
Signature of a conical intersection in the dissociative photoionization of formaldehyde†

Abstract
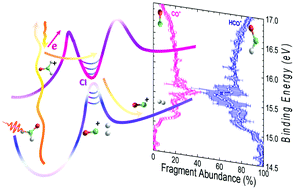
The valence-shell photoionization of formaldehyde is investigated by means of combining Photo-Electron Photo-Ion COincidence (PEPICO) experiments and ab initio calculations. The formation of three ion fragments: HCO+, CO+ and H+2, via dissociative photoionization following excitation at 17 eV is discussed. The experimental results consisting of electron–ion kinetic energy correlation diagrams for the corresponding coincident events, i.e. (HCO+, e−), (CO+, e−) and (H+2, e−), as well as the fragment abundance as a function of the binding energy, are complemented by high level electronic structure calculations including potential energy curves and on-the-fly trajectories. The results are consistent with a main relaxation process via internal conversion into rovibrationally excited levels of the H2CO+ ground state, followed by statistical dissociation, preferentially into HCO+. The analysis of the experimental results reveals nevertheless the signature of a conical intersection controlling the dynamics and favoring dissociation into the molecular channel, CO+ + H2. In addition, the minor formation of the H+2 ion is suggested to occur through a roaming pathway on the cation excited state.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

