
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon dioxide and propane nucleation: the emergence of a nucleation barrier†
Jan
Krohn‡
 ,
Martina
Lippe‡
,
Chenxi
Li§
and
Ruth
Signorell
*
,
Martina
Lippe‡
,
Chenxi
Li§
and
Ruth
Signorell
*
Department of Chemistry and Applied Biosciences, Laboratory of Physical Chemistry, ETH Zürich, Vladimir-Prelog-Weg 2, CH-8093 Zürich, Switzerland. E-mail: rsignorell@ethz.ch; Fax: +41 44 633 13 16; Tel: +41 44 633 46 21
First published on 23rd June 2020
Abstract
We investigate homogeneous gas-phase nucleation of CO2 and C3H8 in the uniform postnozzle flow of Laval expansions in the temperature range of 31.2 K to 62.9 K and 32.0 K to 42.1 K, respectively. Time-dependent cluster size distributions are recorded with mass spectrometry after single-photon ionization with vacuum ultraviolet light. Net monomer–cluster forward rate constants and experimental nucleation rates J are retrieved from the time-dependent cluster size distributions. The comparison of experimental enhancement factors derived from these net forward rates with calculated enhancement factors provides an indication for the transition from barrier-limited to barrierless nucleation. Our data suggest such a transition for CO2, but not for C3H8. The values of J lie in the range from 9 × 1014 cm−3 s−1 to 6 × 1015 cm−3 s−1. For CO2, the comparison of J with a modeled nucleation rate JQM based on quantum chemical calculations of the free energy barrier also hints at a transition from barrierless condensation to barrier-limited nucleation. Furthermore, we address the influence of the carrier gas pressure on the nucleation rate.
1 Introduction
Gas phase nucleation, the first step in the phase transition from the gas phase to the liquid or solid phase, is an important step in technical processes,1 environmental processes2,3 and health science.4,5 Despite its high relevance, nucleation is still poorly understood at a fundamental level. Gas-phase nucleation occurs in a supersaturated parent phase when the supersaturation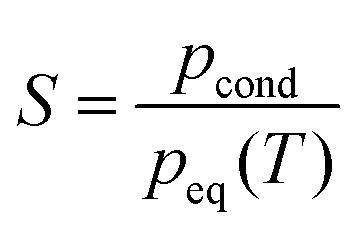 exceeds 1 (pcond is the partial pressure of the condensable and peq(T) is the equilibrium vapor pressure of the condensable at a given temperature). Nucleation processes are often characterized by the nucleation rate. Many different experimental studies have investigated nucleation rates with a variety of methods (see ref. 6–12 and references therein). Often, the number concentrations of particles of the new phase are retrieved from experimental data after nucleation and partial cluster growth have occurred, and classical nucleation theory (CNT) or variants of it are used to extract the nucleation rate. For some cases, it has been shown that the agreement between experimental nucleation rates and CNT predictions is very poor.7–9 Even though the exact reasons for the large deviations between experiments and theory are unknown, the use of bulk properties to describe the molecular process by CNT is a well-known issue. In order to improve the theoretical predictions, empirical corrections13 or microscopic corrections14–16 to CNT are used, but so far none of these approaches have been able to describe nucleation accurately over a wide range of experimental conditions and for different systems. Diemand et al.17 performed large scale molecular dynamics simulations for Ar nucleation, which enabled them to retrieve nucleation rates down to ∼1 × 1017 cm−3 s−1. They demonstrated good agreement with the experiments previously performed by Wyslouzil and coworkers.9 Unfortunately, this computationally very expensive method has major limitations: simulations for more complicated molecular systems and for low nucleation rates are still too costly to be performed.
exceeds 1 (pcond is the partial pressure of the condensable and peq(T) is the equilibrium vapor pressure of the condensable at a given temperature). Nucleation processes are often characterized by the nucleation rate. Many different experimental studies have investigated nucleation rates with a variety of methods (see ref. 6–12 and references therein). Often, the number concentrations of particles of the new phase are retrieved from experimental data after nucleation and partial cluster growth have occurred, and classical nucleation theory (CNT) or variants of it are used to extract the nucleation rate. For some cases, it has been shown that the agreement between experimental nucleation rates and CNT predictions is very poor.7–9 Even though the exact reasons for the large deviations between experiments and theory are unknown, the use of bulk properties to describe the molecular process by CNT is a well-known issue. In order to improve the theoretical predictions, empirical corrections13 or microscopic corrections14–16 to CNT are used, but so far none of these approaches have been able to describe nucleation accurately over a wide range of experimental conditions and for different systems. Diemand et al.17 performed large scale molecular dynamics simulations for Ar nucleation, which enabled them to retrieve nucleation rates down to ∼1 × 1017 cm−3 s−1. They demonstrated good agreement with the experiments previously performed by Wyslouzil and coworkers.9 Unfortunately, this computationally very expensive method has major limitations: simulations for more complicated molecular systems and for low nucleation rates are still too costly to be performed.
The rate-limiting step of nucleation is the formation of the critical cluster (nucleus), which corresponds to the cluster size where the Gibbs free energy reaches a maximum. The usual picture of gas-phase nucleation thus involves overcoming an energy barrier. However, in the limiting case of extremely high supersaturation, nucleation can become barrierless.18–21 Extremely high supersaturations can be achieved either by strongly increasing the concentration of the nucleating species or by substantially lowering the temperature. Alternatively, the energy barrier can also be modified by introducing another substance that provides an alternative nucleation pathway.18,19 The identification of the emergence (or disappearance) of an energy barrier as a function of thermodynamic variables is an important step towards a better molecular-level understanding of the nucleation process.
The transition from barrierless to barrier-limited nucleation can be identified in different ways. One possibility is the direct comparison between experimental nucleation rates and nucleation rates predicted for the gas kinetic limit. The main limitation of this approach arises from the usual approximation that the association rate is given by the collision rate. This only holds in the high pressure limit, i.e. for unit sticking probability. Another method is to utilize the first nucleation theorem to examine the size shift of the critical cluster for different supersaturations at constant temperature,22,23 which can be very challenging experimentally. The emergence (or disappearance) of an energy barrier is also reflected in the change of the relative magnitude of the cluster association and evaporation rates for varying conditions. Hence, the characterization of this change can also be used to probe the transition from barrierless to barrier-limited behavior. To the best of our knowledge, this approach has not yet been applied to experimental data, simply because the evaporation rates of clusters are usually not directly accessible in experiments.
In this work, we study homogeneous gas phase nucleation of weakly bound CO2 and C3H8 clusters at the molecular level in the uniform postnozzle flow of Laval expansions using soft single-photon ionization coupled with time-of-flight mass spectrometry.24–29 These experiments provide cluster size distributions as a function of the temperature, the concentration of the condensable gas and the nucleation time. Experimental nucleation rates are directly determined from these time-dependent, cluster size-resolved data and compared with predicted nucleation rates. Our measurements are performed at high supersaturations (S > 1011), where the region of barrierless nucleation or the transition to barrierless nucleation is reached. In our previous work on H2O nucleation,30 we developed a framework based on the general dynamic equation (GDE)31 to calculate monomer–cluster association rate constants from experimental data under the assumption of negligible evaporation. The latter assumption is valid for H2O clusters at 47.5 K and 87.0 K, and means that H2O condensation is barrierless under these conditions. Here, we apply the same framework to understand CO2 and C3H8 nucleation in a similar temperature range. Since CO2 and C3H8 clusters are more weakly bound than H2O clusters, it is not unlikely that the transition regime to barrier-limited nucleation is reached, in particular for CO2. Instead of extracting actual association rate constants as in the H2O nucleation study, we use the GDE framework to compare the relative magnitude of monomer-cluster association rate constants and cluster evaporation rate constants as we increase the system temperature from ∼30 K to 60 K for CO2. This allows us to identify the emergence of a nucleation barrier. In an attempt to partially overcome the deficiencies of CNT, we also compare our experimental results with density functional theory (DFT) calculations to gain information on the stability of the clusters.
2 Experiment
2.1 Experimental setup
The experimental setup has been described in detail in our previous publications,24–30 and is thus only briefly discussed here. Fig. 1 shows a simplified schematic of our experimental setup. Two pulsed feeding valves with an opening time of 6 μs and a repetition rate of 20 Hz are used to supply the gas mixture to the stagnation volume with stagnation pressure p0 and temperature T0. We use mass flow controllers to regulate the flow of the carrier gas (Ar, PanGas 5.0), the internal standard gas (CH4, Messer 5.5) and the condensable gas (CO2, PanGas 4.5 or C3H8, Linde 3.5). The gas mixture is expanded through the Laval nozzle generating a uniform flow at the nozzle exit with flow temperature TF and pressure pF. The uniform flow can be extended into the postnozzle region (100 mm in length) by matching the background pressure to pF. The Laval nozzle is mounted on a linear translation stage that allows us to change the axial distance l between the nozzle exit and skimmer in steps as small as 1 mm with a positioning error of 25 μm for 100 mm translation. A change in l corresponds to a change in the nucleation time t. Depending on the properties of the expansion, the maximum time span that can be covered is ∼200 μs with a temporal resolution of ∼2 μs. The Mach number M and flow temperature TF are determined from p0 and from the impact pressures pI using the Rayleigh–Pitot equation, which is valid under isentropic flow conditions and for ideal gas behavior (for more information see ref. 24–30). 20 to 40 individual measurements of p0 and pI are typically recorded for a given axial distance. From these measurements, the axially averaged flow temperature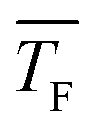 and Mach number
and Mach number 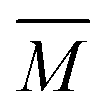 with standard deviations (1σ) are determined. A skimmer (1 mm in diameter) is used to sample the core part of the postnozzle flow. We use single photons at 13.8 eV (89.8 nm) generated by a home-built table-top vacuum ultraviolet (VUV) laser to ionize the clusters. The VUV-photons are generated in a two-color-four-wave mixing process in a krypton expansion at 20 Hz. Single-photon ionization has been proven to be a soft ionization technique also for weakly bound clusters.32–36 The cluster ions are then accelerated using a Wiley McLaren type mass spectrometer with acceleration voltages up to 30 kV and finally detected using a microchannel plate (MCP) detector. The monomer has a 103–4 times higher abundance than the clusters. Therefore, the monomer and the clusters have to be recorded in separate measurements using different experimental settings.28 A deflector electrode located in front of the MCP is used to deflect the monomer during the cluster measurements to avoid saturation effects on the MCP.
with standard deviations (1σ) are determined. A skimmer (1 mm in diameter) is used to sample the core part of the postnozzle flow. We use single photons at 13.8 eV (89.8 nm) generated by a home-built table-top vacuum ultraviolet (VUV) laser to ionize the clusters. The VUV-photons are generated in a two-color-four-wave mixing process in a krypton expansion at 20 Hz. Single-photon ionization has been proven to be a soft ionization technique also for weakly bound clusters.32–36 The cluster ions are then accelerated using a Wiley McLaren type mass spectrometer with acceleration voltages up to 30 kV and finally detected using a microchannel plate (MCP) detector. The monomer has a 103–4 times higher abundance than the clusters. Therefore, the monomer and the clusters have to be recorded in separate measurements using different experimental settings.28 A deflector electrode located in front of the MCP is used to deflect the monomer during the cluster measurements to avoid saturation effects on the MCP.
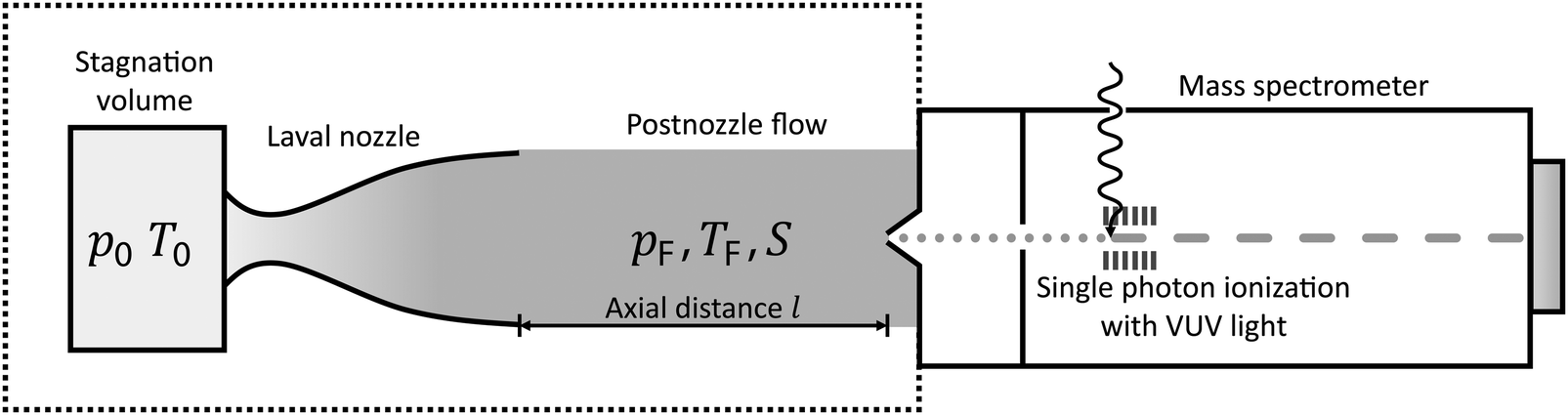 | ||
| Fig. 1 Simplified schematic of the experimental setup with the Laval setup and a mass spectrometer. T0 and p0 are the stagnation temperature and pressure, respectively, and TF and pF are the flow temperature and pressure, respectively. S is the supersaturation. The axial distance l is the distance between the nozzle exit and skimmer. | ||
2.2 Data processing
The determination of the cluster number concentration from the recorded mass spectra is described in our previous publications.28,30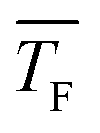 is varied by changing the Laval nozzle and by changing the compositions of the carrier gas (concentrations of Ar and CH4; see Table 1). CH4 gas also serves as an internal standard. The number concentration of CH4, NCH4, is calculated from the ideal gas law. The number concentration of the clusters with n monomer units, Nn, is determined by:
is varied by changing the Laval nozzle and by changing the compositions of the carrier gas (concentrations of Ar and CH4; see Table 1). CH4 gas also serves as an internal standard. The number concentration of CH4, NCH4, is calculated from the ideal gas law. The number concentration of the clusters with n monomer units, Nn, is determined by: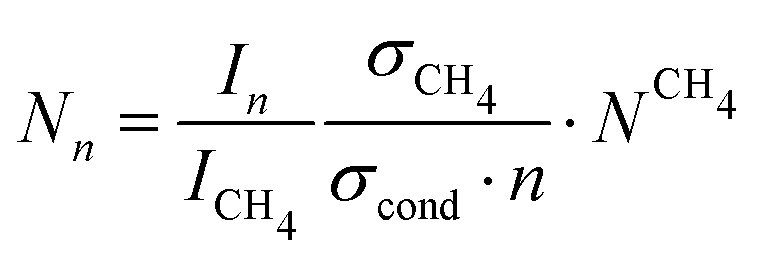 | (1) |
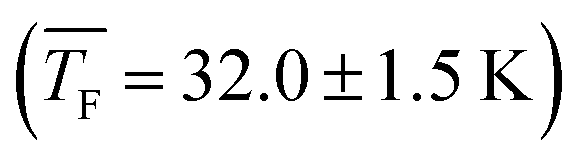 , CH4 forms co-clusters with C3H8. In this case, we use the C3H8 monomer itself as the internal standard for the determination of the cluster number concentrations. This is reasonable as monomer depletion due to cluster formation is negligible under our conditions. We estimate an overall experimental uncertainty of the cluster number concentrations of about a factor of five for all cluster sizes (see ref. 28 and 30).
, CH4 forms co-clusters with C3H8. In this case, we use the C3H8 monomer itself as the internal standard for the determination of the cluster number concentrations. This is reasonable as monomer depletion due to cluster formation is negligible under our conditions. We estimate an overall experimental uncertainty of the cluster number concentrations of about a factor of five for all cluster sizes (see ref. 28 and 30).
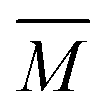 and
and  are the axially averaged Mach number and flow temperature, respectively, and S is the supersaturation
are the axially averaged Mach number and flow temperature, respectively, and S is the supersaturation
| Symbol | 50% CO2 | 10% CO2 | 3.3% CO2 | 1.1% CO2 | 0.12% CO2 | 0.26% C3H8 | 0.035% C3H8 |
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
▲ |
|
|
| Nozzle | Mach 5.2 | Mach 5.2 | Mach 5.2 | Mach 5 | Mach 6 | Mach 5 | Mach 6 |
| p cond/Pa | 13.25 | 2.65 | 0.88 | 0.37 | 0.04 | 0.086 | 0.012 |
| p F/Pa | 26.5 | 26.5 | 26.5 | 33 | 35 | 33 | 35 |
| % CH4 | 7.7 | 23.1 | 15.4 | 1.7 | 1.5 | 0.87 | 0 |
| % Ar | 42.3 | 66.9 | 81.3 | 97.1 | 98.3 | 98.9 | 100 |

|
62.9 ± 1.1 | 56.6 ± 1.3 | 49.3 ± 1.3 | 44.4 ± 1.5 | 31.2 ± 1.3 | 42.1 ± 1.1 | 32.0 ± 1.5 |
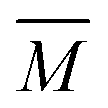
|
3.83 ± 0.06 | 4.16 ± 0.06 | 4.65 ± 0.07 | 4.17 ± 0.08 | 4.89 ± 0.11 | 4.28 ± 0.06 | 4.96 ± 0.11 |
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) S S |
26 | 30 | 38 | 45 | 78 | 44 | 66 |
| J/cm−3 s−1 | 3.6 × 1015 | 4.0 × 1015 | 3.4 × 1015 | 2.7 × 1015 | 1.5 × 1015 | 9.1 × 1014 | 5.8 × 1015 |
| J CNT/cm−3 s−1 | 1.1 × 1022 | 6.5 × 1020 | 1.1 × 1020 | 2.4 × 1019 | 4.7 × 1017 | 8.5 × 1017 | 6.6 × 1016 |
| J MKNT/cm−3 s−1 | 5.9 × 103 | 6.8 × 10−1 | 5.3 × 10−3 | 1.2 × 10−4 | 3.7 × 10−8 | 2.4 × 10−6 | 8.1 × 10−10 |
| J HS/cm−3 s−1 | 1.8 × 1022 | 8.5 × 1020 | 1.1 × 1020 | 2.4 × 1019 | 4.7 × 1017 | 2.2 × 1018 | 6.6 × 1016 |
| J inter/cm−3 s−1 | 5.3 × 1022 | 2.6 × 1021 | 3.6 × 1020 | 7.9 × 1019 | 1.8 × 1018 | 4.2 × 1018 | 1.5 × 1017 |
| J QM/cm−3 s−1 | 5.4 × 1017 | 2.6 × 1016 | 3.5 × 1016 | 3.3 × 1016 | 4.7 × 1016 | — | — |






At early nucleation times t, i.e. when monomer depletion and coagulation can be neglected, the total cluster number concentration summed over all n > nc, Ncluster,tot>nc, provides direct access to the experimental nucleation rate J (see Fig. 5 in ref. 28 and Sections 4.4 and 4.5 in this work):
 | (2) |
Evaporation of a few monomer units upon photoionization cannot be excluded. To estimate the maximum number of monomer units that could be evaporated, let us assume that all excess energy provided by the photons resides in the cluster instead of being converted to the kinetic energy of the photoelectron. For this estimate, we use the vaporization enthalpy of CO2 and C3H8.40–42 In the case of CO2, where ionization takes place very close to the lowest ionization energy, the simple estimate suggests that not even a single monomer unit would be evaporated. In the case of C3H8, for which the excess energy is higher, several monomer units could be evaporated. However, a large part of the excess energy is typically converted into the kinetic energy of the photoelectron and is thus not deposited into the cluster. Therefore, the true number of monomer units being evaporated from C3H8 clusters is likely less than a few molecules. We have tested that our kinetic model yields comparable results for the evaporation of one monomer unit upon ionization compared with no evaporation. Based on these estimates and results, we assume in the following that no monomer units are evaporated.
3 Modeling
3.1 Fit of the net forward rate constant to experimental data
The following describes the model used to extract the net forward rate constants from the experimental data. In eqn (3), we illustrate the kinetic pathways: k1j is the true association rate constant, i.e. for the formation of cluster j + 1 from cluster j. Nj+1 and Nj are the respective number concentrations. Ej+1 is the evaporation rate constant of cluster j + 1. As nucleation proceeds at constant temperature and pressure in our experiments, we assume that k1j and Ej+1 are constant too.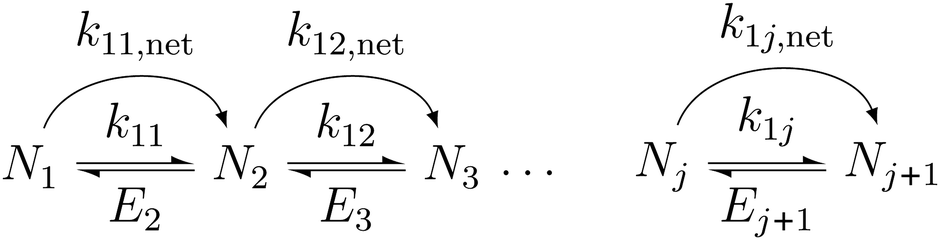 | (3) |
 | (4) |
3.2 Molecular collision model and enhancement factor
For simplicity, monomer–cluster association rate constants are often approximated by hard sphere collision rate constants k1j,HS that are given by:30,43 | (5) |
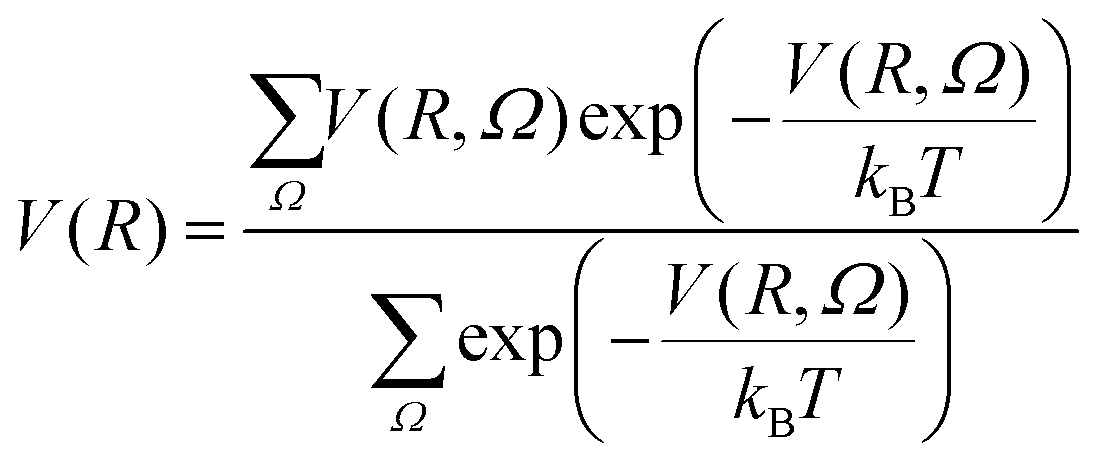 | (6) |
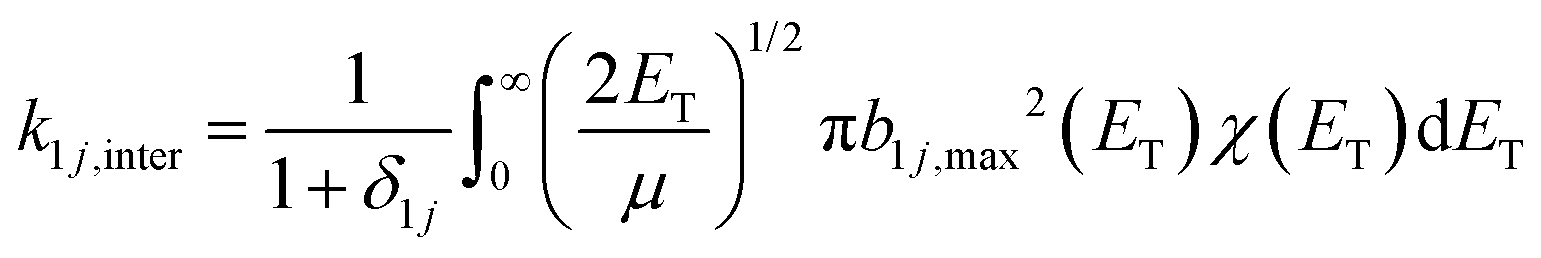 | (7) |
We define two enhancement factors to facilitate the interpretation of our data. The experimental enhancement factor ηexp is the ratio between the net forward rate constant and the hard sphere collision rate constant:
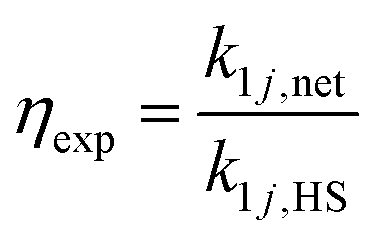 | (8) |
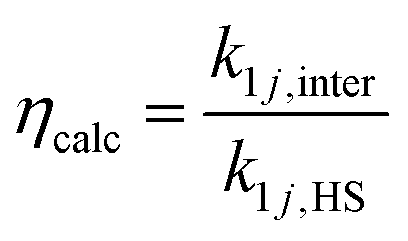 | (9) |
3.3 Nucleation rates
| JHS = N12k11,HS | (10) |
| Jinter = N12k11,inter | (11) |
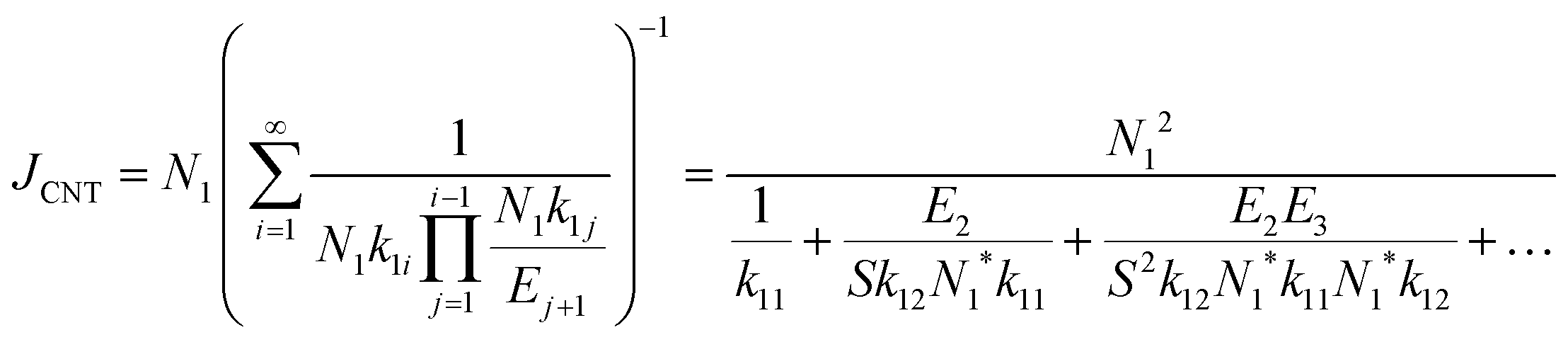 | (12) |
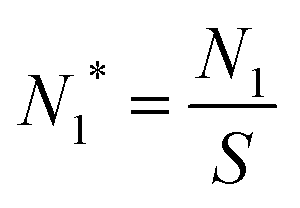 . The equation can be expressed as a function of the Gibbs free energy of formation of cluster j using detailed balance:54,55
. The equation can be expressed as a function of the Gibbs free energy of formation of cluster j using detailed balance:54,55 | (13) |
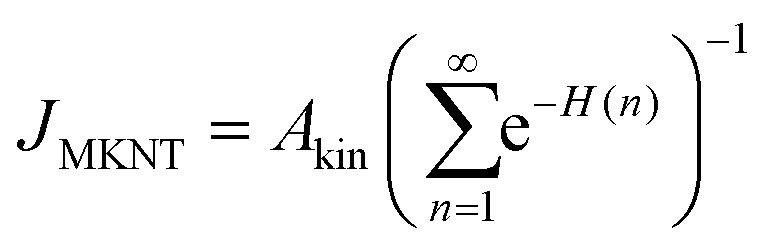 | (14) |
 for a cluster with n molecules and ns surface molecules. The last term in −H(n) includes the dependence of k1n,HS on the cluster size n.
for a cluster with n molecules and ns surface molecules. The last term in −H(n) includes the dependence of k1n,HS on the cluster size n. 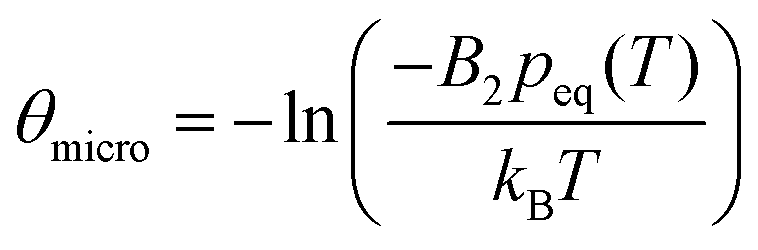 is the microscopic surface tension using the second virial coefficient B2. The kinetic prefactor Akin is calculated as follows:
is the microscopic surface tension using the second virial coefficient B2. The kinetic prefactor Akin is calculated as follows: 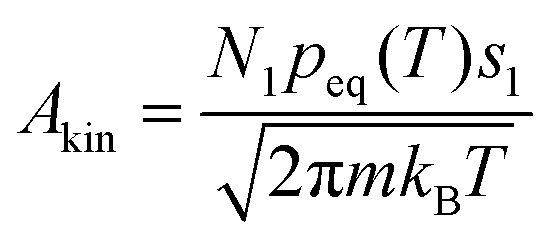 , m is the monomer mass, s1 is the surface area of the monomer and peq is the equilibrium vapor pressure. The formulae used for the thermodynamic properties of CO2 and C3H8 can be found in the ESI.†
, m is the monomer mass, s1 is the surface area of the monomer and peq is the equilibrium vapor pressure. The formulae used for the thermodynamic properties of CO2 and C3H8 can be found in the ESI.†
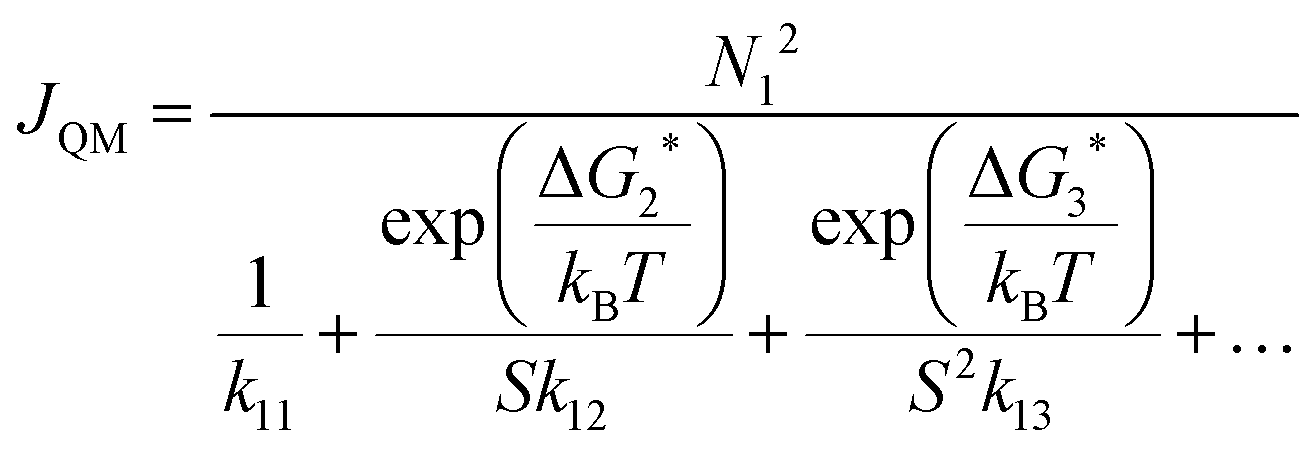 | (15) |
3.4 DFT calculations
The Gaussian09 program package61 was used for all DFT calculations. The starting geometries for the DFT calculations of the CO2 clusters were taken from our previous publication,28 which used the M06-2X functional62 and 6-31+G(d) basis set. For the calculation of JQM (see Section 3.3.4) these were further refined using the same M06-2X functional, but a larger, correlation-consistent basis-set (aug-cc-pVTZ).63 The Gibbs free energy change ΔGj* associated with the reaction j·CO2 → (CO2)j was calculated for all temperatures (Table 1 and Table SI, ESI†) using zero-point and thermal corrections, and applying a scaling factor of 0.956 for the vibrational frequencies.64 As already discussed in ref. 29, Lemke et al.65 showed that the M06-2X functional is appropriate for dispersion bound systems and the extracted energies are consistent with CCSD(T) level calculations. Our calculations are in good agreement with previous studies.65,66 The refined structures and ΔGj* for cluster sizes j = 2–12 are provided in the ESI.†4 Results and discussion
4.1 Mass spectra and kinetic modeling
Fig. 2 shows mass spectra as a function of the nucleation time for a low CO2 concentration (0.12%, a) and a high CO2 concentration (10%, b) (see Table 1 for conditions). The maximal cluster sizes nmax are indicated by arrows in Fig. 2a and b. The spectra are background corrected for clarity. For both conditions, nmax = 1 is observed at the shortest time (44 μs and 36 μs, respectively). At a lower concentration (Fig. 2a), only small clusters are observed even for long nucleation times (maximal cluster size nmax ∼ 40 at t = 196 μs). A similar behavior was observed for water nucleation in our previous publication.28 At a higher concentration (Fig. 2b), very large clusters with several hundred molecules (nmax up to ∼700) are observed at long nucleation times. The explanation for this huge increase in nmax is the higher CO2 content which leads to faster cluster growth. The insets in Fig. 2a and b show zoom-ins of the 0.12% CO2 mass spectrum recorded at 196 μs (red, Fig. 2a) and the 10% CO2 mass spectrum recorded at 45 μs (blue, Fig. 2b). These spectra are chosen for comparison because their nmax values are relatively close. A comparison reveals that the very small clusters (n < 15) are much less intense for the high CO2 concentration (blue trace) than for the low CO2 concentration (red trace). For the larger clusters (n ≥ 15), this difference is less pronounced. The diminished intensity of the very small clusters for 10% CO2 might hint at evaporation of these very small clusters.67 This hypothesis will be evaluated in more detail in Sections 4.3 and 4.4. As already discussed in ref. 29, we observe shell and subshell closings in the CO2 mass spectra for cluster sizes larger than 50–100, which allows us to directly extract the structure of the clusters.68–70 For all our conditions, cuboctahedral structures are observed (no structural change with increasing temperature). Representative mass spectra of C3H8 are shown in the ESI† (Fig. S2). They show a similar behavior as the CO2 mass spectra in Fig. 2a. | ||
| Fig. 2 CO2 mass spectra as a function of the cluster size n and nucleation time t: (a) 0.12% CO2 at a flow temperature of 31.2 K and (b) 10% CO2 at a flow temperature of 56.6 K. Further information can be found in Table 1. The mass spectra for these two temperatures exhibit a distinctly different temporal evolution. The insets show selected mass spectra with similar nmax values: 0.12% CO2 at 196 μs (red, a) and 10% CO2 at 45 μs (blue, b). | ||
4.2 Fit of the net forward rate constants
Using the model in Section 3.1, we can directly fit net forward rate constants k1j,net to the experimental cluster size distributions retrieved from the mass spectra (Fig. 2, see Section 3.1 and the ESI† for fits). The extracted net forward rate constants for CO2 and C3H8 are shown in Fig. 3a and b, respectively. The monomer–monomer net forward rate constant k11,net will be discussed in Section 4.4. Note that k1j,net gives information about the balance of the association rate, determined by k1j, and the evaporation rate, determined by Ej+1, but it cannot provide information about their absolute values. For both substances, CO2 and C3H8, the net forward rate constants generally increase for larger cluster sizes (Fig. 3). There are two possible explanations for this: Firstly, the collision cross section increases with increasing cluster size. Secondly, for smaller clusters the evaporation rate is likely higher and the sticking probability is likely lower. The latter could be attributed to the smaller density of states in smaller clusters. This makes the distribution of the excess energy upon collision less efficient and leads to a lower sticking probability, resulting in a lower forward rate constant for smaller clusters. The retrieved k1j,net values increase with decreasing temperature for both CO2 and C3H8 (Fig. 3). At first sight, this seems counter-intuitive because the hard sphere collision rate constant (eqn (5)) increases with increasing temperature. This observation is further discussed in Section 4.3.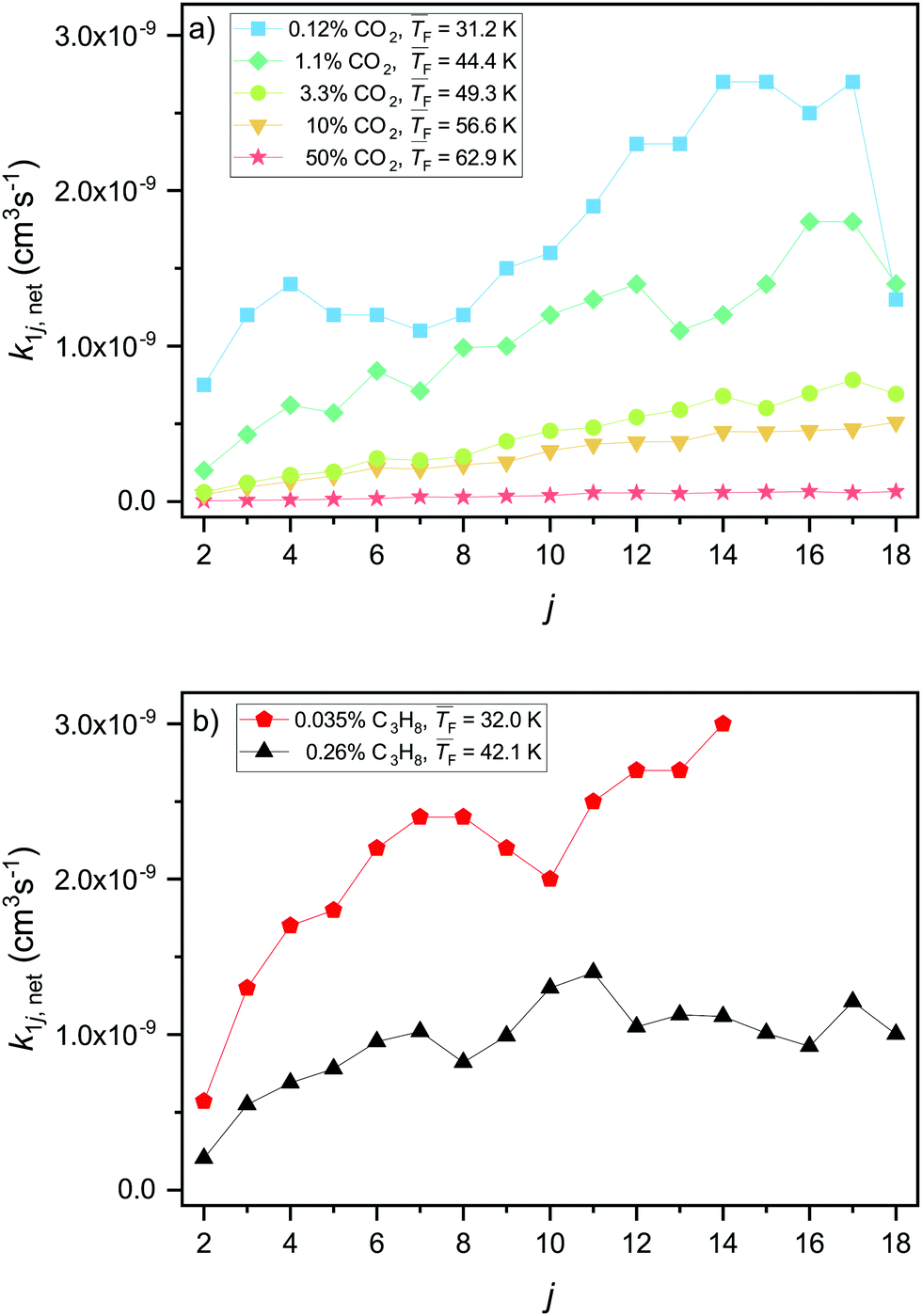 | ||
| Fig. 3 Net forward rate constants k1j,net of CO2 (a) and C3H8 (b) for j ≥ 2. The net forward rate constants combine the contributions from association and evaporation. | ||
4.3 Enhancement factors
The experimental net association rate constant k1j,net (eqn (3)) embodies the net effect of three different processes: (a) monomer–cluster collision (capture), (b) monomer–cluster sticking and (c) cluster evaporation. The enhancement factor ηexp gives the ratio of the aforementioned net forward rate constants and the hard sphere collision rate constants k1j,HS (see Section 3.2). In the limit of unit sticking probability and negligible evaporation (barrierless), k1j,net should be higher than k1j,HS, because of the long-range interactions between the colliding entities, leading to ηexp > 1. A value of ηexp < 1 thus indicates non-unit sticking probability or cluster evaporation. In the following, ηexp is utilized as an indicator to analyze how the nucleation process changes as a function of the temperature by comparison with ηcalc. ηexp is shown in Fig. 4a for CO2 and in Fig. 4d for C3H8. In both cases, ηexp decreases pronouncedly with increasing temperature. Fig. 4b shows the calculated enhancement factor ηcalc for CO2 as described in Section 3.2. A comparison of the temperature dependence of ηcalc and ηexp for two selected cluster sizes is shown in Fig. 4c. The calculations for CO2 (panel b and c) predict only a small decrease in ηcalc with increasing temperature, which is consistent with a T−1/3 dependence of k1j,inter. This is expected for long-range interactions in collisions of non-polar molecules.50 The T−1/3 dependence is indicated in Fig. 4c as a dashed red line. ηexp shows a far stronger temperature dependence, spanning two orders of magnitude for temperatures between 31.2 K and 62.9 K. Averaged over all j, ηexp surpasses ηcalc by only a factor of ∼2.5 at 31.2 K and by only a factor of ∼1.3 at 44.4 K, showing a general good agreement between experiment and calculation for the two lowest temperatures. At the highest temperature, by contrast, ηcalc is up to two orders of magnitude larger than ηexp. Furthermore, ηexp is strongly cluster size dependent at high temperatures, while the model predicts an approximately constant enhancement as a function of j for all temperatures (see Fig. 4b). These differences point towards a fundamental change in the nucleation kinetics with increasing temperature as will be discussed below.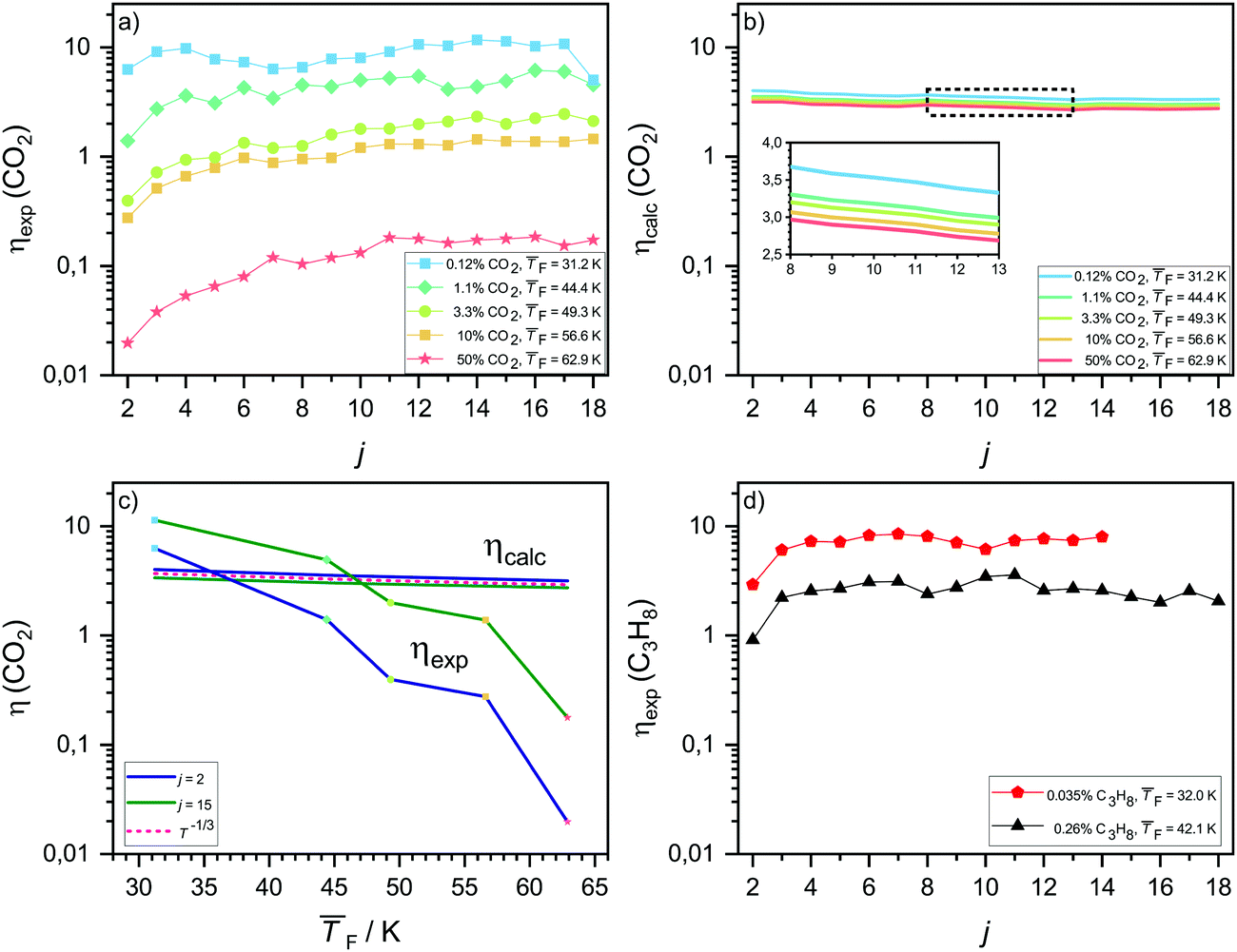 | ||
| Fig. 4 (a) Experimental ηexp and (b) calculated ηcalc enhancement factors for a range of CO2 cluster sizes and five different temperatures. (c) Temperature dependence of CO2 enhancement factors for a small (j = 2) and a larger cluster size (j = 15). (d) ηexp for C3H8 as a function of the cluster size j. | ||
The pronounced increase of ηexp from j = 2 to about j = 6 (Fig. 4a) for all but the coldest temperature indicates a low sticking efficiency and/or high evaporation rate for smaller clusters. With increasing temperature, ηexp falls off rapidly for small cluster sizes and thus increasingly deviates from ηcalc (blue line in Fig. 4c), supporting this interpretation. It is, however, difficult to disentangle possible contributions from the low sticking efficiency and the evaporation rate. As discussed before, compared with larger clusters smaller clusters have fewer degrees of freedom to distribute the excess collision energy, which results in a decreased sticking efficiency. For larger clusters (green line in Fig. 4c) many degrees of freedom are available, making a low sticking efficiency less likely than for small clusters. Therefore, the strong deviation between ηexp and ηcalc even for larger cluster sizes rather hints that increasing evaporation rates are responsible for the observed deviations with increasing temperature. Non-negligible evaporation at higher temperatures implies a change from barrierless nucleation at 31.2 K to barrier-limited nucleation at higher temperatures.
The experimental enhancement for C3H8 (Fig. 4d) is roughly constant for all cluster sizes at a specific flow temperature, except for j = 2 where ηexp is significantly lower. The increase by ∼10 K from 32.0 K to 42.1 K leads to a decreased ηexp by approximately a factor of three averaged over all j. For both temperatures studied for C3H8, we find a similar behavior to that for CO2 at 31.2 K, indicating barrierless nucleation for C3H8 at both temperatures.
In our previous work on water nucleation,30 cluster evaporation was found to be negligible up to 87 K. While negligible evaporation at these low temperatures seems reasonable for strongly hydrogen-bonded water clusters, this is no longer the case for weakly bound CO2 clusters – in agreement with the evidence for non-negligible evaporation for the CO2 clusters described above (see also the further discussions in Section 4.4).
4.4 Nucleation rates of CO2
Fig. 5a shows the total cluster number concentrations for n > 1 as a function of the nucleation time for the five CO2 concentrations examined in this study. The experimental nucleation rates J were determined by linear fitting of these data (eqn (2)). J is shown in Table 1 and Fig. 5b. Except for 50% CO2 and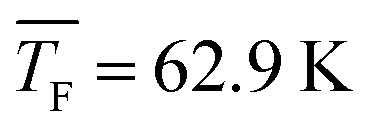 , there seems to be a small increase in J with increasing flow temperature (from 1.5 × 1015 cm−3 s−1 for 0.12% CO2 and
, there seems to be a small increase in J with increasing flow temperature (from 1.5 × 1015 cm−3 s−1 for 0.12% CO2 and 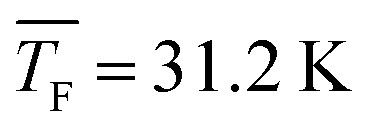 to 4.0 × 1015 cm−3 s−1 for 10% CO2 and
to 4.0 × 1015 cm−3 s−1 for 10% CO2 and 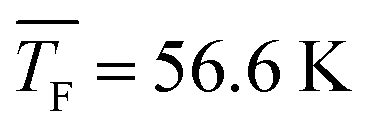 ), which, however, barely exceeds our relative experimental uncertainty. The issue with comparable J values lies in its dependence on the monomer concentration N1 which varies for different conditions (see Table 1). k11,net, by contrast, eliminates this dependence on N1, providing a better comparison between the different conditions. Fig. 5b shows that k11,net decreases strongly by four orders of magnitude in the range from 31.2 K to 62.9 K. Again, this decrease can be attributed to a lower sticking probability between monomers and/or a higher evaporation rate of dimers with increasing temperature. The decreased sticking probability should be most pronounced in monomer–monomer collisions. Even when evaporation rates are negligible and nucleation proceeds barrierless, k11,net can be significantly smaller than k11,HS, indicating that dimer formation has not yet reached the high pressure limit.71
), which, however, barely exceeds our relative experimental uncertainty. The issue with comparable J values lies in its dependence on the monomer concentration N1 which varies for different conditions (see Table 1). k11,net, by contrast, eliminates this dependence on N1, providing a better comparison between the different conditions. Fig. 5b shows that k11,net decreases strongly by four orders of magnitude in the range from 31.2 K to 62.9 K. Again, this decrease can be attributed to a lower sticking probability between monomers and/or a higher evaporation rate of dimers with increasing temperature. The decreased sticking probability should be most pronounced in monomer–monomer collisions. Even when evaporation rates are negligible and nucleation proceeds barrierless, k11,net can be significantly smaller than k11,HS, indicating that dimer formation has not yet reached the high pressure limit.71
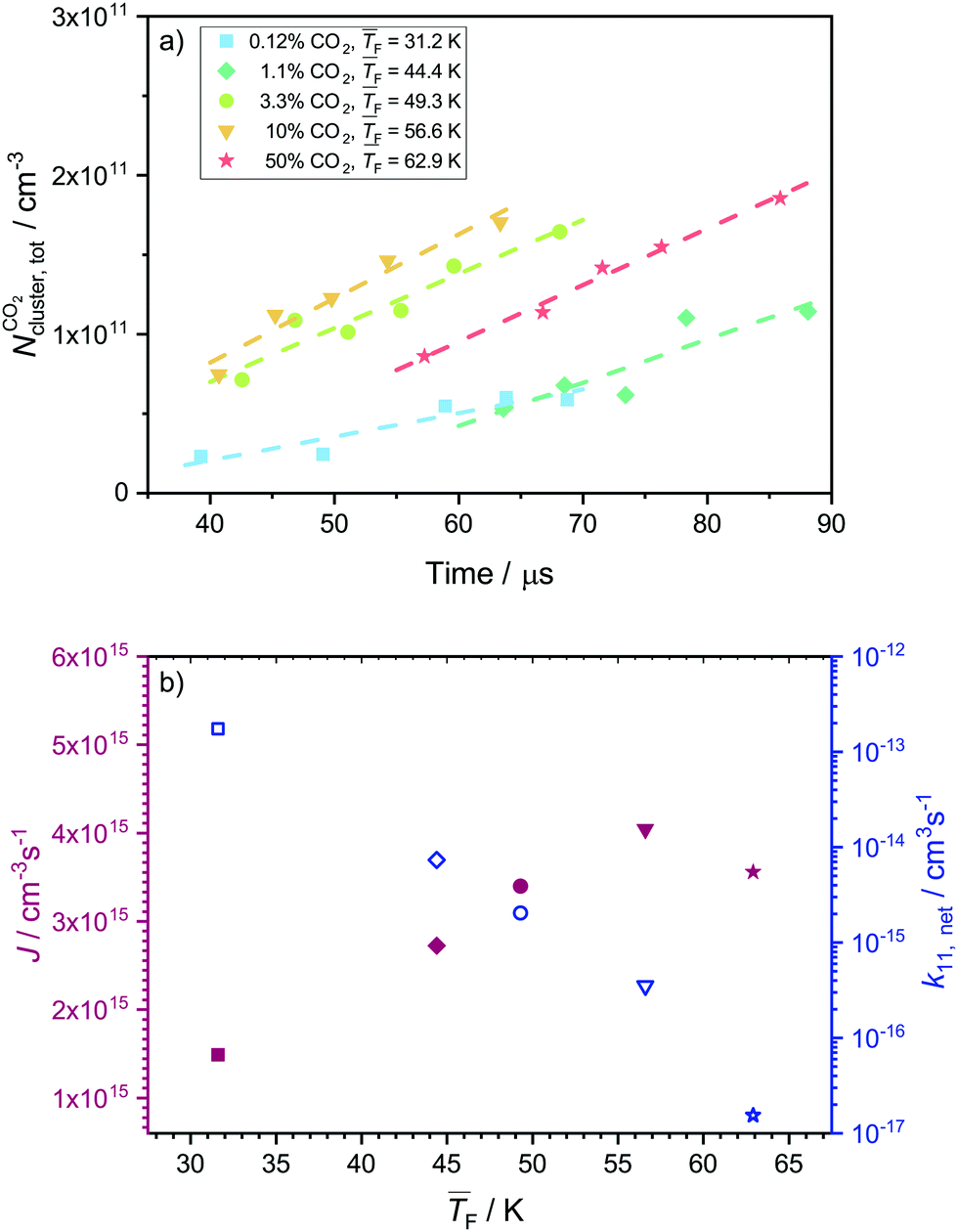 | ||
| Fig. 5 (a) Total cluster number concentration as a function of time for 50%, 10%, 3.3% 1.13% and 0.12% CO2. The linear fits are indicated with dashed lines. The data for 0.12% CO2 were already published in our previous publication.29 (b) Experimental nucleation rate J (full red symbols) and monomer–monomer net forward rate constants k11,net (open blue symbols) extracted from the data shown in panel a. The use of the symbols is consistent in panels a and b. | ||
A direct comparison with previously published nucleation rates is not possible because all previous values were recorded at higher temperatures. At somewhat higher temperatures of >70 K, Wyslouzil and coworkers (Wyslouzil, BE, personal communication) found nucleation rates of ∼1 × 1017 cm−3 s−1, i.e. rates that are ∼2 orders of magnitude higher than the present rates (∼1 × 1015 cm−3 s−1). Such an increase seems reasonable. Note that we cannot find overlap with their conditions because of experimental limitations in t and pF. CO2 nucleation was also studied by Duff72 in the temperature range between 160 K and 190 K and by Lettieri et al.73 in the range between 260 K and 300 K. These measurements lie far outside our experimental conditions and a comparison is thus not possible. For further analysis, we turn to comparisons with results from nucleation theories.
The predictions of the nucleation rate from CNT (Section 3.3.2) and from MKNT (Section 3.3.3) are shown in Table 1. The used thermodynamic properties of CO2 and C3H8 can be found in the ESI.† As expected, CNT yields the hard sphere collision limit JHS or values very close to it, which lies 2 to 7 orders of magnitude above our experimental nucleation rates. MKNT has been shown to provide decent estimates for nucleation rates of Ar in the range of ∼34 K to 53 K, with nucleation rates of ∼1 × 1017 cm−3 s−1 and lnS of up to ∼10 (see ref. 9). In the present study, ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) S varies between ∼26 and ∼78. At such extreme supersaturations, MKNT fails to describe condensation accurately as it gives unreasonably low values. MKNT does not seem to describe the surface free energy of clusters properly for cluster sizes below the bulk coordination number. From the enhancement model outlined in Section 3.2, we can determine Jinter. The enhanced rates result in a nucleation rate which is roughly twice JHS and therefore differs even further from the experimental J. The strong differences between J and Jinter for all conditions support a low sticking probability for monomer–monomer collisions.
S varies between ∼26 and ∼78. At such extreme supersaturations, MKNT fails to describe condensation accurately as it gives unreasonably low values. MKNT does not seem to describe the surface free energy of clusters properly for cluster sizes below the bulk coordination number. From the enhancement model outlined in Section 3.2, we can determine Jinter. The enhanced rates result in a nucleation rate which is roughly twice JHS and therefore differs even further from the experimental J. The strong differences between J and Jinter for all conditions support a low sticking probability for monomer–monomer collisions.
As described in Section 3.3.4, we can use the kinetic framework of CNT and ΔGj* from DFT calculations to calculate JQM. The calculated values for ΔGj* can be found in the ESI.†JQM can be understood as a correction to the capillarity approximation in CNT using cluster formation energies. Cluster evaporation is explicitly considered here by using the method of detailed balance (see Section 3.3.4 and ref. 59), but the high pressure limit is still assumed, leading to a potential overestimation of the nucleation rate. Out of all calculated nucleation rates JQM is the closest to our measured rates. Except for the highest temperature (Table 1), JQM lies only around one magnitude above J, which lies within the experimental uncertainty. Non-unit sticking would lower JQM, and thus bring JQM probably closer to J. However, it cannot completely be excluded that the good agreement between J and JQM could be coincidental because of the uncertainties of JQM arising from the accuracy of the DFT calculations.
The DFT calculations predict negligible evaporation below ∼26 K, but non-negligible evaporation above ∼26 K: at temperatures below ∼26 K, JQM is nearly identical to JHS. This implies a transition from barrierless nucleation at ∼26 K to nucleation in the presence of a barrier above ∼26 K. A sensitivity analysis of JQM with respect to the uncertainty of ΔGj* reveals that a 5% change of ΔGj* results in a change in the nucleation rate of one to two orders of magnitude (see the ESI† for details). This in turn would convert into an uncertainty of the transition temperature (above which JQM < 0.9 × JHS) that ranges from 20.1 K to 30.8 K (see the ESI,† Fig. S7). The uncertainty of this transition region combined with our experimental uncertainties suggests that this region might indeed lie in the temperature range covered by our experiment.
From the DFT calculations, we can also gain information about the size of the critical nucleus. The largest term in the denominator of eqn (15) allows one to determine the critical cluster. At 62.9 K and 56.6 K the critical nucleus is a trimer, while at 49.3 K and 44.4 K a transition to the dimer seems to occur (note that the corresponding terms are very close in magnitude, which impedes a definite assignment). At the lowest temperature of 31.2 K, the critical nucleus is either a dimer or a monomer. These terms are very sensitive to the value of ΔGj*, therefore the uncertainty of the size of the critical nucleus is large, analogous to the uncertainty of the transition temperature. Nevertheless, the calculations show a clear trend towards the emergence of a nucleation barrier at higher temperatures.
4.5 Nucleation rates of C3H8
Fig. 6a and b show the total cluster number concentrations as a function of time and the extracted J and k11,net, respectively, for C3H8. The values of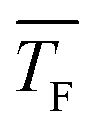 and pF are very similar for 0.12% CO2 and for 0.035% C3H8 as well as for 1.1% CO2 and for 0.26% C3H8. The fact that both C3H8 concentrations are lower demonstrates that C3H8 nucleates more easily than CO2 under otherwise similar conditions. The values of J are of the same order of magnitude as those for CO2 (Table 1). The two data points in Fig. 6b seem to imply a decrease of J with decreasing temperature, which would be opposite to the behavior of CO2 (Section 4.4). However, one needs to keep in mind that a clear trend cannot be extracted from only two data points.
and pF are very similar for 0.12% CO2 and for 0.035% C3H8 as well as for 1.1% CO2 and for 0.26% C3H8. The fact that both C3H8 concentrations are lower demonstrates that C3H8 nucleates more easily than CO2 under otherwise similar conditions. The values of J are of the same order of magnitude as those for CO2 (Table 1). The two data points in Fig. 6b seem to imply a decrease of J with decreasing temperature, which would be opposite to the behavior of CO2 (Section 4.4). However, one needs to keep in mind that a clear trend cannot be extracted from only two data points.
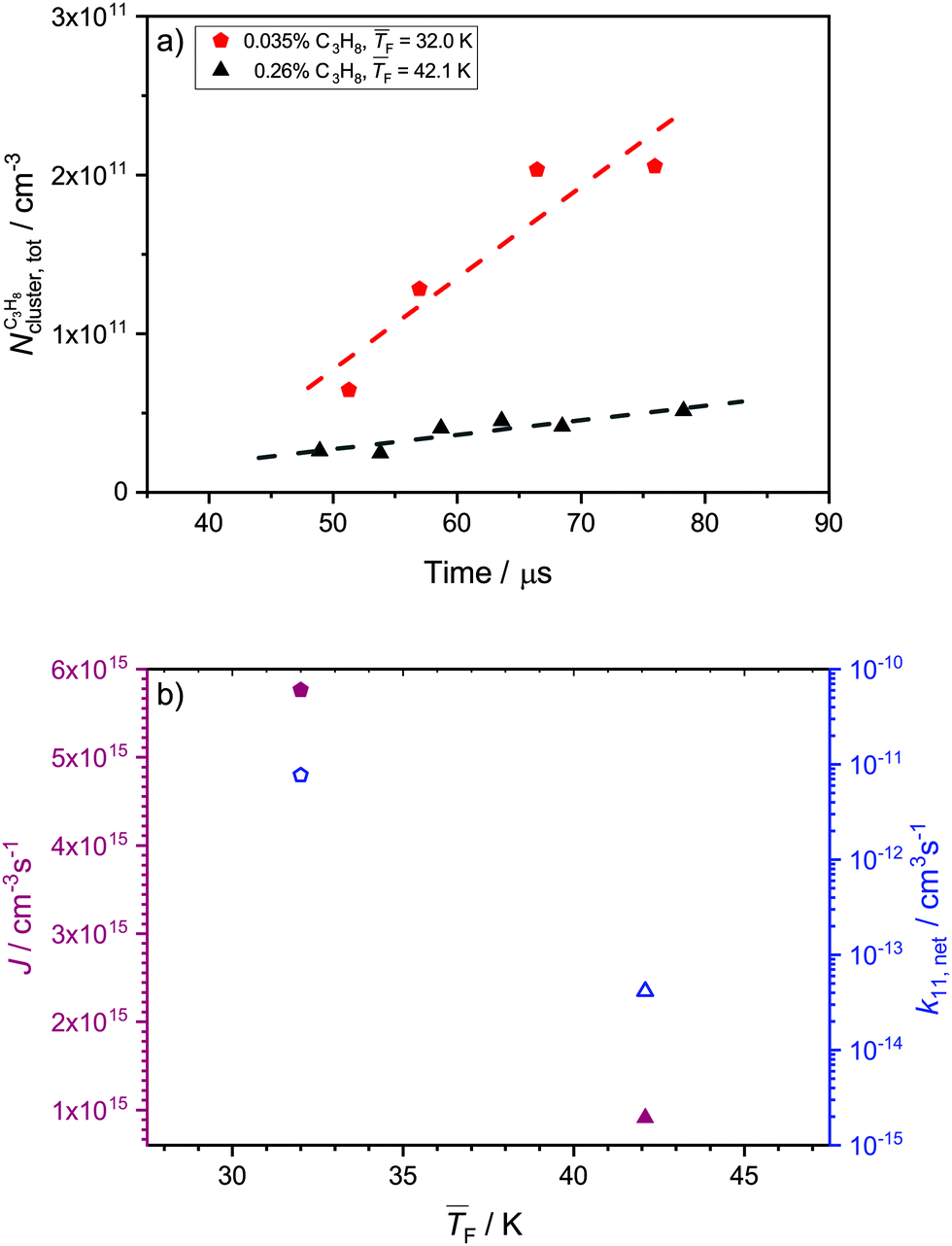 | ||
| Fig. 6 (a) Total cluster number concentration as a function of time for 0.035% and 0.26% C3H8. The linear fits are indicated with dashed lines. (b) Experimental nucleation rates J (full red symbols) and monomer–monomer net forward rate constants k11,net (open blue symbols) extracted from the C3H8 data in panel a as a function of the flow temperature. The data for 0.26% C3H8 are shown as up triangles and those for 0.035% C3H8 as pentagons. | ||
To the best of our knowledge, these are the first experimental nucleation rates for C3H8, so that a comparison with previous experimental data is not possible. However, we can compare the experimental rates with the predictions from the models (see Table 1). As discussed in Section 4.4 for CO2, CNT gives the hard sphere collision limit or values close to it and MKNT gives unreasonably low values. Both theories thus fail in describing J for our conditions for both CO2 and C3H8. Because of the many isomers of C3H8 clusters,52 we do not calculate JQM. Only the monomer–monomer collision rate k11,inter is evaluated, yielding values for Jinter which are about two times larger than JHS. This enhancement of the nucleation rate is comparable to that for CO2, which seems reasonable as both substances form weakly bound clusters. As in the case of CO2, k11,net decreases with increasing temperature and lies far below the theoretical monomer–monomer collision rate constants. This can again be attributed to a lower sticking probability at higher temperatures. In Section 4.3, we found that ηexp > 1 for C3H8, which suggests that nucleation is likely barrierless under both conditions studied here.
4.6 Third body interaction
The carrier gas is expected to have two main influences on nucleation. Firstly, it removes the latent heat which is released during nucleation (thermalization) and secondly, the clusters have to spend volume work to grow in the presence of the carrier gas. Both factors strongly depend on the pressure and type of carrier gas.74 At low absolute pressures, the thermalization effect should dominate leading to a positive pressure effect (increase of the nucleation rate with increasing pressure). At high absolute pressures though, the volume work effect should dominate leading to a negative pressure effect (decrease of the nucleation rate with increasing pressure). Several experimental and theoretical studies have investigated the effect of the total pressure on nucleation.75–79 The findings of the different studies are very ambiguous. Positive, negative and no pressure dependence was found for various substances and experimental conditions.To test the influence of the carrier gas, we change the flow pressure pF while keeping pCO2 and 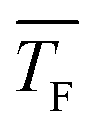 constant (see Table 2). Note that pF can only be changed in a small window because the uniformity of the postnozzle flow has to be maintained. To do so, the carrier gas composition (ratio of Ar to CH4) is slightly varied. Fig. 7 shows the total cluster number concentration as a function of the nucleation time for three different values of pF for CO2. The corresponding nucleation rates J are also indicated in the figure (see also Table 2). The nucleation rate J increases with increasing pF. Assuming a relative error in J of a factor of two, the increase in J is outside our experimental uncertainty for the increase from 22.5 or 30 Pa to 37 Pa. This hints at a positive pressure effect, which appears reasonable in this low pressure regime.74 The arrows in Fig. 7 indicate the onset times of nucleation. A comparison of the onset times shows that the higher the pF, the earlier the nucleation takes place in the postnozzle flow. This indicates that a higher pF accelerates nucleation.
constant (see Table 2). Note that pF can only be changed in a small window because the uniformity of the postnozzle flow has to be maintained. To do so, the carrier gas composition (ratio of Ar to CH4) is slightly varied. Fig. 7 shows the total cluster number concentration as a function of the nucleation time for three different values of pF for CO2. The corresponding nucleation rates J are also indicated in the figure (see also Table 2). The nucleation rate J increases with increasing pF. Assuming a relative error in J of a factor of two, the increase in J is outside our experimental uncertainty for the increase from 22.5 or 30 Pa to 37 Pa. This hints at a positive pressure effect, which appears reasonable in this low pressure regime.74 The arrows in Fig. 7 indicate the onset times of nucleation. A comparison of the onset times shows that the higher the pF, the earlier the nucleation takes place in the postnozzle flow. This indicates that a higher pF accelerates nucleation.
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) ,
, 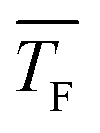 and J are given in the caption of Table 1
and J are given in the caption of Table 1
| Symbol | 1.5% CO2 | 1.13% CO2 | 0.91% CO2 |
|---|---|---|---|
|
|
|
|
|
| Nozzle | Mach 5.2 | Mach 5.2 | Mach 6 |
| p CO2/Pa | 0.338 | 0.338 | 0.338 |
| p F/Pa | 22.5 | 30 | 37 |
| % CH4 | 3.0 | 6.0 | 22.7 |
| % Ar | 95.5 | 92.9 | 76.4 |
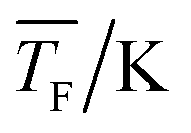
|
42.6 ± 1.1 | 43.0 ± 1.4 | 43.0 ± 1.2 |
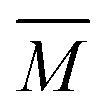
|
4.32 ± 0.07 | 4.35 ± 0.08 | 4.72 ± 0.08 |
| J/cm−3 s−1 | 1.5 × 1015 | 1.8 × 1015 | 1.3 × 1016 |



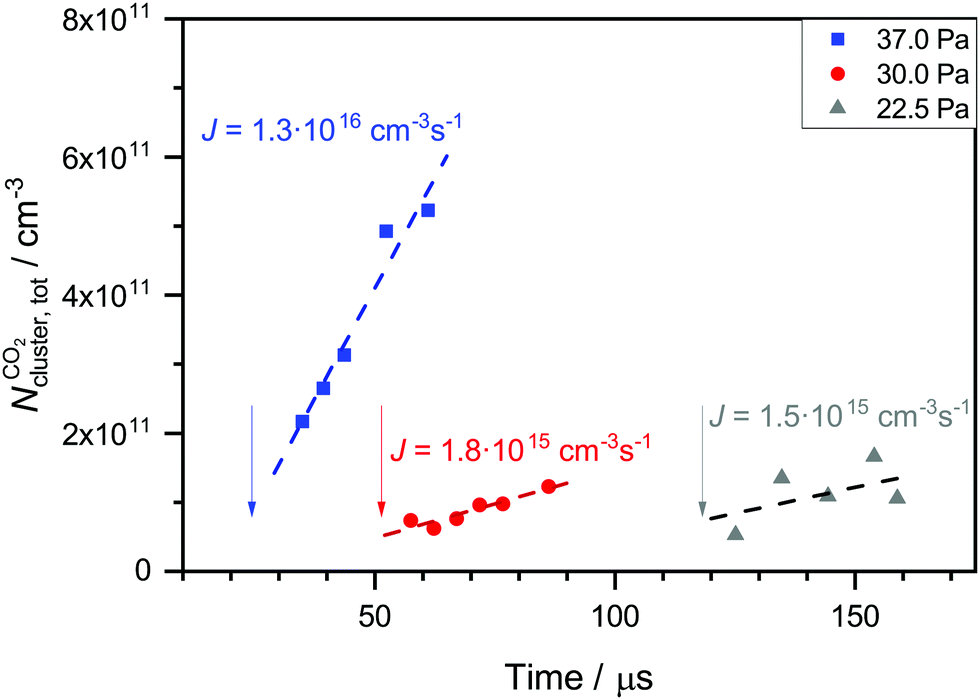 | ||
Fig. 7 Total cluster number concentrations as a function of time t for identical CO2 pressure pCO2 and the same flow temperatures 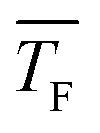 (∼43 K, but three different flow pressures of pF: 22.5 Pa, 30 Pa and 37 Pa (see Table 2)). The arrows indicate the onset of nucleation. The dashed lines correspond to linear fits to the data. J is the nucleation rate. (∼43 K, but three different flow pressures of pF: 22.5 Pa, 30 Pa and 37 Pa (see Table 2)). The arrows indicate the onset of nucleation. The dashed lines correspond to linear fits to the data. J is the nucleation rate. | ||
Since nucleation is also very sensitive to temperature, we have estimated the influence of the small temperature differences (see Table 2) on the value of J by calculating JHS and JQM at slightly different temperatures. A variation in temperature between 41.6 K and 44.4 K corresponds to a change in JHS of about 10% and a change in JQM of around a factor of two. These changes are still too small to explain the observed increase of J from 22.5 or 30 Pa to 37 Pa. Slight variations in temperature or temperature fluctuations seem rather unlikely to be at the origin of the systematic changes in J with pF.
As mentioned above, different carrier gas compositions were used for the measurements in Table 2. The CH4 content increases as pF increases. As discussed in our previous publications28,30 and in the literature,76,77,80–82 the type of carrier gas might also have an influence on the nucleation rate. Dumitrescu et al.81 found better thermalization properties for CH4 compared with Ar. This suggests that a higher CH4 content would also lead to an increase in J. Thus, we cannot completely exclude that the change in the carrier gas composition also contributes to the observed increase in J. In any case, we find an increase in the nucleation rate with increasing flow pressure and changing carrier gas composition.
5 Conclusions
In this work, we investigate homogeneous gas-phase nucleation of CO2 and C3H8 at high supersaturations in the temperature range from ∼31 K to 63 K and ∼32 K to 42 K, respectively. This corresponds to a change in the partial pressure of the condensable by three orders of magnitude. The nucleation rate J and the net forward rate constant k1j,net (the net effect of association and evaporation processes) are directly retrieved from the experimental cluster size distributions recorded as a function of the nucleation time. For CO2, we observe an increase of k1j,net with decreasing flow temperature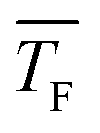 even at high cluster size j, where the sticking probability is most likely close to 1. This result hints at negligible evaporation for low temperature and non-negligible evaporation for high temperature, which implies a transition from barrierless nucleation at low
even at high cluster size j, where the sticking probability is most likely close to 1. This result hints at negligible evaporation for low temperature and non-negligible evaporation for high temperature, which implies a transition from barrierless nucleation at low 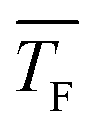 to barrier-limited nucleation at higher
to barrier-limited nucleation at higher 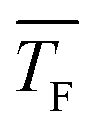 . However, the transition temperature cannot be determined definitively. Quantum-chemical calculations combined with the experimental results further indicate that this transition might lie within the temperature range investigated in the present study. Compared with other nucleation theories (CNT and MKNT), the quantum mechanics based nucleation rate, JQM, yields much better agreement with the experiment. The comparison between CO2 and C3H8 indicates that the nucleation of C3H8 is most likely barrierless for the conditions studied here. Nucleation rate measurements at different pF show a slight increase of J with increasing pF, suggesting that an increase of the flow pressure enhances nucleation. A more detailed description of the nucleation kinetics could be obtained from calculations of association and dissociation rate constants, analogous to those recently presented for H2O in ref. 71, which could pinpoint the aforementioned transition more precisely.
. However, the transition temperature cannot be determined definitively. Quantum-chemical calculations combined with the experimental results further indicate that this transition might lie within the temperature range investigated in the present study. Compared with other nucleation theories (CNT and MKNT), the quantum mechanics based nucleation rate, JQM, yields much better agreement with the experiment. The comparison between CO2 and C3H8 indicates that the nucleation of C3H8 is most likely barrierless for the conditions studied here. Nucleation rate measurements at different pF show a slight increase of J with increasing pF, suggesting that an increase of the flow pressure enhances nucleation. A more detailed description of the nucleation kinetics could be obtained from calculations of association and dissociation rate constants, analogous to those recently presented for H2O in ref. 71, which could pinpoint the aforementioned transition more precisely.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are very grateful to Prof. Barbara Wyslouzil for many stimulating discussions and in particular for suggesting investigation of the pressure dependence of the nucleation rate. We also thank Markus Steger, David Stapfer and Dr Egor Chasovskikh for their help in maintaining the experimental setup. Financial support was provided by the Swiss National Science Foundation (SNF Project No. 200020-172472) and by ETH Zürich.References
- G. P. Sutton and O. Biblarz, Rocket propulsion elements, John Wiley & Sons, 2016 Search PubMed.
- M. Kulmala, I. Riipinen, M. Sipilä, H. E. Manninen, T. Petäjä, H. Junninen, M. Dal Maso, G. Mordas, A. Mirme, M. Vana, A. Hirsikko, L. Laakso, R. M. Harroson, I. Hanson, C. Leung, K. E. J. Lehtinen and V.-M. Kerminen, Science, 2007, 318, 89–92 CrossRef CAS PubMed.
- R. Zhang, Science, 2010, 328, 1366–1367 CrossRef CAS PubMed.
- C. I. Davidson, R. F. Phalen and P. A. Solomon, Aerosol Sci. Technol., 2005, 39, 737–749 CrossRef CAS.
- K.-H. Kim, E. Kabir and S. Kabir, Environ. Int., 2015, 74, 136–143 CrossRef CAS PubMed.
- B. E. Wyslouzil and J. Wölk, J. Chem. Phys., 2016, 145, 211702 CrossRef PubMed.
- K. Iland, J. Wölk, R. Strey and D. Kashchiev, J. Chem. Phys., 2007, 127, 154506 CrossRef PubMed.
- S. Sinha, H. Laksmono and B. Wyslouzil, Rev. Sci. Instrum., 2008, 79, 114101 CrossRef PubMed.
- S. Sinha, A. Bhabhe, H. Laksmono, J. Wölk, R. Strey and B. Wyslouzil, J. Chem. Phys., 2010, 132, 064304 CrossRef.
- L. M. Feldmar, J. Wölk and R. Strey, AIP Conf. Proc., 2013, 15–18 CrossRef CAS.
- D. Ghosh, D. Bergmann, R. Schwering, J. Wölk, R. Strey, S. Tanimura and B. E. Wyslouzil, J. Chem. Phys., 2010, 132, 024307 CrossRef PubMed.
- K. Mullick, A. Bhabhe, A. Manka, J. Wolk, R. Strey and B. E. Wyslouzil, J. Phys. Chem. B, 2015, 119, 9009–9019 CrossRef CAS PubMed.
- J. Wölk, R. Strey, C. H. Heath and B. E. Wyslouzil, J. Chem. Phys., 2002, 117, 4954–4960 CrossRef.
- V. I. Kalikmanov, J. Chem. Phys., 2006, 124, 124505 CrossRef CAS PubMed.
- V. I. Kalikmanov, Nucleation Theory, Springer, Netherlands, Heidelberg, 2013 Search PubMed.
- H. Du, A. B. Nadykto and F. Yu, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 021604 CrossRef PubMed.
- J. Diemand, R. Angélil, K. K. Tanaka and H. Tanaka, J. Chem. Phys., 2013, 139, 074309 CrossRef PubMed.
- J. Merikanto, J. Duplissy, A. Määttänen, H. Henschel, N. M. Donahue, D. Brus, S. Schobesberger, M. Kulmala and H. Vehkamäki, J. Geophys. Res.: Atmos., 2016, 121, 1736–1751 CAS.
- J. Duplissy, J. M. A. Franchin, G. Tsagkogeorgas, J. Kangasluoma, D. Wimmer, H. Vuollekoski, S. Schobesberger, K. Lehtipalo, R. C. Flagan, D. Brus, N. M. Donahue, H. Vehkamäki, J. Almeida, A. Amorim, P. Barmet, F. Bianchi, M. Breitenlechner, E. M. Dunne, R. Guida, H. Henschel, H. Junninen, J. Kirkby, A. Kürten, A. Kupc, A. Määttänen, V. Makhmutov, S. Mathot, T. Nieminen, A. Onnela, A. P. Praplan, F. Riccobono, L. Rondo, G. Steiner, A. Tome, H. Walther, U. Baltensperger, K. S. Carslaw, J. Dommen, A. Hansel, T. Petäjä, M. Sipilä, F. Stratmann, A. Vrtala, P. E. Wagner, D. R. Worsnop, J. Curtius and M. Kulmala, J. Geophys. Res.: Atmos., 2016, 121, 1752–1775 CAS.
- L. Tiszenkel, C. Stangl, J. Krasnomowitz, Q. Ouyang, H. Yu, M. J. Apsokardu, M. V. Johnston and S.-H. Lee, Atmos. Chem. Phys., 2019, 19, 8915–8929 CrossRef CAS.
- C. F. Clement and I. J. Ford, Atmos. Environ., 1999, 33, 489–499 CrossRef CAS.
- Y. Viisanen, R. Strey, A. Laaksonen and M. Kulmala, J. Chem. Phys., 1994, 100, 6062–6072 CrossRef CAS.
- S. Tanimura, H. Pathak and B. E. Wyslouzil, J. Chem. Phys., 2013, 139, 174311 CrossRef PubMed.
- B. Schläppi, J. H. Litman, J. J. Ferreiro, D. Stapfer and R. Signorell, Phys. Chem. Chem. Phys., 2015, 17, 25761–25771 RSC.
- J. J. Ferreiro, T. E. Gartmann, B. Schläppi and R. Signorell, Z. Phys. Chem., 2015, 229, 1765–1780 CAS.
- J. J. Ferreiro, S. Chakrabarty, B. Schläppi and R. Signorell, J. Chem. Phys., 2016, 145, 211907 CrossRef PubMed.
- S. Chakrabarty, J. J. Ferreiro, M. Lippe and R. Signorell, J. Phys. Chem. A, 2017, 121, 3991–4001 CrossRef CAS PubMed.
- M. Lippe, S. Chakrabarty, J. J. Ferreiro, K. K. Tanaka and R. Signorell, J. Chem. Phys., 2018, 149, 244303 CrossRef PubMed.
- M. Lippe, U. Szczepaniak, G.-L. Hou, S. Chakrabarty, J. J. Ferreiro, E. Chasovskikh and R. Signorell, J. Phys. Chem. A, 2019, 123, 2426–2437 CrossRef CAS PubMed.
- C. Li, M. Lippe, J. Krohn and R. Signorell, J. Chem. Phys., 2019, 151, 094305 CrossRef PubMed.
- F. Gelbard and J. H. Seinfeld, J. Colloid Interface Sci., 1979, 68, 363–382 CrossRef CAS.
- B. L. Yoder, J. H. Litman, P. W. Forysinski, J. L. Corbett and R. Signorell, J. Phys. Chem. Lett., 2011, 2, 2623–2628 CrossRef CAS.
- J. H. Litman, B. L. Yoder, B. Schläppi and R. Signorell, Phys. Chem. Chem. Phys., 2013, 15, 940–949 RSC.
- S. Heinbuch, F. Dong, J. J. Rocca and E. R. Bernstein, J. Chem. Phys., 2006, 125, 154316 CrossRef CAS PubMed.
- J. Lengyel, A. Pysanenko, J. Kočišek, V. Poterya, C. C. Pradzynski, T. Zeuch, P. Slavíček and M. Fárník, J. Phys. Chem. Lett., 2012, 3, 3096–3101 CrossRef CAS PubMed.
- J. Lengyel, A. Pysanenko, V. Poterya, J. Kočišek and M. Fárnk, Chem. Phys. Lett., 2014, 612, 256–261 CrossRef CAS.
- K. Kameta, N. Kouchi, M. Ukai and Y. Hatano, J. Electron Spectrosc. Relat. Phenom., 2002, 123, 225–238 CrossRef CAS.
- J. W. Au, G. Cooper and C. E. Brion, Chem. Phys., 1993, 173, 241–265 CrossRef CAS.
- M. Ruberti, R. Yun, K. Gokhberg, S. Kopelke, L. S. Cederbaum, F. Tarantelli and V. Averbukh, J. Chem. Phys., 2013, 139, 144107 CrossRef CAS.
- R. M. Stephenson and S. Malanowski, Handbook of the thermodynamics of organic compounds, Elsevier Science Publishing Co., Inc., 1987 Search PubMed.
- R. Stockbauer and M. G. Inghram, J. Chem. Phys., 1971, 54, 2242–2246 CrossRef CAS.
- W. F. Giauque and C. J. Egan, J. Chem. Phys., 1937, 5, 45–54 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, Inc., 2016 Search PubMed.
- R. Halonen, E. Zapadinsky, T. Kurtén, H. Vehkamäki and B. Reischl, Atmos. Chem. Phys., 2019, 19, 13355–13366 CrossRef CAS.
- A. Kürten, C. Li, F. Bianchi, J. Curtius, A. Dias, N. M. Donahue, J. Duplissy, R. C. Flagan, J. Hakala, T. Jokinen, J. Kirkby, M. Kulmala, A. Laaksonen, K. Lehtipalo, V. Makhmutov, A. Onnela, M. P. Rissanen, M. Simon, M. Sipilä, Y. Stozhkov, J. Tröstl, P. Ye and P. H. McMurry, Atmos. Chem. Phys., 2018, 18, 845–863 CrossRef.
- A. Kürten, T. Jokinen, M. Simon, M. Sipilä, N. Sarnela, H. Junninen, A. Adamov, J. Almeida, A. Amorim, F. Bianchi, M. Breitenlechner, J. Dommen, N. M. Donahue, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, J. Hakala, A. Hansel, M. Heinritzi, M. Hutterli, J. Kangasluoma, J. Kirkby, A. Laaksonen, K. Lehtipalo, M. Leiminger, V. Makhmutov, S. Mathot, A. Onnela, T. Petäjä, A. P. Praplan, F. Riccobono, M. P. Rissanen, L. Rondo, S. Schobesberger, J. H. Seinfeld, G. Steiner, A. Tomé, J. Tröstl, P. M. Winkler, C. Williamson, D. Wimmer, P. Ye, U. Baltensperger, K. S. Carslaw, M. Kulmala, D. R. Worsnop and J. Curtius, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15019–15024 CrossRef PubMed.
- K. Lehtipalo, L. Rondo, J. Kontkanen, S. Schobesberger, T. Jokinen, N. Sarnela, A. Kürten, S. Ehrhart, A. Franchin, T. Nieminen, F. Riccobono, M. Sipilä, T. Yli-Juuti, J. Duplissy, A. Adamov, L. Ahlm, J. Almeida, A. Amorim, F. Bianchi, M. Breitenlechner, J. Dommen, A. J. Downard, E. M. Dunne, R. C. Flagan, R. Guida, J. Hakala, A. Hansel, W. Jud, J. Kangasluoma, V.-M. Kerminen, H. Keskinen, K. Jaeseok, J. Kirkby, A. Kupc, O. Kupiainen-Määttä, A. Laaksonen, M. J. Lawler, M. Leiminger, S. Mathot, T. Olenius, I. K. Ortega, A. Onnela, T. Petäjä, A. Praplan, M. P. Rissanen, T. Ruuskanen, F. D. Santos, S. Schallhart, R. Schnitzhofer, M. Simon, J. N. Smith, J. Tröstl, G. Tsagkogeorgas, A. Tomé, P. Vaattovaara, H. Vehkamäki, A. E. Vrtala, P. E. Wagner, C. Williamson, D. Wimmer, P. M. Winkler, A. Virtanen, N. M. Donahue, K. S. Carslaw, U. Baltensperger, I. Riipinen, J. Curtius, D. R. Worsnop and M. Kulmala, Nat. Commun., 2016, 7, 11594 CrossRef CAS PubMed.
- J. Lengyel, J. Kočišek, V. Poterya, A. Pysanenko, P. Svrčková, M. Fárnk, D. K. Zaouris and J. Fedor, J. Chem. Phys., 2012, 137, 034304 CrossRef CAS PubMed.
- J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS.
- R. D. Levine and R. B. Bernstein, Molecular reaction dynamics and chemical reactivity, Oxford University Press, 1987 Search PubMed.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- S. Tsuzuki, T. Uchimaru, M. Mikami and K. Tanabe, J. Phys. Chem. A, 2002, 106, 3867–3872 CrossRef CAS.
- P. H. McMurry, J. Colloid Interface Sci., 1980, 78, 513–527 CrossRef CAS.
- R. Wegscheider, Monatsh. Chem., 1901, 32, 849–906 CrossRef.
- L. Onsager, Phys. Rev., 1931, 37, 405 CrossRef CAS.
- J. E. McDonald, Am. J. Phys., 1962, 30, 870–877 CrossRef CAS.
- S. L. Girshick and C. Chiu, J. Chem. Phys., 1990, 93, 1273–1277 CrossRef CAS.
- J. Merikanto, E. Zapadinsky, A. Lauri and H. Vehkamäki, Phys. Rev. Lett., 2007, 98, 145702 CrossRef PubMed.
- M. J. McGrath, T. Olenius, I. K. Ortega, V. Loukonen, P. Paasonen, T. Kurtén, M. Kulmala and H. Vehkamäki, Atmos. Chem. Phys., 2012, 12, 2345–2355 CrossRef CAS.
- J. L. Katz and H. Wiedersich, J. Colloid Interface Sci., 1977, 61, 351–355 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian09, Revision A.1, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- NIST Computational Chemistry Comparison and Benchmark Database, ed. R. D. J. Nist III, 2019 Search PubMed.
- K. H. Lemke and T. M. Seward, Chem. Phys. Lett., 2013, 573, 19–23 CrossRef CAS.
- X. Xie, C. Wu, Y. Liu, W. Huang, Y. Deng, Y. Liu, Q. Gong and C. Wu, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 90, 033411 CrossRef.
- P. H. McMurry and C. Li, Aerosol Sci. Technol., 2017, 51, 1057–1070 CrossRef CAS.
- Y. Negishi, T. Nagata and T. Tsukuda, Chem. Phys. Lett., 2002, 364, 127–132 CrossRef CAS.
- O. Ingolfsson and A. M. Wodtke, J. Chem. Phys., 2002, 117, 3721–3732 CrossRef CAS.
- U. Näher, U. Zimmermann and T. P. Martin, J. Chem. Phys., 1993, 99, 2256–2260 CrossRef.
- J. Bourgalais, V. Roussel, M. Capron, A. Benidar, A. W. Jasper, S. J. Klippenstein, L. Biennier and S. D. Le Picard, Phys. Rev. Lett., 2016, 116, 113401 CrossRef CAS PubMed.
- K. M. Duff, PhD thesis, Massachusetts Institute of Technology, 1964.
- C. Lettieri, D. Paxson, Z. Spakovszky and P. Bryanston-Cross, J. Eng. Gas Turbines Power, 2018, 140, 041701 CrossRef.
- J. Wedekind, A.-P. Hyvärinen, D. Brus and D. Reguera, Phys. Rev. Lett., 2008, 101, 125703 CrossRef PubMed.
- D. Brus, V. Ždmal and F. Stratmann, J. Chem. Phys., 2006, 124, 164306 CrossRef PubMed.
- D. Brus, A.-P. Hyvärinen, J. Wedekind, Y. Viisanen, M. Kulmala, V. Ždmal, J. Smolk and H. Lihavainen, J. Chem. Phys., 2008, 128, 134312 CrossRef CAS PubMed.
- M. A. L. J. Fransen, J. Hrubý, D. M. J. Smeulders and M. E. H. van Dongen, J. Chem. Phys., 2015, 142, 164307 CrossRef CAS PubMed.
- D. Kashchiev, J. Chem. Phys., 1996, 104, 8671–8677 CrossRef CAS.
- B. E. Wyslouzil, G. Wilemski, M. G. Beals and M. B. Frish, Phys. Fluids, 1994, 6, 2845–2854 CrossRef CAS.
- D. Barschdorff, Phys. Fluids, 1975, 18, 529–535 CrossRef CAS.
- L. R. Dumitrescu, H. Huinink, D. M. J. Smeulders, J. A. M. Dam and S. V. Gaastra-Nedea, J. Chem. Phys., 2018, 148, 194502 CrossRef PubMed.
- R. H. Heist, J. Ahmed and M. Janjua, J. Phys. Chem., 1995, 99, 375–383 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01771j |
| ‡ These authors contributed equally to this work. |
| § Current Address: School of Environmental Science and Engineering, Shanghai Jiaotong University, Shanghai 200240, China. |
| This journal is © the Owner Societies 2020 |