
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural investigation of doubly-dehydrogenated pyrene cations†
Sanjana
Panchagnula
 *ab,
Jordy
Bouwman
a,
Daniël B.
Rap
c,
Pablo
Castellanos
ab,
Alessandra
Candian
bd,
Cameron
Mackie
b,
Shreyak
Banhatti
e,
Sandra
Brünken
c,
Harold
Linnartz
a and
Alexander G. G. M.
Tielens
b
*ab,
Jordy
Bouwman
a,
Daniël B.
Rap
c,
Pablo
Castellanos
ab,
Alessandra
Candian
bd,
Cameron
Mackie
b,
Shreyak
Banhatti
e,
Sandra
Brünken
c,
Harold
Linnartz
a and
Alexander G. G. M.
Tielens
b
aLaboratory for Astrophysics, Leiden Observatory, Leiden University, PO Box 9513, 2300 RA Leiden, The Netherlands. E-mail: panchagnula@strw.leidenuniv.nl
bLeiden Observatory, Leiden University, PO Box 9513, 2300 RA Leiden, The Netherlands
cRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7, NL-6525ED Nijmegen, The Netherlands
dVan't Hoff Institute for Molecular Sciences, University of Amsterdam, PO Box 94157, 1090 GD Amsterdam, The Netherlands
eI. Physikalisches Institut, Universität zu Köln, Zülpicher Str. 77, 50937 Köln, Germany
First published on 27th July 2020
Abstract
The vibrationally resolved spectra of the pyrene cation and doubly-dehydrogenated pyrene cation (C16H10˙+; Py+ and C16H8˙+; ddPy+) are presented. Infrared predissociation spectroscopy is employed to measure the vibrational spectrum of both species using a cryogenically cooled 22-pole ion trap. The spectrum of Py+ allows a detailed comparison with harmonic and anharmonic density functional theory (DFT) calculated normal mode frequencies. The spectrum of ddPy+ is dominated by absorption features from two isomers (4,5-ddPy+ and 1,2-ddPy+) with, at most, minor contributions from other isomers. These findings can be extended to explore the release of hydrogen from interstellar PAH species. Our results suggest that this process favours the loss of adjacent hydrogen atoms.
1 Introduction
The aromatic infrared bands (AIBs) – a series of broad emission features observed in the near- to mid-infrared spectra of many astronomical sources in interstellar and circumstellar regions – are widely accepted to be caused by polycyclic aromatic hydrocarbons (PAHs).1,2 Free-floating PAH molecules are excited following the absorption of (vacuum) ultraviolet (UV) photons, triggering complex intramolecular vibrational energy redistribution that results in the emission of infrared (IR) radiation. The IR features are most prominent in areas with strong UV radiation fields such as H II regions, reflection and planetary nebulae, and in the diffuse interstellar medium (ISM).2,3In interstellar regions illuminated by strong UV fluxes, interstellar PAHs tend to be photoionised.4,5 In addition, the strong UV flux may lead to additional fragmentation of the formed PAH cations through loss of H, H2, or small hydrocarbons.5,6 Loss of hydrogen from PAHs results in derivatives such as dehydrogenated PAH cations.7,8 Astrochemical models show that the hydrogenation state of a PAH depends on the local interstellar UV radiation field (leading to hydrogen-loss), and the atomic hydrogen density (which drives re-hydrogenation of the dehydrogenated radical).4,7,9 In UV-rich regions or regions of low atomic hydrogen density such as clouds near massive stars, PAHs are predicted to exhibit a high degree of dehydrogenation.4,7 The extent of hydrogen loss also depends strongly on the size of the molecule, with smaller PAHs being subject to more dehydrogenation.5,9 While hydrogen-loss is the dominant fragment channel, small PAHs (<16 carbon atoms) may exhibit a (minor) carbon-loss channel. However, for pericondensed PAHs, including pyrene, loss of C2H2 is inhibited because a key transition state is too high in energy.10 Indeed, experiments reveal that for PAHs in the astrophysically-relevant size range (50–100 carbon atoms), fragmentation is dominated by hydrogen-loss until most – if not all – hydrogen atoms are lost.8,10,11 For completeness, we mention that in regions with high atomic hydrogen density and low UV fields, interstellar PAHs can be superhydrogenated (i.e. some or all of the edge carbon atoms have acquired an additional hydrogen atom).7,9 Here, we focus on dehydrogenated PAHs as they are particularly relevant in regions of massive star formation.7
Precision measurements of the AIBs (strongest at 3.3, 6.2, 7.7, 8.6 and 11.2 μm) reveal subtle changes in band profiles and peak positions.12,13 These changes must have a molecular origin and reflect that PAHs in space can exist in several forms14,15 with varying charge states,16,17 possibly as fragments of larger precursors18 or with hetero-atom substitutions in the carbon skeleton.19 Dehydrogenated PAHs in particular have gained interest from the astronomical community as they may be involved as by-products in the formation of molecular hydrogen in regions illuminated by strong UV fields,20,21 and in the first step towards the conversion of PAHs to cages and fullerenes in space.11 The exact mechanism by which hydrogen is formed from aromatic molecules has not yet been experimentally determined, and the structures of the resulting dehydrogenated PAHs are also unknown.
Experimental challenges such as low molecular densities in molecule-specific detection methods have made it difficult to measure and assign the mid-IR spectra of large PAH cations and radicals in the gas phase, particularly when investigating charged fragmentation products. Action spectroscopy has been widely applied to overcome these challenges.6,22 Infrared multiphoton dissociation (IRMPD) spectroscopy using strong infrared light sources such as free electron lasers in combination with ion traps offers an effective spectroscopic tool capable of recording spectra using very low molecular abundances; ion traps are typically filled with only a few thousand ions.23 However, IRMPD also comes with a number of disadvantages, most notably a non-linear response that shifts band positions and alters relative band intensities.24,25
Recent advancements in cryogenic techniques are one means of addressing these issues. Infrared predissociation (IRPD) spectroscopy uses the rare gas tagging messenger technique in combination with tunable infrared radiation to provide spectroscopic information on the cold ion-rare gas complex following a single-photon absorption process. For IRPD, observed intensities more closely resemble calculated absorption cross-sections than in the case of IRMPD.26 This predissociation technique, using IR and NIR photons, has recently been applied to successfully study the spectra of a large number of PAH, PAH photofragment, and fullerene cations.27–30
Pyrene (Fig. 1) is a highly stable and commercially available species representing the smallest pericondensed PAH. It has therefore been the subject of many experimental studies as a model system for PAHs when studying their physical chemistry. While small compared to the astrophysically-relevant size range, pyrene exhibits several of the characteristic hydrocarbon edge sites present in interstellar PAHs (Fig. 1) and as such it has become a prototypical species in astronomical model studies of PAHs. The first measurement of IR emission from a gaseous PAH cation (generated by electron impact ionisation) was achieved with pyrene; the spectrum showed relative intensities consistent with astrophysical observations of the AIBs, as well as unidentified features that may be attributed to dehydrogenated pyrene species.31 The IR absorption features of the pyrene cation have been recorded in matrix isolation experiments and assigned on the basis of theory work.32 These have triggered IRMPD work23 onto which the present work extends.
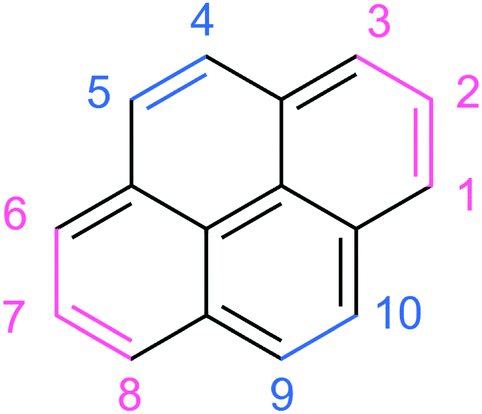 | ||
| Fig. 1 Chemical structure of pyrene. The hydrogen-numbering system shown here is employed when labelling isomers of the doubly-dehydrogenated pyrene cation. Blue shows duo hydrogen sites, and pink shows trio hydrogen sites. | ||
In this work we present the IRPD spectra of the pyrene cation (Py+; C16H10˙+) and its doubly-dehydrogenated cation (ddPy+; C16H8˙+) recorded using the neon-tagging messenger technique. We explore the dehydrogenation process resulting in ddPy+ and, with the aid of theoretical computations, propose the most likely mixture of isomeric candidates that constitute the ddPy+ sample.
2 Methods
2.1 Experimental
The infrared spectra of neon-tagged pyrene (Ne⋯C16H10˙+, m/z = 222) and doubly-dehydrogenated pyrene (Ne⋯C16H8˙+, m/z = 220) were recorded using the cryogenic 22-pole ion trap instrument FELion at the Free Electron Laser for Infrared Experiments Laboratory (FELIX, Radboud University).33 A detailed description of the FELion end-station is provided elsewhere,26 and here only a brief description of the apparatus and information specific to the target ions is given. One major advantage of this method, compared to the previously applied IRMPD, is that intensity ratios of recorded spectra do not suffer from a highly non-linear multi-photon response and allow for better comparisons with theoretical predictions.A solid pyrene sample (Sigma-Aldrich, 99%) was heated to reach a vapour pressure of 1–2 × 10−5 mbar and dissociatively ionised by electron impact ionisation at electron energies of 20–40 eV in an ion source. A 100 ms long pulse of the resulting cations (Py+ or ddPy+) was extracted from the source and filtered for the mass of interest by a first quadrupole mass selector (mass spectrum given in Fig. S1, ESI†). The ions were guided into the 22-pole ion trap which was maintained at a fixed temperature of 6.5 K by means of a closed-cycle helium refrigerator. They were then cooled close to the ambient temperature by a 140 ms long 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 Ne:He gas mixture pulse at a number density of ∼1015 cm−3 provided by a pulsed piezo valve. The buffer gas inlet was triggered 20 ms before the ions were admitted into the trap, and lasted for 20 ms after the ion pulse. Under these conditions, around 10% of the primary ions (Py+ or ddPy+) formed weakly bound complexes with neon.
:3 Ne:He gas mixture pulse at a number density of ∼1015 cm−3 provided by a pulsed piezo valve. The buffer gas inlet was triggered 20 ms before the ions were admitted into the trap, and lasted for 20 ms after the ion pulse. Under these conditions, around 10% of the primary ions (Py+ or ddPy+) formed weakly bound complexes with neon.
The so-formed complexes were typically stored in the trap for 1.6 s, and during this time irradiated by several laser pulses of the defocused IR radiation provided by the free electron laser FEL-2 operated in the 5.6–18 m (550–1800 cm−1) range at a repetition rate of 10 Hz. FEL-2 delivered up to 30 mJ per macropulse into the 22-pole trap, and the FEL was optimised for narrow bandwidths of full-width-at-half-maximum (FWHM) of 0.5–1% of the used wavelength.
An IRPD spectrum was recorded by mass-selecting and counting the target PAH–neon complex ions and varying the laser wavelength. The absorption of a resonant IR photon caused the complex to dissociate and led to a depletion in the number of complex ions compared to the baseline number observed off-resonance. To account for saturation, varying baseline, laser pulse energy, and pulse numbers, the signal was normalised prior to averaging, yielding the intensity in units of relative cross section per photon. Three iterations were averaged for every data point and the spectra were binned to a resolution of 1 cm−1.
2.2 Theoretical
The density functional theory (DFT) quartic force field of Py+ was obtained with the Gaussian1634 software package. The B3LYP35,36 hybrid functional was used in conjunction with the polarized double-ζ basis set N07D,37 which has been established to work well for open-shell systems.38 The resulting calculated force field was supplied as an input for SPECTRO 8 to determine the anharmonic spectrum of the molecule.18Vibrational harmonic spectra of all possible ddPy+ isomers were predicted by DFT computations. Geometry optimisations were performed at the B3LYP/6-311++G(2d,p) level of theory and the calculated harmonic frequencies were uniformly scaled by 0.983 to account for anharmonicity.39 This empirically-determined scaling factor is in-line with the suggested scaling factor of 0.982 for PAH IR spectra computed with the chosen basis set at the B3LYP level of theory.40 All modes were convolved using a Gaussian profile with FWHM of 8 cm−1 to facilitate comparison with the experimental spectra.
The B3LYP/6-311++G(2,dp) level of theory was chosen as it is particularly well-suited to vibrational frequency studies. For consistency, we also explored the potential energy surfaces connecting the ddPy+ isomers using the same level of theory. As this functional is not optimised for the study of reaction energies and barriers, we have repeated these calculations using the more accurate range-separated functional wB97XD with the 6-311++G(2d,p) basis set for several key isomers and transition states.
3 Results and discussion
3.1 Pyrene cation, Py+
The black trace in Fig. 2 shows the IRPD spectrum recorded for neon-tagged Py+. For comparison, the theoretical anharmonic and harmonic spectra convolved with a Gaussian profile at a FWHM of 8 cm−1 are plotted in blue and pink, respectively (Fig. 2). The calculated band positions are represented by sticks. The experimental spectrum obtained in this work represents a clear improvement on the gas-phase IRMPD spectrum of Py+ available from literature,23 showing better signal-to-noise ratio, narrower linewidths, and well-resolved features. Peak positions and band assignments are listed in Table 1 along with unpublished gas-phase IRMPD data recorded by our group (experimental details58 and data are provided in Fig. S2 and S3, ESI†), and matrix data available from the literature.32 The band positions derived here are consistent with those from the IRMPD study, and improved FWHM values as low as 4 cm−1 are realised for isolated absorptions.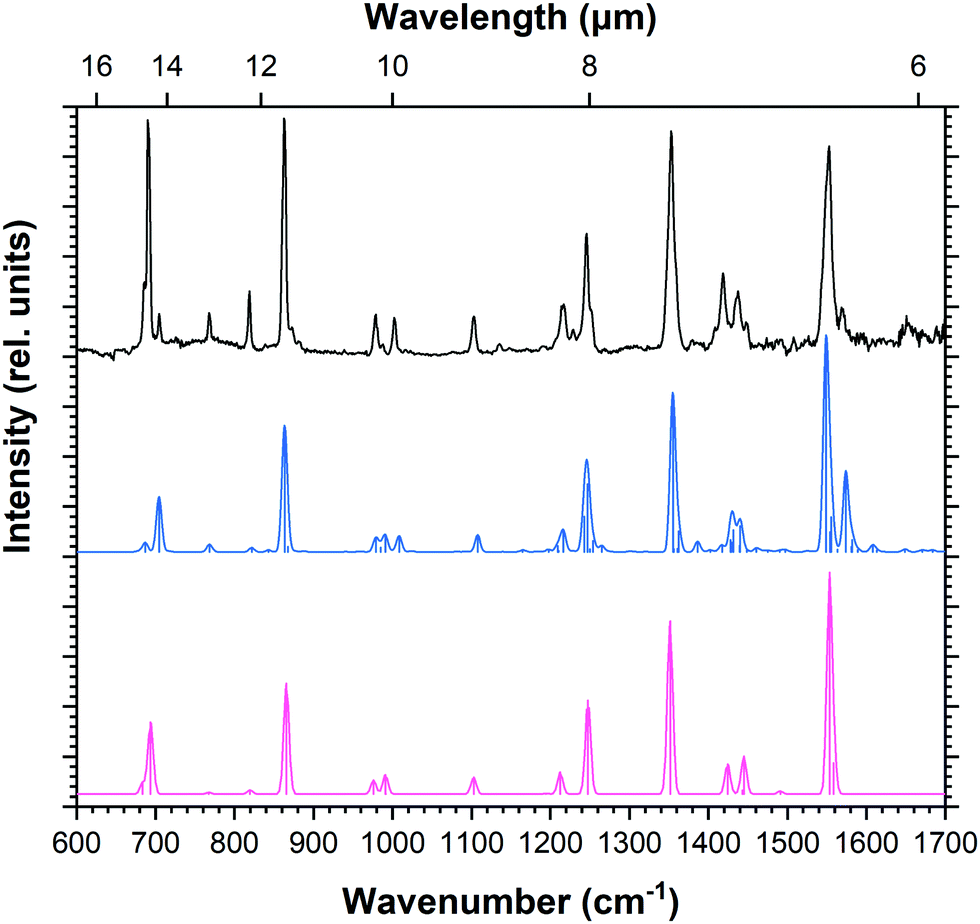 | ||
| Fig. 2 IRPD spectrum of Py+ (black trace) with the calculated anharmonic spectrum (blue), and the calculated harmonic spectrum scaled by 0.983 (pink). | ||
a
| IRPD (this work) | IRMPDb | Argon matrixc | Emissiond | Harm. DFTe | Anh. DFT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ν vib | FWHM | I rel | ν vib | I rel | ν vib | I rel | ν vib | I rel | ν vib | I abs | ν vib | I abs |
| a Frequencies are given in cm−1 and calculated intensities (Iabs) are given in km mol−1. In the case of anharmonic calculations, only modes with relative intensities higher than 3% are reported. b See Fig. S2 and S3, ESI. c Hudgins et al.32 d Kim et al.31 e No frequency scaling is used. | ||||||||||||
| 686 | 8.7 | 0.58 | 694 | 7.8 | 687 | 7.1 | ||||||
| 691 | 3.4 | 1.00 | 680 | 0.15 | 690 | 0.23 | 691 | 43.3 | 704 | 43.3 | ||
| 705 | 4.6 | 0.11 | ||||||||||
| 768 | 3.9 | 0.13 | 777 | 6.0 | 768 | 5.7 | ||||||
| 819 | 3.5 | 0.24 | 822 | 3.3 | ||||||||
| 843 | 1.5 | |||||||||||
| 864 | 5.1 | 0.95 | 854 | 0.56 | 861 | 0.27 | 871 | 102.8 | 863 | 97.9 | ||
| 872 | 10.0 | 0.11 | 868 | 0.04 | 868 | 4.1 | ||||||
| 954 | 0.07 | |||||||||||
| 978 | 6.0 | 0.95 | 989 | 0.13 | 977 | 0.10 | 993 | 11.1 | 979 | 10.8 | ||
| 987 | 4.3 | 0.24 | 985 | 3.4 | ||||||||
| 991 | 13.0 | |||||||||||
| 1002 | 5.2 | 0.15 | 1001 | 14.6 | 1008 | 12.3 | ||||||
| 1103 | 5.7 | 0.16 | 1095 | 0.05 | 1102 | 0.09 | 1123 | 13.5 | 1108 | 13.3 | ||
| 1136 | 7.5 | 0.15 | ||||||||||
| 1164 | 1.2 | |||||||||||
| 1212 | 0.63 | 1189 | 0.04 | 1189 | 0.01 | 1198 | 2.0 | |||||
| 1210 | 10.4 | 0.05 | 1209 | 4.6 | ||||||||
| 1216 | 7.3 | 0.18 | 1216 | 0.13 | 1209 | 0.10 | 1237 | 17.4 | 1216 | 16.9 | ||
| 1229 | 10.9 | 0.07 | 1235 | 1.00 | 1229 | 6.9 | ||||||
| 1244 | 12.7 | 0.83 | 1245 | 0.38 | 1243 | 0.18 | 1243 | 27.8 | ||||
| 1246 | 8.8 | 0.44 | 1247 | 53.4 | ||||||||
| 1250 | 2.1 | |||||||||||
| 1252 | 0.51 | 1255 | 0.05 | 1265 | 75.9 | 1254 | 8.8 | |||||
| 1345 | 1.25 | 1265 | 5.0 | |||||||||
| 1353 | 10.2 | 0.86 | 1378 | 0.49 | 1357 | 1.12 | 1343 | 1.20 | 1385 | 138.3 | 1355 | 121.0 |
| 1356 | 2.1 | |||||||||||
| 1361 | 2.6 | |||||||||||
| 1362 | 0.18 | 1366 | 0.22 | 1362 | 16.4 | |||||||
| 1386 | 8.1 | |||||||||||
| 1408 | 5.8 | 0.07 | 1405 | 0.65 | 1403 | 0.53 | 1402 | 1.2 | ||||
| 1419 | 8.9 | 0.29 | 1421 | 0.18 | 1418 | 0.46 | 1418 | 5.0 | ||||
| 1426 | 0.21 | 1428 | 9.3 | |||||||||
| 1437 | 12.0 | 0.22 | 1440 | 0.11 | 1429 | 28.6 | ||||||
| 1438 | 5.0 | |||||||||||
| 1449 | 4.5 | 0.10 | 1467 | 27.0 | 1440 | 20.4 | ||||||
| 1461 | 3.0 | |||||||||||
| 1475 | 1.2 | |||||||||||
| 1490 | 1.4 | |||||||||||
| 1497 | 1.5 | |||||||||||
| 1551 | 9.7 | 0.82 | 1538 | 1.00 | 1553 | 1.00 | 1553 | 1.00 | 1586 | 181.4 | 1549 | 159.1 |
| 1554 | 15.7 | |||||||||||
| 1555 | 4.8 | |||||||||||
| 1555 | 27.4 | |||||||||||
| 1564 | 2.0 | |||||||||||
| 1570 | 9.7 | 0.14 | 1591 | 28.6 | 1574 | 62.6 | ||||||
| 1581 | 4.0 | |||||||||||
| 1582 | 9.5 | |||||||||||
| 1589 | 1.7 | |||||||||||
| 1608 | 5.1 | |||||||||||
| 1613 | 1.3 | |||||||||||
| 1649 | 1.9 | |||||||||||
| 1671 | 1.2 | |||||||||||
| 1684 | 1.4 | |||||||||||
IRMPD is a non-linear process in which several photons are required to reach the dissociation threshold. The IRPD technique is a linear process where only one photon is needed to dissociate the complex, since the binding energy of the neon atom is typically only a few hundred wavenumbers. This allows weaker bands to be detected. Most notably, the C–H in-plane bending modes at 978 and 1103 cm−1 are prominent in the IRPD spectrum whereas in the IRMPD spectrum only the former could be observed as a weak band. Similarly for the C–H out-of-plane bending modes; while only the bands at 690 and 861 cm−1 were observed in the IRMPD data, additional bands were recorded in the IRPD spectrum. The band positions appearing in our IRPD and previous argon matrix data show excellent agreement (deviations ≤4 cm−1).32
DFT computations for Py+ resulted in a 2B1g electronic ground state with a D2h structure. Band positions resulting from (unscaled) harmonic and anharmonic DFT calculations are listed in Table 1. All fundamental and combination modes (νi + νj) with relative intensities higher than 3% are reported. The spectroscopic assignment of all IRPD bands to calculated anharmonic frequencies is straightforward. The agreement between the anharmonic frequencies and the experimental data is excellent despite a minor blue-shift of 2–10 cm−1 in the calculations. While the two calculated spectra appear remarkably similar after scaling the harmonic spectra by an empirically-determined factor of 0.983, the anharmonic calculations reveal weak bands (that are absent in the harmonic calculations) which contribute to most features such as the 1408 cm−1 band due to the combination of ν53 (704 cm−1) and ν54 (687 cm−1). The convolved line shapes for the harmonic and anharmonic spectra are similar enough that one can draw an equally convincing match to the observed spectrum from either calculation.
The recorded vibrational spectra presented in this study show no unaccounted bands, leading us to the conclusion that no other electronically-excited states are present. The precise effect of neon-coordination on the IRPD spectrum of Py+ is unknown but is expected to be small (a few cm−1).41 Theoretical calculations of the various neon tag coordinations are, given the good match between the DFT calculated spectrum of the untagged pyrene cation and the experimental data, out of the scope of this work.
3.2 Doubly-dehydrogenated pyrene cation, ddPy+
IRPD spectroscopy was also employed to record the mid-IR spectrum of the doubly-dehydrogenated pyrene radical cation (Fig. 3). There are a greater number of features, several of which appear broader than those recorded for Py+. The doubly-dehydrogenated form of pyrene has many more active modes than the fully hydrogenated pyrene cation due to the loss of symmetry. This leads to apparently broader structures that in reality consist of multiple overlapping bands. The spectrum also shows greater variation between the recorded and predicted intensities of the bands when compared to the pyrene molecule, as will be discussed later. This is likely a result of saturation of the features which causes weaker modes to appear more intense. The spectrum is fitted using multiple Gaussian components with typical FWHM values of 10 cm−1, giving a total of 39 bands listed in Table 3.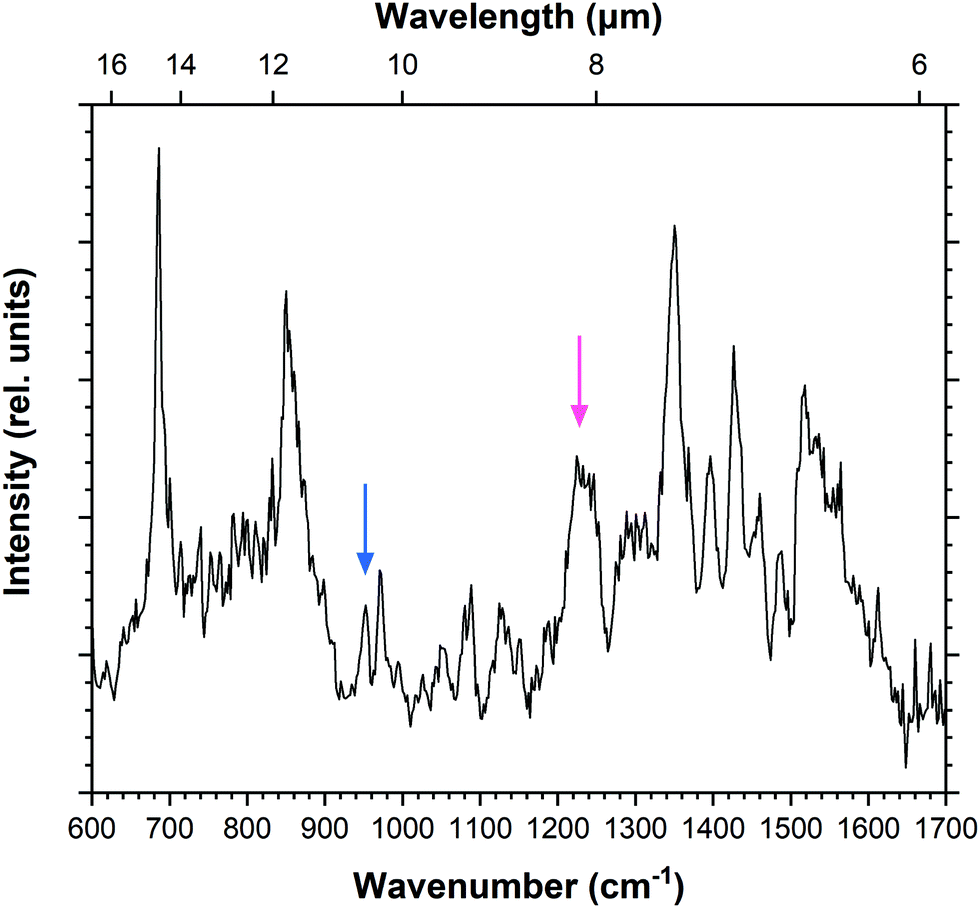 | ||
| Fig. 3 IRPD spectrum of ddPy+. Arrows highlight bands at 952 cm−1 (blue) and 1245 cm−1 (pink) which are relevant for saturation–depletion measurements. | ||
In order to assign the experimental vibrational spectrum of the ddPy+ species, DFT structure optimisations and harmonic frequency calculations were performed on fourteen possible isomers in the doublet state using the B3LYP/6-311++G(2d,p) level of theory. The relative energies of the fourteen ddPy+ isomers are listed in Table 2. The good agreement in peak frequency between the shifted harmonic DFT calculations for Py+ gives confidence that scaled harmonic calculations for ddPy+ will suffice. While all isomers with π14⋯π1σ1σ1 configuration have approximately the same relative energies (0.14 eV range), the most stable isomers are the three with a π14⋯π1σ2 configuration: 4,5-ddPy+, 1,2-ddPy+, and 1,3-ddPy+ (shown in Fig. 4). The numbers here refer to the positions where the H-atoms are missing (see Fig. 1). Further analysis using the range-separated wB97XD/6-311++G(2d,p) level of theory reveals that the relative energies of these species are essentially identical (see Table S0, ESI†).
| Isomer | Config. (π14⋯) | EGS | Symm. | ΔEb |
|---|---|---|---|---|
| a Isomers are sorted in decreasing order of stability with hydrogen-loss sites numbered according to Fig. 1. b Energy difference with respect to the most stable isomer, 4,5-ddPy+. | ||||
| 4,5-ddPy+ | π1σ2 | 2B1 | C 2v | 0.00 |
| 1,2-ddPy+ | π1σ2 | 2A′′ | C s | 0.05 |
| 1,3-ddPy+ | π1σ2 | 2A2 | C 2v | 0.28 |
| 1,5-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.08 |
| 1,8-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.11 |
| 2,7-ddPy+ | π1σ1σ1 | 2Au | D 2h | 1.11 |
| 2,4-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.14 |
| 2,5-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.14 |
| 1,4-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.15 |
| 1,7-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.16 |
| 4,9-ddPy+ | π1σ1σ1 | 2Au | C 2h | 1.16 |
| 4,10-ddPy+ | π1σ1σ1 | 2B1 | C 2v | 1.17 |
| 1,9-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.19 |
| 1,10-ddPy+ | π1σ1σ1 | 2A′′ | C s | 1.22 |
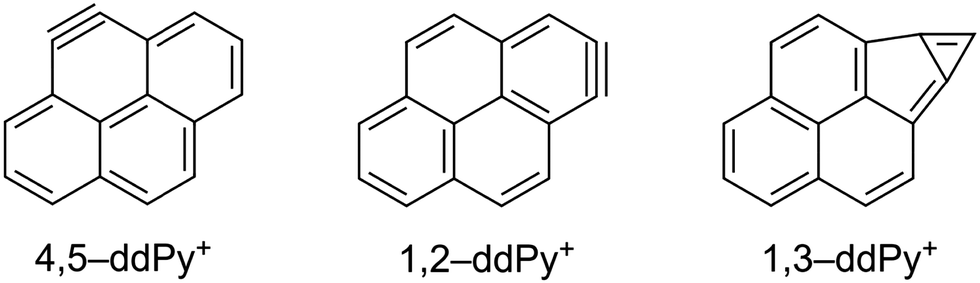 | ||
| Fig. 4 Chemical structures of 4,5-ddPy+, 1,2-ddPy+, and 1,3-ddPy+. | ||
For these three isomers, two hydrogen atoms are removed from adjacent or nearby carbon atoms. In the case of hydrogen removal from adjacent carbon atoms (4,5-ddPy+ and 1,2-ddPy+), the dehydrogenated carbons become sp-hybridised and an additional π-bond is formed, creating a triple bond as the lowest energy structure in a doublet state. With 1,3-ddPy+, all carbon atoms remain sp2-hybridised and an additional σ-bond is formed between carbons 1 and 3, transforming a 6-membered carbon ring into a cyclopropenyl unit (a bicyclic structure with a 5-membered ring and a 3-membered ring) which has been shown to stabilise cationic molecules.42 As an aside, we note that this latter species is even better stabilised as a dicationic molecule with charges delocalised separately in a phenalene cation and a cyclopropenyl cation.42
Computed vibrational spectra of the six most stable ddPy+ isomers plotted together with the experimental ddPy+ spectrum are shown in Fig. 5. The calculated vibrational transitions of these isomers are listed in Table 3 along with the experimentally recorded ddPy+ absorption bands. A scaling factor of 0.983 that was derived from the Py+ spectrum is applied to the computations. Because of the structural similarity of the isomers, they generally exhibit only small changes between the positions and relative intensities of the main transitions.
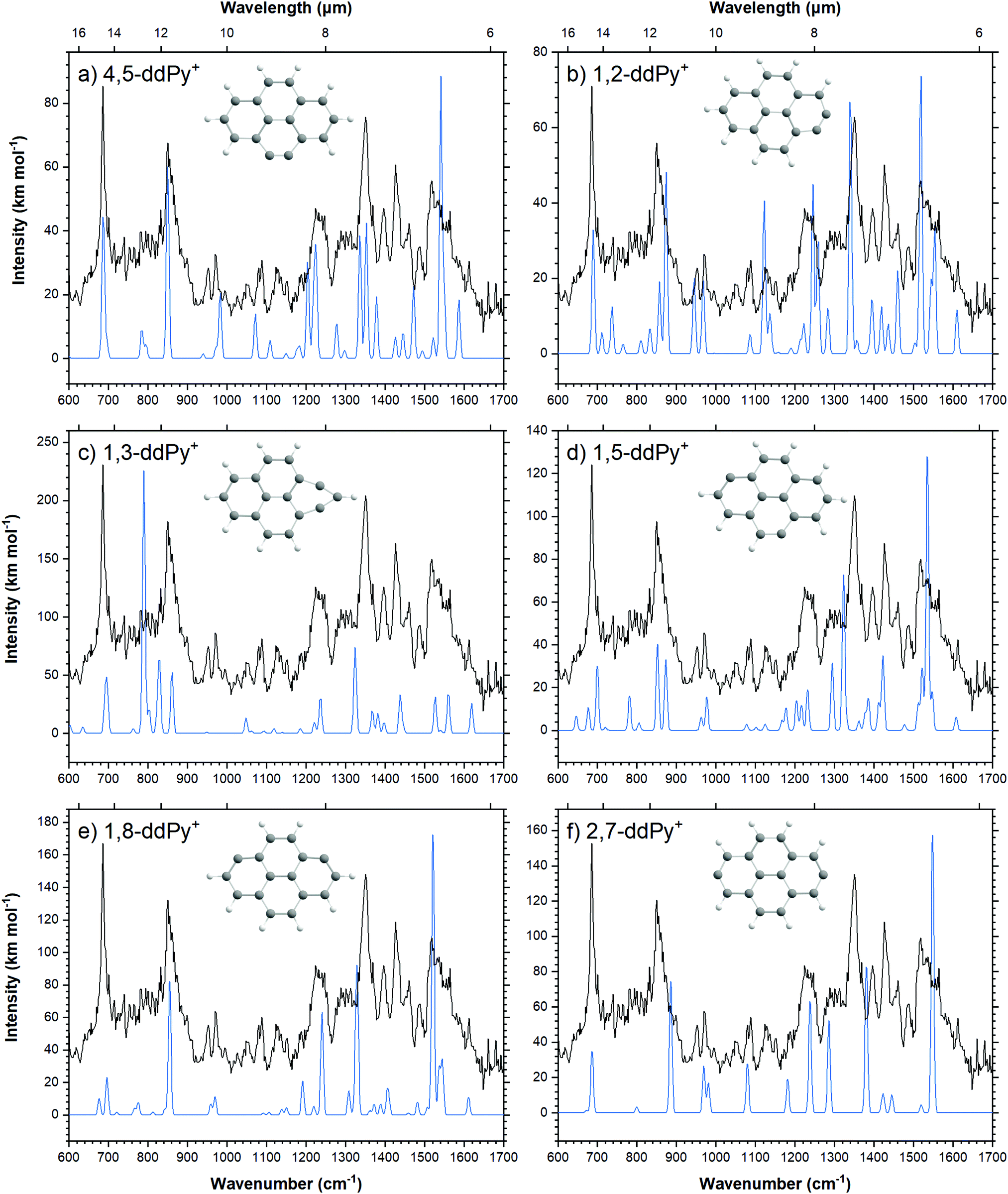 | ||
| Fig. 5 Calculated spectra (blue) of the six most stable isomers of ddPy+ (see Table 3) performed with B3LYP/6-311++G(2d,p), scaled by 0.983. The recorded spectrum is shown in black. | ||
| IRPD (this work) | DFT calculations | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4,5-ddPy+ | 1,2-ddPy+ | 1,3-ddPy+ | 1,5-ddPy+ | 1,8-ddPy+ | 2,7-ddPy+ | |||||||||
| ν vib | FWHM | I rel | ν vib | I abs | ν vib | I abs | ν vib | I abs | ν vib | I abs | ν vib | I abs | ν vib | I abs |
| a Frequencies (νvib and FWHM) are given in cm−1 and calculated intensities (Iabs) are given in km mol−1. | ||||||||||||||
| 580 | 4.5 | 0.10 | ||||||||||||
| 586 | 3.1 | 0.11 | 591 | 5.5 | ||||||||||
| 600 | 5.2 | 0.12 | 599 | 1.0 | 595 | 2.6 | 602 | 7.1 | ||||||
| 686 | 8.9 | 1.00 | 686 | 44.3 | 688 | 32.9 | 686 | 34.7 | ||||||
| 700 | 9.7 | 0.36 | 695 | 48.3 | 700 | 23.0 | ||||||||
| 714 | 11.6 | 0.23 | 711 | 5.7 | ||||||||||
| 730 | 16.0 | 0.19 | 737 | 12.5 | ||||||||||
| 764 | 4.2 | 0.09 | 763 | 2.3 | 764 | 3.8 | ||||||||
| 782 | 5.2 | 0.16 | 786 | 8.6 | 781 | 16.1 | ||||||||
| 794 | 10.4 | 0.17 | 794 | 4.4 | ||||||||||
| 800 | 3.6 | 0.10 | 797 | 3.5 | 800 | 3.4 | ||||||||
| 810 | 13.9 | 0.22 | 808 | 3.4 | ||||||||||
| 828 | 9.7 | 0.16 | 829 | 63.0 | ||||||||||
| 832 | 2.5 | 0.13 | 834 | 6.6 | ||||||||||
| 850 | 20.3 | 0.88 | 849 | 60.2 | 857 | 19.1 | 852 | 40.2 | ||||||
| 874 | 9.7 | 0.33 | 874 | 48.2 | 873 | 32.8 | ||||||||
| 954 | 12.4 | 0.16 | 946 | 20.0 | 958 | 6.8 | ||||||||
| 970 | 9.1 | 0.25 | 968 | 19.3 | 977 | 15.5 | 970 | 11.1 | 969 | 26.5 | ||||
| 1054 | 17.3 | 0.09 | 1048 | 13.2 | ||||||||||
| 1080 | 11.4 | 0.17 | 1071 | 14.0 | ||||||||||
| 1088 | 6.7 | 0.20 | 1085 | 5.2 | 1090 | 1.8 | ||||||||
| 1129 | 9.6 | 0.16 | 1122 | 40.6 | 1125 | 2.9 | ||||||||
| 1150 | 8.5 | 0.13 | 1149 | 1.6 | 1150 | 4.4 | ||||||||
| 1188 | 14.1 | 0.13 | 1184 | 4.0 | 1186 | 4.1 | 1191 | 21.1 | ||||||
| 1225 | 23.4 | 0.52 | 1224 | 35.8 | 1222 | 8.0 | 1220 | 9.3 | ||||||
| 1252 | 15.7 | 0.48 | 1245 | 45.0 | ||||||||||
| 1350 | 14.4 | 0.83 | 1352 | 42.5 | ||||||||||
| 1371 | 12.9 | 0.45 | 1378 | 19.3 | ||||||||||
| 1396 | 16.6 | 0.45 | 1393 | 14.3 | 1395 | 8.2 | ||||||||
| 1426 | 12.4 | 0.69 | 1427 | 6.7 | 1419 | 12.6 | 1422 | 35.1 | 1422 | 10.8 | ||||
| 1430 | 5.7 | 0.46 | 1436 | 7.89 | ||||||||||
| 1460 | 7.1 | 0.38 | 1459 | 22.1 | ||||||||||
| 1486 | 11.2 | 0.21 | 1472 | 22.5 | 1473 | 22.5 | 1482 | 7.8 | ||||||
| 1496 | 1.7 | 0.14 | 1492 | 2.3 | 1512 | 12.6 | ||||||||
| 1516 | 15.2 | 0.64 | 1521 | 6.5 | 1519 | 73.7 | 1522 | 29.2 | 1520 | 172.5 | 1519 | 4.5 | ||
| 1539 | 20.4 | 0.57 | 1541 | 88.5 | 1544 | 19.8 | 1536 | 28.3 | ||||||
| 1561 | 14.4 | 0.50 | 1559 | 33.1 | ||||||||||
| 1588 | 13.9 | 0.25 | 1586 | 18.5 | 1587 | 18.4 | ||||||||
| 1613 | 6.0 | 0.23 | 1610 | 11.6 | 1619 | 25.9 | 1610 | 10.6 | ||||||
For all fourteen considered ddPy+ isomers, the computed IR spectra show strong absorptions at the characteristic C–H out-of-plane bending modes at ∼680 and ∼840 cm−1. The C–H in-plane bending modes at ∼1200 and ∼1300 cm−1 are also visible, as is the strong, broad C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ring stretch at ∼1500 cm−1. 1,2-ddPy+ has a strong C–H in-plane bending mode at 1140 cm−1 that the other isomers do not exhibit. Likewise, 1,3-ddPy+ has a unique transition at 790 cm−1 originating from an in-plane deformation mode involving the triangular structure formed by the carbon bonds. This is the strongest transition for this isomer. A similar bicyclic structure was determined as the most stable isomer for the C6H4+ radical (m-benzyne) cation with CASPT2, and DFT, CCSD(T) and MP2 calculations,43,44 supporting that the cyclopropenyl deformation is real and not simply an artefact of the computational methods used. 1,3-ddPy+ was also investigated with the BP86 and wB97XD functionals using the same 6-311++G(2d,p) basis set to ensure that the 790 cm−1 band was real. The corresponding band appears at 721 cm−1 when scaled by 0.983 for BP86, and 864 cm−1 when scaled by an empirical scaling factor of 0.973 (see Fig. S6, ESI†) for wB97XD. This underlines that the calculated band position of the cyclopropenyl vibrational mode is rather sensitive to the choice of computational method (with a shift of 69 cm−1 in B3LYP vs. BP86, and 74 cm−1 in B3LYP vs. wB97XD), but all methods agree that this mode should have a very strong intensity. It follows that the 1,3-ddPy+ isomer would produce a single, very intense band in the vicinity of 864 cm−1, however such a band is conspicuously absent over the relevant frequency range (721–864 cm−1) indicated by these different calculations. Therefore, the absence of this distinct band implies that 1,3-ddPy+ may not be an abundant species in our ddPy+ spectrum, which hints at even lower abundances of isomers with higher energy configurations.
C ring stretch at ∼1500 cm−1. 1,2-ddPy+ has a strong C–H in-plane bending mode at 1140 cm−1 that the other isomers do not exhibit. Likewise, 1,3-ddPy+ has a unique transition at 790 cm−1 originating from an in-plane deformation mode involving the triangular structure formed by the carbon bonds. This is the strongest transition for this isomer. A similar bicyclic structure was determined as the most stable isomer for the C6H4+ radical (m-benzyne) cation with CASPT2, and DFT, CCSD(T) and MP2 calculations,43,44 supporting that the cyclopropenyl deformation is real and not simply an artefact of the computational methods used. 1,3-ddPy+ was also investigated with the BP86 and wB97XD functionals using the same 6-311++G(2d,p) basis set to ensure that the 790 cm−1 band was real. The corresponding band appears at 721 cm−1 when scaled by 0.983 for BP86, and 864 cm−1 when scaled by an empirical scaling factor of 0.973 (see Fig. S6, ESI†) for wB97XD. This underlines that the calculated band position of the cyclopropenyl vibrational mode is rather sensitive to the choice of computational method (with a shift of 69 cm−1 in B3LYP vs. BP86, and 74 cm−1 in B3LYP vs. wB97XD), but all methods agree that this mode should have a very strong intensity. It follows that the 1,3-ddPy+ isomer would produce a single, very intense band in the vicinity of 864 cm−1, however such a band is conspicuously absent over the relevant frequency range (721–864 cm−1) indicated by these different calculations. Therefore, the absence of this distinct band implies that 1,3-ddPy+ may not be an abundant species in our ddPy+ spectrum, which hints at even lower abundances of isomers with higher energy configurations.
On the basis of the theoretical isomeric spectra it is evident that no single isomer is capable of explaining the full measured ddPy+ spectrum, and instead a mixture of isomers is required. Three possible mixtures of isomers were considered and compared with the experimental spectrum to gain further insight:
(A) A 1:1 mixture of 4,5-ddPy+:1,2-ddPy+
(B) A 1:1:0.5 mixture of 4,5-ddPy+:1,2-ddPy+:1,3-ddPy+
(C) A homogeneous mixture of all isomers listed in Table 2
A visual comparison of these mixtures is given in Fig. S7–S9 in the ESI.† In general, all three mixtures show a very comparable and rich spectrum and all of the calculated bands show counterparts in the recorded spectrum except for the strong band in the 721–864 cm−1 range. This band, coming exclusively from the 1,3-ddPy+ isomer, is clearly visible in an unweighted mixture of all fourteen isomers as well as the weighted 1:1:0.5 mixture with the three lowest energy isomers, but is not present in the measured spectrum. Although the 1:1:0.5 mixture reproduces the experimental spectrum well, suggesting that a 20% contribution of 1,3-ddPy+ is plausible, the 1:1 mixture of only 4,5-ddPy+ and 1,2-ddPy+ (Fig. 6) provides a better match to the measured spectrum given the absence of the 721–864 cm−1 band. Given that all remaining isomers are relatively much higher in energy, it is reasonable to assume that they do not substantially contribute to the overall isomeric mixture.
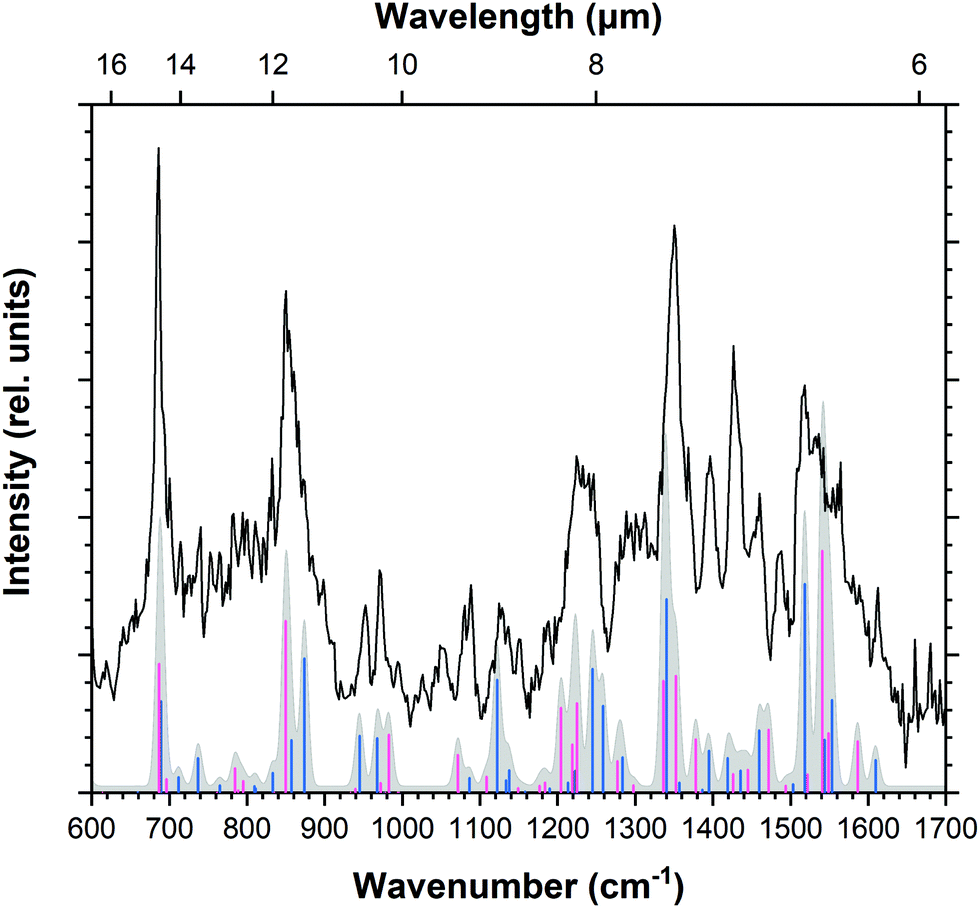 | ||
| Fig. 6 IRPD spectrum of ddPy+ (black trace) with B3LYP/6-311++G(2d,p) calculated spectra of 4,5-ddPy+ (pink sticks) and 1,2-ddPy+ (blue sticks) plotted over the Gaussian convolved spectrum (FWHM of 8 cm−1) of the 1:1 mixture (A) of both isomers (grey). | ||
Close inspection of the spectra reveals that, in mixture A, the relative intensities of the bands between 1200 and 1300 cm−1 corresponding to the C–H in-plane bending modes more closely follow those observed in the recorded spectrum. This mixture has clear counterparts to all the observed bands in the recorded spectrum. Mixtures B and C show a transition present at 1128 cm−1. DFT calculations predict this as a C–H in-plane bending mode for 1,2-ddPy+. This transition is almost entirely imperceptible in mixture C, reflecting the low abundance of this isomer in this mixture. Its detection in the experimental spectrum suggests that, at the very least, 1,2-ddPy+ must be a dominant isomer in the ion mixture.
A complementary saturation–depletion measurement was conducted to attempt quantification of branching over the various ddPy+ isomers. For this, the relative peak depletion signal is recorded as a function of deposited energy (i.e. number of laser pulses applied to the ion cloud) at a wavelength coincident with the centre of an isomer-specific vibrational band. The relative proportion of ion counts at a resonant frequency and an off-resonant frequency reveals the fraction of absorbing ions at this wavelength. The ion counts are determined at the same storing time and laser power to account for the effects of ion losses from the trap by non-radiative processes as well as heating of the ion trap by laser irradiation. The energy can be varied by changing the irradiation time (equivalent to the storage time in the trap, which can be extended to tens of seconds) and/or the light intensity. A similar method has been described elsewhere.26,27,45
Guided by scaled harmonic DFT calculated modes, a vibrational band unique to both 4,5-ddPy+ and 1,2-ddPy+ was chosen as the target for the saturation–depletion study. The resulting scan and its analysis are given in Fig. S10, ESI.† The band at 1245 cm−1 (indicated with a pink arrow in Fig. 3) was predicted to originate from the C–H in-plane modes of both isomers, and showed a maximum saturation of 80%, i.e.4,5-ddPy+ and 1,2-ddPy+ make up a total fraction of 80% of the ion population (as shown in the ESI†).
An additional scan was then carried out over the C–H out-of-plane mode over 952 cm−1 (marked by a blue arrow in Fig. 3), a band characteristic to the 1,2-ddPy+ isomer. This scan was performed using a high laser power (15 mJ per pulse) and trapping the target ions for longer (5.7 s). At this high power, the laser saturates the band completely, allowing the percentage of the isomer that is present in the mixture to be determined. The band was saturated to 40%, revealing the relative proportion of the 1,2-ddPy+ isomer. This, in combination with the saturation–depletion study, is consistent with the conclusion that the 4,5-ddPy+ and 1,2-ddPy+ dominate at equal abundances in the isomeric mixture. This reflects the importance of kinetics in the fragmentation process.
The loss of two hydrogen atoms upon ionisation of pyrene occurs after the migration of one hydrogen atom to its neighbouring carbon, creating an aliphatic CH2 group at this site.21,46 Following this, the two hydrogens are lost either sequentially (H + H) or as molecular hydrogen (H2). The bond dissociation energy for the first hydrogen atom removal was found to be 4.8 eV, regardless of its position.21 For the second dehydrogenation step, 3.7 eV is required to remove the hydrogen atom from an adjacent position, and 4.8 eV for all other positions. This decrease in energy for the second hydrogen-loss from an adjacent position is due to the spin pairing of the involved carbon atoms, which creates a triple bond to stabilise the lowest energy structure. This complements the results from other studies well.21,47 More energy is required for the hydrogen to roam to non-adjacent carbon atoms, thus limiting the double dehydrogenation process to form predominantly 4,5-ddPy+ and 1,2-ddPy+. This can be qualitatively understood as reflecting the formation of a new bond in these isomers. 1,3-ddPy+ can form when the hydrogen at position 1 (Fig. 1) moves to position 2 and then position 3, creating a CH2 unit which then undergoes double-dehydrogenation. A CH2 unit is also formed when the hydrogen is at position 2. Dehydrogenation at this stage is energetically more favourable than roaming further to position 3, leading to the formation of 1,2-ddPy+ instead. Experiments have also shown that the barrier for a hydrogen atom to roam from one ring to another (via a tertiary carbon) is much higher than to roam within a ring.21 The measured IR spectrum is consistent with these theory-based expectations that the 4,5-ddPy+ and 1,2-ddPy+ species will dominate the mixture, while the 1,3-ddPy+ isomer is at most a minor contributor.
To elucidate the dissociation process and the resulting isomeric distribution, the potential energy surface (PES) connecting the isomers was explored (Fig. 7). The geometries of all transition and intermediate states along with the wB97XD/6-311++G(2d,p) values of the most stable isomers and the transition states connecting these are available in Fig. S11 and S12, ESI.† It should be noted that following the loss of the second hydrogen atom, ddPy+ may still have enough internal energy to allow for hydrogen-roaming, offering pathways for other isomers to form as well. This scrambling is affected by the barriers involved, with the lowest barrier (between 1,2-ddPy+ and 1,3-ddPy+) (TS8, Fig. 7) being 2.90 eV at the B3LYP/6-311++G(2d,p) level. This barrier is slightly lower at the wB97XD/6-311++G(2d,p) level (see Table S0, ESI†). All other roaming transitions have much larger barriers as movement of hydrogen from one ring to the next involves bridging a carbon atom: 3.45 eV (TS1, Fig. 7) and 3.86 eV (TS9, Fig. 7). Since the energy of the ionising electrons (20–40 eV) is well above the appearance energy for this system, in principle, all barriers can be easily overcome if all of this energy were transferred.48 However, the recorded spectra suggest that only 20% of the ion population may exist as isomers other than 4,5-ddPy+ and 1,2-ddPy+. Therefore we conclude that after the formation of these two isomers, the remaining internal energy in ddPy+ does not allow for efficient hydrogen-roaming.
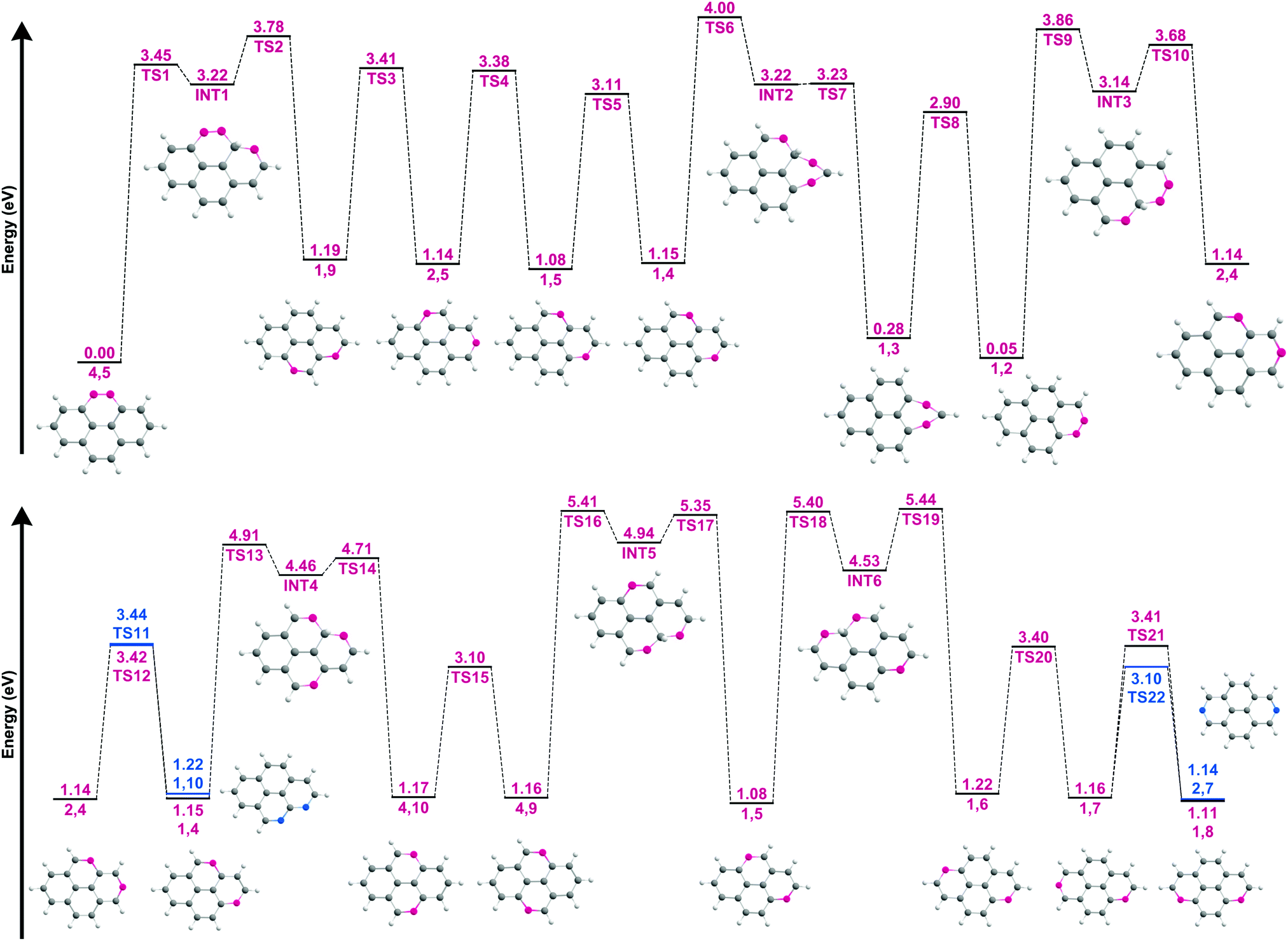 | ||
| Fig. 7 Potential energy surface for the hydrogen walk between the fourteen ddPy+ isomers listed in Table 2. The isomers are characterised by their missing hydrogen atoms (shown in pink and blue) using the labelling system shown in Fig. 1. The corresponding structures for the isomers and their intermediate states (INT) are shown. Energies are given with respect to that of the most stable isomer (4,5-ddPy+). Transition states are labelled as TS. Blue is used to distinguish pathways to an additional isomer from a single species. | ||
From a spectroscopic perspective, the reduced intensity around the highly characteristic band of 1,3-ddPy+ at 790 cm−1 points to a low abundance of this isomer. This, coupled with the energetics of the dissociation process (which favours the loss of directly adjacent hydrogens), is fully consistent with a picture in which the ddPy+ mixture is dominated by 4,5-ddPy+ and 1,2-ddPy+.
4 Astronomical implications
The recorded ddPy+ spectrum extends the existing literature. While there is no strong signature of dehydrogenated PAHs in the studied frequency range of this work, we expect that the characteristic C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch may allow unambiguous detection of partially dehydrogenated PAHs in space. Unscaled harmonic DFT calculations predict this mode at 2026 cm−1 for 4,5-ddPy+ and 1945 cm−1 for 1,2-ddPy+. Future studies would benefit from exploring this mode in further detail.
C stretch may allow unambiguous detection of partially dehydrogenated PAHs in space. Unscaled harmonic DFT calculations predict this mode at 2026 cm−1 for 4,5-ddPy+ and 1945 cm−1 for 1,2-ddPy+. Future studies would benefit from exploring this mode in further detail.
The present work adds insight on small PAHs involved in the formation of H2 in space. Molecular hydrogen is the most abundant molecule in the interstellar medium. It is generally accepted that, in diffuse clouds, H2 is formed on cold (10–15 K) dust grain surfaces through the recombination of hydrogen atoms in a Langmuir–Hinshelwood mechanism.49,50 This is supported by laboratory experiments.51,52 However, H2 is also abundant in regions close to luminous stars known as photodissociation regions (PDRs) where the dust temperature is much higher (30–75 K) and the residence time of atomic hydrogen on a grain surface is too short to allow efficient H2 formation.52 In fact, analysis of the data for PDRs suggests that the H2 formation rate is actually much higher here than in diffuse clouds,53 suggesting an efficient H2 formation process that is active in regions with abundant UV light. IR observations show that IR emission of UV-pumped PAH species is very bright in these regions and, as reviewed by Wakelam et al.,54 it is therefore likely that PAHs play a role in the H2 formation process. Given the high UV fields in PDRs, H2 formation is thought to be triggered by UV absorption,7 which can lead to dissociative ionisation with H2 as well as H-loss channels.21,55,56
Recent work by Castellanos et al. addresses the dehydrogenation processes of a number of small interstellar PAHs in detail.21,57 They found that PAH size and edge topology influence the preferred hydrogen-loss channel, with smaller PAHs such as pyrene losing hydrogen atoms sequentially (H + H) and larger ones as molecular hydrogen (H2). Their analysis revealed that hydrogen-roaming is an important process in the fragmentation of highly excited PAHs, converting aromatic hydrogen in sp2 sites into CH2 sp3 sites from which either H or H2 can be lost. The formation of aliphatic-like side groups is critical in fragmentation and sets the balance of the competition between H- and H2-loss. Additionally, their analysis revealed that hydrogen-roaming from one ring to an adjacent ring of the PAH is inhibited.57
Our study on dehydrogenated pyrene supports this analysis. The IR spectrum of doubly-dehydrogenated pyrene shows that the two hydrogens atoms are lost from the same ring. Hence, roaming from one ring to the next one followed by hydrogen-loss and the creation of two rings each with a radical site is not important. This process of creating an aliphatic sp3 site provides a viable channel for the loss of H2 in an interstellar environment. It is yet to be shown that hydrogen-loss in a concerted fashion from PAHs can be important in PDRs. An interesting follow-up study would be to explore the doubly-dehydrogenated states of astronomically-sized PAHs (≃50 carbon atoms), and to further investigate the effects of edge topology on hydrogen loss to gain an insight into the photoproducts of PAHs in regions with high UV radiation. Of course, the study of such larger PAHs would come with additional challenges, the foremost being an even greater number of possible isomers with energetically low-lying geometries.
5 Conclusions
The IR spectrum of the pyrene radical cation has been recorded using the neon-tagging pre-dissociation technique. The measured band positions are shown to compare well with the anharmonic calculations. The spectrum of Py+ presented here adds spectral information and accuracy to existing data. All bands having DFT-predicted intensities larger than 10 km mol−1 have now been detected in the gas phase. With the application of a suitable scaling factor to account for anharmonicity, the overall profile of the convolved harmonic spectrum is similar to that of the anharmonic one, and the scaled harmonic calculations are thus shown to provide a good benchmark for the isomeric assignments in the ddPy+ spectrum. The good fit between the experimental and calculated spectra indicates that the influence of the neon-tag need not be calculated as it is negligible for these larger systems.The first experimental IR spectrum of ddPy+ is presented. Based on a comparison with the scaled harmonic DFT calculations shown here, the recorded spectrum cannot be reproduced with only one isomer but a fair reproduction is realised by combining the spectral features of two isomers: 4,5-ddPy+ and 1,2-ddPy+. This is supported by depletion studies that reveal that the spectra are dominated at the 80% level by these two isomers. In addition, there is no evidence for the presence of the very strong mode of 1,3-ddPy+, the next most stable isomer, in the 721–864 cm−1 range. Theoretical calculations support that hydrogen-loss from the same aromatic ring is favoured over hydrogen-loss from separate rings. The dehydrogenation process at a trio-site is initiated by roaming of a hydrogen to form a CH2 group at either position 2 or 3 (Fig. 1), resulting in 1,2-ddPy+ rather than the energetically equally favoured 1,3-ddPy+. Formation of the latter cannot be fully excluded but likely will not contribute to the overall signal by more than 20%. The data show no unique signatures that allow clear comparison with astronomical IR data on PAH sources, but allow to conclude, in agreement with earlier work, that the dissociative ionisation of a small PAH like pyrene may contribute to interstellar H2 formation.
The work presented here illustrates a number of advantages of neon-tagging IRPD spectroscopy. It is possible to record high resolution gas-phase spectra of ions that are difficult to study using alternate methods, particularly at low molecular abundances. However, this experimental method is mass-selective and not isomer-selective. As a consequence, the spectroscopic investigation of fragment species is only possible with complementary theoretical data, as shown here. Similar studies on larger PAHs will be very useful, linking the structural findings derived for pyrene to astronomically-relevant PAHs. Future progress in such fragmentation studies is reliant on the combined advancement of highly sensitive instrumentation and theoretical quantum chemistry in computing anharmonic spectra of large aromatic molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SP and SBa acknowledge the European Union and Horizon 2020 funding awarded under the Marie Skłodowska-Curie action to the EUROPAH consortium (grant number 722346). JB acknowledges the Netherlands Organisation for Scientific Research (Nederlandse Organisatie voor Wetenschappelijk Onderzoek, NWO) for a VIDI grant (grant number 723.016.006). AC acknowledges NWO for a VENI grant (grant number 639.041.543). This work was supported by NWO Exact and Natural Sciences for the use of supercomputer facilities (grant numbers 16638, 17676, and SH-362-15). We thank Aravindh Nivas Marimuthu for support with the data analysis, and Olivier Burggraaff for helping create Fig. 7. The authors greatly appreciate the experimental support provided by the FELIX team, and acknowledge the NWO for the support of the FELIX Laboratory. We thank the Cologne Laboratory Astrophysics group for providing the FELion ion trap instrument for the current experiments and the Cologne Center for Terahertz Spectroscopy (core facility, Deutsche Forschungsgemeinschaft grant SCHL 341/15-1) for supporting its operation. We thank the two anonymous reviewers for their thorough and constructive reviews.References
- L. J. Allamandola, A. G. G. M. Tielens and J. R. Barker, Interstellar polycyclic aromatic hydrocarbons – The infrared emission bands, the excitation/emission mechanism, and the astrophysical implications, Astrophys. J., Suppl. Ser., 1989, 71, 733–775 CrossRef CAS PubMed.
- A. G. G. M. Tielens, Interstellar polycyclic aromatic hydrocarbon molecules, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- B. J. Hrivnak, T. R. Geballe and S. Kwok, A study of the 3.3 and 3.4 μm emission features in proto-planetary nebulae, Astrophys. J., 2007, 662, 1059–1066 CrossRef CAS.
- J. Montillaud, C. Joblin and D. Toublanc, Evolution of polycyclic aromatic hydrocarbons in photodissociation regions: Hydrogenation and charge states, Astron. Astrophys., 2013, 552, A15 CrossRef.
- T. Allain, S. Leach and E. Sedlmayr, Photodestruction of PAHs in the interstellar medium: II. Influence of the states of ionization and hydrogenation, Astron. Astrophys., 1996, 305, 616–630 CAS.
- J. Bouwman, A. J. De Haas and J. Oomens, Spectroscopic evidence for the formation of pentalene+ in the dissociative ionization of naphthalene, Chem. Commun., 2016, 52, 2636–2638 RSC.
- H. Andrews, A. Candian and A. G. G. M. Tielens, Hydrogenation and dehydrogenation of interstellar PAHs: Spectral characteristics and H2 formation, Astron. Astrophys., 2016, 595, A23 CrossRef.
- J. Zhen, S. R. Castillo, C. Joblin, G. Mulas, H. Sabbah, A. Giuliani, L. Nahon, S. Martin, J. P. Champeaux and P. M. Mayer, VUV photo-processing of PAH cations: Quantitative study on the ionization versus fragmentation processes, Astrophys. J., 2016, 822, A113 CrossRef PubMed.
- V. Le Page, T. P. Snow and V. M. Bierbaum, Hydrogenation and Charge States of Polycyclic Aromatic Hydrocarbons in Diffuse Clouds. II. Results, Astrophys. J., 2003, 584, 316–330 CrossRef CAS.
- B. J. West, L. Lesniak and P. M. Mayer, Why Do Large Ionized Polycyclic Aromatic Hydrocarbons Not Lose C2H2?, J. Phys. Chem. A, 2019, 123, 3569–3574 CrossRef CAS PubMed.
- J. Zhen, P. Castellanos, D. M. Paardekooper, H. Linnartz and A. G. G. M. Tielens, Laboratory formation of fullerenes from PAHs: Top-down interstellar chemistry, Astrophys. J., Lett., 2014, 797, L30 CrossRef CAS.
- E. Peeters, S. Hony, C. van Kerckhoven, A. G. G. M. Tielens, L. J. Allamandola, D. M. Hudgins and C. W. Bauschlicher, The rich 6 to 9 μm spectrum of interstellar PAHs, Astron. Astrophys., 2002, 390, 1089–1113 CrossRef CAS.
- B. van Diedenhoven, E. Peeters, C. van Kerckhoven, S. Hony, D. M. Hudgins, L. J. Allamandola and A. G. G. M. Tielens, The profiles of the 3-12 micron polycyclic aromatic hydrocarbon features, Astrophys. J., 2004, 611, 928–939 CrossRef CAS.
- C. W. Bauschlicher, Jr., E. Peeters and L. J. Allamandola, The infrared spectra of very large, compact, highly symmetric, Polycyclic Aromatic Hydrocarbons (PAHs), Astrophys. J., 2008, 678, 316–327 CrossRef.
- A. Candian, T. H. Kerr, I. O. Song, J. McCombie and P. J. Sarre, Spatial distribution and interpretation of the 3.3 μm PAH emission band of the Red Rectangle, Mon. Not. R. Astron. Soc., 2012, 426, 389–397 CrossRef CAS.
- C. Joblin, A. G. G. M. Tielens, T. R. Geballe and D. H. Wooden, Variations of the 8.6 and 11.3 μm emission bands within NGC 1333: Evidence for polycyclic aromatic hydrocarbon cations, Astrophys. J., 1996, 460, L119–L122 CAS.
- G. C. Sloan, T. L. Hayward, L. J. Allamandola, J. D. Bregman, B. DeVito and D. M. Hudgins, Direct spectroscopic evidence for ionized polycyclic aromatic hydrocarbons in the interstellar medium, Astrophys. J., 1999, 513, L65–L68 CrossRef CAS PubMed.
- C. J. Mackie, A. Candian, X. Huang, E. Maltseva, A. Petrignani, J. Oomens, W. J. Buma, T. J. Lee and A. G. G. M. Tielens, The anharmonic quartic force field infrared spectra of three polycyclic aromatic hydrocarbons:Naphthalene, anthracene, and tetracene, J. Chem. Phys., 2015, 143, A224314 CrossRef PubMed.
- D. M. Hudgins, C. W. Bauschlicher, Jr. and L. J. Allamandola, Variations in the peak position of the 6.2 μm interstellar emission feature: A tracer of N in the interstellar polycyclic aromatic hydrocarbon population, Astrophys. J., 2005, 632, 316–332 CrossRef CAS.
- M. H. Vuong and B. H. Foing, Dehydrogenation of polycyclic aromatic hydrocarbons in the diffuse interstellar medium, Astron. Astrophys., 2000, 363, L5–L8 CAS.
- P. Castellanos, A. Candian, J. Zhen, H. Linnartz and A. G. G. M. Tielens, Photoinduced polycyclic aromatic hydrocarbon dehydrogenation: The competition between H- and H2-loss, Astron. Astrophys., 2018, 616, A166 CrossRef.
- A. J. De Haas, J. Oomens and J. Bouwman, Facile pentagon formation in the dissociation of polyaromatics, Phys. Chem. Chem. Phys., 2017, 19, 2974–2980 RSC.
- J. Oomens, A. J. A. van Roij, G. Meijer and G. von Helden, Gas-phase infrared photodissociation spectroscopy of cationic polyaromatic hydrocarbons, Astrophys. J., 2000, 542, 404–410 CrossRef CAS.
- J. Oomens, B. G. Sartakov, G. Meijer and G. von Helden, Gas-phase infrared multiple photon dissociation spectroscopy of mass-selected molecular ions, Int. J. Mass Spectrom., 2006, 254, 1–19 CrossRef CAS.
- P. Parneix, M. Basire and F. Calvo, ccurate modeling of infrared multiple photon dissociation spectra: The dynamical role of anharmonicities, J. Phys. Chem. A, 2013, 117, 3954–3959 CrossRef CAS PubMed.
- P. Jusko, S. Brünken, O. Asvany, S. Thorwirth, A. Stoffels, L. van der Meer, G. Berden, B. Redlich, J. Oomens and S. Schlemmer, The FELion cryogenic ion trap beam line at the FELIX free-electron laser laboratory: Infrared signatures of primary alcohol cations, Faraday Discuss., 2019, 217, 172–202 RSC.
- P. Jusko, A. Simon, S. Banhatti, S. Brünken and C. Joblin, Direct Evidence of the Benzylium and Tropylium Cations as the Two Long-Lived Isomers of C7H7+, ChemPhysChem, 2018, 19, 3182–3185 CrossRef CAS PubMed.
- P. Jusko, A. Simon, G. Wenzel, S. Brünken, S. Schlemmer and C. Joblin, Identification of the fragment of the 1-methylpyrene cation by mid-IR spectroscopy, Chem. Phys. Lett., 2018, 698, 206–210 CrossRef CAS PubMed.
- E. K. Campbell, M. Holz, D. Gerlich and J. P. Maier, Laboratory confirmation of C60+ as the carrier of two diffuse interstellar bands, Nature, 2015, 523, 322–323 CrossRef CAS PubMed.
- E. K. Campbell, M. Holz, J. P. Maier, D. Gerlich, G. A. H. Walker and D. Bohlender, Gas phase absorption spectroscopy of C60+ and C70+ in a cryogenic ion trap: Comparison with astronomical measurements, Astrophys. J., 2016, 822, 17 CrossRef.
- H. S. Kim and R. J. Saykally, Single-photon infrared emission spectroscopy of gaseous polycyclic aromatic hydrocarbon cations: A direct test for proposed carriers of the unidentified infrared emission bands, Astrophys. J., Suppl. Ser., 2002, 143, 455–467 CrossRef CAS.
- D. M. Hudgins and L. J. Allamandola, Infrared spectroscopy of matrix-isolated polycyclic aromatic hydrocarbon cations. 2. The members of the thermodynamically most favorable series through coronene, J. Phys. Chem., 1995, 99, 3033–3046 CrossRef CAS PubMed.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, The Free-Electron-Laser user facility FELIX, Infrared Phys. Technol., 1995, 36, 297–308 CrossRef CAS.
- M. J. Frisch, et al., Gaussian09, 2009 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- V. Barone, P. Cimino and E. Stendardo, Development and validation of the B3LYP/N07D computational model for structural parameter and magnetic tensors of large free radicals, J. Chem. Theory Comput., 2008, 4, 751–764 CrossRef CAS PubMed.
- V. Barone, M. Biczysko and J. Bloino, Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation, Phys. Chem. Chem. Phys., 2014, 16, 1759–1787 RSC.
- M. P. Andersson and P. Uvdal, New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis Set 6-311+G(d,p), J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- C. W. Bauschlicher, A. Ricca, C. Boersma and L. J. Allamandola, The NASA Ames PAH IR spectroscopic database: Computational version 3.00 with updated content and the introduction of multiple scaling factors, Astrophys. J., Suppl. Ser., 2018, 234, 32 CrossRef.
- S. Brünken, F. Lipparini, A. Stoffels, P. Jusko, B. Redlich, J. Gauss and S. Schlemmer, Gas-Phase Vibrational Spectroscopy of the Hydrocarbon Cations l-C3H+, HC3H+, and c-C3H2+: Structures, Isomers, and the Influence of Ne-Tagging, J. Phys. Chem. A, 2019, 123, 8053–8062 CrossRef PubMed.
- G. Trinquier, A. Simon, M. Rapacioli and F. X. Gadéa, PAH chemistry at eV internal energies. 2. Ring alteration and dissociation, Mol. Astrophys., 2017, 7, 37–59 CrossRef.
- J. Fulara, A. Nagy, K. Filipkowski, V. S. Thimmakondu, J. F. Stanton and J. P. Maier, Electronic transitions of C6H4+ isomers: Neon matrix and theoretical studies, J. Phys. Chem. A, 2013, 117, 13605–13615 CrossRef CAS PubMed.
- P. Bera, R. Peverati, M. Head-Gordon and T. J. Lee, Hydrocarbon growth via ion-molecule reactions: Computational studies of the isomers of C4H2+, C6H2+ and C6H4+ and their formation paths from acetylene and its fragments, Phys. Chem. Chem. Phys., 2015, 17, 1859–1869 RSC.
- J. Jašík, D. Gerlich and J. Roithová, Two-color infrared predissociation spectroscopy of C6H62+ isomers using helium tagging, J. Phys. Chem. A, 2015, 119, 2532–2542 CrossRef PubMed.
- T. Chen, M. Gatchell, M. H. Stockett, R. Delaunay, A. Domaracka, E. R. Micelotta, A. G. G. M. Tielens, P. Rousseau, L. Adoui, B. A. Huber, H. T. Schmidt, H. Cederquist and H. Zettergren, Formation of H2 from internally heated polycyclic aromatic hydrocarbons: Excitation energy dependence, J. Chem. Phys., 2015, 142, 144305 CrossRef CAS PubMed.
- B. West, F. Useli-Bacchitta, H. Sabbah, V. Blanchet, A. Bodi, P. M. Mayer and C. Joblin, Photodissociation of pyrene cations: Structure and energetics from C16H10+ to C14+ and almost everything in between, J. Phys. Chem. A, 2014, 118, 7824–7831 CrossRef CAS PubMed.
- H. W. Jochims, E. Ruhl, H. Baumgartel, S. Tobita and S. Leach, Size effects on dissociation rates of polycyclic aromatic hydrocarbon cations: Laboratory studies and astrophysical implications, Astrophys. J., 2019, 420, 307–317 CrossRef.
- D. Hollenbach and E. E. Salpeter, Surface recombination of hydrogen molecules, Astrophys. J., 1971, 163, 155 CrossRef CAS.
- B. D. Savage, J. F. Drake, W. Budich and R. C. Bohlin, A survey of interstellar molecular hydrogen. I, Astrophys. J., 1977, 216, 291 CrossRef CAS.
- V. Pirronello, O. Biham, C. Liu, L. Shen and G. Vidali, Efficiency of molecular hydrogen formation on silicates, Astrophys. J., 1997, 483, L131–L134 CrossRef CAS.
- G. Vidali, H2 formation on interstellar grains, Chem. Rev., 2013, 113(12), 8762–8782 CrossRef CAS PubMed.
- E. Habart, F. Boulanger, L. Verstraete, C. M. Walmsley and G. Pineau des Forêts, Some empirical estimates of the H2 formation rate in photon-dominated regions, Astron. Astrophys., 2004, 414, 531–544 CrossRef CAS.
- V. Wakelam, E. Bron, S. Cazaux, F. Dulieu, C. Gry, P. Guillard, E. Habart, L. Hornekær, S. Morisset, G. Nyman, V. Pirronello, S. D. Price, V. Valdivia, G. Vidali and N. Watanabe, H2 formation on interstellar dust grains: The viewpoints of theory, experiments, models and observations, Mol. Astrophys., 2017, 9, 1–36 CrossRef.
- C. Lifshitz, Nergetics and dynamics through time-resolved measurements in mass spectrometry: Aromatic hydrocarbons, polycyclic aromatic hydrocarbons and fullerenes, Int. Rev. Phys. Chem., 1997, 16, 113–139 Search PubMed.
- C. Joblin, G. Wenzel, S. Rodriguez Castillo, A. Simon, H. Sabbah, A. Bonnamy, D. Toublanc, G. Mulas, M. Ji, A. Giuliani and L. Nahon, Photo-processing of astro-PAHs.arXiv e-prints 1994, 420, arXiv e-prints 1994, 420, arXiv:1912.03137v1.
- P. Castellanos, A. Candian, H. Andrews and A. G. G. M. Tielens, Photoinduced polycyclic aromatic hydrocarbon dehydrogenation: Molecular hydrogen formation in dense PDRs, Astron. Astrophys., 2018, 616, A167 CrossRef.
- J. Bouwman, P. Castellanos, M. Bulak, J. Terwisscha van Scheltinga, J. Cami, H. Linnartz and A. G. Tielens, Effect of molecular structure on the infrared signatures of astronomically relevant PAHs, Astron. Astrophys., 2019, 621, A80 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02272a |
| This journal is © the Owner Societies 2020 |