
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural dynamics of an iron molybdate catalyst under redox cycling conditions studied with in situ multi edge XAS and XRD†
Abhijeet
Gaur
 *ab,
Matthias
Stehle
a,
Kristian Viegaard
Raun
c,
Joachim
Thrane
c,
Anker Degn
Jensen
c,
Jan-Dierk
Grunwaldt
ab and
Martin
Høj
*c
*ab,
Matthias
Stehle
a,
Kristian Viegaard
Raun
c,
Joachim
Thrane
c,
Anker Degn
Jensen
c,
Jan-Dierk
Grunwaldt
ab and
Martin
Høj
*c
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Karlsruhe, D-76131, Germany. E-mail: abhijeet.gaur@kit.edu; Tel: +49 721 608 44320
bInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology (KIT), Karlsruhe, D-76131, Germany
cDepartment of Chemical and Biochemical Engineering, Technical University of Denmark (DTU), Kgs. Lyngby, DK-2800, Denmark. E-mail: mh@kt.dtu.dk; Tel: +45 452 52842
First published on 5th May 2020
Abstract
The structural dynamics and phase transformations of an iron molybdate catalyst with excess molybdenum trioxide (Mo/Fe = 2.0) were studied during redox cycling of the catalyst using in situ multi-edge X-ray absorption spectroscopy (XAS) at the Mo K-edge (transmission mode) and Fe K-edge (fluorescence mode) in combination with X-ray diffraction (XRD). X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analysis showed that heating under reducing conditions with methanol up to 400 °C produced MoO2 and FeMoO4. Linear combination fitting (LCF) analysis showed that iron was reduced completely, while molybdenum remained partly oxidized (60% as Mo(VI)). Complementary in situ XRD also supported the phase transformation due to reduction of Fe2(MoO4)3 and MoO3 to FeMoO4 and MoO2. Subsequent heating under oxidative conditions from 200 to 500 °C transformed the catalyst into its initial state via Fe2O3 and extra MoO3 as intermediate phases. This underlines the segregation and iron enrichment during redox cycling. MoO3 volatilization, observed under industrial reaction conditions of a methanol and oxygen containing atmosphere, causes this segregation to be permanent. Complete regeneration could only be achieved at 500 °C, which is significantly higher than industrial reaction temperatures. Overall, multi edge in situ XAS along with complementary XRD was found to be an ideal tool for tracing the different amorphous and crystalline phases present during redox cycling of the catalyst.
1. Introduction
Iron molybdate with an excess of molybdenum oxide (denoted FeMo throughout the paper) is an industrially important catalyst for the selective oxidation of methanol to formaldehyde, which has led to a number of fundamental studies concerning its structure, activity, mechanism, stability and deactivation.1–6 To improve the performance of a catalyst on a knowledge-based approach, it is critical to understand the structure and structural dynamics of the catalyst under reaction conditions. Commercial iron molybdate catalysts are typically formulated with an excess of molybdenum(VI)oxide, since Mo is known to volatilize from the reactor inlet and hot spot7 during the reaction, leaving an unselective Fe rich surface.8 The FeMo system contains multiple distinct phases including bulk α-MoO3 and Fe2(MoO4)3, as well as a surface layer of MoOx on the Fe2(MoO4)3 surface.9 Hence, it is difficult to assess the nature of the active sites and role of Fe2(MoO4)3, specifically whether it merely acts as a support for the active MoOx or whether it has its own inherent activity. The catalytic behavior of FeMo catalysts was observed to be mainly dependent on their Mo/Fe atomic ratio.2,10 The most active phase composition of the bulk iron molybdate catalyst was proposed to have the stoichiometric Mo/Fe ratio of 1.53 and the function of excess MoO3 is speculated to maintain the active phase of the catalyst at this ratio. The activity and selectivity of bulk iron molybdate catalysts for methanol oxidation was examined as a function of Mo/Fe ratios from 0.02 to 4,2 revealing that Mo/Fe ratios greater than 1.5 are required for high catalytic activity and selectivity to formaldehyde. The presence of an octahedral Mo oxide overlayer has been reported,1 which corresponds to one monolayer. Various groups have proposed that this overlayer structure is the fundamental, active surface for the selective oxidation of methanol to formaldehyde.1,9,11 The presence of two terminal oxygen atoms double bonded to Mo in such coordination allows the reacting methanol molecules to be bonded simultaneously at two points to the surface. The formation of several molybdenum oxy-methoxy compounds was also reported12 and it was argued that hydrogen abstraction of the methanol hydroxyl group produces methoxy species that are intermediates in formaldehyde formation. The role of Fe in iron molybdate catalysts would be to favor the transfer of O2 and H2O between the surface and the gas phase and to facilitate re-oxidation of reduced Mo.11 Furthermore, oxygen atoms from terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups were argued to be crucial for the activity and selectivity of MoO3.13–16 Several samples revealed a co-existence of various MoOx phases and similar qualitative phase compositions existed for different Mo/Fe ratios which does not allow to draw any conclusions about a correlation between a certain MoO3 phase and its catalytic behavior.
O groups were argued to be crucial for the activity and selectivity of MoO3.13–16 Several samples revealed a co-existence of various MoOx phases and similar qualitative phase compositions existed for different Mo/Fe ratios which does not allow to draw any conclusions about a correlation between a certain MoO3 phase and its catalytic behavior.
In order to predict and model the performance of catalysts, their electronic and structural properties need to be studied at an atomic scale as they strongly influence the catalytic properties. Under dynamic conditions (temperature, pressure, gas atmosphere, etc.) catalysts can be chemically transformed, giving rise to changes in their initial surface composition correlated to bulk structure, morphology and particle size. Thus, complementary techniques have been used for detailed studies of the structural, electronic and chemical properties of different types of catalysts under these conditions. The combination of X-ray absorption spectroscopy (XAS) with complementary techniques can provide in situ and operando chemical, structural, and electronic insights into the catalysts and other involved species to holistically understand the catalytic process. XAS coupled with X-ray diffraction (XRD) is beneficial since the nature of crystallite phases can be determined by XRD whereas XAS is useful for determining the oxidation state and coordination geometry of the metal atoms. In order to evaluate the potential of the multiple-technique approach and usefulness of different in situ/operando reactors at the synchrotron facilities, the behavior of an Fe2(MoO4)3 catalyst under redox conditions was discussed in the literature.17–19 During reduction in MeOH, due to continuous removal of oxygen, pseudo-molybdate and a MoC type phase were observed, leading to the breakdown of the catalyst to separate oxides (low Mo/Fe ratio of 1.5).17 The rate of re-oxidation was found to increase with increasing Mo concentration. Along with other observations, this clarified the role of MoO3 excess in the as-prepared catalyst, which is to maintain the stability of the active phase and to replenish any evaporated molybdenum.18
In a recent study20 on the same catalytic system, we have shown that during its evolution with time on stream for selective oxidation of methanol to formaldehyde, the FeMo catalyst undergoes strong structural changes due to evaporation of MoO3. In this study we now probe the structural changes of Mo and Fe related crystalline and amorphous phases during redox cycling using in situ multi-edge XAS at the Mo K- and Fe K-edges combined with complementary XRD. Using redox cycling in methanol and under oxidizing conditions insight into the structural dynamics can be received. The combination of multi-edge XAS and XRD allows for investigating the effect of over-reduction followed by re-oxidation of the catalyst, corresponding to one cycle of the selective oxidation of methanol to formaldehyde. For this purpose we exploited the advanced beamline set-ups available at the synchrotron radiation sources which allow obtaining high quality bulk structural data by XAS and XRD from catalysts under dynamic conditions when simultaneously using multiple techniques.
2. Experimental
The catalyst was prepared by hydrothermal synthesis with special interest paid to the precise adjustment of the atomic molybdenum to iron ratio.7 The Mo/Fe ratio was 2.0, which resulted in an active and selective catalyst in a broad temperature range, typically 250–400 °C, at a feed concentration of 5 vol% CH3OH with about 10 vol% O2.7 The catalyst was calcined at 535 °C in air, resulting in a BET surface area of 4.7 m2 g−1 and Rietveld refined particles size of 133 nm for the MoO3 phase and 375 nm for the Fe2(MoO4)3 phase. The catalyst was crushed and ground with silica in a 1/5 catalyst/silica weight ratio. This mixture was pressed as a pellet, crushed and sieved to 100–150 μm. Combined XAS/XRD studies during temperature programmed experiments were conducted at the SNBL beamline BM31 at ESRF, Grenoble, France.21Fig. 1 shows the schematics of the advance experimental setup used for the in situ multi-edge XAS/XRD experiments. As optics requirements for high quality XAS and XRD are quite different, the beamline is equipped with a channel-cut monochromator (CCM) for a high-resolution powder diffraction and a second double crystal monochromator (DCM) for XAS (interchanged within 10 s).21 Measurements were performed by means of a fixed-bed micro-reactor based on quartz glass capillaries22 for mimicking optimal heat and mass transfer.23,24 The catalyst in the form of powder was loaded in a glass capillary with inner diameter of 1.5 mm with the total bed length inside the capillary of 1 cm and the catalyst was fixed from both sides by quartz wool. XAS was performed at the Fe K-edge (7.112 keV) in fluorescence mode and at the Mo K-edge (20.000 keV) in transmission mode quasi-simultaneously by using the fast edge changing facility at the beamline. A molybdenum foil was measured simultaneously as a reference compound for energy calibration. The measurements were conducted in a loop that consisted of two Mo K-edge scans (4 min each), three Fe K-edge scans (5 min each) and one XRD pattern (1 min). Switching between Fe K- and Mo K-energies took approximately 30 s and switching from XAS to XRD monochromator and detector took 60 s.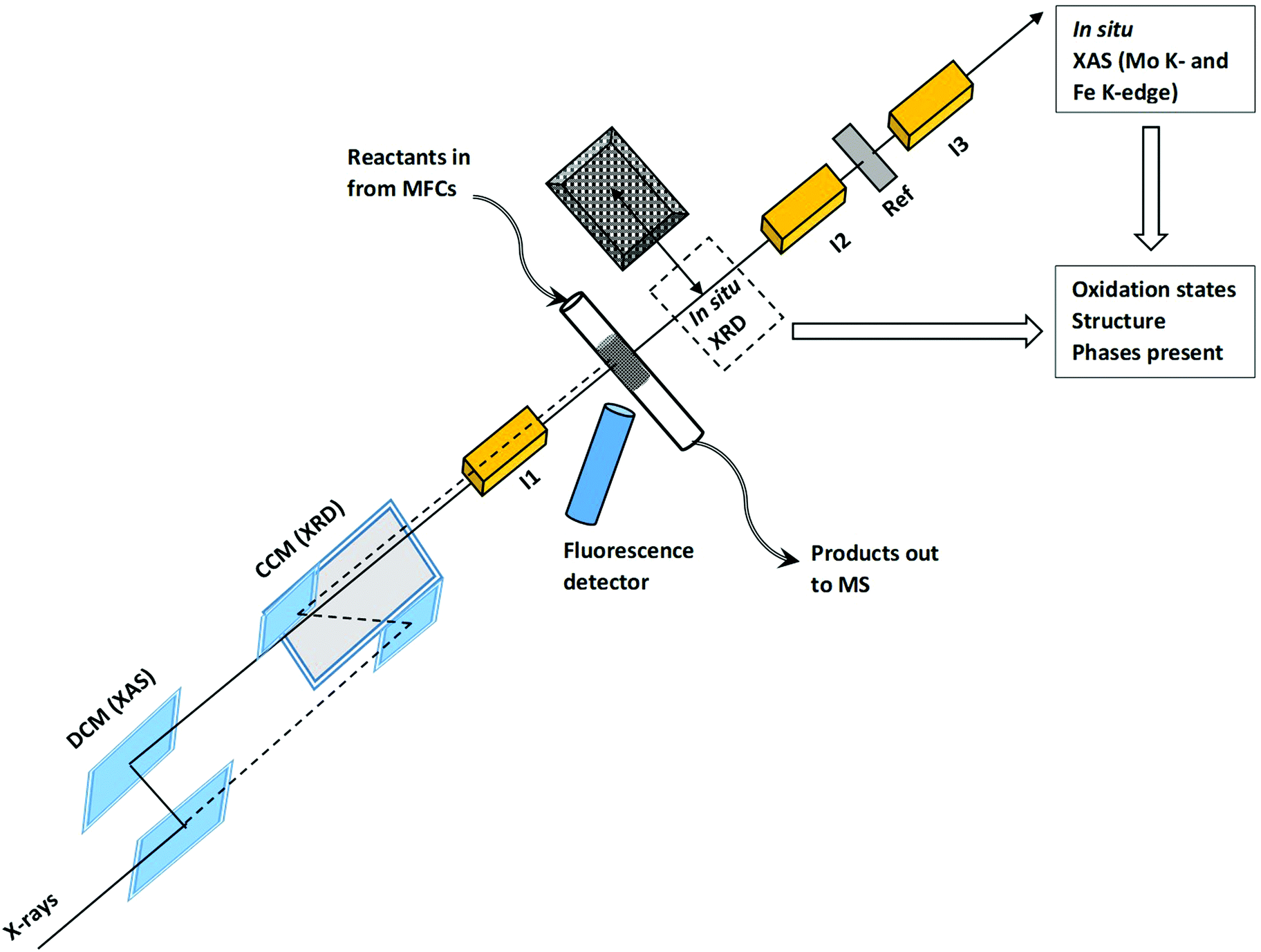 | ||
| Fig. 1 Schematic setup for in situ XAS/XRD experiments performed at the unique setup at SNBL beamline BM31, ESRF. Double crystal monochromator (DCM), channel-cut monochromator (CCM), ionization chambers (I1, I2 and I3), reference foil (Ref), mass flow controllers (MFCs) and mass spectrometer (MS). | ||
The overview of the experimental procedure followed during in situ experiments is given in the ESI,† Fig. S1. The catalyst was first heated to 100 °C in a flow of 10% O2/He. Then the flow was changed to 5% MeOH/He and the sample was heated to 400 °C at 1 °C min−1 for temperature programmed reduction (TPR) and was kept at 400 °C for 1 hour. The sample was cooled to 200 °C after which the flow was changed back to 10% O2/He. In the next step, the sample was heated to 500 °C at 1 °C min−1 for temperature programmed oxidation (TPO) and after a stabilization period of 1 hour finally cooled to 100 °C.
XAS data were treated with the software package IFEFFIT.25 The spectra were energy calibrated, normalized, and background subtracted in Athena. Linear combination fitting (LCF) of XANES spectra was performed in a range of −20 to +50 eV at the Mo K- and Fe K-edges, respectively. EXAFS fitting was performed in Artemis by using model structures obtained from references of Fe2(MoO4)3 and MoO3, in the case of Mo. At the Fe K-edge, spectra were only measured in the XANES region due to high noise in the EXAFS region. Fittings at the Mo K-edge were performed in R – as well as k-space for determining the structural parameters which included energy shift of the path (ΔE0), change in the half path length (ΔR), amplitude reduction factor (S02), number of identical paths (N) and relative mean-square displacement of the atoms included in the path (Debye–Waller factor, σ2). Principal component analysis (PCA) at the Fe K-edge was performed using XANES spectra in the range of −20 to +80 eV using the PCA subroutine of Demeter.25
XRD patterns were recorded with a CMOS-Dexela 2D detector at a short X-ray wavelength of 0.4934 Å (25.13 keV) so that the penetration of X-rays is optimal. For each diffractogram, five bright and additionally five dark images for background subtraction were recorded in the 2θ range of 2–35° with a data point spacing of approximately 0.01° and subsequently averaged. The sample to detector distance was calibrated with a LaB6 reference. For XRD data treatment, the diffractograms were normalized to the most intense reflection of the SiO2 dilutant, and a normalized diffractogram of only the dilutant measured at the same temperature was subtracted using an in-house Matlab script.
3. Results and discussion
3.1 Initial state and temperature programmed reduction (TPR)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 007 eV and another feature B at 20027 eV. The pre-edge peak A is attributed to the dipole-forbidden/quadrupole-allowed 1s–4d transition, which is associated primarily with tetrahedral geometry. The feature is also present but to weaker extent in distorted octahedral geometry. Feature B at 20027 eV is assigned to the dipole-allowed 1s–5p transition, a characteristic feature of Mo species with octahedral/distorted octahedral geometry.26 A comparison of the XANES spectrum of the FeMo catalyst with MoO3 showed that the catalyst had a slightly stronger feature A than for MoO3 and weaker intensity for peak B. Also, the intensity of peak A for the catalyst was considerably lower compared to the reference spectrum of Fe2(MoO4)3.27 This showed that in the FeMo catalyst, Mo had both tetrahedral (Fe2(MoO4)3 nature) as well as octahedral geometry (MoOx nature, i.e., molybdenum oxide with an average oxidation state between Mo(IV) to Mo(VI)).
007 eV and another feature B at 20027 eV. The pre-edge peak A is attributed to the dipole-forbidden/quadrupole-allowed 1s–4d transition, which is associated primarily with tetrahedral geometry. The feature is also present but to weaker extent in distorted octahedral geometry. Feature B at 20027 eV is assigned to the dipole-allowed 1s–5p transition, a characteristic feature of Mo species with octahedral/distorted octahedral geometry.26 A comparison of the XANES spectrum of the FeMo catalyst with MoO3 showed that the catalyst had a slightly stronger feature A than for MoO3 and weaker intensity for peak B. Also, the intensity of peak A for the catalyst was considerably lower compared to the reference spectrum of Fe2(MoO4)3.27 This showed that in the FeMo catalyst, Mo had both tetrahedral (Fe2(MoO4)3 nature) as well as octahedral geometry (MoOx nature, i.e., molybdenum oxide with an average oxidation state between Mo(IV) to Mo(VI)).
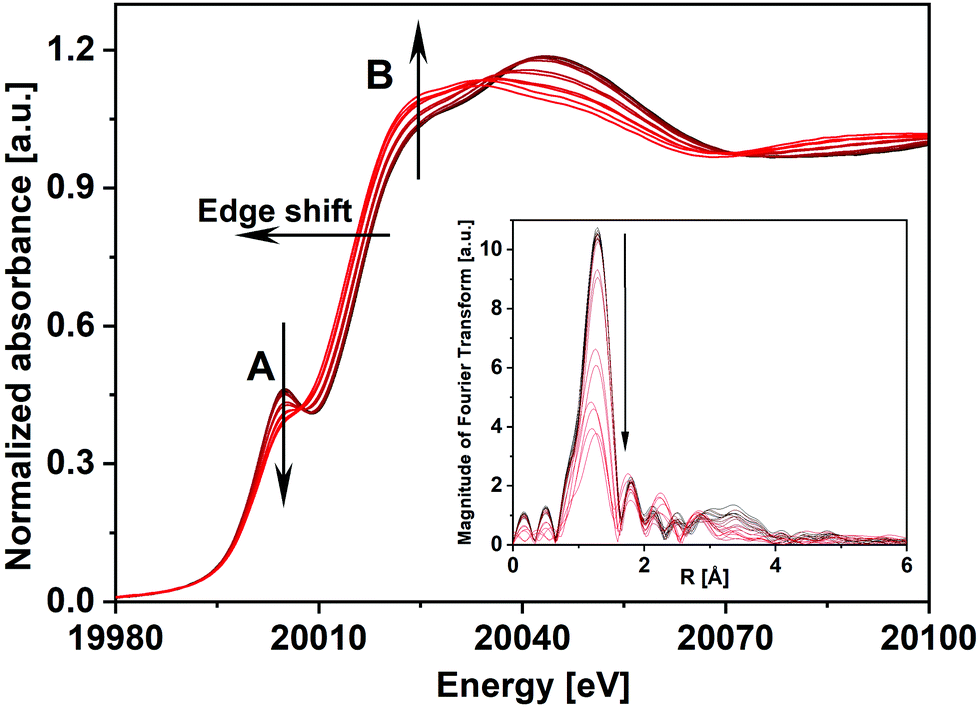 | ||
| Fig. 2 In situ Mo K-edge XANES spectra and corresponding k3-weighted Fourier transformed EXAFS spectra (inset) of the FeMo catalyst during TPR under 5% MeOH/He. Arrows point towards increasing temperature. | ||
As the initial state of the FeMo catalyst at 100 °C indicated mixed Mo(VI) phases, linear combination fitting (LCF) of the XANES spectrum was performed with standards Fe2(MoO4)3 and α-MoO3. This showed 76% contribution from ferric molybdate and 24% contribution from molybdenum trioxide. This is in agreement with the composition of the fresh catalyst as determined by XRD containing 83 wt% Fe2(MoO4)3 and 17 wt% α-MoO3, which corresponds to 22 mol% of the Mo bound as MoO3 and 78 mol% as Fe2(MoO4)3.7
The XANES features of the spectra during TPR (Fig. 2) showed that the edge position shifted to lower energies, from 20012.3 to 20010.2 eV, revealing partial reduction of Mo. The decrease in intensity of peak A and simultaneous increase in the intensity of feature B showed a rise in the octahedral nature of Mo centers, corresponding to Fe2(MoO4)3 being decomposed to FeMoO4 and thereby forming MoOx species e.g. MoO2 and intermediate MoOx (x = 2–3) species. However, it can be seen that after TPR, peak A was still present showing that Mo(VI) was not completely reduced to Mo(IV), which does not have this feature. Since MoO2 has a strictly octahedral field, the 1s–4d transition is forbidden and no pre-edge peak would be observed.28 The corresponding Fourier transformed EXAFS spectra during TPR are shown in the inset of Fig. 2 where in the initial state of the catalyst a high amplitude of the first peak (Mo–O1) indicated strong tetrahedral nature due to a high content of Fe2(MoO4)3. Metal backscattering at ∼3 Å (Fe) additionally indicated the presence of Fe2(MoO4)3.26 The fitting results for the initial state of the catalyst at 100 °C in 10% O2/He (see ESI,† Table S1) also supported the presence of multiple Mo phases (see also ESI,† Fig. S3(a)). At about 290 °C, this metal backscattering disappeared due to the reduction of Fe2(MoO4)3 (Fig. S3(b), ESI†). In the final state during TPR at 350 °C, the amplitude of the Mo–O1 contribution was highly reduced showing the presence of disordered Mo oxide phases (Fig. S3(c), ESI†).
In order to determine the contribution of different phases during the TPR, LCF was performed using standard spectra of Fe2(MoO4)3, MoO3 and MoO2. FeMoO4 was not available as reference due to unavailability of this compound commercially and its instability during synthesis. Also, at the Mo K-edge weak XANES features present in the FeMoO4 phase were found to be overlapping with strong features of the Mo(VI) phases Fe2(MoO4)3 and MoO3 (for details see ESI,† Fig. S4). Thus, the formation of FeMoO4, and associated reduction of iron, is measured and discussed more clearly using Fe K-edge XANES in Section 3.1.2. It is worth mentioning here that PCA cannot be applied at Mo K-edge due to the unavailability of set of proper standard spectra. The contributions of the different phases obtained from LCF analysis of the XANES spectra at the Mo K-edge during TPR are shown in Fig. 3(a). Reduction of Mo started at approximately 270 °C and the contribution from MoO2 was observed at the expense of an initial decrease in MoO3 content. In the final state at 400 °C, contributions from Fe2(MoO4)3, MoO3 and MoO2 were observed to be 43%, 20% and 37%, respectively. Note that MoO3 and Fe2(MoO4)3 together represent the Mo(VI) present as FeMoO4 after reduction, see also Fe K-edge XANES and XRD results in Section 3.1.2. Thus, at 400 °C, about 40% of the Mo(VI) species were reduced to Mo(IV) species, represented by MoO2. Some MoOx (x = 2–3) may also be presented by a combined contribution from MoO3 and MoO2 as it is hard to detect these species by their XANES fingerprint. More than 50% of the Mo was still present in oxidation state VI, which is in agreement with a previous in situ catalytic study9 at 250 °C, which showed that during formaldehyde production only Mo(VI) was present throughout the experiment, indicating that gas phase oxygen transfer to molybdenum is rapid under reaction conditions at temperatures near the lower range of industrial conditions (typically 220 to 350 °C).
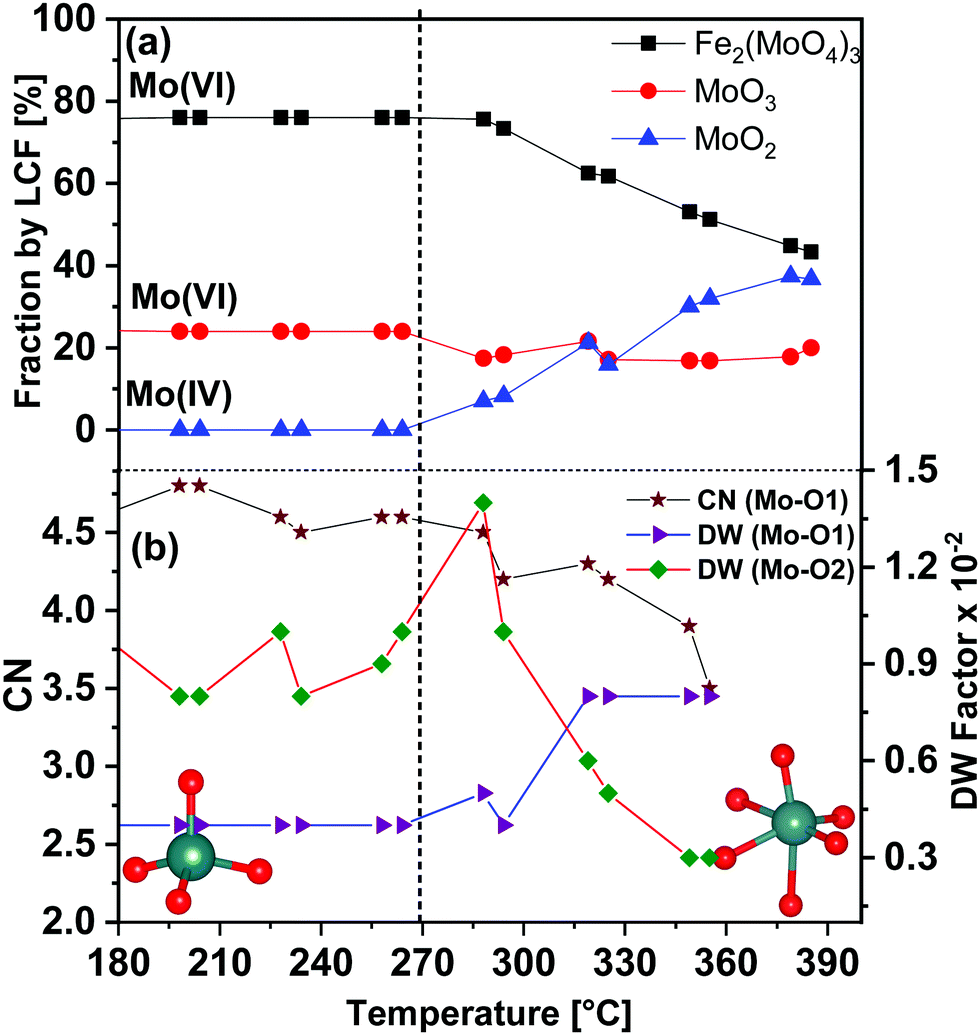 | ||
| Fig. 3 (a) Mo K-edge XANES linear combination fitting (LCF) results for the FeMo catalyst during heating under 5% MeOH/He employing XANES spectra of the reference compounds Fe2(MoO4)3, MoO3 and MoO2. (b) Variation in the selected parameters during TPR as determined from EXAFS fitting analysis in R space. Dashed line indicates the temperature at which Mo reduction started. The insets show the transition from tetrahedral to octahedral coordination geometry of Mo during TPR, Mo (blue spheres) and O (red spheres). CN of Mo–O2 bonds were kept fixed to 1. | ||
Corresponding EXAFS spectra were analyzed by performing fitting in R-space and variation of selected obtained parameters are given in Fig. 3(b). Initially at 100 °C, the CN of Mo–O1 was found to be 5.0 (±0.4) at 1.76 Å, which indicates strong contribution from the tetrahedral Mo centers present in Fe2(MoO4)3. A considerable decrease in the CN of Mo–O1 was observed at approximately 290 °C (see also Fig. S3(b, c) and Table S2, ESI†). Also, the DW factor for Mo–O1 started increasing in parallel with the reduction process and was almost doubled at the end of TPR, showing higher disorder in these bonds. Conversely, the DW factor for the longer Mo–O2 bonds (axial position), which corresponds to octahedral Mo centers, decreased at higher temperature showing stronger axial bonds due to an increase in octahedral nature of Mo centers present in MoOx species (the CN of each of Mo–O2 bond were fixed to 1). Hence, with increasing temperature, a decrease in Mo oxidation state and an increase in octahedral nature of Mo centers were observed. These structural changes point towards formation of MoO2 and as well as MoOx species during TPR (see also Fe K-edge XANES and XRD results in Section 3.1.2).
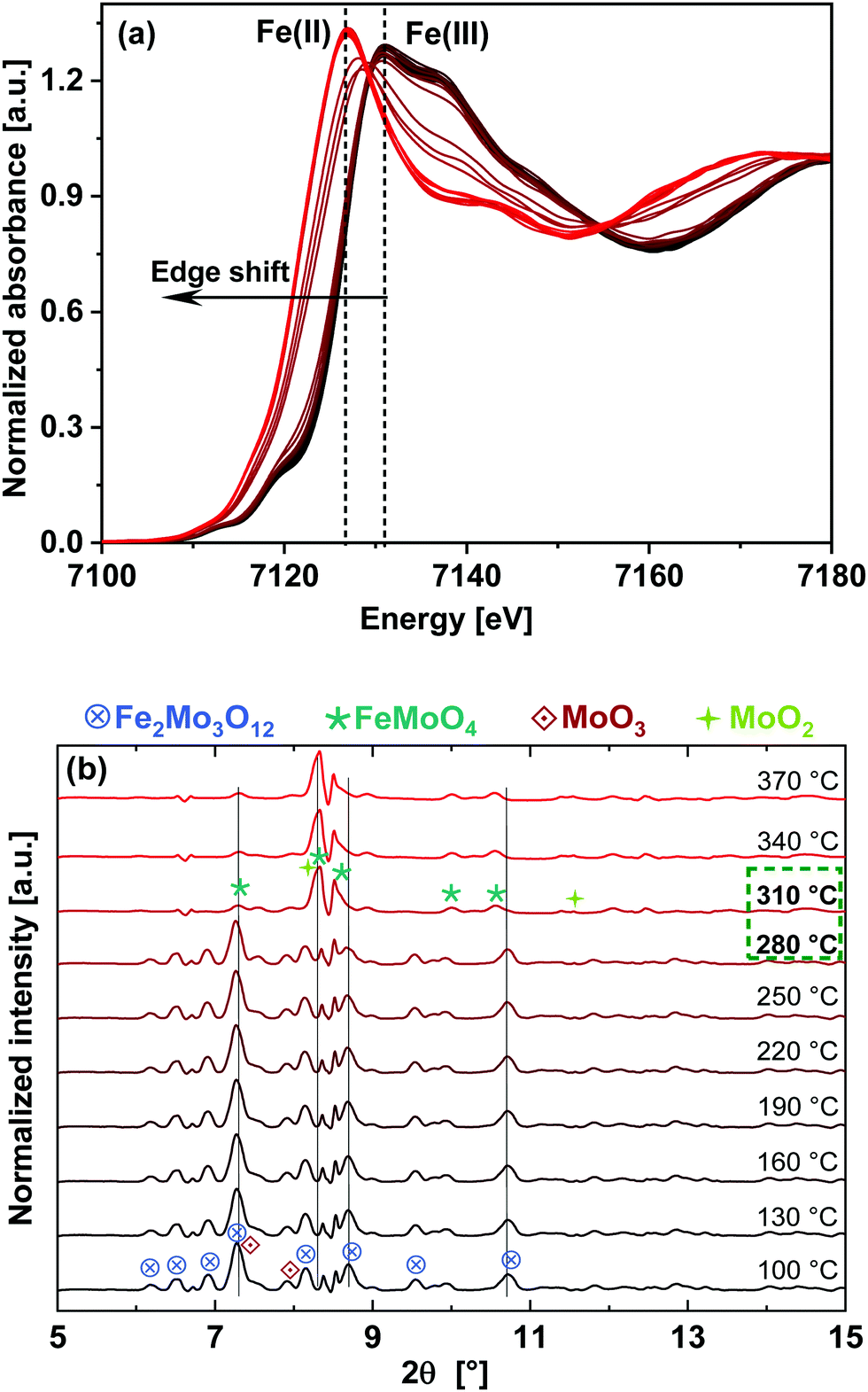 | ||
| Fig. 4 (a) In situ Fe K-edge XANES spectra for the catalyst during heating in 5% MeOH/He (TPR). Dashed lines indicate the energy positions of the white line peak for oxidized and reduced Fe phases. The arrow points towards increasing temperature. (b) In situ XRD at 0.4934 Å (25.13 keV) during TPR in 5% MeOH/He. Phase transition to FeMoO4 can be observed by the increasing intensity of the reflections at 8.32 and 8.50°. | ||
PCA uses matrix operations on an experimental spectra data matrix, to give two main outputs in the form of eigenvectors and eigenvalues, thereby providing the minimum number of significant components required to satisfactorily regenerate the data matrix.30 Thus, absorbance in a set of spectra can be mathematically modelled as a linear sum of individual components, called factors, which correspond to each one of the species present in the sample, plus noise. The PCA result at 275 °C is given as an example in the ESI,† Fig. S5 and Table S3. The figure shows the different components weighted by their eigenvalues where the first two components clearly dominate the spectra and the contribution from the third component is also significant. The fourth component appears to be noise, indicating that 3 components were required to fit the data. Thus, both Fe2O3 and FeO were evaluated as standards during LCF, but Fe2O3 gave better fit results, confirming its presence as intermediate phase during TPR (200–300 °C).
Results obtained from LCF analysis performed using these XANES spectra are given in the ESI,† Fig. S6. It was observed that the reduction of Fe starts at approximately 270 °C, parallel to reduction of Mo. Fe(ox) is the initial state which mainly corresponds to Fe2(MoO4)3 and Fe(red) is the final state after TPR, which have been taken as the standards for LCF analysis. The final reduced state of Fe, having features similar to FeMoO4,29 was observed at 330 °C, showing faster reduction of Fe compared to Mo. These results, combined with the Mo K-edge results, indicate that at the end of TPR at 400 °C, the phases present were Fe2(MoO4)3, FeMoO4, and MoO2 (possibly including intermediate phases MoOx), which confirm the presence of both Mo(IV) and Mo(VI) species.
Due to the fast interchange between the monochromators, DCM for XAS and CCM for XRD, the XAS measurements at the Mo K- and Fe K-edges were further supported by quasi-simultaneously recorded XRD patterns of the FeMo catalyst during TPR under 5% MeOH/He, as shown in Fig. 4(b). An XRD pattern characteristic for Fe2(MoO4)3 (most intense reflection at 2θ = 7.32° and other significant reflections at 6.93, 7.55–7.63, 7.92, 8.16, 8.73–8.82°) at the beginning of the TPR was observed, which disappeared almost completely at 310 °C and reflections corresponding to FeMoO4 appeared (reflections at 8.32 and 8.50°). Furthermore, at the end of TPR a weak reflection from MoO2 was also observed at 11.6°. In the initial state, reflections from α-MoO3 were observed as well (reflections at 7.43 and 8.16–8.23°), partly overlapping with the reflections of Fe2(MoO4)3, which disappeared as well. Thus, in situ XRD confirms that the phase transformation of Fe2(MoO4)3 to FeMoO4 started at around 280 °C, in agreement with XAS results at the Mo K- and Fe K-edges.
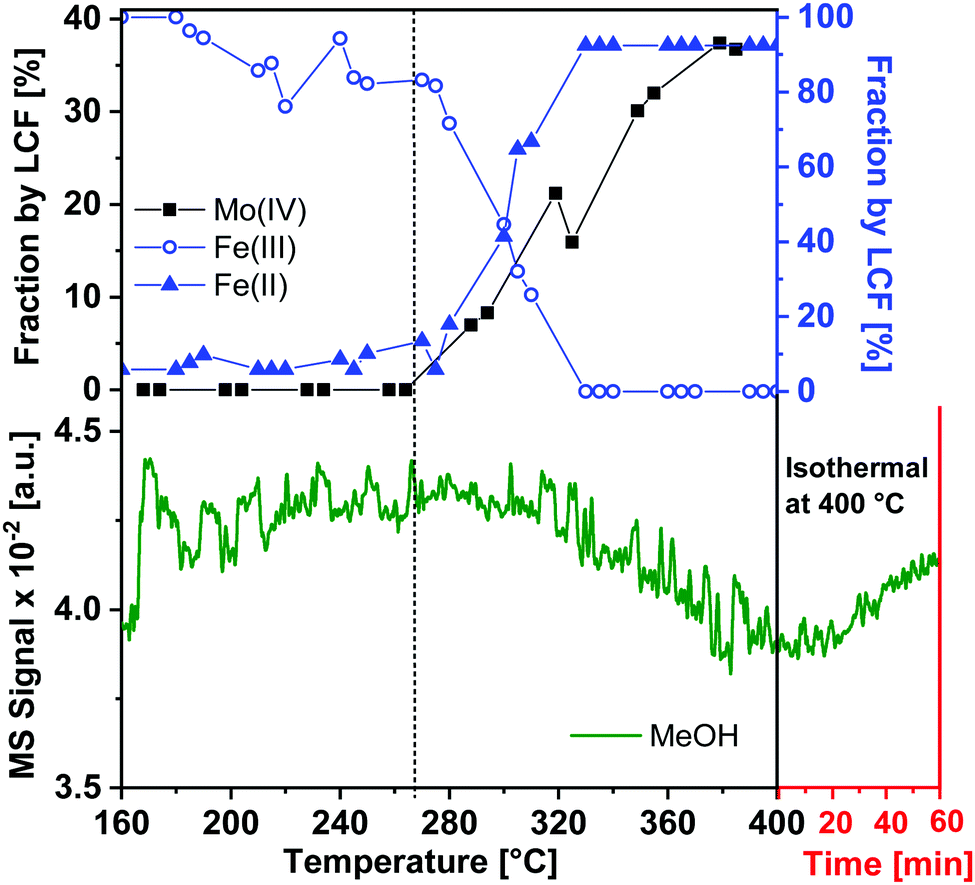 | ||
| Fig. 5 Fraction of Mo(IV) (as determined from Mo K-edge LCF), Fe(III) and Fe(II) (from Fe K-edge LCF) as function of temperature with MS trace of MeOH overlaid during heating (100 to 400 °C at 1 °C min−1) under reducing conditions of 5% MeOH/He. The extended X-axis (in red) shows the isothermal period at 400 °C for the next 60 minutes. The dashed line indicates the start of reduction of Mo as well as Fe species. | ||
Though the MS-traces m/z = 29 and 18 corresponding to the formation of HCHO and H2O, respectively, have also been observed as shown in the ESI,† Fig. S7, the MS was slow at pumping down from the previous experiment performed. Hence there was remarkably high background signal of HCHO and H2O during the TPR up to about 300 °C. Nevertheless, it can be observed from Fig. S7 (ESI†) that during heat up the concentration of MeOH decreased and those of HCHO and H2O increased, as expected, and during the isothermal period, when the reduction was complete, the concentration of MeOH increased and those of HCHO and H2O decreased, both with some delay due to the MS pumping down.
Hence, using in situ XAS/XRD with MS measurements of the gas phase, it can be concluded that during heating under reducing conditions, Fe2(MoO4)3 and MoO3 were observed to reduce thereby forming FeMoO4 and MoO2 which is in agreement with the proposed reactions (1) and (2).
| Fe2(MoO4)3 + CH3OH → 2FeMoO4 + MoO3 + HCHO + H2O | (1) |
| MoO3 + CH3OH → MoO2 + HCHO + H2O | (2) |
3.2 Heating under oxidizing conditions (TPO)
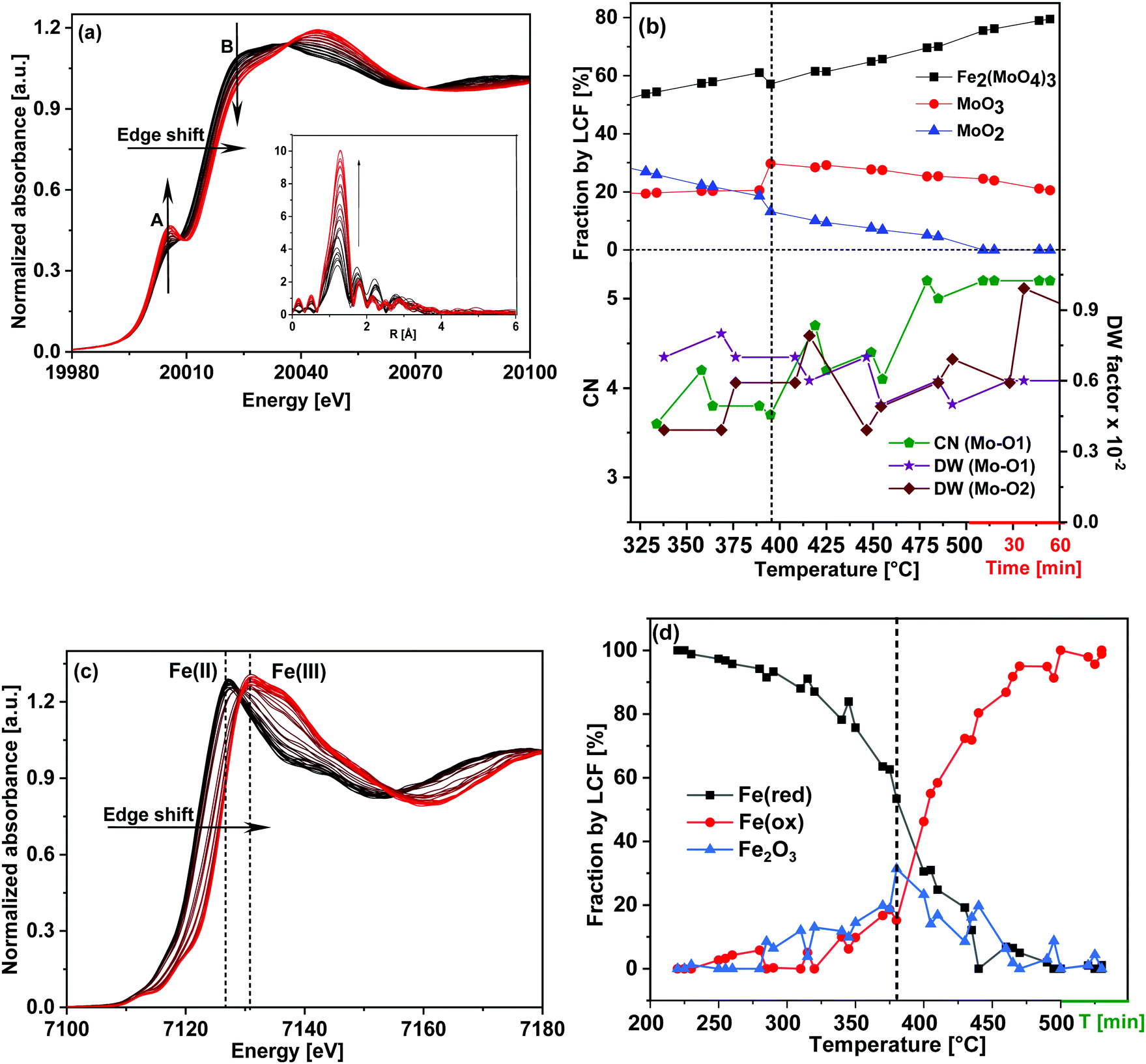 | ||
| Fig. 6 For the FeMo catalyst during heating under oxidizing conditions 10% O2/He (TPO): (a) Mo K-edge XANES spectra, (b) corresponding XANES LCF results and variation in the selected parameters as determined from EXAFS analysis. At the X-axis, the red line after 500 °C represents the isothermal period, (c) Fe K-edge XANES spectra and (d) corresponding XANES LCF results. In (a) and (c) arrows point towards increasing temperature. The vertical dashed lines in (b) and (d) indicate the temperature ∼390 °C at which a sudden increase in MoO3 and Fe2O3 content was observed, respectively. | ||
LCF analysis of the XANES spectra using the same standards as during TPR and variations of the parameters obtained from EXAFS analysis are shown in Fig. 6(b) in the temperature range 320–500 °C (for the complete LCF results from 200–500 °C please see ESI,† Fig. S9). The initial composition before TPO at 200 °C was 39% Fe2(MoO4)3, 20% MoO3 and 41% MoO2 which is similar to the composition of the final state after TPR (43% Fe2(MoO4)3, 20% MoO3 and 37% MoO2) within the margin of error of LCF and considering the temperature difference.
Oxidation of Mo began as soon as the temperature started to rise which is evident from the decreasing MoO2 and increasing Fe2(MoO4)3 contributions, showing that overall MoO2 combined with FeMoO4 to give Fe2(MoO4)3. The content of MoO3 was quite stable until 380 °C, but in the range of 380–400 °C a sharp increase was observed (Fig. 6(b)), indicating formation of MoO3 as well.
At the same temperature, the EXAFS parameters (Fig. 6(b)) showed a decrease in the DW factor of Mo–O1 and an increase in that of Mo–O2 supporting formation of octahedral Mo centers present in MoO3. The contribution from MoO2 continuously decreased and became negligible at around 500 °C, showing that almost all of Mo was in the Mo(VI) state. After the sudden increase at approximately 390 °C, the contribution from MoO3 fell back to 21%, slightly lower than what was present initially before TPO (24%). The remaining 80% of Mo(VI) was found to be Fe2(MoO4)3, which is slightly higher compared to the initial composition of the catalyst at 100 °C before TPR (76%). It is possible that some MoO3 formed volatile species and evaporated during the TPR, similar to the evaporation clearly observed under reaction conditions for methanol oxidation.20 EXAFS analysis also confirmed this conclusion since the CN of Mo–O1 at 1.77 Å increased and the corresponding DW factor decreased, showing a rise in tetrahedral nature of Mo. Similarly, the DW factor of the Mo–O2 (axial) concurrently increased, showing a decrease in octahedral nature of Mo centers (the CN of both Mo–O2 bonds were kept fixed to 1).
Fig. 6(c) shows the Fe K-edge XANES spectra during heating under oxidizing conditions. PCA was performed to confirm the number components required to fit the spectra and the results obtained from PCA analysis for Fe K-edge XANES spectrum at 380 °C is given as an example in ESI,† Fig. S10 and Table S4. In this case also, the first two components clearly dominate the spectra, with a significant contribution from the third component and the fourth component appears to be noise, indicating that 3 components were required to fit the data. Thus, Fe2O3 was also employed as standard during LCF, which gives better fit results than FeO, confirming its presence as intermediate phase during TPO (300–450 °C). LCF analysis shown in Fig. 6(d) showed that oxidation of Fe effectively started at 280 °C. The contribution from Fe2O3 was found to be high (around 30%) between 380–410 °C. In this temperature range LCF at the Mo K-edge also showed a high presence of MoO3 pointing towards FeMoO4 reacting with O2 to give Fe2O3 and MoO3 (eqn (3)) in competition with the reactions regenerating the catalyst (eqn (4) and (5)).31 However, like the excess MoO3, the Fe2O3 phase was an intermediate and the contribution was found to decrease at higher temperature. At 500 °C Fe was found mostly as Fe2(MoO4)3, due to recombination of Fe2O3 and MoO3 by solid state reaction (eqn (6)). These redox dynamics explain the formation of Fe2O3 observed under reaction conditions for selective oxidation of methanol at 380 to 400 °C, where MoO3 evaporation competes with the recombination and with extended time on stream MoO3 eventually evaporates completely.7
Corresponding in situ XRD results are given in Fig. 7. They also show the presence of FeMoO4 and MoO2 initially, as observed at the end of TPR. During TPO, a phase transition was observed between 380–410 °C where higher presence of MoO3 (by LCF at Mo K-edge data) and Fe2O3 (by LCF at Fe K-edge data) were observed by XANES. Only Fe2(MoO4)3 and MoO3 were observed as oxidation products by XRD, which agrees with the overall re-oxidation and recombination reactions given in eqn (3), (4) and (6). The Fe2O3 detected by LCF of the XANES data was an intermediate, possibly an amorphous, phase and thus was not detected by XRD.
| 2FeMoO4 + 1/2O2 → Fe2O3 + 2MoO3 | (3) |
| MoO2 + 1/2O2 → MoO3 | (4) |
| 2FeMoO4 + MoO3 + 1/2O2 → Fe2(MoO4)3 | (5) |
| Fe2O3 + 3MoO3 → Fe2(MoO4)3 | (6) |
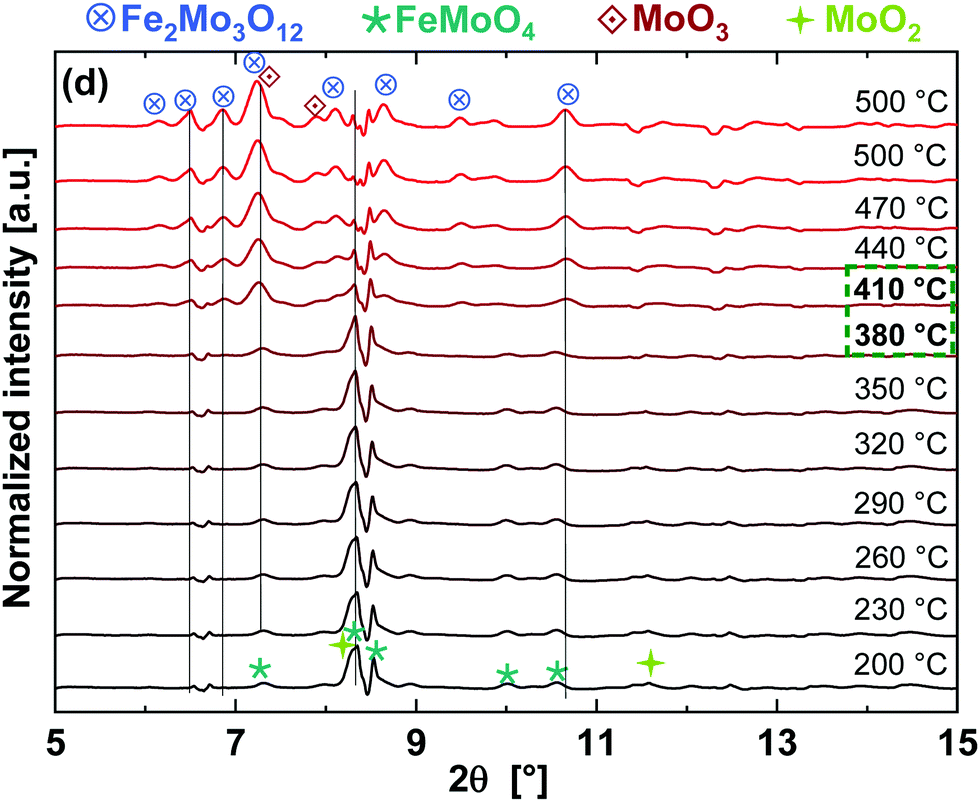 | ||
| Fig. 7 XRD patterns at different temperatures during TPO (at 25.23 eV corresponding to λ = 0.4934 Å). | ||
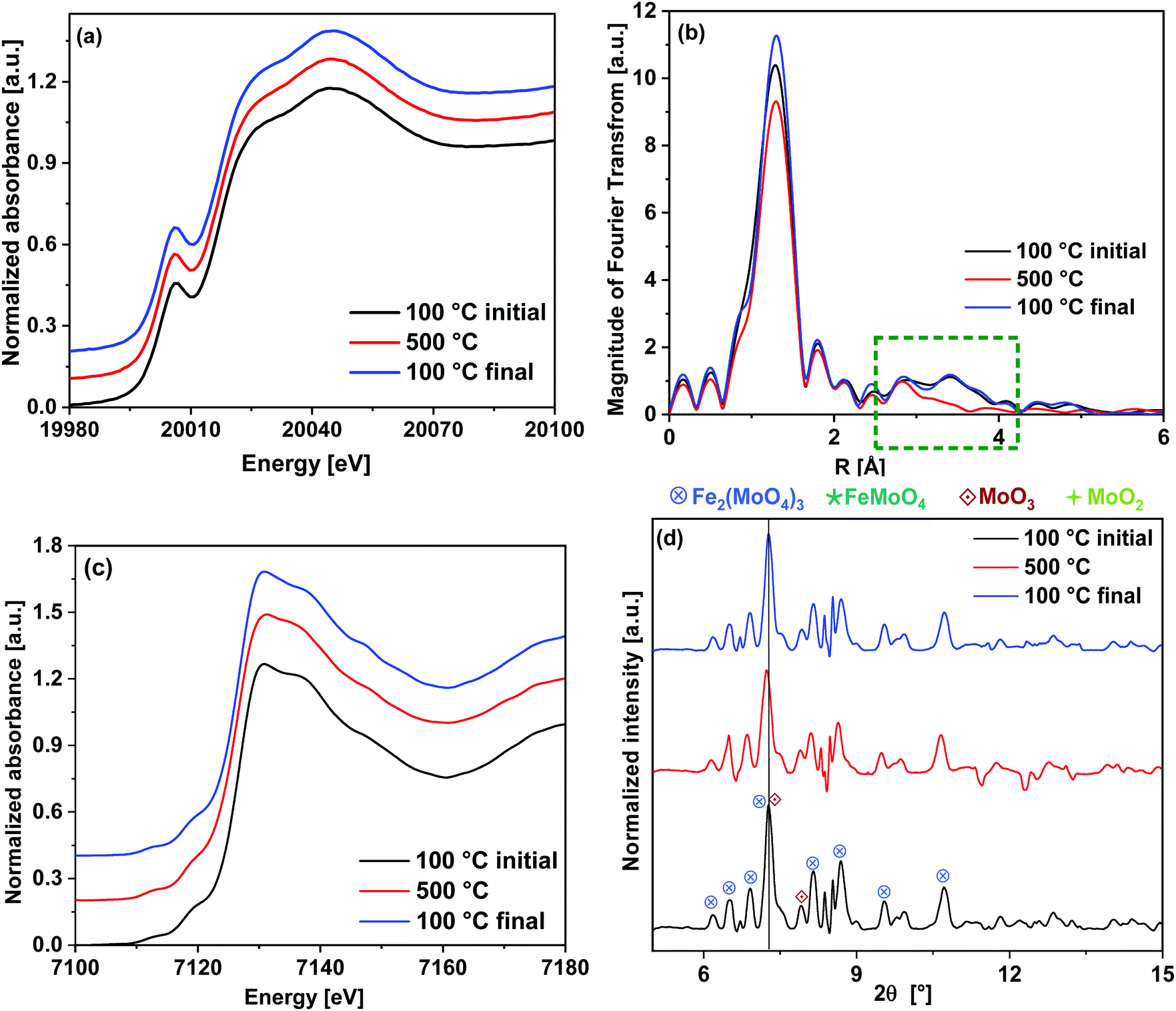 | ||
| Fig. 8 Comparison of the initial state of the catalyst measured at 100 °C, the state at 500 °C after TPO and the final state after cool-down to 100 °C under 10% O2/He: (a) Mo K-edge XANES (b) corresponding k3-weighted FT EXAFS spectra with marked region 2.5–4.0 Å. (c) Fe K-edge XANES and (d) XRD patterns with the assignment of phases. | ||
In Fig. 8(b), the FT EXAFS spectra are given and Table 1 shows the fitting parameters (see also ESI,† Fig. S3(a, e and f)). The major difference was observed in the FT spectra in the region of 2.5–4.0 Å where there are backscattering peaks due to Mo–Fe and Mo–Mo contributions. The Mo–Mo peak at 3.7 Å completely disappeared at 500 °C after the redox cycling, which indicates high distortions in the Mo–Mo metal shells related to the MoO3 phase. By cooling down to 100 °C, this backscattering peak was observed to be regenerated indicating that the catalyst returns to its crystalline phase. At 500 °C, the CN of the first peak (Mo–O1) was comparatively higher than its value at 100 °C in the initial as well as the final state, which indicate stronger tetrahedral nature of Mo coordination at higher temperature. In case of the longer axial Mo–O2 bonds, a considerable increase in the DW factor was observed between the initial and final states at 100 °C. This indicates loss of octahedral character of the Mo centers during the redox cycle, corresponding to evaporation of some of the excess MoO3. However, changes in the MoO3 crystallite particle size or shape during the redox cycling may also occur. As the CN of Mo–O2 bonds were kept fixed to the value of 1, the DW factor obtained from the fitting corresponds to the strength of these bonds.
| Path | 100 °C (initial) | 500 °C | 100 °C (final) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| R [Å] | CN | σ 2 [Å−2] | R [Å] | CN | σ 2 [Å−2] | R [Å] | CN | σ 2 [Å−2] | |
| a Fixed during the fitting. | |||||||||
| Mo–O1 | 1.76 ± 0.01 | 5.0 ± 0.5 | 0.0046 ± 0.0006 | 1.77 ± 0.02 | 5.2 ± 0.4 | 0.0054 ± 0.0007 | 1.77 ± 0.02 | 4.3 ± 0.4 | 0.0030 ± 0.0006 |
| Mo–O2 | 2.04 ± 0.06/ 2.36 ± 0.06 | 1a/1a | 0.0084 ± 0.0051 | 2.04 ± 0.07/ 2.37 ± 0.07 | 1a/1a | 0.0084 ± 0.0031 | 1.99 ± 0.06/ 2.31 ± 0.06 | 1a/1a | 0.0122 ± 0.0063 |
| Mo–Fe | 3.50 ± 0.02 | 1a | 0.0064 ± 0.0025 | 3.5 ± 0.06 | 1a | 0.0074 ± 0.0031 | 3.50 ± 0.02 | 1a | 0.0064 ± 0.0021 |
| Mo–Mo | 3.73 ± 0.03 | 1a | 0.0064 ± 0.0025 | — | 3.7 ± 0.03 | 1a | 0.0064 ± 0.0021 | ||
4. Conclusions
We have shown that the advanced setup at the synchrotron facilities makes it possible to obtain bulk structural as well as chemical information on the catalyst system under investigation. The combination of transmission XAS at the Mo K-edge, fluorescence XAS at the Fe K-edge and XRD at high energy proved to be a successful tool for detection of individual crystalline and amorphous phases in situ during the different stages of redox cycling of an iron molybdate catalyst with excess molybdenum trioxide, thereby providing important information about the phase transformations. The simplified reducing/oxidizing conditions employed in this study are needed to gain deeper insight into the catalytic system and catalytic cycle. Subsequent XAS measurements at the Mo K- and Fe K-edges showed that reduction of Mo as well as Fe started at ∼270 °C and at 400 °C MoO2 and FeMoO4 were observed as reduced phases. Complete reduction of Fe(III) to Fe(II), forming FeMoO4, was observed at 330 °C whereas 60% Mo was still present as Mo(VI) after reduction at 400 °C. Quasi-simultaneously recorded in situ XRD at high energy confirmed that the phase transition from Fe2(MoO4)3 and MoO3 to FeMoO4 and MoO2 occurred at about 280–300 °C. Online gas phase analysis recorded simultaneously showed that the concentration of methanol decreased parallel to reduction of Fe and Mo observed by XAS and XRD, confirming the reaction between gas and solid.Subsequent heating in an oxidative atmosphere from 200 to 500 °C caused Mo and Fe to reoxidize thereby forming Fe2(MoO4)3. Intermediates phases Fe2O3 and excess MoO3 were also formed in parallel reactions, but they recombined above 400 °C to give Fe2(MoO4)3, which was observed to be the most dominant phase at 500 °C. XRD confirmed that the main transition occurred at about 380–400 °C, at which rise in the MoO3 and Fe2O3 contributions were observed by XANES LCF.
The reduction/re-oxidation dynamics observed in this study, particularly the formation of FeMoO4 and MoO2 during reduction and Fe2O3 during re-oxidation, shows the segregation of iron and molybdenum (forming FeMoO4 and Fe2O3) observed under industrial reaction conditions, where the concurrent MoO3 evaporation hinders recombination to form Fe2(MoO4)3, which results in catalyst deactivation and selectivity loss by iron enrichment. In the future, it would be helpful to receive complementary insight by more surface sensitive methods, typically only possible under quasi in situ conditions, or using samples with higher surface area that would enhance the sensitivity of XAS for the surface.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Independent Research Fund Denmark for the financial support (grant no. DFF–4184-00336). We are grateful to ESRF for providing beamtime at the Swiss-Norwegian beamline BM31 for combined XAS/XRD studies and, in particular, to Dr Hermann Emerich and Dr Wouter van Beek for their help and technical support during XAS experiments. We thank Marc-André Serrer (KIT) for his support during the beamtime and the HGF for support in the programs “storage and crosslinked infrastructures” (SCI) and “science and technology of nanosystems” (STN). We also thank KIT's synchrotron radiation source (operated by KIT-IBPT) for ex situ XAS measurements conducted at the CAT-ACT beamline.References
- M. Bowker, R. Holroyd, A. Elliott, P. Morrall, A. Alouche, C. Entwistle and A. Toerncrona, The Selective Oxidation of Methanol to Formaldehyde on Iron Molybdate Catalysts and on Component Oxides, Catal. Lett., 2002, 83(3), 165–176 CrossRef CAS.
- M. P. House, A. F. Carley, R. Echeverria-Valda and M. Bowker, Effect of Varying the Cation Ratio within Iron Molybdate Catalysts for the Selective Oxidation of Methanol, J. Phys. Chem. C, 2008, 112(11), 4333–4341 CrossRef CAS.
- K. Routray, W. Zhou, C. J. Kiely, W. Grünert and I. E. Wachs, Origin of the synergistic interaction between MoO3 and iron molybdate for the selective oxidation of methanol to formaldehyde, J. Catal., 2010, 275(1), 84–98 CrossRef CAS.
- L. J. Burcham, L. E. Briand and I. E. Wachs, Quantification of Active Sites for the Determination of Methanol Oxidation Turn-over Frequencies Using Methanol Chemisorption and in Situ Infrared Techniques. 1. Supported Metal Oxide Catalysts, Langmuir, 2001, 17(20), 6164–6174 CrossRef CAS.
- A. P. V. Soares, M. F. Portela and A. Kiennemann, Methanol Selective Oxidation to Formaldehyde over Iron-Molybdate Catalysts, Catal. Rev., 2005, 47(1), 125–174 CrossRef CAS.
- M. P. House, A. F. Carley and M. Bowker, Selective oxidation of methanol on iron molybdate catalysts and the effects of surface reduction, J. Catal., 2007, 252(1), 88–96 CrossRef CAS.
- K. V. Raun, L. F. Lundegaard, J. Chevallier, P. Beato, C. C. Appel, K. Nielsen, M. Thorhauge, A. D. Jensen and M. Høj, Deactivation behavior of an iron-molybdate catalyst during selective oxidation of methanol to formaldehyde, Catal. Sci. Technol., 2018, 8(18), 4626–4637 RSC.
- C. Brookes, P. P. Wells, N. Dimitratos, W. Jones, E. K. Gibson, D. J. Morgan, G. Cibin, C. Nicklin, D. Mora-Fonz, D. O. Scanlon, C. R. A. Catlow and M. Bowker, The Nature of the Molybdenum Surface in Iron Molybdate. The Active Phase in Selective Methanol Oxidation, J. Phys. Chem. C, 2014, 118(45), 26155–26161 CrossRef CAS.
- C. Brookes, M. Bowker, E. K. Gibson, D. Gianolio, K. M. H. Mohammed, S. Parry, S. M. Rogers, I. P. Silverwood and P. P. Wells, In situ spectroscopic investigations of MoOx/Fe2O3 catalysts for the selective oxidation of methanol, Catal. Sci. Technol., 2016, 6(3), 722–730 RSC.
- T. H. Kim, B. Ramachandra, J. S. Choi, M. B. Saidutta, K. Y. Choo, S.-D. Song and Y.-W. Rhee, Selective Oxidation of Methanol to Formaldehyde Using Modified Iron-Molybdate Catalysts, Catal. Lett., 2004, 98(2), 161–165 CrossRef CAS.
- M. Rellán-Piñeiro and N. López, The Active Molybdenum Oxide Phase in the Methanol Oxidation to Formaldehyde (Formox Process): A DFT Study, ChemSusChem, 2015, 8(13), 2231–2239 CrossRef PubMed.
- E. M. McCarron III and A. W. Sleight, Oxy-Methoxy Compounds of Molybdenum (VI) and their Relationship to the Selective Oxidation of Methanol over Molybdate Catalysts, Polyhedron, 1986, 5, 129–139 CrossRef.
- J. Ziolkowski, Advanced Bond-Strength Model of Active Sites on Oxide Catalysts, J. Catal., 1983, 84, 317–332 CrossRef CAS.
- R. K. Grasselli, Fundamental principles of selective heterogeneous oxidation catalysis, Top. Catal., 2002, 21, 79–88 CrossRef CAS.
- W. E. Farneth, F. Ohuchi, R. H. Staley, U. Chowdry and A. W. Sleight, Mechanism of Partial Oxidation of Methanol Over MoO3 as Study by Temperature-Programmed Desorption, J. Phys. Chem., 1985, 89, 2493 CrossRef CAS.
- J. M. Tatibouët, Methanol oxidation as a catalytic surface probe, Appl. Catal., A, 1997, 148(2), 213–252 CrossRef.
- M. G. O’Brien, A. M. Beale, S. D. M. Jacques and B. M. Weckhuysen, A Combined Multi-Technique In Situ Approach Used to Probe the Stability of Iron Molybdate Catalysts During Redox Cycling, Top. Catal., 2009, 52(10), 1400–1409 CrossRef.
- S. D. M. Jacques, O. Leynaud, D. Strusevich, P. Stukas, P. Barnes, G. Sankar, M. Sheehy, M. G. O’Brien, A. Iglesias-Juez and A. M. Beale, Recent progress in the use of in situ X-ray methods for the study of heterogeneous catalysts in packed-bed capillary reactors, Catal. Today, 2009, 145(3), 204–212 CrossRef CAS.
- M. G. O’Brien, A. M. Beale, S. D. M. Jacques, M. Di Michiel and B. M. Weckhuysen, Closing the operando gap: The application of high energy photons for studying catalytic solids at work, Appl. Catal., A, 2011, 391(1), 468–476 CrossRef.
- A. Gaur, M. Schumann, K. V. Raun, M. Stehle, P. Beato, A. D. Jensen, J. D. Grunwaldt and M. Hoj, Operando XAS/XRD and Raman Spectroscopic Study of Structural Changes of the Iron Molybdate Catalyst during Selective Oxidation of Methanol, ChemCatChem, 2019, 11(19), 4871–4883 CrossRef CAS.
- W. van Beek, O. V. Safonova, G. Wiker and H. Emerich, SNBL, a dedicated beamline for combined in situ X-ray diffraction, X-ray absorption and Raman scattering experiments, Phase Transitions, 2011, 84(8), 726–732 CrossRef CAS.
- J.-D. Grunwaldt, N. v. Vegten and A. Baiker, Insight into the structure of supported palladium catalysts during the total oxidation of methane, Chem. Commun., 2007,(44), 4635–4637 RSC.
- J.-D. Grunwaldt and B. S. Clausen, Combining XRD and EXAFS with on-Line Catalytic Studies for in situ Characterization of Catalysts, Top. Catal., 2002, 18(1), 37–43 CrossRef CAS.
- J. D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, X-ray absorption spectroscopy under reaction conditions: suitability of different reaction cells for combined catalyst characterization and time-resolved studies, Phys. Chem. Chem. Phys., 2004, 6(11), 3037–3047 RSC.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541 CrossRef CAS PubMed.
- A. M. Beale, S. D. M. Jacques, E. Sacaliuc-Parvalescu, M. G. O’Brien, P. Barnes and B. M. Weckhuysen, An iron molybdate catalyst for methanol to formaldehyde conversion prepared by a hydrothermal method and its characterization, Appl. Catal., A, 2009, 363(1), 143–152 CrossRef CAS.
- C. Brookes, P. P. Wells, G. Cibin, N. Dimitratos, W. Jones, D. J. Morgan and M. Bowker, Molybdenum Oxide on Fe2O3 Core–Shell Catalysts: Probing the Nature of the Structural Motifs Responsible for Methanol Oxidation Catalysis, ACS Catal., 2014, 4(1), 243–250 CrossRef CAS.
- M. Lv, W. Xie, S. Sun, G. Wu, L. Zheng, S. Chu, C. Gao and J. Bao, Activated-carbon-supported K–Co–Mo catalysts for synthesis of higher alcohols from syngas, Catal. Sci. Technol., 2015, 5(5), 2925–2934 RSC.
- L.-b. Wu, L.-h. Wu, W.-m. Yang and A. I. Frenkel, Study of the local structure and oxidation state of iron in complex oxide catalysts for propylene ammoxidation, Catal. Sci. Technol., 2014, 4(8), 2512–2519 RSC.
- A. Gaur and B. D. Shrivastava, Speciation using X-ray absorption fine structure (XAFS), Rev. J. Chem., 2015, 5(4), 361–398 CrossRef CAS.
- N. Pernicone, Deactivation of Fe–Mo oxide catalyst in industrial plant and simulation tests on laboratory scale, Catal. Today, 1991, 11(1), 85–91 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Overview of the experimental procedure, XANES spectrum of the initial state of the FeMo catalyst and the structural parameters obtained from EXAFS fitting, EXAFS fitting curves at the Mo K-edge for the FeMo catalyst during different stages of redox cycling, structural parameters obtained from EXAFS fitting for intermediate phases during TPR and TPO, PCA and LCF results at the Fe K-edge during TPR, MS traces of MeOH, HCHO and H2O during TPR and isothermal period, in situ XANES spectra at the Mo K- and Fe K-edge and XRD during cooling down to 200 °C after TPR and switching to oxidizing conditions, complete LCF results at the Mo K-edge during TPO and PCA results at the Fe K-edge during TPO. See DOI: 10.1039/d0cp01506g |
| This journal is © the Owner Societies 2020 |