
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFB-REDA: fragment-based decomposition analysis of the reorganization energy for organic semiconductors†
Kun-Han
Lin
 and
Clémence
Corminboeuf
*
and
Clémence
Corminboeuf
*
Laboratory for Computational Molecular Design, Institute of Chemical Sciences and Engineering and National Centre for Computational Design and Discovery of Novel Materials (MARVEL), École Polytechnique Fedérale de Lausanne, 1015 Lausanne, Switzerland. E-mail: clemence.corminboeuf@epfl.ch
First published on 12th May 2020
Abstract
We present a fragment-based decomposition analysis tool (FB-REDA) for the reorganisation energy (λ). This tool delivers insights on how to rationally design low-λ organic semiconductors. The contribution of the fragment vibrational modes to the reorganization energy is exploited to identity the individual contributions of the molecular building blocks. The usefulness of the approach is demonstrated by offering three strategies to reduce the reorganization energy of a promising dopant-free hole transport material (TPA1PM, λ = 213 meV). A reduction of nearly 50% (TPD3PM, λ = 108 meV) is achieved. The proposed design principles are likely transferable to other organic semiconductors exploiting common molecular building blocks.
Introduction
Organic semiconductors based on small molecules are exploited in various optoelectronic applications owing to their well-defined chemical structures (monodispersity), low cost, flexibility and low-temperature and solution processability.1,2 In the spirit of “molecular LEGOs,”3,4 their properties are easily tuneable with the charge mobility being the most relevant target5–11 for rational design. Relevant examples include the incorporation of hole transport materials with high hole mobility into photovoltaics or organic light-emitting diodes with low hole–electron recombination rate.12–14The reorganization energy (λ) is one of the key parameters for determining the efficiency of charge transport in both the hopping and band transport (related to electron–phonon coupling term) regime.15,16 Organic semiconductors with smaller reorganization energy (assuming the other parameters fixed) generally show a higher charge mobility.17 Therefore, understanding the relationship between molecular structures and reorganization energy is the key to a successful rational design.2,18
A molecule's reorganization energy is generally computed using Nelsen's 4-point method or the normal mode (NM) analysis.17,19,20 In order to assess the role played by each molecular building block, it is insightful to decompose the overall reorganization energy into the individual molecular fragment contribution (λfrag). However, analysis tools giving direct access to the fragment contribution are still lacking. There are currently two existing analysis that serve to evaluate the atomic or fragment contributions to λ or the vibronic coupling constant (V).
The first approach, developed by Shuai, Coropceanu et al. based on the NM analysis, expresses the displacements along the normal modes in terms of molecular internal coordinates.21,22 The total reorganization energy is then equal to a sum of internal coordinate contributions (see eqn (S1) in the ESI†). Using this approach, the authors were able to demonstrate the influence of substituents and substitution sites on the overall reorganization energy of indolo [3,2-b]carbazole derivatives. While such an analysis tool is useful to identify the substituent leading to the lowest λ, it is less convenient to evaluate the fragment contributions to λ in a “molecular LEGOs” framework. The reorganization energy in the internal coordinate representation contains cross terms involving two different internal coordinates, which are not necessarily small in comparison to the square terms (see eqn (S1) in the ESI†). However, there is no trivial way to divide these cross terms into internal coordinate contributions or to partition internal coordinates involving more than one fragment.
The second method developed by Sato et al., is associated with the concept of atomic vibronic coupling constant (AVCC).23,24 They investigated the chain-length dependence of the reorganization energy for oligofluorenes and oligothiophenes by evaluating the change in local degree of vibronic coupling when increasing the length of the oligomer. Because the sum of all AVCC is equal to the total vibronic coupling constant (see eqn (S2) in the ESI†), the fragment vibronic coupling constants can be defined as a sum over all the AVCC of the corresponding atoms within a fragment. However, given that the total reorganization energy is proportional to the square of the total vibronic coupling constant, the representation of λ in terms of AVCC inevitably leads to cross terms involving two different atomic vibronic constants (coupled contributions from different atoms).
This work proposes an alternative strategy to decompose the reorganization energy into local contributions. The approach is inspired by the idea of Huix-Rotllant et al.,25 who decomposed the molecular normal modes in terms of local fragment modes. Instead of first decomposing λ into internal coordinates or atomic contributions, we avoid the generation of cross terms by computing the fragment contributions directly from the fragment modes. The equations and associated details are discussed in the Methodology section.
As a proof of concept, we use the proposed fragment reorganization decomposition analysis to identify organic hole transport materials (HTM) with low reorganization energy. We start with TPA1PM26 that has proved to be an efficient dopant-free HTM good for defect-passivation. The molecule (shown in Fig. 1(a)) is composed of a triphenylamine core (TPA), a phenyl-substituted carbazole arm (PCZ) and methoxy (M) substituent groups. The chosen strategy for reducing the reorganization energy is established based on the fragment-based decomposition analysis. Three molecular design approaches were successively adopted and their effects on the total hole reorganization energy (λtot) and the fragment contributions (divided into core (λcore), arm (λarm) and substituent (λsub) contributions) are discussed. The three strategies are: (1) introducing multi-arm, (2) non-covalent lock and (3) core rigidification. The usefulness of the computational approach is demonstrated by the reduction by half of the reorganization energy compared to TPA1PM (from 213 to 108 meV).
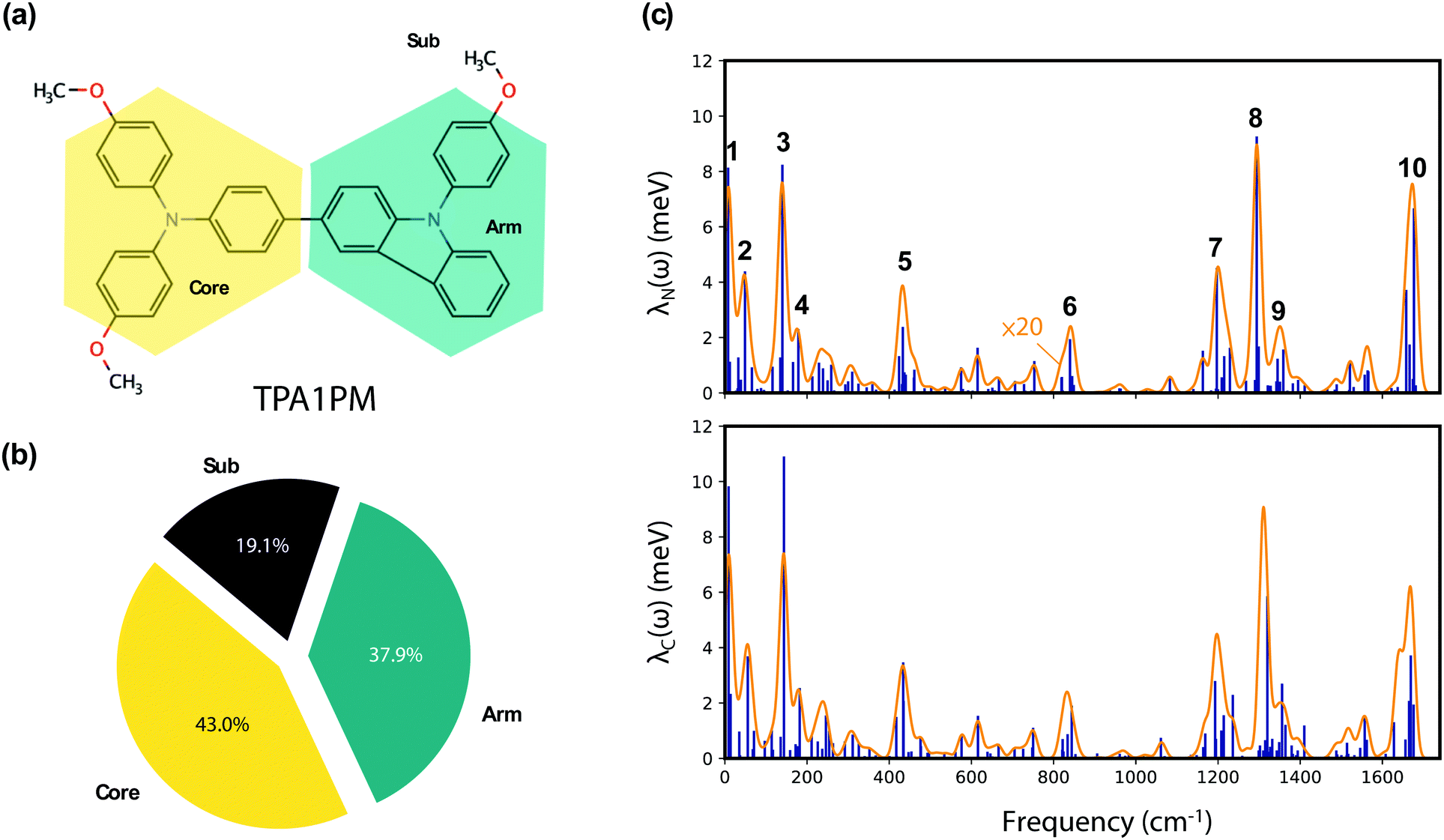 | ||
| Fig. 1 (a) Illustration of the decomposition of TPA1PM into Core, Arm and Sub fragments. (b) The ratio of the fragment to the total reorganization energy for each fragment. (c) The spectral density plot, blue lines are λNi/λCj and orange lines are λN(ω) and λC(ω) with intensity multiplied by 20. The 10 most important peaks are indexed. | ||
Methodology
Reorganization energy
There are two computational ways to evaluate the reorganization energy. The first method is known as Nelsen's 4-point method, where λ is calculated using four points on the adiabatic potential energy surfaces of the neutral and cation state of a molecule. The reorganization energy can be represented as:| λ = λN + λC = (EcN − EnN) + (EnC − EcC) | (1) |
 | (2) |
| ΔQNi = (CNi)Tm1/2(RC − RN) |
| ΔQCj = (CCj)Tm1/2(RN − RC) | (3) |
 | ||
| Scheme 1 Sketch of the potential energy surface of the neutral and cationic molecular states. The relationship between the four points (EnN, EcN, EcC and EnC), λN, λC and ΔQ on the potential energy surfaces is depicted. | ||
Fragment contributions
Within the NM method, the reorganization energy can be further partitioned into individual fragment contributions via the fragment modes analysis. Following the protocol proposed by Huix-Rotllant and Ferré,25 the modes for each fragment are obtained by first partitioning the total mass-weighted Hessian matrix Htot into fragment submatrices Hf. The eigenvectors of the fragment modes Cf are then obtained by diagonalizing these submatrices. These eigenvectors form a complete orthonormal basis set to expand the total normal modes: | (4) |
For convenience, the following derivation is based on λN, but the conclusion is the same for λC. Inserting eqn (3) and (4) into eqn (2) leads to an expression for λN in which the diagonal terms comes from the same fragment mode (di1ΔqNf,1)2 whereas the non-diagonal terms originates from two different fragment modes (di1di2ΔqNf,1ΔqNf,2) (see eqn (S5) and (S6), ESI†). The difficulty associated with how to deal with the non-diagonal terms, is similar to the approaches mentioned in the introduction. To avoid the non-diagonal terms, we thus define the contribution of the mode j of the fragment k as follow:
 | (5) |
 | (6) |
 | (7) |
In addition to the fragment reorganization energy, we also define the fragment mode reorganization energy (λjk), to identify crucial fragment modes:
 | (8) |
Computational details
Geometry optimization, electronic structure and frequency computations of the neutral and cationic molecules were performed at the B3LYP-30,31D3BJ32 level in conjunction with the 6-31G(d,p) basis set. All DFT computations were performed with Gaussian16.33 The NM analysis was performed using our in-house code, DUSHIN20 and Gaussian16 in tandem.To evaluate the validity of harmonic approximation, the reorganization energies were computed using both the 4-point method and the NM analysis (Fig. S1, ESI†). Our results show that the reorganization energies obtained from these two methods are very similar, justifying the use of the harmonic approximation.
Results and discussion
Analysis of TPA1PM
We first compute the reorganization energy of the parent TPA1PM molecule (211 meV) prior to its decomposition into three fragments: (1) a TPA core (Core), (2) a PCZ arm (Arm), and (3) three methoxy substituents (Sub), as shown in Fig. 1(a). The fragment reorganization energy (in meV) of the core (91), Arm (80) and Sub (40) are obtained viaeqn (6). The λfrag/λtot ratio for each fragment is represented in the pie chart (Fig. 1(b)). The core contributes the most (43.0%) to total reorganization energy. The contribution from the arm is slightly less significant (37.9%), with Sub contributing the least (19.1%).The normal modes that are dominating the reorganization energy are plotted in Fig. 1(c) with λNi/λCj representing the contribution for each normal mode i and j. Given the large number of normal modes in each system, we define the spectral density λN/C(ω) to facilitate the analysis:34,35
 | (9) |
The 10 most contributing peaks in λN(ω) are shown in Fig. 1(c) and the composition in terms of their fragment modes is shown in Table S1 (ESI†). Overall, several fragment modes recurrently contribute to these 10 important peaks and those with λjk > 1.2 meV (10 cm−1) are listed in Table S2 (ESI†). More specifically, the two most relevant fragment modes correspond to in-plane bond-stretching and angle-bending in the Core (Fig. 2). Considering the fact that the Core is the largest contributor to the reorganization energy, an efficient strategy to reduce λ consist in spreading the HOMO away from the TPA core. As shown in our former work,17 this objective can be achieved by introducing additional PCZ arms.
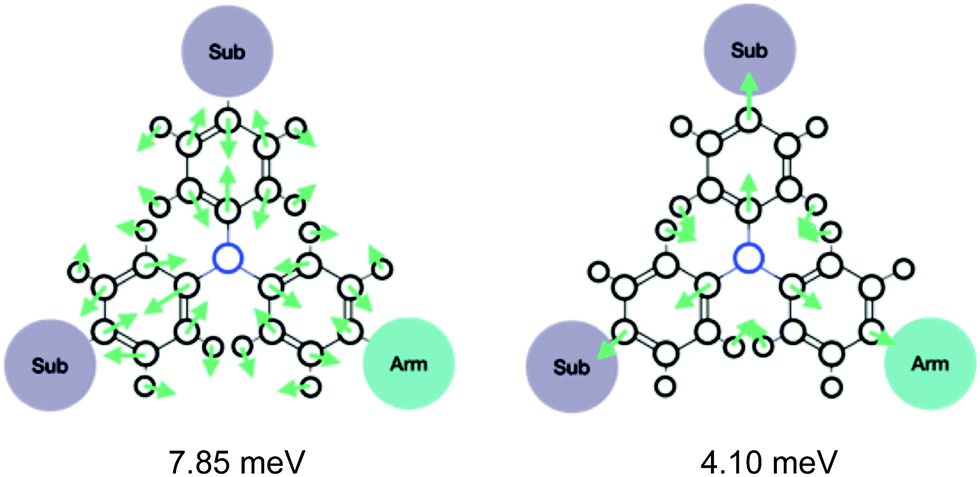 | ||
| Fig. 2 Illustration of the two most contributing fragment modes and their corresponding fragment reorganization energies in TPA1PM. | ||
Introducing multi-arm
The first strategy to reduce the reorganization energy of TPA1PM is thus to increase the number of PCZ arms, as exemplified in Fig. 3(a). Given that the methoxy groups serve as a Lewis base passivating defects in the perovskite layer,26 the number of methoxy substituents are kept constant. They are inserted either directly on the core or on the arms depending on their number. | ||
| Fig. 3 (a) The TPA1PM, TPA2PM and TPA3PM molecules and their fragments. (b) Total reorganization energy, fragment reorganization energy, (c) contribution to the HOMO from the NAO analysis and (d) the ratio of λfrag to λtot of each fragment. (e) Spectral density plots for λfrag and λtot for TPA1PM, TPA2PM and TPA3PM. | ||
The design strategy is efficient as the total reorganization energy decreases by up to ∼20% with increasing the number of PCZ arms, from 213, 173 to 156 meV (Fig. 3(b)). As expected, the highest occupied molecular orbitals (HOMO) spread through the PCZ arms (Fig. S2, ESI†). The contribution from Core and Sub to the HOMO (evaluated using natural atomic orbital analysis (NAO) Fig. 3(c)) decreases from ∼80% to ∼72% for Core and from ∼7% to ∼0% for Sub respectively, while that of Arm increases. These trends are in line with the decrease of λcore and λsub, which contrasts with the λarm increase from 80 to 101 meV. Overall, it is clear that the redistribution of the HOMO is the dominant factor affecting λ and that the largest contribution comes from the arms Fig. 3(d).
The spectral density of the Total, Core, Arm and Sub contribution plotted in Fig. 3(e) illustrates how the number of arms alter the fragment contribution. The overall features of λtot(ω) are very similar for the three molecules with the exception of an additional peak (marked with*) appearing in the low frequency range of the TPA3PM spectrum, (composition shown in Table S3, ESI†).
The 1st peak originates from three degenerated (by symmetry) Arm fragment modes, featuring inter-fragment rotation between each Arm and the TPA Core (see Fig. 4(a)). As the number of arms increases, a larger number of “arm” atoms contribute to the normal mode, leading to a larger ΔQNi. Yet, the eigen frequency of this mode changes negligibly upon the addition of arms, leading to a larger λNi in line with eqn (2). Except for the first peak, the intensity of nearly all peaks decreases as the number of arm increases. The 5th and 6th peaks that remain unchanged between TPA1PM and TPA3PM constitute another exception.
 | ||
| Fig. 4 Illustration of (a) the inter-fragment rotational modes present in the 1st peak and (b) the two dominant Core fragment modes present in the 5th and the 6th peak of TPA3PM. (a and c) The two most contributing fragment modes in TPA3PM. | ||
Unsurprisingly, the decrease of most peaks in λtot(ω) comes from the fact that the relevant normal modes involve the Core and Sub fragments whose contribution is reduced. The main fragment modes present in the unchanged 5th and 6th peaks feature an out-of-plane torsion of benzene rings (Fig. 4(b)) that negligibly change across the TPA1PM to TPA3PM series. The multi-arm strategy is thus unable to efficiently reduce the vibronic coupling caused by this modes.
The fragment modes with λjk > 1.2 meV (10 cm−1) for TPA3PM (given in Table S4, ESI†) show that the two most important contributions comes from the Arm and Sub fragments and correspond to inter-fragment rotational motions (Fig. 4(a and c)). Interestingly, both these modes are present in the 1st peak via their coupling with the Arm and Core rotations around the C–C bridging bond. These results suggest that freezing the relative rotational motion between the Core and the Arm can further reduce λ by elimination of the 1st peak. Taking TPA3PM as our new starting point, the second strategy is thus to quench this inter-fragment rotational motion by introducing “non-covalent locks” that force the planarity between the benzene in the TPA Core and the carbazole in the PCZ Arm.36 We expect that locking the out-of-plane mode will partially eliminate the contribution from the 1st peak (see next section).
An alternative strategy consists in reducing the contribution from the 5th and 6th peaks that are not affected by the number of arms. As discussed previously, the corresponding Core fragment modes correspond to an out-of-plane torsion in three benzene groups (Fig. 4(b)). This type of vibrational motion can be significantly restricted by introducing covalent bond between nearby benzene moieties. An example would be to substitute TPA by PCZ or indolo[3,2,1-jk]carbazole moiety (ICZ). This alternative will be discussed in the “Core Rigidification” section.
Non-covalent Lock
The non-covalent locks are introduced to restrict the inter-moiety rotational motion between the Core and Arm in order to further reduce λ. In Fig. 5(a), the benzene rings of the core are replaced by 1 or 3 pyrimidine rings able to form 2 pairs of N⋯H noncovalent interaction for each pyrimidine-carbazole pair. | ||
| Fig. 5 (a) The TPA3PM, MPD3PM and TPD3PM molecules and their fragments. (b) Total reorganization energy, fragment reorganization energy, (c) the ratio of λfrag to λtot of each fragment, and (d) spectral density plots for λfrag and λtot for TPA3PM, MPD3PM and TPD3PM. | ||
The total reorganization energy decreases with increasing the number of pyrimidine in the Core (MPD3PM and TPD3PM), from 156, 144 to 108 meV (Fig. 5(b)). A further ∼31% reduction in reorganization energy is achieved, demonstrating the efficiency of this strategy. The overall fragment contributions, λcore, λarm and λsub decrease with increasing the number of pyrimidine. By restricting the contribution to the first peak, one expects the reduction in λarm and λsub. However, λcore also decreases from 36, 35 to 30 meV. Since all the fragment reorganization energies decrease, the λfrag to λtot ratio for each fragment of TPD3PM does not change significantly compared to TPA3PM (Fig. 5(c)). The arms are still the largest contributors (62.9%) followed by Core (28.3%) and Sub (8.8%).
The spectral density in Fig. 5(d) provides several explanations to the observed trends: first, the 1st peak indeed decreases with increasing the number of pyrimidine and almost disappear in the TPD3PM spectra of λtot(ω), λarm(ω) and λsub(ω). Interestingly, the 2nd peak, although less intense, follows a similar trend. The fragment mode analysis indicates that the 2nd peak contains an arm-based fragment modes associated with inter-moiety bending motions (see Table S3, ESI†). Those are also being restricted by the non-covalent locker.
Finally, the unexpected decrease in λcore is shown to essentially arises from the significant reduction of the 5th peak (in λcore(ω)). As mentioned earlier, the 5th peak of TPA3PM is dominated by the benzene out-of-plane torsion. When the pyrimidine rings are introduced, the motion is attenuated by the intramolecular non-covalent interactions, (Fig. 6). The 5th peak of λarm(ω) follows a very similar trend to that of λcore(ω) owing to the key Arm fragment mode featuring analogue out-of-plane torsional motion of the carbazole in the PCZ Arm, as shown in Table S3 (ESI†). This movement is also restricted by the non-covalent locks (Fig. 6).
 | ||
| Fig. 6 The fragment modes of TPD3PM features out-of-plane torsions involving atoms in the non-covalent interaction pairs (only the motion of non-H atoms near the non-covalent interaction pairs is shown with the green arrows). | ||
Overall, this section demonstrates that the non-covalent lockers reduce the contribution from the fragment modes dominated by out-of-plane inter-moieties motions involving the Core and Arm fragments. This strategy can be applied to other systems. For instance, Malagoli and Brédas showed that the reorganization energy of N,N′,-diphenyl-N,N′-bis(3-methylphenyl)-(1,1′-biphenyl)-4,4′-diamine (TPD), a widely used hole transport material for organic light-emitting diodes, is more than twice of that of its monomer (TPA).37 They found that λ of TPD is dominated by the central biphenyl fragment, which may be due to the huge change in the inter-ring dihedral angle between neutral and cation ground state geometry. We anticipate the reorganization energy of TPD to be largely reduce upon introduction of non-covalent locks between the two phenyl groups.
Core rigidification
As mentioned in the “Multi-arm” section, the alternative strategy to reduce the reorganization energy of TPA3PM is to restrict the out-of-plane torsion of the benzene moieties in the TPA Core. To achieve this goal, the core can be “rigidified” by introducing covalent bonds between the neighboring benzene moieties transforming TPA into to a PCZ (1 bond) or a ICZ Core (2 bonds) shown in Fig. 8(a).Overall, the core rigidification is relatively efficient and leads to up a 28% reduction in total reorganization energy from 156 to 112 meV (Fig. 8(b)). As expected, the value of λcore goes down but more surprisingly λarm and λsub also decreases upon replacement of TPA by ICZ. Given that the reorganization energy of every fragment decreases, the λfrag to λtot ratios of TPD3PM is very similar to those of TPA3PM (Fig. 8(c)).
The spectral density of λtot(ω) and λcore(ω) (Fig. 8(d)) indicates that the intensity of the 5th and 6th peaks decreases from TPA to ICZ. In addition, the * and 3rd peaks in λcore(ω) decreases upon core-rigidification. Since their dominant Core modes involve relative motions between the TPA benzene rings (Fig. 7), it is not surprising that the contribution from these modes is attenuated by the core-rigidification.
 | ||
| Fig. 7 The main Core fragment modes of TPA3PM present in the * and the 3rd peaks. | ||
 | ||
| Fig. 8 (a) The TPA3PM, PCZ2PM and ICZ3PM molecules and their fragments. (b) Total reorganization energy, fragment reorganization energy, (c) the ratio of λfrag to λtot of each fragment, and (d) spectral density plots for λfrag and λtot for TPA3PM, PCZ3PM and ICZ3PM. | ||
The unexpected decrease in λarm and λsub originates from the reduced contributions in the low-frequency region, ω < 200 cm−1. In the case of λarm(ω), this region features relative motions (rotational and translational) between Arms and Core shown in Table S3 (ESI†). Given that ICZ3PM undergoes less geometrical reorganization in relevant internal coordinates (defined in Fig. S3, ESI†) upon oxidation, ΔQ and thus the reorganization energy are smaller (see Table S5, ESI†). The 1st peak in λsub(ω) decreases upon binding covalently the benzene rings because of the smaller dihedral angle change between the Core and Arm in ICZ3PM, which cause the reduction of the contribution from the Sub rotational modes to which they are coupled (vide supra).
This section demonstrated that ICZ is a promising building block in organic semiconductors if the goal is to lower the reorganization energy. Interestingly, a recent work by Jiang, Wang and collaborators concluded that perovskite solar cells exploiting ICZ-based HTMs exhibited outstanding performance.38 As a building block, the use of the ICZ core is, however, scarce and our work calls for a more systematic analysis of the performance of HTMs for PSCs involving this core fragment.
Conclusions
We developed a fragment-based decomposition analysis tool for the reorganization energy and demonstrated its usefulness by rationally designing low λ organic HTMs. Starting from the promising dopant-free TPA1PM HTM composed of triphenyl amine and carbazole building blocks, three design strategies were adopted to lower the reorganization energy of TPA1M (λ = 213 meV): multi-arm, non-covalent lock, and core rigidification. The introduction of multiple arms reduces λ by extending the π-conjugation away from the core. The lower λ of TPA3PM (156 meV) originates from diminishing the contributions from the TPA core that arises from two types of modes: (1) the Arm/Core rotational motions around the C–C bridge and (2) the out-of-plane torsions of the core benzene rings. We attenuated the former by introducing non-covalent locks at the Core and Arm bridging point. The intramolecular non-covalent interactions constrain the reorganization of the nuclei, which allows for a ∼31% reduction in the λ of TPD3PM (108 meV). On the other hand, the out-of-plane motion of the benzene rings is constrained by the insertion of covalent bonds between the benzene rings, which leads to a nearly 28% decrease in λ (ICZ3PM, 112 meV). Given the extensive use of triphenylamine and carbazole building blocks in organic semiconductors, we expect the two types of aforementioned fragment modes to largely contribute to their organization energies. For this reason the proposed strategies should be transferable to a variety of systems, such as, for instance, TPD (a common HTM for organic light emitting diodes). As such, we believe that the precise insight brought by our computational analysis tools has significant potential for facilitating and accelerating the search for novel low-λ organic semiconductors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C.C. and K.-H. L. thank EPFL for financial support.References
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51(9), 2020–2067 CrossRef CAS PubMed.
- G. Gryn’Ova, K. H. Lin and C. Corminboeuf, J. Am. Chem. Soc., 2018, 140(48), 16370–16386 CrossRef PubMed.
- C. Kunkel, C. Schober, J. T. Margraf, K. Reuter and H. Oberhofer, Chem. Mater., 2019, 31(3), 969–978 CrossRef CAS.
- S. Vela, C. Krüger and C. Corminboeuf, Phys. Chem. Chem. Phys., 2019, 21(37), 20782–20790 RSC.
- K.-H. Lin, A. Prlj and C. Corminboeuf, J. Phys. Chem. C, 2017, 121(39), 21729–21739 CrossRef CAS.
- Y. Yamashita, Sci. Technol. Adv. Mater., 2009, 10(2), 024313–024321 CrossRef PubMed.
- H. Sun, J. Gerasimov, M. Berggren and S. Fabiano, J. Mater. Chem. C, 2018, 6(44), 11778–11784 RSC.
- B. Kippelen and J. L. Brédas, Energy Environ. Sci., 2009, 2(3), 251–261 RSC.
- I. Yavuz, J. B. Lin and K. N. Houk, Phys. Chem. Chem. Phys., 2019, 21(2), 901–914 RSC.
- N. R. Tummala, S. A. Elroby, S. G. Aziz, C. Risko, V. Coropceanu and J. L. Brédas, J. Phys. Chem. C, 2016, 120(31), 17242–17250 CrossRef CAS.
- J. Guo, Y. Zhang, W. Cai, Z. Zhang, R. He, W. Shen and M. Li, Mater. Chem. Phys., 2020, 240, 122058 CrossRef CAS.
- L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55(47), 14522–14545 CrossRef PubMed.
- S. Shahnawaz, S. Sudheendran Swayamprabha, M. R. Nagar, R. A. K. Yadav, S. Gull, D. K. Dubey and J. H. Jou, J. Mater. Chem. C, 2019, 7(24), 7144–7158 RSC.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5(4), 1348–1373 RSC.
- C. Schober, K. Reuter and H. Oberhofer, J. Phys. Chem. Lett., 2016, 7(19), 3973–3977 CrossRef CAS PubMed.
- H. Oberhofer, K. Reuter and J. Blumberger, Chem. Rev., 2017, 117(15), 10319–10357 CrossRef CAS PubMed.
- K. H. Lin, A. Prlj, L. Yao, N. Drigo, H. H. Cho, M. K. Nazeeruddin, K. Sivula and C. Corminboeuf, Chem. Mater., 2019, 31(17), 6605–6614 CrossRef CAS.
- G. Gryn’ova and C. Corminboeuf, J. Phys. Chem. Lett., 2016, 7(24), 5198–5204 CrossRef PubMed.
- S. F. Nelsen, S. C. Blackstock and Y. Kim, J. Am. Chem. Soc., 1987, 109(3), 677–682 CrossRef CAS.
- J. R. Reimers, J. Chem. Phys., 2001, 115(20), 9103–9109 CrossRef CAS.
- H. Geng, Y. Niu, Q. Peng, Z. Shuai, V. Coropceanu and J. L. Brédas, J. Chem. Phys., 2011, 135, 10 CrossRef PubMed.
- Z. Shuai, H. Geng, W. Xu, Y. Liao and J. M. André, Chem. Soc. Rev., 2014, 43(8), 2662–2679 RSC.
- T. Sato, M. Uejima, N. Iwahara, N. Haruta, K. Shizu and K. Tanaka, J. Phys.: Conf. Ser., 2013, 428, 1 CrossRef.
- M. Uejima, T. Sato, K. Tanaka and H. Kaji, Phys. Chem. Chem. Phys., 2013, 15(33), 14006–14016 RSC.
- M. Huix-Rotllant and N. Ferré, J. Chem. Theory Comput., 2016, 12(10), 4768–4777 CrossRef CAS PubMed.
- S. J. Park, S. Jeon, I. K. Lee, J. Zhang, H. Jeong, J. Y. Park, J. Bang, T. K. Ahn, H. W. Shin, B. G. Kim and H. J. Park, J. Mater. Chem. A, 2017, 5(25), 13220–13227 RSC.
- H. Geng, Y. Niu, Q. Peng, Z. Shuai, V. Coropceanu and J. L. Brédas, J. Chem. Phys., 2011, 135, 10 CrossRef PubMed.
- O. Kwon, V. Coropceanu, N. E. Gruhn, J. C. Durivage, J. G. Laquindanum, H. E. Katz, J. Cornil and J. L. Brédas, J. Chem. Phys., 2004, 120(17), 8186–8194 CrossRef CAS PubMed.
- V. Coropceanu, M. Malagoli, D. A. da Silva Filho, N. E. Gruhn, T. G. Bill and J. L. Brédas, Phys. Rev. Lett., 2002, 89(27), 1–4 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- X. Xie, A. Santana-Bonilla, W. Fang, C. Liu, A. Troisi and H. Ma, J. Chem. Theory Comput., 2019, 15(6), 3721–3729 CrossRef CAS PubMed.
- J. Bednarska, R. Zaleśny, W. Bartkowiak, B. Ośmiałowski, M. Medved and D. Jacquemin, J. Chem. Theory Comput., 2017, 13(9), 4347–4356 CrossRef CAS PubMed.
- Y. Cheng, Y. Qi, Y. Tang, C. Zheng, Y. Wan, W. Huang and R. Chen, J. Phys. Chem. Lett., 2016, 7(18), 3609–3615 CrossRef CAS PubMed.
- M. Malagoli and J. L. Brédas, Chem. Phys. Lett., 2000, 327(1–2), 13–17 CrossRef CAS.
- X. J. Ma, X. D. Zhu, K. L. Wang, F. Igbari, Y. Yuan, Y. Zhang, C. H. Gao, Z. Q. Jiang, Z. K. Wang and L. S. Liao, Nano Energy, 2019, 63, 103865 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on fragment-based decomposition analysis, fragment mode composition analysis and neutral and cationic ground state molecular structures. See DOI: 10.1039/d0cp01722a |
| This journal is © the Owner Societies 2020 |