiedemann, M. Fandrich and M. Gorlach, J. Mol. Biol., 2016, 428, 268–273 CrossRef CAS PubMed.
- A. K. Paravastu, A. T. Petkova and R. Tycko, Biophys. J., 2006, 90, 4618–4629 CrossRef CAS PubMed.
- A. T. Petkova, R. D. Leapman, Z. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS PubMed.
- D. S. Wishart and B. D. Sykes, Methods Enzymol., 1994, 239, 363–392 CAS.
- H. A. Scheidt, I. Morgado, S. Rothemund and D. Huster, J. Biol. Chem., 2012, 287, 2017–2021 CrossRef CAS PubMed.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef PubMed.
- W. M. Tay, D. Huang, T. L. Rosenberry and A. K. Paravastu, J. Mol. Biol., 2013, 425, 2494–2508 CrossRef CAS.
- A. Potapov, W. M. Yau, R. Ghirlando, K. R. Thurber and R. Tycko, J. Am. Chem. Soc., 2015, 137, 8294–8307 CrossRef CAS.
- H. A. Scheidt, I. Morgado and D. Huster, J. Biol. Chem., 2012, 287, 22822–22826 CrossRef CAS PubMed.
- C. Lendel, T. Sparrman, M. Mayzel, C. E. Andersson, G. Karlsson and T. Härd, ChemistrySelect, 2016, 1, 5850–5853 CrossRef CAS.
- B. K. Maity, A. K. Das, S. Dey, U. K. Moorthi, A. Kaur, A. Dey, D. Surendran, R. Pandit, M. Kallianpur, B. Chandra, M. Chandrakesan, S. Arumugam and S. Maiti, ACS Chem. Neurosci., 2019, 10, 2498–2509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02307h |
| This journal is © the Owner Societies 2020 |