
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate water exchange in the S2 state of photosystem II is dependent on the conformation of the Mn4Ca cluster
Casper
de Lichtenberg
 a and
Johannes
Messinger
*ab
a and
Johannes
Messinger
*ab
aDepartment of Chemistry, Umeå University, Linnaeus väg 6 (KBC huset), SE-901 87, Umeå, Sweden. E-mail: johannes.messinger@kemi.uu.se
bMolecular Biomimetics, Department of Chemistry – Ångström, Uppsala University, POB 523, SE-75120 Uppsala, Sweden
First published on 27th April 2020
Abstract
In photosynthesis, dioxygen formation from water is catalyzed by the oxygen evolving complex (OEC) in Photosystem II (PSII) that harbours the Mn4Ca cluster. During catalysis, the OEC cycles through five redox states, S0 to S4. In the S2 state, the Mn4Ca cluster can exist in two conformations, which are signified by the low-spin (LS) g = 2 EPR multiline signal and the high-spin (HS) g = 4.1 EPR signal. Here, we employed time-resolved membrane inlet mass spectrometry to measure the kinetics of H218O/H216O exchange between bulk water and the two substrate waters bound at the Mn4Ca cluster in the SLS2, SHS2, and the S3 states in both Ca-PSII and Sr-PSII core complexes from T. elongatus. We found that the slowly exchanging substrate water exchanges 10 times faster in the SHS2 than in the SLS2 state, and that the SLS2 → SHS2 conversion has at physiological temperature an activation barrier of 17 ± 1 kcal mol−1. Of the presently suggested SHS2 models, our findings are best in agreement with a water exchange pathway involving a SHS2 state that has an open cubane structure with a hydroxide bound between Ca and Mn1. We also show that water exchange in the S3 state is governed by a different equilibrium than in S2, and that the exchange of the fast substrate water in the S2 state is unaffected by Ca/Sr substitution. These findings support that (i) O5 is the slowly exchanging substrate water, with W2 being the only other option, and (ii) either W2 or W3 is the fast exchanging substrate. The three remaining possibilities for O–O bond formation in PSII are discussed.
Introduction
Plants, algae and cyanobacteria harvest photons of visible light to convert solar light into chemical energy in a process known as oxygenic photosynthesis. The key reactions of this process are the extraction of electrons and protons from water and the reduction of carbon dioxide to carbohydrates. The final products, molecular oxygen and biomass, are essential for most life on Earth. Water oxidation to molecular oxygen is performed at the Mn4Ca cluster of the oxygen-evolving complex (OEC) that resides within the transmembrane pigment–protein complex Photosystem II (PSII).1–5 Driven by light-induced charge separations in the reaction center of PSII, the OEC cycles through five intermediate states, S0 through S4, where the subscript indicates the number of oxidizing equivalents stored.6 The S4 state is highly reactive and converts within milliseconds into the S0 state, releasing O2 and rebinding one ‘substrate water’ (the term is used independent of the protonation state).7 If left in the dark, the OEC will eventually relax into the dark-stable S1 state.8The lowest oxidation state of the Mn4Ca cluster in the water splitting cycle, the S0 state, was shown by 55Mn-ENDOR spectroscopy to have the oxidation states Mn(III,III,III,IV).9 These overall ‘high’ oxidation states were recently confirmed by photoactivation experiments.10 Each forward S-state transition involves the oxidation of one Mn(III) ion to Mn(IV) until Mn(IV,IV,IV,IV) is reached in the S3 state.3,11–19 The S3 → S4 transition remains poorly understood and is suggested to lead to the formation of either an oxyl-radical, Mn(V,IV,IV,IV) or Mn(VII,III,III,III).11,20–22
The structure of the Mn4Ca cluster was first reported at high resolution (1.9 Å and 1.95 Å) for the S1 state,23,24 and recently also at resolutions between 2.0 Å and 2.1 Å for all S-states, except S4.3 (see also ref. 25). The S0, S1 and S2 states have similar structures, except that in S0 the Mn3–Mn4 distance is longer indicating that the O5 bridge is protonated.9,19,26 This structure, often referred to as open cubane or A-type structure, is depicted schematically as SA2 state in Scheme 1. The Ca ion and four Mn ions are connected by five oxo-bridges, and Ca and Mn4 bind two terminal water ligands each (W1–W4; W2 may be a hydroxo ligand).27–30 The remaining coordination sites of the Mn4Ca cluster are completed by bridging carboxy ligands supplied by the D1 and CP43 proteins of PSII (not shown). The only exception is the Mn1 site, which features one histidine ligand and is five-coordinate.3,11,31,32
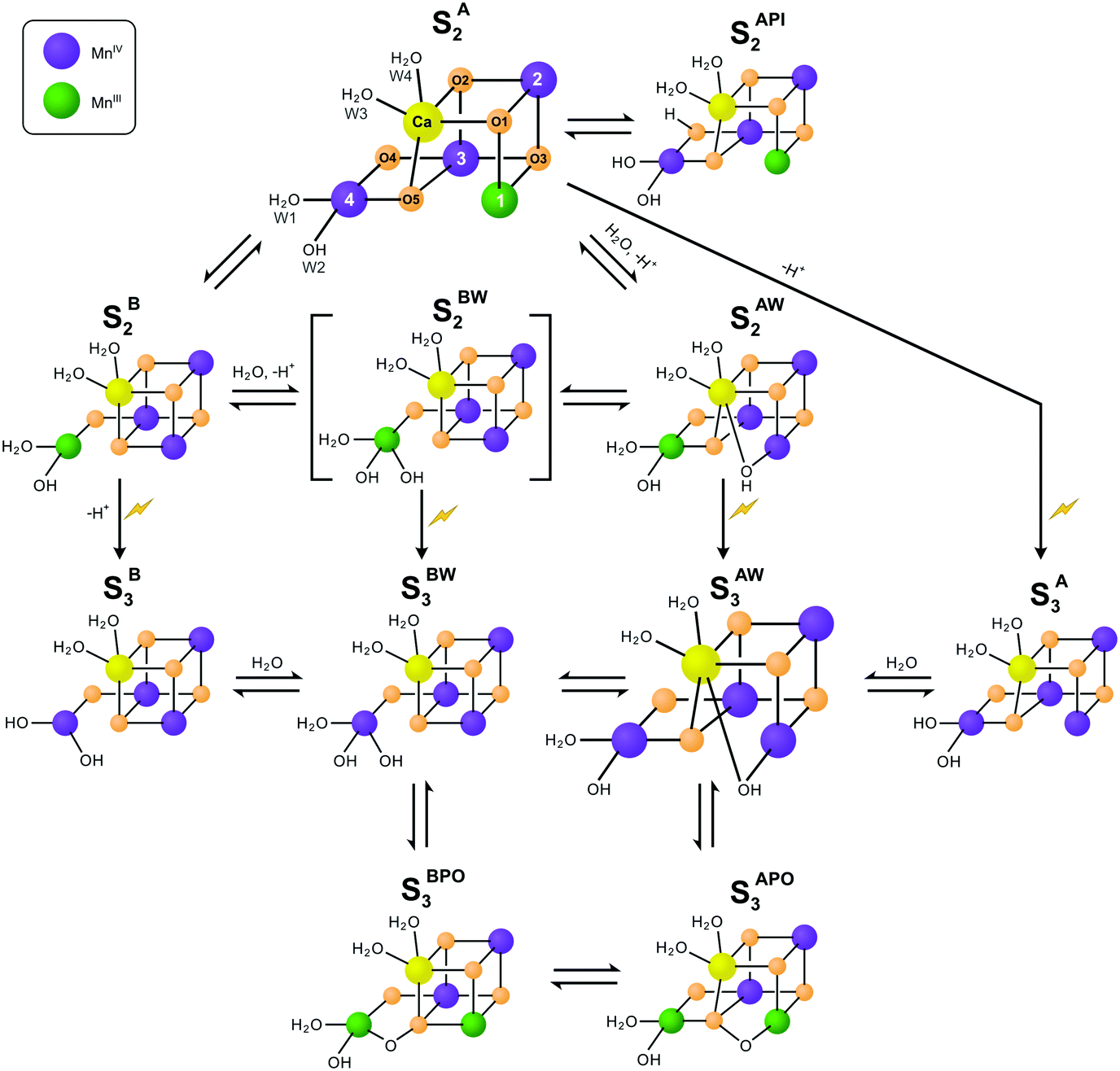 | ||
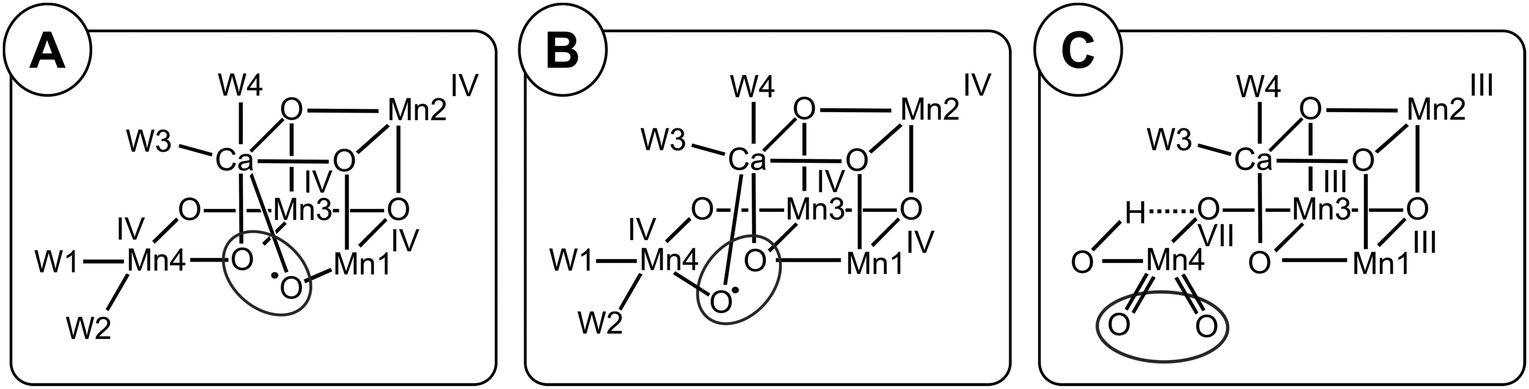
| Scheme 1 Isomers of the S2 and S3 states in photosystem II. The structures of the SA2 and SAW3 states were determined by X-ray crystallography; however, it remains controversial if the newly added oxygen bridge between Ca and Mn1 is an OH (as shown) or rather an oxo or oxyl.3,25 Similarly, W2 may be a water instead of a hydroxide.27–30 All other states are proposed on the basis of EPR spectroscopy and DFT calculations. Open cubane structures are labelled A, while closed cubane structures are signified with B. W indicates an additional hydroxo group, while PI signifies a proton isomer and PO the formation of a peroxidic intermediate. SA2 has been assigned to the SLS2 state, while there are three proposals for the SHS2 state: the closed cube SB2 state,40 the hydroxo bound SAW2 state41 (see ref. 39 and 43 for related proposals), and the proton shift isomer SAPI2.42 The SBW2 state is shown in brackets since it is a proposed intermediate in the S2 → S3 transition55,98 and for water exchange;81 please note that the position of the Mn(III) valence and protonation states of oxygen ligands and bridges differ among the various suggestions. Evidence for the SB3 or SA3 states comes from EDNMR experiments indicating the presence of a five coordinate Mn(IV) ion under conditions preventing water binding.55 The peroxide bound S3 states are consistent with early proposals by Renger,44 and recent DFT calculations by the Yamaguchi group.57 Labelling of atoms referred to in the text is provided in the SA2 structure. Mn(IV) ions are shown in purple, Mn(III) in green, Ca in yellow and oxygen in orange. Transitions from one structure to the next may involve multiple steps. | ||
While this SA2 structure is the only one observed by crystallography, there is experimental and computational evidence for at least one additional conformation in the S2 state. The S2 state features two EPR signals at cryogenic temperatures: the low spin (S = 1/2) S2 EPR multiline signal (SLS2) and the high-spin (S = 5/2) g = 4.1 signal (SHS2).33–35 At near neutral pH values, the SLS2 state is clearly dominant, and its structure is that of SA2 (Scheme 1). The energy difference between the two S2 states and the transition state barrier between them are small enough so that the normally less stable SHS2 state can be enriched in many ways, for example by illumination of S1 state samples at 130–140 K or by exposing the SLS2 state to high pH (8.3–9.0), IR illumination or fluoride addition.34,36–38 It is not clear if all SHS2 states have the same structure, since slightly different g values in the range of 4.1–4.7 have been reported for the various conditions.39 In absence of a crystal structure for the SHS2 state, the SB2, SAPI2 and SAW2 structures have been proposed to give rise to the g = 4 signal, where W indicates that an additional hydroxide is bound to the Mn4Ca cluster, and PI signifies a proton isomer (Scheme 1).27–30,40–43
Among these, the closed cubane SB2 state is the most prevalent suggestion for the SHS2 state. The SB2 state may be reached from the open cubane SA2 state by moving the central O5 bridge away from Mn4 so that it instead forms a bond with Mn1. This structural change makes Mn4 five- and Mn1 six-coordinate, and is suggested to be accompanied by a valence swap between Mn4 and Mn1.30,40,45,46 However, EXAFS experiments of samples in the SHS2 state generated by 140 K illumination of S1 state samples result in Mn–Mn distances that are inconsistent with the SB2 state structure.47,48 It was recently argued that the SB2 state also does not provide a rational for many of the treatments leading to the SHS2 state formation.42 The latter study instead proposes a proton isomer of the SA2 state as SHS2 state (SAPI2 in Scheme 1).
Since the high-pH induced SHS2 state, in contrast to the SLS2 state, can be advanced down to 77 K to the SAW3 state, it was alternatively proposed that the SHS2 state may already have the ‘water’ bound (SAW2) that should otherwise insert during the S2 → S3 transition (see below).39,41,43 It is noted that the Mn–Mn distances of the SAW2 state are likely also not in line with the above discussed EXAFS data of the SHS2 state generated by 140 K illumination.47 The light-induced formation of the S3 state from S2 involves significant structural changes that include the binding of a water molecule in form of an additional oxo/hydroxo or oxyl bridge between Ca and Mn1 (SAW3 in Scheme 1).3,11,13,25 Even so, the precise molecular sequence for the formation of this sixth bridge remains controversial. Both the rotation of the Ca-bound W3 ligand towards Mn1, and the addition of W3 or a protein ligated water to Mn4 in combination with a pivot or carrousel rearrangement of W1, W2 and O5 have been proposed.32,45,49,50 Thus, starting from the SA2 state, many different pathways can be envisioned for the formation of this most stable form of the S3 state (SAW3 in Scheme 1). The structure of the SAW3 state is well-characterized by X-ray crystallography and EPR spectroscopy.3,13,25
Importantly, EPR experiments additionally indicate the presence of an EPR-silent form of the S3 state that under IR illumination converts into the EPR-detectable S2YZ˙ state.36,39,51 This indicates that the Mn4Ca cluster and D1-Tyr161 (YZ), the electron donor to P680˙+, are in a delicate redox equilibrium in the S3 state.52–54 The EPR silent S3 state has been tentatively assigned to the SB3 or SBW3 structures.39 Additionally, a recent EDNMR study of the S3 state identified a signal indicative of a five-coordinate Mn(IV) ion within the either the SA3 or SB3 structure.55 Furthermore, peroxidic states (SAPO3, SBPO3) have been proposed to exist in the S3 state.56–58
For determining the mechanism of O–O bond formation, which occurs during the S3 → S4 → S0 transition, it is crucial to identify the two substrate waters. Presently, the only technique able to probe the binding sites of substrate water in PSII is time-resolved membrane inlet mass spectrometry (TR-MIMS).7,59,60 This method utilizes a rapid increase in H218O concentration of the bulk water to determine the exchange rates of the two bound substrates by measuring the isotopic composition of O2 generated after various incubation times (Fig. 1).
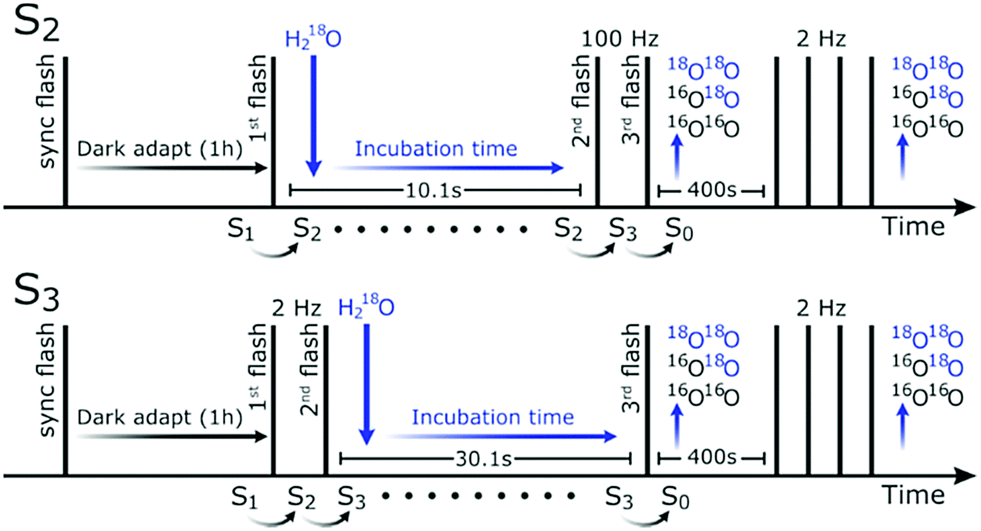 | ||
| Fig. 1 Flash and injection scheme for TR-MIMS measurements in the S2-state (top) and the S3-state (bottom). Vertical lines indicate saturating flashes and the downward pointing arrows indicate injection of 18O-labelled water. The first flash was given to synchronize the samples in the S1YoxD state, while the final group of four flashes is employed for normalization. | ||
TR-MIMS measurements show that the two substrate waters exchange with different rates. The slow exchanging substrate water (Ws) is bound in all S-states. Its exchange rate slows 500-fold from S0 to S1, increases 100-fold upon S2 formation and remains about the same in the S3 state despite the above described complexity of the S2 → S3 transition.7,60 Importantly, Ws exchange is in all S states about 5–10 times faster in samples in which the natural Ca co-factor of the Mn4Ca cluster is replaced by Sr (Sr-PSII).61
In the S2 and S3 states, TR-MIMS measurements can also resolve the exchange of the faster exchanging substrate water, Wf.62,63 This shows that both substrates are bound to the OEC in the S2 state. Since protein- or Ca-ligated water molecules generally exchange at rates too fast for the present MIMS approach, this result indicates that Wf is Mn-bound in the S2 state and that the new water molecule binding in the S2 → S3 transition is not a substrate in the ongoing S-state cycle. Consequently, we suggested W2 as candidate for Wf.7,60,64 As noted in these studies, this conclusion does not hold if the exchange of Wf is limited by the diffusion of water through the protein channels.
Together with structural information available at the time,26,65 the TR-MIMS data led to the proposal that the central oxygen bridge between Ca and two Mn ions, now referred to as O5, is the slow exchanging substrate Ws that forms the O–O bond with W2 in a SB2 like conformation.64 A related, more detailed proposal for the mechanism of water oxidation that involves a similar O–O bond formation mechanism, but utilizes a SAW3 like conformation, was later made on the basis of DFT calculations.66 Importantly, subsequent advanced EPR measurements have demonstrated that O5 exchanges fast enough with bulk water to be compatible with Ws exchange kinetics observed by TR-MIMS.67 Nevertheless, W2, W3 and O4 have been suggested by other groups to be the slow substrate water instead of O5.68–70
Up to now, all TR-MIMS measurements were performed under conditions where the open cubane states are predominant. To compare the substrate water exchange rates in the two structural forms of the S2 state, we followed in this study a recently developed protocol for enriching PSII core preparations from Thermosynechococcus elongatus (T. elongatus) in either the SLS2 or the SHS2 state.38 The data presented below provide unique insights into the pathway of substrate water exchange and the binding sites of Wf and Ws.
Experimental procedure
Photosystem II core preparation
The T. elongatus ΔpsbA1ΔpsbA2 deletion mutant71,72 was grown in Ca- or Sr-containing buffers, and the PSII core preparations were isolated and purified as described previously.73,74 After preparation, the PSII cores were washed with an aqueous solution of 1 M betaine, 15 mM CaCl2 and 15 mM MgCl2, in an Amicon Ultra-15 centrifugal filter unit (cut-off 100 kDa) until the estimated residual MES concentration was smaller than 1 μM. Finally, the samples were frozen in liquid nitrogen until used.Time-resolved membrane inlet mass spectrometry
For TR-MIMS measurements, an isotope ratio mass spectrometer (Finnigan Delta plus XP) was used. The spectrometer was connected to a membrane inlet rapid mixing cell (volume of 165 μl) via a steel pipe that runs through a cooling trap containing ethanol/dry ice.59,62For each measurement, an aliquot of PSII cores was thawed on ice and then diluted 10-fold into an unbuffered solution containing 15 mM CaCl2, 15 mM MgCl2 and 1 M Betaine. To fully oxidize tyrosine D, the samples were then exposed to a saturating xenon-flash (full width at half maximum ≈5 μs), followed by 60 min dark adaptation at 20 °C, during which the sample was loaded into the MIMS chamber. Five minutes before the measurements, the pH was adjusted by injecting 8 μl of 1 M buffer (see below) containing 2 mM of the artificial electron acceptor 2,6-dimethyl-1,4-benzoquinone (DMBQ), from a 50 mM stock solution in dimethyl sulfoxide. The final concentrations were 0.29 mg of Chl ml−1, in 50 mM buffer (MES pH 6.0, TAPS pH 8.3 for Sr-PSII or TAPS pH 8.6 for Ca-PSII) and 100 μM DMBQ. The slightly lower pH used for Sr-PSII was chosen to ensure the integrity of the Sr-PSII samples. We note that the SLS2 to SHS2 conversion was nearly complete at this pH for Sr-PSII, while pH 8.6 was required to achieve a similar conversion in Ca-PSII.38 For measurements with ammonia, a final concentration of 50 mM NH4Cl was employed.
Rapid enrichment of the sample with H218O was achieved by means of a modified gas-tight syringe (Hamilton CR-700-200) that was driven by air-pressure via a fast-switching solenoid valve (kinj = 170 s−1 based on fluorescence rise after injections of fluorescein; see also ref. 59, 63 and 75). To minimize artifacts from dissolved oxygen, the syringe was loaded with 97% H218O in a N2-filled glove box. The H218O was further deoxygenated with a mixture of glucose/glucose oxidase (Sigma Aldrich, A. niger) and catalase (Sigma Aldrich, B. taurus).59
The measurement consisted of a series of saturating flashes and a single injection as shown in Fig. 1. After synchronization in the S1YoxD state, the PSII samples were illuminated with one (S2) or two (2 Hz; S3) saturating flash(es) to advance the majority of the centers from the S1 state to the desired S-state. This step was followed by a fixed delay (10.1 s for the S2 and 30.1 s for the S3 state) before the O2-generating flash(es) were given (two at 100 Hz for S2, and one in case of S3) that advanced the enzyme via the S4 state to the S0 state. At various times ti before the O2-generating flash(es), the H218O was injected into the PSII sample resulting in the reported incubation times. After a delay of 400 s, which allowed all signals to return to baseline levels, the PSII samples were exposed to four more flashes given at 2 Hz. This signal was used for normalization, and for determining the relative flash-induced oxygen evolution activity, which was, at pH 8.6, 50% of that at pH 6.0, independent of the ammonia addition.
The mass-to-charge ratios m/z 34 and m/z 36 were monitored for determination of the flash-induced O2-production in PSII, while m/z 40 (Ar) was recorded as a reference. The H218O enrichment after complete mixing was calculated from the m/z 34/36 ratio of the four normalizing flashes to be ≈20%.59,62 Data points recorded at short incubation times that approached the mixing time were corrected for the change in isotopic enrichment and PSII concentration as described previously.59,62,75
Kinetic modelling of substrate exchange
Exchange rates (kf, ki, ks) were determined by simultaneous fitting of the corrected 16,18O2 and 18,18O2 data to eqn (1) and (2).59,62 The pre-exponential a represents the ratio between fast and slow exchange in the 16,18O2 data. It was calculated from the initial H218O enrichment (αin; 0.07%), which was determined slightly higher than natural abundance due to a small leakage from the syringe tip, and the final (αf) H218O enrichment using eqn (3). The pre-exponential b represents the ratio between two distinct populations of slowly exchanging substrate waters. b was determined from an initial separate fit of the normalized 36O2 yield to eqn (2). Both parameters, a and b, were held constant in the final global fit of the m/z 34 and 36 data.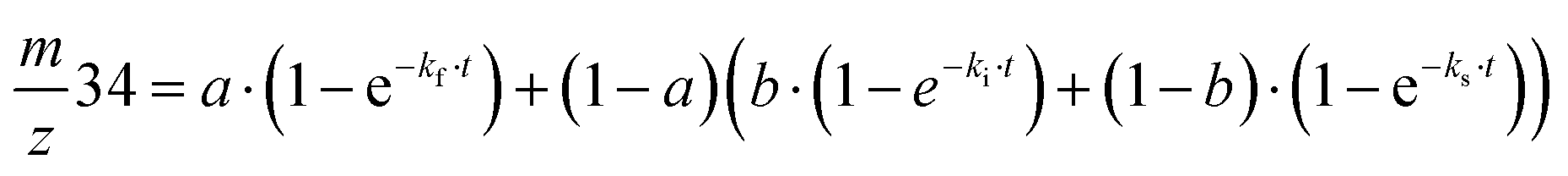 | (1) |
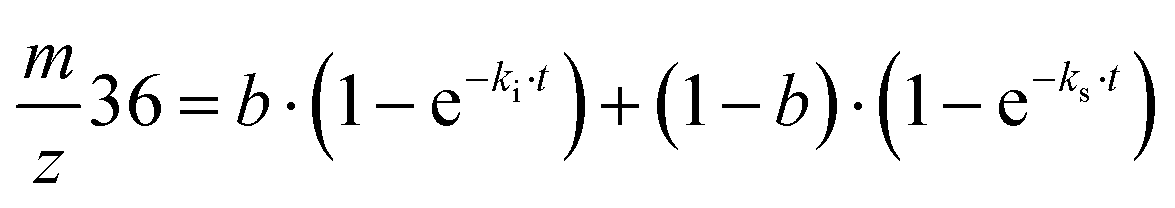 | (2) |
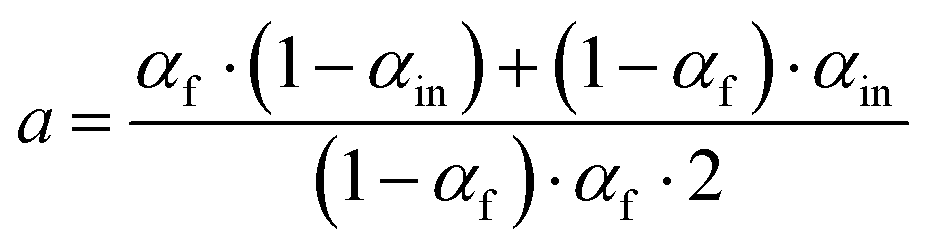 | (3) |
Activation energies were calculated according to the transition state theory:
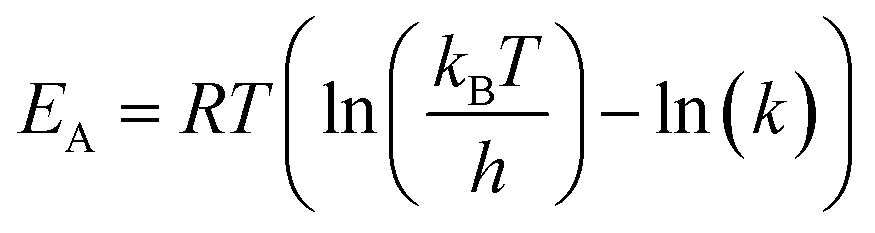 | (4) |
Results
Fig. 2 shows the results of the substrate water exchange experiments in the S2 state of PSII core samples from T. elongatus containing the natural Ca cofactor in the OEC (Ca-PSII) or instead Sr (Sr-PSII). Each dot represents the normalized flash-induced yield of dioxygen produced after the exchange of one (16,18O2; m/z 34) or both (18,18O2; m/z 36) substrate waters during a discrete time of incubation with H218O enriched water. For Ca-PSII at pH 6.0, the typical biphasic rise of the m/z 34 signal was observed (Fig. 2A, black points). The biphasic nature of this signal reflects the independent exchange of the two substrate waters, Wf and Ws, with bulk water at rates kf and ks.7,59,62 The corresponding rise of the 18,18O2 signal at m/z 36 (Fig. 2B, black dots), which requires the exchange of both substrate water molecules, shows the previously reported monophasic rise with rate ks, corresponding to the slow component of the m/z 34 rise in Fig. 2A. These results are consistent with a single conformation (SLS2 = SA2) for the S2 state under these conditions, which is in line with EPR spectroscopy performed previously under the same conditions on the same type of samples.38 The data were thus fit employing eqn (1) and (2), using only two kinetic components (b was set to zero).59,62 The parameter of the best fits (solid lines in Fig. 2A and B) are given in Table 1. The results are fully consistent with previous measurements.63,76,77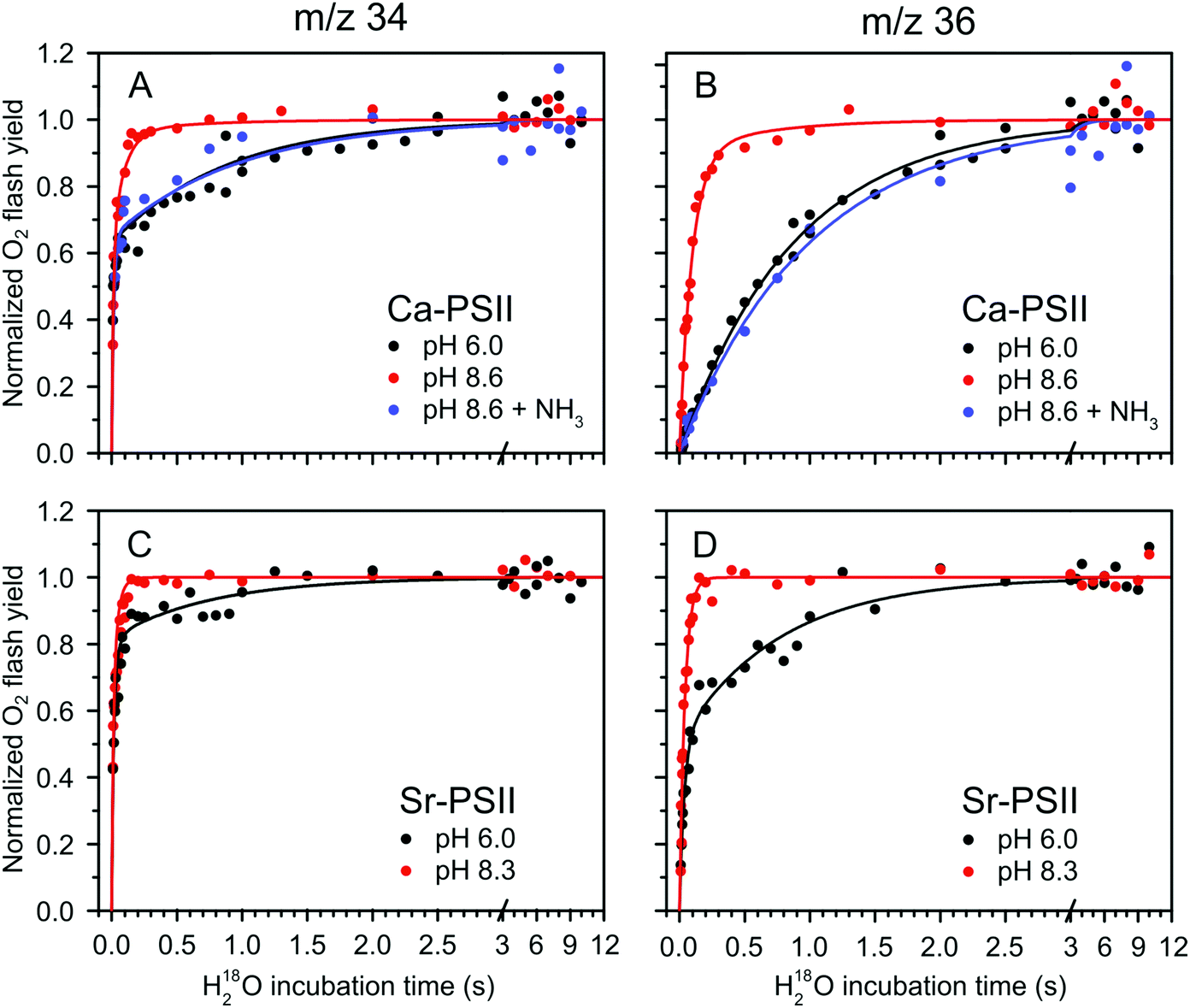 | ||
| Fig. 2 H218O substrate exchange of Ca-PSII (A) and (B) and Sr-PSII (C) and (D) in the S2-state. (A) and (C) represent the normalized flash yields of single-labelled dioxygen (m/z 34), while (B) and (D) represent the normalized flash yields of double-labelled dioxygen (m/z 36). Black dots represent measurements performed at pH 6.0, while red dots are data from measurements taken at pH 8.6 for Ca-PSII and at pH 8.3 for Sr-PSII. Blue dots signify the results of experiments with Ca-PSII at pH 8.6 in presence of 50 mM NH4Cl. Lines are fits according to eqn (1)–(3), of which the parameters are given in Table 1. | ||
| Sample | S state | Conditions | W f k f (s−1) | W s k i (s−1) | W s k s (s−1) | a | b | |
|---|---|---|---|---|---|---|---|---|
| Ca-PSII | This study | S2 | pH 6.0 | 102 ± 9 | — | 1.14 ± 0.03 | 0.63 | 0 |
| This study | S2 | pH 8.6 | 75 ± 7 | 10.5 ± 0.6 | 1.6 ± 0.9 | 0.63 | 0.9 | |
| This study | S2 | pH 8.6 + NH3 | 64 ± 18 | — | 1.02 ± 0.09 | 0.65 | 0 | |
| Ref. 77 | S3 | pH 6.5 | 40 ± 4 | — | 0.69 ± 0.06 | 0.65 | 0 | |
| This study | S3 | pH 8.6 | 19.5 ± 2.2 | — | 0.25 ± 0.01 | 0.65 | 0 | |
| Sr-PSII | This study | S2 | pH 6.0 | 85 ± 10 | 29.7 ± 3.2 | 1.5 ± 0.1 | 0.63 | 0.49 |
| This study | S2 | pH 8.3 | 76 ± 7 | 24.3 ± 1.1 | — | 0.65 | 1.0 |
The TR-MIMS data for Sr-PSII at pH 6.0 are displayed in Fig. 2C and D (black points). It is clearly seen in Fig. 2D that the rise of the m/z 36 signal was biphasic, with the two phases having comparable amplitudes: a slow phase with a rate ks similar to that seen in Ca-PSII, and a 20–30 times faster phase, designated here as the intermediate phase, with rate ki (eqn (2) and Table 1). Such a biphasic behavior of the m/z 36 data was not reported previously. It can best be rationalized by the presence of two distinct forms of the S2 state in Sr-PSII that are in slow equilibrium at room temperature. The proposed presence of two conformations in Sr-PSII is in agreement with recent low temperature EPR data showing that the SLS2 and SHS2 EPR signals coexist under these conditions (yet SHS2 being present at lower ratio).38
Given the observation of two rates for Ws exchange, the m/z 34 rise was fit with three kinetic components according to eqn (1). The resulting Wf exchange rate for the S2 state was found to be similar to that measured in Ca-PSII at the same pH value.
At pH 8.6/pH 8.3, a strong acceleration in the exchange rate of the slow substrate was observed for both Ca-PSII and Sr-PSII samples (red dots and lines in Fig. 2). This rate was similar to ki observed at pH 6.0 in Sr-PSII, and accounted for 90% of the rise of the m/z 36 signal in the Ca-PSII samples, and for 100% in case of the Sr-PSII samples. This is fully consistent with the near total conversion of the multiline signal into the g = 4 signal observed by EPR under these conditions.38Table 1 shows that the rate constants kf and ks are essentially insensitive to pH and Ca/Sr substitution. In Sr-PSII, ki is also essentially unaffected by pH, but the ki of Sr-PSII is larger by a factor of 2–3 compared to that of Ca-PSII.
It was recently demonstrated that addition of ammonia to PSII core complexes from T. elongatus in a pH 8.6 buffer leads to the quasi quantitative formation of the ammonia modified S2 multiline signal (SLS2) at the expense of the g = 4 signal (SHS2).38 Employing this treatment we tested whether the accelerated water exchange is caused by the high pH or instead is related to the different structures of the Mn4Ca cluster in the SLS2 and SHS2 states (blue dots and lines in top panels of Fig. 2). It can be seen that ammonia addition essentially reverted the rates to those seen at pH 6.0 (black data points). This strongly suggests that the water exchange rates observed are a direct consequence of the conformation of the Mn4Ca cluster, rather than pH. In agreement with this conclusion, ammonia had very little effect on the substrate exchange kinetics at lower pH, where basically only the SA2 state was present in Ca-PSII samples.76
In contrast to the slowly exchanging substrate water Ws, only minor variations were observed for the exchange rate of the fast exchanging substrate Wf in the S2 state under all conditions (Fig. 2A, C and Table 1). The lack of any significant effect of the substitution of Ca by Sr in the S2 state is especially notable, and indicates that W3 is either not a substrate (that possibly binds as Ox/O6 in the S3 state), or its exchange at the Ca site is limited by factors other than breaking the bond to Ca/Sr, such as for example the diffusion of bulk water through water channels.
Fig. 3 shows the substrate water exchange in the S3 state in Ca-PSII core complexes of T. elongatus at pH 8.6. These experiments revealed that the rate of Ws exchange in the S3 state is well described by a monophasic rise (red dots and line in Fig. 3). In stark contrast to S2, ks was in the S3 state slower at pH 8.6 than observed previously at neutral pH (dashed black line in Fig. 3).55,78 This results in a 6-fold difference between the slow substrate water exchange rates of the S2 and S3 states at pH 8.6 (Table 1), indicating that the substrate exchange in the S2 and S3 states is governed by different exchange mechanisms and rate limiting steps. Thus, the previously found near identical exchange rate for these two S-states appears to be coincidental.
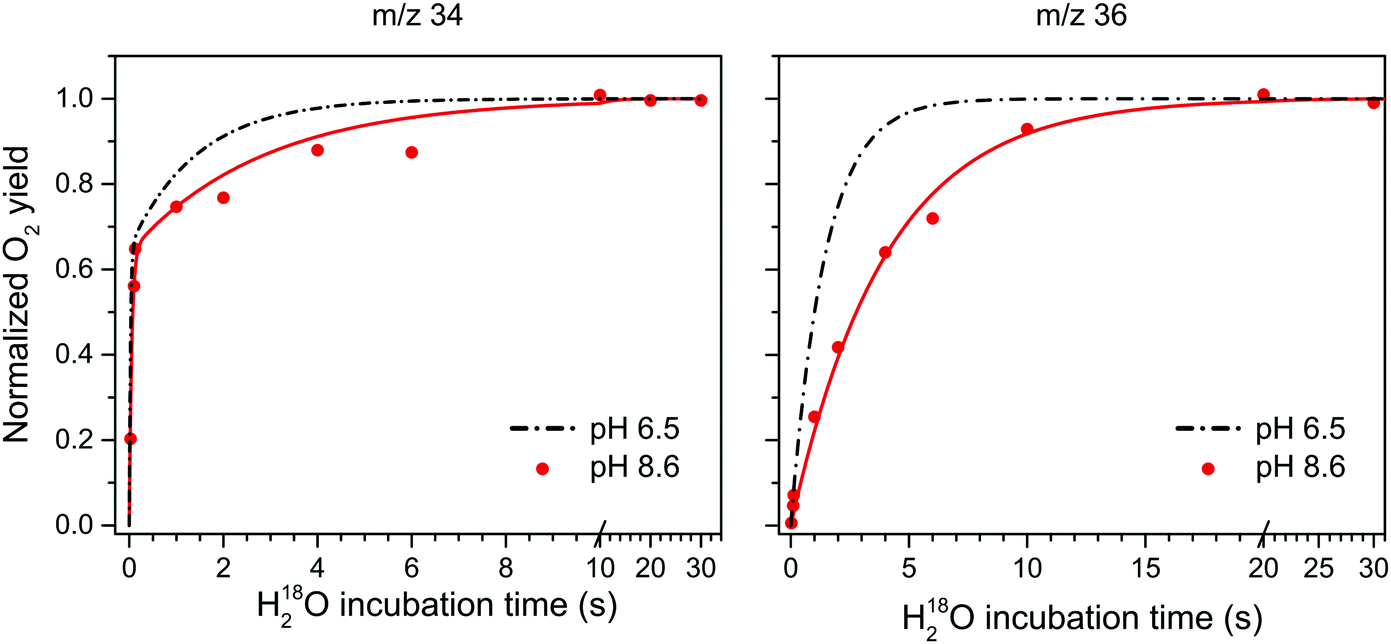 | ||
| Fig. 3 H218O substrate exchange of Ca-PSII at pH 8.6 in the S3 state. Red dots represent the results from single time points. Red lines are fits according to eqn (1)–(3,) while the black dashed lines represent simulated exchange rates based on literature values of exchange in similar preps and conditions, but at pH 6.5.77 The fitted exchange rates are listed in Table 1. | ||
Mechanistic and energetic analysis
The fact that two different rates of Ws exchange were measured under the same conditions (Fig. 2C) implies that the equilibrium between the two S2 state conformations has a similar or higher barrier than substrate water exchange. Thus, two possibilities exist: (I) there are two independent exchange pathways for Ws in the SLS2 and SHS2 states, of which the SHS2 exchange has a lower barrier (exchange pathway I in Fig. 4), or (II) the SLS2 conformation has to convert into the SHS2 conformation so that water exchange can occur (pathway II in Fig. 4). In these two schemes, the rate ks corresponds to the exchange of Ws that starts from the SLS2 conformation; it thus reports either on the activation energy for the exchange process starting from this structure (pathway I), or on the energetic barrier for reaching the SHS2 conformation (pathway II). Since ks is nearly pH and Ca/Sr independent (Table 1), it must be the energy difference between the SLS2 and SHS2 conformations that changes at high pH. Furthermore, as the HS state is stabilized at high pH, it is likely that a deprotonation is involved in the SLS2 → SHS2 conversion, as suggested previously.38 By contrast, the rate ki signifies in both pathways the Ws exchange rate starting from the SHS2 conformation. This rate is also nearly pH independent, but ki is about three-fold larger in Sr-PSII than in Ca-PSII.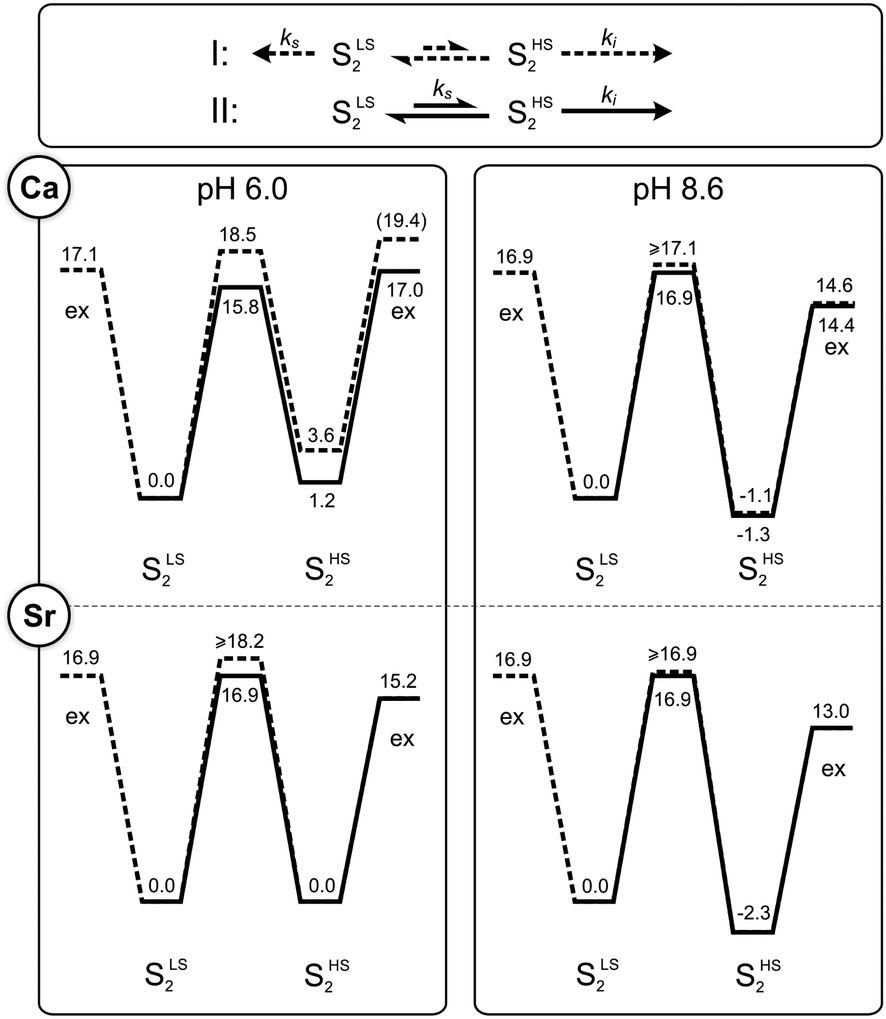 | ||
| Fig. 4 Kinetic models (top panel) and energy diagrams (lower panels) for the exchange (‘ex’) of the slow substrate water Ws in the SLS2 and SHS2 conformations of photosystem II. The barriers were calculated from the rates listed in Table 1 using transition state theory (eqn (4)). They are given in kcal mol−1. Dashed lines correspond to pathway I, where SLS2 and SHS2 exchange independently, while solid lines represent the sequential pathway II, in which the SLS2 conformation has to convert first into the SHS2 conformation before water exchange can occur. Where lines overlap, only the solid line is visible. The length of the arrows in the top panel correspond to the rates of Ws exchange in Ca-PSII at pH 6.0. | ||
Employing the Eyring equation (eqn (4)), energy diagrams for the two exchange pathways were established for Ca- and Sr-PSII at both pH regimes (Fig. 4). The energy diagrams shown are not unique in all aspects, but rather the simplest ones we could conceive to explain our findings with minimal variations of parameters. As such, the relative energy levels of SLS2 and SHS2 were adjusted to reflect the percentages of centers undergoing intermediate and slow water exchange as reflected in the m/z = 36 data.
In the sequential exchange pathway II, shown in black lines in Fig. 4, the energy of the SHS2 conformation is by 1.2 kcal mol−1 higher than that of the SLS2 conformation in Ca-PSII at pH 6. This value is highly similar to that determined by previous DFT calculations that were based on the proposal that the SHS2 conformation attains the SB2 structure.40,45 Substitution of Ca by Sr makes the two conformations of the S2 state iso-energetic at pH 6, while the increase of pH to 8.3/8.6 stabilizes the SHS2 state by 2.3–2.5 kcal mol−1 in both samples. Within the sequential exchange pathway, ks is a direct measure of the activation energy of the SLS2 → SHS2 transition. A value of 15.8 kcal mol−1 was found for Ca-PSII at pH 6, while it was 16.9 kcal mol−1 under all other conditions tested here. This is higher than estimated in two previous DFT studies that modeled the HS to LS conversion to be a shift of O5 between Mn4 and Mn1 (6–11 kcal mol−1 for Ca and Sr).40,45,79 It is also more than twice the value (6.5 kcal mol−1) derived in one EPR study that followed the rate of SHS2 to SLS2 conversion in a temperature range between 150–170 K (see also ref. 100).80 However, the value is rather similar to the barrier (17.6 kcal mol−1) calculated by Siegbahn for the exchange of O5 in the S2 state.81
Similar energy levels and barriers were obtained when examining the alternative parallel exchange pathway I (dashed lines in Fig. 4). The main difference is that the barrier between SLS2 and SHS2 must be higher to block water exchange of centers in the SLS2 state via the SHS2 route.
Discussion
In this study, we examined the exchange rates of the two substrate water molecules in the SLS2 and SHS2 conformations of PSII-core preparations of T. elongatus by pH shifts, ammonia addition and Ca/Sr substitution. We report for the first time that the slowly exchanging substrate water, Ws, equilibrates 10 times faster in the SHS2 state than in the SLS2 (SA2) state. While we employed a pH shift for switching between the two conformations of the S2 state,38 we excluded that the observed changes in rates are a consequence of the different proton concentrations by adding ammonia, which was previously shown to stabilize the SLS2 configuration at high pH by directly binding to Mn.38,76,82–84 We also discovered that at alkaline pH the slow substrate water no longer exchanges with similar rate in the S2 and S3 states, and that the exchange rate of the fast exchanging substrate water is not only insensitive to Ca/Sr substitution in the S3 state, as reported previously, but also in the S2 state.Below we discuss these three new findings in detail on the basis of present structural knowledge about the Mn4Ca cluster and with regard to the only detailed exchange pathway that has been proposed thus far for O5. The aim of the discussion is to both gain an improved understanding of the mechanism of substrate water exchange, and to scrutinize the presently favored assignments of Ws to O5 and of Wf and W2 or W3. This task is complicated by the fact that there is an ongoing vivid discussion regarding the structure of the SHS2 state, with no less than 3 different proposals. This uncertainty in the field necessitates to discuss a variety of options. After identifying the assignments for the substrates consistent with our present and previous data, we formulate consequences for current proposals for the mechanism of water oxidation in PSII.7,11,63,64,85–87
General considerations
Water exchange can follow an associative or dissociative pathway. In the former, a new water molecule binds first before the original water molecule is released into the bulk, while in the latter, the coordinated water molecule dissociates before a new water can bind. Ligand exchange rates are known to slow down with increasing metal oxidation state, and Mn(IV) is generally seen as being exchange inert, while in case of Mn(III) at least the water bound along the Jahn–Teller (JT) axis should be readily exchangeable.60,81,88–94 If water is bound in a deprotonated form, it needs to be protonated, and bridging oxygen's need additionally be brought into a terminal position before exchange with bulk water can occur. This implies that the exchange of O5 is a complex process that requires conformational changes of the Mn4Ca cluster, likely involving a number of the proposed structures summarized in Scheme 1.Evaluation of O5 as the slowly exchanging substrate water Ws
On the basis of substrate water exchange experiments64 and theoretical calculations,11 it was postulated that O5 is the slowly exchanging substrate Ws. The rational for the experimental assignment was twofold: firstly, the exchange rate of Ws is dependent on both Ca/Sr substitution and S-state; thus Ws was suggested to be a bridge between Mn and Ca.61,64 Secondly, this bridge was assigned to O5,64 because EPR and EXAFS data are indicative of its deprotonation during the S0 → S1 transition,9,11,18,19,26,27 matching the 500-fold decrease in substrate exchange rate between S0 and S1.95 Subsequent EDNMR experiments have confirmed that O5 exchanges with bulk water within 15 s in the S1 state,67 which is unusually fast for a μ-oxo bridge,92 and this finding makes O5 a candidate for Ws. However, a definitive assignment needs to await a higher time resolution that allows matching the EDNMR-based O5 exchange kinetics with those obtained for Ws by TR-MIMS.In 2013, Siegbahn proposed a mechanism for the exchange of O5 with bulk water.81 Starting from the SA2 conformation (2A-1), the first step is the binding of a bulk water molecule (marked blue in Scheme 2A) to the open coordination site of Mn1. This step, which has a calculated barrier of 17.6 kcal mol−1 and is thus rate limiting for the O5 exchange,81 results in a structure (2A-2) resembling SAW2, but with one additional proton on O5 and swapped oxidation states. Next, the newly inserted hydroxo swings into the O5 binding site and O5H becomes a terminal ligand of Mn4 (2A-3). The new bridging OH transfers its proton to form a fully protonated terminal O5 ligand on Mn4(IV). After a valence swap between Mn4 and Mn3 the Mn4(III)–O5H2 conformation is reached (2A-4; a SBW2 like structure with one additional proton) that allows O5 to exchange with bulk water, presumably via a dissociative mechanism. Thereafter, this multi-step sequence reverses to yield back the SA2 state, but with O5 exchanged from 16O to 18O.
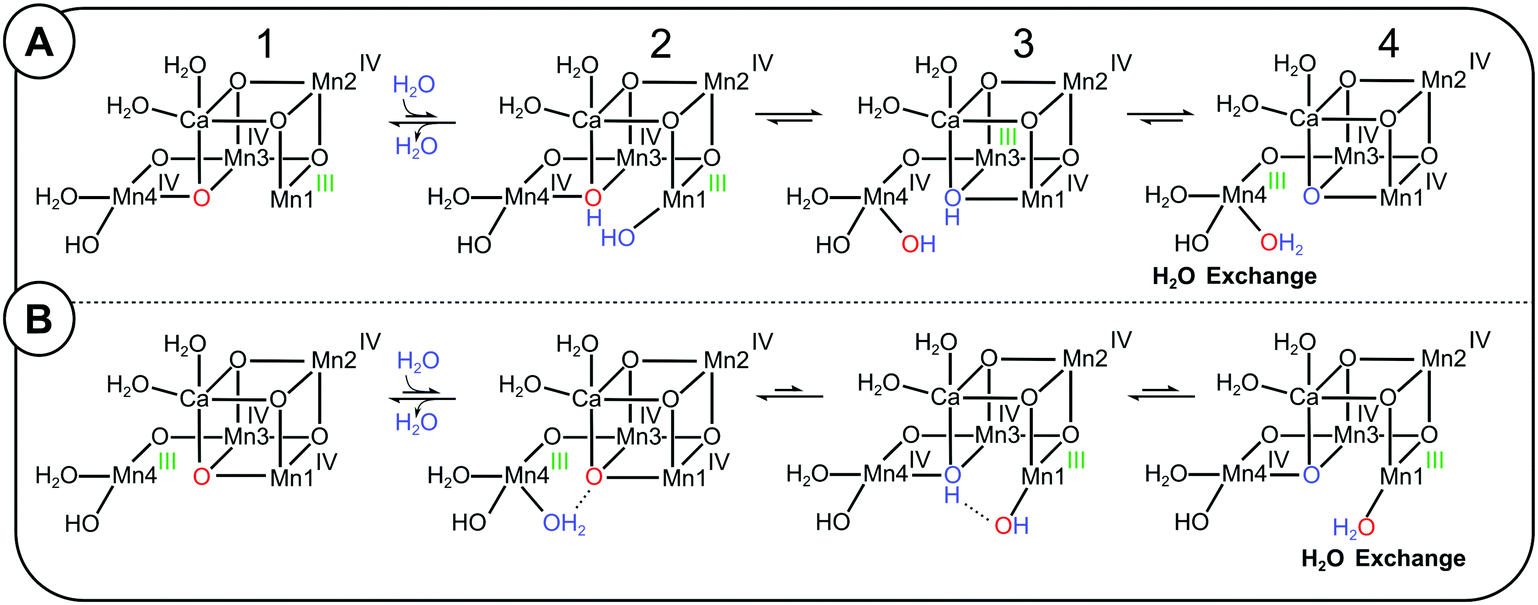 | ||
| Scheme 2 Possible exchange pathways for O5 starting from the SLS2 state (panel A) and the SHS2 state (panel B). (A) Mn4 site O5 exchange mechanism (redrawn after ref. 81). A bulk water or W3 (blue) binds to Mn1 in the SA2 conformation, leading to a valence flip between Mn4 and Mn3, and the transfer of one proton from the new water to O5 (red). The final conformation has a water-bound SB2-type structure, in which Mn4 has the oxidation state III (green), allowing the exchange of O5 before returning to the SA2 conformation by reversing the sequence. (B) Proposal of a Mn1-site O5 exchange pathway starting from the SB2 conformation. A water (blue) binds to the five coordinated Mn4(III) in the SB2 conformation, which induces a proton transfer and valence flip that leads to the formation of a water-bound SA2 conformation, in which O5 is bound to the five-coordinated Mn1(III) site, where water exchange may occur. | ||
It is important to note that the SBW2 conformation reached via this Mn4-site exchange pathway is fundamentally different from the SB2 conformation formed via the SA2 ↔ SB2 equilibrium proposed by the Pantazis, Guidoni and Yamaguchi groups.32,40,96,97 The important difference is that the original O5 (red) is bound terminally to Mn4(III), and not as a μ3-oxo between Ca, Mn3 and Mn1 (Scheme 1). Therefore, the SB2 conformation is not an intermediate of Siegbahn's Mn4-site exchange mechanism of O5.
Thus, if the frequently accepted proposals that, firstly, SB2 is the structure of the SHS2 conformation and, secondly, the Mn4-site exchange mechanism describes the exchange of Ws are both correct, then it follows that the 10-fold faster exchange of Ws in the SHS2 conformation cannot be understood within a sequential exchange mechanism in which SA2 converts first into SB2 before water exchange can take place (pathway II in Fig. 4). Accordingly, a separate pathway starting from the SHS2 state must be considered that can explain the 10-fold faster Ws exchange in this conformation (Scheme 2B; Mn1-site O5 exchange pathway). The first step is water binding to Mn4, which induces a flip of bonds and charges akin to the pivot and carousel mechanisms describing water binding during the S2 → S3 transition (2B-1 to 2B-4).7,32,49,98 This is essentially the reverse of the Mn4-site exchange pathway (Scheme 2A), and places O5 in a SAW2 like structure into a terminal position at Mn1(III), where it may exchange with bulk water, possible via Ca. However, since it is not obvious why this pathway would have a lower barrier than the Mn4 exchange pathway, we presently disfavor this option.
Looking at the two other structural proposals for the SHS2 state (Scheme 1), it is noted that the SAW2 conformation may provide an explanation for the faster exchange of O5 in the SHS2 state, since it resembles the first intermediate of the Mn4 exchange pathway (2A-2; note the different oxidation state assignments and the extra proton). Indeed, the energy barrier determined here for the SLS2 → SHS2 conversion is with 16 to 17 kcal mol−1 similar to that calculated by Siegbahn for the first step of the Mn4-site exchange pathway for O5 (17.6 kcal mol−1).81 Similar values for water binding to Mn1 (in the S2 → S3 transition) were obtained by Guidoni and Pantazis.32,99 By contrast, the theoretical estimates for SA2 → SB2 (6–11 kcal mol−1) are significantly lower,40,45,46 as are previous experimental determinations of the barrier for the SHS2 to SLS2 conversion that gave values of 6.7 ± 0.5 kcal mol−1 and 7.9 ± 1.4 kcal mol−1, respectively.80,100 These previous experimental barriers were obtained by generating the SHS2 state from S1 by illumination at 130–135 K in spinach PSII membrane fragments, and measuring the temperature dependence of the conversion of the g = 4.1 signal into the S2 multiline signal in the temperature range of 150–170 K. Thus, the experimental conditions are highly different from the ones in the present study, where for the first time this barrier was determined at physiological temperatures that facilitate protonation state and structural changes, including water binding. By contrast, such changes are inhibited in T. elongatus PSII samples at cryogenic temperatures, as indicated by the experiments by Boussac, in which he needed to warm the samples to room temperature for a few seconds to allow the conversion of the SLS2 state into the SHS2 state after a 200 K illumination at alkaline pH.38 As such it seems likely that the SHS2 signal obtained at cryogenic conditions has a different structure and hence a different barrier for the conversion of the SHS2 state into the SLS2 state than found here at physiological temperature. Alternatively, the discrepancies to the earlier experimental data are due to species differences.
Since the assignments of the Ws exchange rates to the SHS2 and SLS2 states is solid, and the conversion of these rates into energetic barriers is straight forward, we regard our determination of the energetic barrier to be relevant for T. elongatus PSII core preparations at physiological temperatures, and to be a strong support for (i) the Mn4 exchange pathway for O5 proposed previously based on DFT calculations,81 and (ii) the identification of Ws as O5. It is also in line with the idea that the high pH induced SHS2 state has an SAW2 like structure,39,41,43,47 but other water/hydroxide-bound conformations as for example SBW2 cannot be excluded. SBW2 is similar to intermediate 2A-3 (Scheme 2A), which was calculated to have a total energy 4.6 kcal above SA2,81 thus not too far from the level expected for SHS2 (Fig. 4). Additional constraints for structure and oxidation states of the SHS2 state comes from a recent report of Mino and Nagashima, in which they utilized the orientation dependence of the SHS2 EPR signal to identify that (i) Mn4 is the only Mn(III) ion in the SHS2 state, and (ii) there needs to be a strong coupling (short distance) between Mn4 and Mn3 to simulate their data within a four-spin coupling scheme.101
Exchange of O5 in the S3 state
For the S3 state, Siegbahn proposed that water exchange requires the back-donation of one electron from YZ to the Mn4Ca cluster in order to reduce one of the four Mn(IV) ions to Mn(III),81 which would allow S2-type water exchange. In this S2YZ˙ state, the Mn4Ca cluster would likely reside in the SAW2 structure, and could thus exchange O5 with the rate ki. If one then assumes that the transition state for the reduction of the Mn4Ca cluster by YZ has a similar barrier to the water exchange starting from SA2, this would resolve a major criticism of Siegbahn's Mn4-site exchange proposal for O5. This criticism relates to the experimental finding that, at neutral pH, Ws exchanges in the S2 and S3 states with very similar rates, while the equilibrium between S3YZ and S2YZ˙ would be expected to slow down the O5 exchange in the S3 state, given that the S2YZ˙ population must be very low, as this state has not been experimentally observed at neutral pH.The situation is, however, very different at pH 8.6. Here, the S2YZ˙ state is clearly observed by EPR and hence significantly populated.102 Thus, one may expect that substrate water exchange in the S3 state at pH 8.6 should occur fast (with rate ki) in a significant fraction of centers, resulting in a bi-phasic exchange curve as observed in Fig. 2D for the S2 state. By contrast, a monophasic exchange was observed at high pH for the S3 state samples (Fig. 3), which occurred with a rate that was 6-fold slower than that in the S2 state, and also clearly retarded relative to comparable S3 state data obtained previously at neutral pH,55,77,103 see also ref. 75.
The recent experimental evidence for the SB3 conformation55 allows proposing an alternative exchange pathway for O5 in the S3 state. As shown in Scheme 1, SB3 may be reached from the dominant SAW3 conformation via SBW3. After the loss of the O5–water molecule, a new water may bind leading to the re-formation of SAW3 containing a new O5.
Thus, also the new S3 state substrate water exchange data are consistent with O5 being the slowly exchanging substrate water Ws.
W2 as possible alternative assignment for Ws
We evaluated the structures of the Mn4Ca-cluster to see if Ws = O5 is the only option to explain our data. One possible alternative was found assuming that the SA2 and SB2 structures proposed by Pantazis represent the SLS2 and SHS2 state, respectively.40 While we presently favor that the SHS2 state has a SAW2 like structure under our experimental conditions, we discuss this option since it emphasizes the importance of a unique structural resolution of the SHS2 state for deriving the mechanism of water oxidation.Aside from the different position of O5 in the SA2 and SB2 structures, the major difference between the two S2 state conformations is the position of the five-coordinate Mn(III) ion. Since a five-coordinate Mn(III) ion should promote rapid water exchange via an associative exchange pathway, water ligands bound to the five-coordinate Mn(III) ion in the SHS2 state may constitute Ws. Mn4 has two terminal water derived ligands: W1, which is proposed to bind as fully protonated water molecule hydrogen bonded to D1-Asp61, and W2, which may be bound as a water or hydroxide.27–30,104 We have previously excluded W1 from being Ws as it can be replaced by ammonia in the S2 state with only minor effects on the substrate exchange rates (at pH 7.5).76 This leaves W2 as a possible alternative candidate for Ws.
W2, here assumed to be a hydroxide, can be expected to exchange in the SHS2 = SB2 state readily with bulk water (lower panel in Scheme 3) since Mn4(III) is placed near the exit of two proposed water channels (O4 and Cl channels) and is also in reach of W3 that is connected to the O1 water channel.3,105–112 Water exchange in the SLS2 = SA2 state would then occur via the equilibrium with the SB2 conformation (Scheme 3 and pathway II in Fig. 4).
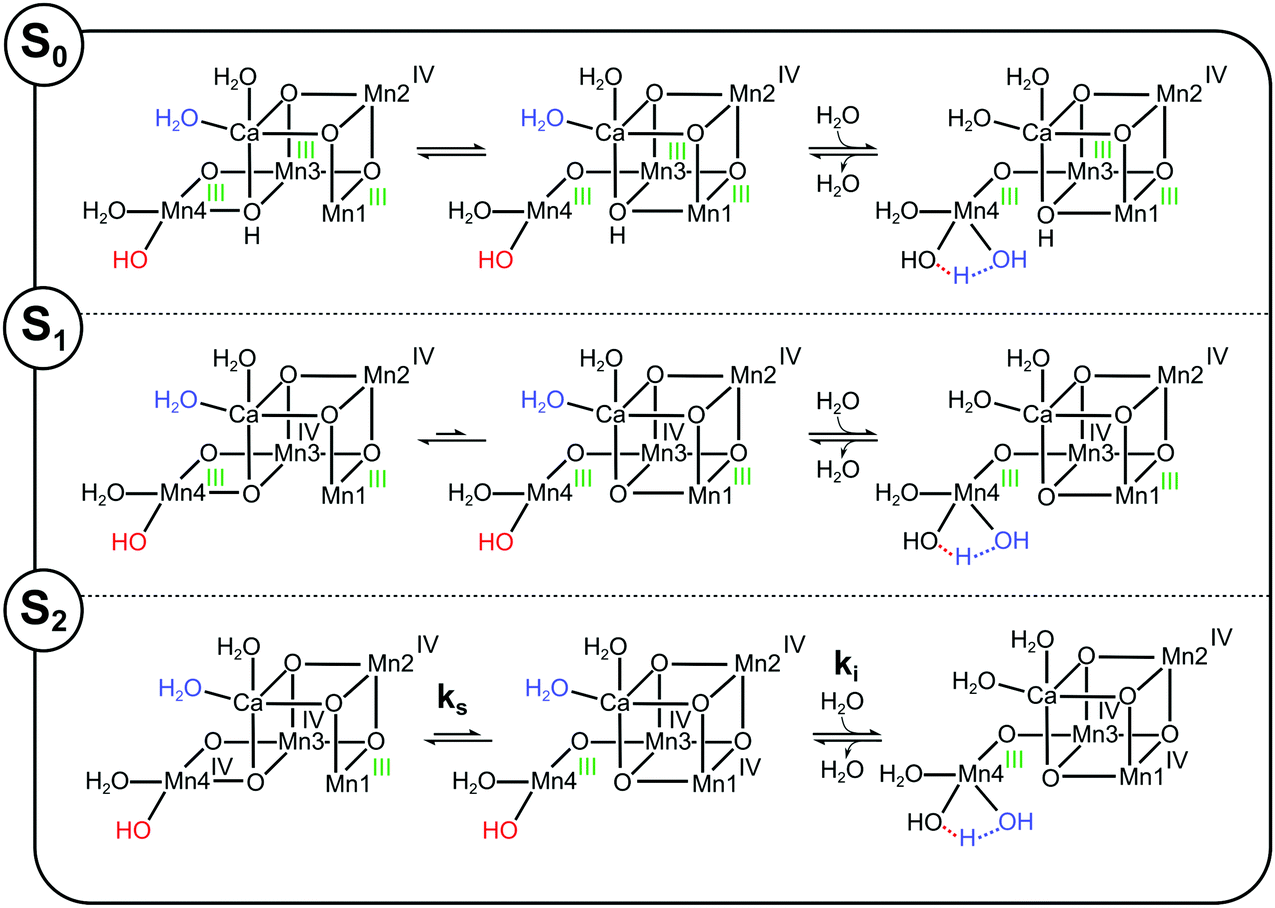 | ||
| Scheme 3 Proposed substrate exchange mechanisms for the S0, S1 and S2 states assuming that W2 (labelled red) is the slow substrate water and that SHS2 has the SB2 structure. Mn oxidation states are labelled green for oxidation state III and black for oxidation state IV. In all three S-states the generally more stable SA2 conformation (the energy difference in S0 is proposed to be small) must first convert into the SB2 conformation before a water molecule (blue), here suggested to be W3, binds to Mn4. This water donates a proton to W2, which then detaches, after which the Mn4Ca cluster returns to the SA2 conformation with the new water in the W2 position. | ||
To scrutinize the alternative W2 proposal, we used the S-state dependence of Ws exchange. For this, we extended pathway II (Fig. 4) to the other S-states by proposing, in line with previous suggestions,7,56 as well as experimental data and theoretical calculations,29,30,38,110,113 that all S-states can exist in A- and B-type conformations, and that the barriers between these conformations are S-state dependent. In addition, we assume that W2, if bound to a non-JT-axis at a six coordinated Mn(III) ion, exchanges much slower as compared to when it is bound to a five-coordinated Mn(III) ion.
In the S0 state of Ca-PSII, where all Mn ions ligating O5 are in oxidation state (III) and O5 is protonated, the energy difference between the A and B structures and the transition state barrier between them should be small (long arrows of equal length in Scheme 3). Consequently, the SB0 form, containing a 5-coordinated Mn4(III) site, should be easily attainable, resulting in the fastest Ws exchange of all S-states. Consistent with the idea that Ws exchange occurs in the S0, S1 and S2 states at a five-coordinated Mn(III) site, and that the barrier for reaching this state is low in the S0 state, the rate ks in the S0 state is with about 10–20 s−1 (in spinach)75,95 nearly identical to the rate ki measured here for the water exchange in the SHS2 state (Table 1).
Oxidation of Mn3 during the S0 → S1 transition strongly stabilizes the SA1 state, making it the clearly dominant conformation. Thus, the exchange rate measured for Ws in the S1 state, which is about 500 times slower than in the S0 state, may either reflect the exchange of W2 at the six-coordinate Mn4(III) ion, or the barrier for reaching the SB1 conformation, in which W2 is bound to a five-coordinated Mn4(III) ion facilitating rapid exchange.
Further oxidation of the Mn4Ca cluster into the S2 state is expected to increase the exchange rate of W2, since the B-type state can now be stabilized by locating the additional oxidizing equivalent on Mn1. Thus, the barrier for reaching the fast exchanging SB2 state can be assumed to be lower than in the S1 state, explaining the 100-fold faster ks exchange rate. In the S3 state, water exchange can then occur as described above, either by the reduction of the Mn4Ca into the S2 state by YZ, or via the five-coordinate Mn4(IV) site of the SB3 state. The main shortcoming of this proposal is the mismatch of the activation barriers described above, that in our view favors the assignment of SHS2 to a water/hydroxide bound conformation of the S2 state, such as SAW2. We anticipate that all the arguments above are exactly the same if W2 were a water molecule instead of a hydroxide.
O4 as possible alternative assignment for Ws
The SAPI2 structure for the SHS2 state involves the protonation of O4. Thus, if O4 were Ws, this would likely result in a faster exchange of Ws in the SHS2 state as compared to the SLS2 = SA2 state. Indeed, O4 was recently suggested to form the O–O bond with an H-bonded water molecule.69,70 However, we were not able to propose a scheme for the exchange of O4 that appeared consistent with the water exchange data. Additionally, the assignment of Ws to O4 would be in conflict with the EDNMR assignment of O5 as the only exchangeable oxo bridge of the Mn4Ca cluster,67 and with the recent polarized EPR data of the SHS2 state.101The fast exchanging substrate Wf
The most significant finding of this study regarding the exchange of Wf in the S2 state is the invariance of kf towards the substitution of Ca by Sr. This is important, since in recent proposals for the S2 → S3 transition it is frequently assumed that W3 is Wf, which would be bound to Ca in the S0, S1 and S2 states, but to Mn1 or Mn4 in the S3 state. The lack of Ca/Sr dependence in the present S2 state data thus disfavors that W3 is a substrate. However, this option cannot be excluded until firm data for the rate of diffusion of substrate water to the catalytic site are obtained. While it generally would be assumed to be unlikely that water access is limiting the fast water exchange, it cannot be excluded a priori since present calculations indicate that all channels have barriers in the range of 10 kcal mol−1,109,114 and a NMR proton relaxation study indicates a distance of 10 Å from the spin center of the Mn4Ca cluster to the protons that rapidly exchange with the protons of bulk water.115In case that there are no significant access barriers for Wf exchange, the previously proposed W2 assignment remains the best option for Wf, and the reported low barrier of the SA2 ↔ SB2 equilibrium may provide the means for fast W2 exchange (similar to Scheme 3).
Given these presently equally likely options for Wf (in case Ws = O5), detailed mutational studies aiming to increase or decrease the access of water through the known channels connecting the OEC with bulk water will be needed for a final decision. Such experiments are beyond the scope of the present study.
Possible mechanisms of water oxidation in PSII
The energetics for Ws exchange determined here for samples in the SLS2 state agree well with those calculated by Siegbahn for the O5 exchange starting from the SA2 state, and thereby strongly support the earlier assignment of Ws to O5 by Messinger and Siegbahn.64,66 The present data favor that a SAW2-like conformation is both an intermediate in the exchange of O5 and the structure of the SHS2 state (Fig. 5A). However, in case that the SHS2 state adopts the closed cubane conformation (SB2), we are unable to exclude W2 as the slow substrate water, since a consistent proposal for water exchange could be made for both O5 and W2 (Fig. 5B and C). For a final assignment further studies will be required, such as 17O-EDNMR experiments with high-enough time and spectral resolution to allow monitoring the time course of 17O/16O-exchange of both W2 and O5,67 thus allowing the comparison of the W2 and O5 exchange rates with those of Ws determined by TR-MIMS experiments. In addition, obtaining room temperature crystal structures of the high pH SHS2 state would allow removing the remaining uncertainties. With regard to Wf, both W2 and W3 remain options until the possible role of water accessibility to the catalytic site on the rate of fast water exchange is clarified by mutational studies in combination with substrate water exchange. By contrast, all other options can be excluded.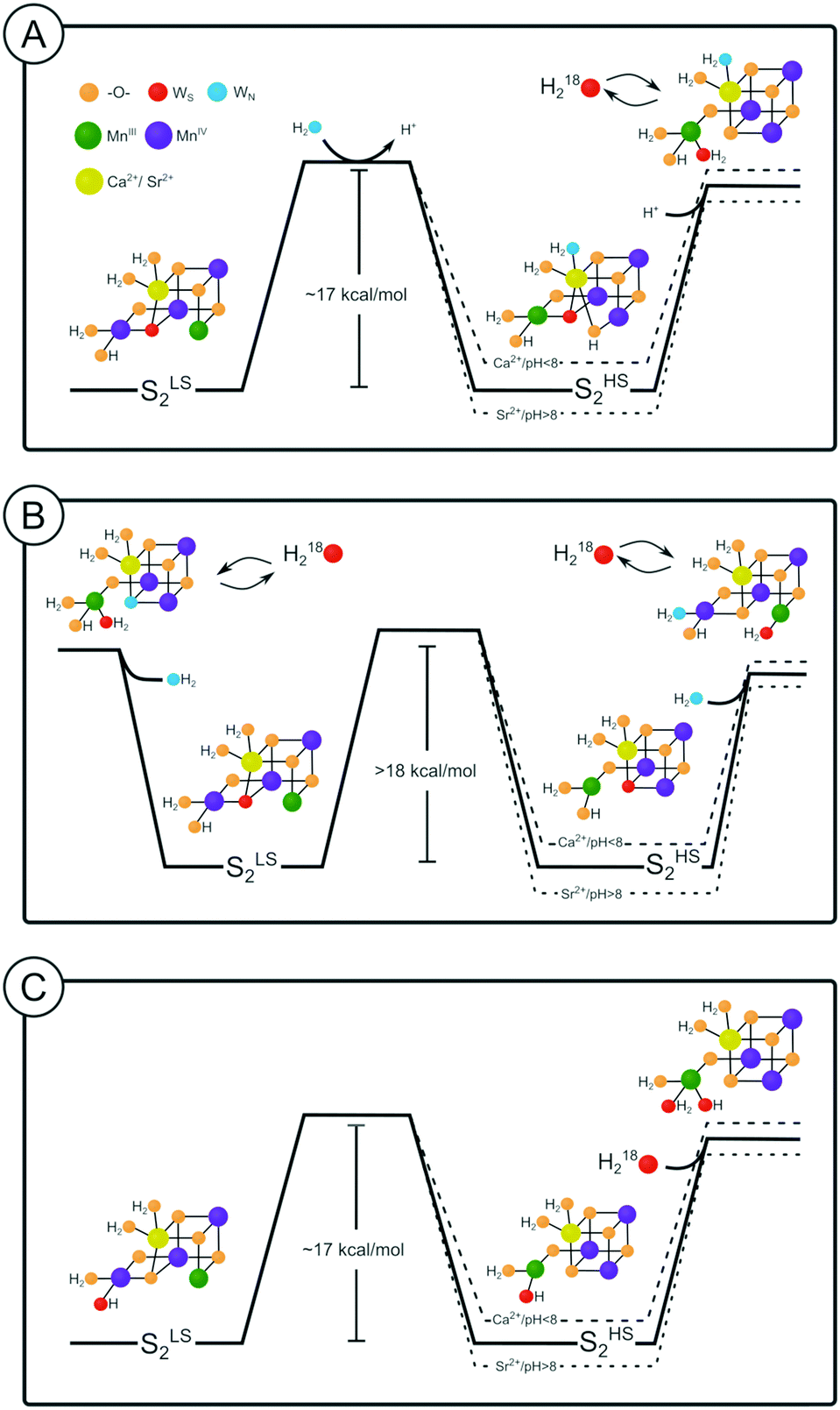 | ||
| Fig. 5 Possible substrate water exchange pathways in the S2 state. The energy diagram shown as solid line indicates the case for Sr-PSII at pH 6. Dashed lines indicate changes in the relative energies of SLS2 and SHS2 due to pH and/or substitution of Ca by Sr. | ||
On that basis, O–O bond formation mechanisms via radical coupling involving O5 as Ws are strongly favored by our new results and may occur in either an A-type or B-type conformation of the Mn4Ca cluster (Scheme 4A and B).7,11,12,63,64,66,85,116,117 The only difference would be that the radical coupling in the SBW3 state would require first a structural rearrangement starting from SAW3, possibly in line with the lag phase observed after the S3YZ˙ formation.118,119 Interestingly, the origin of the two substrate oxygen's would vary depending on the water insertion pathway.
 | ||
| Scheme 4 Possible O–O bond formation pathways in photosystem II. (A) A-type radical coupling,12,65 (B) B-type radical coupling63,64,85 and (C) geminal coupling at Mn4.22 For further details, see text. | ||
Assuming that the S2 → S3 transition would involve W3 binding to Mn1 or Mn4,3,45,50,120,121 then in both cases the O–O bond would be formed between the former W3 and O5, but the origin of oxygen's in the O5 and Ox/O6 positions would be swapped depending on the insertion site. If, however, water is inserted during the S2 → S3 transition via the pivot mechanism,32 then mechanisms A and B (Scheme 4) would involve O–O bond formation between W2 and O5.7,13,64
By contrast, if Ox/O6 originates from W3, but is not a substrate, W3 would be ‘parked’ in the S3 state between Ca and Mn1 to replace O5 during the S3 → S4 → S0 transition, while the O–O bond would be formed between W2 and O5 via geminal coupling at the Mn4 site (Scheme 4C). Geminal coupling at this site was proposed first by Kusunoki on the basis of DFT calculations that resulted in a B-type structure for the Mn4Ca cluster.113 The proposal was further inspired by his analysis of substrate water exchange data from Hillier and Wydrzynski.60 He suggested that, in the S3 state, there is a significant correlation between the exchange of Wf and Ws, indicating that both must be bound at the same Mn ion to allow them to swap places. At the time the proposal was made, water addition to the Mn4Ca cluster during the S2 → S3 transition was not established, and thus the different S-state dependence of Wf and Ws seemed to exclude this idea. In addition, the demonstration that ammonia binds to the W1 site in the S2 state without significantly affecting the exchange of the two substrate waters argued against this proposal.76,122 However, in the light of the recent data suggesting the binding of W3 to Mn1 during SAW3 formation, geminal coupling is now consistent with present experimental results. Zhang and Sun recently proposed that this type of O–O bond formation involves the transient formation of a Mn4(VII) species obtained via disproportionation within the Mn4Ca cluster.22
While we cannot yet distinguish between the three options displayed in Scheme 4, we exclude nucleophilic attack of a Ca-bound (W3) water onto W2,59,65,94,123–126 since W3 would need to serve a dual role: firstly it would need to fill the open coordination site of Mn1 for preloading the new O5, and secondly the successor water ligand at the W3 site would need to be the fast exchanging substrate Wf. Notably, in case of the pivot pathway for filling the Ox/O6 site, both nucleophilic attack of W3 onto W2, and geminal coupling at the Mn4 site are excluded. Thus clarifying the pathway for water insertion in the S2 → S3 transition is another requirement for deriving at an experimentally confirmed mechanism. Such studies are ongoing in the field, so that the discussion provided here can serve as blueprint for identifying the substrate once this independent problem is solved.
Conclusion
Substrate water exchange experiments provide a unique and independent view on the water oxidation mechanism. In this study, we have advanced this approach significantly by providing unique new experimental results. By combining these new data with emerging knowledge about the structures of various conformers of each S-state, together with earlier DFT calculations regarding O5 exchange, we have derived molecular interpretations of substrate water binding and its exchange with bulk water not previously attainable. The present analysis along with future investigations of the temperature dependence for the barrier between LS and HS states, provides the basis for the interpretation of ongoing TR-MIMS experiments utilizing point mutations, Ca/Sr-exchange and H/D-labelling, which together with other outlined experiments have the potential to resolve the mechanism of water oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was initiated together with Alain Boussac who provided the Ca-PSII and Sr-PSII samples and contributed to the experimental design. We acknowledge the discussions with Vittal Yachandra and Dimitrios Pantazis regarding the nature of the SHS2 state, and Dmitry Shevela for assistance with graphical representations for this manuscript. Financial support for this project was provided by Vetenskapsrådet (grant number: 2016-05183).References
- J.-R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- N. Nelson and W. Junge, Annu. Rev. Biochem., 2015, 84, 659–683 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
- D. A. Pantazis, ACS Catal., 2018, 8, 9477–9507 CrossRef CAS.
- W. Junge, Q. Rev. Biophys., 2019, 52, 1–17 CrossRef PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- N. Cox and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1020–1030 CrossRef CAS PubMed.
- J. Messinger and G. Renger, in Primary Processes of Photosynthesis, Part 2, ed. G. Renger, The Royal Society of Chemistry, Cambridge, UK, 2008, pp. 291–349 Search PubMed.
- L. V. Kulik, B. Epel, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2007, 129, 13421–13435 CrossRef CAS PubMed.
- M. H. Cheah, M. Zhang, D. Shevela, F. Mamedov, A. Zouni and J. Messinger, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 141 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- I. Zaharieva, P. Chernev, G. Berggren, M. Anderlund, S. Styring, H. Dau and M. Haumann, Biochemistry, 2016, 55, 4197–4211 CrossRef CAS PubMed.
- M. Askerka, G. W. Brudvig and V. S. Batista, Acc. Chem. Res., 2017, 50, 41–48 CrossRef CAS PubMed.
- N. Schuth, I. Zaharieva, P. Chernev, G. Berggren, M. Anderlund, S. Styring, H. Dau and M. Haumann, Inorg. Chem., 2018, 57, 10424–10430 CrossRef CAS PubMed.
- H. Dau, L. Iuzzolino and J. Dittmer, Biochim. Biophys. Acta, Bioenerg., 2001, 1503, 24–39 CrossRef CAS.
- M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke and H. Dau, Biochemistry, 2005, 44, 1894–1908 CrossRef CAS PubMed.
- W. Hillier and J. Messinger, in Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase, ed. T. J. Wydrzynski, K. Satoh and J. A. Freeman, Springer, Netherlands, Dordrecht, 2005, pp. 567–608 Search PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Dalton Trans., 2018, 47, 14381–14387 RSC.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-r. Shen, Nature, 2014, 517, 99–103 CrossRef PubMed.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L.-J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J.-R. Shen, Science, 2019, 366, 334 CrossRef CAS PubMed.
- J. H. Robblee, J. Messinger, R. M. Cinco, K. L. McFarlane, C. Fernandez, S. A. Pizarro, K. Sauer and V. K. Yachandra, J. Am. Chem. Soc., 2002, 124, 7459–7471 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- S. Nakamura and T. Noguchi, Proc. Natl. Acad. Sci. U. S. A., 2016, 201607897, DOI:10.1073/pnas.1607897113.
- D. Narzi, G. Mattioli, D. Bovi and L. Guidoni, Chem. – Eur. J., 2017, 23, 6969–6973 CrossRef CAS PubMed.
- K. Miyagawa, H. Isobe, T. Kawakami, M. Shoji, S. Yamanaka, M. Okumura, T. Nakajima and K. Yamaguchi, Chem. Phys. Lett., 2019, 734, 136731 CrossRef CAS.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821–825 CrossRef CAS PubMed.
- M. Retegan, V. Krewald, F. Mamedov, F. Neese, W. Lubitz, N. Cox and D. A. Pantazis, Chem. Sci., 2016, 7, 72–84 RSC.
- G. C. Dismukes and Y. Siderer, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 274–278 CrossRef CAS PubMed.
- J. L. Casey and K. Sauer, Biochim. Biophys. Acta, Bioenerg., 1984, 767, 21–28 CrossRef CAS.
- J. L. Zimmermann and A. W. Rutherford, Biochemistry, 1986, 25, 4609–4615 CrossRef CAS.
- A. Boussac, J. J. Girerd and A. W. Rutherford, Biochemistry, 1996, 35, 6984–6989 CrossRef CAS PubMed.
- T. S. Kuntzleman and A. Haddy, Photosynth. Res., 2009, 102, 7 CrossRef CAS PubMed.
- A. Boussac, I. Ugur, A. Marion, M. Sugiura, V. R. I. Kaila and A. W. Rutherford, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 342–356 CrossRef CAS PubMed.
- A. Boussac, Biochim. Biophys. Acta, Bioenerg., 2019, 1860, 508–518 CrossRef CAS PubMed.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2018, 20, 22926–22931 RSC.
- T. A. Corry and P. J. O’Malley, J. Phys. Chem. Lett., 2019, 5226–5230 CrossRef CAS PubMed.
- Y. Pushkar, A. K. Ravari, S. C. Jensen and M. Palenik, J. Phys. Chem. Lett., 2019, 10, 5284–5291 CrossRef CAS PubMed.
- G. Renger, Photosynthetica, 1987, 21, 203–224 CAS.
- D. Bovi, D. Narzi and L. Guidoni, Angew. Chem., Int. Ed., 2013, 52, 11744–11749 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, S. Yamanaka, Y. Umena, K. Kawakami, N. Kamiya, J.-R. Shen and K. Yamaguchi, Dalton Trans., 2012, 41, 13727 RSC.
- R. Chatterjee, G. Han, J. Kern, S. Gul, F. D. Fuller, A. Garachtchenko, I. D. Young, T.-C. Weng, D. Nordlund, R. Alonso-Mori, U. Bergmann, D. Sokaras, M. Hatakeyama, V. K. Yachandra and J. Yano, Chem. Sci., 2016, 7, 5236–5248 RSC.
- R. Chatterjee, L. Lassalle, S. Gul, F. D. Fuller, I. D. Young, M. Ibrahim, C. de Lichtenberg, M. H. Cheah, A. Zouni, J. Messinger, V. K. Yachandra, J. Kern and J. Yano, Physiol. Plant., 2019, 166, 60–72 CrossRef CAS PubMed.
- M. Askerka, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2015, 54, 5783–5786 CrossRef CAS PubMed.
- I. Ugur, A. W. Rutherford and V. R. I. Kaila, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 740–748 CrossRef CAS PubMed.
- N. Ioannidis and V. Petrouleas, Biochemistry, 2000, 39, 5246–5254 CrossRef CAS PubMed.
- V. Petrouleas, D. Koulougliotis and N. Ioannidis, Biochemistry, 2005, 44, 6723–6728 CrossRef CAS PubMed.
- K. G. V. Havelius, J.-H. Su, Y. Feyziyev, F. Mamedov and S. Styring, Biochemistry, 2006, 45, 11 CrossRef PubMed.
- A. Boussac, M. Sugiura, T.-L. Lai and A. W. Rutherford, Philos. Trans. R. Soc., B, 2008, 363, 1203–1210 CrossRef CAS PubMed.
- M. Chrysina, E. Heyno, Y. Kutin, M. Reus, H. Nilsson, M. M. Nowaczyk, S. DeBeer, F. Neese, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2019, 166, 6 Search PubMed.
- G. Renger, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1164–1176 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, J. R. Shen and K. Yamaguchi, Inorg. Chem., 2016, 55, 502–511 CrossRef CAS PubMed.
- T. A. Corry and P. J. O’Malley, J. Phys. Chem. Lett., 2018, 9, 6269–6274 CrossRef CAS PubMed.
- J. Messinger, M. Badger and T. Wydrzynski, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3209–3213 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Coord. Chem. Rev., 2008, 252, 306–317 CrossRef CAS.
- G. Hendry and T. Wydrzynski, Biochemistry, 2003, 42, 6209–6217 CrossRef CAS PubMed.
- W. Hillier, J. Messinger and T. Wydrzynski, Biochemistry, 1998, 37, 16908–16914 CrossRef CAS PubMed.
- H. Nilsson, T. Krupnik, J. Kargul and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1257–1262 CrossRef CAS PubMed.
- J. Messinger, Phys. Chem. Chem. Phys., 2004, 6, 4764–4771 RSC.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Chem. – Eur. J., 2006, 12, 9217–9227 CrossRef CAS PubMed.
- L. Rapatskiy, N. Cox, A. Savitsky, W. M. Ames, J. Sander, M. M. Nowaczyk, M. Rögner, A. Boussac, F. Neese, J. Messinger and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 16619–16634 CrossRef CAS PubMed.
- D. J. Vinyard, S. Khan and G. W. Brudvig, Faraday Discuss., 2015, 185, 37–50 RSC.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L.-J. Yu, T. Sakamoto, T. Motomura, J.-H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J.-R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- K. Kawashima, T. Takaoka, H. Kimura, K. Saito and H. Ishikita, Nat. Commun., 2018, 9, 1247 CrossRef PubMed.
- M. Sugiura and Y. Inoue, Plant Cell Physiol., 1999, 40, 1219–1231 CrossRef CAS PubMed.
- M. Sugiura, A. Boussac, T. Noguchi and F. Rappaport, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 331–342 CrossRef CAS PubMed.
- N. Ishida, M. Sugiura, F. Rappaport, T.-L. Lai, A. W. Rutherford and A. Boussac, J. Biol. Chem., 2008, 283, 13330–13340 CrossRef CAS PubMed.
- M. Sugiura, Y. Ozaki, M. Nakamura, N. Cox, F. Rappaport and A. Boussac, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1922–1931 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Phys. Chem. Chem. Phys., 2004, 6, 4882–4889 RSC.
- M. Perez Navarro, W. M. Ames, H. Nilsson, T. Lohmiller, D. A. Pantazis, L. Rapatskiy, M. M. Nowaczyk, F. Neese, A. Boussac, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15561–15566 CrossRef PubMed.
- H. Nilsson, F. Rappaport, A. Boussac and J. Messinger, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- W. Hillier, I. McConnell, S. Singh, R. Debus, A. Boussac and T. Wydrzynski,Photosynthesis. Energy from the Sun: 14th International Congress on Photosynthesis, Springer, Dordrecht, 2008, pp. 427–430 Search PubMed.
- F. Pitari, D. Bovi, D. Narzi and L. Guidoni, Biochemistry, 2015, 54, 5959–5968 CrossRef CAS PubMed.
- D. J. Vinyard, S. Khan, M. Askerka, V. S. Batista and G. W. Brudvig, J. Phys. Chem. B, 2017, 121, 1020–1025 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135, 9442–9449 CrossRef CAS PubMed.
- W. F. Beck, J. C. De Paula and G. W. Brudvig, J. Am. Chem. Soc., 1986, 108, 4018–4022 CrossRef CAS.
- R. D. Britt, J.-L. Zimmermann, K. Sauer and M. P. Klein, J. Am. Chem. Soc., 1989, 111, 3522–3532 CrossRef CAS.
- M. C. W. Evans, R. J. Ball and J. H. A. Nugent, FEBS Lett., 2005, 579, 3081–3084 CrossRef CAS PubMed.
- X. Li and P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2015, 17, 12168–12174 RSC.
- M. Shoji, H. Isobe, Y. Shigeta, T. Nakajima and K. Yamaguchi, J. Phys. Chem. B, 2018, 122, 6491–6502 CrossRef CAS PubMed.
- M. Shoji, H. Isobe and K. Yamaguchi, Chem. Phys. Lett., 2019, 714, 219–226 CrossRef CAS.
- W. Hillier and T. Wydrzynski, Biochemistry, 2000, 39, 4399–4405 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Biochim. Biophys. Acta, Bioenerg., 2001, 1503, 197–209 CrossRef CAS.
- M. Lundberg, M. R. A. Blomberg and P. E. M. Siegbahn, Theor. Chem. Acc., 2003, 110, 130–143 Search PubMed.
- L. Helm and A. E. Merbach, Chem. Rev., 2005, 105, 1923–1960 CrossRef CAS PubMed.
- R. Tagore, H. Chen, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2006, 128, 9457–9465 CrossRef CAS PubMed.
- R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 2193–2203 CrossRef CAS PubMed.
- E. M. Sproviero, M. B. Newcomer, J. A. Gascón, E. R. Batista, G. W. Brudvig and V. S. Batista, Photosynth. Res., 2009, 102, 455–470 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Biochemistry, 2000, 39, 4399–4405 CrossRef CAS PubMed.
- M. Capone, D. Bovi, D. Narzi and L. Guidoni, Biochemistry, 2015, 54, 6439–6442 CrossRef CAS PubMed.
- M. Shoji, H. Isobe and K. Yamaguchi, Chem. Phys. Lett., 2015, 636, 172–179 CrossRef CAS.
- M. Askerka, J. Wang, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2016, 55, 981–984 CrossRef CAS PubMed.
- M. Capone, D. Narzi, D. Bovi and L. Guidoni, J. Phys. Chem. Lett., 2016, 7, 592–596 CrossRef CAS PubMed.
- J. C. de Paula, W. F. Beck, A.-F. Miller, R. B. Wilson and G. W. Brudvig, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 3635–3651 RSC.
- H. Mino and H. Nagashima, J. Phys. Chem. B, 2020, 124, 128–133 CrossRef CAS PubMed.
- P. Geijer, F. Morvaridi and S. Styring, Biochemistry, 2001, 40, 10881–10891 CrossRef CAS PubMed.
- W. Hillier, I. Mcconnell, S. Singh, R. Debus, A. Boussac and T. Wydrzynski,Photosynthesis. Energy from the Sun: 14th International Congress on Photosynthesis, Springer, Dordrecht, 2008, pp. 427–430 Search PubMed.
- W. Ames, D. A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz and F. Neese, J. Am. Chem. Soc., 2011, 133, 19743–19757 CrossRef CAS PubMed.
- J. W. Murray and J. Barber, J. Struct. Biol., 2007, 159, 228–237 CrossRef CAS PubMed.
- F. M. Ho and S. Styring, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 140–153 CrossRef CAS PubMed.
- F. M. Ho, Photosynth. Res., 2008, 98, 503–522 CrossRef CAS PubMed.
- J. W. Murray, K. Maghlaoui, J. Kargul, M. Sugiura and J. Barber, Photosynth. Res., 2008, 98, 523–527 CrossRef CAS PubMed.
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1671–1678 CrossRef CAS PubMed.
- M. Shoji, H. Isobe, S. Yamanaka, Y. Umena, K. Kawakami, N. Kamiya, J.-R. Shen, T. Nakajima and K. Yamaguchi, Adv. Quantum Chem., 2015, 70, 325–413 CrossRef CAS.
- M. Retegan and D. A. Pantazis, Chem. Sci., 2016, 7, 6463–6476 RSC.
- M. Retegan and D. A. Pantazis, J. Am. Chem. Soc., 2017, 139, 14340–14343 CrossRef CAS PubMed.
- M. Kusunoki, J. Photochem. Photobiol., B, 2011, 104, 100–110 CrossRef CAS PubMed.
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1148–1155 CrossRef CAS PubMed.
- G. Han, Y. Huang, F. H. M. Koua, J.-R. Shen, P.-O. Westlund and J. Messinger, Phys. Chem. Chem. Phys., 2014, 16, 11924 RSC.
- P. E. M. Siegbahn, J. Photochem. Photobiol., B, 2011, 104, 94–99 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- F. Rappaport, M. Blanchard-Desce and J. Lavergne, Biochim. Biophys. Acta, Bioenerg., 1994, 1184, 178–192 CrossRef CAS.
- M. Haumann, P. Liebisch, C. Müller, M. Barra, M. Grabolle and H. Dau, Science, 2005, 310, 1019 CrossRef CAS PubMed.
- C. J. Kim and R. J. Debus, Biochemistry, 2017, 56, 2558–2570 CrossRef CAS PubMed.
- H. Sakamoto, T. Shimizu, R. Nagao and T. Noguchi, J. Am. Chem. Soc., 2017, 139, 2022–2029 CrossRef CAS PubMed.
- P. H. Oyala, T. A. Stich, R. J. Debus and R. D. Britt, J. Am. Chem. Soc., 2015, 137, 8829–8837 CrossRef CAS PubMed.
- V. L. Pecoraro, M. J. Baldwin, M. T. Caudle, W.-Y. Hsieh and N. A. Law, Pure Appl. Chem., 1998, 70, 925–929 CAS.
- E. M. Siegbahn and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 10 CrossRef.
- J. S. Vrettos, J. Limburg and G. W. Brudvig, Biochim. Biophys. Acta, Bioenerg., 2001, 1503, 229–245 CrossRef CAS.
- J. Barber, Q. Rev. Biophys., 2016, 49, 1–21 CrossRef PubMed.
| This journal is © the Owner Societies 2020 |