
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular dynamics simulation of soot formation during diesel combustion with oxygenated fuel addition
Cheng
Chen
and
Xi
Jiang

School of Engineering & Materials Science, Queen Mary University of London, London E1 4NS, UK. E-mail: xi.jiang@qmul.ac.uk
First published on 24th August 2020
Abstract
This study investigates the soot formation process of diesel combustion using molecular dynamics simulations with reactive force fields by examining the effects of five oxygenated additives on diesel soot reduction. The newly improved ReaxFF CHO2016 parameters are employed due to their feasibility in modelling the kinetics of large hydrocarbon fuels and describing the chemistry of carbon condensed phases. The detailed pathways of soot formation examined include the thermal decomposition of fuels, the formation of aromatic rings, the mechanism of nucleation, and the mass growth of nascent soot. The morphological developments of the soot formation are obtained, together with quantities indicating the physical and chemical properties, such as mass, size, C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H ratio and aliphatic-to-aromatic C:H ratio. The role of aliphatic-substituted aromatics in nascent soot coalescence and the appearance of aliphatic side chains in soot structures are identified. The concentrations of C-atoms in nascent soot particles contributed from oxygenates blended with diesel are quantified and visualized for the first time. The effects of the molecular structure of the oxygenated additives, i.e. the existence of esters, alcohols, carbonyl groups and ethers, on soot precursor mitigation are elucidated by evaluating the early formation of CO and CO2 quantitatively during the thermal decomposition.
:H ratio and aliphatic-to-aromatic C:H ratio. The role of aliphatic-substituted aromatics in nascent soot coalescence and the appearance of aliphatic side chains in soot structures are identified. The concentrations of C-atoms in nascent soot particles contributed from oxygenates blended with diesel are quantified and visualized for the first time. The effects of the molecular structure of the oxygenated additives, i.e. the existence of esters, alcohols, carbonyl groups and ethers, on soot precursor mitigation are elucidated by evaluating the early formation of CO and CO2 quantitatively during the thermal decomposition.
1 Introduction
Airborne particulate matter such as soot is of major environmental concern and can affect public health. Diesel combustion is known to be the main contributor to soot emission. Among various advanced control strategies and combustion techniques developed to reduce soot emission from diesel combustion, modifying fuel composition by blending the fuel with renewable oxygenated additives has drawn particular attention due to the potential of tackling the problem both ecologically and economically.1 Although previous work has shown that the oxygen contents in fuel composition have a major influence on soot production (when the oxygen content of the fuel reached 25–30% by mass, all soot emissions virtually vanished2), the chemical mechanisms involved in the soot reduction process are not well understood because of the complicated fuel molecular structure and the lack of in-depth kinetics studies. There has been no clear agreement on the impact of the oxygenated functionalities, e.g., alcohols, esters, and carbonyl groups, on the soot reduction process, nor on the mechanisms involved.3 This motivates the present study to reveal the effect of the specific molecular structure of oxygenates on diesel soot suppression.Understanding the soot formation mechanism and thereby reducing soot emissions from combustion devices have always been a challenge. It is acknowledged that soot nucleation defined as the transition from gaseous molecular precursors to condensed matter, yielding carbonaceous nanoparticles, is a key step but mostly unknown.4 Critical gaps remain between the experimental observation and mechanistic description.5 The widely reported soot nucleation pathway involving the dimerization of polycyclic aromatic hydrocarbons (PAHs) into stacks via van der Waals forces is debatable.6 It fails to explain, for example, the existence of a large amount of aliphatic C–H bonds in the shell of nascent soot.7 The irreversible process of PAH dimerization, with pyrene (C16H10) recognized as the smallest building block, is not thermodynamically favoured.8 The binding forces (e.g., electrostatic, dispersive and π–π stacking interaction forces) of dimers with moderate-sized PAHs are too weak to allow the PAH clusters to survive in the environment above their boiling or sublimation temperatures. The species as large as ovalene (C32H14), hexcoronene (C42H18) or circumcoronene (C54H18) may generate dimers capable of surviving at flame temperature. The concentration of these PAH species in the gas phase during combustion is low, which is contradictory to the phenomenon that nascent soot was observed before the concentration peaks of these PAH species.9 The hypothesis of the dimerization of PAHs with a larger size in the soot nucleation process therefore appears highly unlikely. It is speculated that some stronger interactions are prerequisite for soot nucleation, such as covalent bonds combining PAH radicals and aryl radicals, but the existing theories are still inadequate to explain the rapidly persistent mass growth and the gas-to-solid nucleation process in the post flame region.5 In general, although the conceptual pathways of nucleation in the existing theory are feasible and important, chemical pathways besides physical stacking are needed to achieve a comprehensive and accurate understanding of soot formation.
The availability of modelling/simulation tools at the atomistic level may shed light on these unsolved issues. The multiscale model for soot inception developed by Violi et al.,10 integrating kinetic Monte Carlo and molecular dynamics (MD), allows the extension of the accessible time scale by orders of magnitude while retaining the full atomistic details. Application of this model to acetylene and benzene flames showed that the reaction pathways concerning the growth of incipient soot particles are dependent on the molecular structure of the fuel. Some indicators were employed to evaluate the properties of the three-dimensional structure of nascent soot particles including physical properties (e.g., porosity, density, radial distribution function and sphericity) and chemical properties (e.g., C:H ratio, aromatic moieties and number of cross-links).11,12 MD simulation with the OPLS-AA force field13,14 was used to quantify the impact of the mass, symmetry and morphology of peri-condensed aromatic hydrocarbons (PCAHs), aromatic aliphatically linked hydrocarbons (AALHs) and aliphatic substituted PAHs on the stability of the molecular dimers. Meanwhile, the contributions from potential energy and entropy were taken into consideration simultaneously by using the free energy methods to describe the process. It was understood that the attached aliphatics increased the nucleation rate greatly due to the collision energy accommodation enhancement provided by the added internal degrees of freedom. Modelling work was also performed by using replica exchange MD simulation to investigate the molecular size distributions of the heterogeneous PAH clusters in a core–shell model.15 It was found that larger PAHs are stacked at the core while small PAHs are located on the cluster surface. Of the two main factors, the strength of intermolecular interactions played a more critical role in determining cluster partition than the molecular size difference. Another method for modelling the combustion and soot formation process is reactive force field (ReaxFF) based MD simulations. For example, Mao et al.16–18 found that physical dimerization depended on both the temperature and PAH size while all the PAHs became chemically active at temperatures above 2500 K and generated fullerene-like soot particles. It is recognized that MD-type modelling/simulation will likely lead to high-fidelity soot models capable of simultaneously resolving particle size distribution, chemical composition, and morphology.5,10,16
The improved CHO2016 parameters in ReaxFF have been successfully used to investigate the combustion kinetics of conventional fuels like syngas, ethanol, JP-10, and toluene.19–21 Most importantly, the CHO2016 description is also trained from C2013 parameters that are able to describe the chemistry of aromatic hydrocarbons properly in condensed and gaseous phases simultaneously.16,19,22 Therefore, we perform ReaxFF MD simulation in a single force field without any predefined radicals or reaction pathways to reveal the role of fuel molecular structure on soot formation, which was previously inaccessible using existing computational chemistry methods.
2 Simulation methodology
2.1 Simulation setup
In this study, a recently proposed 5-component diesel surrogate fuel by Qian et al.23 is employed to simulate diesel soot formation, which can accurately reproduce the gaseous emissions of the target diesel fuel under actual engine combustion conditions. The surrogate fuel shown in Fig. 1(a) includes 21.6% n-hexadecane (n-HXD, C16H34), 15.5% n-octadecane (n-OTD, C18H38), 26.0% heptamethylnonane (HMN, C16H34), 20.7% 1-methylnaphthalene (1-MNT, C11H10) and 16.2% decalin (C10H18) by mol. Five representative oxygenates, i.e., dimethyl carbonate (DMC, C3H6O3), cyclopentanone (CPO, C5H8O), methyl butanoate (MB, C5H10O2), dimethoxymethane (DMM, C3H8O2) and ethanol (C2H6O), shown in Fig. 1(b), are selected to blend with diesel surrogates so as to evaluate the effects of ketones, esters, ethers and alcohols on soot precursor mitigation.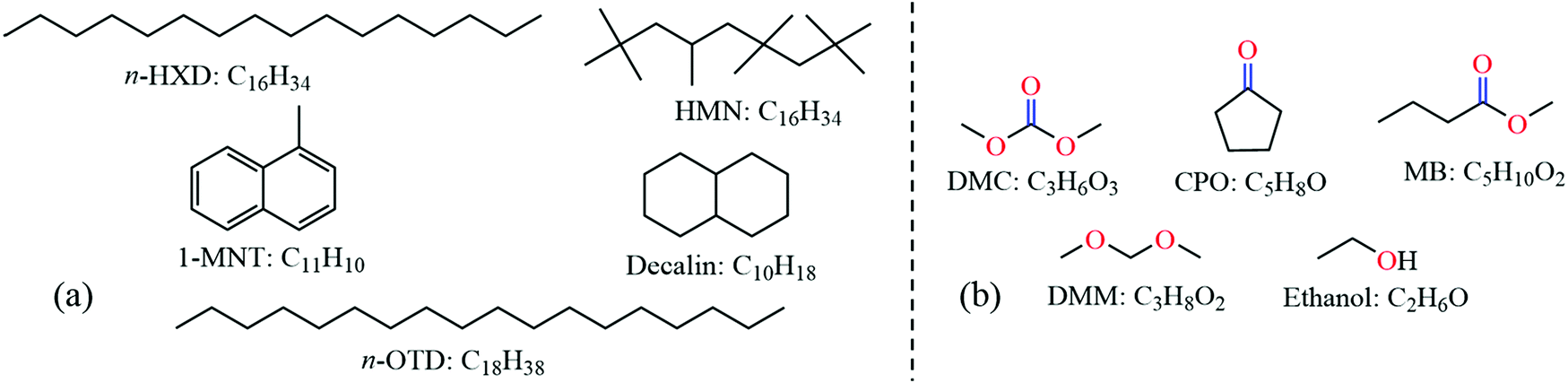 | ||
| Fig. 1 Molecular structures and chemical formulas of (a) the diesel surrogate fuel and (b) oxygenated fuel additives. | ||
The reactive systems studied are shown in Table 1. System 1 is established as a baseline containing 149 pure diesel surrogate molecules with 150 oxygen molecules to model the fuel-rich combustion. To study the effect of fuel additives, oxygenated fuel molecules are doped into the simulation boxes containing the diesel model. A rule is adopted in which systems 2–6 are set up by adjusting the number of diesel molecules in the box so that these systems have approximately the same carbon and oxygen numbers as system 1. This could highlight the influence of the molecular structure of the additive instead of the amount of oxygen. The density of system 1 is set to be 0.1 g cm−3, and the edge length of the cubic box is 82.5 Å. The size of the simulation boxes in systems 2–6 is adjusted accordingly to have the same density as system 1.
| System no. | n-HXD | HMN | 1-MNT | Decalin | n-OTD | Additives |
|---|---|---|---|---|---|---|
| 1 | 32 | 39 | 31 | 24 | 23 | O2: 150 |
| 2 | 27 | 34 | 27 | 21 | 20 | DMC: 100 |
| 3 | 9 | 12 | 9 | 7 | 7 | CPO: 300 |
| 4 | 25 | 31 | 24 | 19 | 18 | DMM: 150 |
| 5 | 21 | 25 | 20 | 16 | 15 | MB: 150 |
| 6 | 23 | 28 | 22 | 17 | 17 | Ethanol: 300 |
2.2 Simulation details
The simulation is performed using the constant temperature canonical ensemble, i.e. the NVT ensemble. The periodic boundary conditions are imposed in all three directions. Before the production run, every system undergoes energy minimization and equilibration at 1000 K for 50 ps. After the equilibration, a series of reactive NVT MD simulations for each system is performed for 2 ns at 3000 K. The system temperature is controlled by a Nosé–Hoover thermostat algorithm with a damping constant of 25 fs. In ReaxFF simulations, employing high temperature and pressure to facilitate the collision and accelerate the simulation process is a common strategy.19,20,24,25 A time step of 0.25 fs is adopted for both the equilibrating and reactive simulations. The species information is recorded every 100 fs while dynamic trajectory and bonding information is dumped every 250 fs. For intermediate and product recognition analysis, a 0.3 bond order cut-off is chosen to capture all the chemical reaction pathways including those with short-lived intermediates.21 All the simulations are carried out with the REAXC package in the LAMMPS platform26 with force field parameters modified accordingly. The reaction pathways are analysed using Chemical Trajectory AnalYzer scripts.27 The visualisation of simulation results is carried out using Visual Molecular Dynamics software.283 Results and discussion
3.1 Incipient soot formation mechanism of diesel combustion
Force field parameters play a key role in the capability of MD simulations. Between the two sets of widely used reactive force field parameters for combustion simulation, CHO2016 retains the capability of CHO2008 for predicting complex hydrocarbon pyrolysis and oxidation reactions but has taken into account more chemical pathways with improved accuracy19 that are important for soot formation. The feasibility of the two sets of parameters in predicting particle growth during the soot inception process is evaluated first, as shown in Fig. 2. The carbon number of the largest particle in CHO2016 can be as large as 1000 at the subsequent steady mass growth stage after the initial fuel decomposition, whereas CHO2008 fails to predict the particle growth with the carbon number remaining at an extremely low level. After 1.6 ns, the C:H ratio of the largest molecule in CHO2016 starts to increase and becomes much higher than that of CHO2008, which is as one would have expected, indicating that CHO2016 is able to describe the continuous dehydrogenation reasonably during soot inception. As a consequence, CHO2016 parameters are employed in the following simulations.
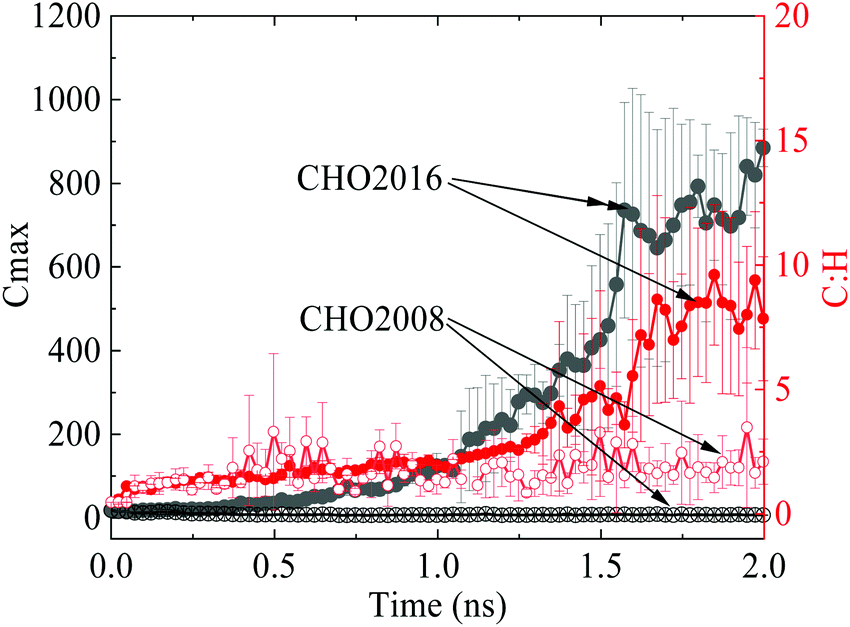 | ||
| Fig. 2 Evaluation of CHO2008 and CHO2016 force field parameters in predicting diesel incipient soot formation in terms of the number of carbon atoms in the largest molecule and the C:H ratio of the largest molecule, where the solid cycle represents CHO2016 while the hollow cycle represents CHO2008. | ||
The time evolutions of the regular products of diesel combustion, e.g., CO, CO2, H2O and formaldehyde (CH2O), are shown in Fig. 3(a), and those of the typical C1–C3 intermediates like the methyl radical (CH3), ethylene (C2H4), ethyne (C2H2), vinyl (C2H3) and propargyl (C3H3) are displayed in Fig. 3(b). Visualisation and identification of the representative molecular structures are captured and presented in Fig. 4. In fuel-rich combustion, O2 is consumed at an extremely high rate at the initial stage accompanying the acute increase of CO molecules. The formation of CO2 occurs around 1.8 ps later compared to CO because of its dependence on the production of C1 species like CO, HCO, and CH2O. Only around 6CO2 molecules are produced, which is a rather small volume compared with CO. The amount of CO2 molecules remained almost constant in this period, manifesting that no CO has been further converted into CO2. H2O production takes place earlier than CO2 production in an appreciable quantity. The formaldehyde molecule is also found to be an important intermediate and it reaches a maximum number of 92 at 29.1 ps. The existence of the formaldehyde peak is a feature of the low-temperature combustion of large alkanes. At temperatures as high as 3000 K, it is unstable and would decompose to CO easily, which explains the steady increase of CO at the late stage.
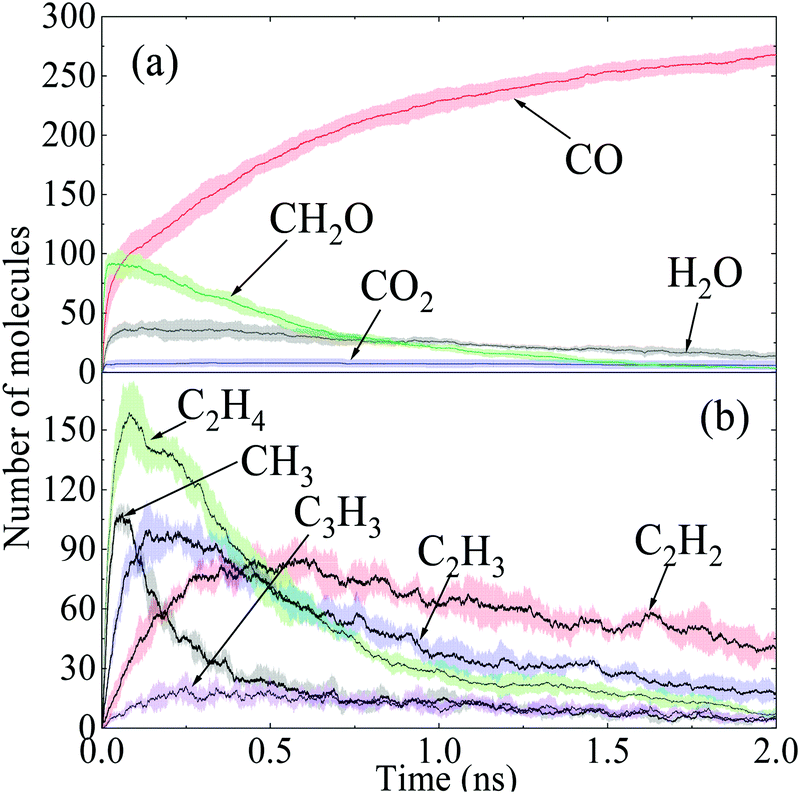 | ||
| Fig. 3 Time evolutions of major intermediates and products in diesel combustion, where the shaded area indicates the average standard deviations of five repeated simulations. | ||
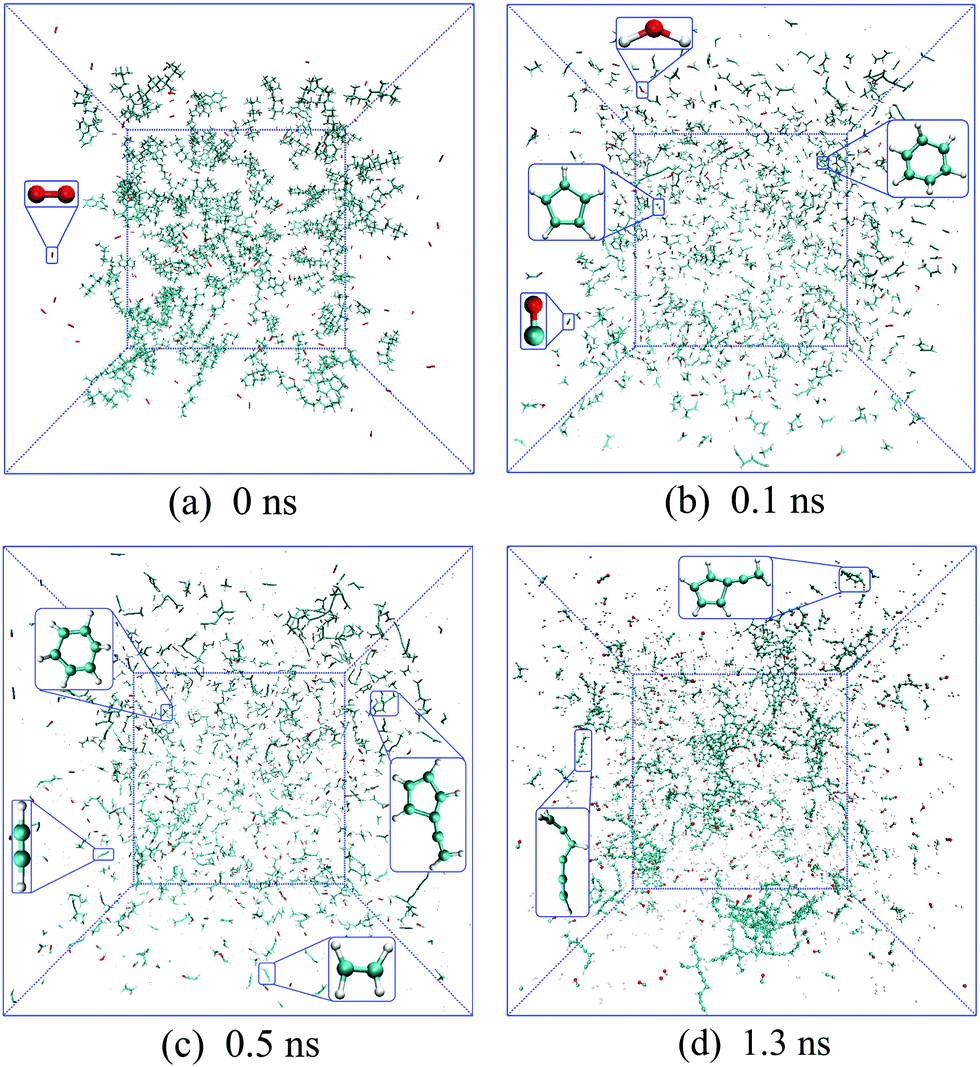 | ||
| Fig. 4 Time evolutions of incipient soot formation of diesel combustion, where C, H and O atoms are represented in cyan, silver and red, respectively. | ||
In Fig. 3(b), the number of ethylene molecules decreases immediately after reaching its maximum at around 80.8 ps while the peaks of ethyne and the vinyl radical are relatively inconspicuous. Ethyne, the vinyl radical and the propargyl radical can persist throughout the whole process, whereas the methyl radical and ethylene deplete almost completely at the late stage. This is mainly because paraffins like n-HXD and n-OTD accounting for the largest proportion of the diesel composition would experience H-abstraction and consecutive unimolecular C–C fission to produce alkyl radicals like C4H9, C7H15, C8H17 and C13H27, releasing ethylene at the same time.29 A series of β-scission reactions of these C4–C13 fragments takes place subsequently to form alkenes. HMN tends to produce more of the large alkenes as it is more likely to dissociate to alkyl radicals with substitution at the β C-atom. Ethylene mainly reacts into ethyne and the vinyl radical while the propargyl radical is produced from the dehydrogenation of propene via a series of H-abstractions with H-atom as the primary abstractor.30 The propargyl radical is a kind of resonantly-stabilized radical (RSR), which would undergo decomposition and oxidation at much lower rates than nonresonant radicals do. These C2 and C3 unsaturated hydrocarbons play crucial roles in cyclopentadienyl and benzene formation whilst the specific pathways vary considerably. Another observation is that the large aliphatics are repolymerized by a wide variety of these precursors and alkenes after the depletion of the initial diesel alkanes, and the relevant molecular structures are captured and illustrated in Fig. 4(d).
Hydrocarbon combustion is triggered either by unimolecular C–C bond dissociation or the oxidative reaction via the abstraction of a hydrogen free radical from the substrate. In this study, the earliest reaction of 1-MNT detected is the H-abstraction reaction by O2 through cleavage of the C–H bond in the methyl group at 0.3 ps, producing the hydroxyl radical and the corresponding 1-naphthylmethyl radical. This is ascribed to the relatively low dissociation energy of the methyl C–H bond, i.e., 85.1 kcal mol−1.31 The aromatics are generally stable and can survive a long time before the ring-opening reactions. In this period, it is possible to break the methyl-aryl C–C bond and the aryl C–H bond. These decomposition reactions would favour the formation of the 1-MNT radical and naphthalene radical. Moreover, the 2-methylindene radical with a pentagonal ring is also observed. The early formation of these aryl groups, with molecular structures and highlighted radical sites displayed in Fig. 5(a), was also observed experimentally by Shaddix et al.32 They are regarded as potential building blocks with high propensity to produce nascent soot by reacting with additional benzenoid rings. Thus, the soot precursor formation mechanism is closely correlated with the fuel molecular structures, particularly the large hydrocarbons existing in practical fuels that can decompose in a wide range of pathways.
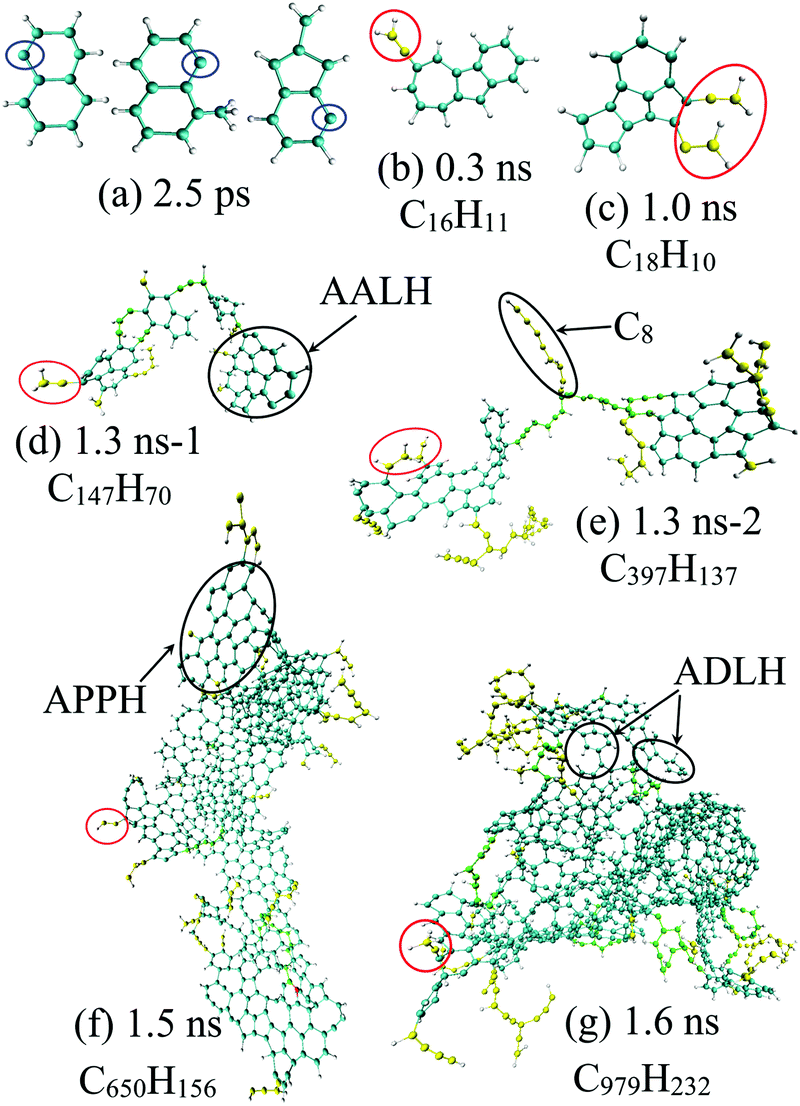 | ||
| Fig. 5 Snapshots of the enlarged representative carbonaceous nanoparticle morphology during diesel combustion, where aliphatic side chains are coloured in yellow with direct C2H2 or C2H3 addition highlighted with a red ellipse, while aliphatic links between aromatic moieties in nascent soot are highlighted in green. | ||
In this study, the initial mass growth is found to be triggered by the addition of C2H2 or C2H3 to cyclopentadienyl and benzene. The newly generated RSRs are constructed by five-, six- or seven-membered carbon rings substituted with unsaturated aliphatics, as shown in Fig. 4(c) and 5(b). More recently, the aliphatic-substituted aromatics in nascent soot particles were observed and visualized directly via high-resolution atomic force microscopy.33 The small aliphatics would connect the RSRs via a chemical covalent bond, dimerizing into cross-linked three-dimensional AALH structures, as shown in Fig. 5(d). The unstable AALH structures further evolve into a peri-condensed plane through rearrangement and ring closure,9 leading to aromatic peri-condensed polycyclic hydrocarbon (APPH) structures at 1.5 ns. An aromatic directly linked hydrocarbon (ADLH) is also observed at around 1.6 ns. In this period, addition reactions also take place in the hydrocarbon flame, generating the elongated aliphatic side chains, e.g., C8 alkene, as shown in Fig. 5(e). The flat size of incipient soot can have a length of 6.03 nm, as shown in Fig. 5(f). At high temperature, it would undergo cyclization and condensation, resulting in a branched fullerene/ellipsoid structure. The morphology of the nascent soot at 1.6 ns is an amorphous structure, around 3.7 nm in diameter and 998 amu in mass, rich in aliphatics, with a C:H ratio of 6–10. The ratio of C–H groups present in aliphatics to that in aromatic rings is around 1.2 at 1.6 ns and 2.3 at 2 ns, respectively. In addition, it can be assumed that aliphatic-substituted aromatics can also act as building blocks to stack into the outer shell when the temperature becomes somewhat lower. RSR moieties on shell surfaces could provide sites for small hydrocarbons in flames to attach. This pathway could potentially explain recent experimental results showing that the soot shell surface is abundant with alkyl or alkenyl functionalities in the range of C2–C8.5
3.2 Soot formation during diesel combustion with blending of oxygenates
In this study, the blending ratio was configured properly to unify the oxygen contents and the overall carbon quantity to highlight the effect of oxygenate molecular structure on the soot formation process. Some insights into the soot inception of diesel with oxygenates can be gained from the detailed morphological development comparison, as shown in Fig. 6 at the time instants of 1.2 ns and 2 ns, respectively. The C-atoms contributed from the oxygenates are quantified and highlighted in the nascent soot. | ||
| Fig. 6 Morphological snapshots of nascent soot at 1.2 ns (above the dashed line) and final soot particles at 2 ns (below the dashed line) in diesel combustion with various additives, from left to right: (a) DMC, (b) CPO, (c) DMM, (d) MB and (e) ethanol, where the numbers of carbon atoms contributed from oxygenated fuel additives are given in purple. | ||
In Fig. 6, both the morphology of the nascent soot and the number of C-atoms from oxygenates vary in different cases, among which C-atoms from DMC only account for 5.9% (calculated from 18/304) and 5.7% (from 46/814) at 1.2 ns and 2 ns, respectively, as shown in Fig. 6(a), while almost 73.8% and 67.0% of the C-atoms are from CPO at 1.2 ns and 2 ns, as shown in Fig. 6(b). This is mainly attributed to the initial configuration, which was determined by the C:O ratio of the oxygenate molecular structures. It is more meaningful to compare the C-atom ratio originating from oxygenated additives in nascent soot with the corresponding initial stoichiometric configuration to evaluate the sooting propensity of oxygenates. For the cases of diesel blending with DMC and DMM, the C-atom contribution ratios at 2 ns are 5.7% and 12.8%, respectively, both of which are much smaller than the C-atom ratio between oxygenates and fuel mixtures in the initial box, which are 14.0% and 21.2%, respectively. This means that DMC and DMM are much less likely to form the nascent soot compared with diesel surrogate molecules. The ratios for cases blending with CPO and ethanol are 67.0% and 23.5% in nascent soot, both of which are lower but at almost the same level as the initial ratio, i.e., 70.4% and 28.1%, respectively. It is thus indicated that CPO and ethanol have similar sooting propensities to diesel. This is also the case for fuel mixtures blending with MB, where 35.1% of the C-atoms are from MB in the initial box but the contribution is 32.3% in the final nascent soot. These observations can be ascribed to the consecutive C–C bond in the molecular structures of CPO, MB and ethanol, which would lead to soot formation. However, the C-atoms in DMC and DMM are separate and each of them is bound to a different O-atom, avoiding significant levels of the products such as ethylene, acetylene or unsaturated radicals, which are known to be PAH precursors.
3.3 Quantification of oxygenate effectiveness in soot precursor mitigation
The effectiveness of oxygenates in soot suppression is not well understood and is attracting increasing interest.34–36 In the kinetics modelling performed by Westbrook et al.2 for the premixed fuel-rich combustion of n-heptane and oxygenate blends, it was observed that all oxygenates tested reduced the emission of soot precursors, but with different effectiveness. In the previous experimental study by Farooq et al.,34 the effectiveness was represented by the number of C-atoms captured by CO and CO2. The time histories of CO and CO2 for the thermal decomposition of oxygenates blended with diesel are presented in Fig. 7. The capability of ReaxFF in capturing chemical reaction pathways and predicting Arrhenius parameters quantitatively has been validated extensively against experiments and continuum methods.19,20 It is a common practice to normalize the absolute rate constants using the slowest reaction obtained from ReaxFF simulations, that is, the rate constant of the slowest reaction is set to be 1.24 The thermal pyrolysis pathways of the oxygenates and the relative kinetic rates are summarized in Table 2.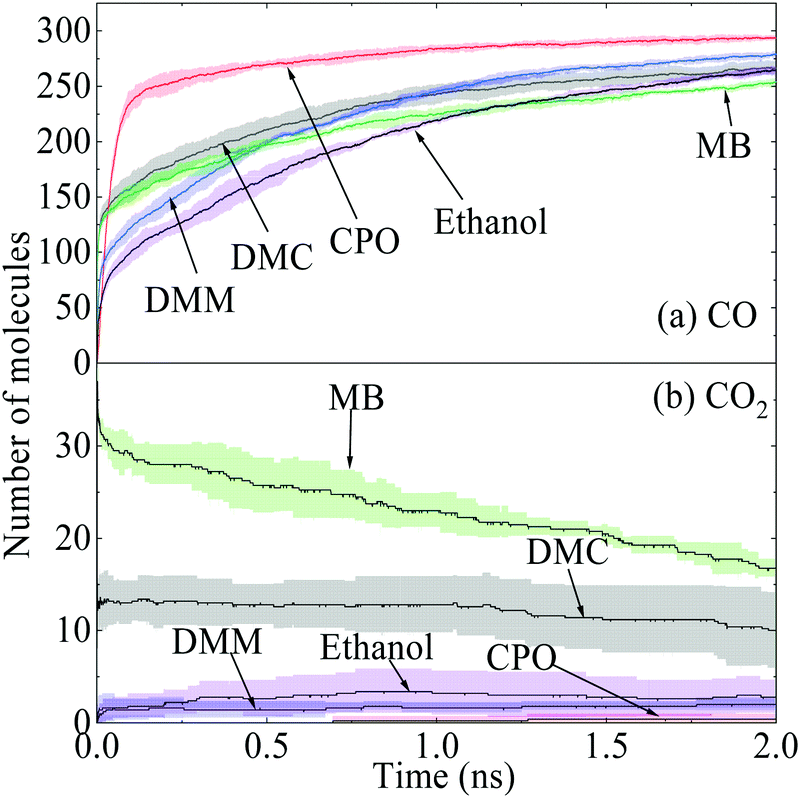 | ||
| Fig. 7 Time evolutions of (a) CO and (b) CO2 for diesel blending with various additives at 3000 K. | ||
| ID | Reaction | Relative rate |
|---|---|---|
| R1 | DMC → CH3O˙ + CH3O(C˙O) | (1.01 ± 0.09) × 104 |
| R2 | DMC → CH3˙ + CH3O(CO)O˙ | (2.44 ± 0.14) × 102 |
| R3* | CPO → CO + CH2˙(CH2)2CH2˙ | (7.99 ± 0.73) × 103 |
| R4 | CPO → CH2˙CH2˙ + (C˙O)CH2CH2˙ | (1.84 ± 0.12) × 103 |
| R5 | CPO → CH2˙CH2CH2˙ + (C˙O)CH2˙ | (4.66 ± 0.24) × 102 |
| R6 | DMM → CH3˙ + CH3OCH2O˙ | (5.22 ± 0.31) × 103 |
| R7 | DMM + HO˙ → H2O + CH2˙OCH2OCH3 | (1.27 ± 0.16) × 103 |
| R8 | DMM + CH3O˙ → CH3OCH˙OCH3 + HOCH3 | (2.50 ± 0.17) × 101 |
| R9 | MB → CH3O˙ + CH3(CH2)2(C˙O) | (1.78 ± 0.15) × 103 |
| R10 | MB → CH3˙ + CH3(CH2)2(CO)O˙ | (5.00 ± 0.27) × 102 |
| R11 | MB → CH3CH2CH2˙ + CH3O(C˙O) | (1.54 ± 0.11) × 102 |
| R12 | MB → CH3O(CO)CH2˙ + CH3CH2˙ | (2.51 ± 0.15) × 101 |
| R13 | MB + H˙ → CH3CH2CH˙(CO)OCH3+ H2 | (3.77 ± 0.20) × 100 |
| R14 | Ethanol → ˙OH + CH3CH2˙ | (4.71 ± 0.41) × 101 |
| R15 | Ethanol → CH2˙OH + CH3˙ | (9.39 ± 0.51) × 100 |
| R16 | Ethanol + ˙OH → H2O + CH3CH2O˙ | (0.99 ± 0.09) × 100 |
| R17 | Ethanol → ˙H + CH2˙CH2OH | (4.02 ± 0.23) × 100 |
Among the additives, CPO shows the best performance with 294 oxygen atoms used to fix C-atoms while MB has the lowest quantity. This is because the scission of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds in the CPO carbonyl group hardly occurs even at 3000 K, and double bonds are maintained all the way to CO formation through unimolecular decomposition, i.e., R3*, with a rapid kinetic rate. This also explains the steep increase of CO in the initial stage of Fig. 7(a) before the turning point at around 0.12 ns. In contrast, MB has the lowest plateau value for CO but with a fair amount of CO2, as demonstrated in Fig. 7(b), indicating that the CO2 moieties in esters tend to result in the direct formation of CO2 molecules considerably. In this way, two O-atoms together captured only one C-atom from the species pool. The oxygen initially present in esters is therefore less effective. As a carbonate ester, the early formation of CO2 was also observed in DMC. Specifically, pyrolysis of DMC and MB by R1 and R11, respectively, can produce a considerable amount of methoxycarbonyl radical, i.e., CH3O(C˙O), which has two competitive decomposition pathways to produce CO2 or CO + CH3O. However, both CH3O(CO)O˙ in DMC by R2 and CH3(CH2)(CO)O˙ in MB by R10 are more likely to produce CO2 directly via β-scission.34
O double bonds in the CPO carbonyl group hardly occurs even at 3000 K, and double bonds are maintained all the way to CO formation through unimolecular decomposition, i.e., R3*, with a rapid kinetic rate. This also explains the steep increase of CO in the initial stage of Fig. 7(a) before the turning point at around 0.12 ns. In contrast, MB has the lowest plateau value for CO but with a fair amount of CO2, as demonstrated in Fig. 7(b), indicating that the CO2 moieties in esters tend to result in the direct formation of CO2 molecules considerably. In this way, two O-atoms together captured only one C-atom from the species pool. The oxygen initially present in esters is therefore less effective. As a carbonate ester, the early formation of CO2 was also observed in DMC. Specifically, pyrolysis of DMC and MB by R1 and R11, respectively, can produce a considerable amount of methoxycarbonyl radical, i.e., CH3O(C˙O), which has two competitive decomposition pathways to produce CO2 or CO + CH3O. However, both CH3O(CO)O˙ in DMC by R2 and CH3(CH2)(CO)O˙ in MB by R10 are more likely to produce CO2 directly via β-scission.34
Unimolecular decomposition plays a leading role in the pyrolysis of oxygenates while H-abstraction by a H-atom, or a methoxy or hydroxyl radical also accounts for a certain proportion of the consumption of oxygenates. Relevant pathways in DMM and ethanol produce H2O, which affects the effectiveness of the O-atom in combining C-atoms to some extent and should not be ignored. From Table 2, it can be summarized that although oxygenates decompose primarily into oxygenated intermediates, they also produce methyl, ethyl, and propyl radicals as well as some biradicals, which in turn are likely to favour soot precursor production. For example, three of the MB pyrolysis pathways produce alkyl radicals, whereas in the case of DMC and DMM, only one methyl radical formation pathway is observed. In this manner, the consumption patterns of the additive can be expressed in two aspects, removing C-atoms from the soot precursor pool via combination with O-atoms in functional groups while contributing to soot precursor formation due to the alkyl chains simultaneously.
4. Conclusion
The evolutions of regular emission products and unsaturated PAH precursors of diesel combustion are examined qualitatively and quantitatively using ReaxFF MD simulation. Cyclopentadienyl and benzene, generated from the precursors, tend to react into PAHs with five-, six- and seven-membered carbon rings via C2H2 or C2H3 addition. The substituted radicals favour the subsequent continuous mass growth by reacting with other resonantly-stabilized radicals and saturated/unsaturated aliphatic species, producing the aromatic aliphatically linked hydrocarbons. In the ReaxFF simulation, these larger hydrocarbon clusters undergo coalescence through aliphatic chain connection. This process results in rapid mass growth and eventually leads to the condensed nascent soot particle, which is characterised by an amorphous structure, around 3.7 nm in diameter and 998 amu in mass, a C:H ratio in the range of 6–10, and being rich in aliphatics, with an aliphatic-to-aromatic C:H ratio of 2.3.
The effects of oxygenated molecular structures on soot formation are quantified and reflected in two aspects: removing C-atoms from the soot precursor pool through combination with the O-atoms, and contributing to soot precursor formation due to the existence of C–C bonds simultaneously. Regarding the soot formation propensity, the C-atoms from DMC and DMM are less likely than diesel to appear in incipient soot particles, while MB, CPO and ethanol show similar tendencies to that of diesel. The CO double bond in the carbonyl group is sufficiently stable to survive under the high-temperature condition and favours the formation of CO while the production of CO2 in DMC and MB is mainly due to C–O fission between the ester moiety and the methyl group and the subsequent β-scission. The effect of oxygenated additives on the sooting propensity of diesel blends is the consequence of interactions of the oxygenated moiety and the aliphatics at the atomistic level.
Conflicts of interest
There are no conflicts of interest to declare.References
- A. K. Agarwal, J. G. Gupta and A. Dhar, Potential and challenges for large-scale application of biodiesel in automotive sector, Prog. Energy Combust. Sci., 2017, 61, 113–149 CrossRef.
- C. K. Westbrook, W. J. Pitz and H. J. Curran, Chemical kinetic modeling study of the effects of oxygenated hydrocarbons on soot emissions from diesel engines, J. Phys. Chem. A, 2006, 110, 6912–6922 CrossRef.
- P. Pepiot-Desjardins, H. Pitsch, R. Malhotra, S. Kirby and A. L. Boehman, Structural group analysis for soot reduction tendency of oxygenated fuels, Combust. Flame, 2008, 154, 191–205 CrossRef.
- X. Mercier, O. Carrivain, C. Irimiea, A. Faccinetto and E. Therssen, Dimers of polycyclic aromatic hydrocarbons: the missing pieces in the soot formation process, Phys. Chem. Chem. Phys., 2019, 21, 8282–8294 RSC.
- H. Wang, Formation of nascent soot and other condensed-phase materials in flames, Proc. Combust. Inst., 2011, 33, 41–67 CrossRef.
- H. Michelsen, Probing soot formation, chemical and physical evolution, and oxidation: a review of in situ diagnostic techniques and needs, Proc. Combust. Inst., 2017, 36, 717–735 CrossRef.
- J. P. Cain, J. Camacho, D. J. Phares, H. Wang and A. Laskin, Evidence of aliphatics in nascent soot particles in premixed ethylene flames, Proc. Combust. Inst., 2011, 33, 533–540 CrossRef.
- B. Zhao, Z. Yang, Z. Li, M. V. Johnston and H. Wang, Particle size distribution function of incipient soot in laminar premixed ethylene flames: effect of flame temperature, Proc. Combust. Inst., 2005, 30, 1441–1448 CrossRef.
- M. Keller, T. de Bruin, M. Matrat, A. Nicolle and L. Catoire, A Theoretical Multiscale Approach to Study the Initial Steps Involved in the Chemical Reactivity of Soot Precursors, Energy Fuels, 2019, 33, 10255–10266 CrossRef.
- A. Violi, A. Kubota, T. Truong, W. Pitz, C. Westbrook and A. Sarofim, A fully integrated kinetic Monte Carlo/molecular dynamics approach for the simulation of soot precursor growth, Proc. Combust. Inst., 2002, 29, 2343–2349 CrossRef.
- A. Violi, Modeling of soot particle inception in aromatic and aliphatic premixed flames, Combust. Flame, 2004, 139, 279–287 CrossRef.
- A. Violi and A. Venkatnathan, Combustion-generated nanoparticles produced in a benzene flame: a multiscale approach, J. Chem. Phys., 2006, 125, 054302 CrossRef.
- P. Elvati and A. Violi, Thermodynamics of poly-aromatic hydrocarbon clustering and the effects of substituted aliphatic chains, Proc. Combust. Inst., 2013, 34, 1837–1843 CrossRef.
- J. S. Lowe, J. Y. Lai, P. Elvati and A. Violi, Towards a predictive model for polycyclic aromatic hydrocarbon dimerization propensity, Proc. Combust. Inst., 2015, 35, 1827–1832 CrossRef CAS.
- K. Bowal, J. W. Martin and M. Kraft, Partitioning of polycyclic aromatic hydrocarbons in heterogeneous clusters, Carbon, 2019, 143, 247–256 CrossRef CAS.
- Q. Mao, A. C. T. van Duin and K. H. Luo, Formation of incipient soot particles from polycyclic aromatic hydrocarbons: a ReaxFF molecular dynamics study, Carbon, 2017, 121, 380–388 CrossRef CAS.
- Q. Mao, J. Zhou, K. H. Luo and A. C. T. van Duin, Atomistic insights into the dynamics of binary collisions between gaseous molecules and polycyclic aromatic hydrocarbon dimers, Phys. Chem. Chem. Phys., 2019, 21, 3849–3856 RSC.
- Q. Mao, D. Hou, K. H. Luo and X. You, Dimerization of polycyclic aromatic hydrocarbon molecules and radicals under flame conditions, J. Phys. Chem. A, 2018, 122, 8701–8708 CrossRef CAS.
- C. Ashraf and A. C. T. van Duin, Extension of the ReaxFF combustion force field toward syngas combustion and initial oxidation kinetics, J. Phys. Chem. A, 2017, 121, 1051–1068 CrossRef CAS.
- C. Ashraf, S. Shabnam, A. Jain, Y. Xuan and A. C. T. van Duin, Pyrolysis of binary fuel mixtures at supercritical conditions: a ReaxFF molecular dynamics study, Fuel, 2019, 235, 194–207 CrossRef CAS.
- X. Z. Jiang, M. Feng, W. Zeng and K. H. Luo, Study of mechanisms for electric field effects on ethanol oxidation via reactive force field molecular dynamics, Proc. Combust. Inst., 2019, 37, 5525–5535 CrossRef CAS.
- S. G. Srinivasan, A. C. T. van Duin and P. Ganesh, Development of a ReaxFF potential for carbon condensed phases and its application to the thermal fragmentation of a large fullerene, J. Phys. Chem. A, 2015, 119, 571–580 CrossRef.
- Y. Qian, L. Yu, Z. Li, Y. Zhang, L. Xu and Q. Zhou, et al., A new methodology for diesel surrogate fuel formulation: bridging fuel fundamental properties and real engine combustion characteristics, Energy, 2018, 148, 424–447 CrossRef CAS.
- Z. Chen, W. Sun and L. Zhao, Combustion mechanisms and kinetics of fuel additives: a ReaxFF molecular simulation, Energy Fuels, 2018, 32, 11852–11863 CrossRef CAS.
- S. Shabnam, Q. Mao, A. C. T. van Duin and K. H. Luo, Evaluation of the effect of nickel clusters on the formation of incipient soot particles from polycyclic aromatic hydrocarbons via ReaxFF molecular dynamics simulations, Phys. Chem. Chem. Phys., 2019, 21, 9865–9875 RSC.
- S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- M. Döntgen, M.-D. Przybylski-Freund, L. C. Kröger, W. A. Kopp, A. E. Ismail and K. Leonhard, Automated discovery of reaction pathways, rate constants, and transition states using reactive molecular dynamics simulations, J. Chem. Theory Comput., 2015, 11, 2517–2524 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- Z. Chen, W. Sun and L. Zhao, Initial mechanism and kinetics of diesel incomplete combustion: ReaxFF molecular dynamics based on a multicomponent fuel model, J. Phys. Chem. C, 2019, 123, 8512–8521 CrossRef CAS.
- C. S. McEnally, L. D. Pfefferle, B. Atakan and K. Kohse-Höinghaus, Studies of aromatic hydrocarbon formation mechanisms in flames: progress towards closing the fuel gap, Prog. Energy Combust. Sci., 2006, 32, 247–294 CrossRef CAS.
- S. V. Kalpathy, N. B. Poddar, S. P. Bagley and M. J. Wornat, Reaction pathways for the growth of polycyclic aromatic hydrocarbons during the supercritical pyrolysis of n-decane, as determined from doping experiments with 1-and 2-methylnaphthalene, Proc. Combust. Inst., 2015, 35, 1833–1841 CrossRef CAS.
- C. Shaddix, K. Brezinsky and I. Glassman, Analysis of fuel decay routes in the high-temperature oxidation of 1-methylnaphthalene, Combust. Flame, 1997, 108, 139–157 CrossRef CAS.
- F. Schulz, M. Commodo, K. Kaiser, G. De Falco, P. Minutolo and G. Meyer, et al., Insights into incipient soot formation by atomic force microscopy, Proc. Combust. Inst., 2019, 37, 885–892 CrossRef CAS.
- A. Farooq, W. Ren, K. Y. Lam, D. F. Davidson, R. K. Hanson and C. K. Westbrook, Shock tube studies of methyl butanoate pyrolysis with relevance to biodiesel, Combust. Flame, 2012, 159, 3235–3241 CrossRef CAS.
- W. Sun, C. Huang, T. Tao, F. Zhang, W. Li and N. Hansen, et al., Exploring the high-temperature kinetics of diethyl carbonate (DEC) under pyrolysis and flame conditions, Combust. Flame, 2017, 181, 71–81 CrossRef CAS.
- W. Sun, T. Tao, H. Liao, N. Hansen and B. Yang, Probing fuel-specific reaction intermediates from laminar premixed flames fueled by two C5 ketones and model interpretations, Proc. Combust. Inst., 2019, 37, 1699–1707 CrossRef CAS.
| This journal is © the Owner Societies 2020 |