
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando potassium K-edge X-ray absorption spectroscopy: investigating potassium catalysts during soot oxidation†
Catherine J.
Davies
a,
Alexander
Mayer
b,
Jess
Gabb
a,
Jake M.
Walls
 b,
Volkan
Degirmenci
c,
Paul B. J.
Thompson
de,
Giannantonio
Cibin
f,
Stan
Golunski
a and
Simon A.
Kondrat
*b
b,
Volkan
Degirmenci
c,
Paul B. J.
Thompson
de,
Giannantonio
Cibin
f,
Stan
Golunski
a and
Simon A.
Kondrat
*b
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
bDepartment of Chemistry, Loughborough University, Loughborough, Leicestershire LE113TU, UK. E-mail: s.kondrat@lboro.ac.uk
cThe School of Engineering, University of Warwick, Coventry, CV4 7AL, UK
dXMaS, UK CRG, ESRF, 71 Avenue des Martyrs, 38043 Grenoble, France
eDepartment of Physics, University of Liverpool, Oliver Lodge Laboratory, Liverpool L69 7ZE, UK
fDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK
First published on 2nd July 2020
Abstract
The chemical and structural nature of potassium compounds involved in catalytic soot oxidation have been studied by a combination of temperature programmed oxidation and operando potassium K-edge X-ray absorption spectroscopy experiments. These experiments are the first known operando studies using tender X-rays (∼3.6 keV) under high temperature oxidation reaction conditions. X-ray absorption near edge structure analysis of K2CO3/Al2O3 catalysts during heating shows that, at temperatures between 100 and 200 °C, potassium species undergo a structural change from an initial hydrated K2CO3·xH2O and KHCO3 mixture to well-defined K2CO3. As the catalyst is heated from 200 °C to 600 °C, a feature associated with multiple scattering shifts to lower energy, indicating increased K2CO3 dispersion, due to its mobility at high reaction temperature. This shift was noted to be greater in samples containing soot than in control experiments without soot and can be attributed to enhanced mobility of the K2CO3, due to the interaction between soot and potassium species. No potassium species except K2CO3 could be defined during reactions, which excludes a potential reaction mechanism in which carbonate ions are the active soot-oxidising species. Simulations of K-edge absorption near edge structures were performed to rationalise the observed changes seen. Findings showed that cluster size, unit cell distortions and variation in the distribution of potassium crystallographic sites influenced the simulated spectra of K2CO3. While further simulation studies are required for a more complete understanding, the current results support the hypothesis that changes in the local structure on dispersion can influence the observed spectra. Ex situ characterisation was carried out on the fresh and used catalyst, by X-ray diffraction and X-ray photoelectron spectroscopy, which indicated changes to the carbonate species, in line with the X-ray absorption spectroscopy experiments.
Introduction
Alkali metal compounds, such as potassium, have a long-established role in heterogeneous catalysis as promotors1–6 or act as catalytic poisons.7–10 These properties are attributed to different alkali-adsorbate interactions; such as (1) the interaction of an alkali-surface electrostatic field and the adsorbate,11 (2) long range interactions mediated by surface electrons12 or (3) the direct orbital overlap between the alkali cation and the adsorbate to provide a lower transition state for a given reaction.13–15 In some reactions, such as the gasification and oxidation of elemental carbon,16–18 alkali compounds alone show reactivity. In addition to the complexity of molecular level mechanisms, other considerations around the role of alkali compounds should be considered, such as their local structure, stability and surface mobility.Many mechanistic studies of alkali species in catalysis have used ultra-high vacuum (UHV) experiments, with single crystal catalyst models.10,12,14,19 These informative studies have helped address fundamental surface science discussed above. Yet due to the much-discussed pressure and materials gaps, alternative experiments using working catalysts require alternative approaches. X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) of prior and post reaction powdered catalysts, more closely related to applied catalytic materials have been reported.20–23
Operando spectroscopy, which is the spectroscopic study of a catalyst while simultaneously measuring its catalytic performance, could provide an opportunity to observe alkali compound speciation and dispersion during a catalytic reaction. However, this is not without significant technical challenge. Alkali compounds are usually present as a low weight fraction of poorly crystalline surface species on a matrix of crystalline metals and metal oxides, making specific observations of their properties challenging when using many characterisation techniques. XAS at the K-edge energies of alkali promotors allows for direct measurement of these compounds’ local structure.
With the aim of understanding alkali species during a particularly challenging catalytic reaction, under realistic conditions, we carried out potassium K-edge XAS of a 10 wt% K/α-Al2O3 catalyst performing soot oxidation. To our knowledge, this is the first attempt of operando potassium K-edge XAS study reported. There exists only two other in situ studies reported to date: (1) the potassium promoted cobalt Fischer–Tropsch catalysts reported by Huffman et al.,24 and (2) the evolution of calcium or potassium species during the pyrolysis of lignite chars by Huggins et al.25 The tender energy of the potassium K-edge makes operando experiments, where reactant and gases are contained within a reaction vessel, viable. Also, potassium species are active for soot oxidation in the absence of other catalytic species, such as Pt, enabling a simple supported potassium catalyst to be used. Further, the oxidation of soot is a well-researched topic with significant application in the automotive industry. In particular, the passive regeneration of diesel particulate filters, by combustion of the soot, requires the porous ceramic filter material to be coated in a catalyst to lower combustion temperatures to within the range of engine exhaust-gas temperatures.26 Several different catalyst formulations including noble metals and various metal oxides have been reported, with the addition of alkali promotors substantially lowering the onset temperature for soot combustion.26–31
These alkali compounds are hypothesised to form high surface area oxides and superoxide species, which are key intermediates in the oxidation reaction.32–34 The electro-positivity of alkali metal and alkaline earth compounds correlates with lowering of reaction temperatures, suggesting that electron donation from the alkali weakens C–C bonds.35 In an alternative mechanism (which owes much to the routes proposed for carbon gasification), alkali-metals are assumed to form carbonates as the thermodynamically stable species under exhaust gas conditions.34,36 If elemental carbon comes into contact with an alkali metal carbonate, it can reduce the carbonate to the metal oxide and/or superoxide (e.g. as shown for potassium in eqn (1)), which in turn can transform back to the carbonate by reaction with CO2 from the exhaust gas (eqn (2)). In a fuel-lean exhaust gas (as produced by a diesel engine), the CO formed in eqn (1) can immediately be catalytically oxidised to CO2.
| Soot oxidation: K2CO3 + C → K2O + 2CO | (1) |
| Regeneration of active species: K2O + CO2 → K2CO3 | (2) |
A further characteristic found to be important, when alkali metal compounds are used as catalyst promoters, is their mobility on the catalyst surface, which increases the soot-catalyst contact.33,35 While potassium compounds have been reported to be among the most reactive under loose contact conditions, the exact nature of the catalytic species and its dispersion under reaction conditions is not known. Therefore, the reported operando potassium K-edge XAS study is aimed to shed light on the nature and dispersion of potassium species in soot oxidation and, more generally, to present a proof of concept for operando experiments to provide insight into alkali compounds in catalysis. Our findings from these operando experiments have been supported by more conventional application of ex situ XRD and XPS before and after reaction with soot.
Experimental
Catalyst preparation
K2CO3 (0.3538 g, Aldrich, 99.995%) was dissolved in deionised water (5 ml) and heated with stirring in an oil bath at 80 °C. α-Al2O3 (1.80 g, Alfa Aesar, 99.9%) was added, the temperature increased to 110 °C and stirred for 30 min until a thick paste was obtained. The paste was dried overnight (16 h) at 110 °C prior to calcination in static air for 5 h at 500 °C, heated at a ramp rate of 10 °C/min. The catalyst is referred to as K/α-Al2O3 throughout.Catalyst testing
Soot oxidation light off temperature was tested using thermal-gravimetric analysis (TGA) on a Setaram TGA-DTA/DSC instrument. Catalyst was mixed in ‘loose contact’ (to mimic the degree of catalyst-soot contact in a catalytic soot filter as it begins to trap soot particulate from an exhaust gas26) with carbon black (which has been determined to be a representative model of the elemental carbon component of diesel soot26), by stirring together in a vial for approx. 1 min, in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 catalyst to soot ratio by mass. The mixture was heated to 900 °C at a rate of 5 °C min−1 in an air atmosphere.
:1 catalyst to soot ratio by mass. The mixture was heated to 900 °C at a rate of 5 °C min−1 in an air atmosphere.
Characterisation
XRD was carried out on a Panalytical X’Pert Pro diffractometer with Ni-filtered Cu Kα radiation, at 40 mA and 40 kV, with 20° < 2θ < 80°. The phases present were confirmed by matching patterns to the ICSD. XPS was carried out using a Kratos AXIS ULTRA-DLD spectrometer, using monochromatic Al-Kα radiation (photon energy = 1486.6 eV), under ultra-high vacuum. The data obtained was achieved at a pass energy of 40 eV, and the analysis of the data from the scans was carried out in CasaXPS™ software, with all peaks calibrated to the C(1s) which has a binding energy of 284.7 eV.X-ray Absorption Near Edge Structure (XANES) experiments were performed on beamline BM28 (XMaS) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Data were collected at the Potassium K-Edge in fluorescence mode both in operando and ex situ measurements using silicon drift diode detectors. Ex situ samples were adhered to carbon tape, while operando measurements were carried out using a custom-made stainless-steel controlled atmosphere cell (shown in Scheme S1, ESI†). The cell was heated with two fire rods controlled by a temperature controller. A thermocouple was placed close to the catalyst sample. Typically, a predetermined amount of finely ground sample was pressed in a stainless-steel holder and placed in the cell. Carbon foils were held between two high-purity carbon spacers. Gases were delivered by thermal mass flow controllers (Bronkhorst) and the outlet line was connected to a Hiden QGA mass spectrometer. The operando experiments were designed to reproduce as closely as possible the protocol (above) used for catalyst testing, which in turn was intended to mimic the catalyst–soot interactions in a catalytic filter. Mixtures of 10:1 catalyst to soot (by mass) were loosely mixed and pressed (with minimum compression) into a coherent pellet and placed into the cell. Given that the proposed experiment was to study soot combustion, there was a clear concern that the graphite windows would also combust. However, physical separation of the catalyst/soot pellet, coupled with the intimate contact of the catalyst and reactant, provided a sufficient operating temperature window (up to 600 °C) to observe soot oxidation. A 15 ml min−1 gas flow of He (99.99%, Air Liquide) or 20% O2 balanced with He (Air Liquide) was used to purge the reaction cell and the outlet was continuously monitored by mass spectrometer. The sample temperature was then stabilised at 30 °C for 20 min before being heated at 2 °C min−1 to 600 °C. XANES measurements were obtained sequentially throughout the heating ramp, with the temperature being recorded. Confidence of measurement errors were estimated to be ±0.1 eV of reported positions.
XANES simulation
XANES simulations were performed with FEFF9.05 code37,38 starting from cif structures (ICSD Collection Code 66943 and ICSD Collection Code 2074). Simulations were run on the K K edge using self-consistent field (SCF) molecular electron densities calculated over a cluster including 31 atoms. The Hedin–Lundqvist self-energy has been used for calculation of the exchange correlation potential with a correction to K1 and K2 of −2.269 and −1.826 eV on the Fermi energy. The full multiple scattering contributions have been calculated using a sphere including 6 to 223 atoms. The energy resolution was set to 0.3 eV near the edge and 0.05 Å−1 in the near EXAFS region up to 5 Å−1. S02 amplitude has been fixed to 1. When comparing simulations to experimental data the energy was shifted by 4 eV in alignment with feature A of the experimental data.Results and discussion
Catalyst testing and ex situ characterisation
Prior to testing within the operando XAFS cell, the catalytic performance of K/α-Al2O3 was studied by performing soot oxidation via TGA and characterised using ex situ XRD and XPS. Fig. 1 shows the derivative of the mass loss (dTG) for soot alone, soot mixed with α-Al2O3 and soot mixed with K/α-Al2O3. In the absence of any additional material, the combustion of soot began at 500 °C and, following a slow induction, gave rise to a sharp non-symmetrical peak with a maximum at 625 °C. The non-symmetrical peak shape closely resembles that observed during combustion of real diesel soot,26 though the position of the peak in Fig. 1 is about 30 °C lower. When mixed with α-Al2O3, the position of the soot combustion peak was shifted to a slightly higher temperature, so that it now reached its maximum at 640 °C. This is consistent with the α-Al2O3 having a diluting effect, which inhibits heat transfer between the carbon particles in the soot sample. However, when soot was mixed with K/α-Al2O3, the profile of the combustion peak changed markedly and was shifted to lower temperature, confirming current literature which shows that the presence of a potassium compound provides oxidation pathways with lower activation energy.33,35 A multiple peak in the dTG of K/α-Al2O3 and soot suggests that several soot oxidation processes may be taking place via different mechanisms. Comparison of TGA profiles of K/α-Al2O3 in the presence and absence of soot (Fig. 1b) showed that mass losses between 325 and 700 °C are exclusively associated with soot and not catalyst decomposition. Therefore, the onset of the first soot oxidation event can be shown to occur at ca. 325–350 °C. Such low onset temperatures for soot oxidation are in line with those previously reported by Matarrese et al. of 300–350 °C.33 These initial soot oxidation tests confirmed, therefore, that this K/α-Al2O3 formulation has potential for application in a catalytic soot filter for the passive control of particulate on diesel passenger cars, where the exhaust-gas temperature rarely exceeds 350 °C. The main oxidation process for K/α-Al2O3 occurred at 540 °C, 100 °C lower than for α-Al2O3. An indication of the rate of reaction can be determined from the magnitude of dTG peaks, with the maximum rate of soot decomposition between α-Al2O3 and main feature in K/α-Al2O3 being comparable, despite the difference in reaction temperature. TGA analysis also showed a mass loss event starting at 700–750 °C in the catalyst-containing sample, contributing to a loss of 2.5% of sample mass, which could be attributed to either the volatilisation of a potassium compound or its decomposition. TGA/DTA analysis of bulk K2CO3 shows that it decomposes at 900 °C, the maximum temperature of our experiment, suggesting that the observed mass loss from 750 °C is predominantly due to volatilisation.39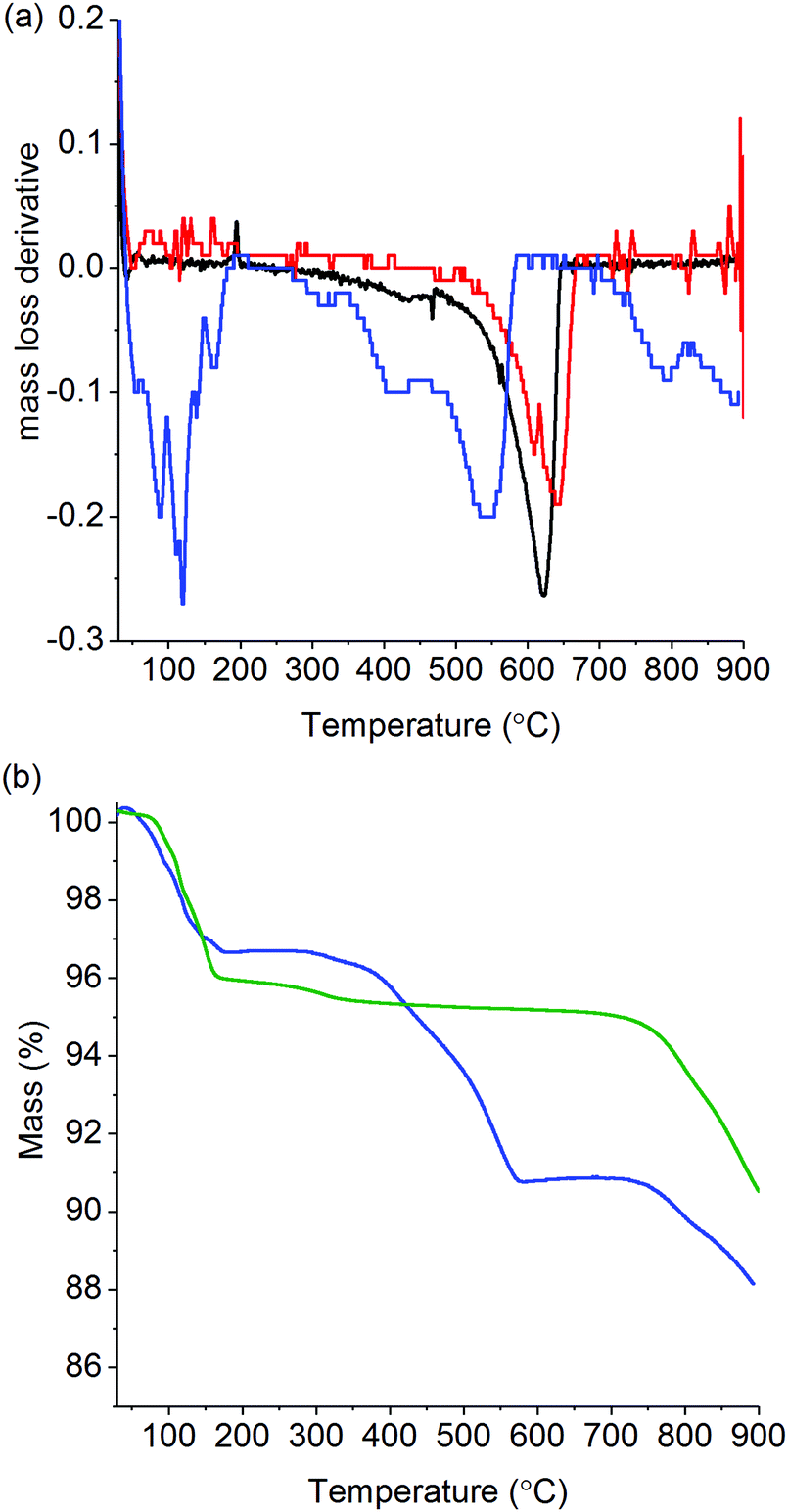 | ||
| Fig. 1 Thermogravimetric analysis of soot oxidation. Experiments performed under an air atmosphere with a ramp rate of 5 °C min−1. (a) Derivative of mass losses of soot (black line), α-Al2O3 + soot (red line) and K/α-Al2O3 + soot (blue line). (b) Thermogravimetric profile of K/α-Al2O3 + soot decomposition (blue line) and K/α-Al2O3 (green line). | ||
XRD patterns of α-Al2O3 and K/α-Al2O3, both before and after soot oxidation (Fig. S1, ESI†) show reflections associated with α-Al2O3. Fresh K/α-Al2O3 had additional reflections, attributed to KHCO3 and hydrated K2CO3·xH2O, which are no longer present after reaction. A series of broad ill-defined reflections that could not be attributed to any anticipated crystalline potassium carbonate or hydroxide phases were present post reaction. Potentially, these reflections could be associated with trace formation of potassium aluminate phases and K β-Al2O3 which have been shown to form between 800–1000 °C.40 Although potassium aluminate phases are reported to be unstable in the presence of atmospheric moisture and phase separate within days.41 The loss of crystalline K2CO3 could be due a range of single or multiple causes; namely, the decomposition of the carbonate to an alternative poorly crystalline compound, the formation of K β-Al2O3, dispersion of the K2CO3 across the Al2O3 surface to form non-crystalline surface species or sufficient volatilisation to remove potassium species from the catalyst entirely.
XPS of K/α-Al2O3 and α-Al2O3 both before and after use for soot oxidation can help determine if the potassium species has changed during reaction and/or is being lost from the Al2O3 surface. Quantification of surface composition as determined by XPS (Table 1) showed that for both materials there are signals associated with K, Al, C and O in addition to Na+ impurity in the support. After reaction, the Na+ surface concentration increased due to the migration from the bulk structure of the support to the surface. On bare α-Al2O3 the presence of carbon is due to adventitious surface adsorbed species. After reaction with soot, slight increases in Al and O concentrations and a decrease in carbon indicate that some of this adventitious carbon is lost during the reaction. In K/α-Al2O3 the Al surface concentration decreases by 50% after reaction with soot, along with a small decrease in O, a corresponding increase in C and a 85% increase in the surface K concentration. These changes in surface concentration support the theory that a mobile potassium species has become dispersed over the Al2O3 during reaction, accounting for the decrease in Al and increase in K. It further suggests that the formation of bulk potassium aluminate phases has not occurred to a significant extent, as a drop in surface potassium concentration would be expected, through its migration into the sample bulk. While, in the case of γ-Al2O3 surface potassium species can migrate deep into the pore network, using α-Al2O3 ensures that this migration can be discounted.
| Sample | Concentration based on XPS peak area (at%) | ||||
|---|---|---|---|---|---|
| Na (1s) | O (1s) | C (1s) | Al (2s) | K (2p3/2) | |
| K/α-Al2O3 | 0.1 | 58.1 | 20.4 | 27.4 | 4.0 |
| K/α-Al2O3 after soot oxidation | 0.1 | 46.1 | 33.3 | 13.0 | 7.4 |
| α-Al2O3 | 0.1 | 42.2 | 33.1 | 24.7 | — |
| α-Al2O3 after soot oxidation | 2.5 | 45.3 | 27.1 | 25.1 | — |
Spectra from the K2p/C1s region of K/α-Al2O3, before and after soot oxidation, are shown in Fig. S2 (ESI†). The positions of the K2p1/2 and K2p3/2 binding energies are comparable in both samples, at 292.5/292.7 eV and 295.2/295.5 eV, and match values reported in the literature, which was expected given that it is known to be insensitive to chemical environment.42 The C1s spectra of both samples were fitted with 3 peaks, 2 associated with adventitious carbon at 287.6 and 285.2 eV and a peak at ca. 289 eV attributed to a carbonate anion. Indeed, the C1s associated with carbonate in the fresh K/α-Al2O3 at 288.6 eV is comparable with that observed by Kantschewa et al. for K2CO3/γAl2O3 at a binding energy of 289.6 eV (when normalised to C1s).42 After reaction with soot, a notable shift in this C1s binding energy to 289.4 eV was seen, which while still within the range anticipated for a carbonate species, indicates that the compound is not bulk K2CO3. The shift could, speculatively, be interpreted as being due to a reduction in particle size of K2CO3 on dispersion over the α-Al2O3.
The O1s spectra for K/α-Al2O3 before and after use are shown in Fig. S3 (ESI†). As noted previously by Kantschewa et al. the feature must comprise of multiple components, including O associated with carbonate, surface hydroxyl species and surface O2− from Al2O3.42 The authors attributed a O1s binding energy of 532 eV to pure K2CO3, 532.2 eV to surface hydroxyl groups and 530.3 eV to lattice oxygen in γ-Al2O3. Interestingly, the unfitted O1s spectra of K/α-Al2O3 shifted from 531.5 eV to a higher binding energy of 521.5 eV after reaction with soot, strongly indicating that the surface coverage of carbonate, relative to lattice oxygen has increased significantly.
In summary, findings from TGA, ex situ XRD and XPS provide initial understanding of the structural changes of potassium speciation before and after reaction. Crystalline and poorly dispersed K2CO3·xH2O and KHCO3 are present on the fresh catalyst. Given that KHCO3 decomposes at ca. 150 °C it cannot have been within the catalyst directly on preparation, as the material was calcined to 500 °C, but must have formed by exposure to atmospheric CO2 and H2O during storage of the catalyst. After reaction with soot, the surface concentration of potassium increased dramatically and can be attributed to being predominantly associated with a surface distributed potassium carbonate. TGA analysis also indicated that at above 750 °C potassium carbonate decomposition or volatilisation began. Clearly, the reactive potassium species are highly mobile at soot decomposition temperatures and aid soot-catalyst contact to the benefit of catalytic performance. The findings cannot, however, identify intermediate species present during the reaction or provide the detail required to correlate structural changes directly with catalytic properties. The sensitivity of alkali metal compounds to moisture and CO2 further suggest that species present ex situ may be quite different to those under reaction conditions. Hence, using these ex situ findings as a guide, an operando potassium K-edge XANES of K/α-Al2O3 during soot oxidation was performed.
Operando potassium K-edge X-ray absorption spectroscopy
Fig. 2 shows the potassium K-edge XANES spectra of K/α-Al2O3 at room temperature alongside potassium standards of carbonate compounds (K2CO3 and KCHO3), a carboxylic acid (KCH3CO2), the superoxide KO2 and a hydroxide compound (KOH·xH2O). Sugiura and Muramatsu provide one of the few XANES studies of an extensive range of potassium compounds reported in the literature.43 The authors, in conjunction with the work of Dutta and co-workers,44 assign potassium K-edge XANES features as follows; pre-edge features assigned to K+ 1s → 3d and/or 1s → 4s forbidden transitions (denoted bands Ai and A respectively), the predominant 1s → 4p main transition (denoted band B), which can split into two features under the influence of a crystal ligand field (denoted band Bi), and higher energy bands associated with multiple scattering processes (band C). The first principles understanding, and assignment of these bands, is limited and based on atomic approaches, where the assumption is made that as potassium is highly ionic, ligand coordination can be treated by ligand fields. Despite this, the XANES of K/α-Al2O3, when simply compared with the potassium standards, most visibly matched that of K2CO3 and KHCO, in broad agreement with ex situ XRD and XPS data.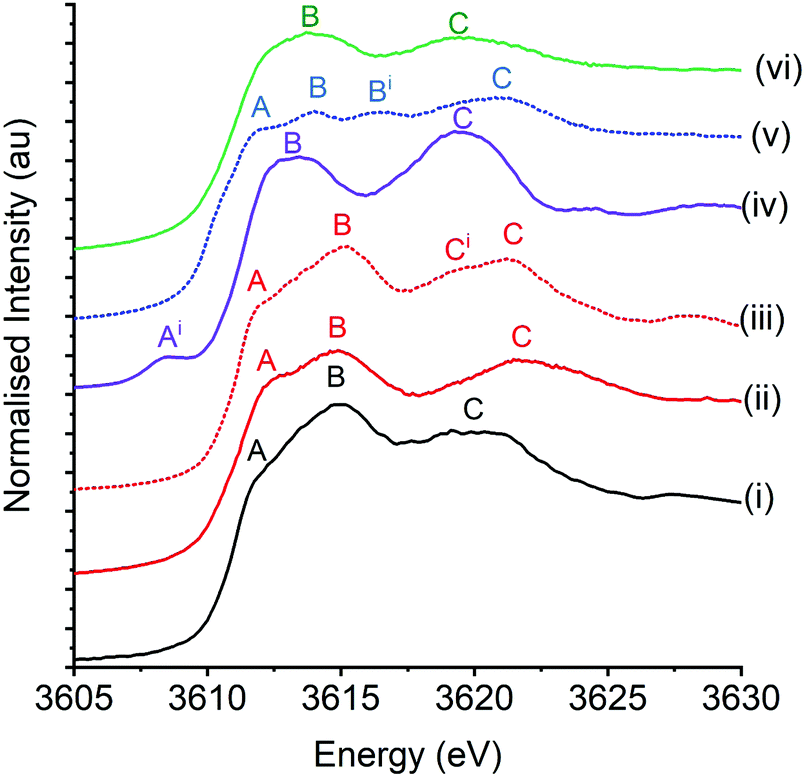 | ||
| Fig. 2 Potassium K-edge XANES of K/α-Al2O3 and standards. (i) K/α-Al2O3 (ii) K2CO3, (iii) KHCO3, (iv) K2O, (v) K(CH3CO2) and (vi) KOH·xH2O. Features; Ai is 1s → 3d, A is 1s → 4s, B and Bi is 1s → 4d and C/Ci is multiple scattering resonances. | ||
The energy positions of the XANES features of K/α-Al2O3 with the K2CO3 and KHCO3 standards are shown in Table 2. The positioning in each material of features A and B were similar, with the catalyst's B feature being in between the 0.4 eV difference seen for the two standards. However, the multiple scattering resonance band C was notably different between samples. The broad feature seen in K/α-Al2O3, which has been assigned as a single feature as opposed to two overlapping features, was 2.1 eV lower in energy compared with K2CO3. Jacobs et al. reported shifts in the position of multiple scattering resonance features of K2CO3 dispersed on SiO2 compared to their bulk structures.20 The feature being sharper and at a higher energy in bulk compounds, compared to that seen in potassium compounds with reduced long-range order that are on a support. Therefore the 2.1 eV decrease in position C seen in K/α-Al2O3 could be attributed to reduced long-range order of dispersed K2CO3, relative to bulk K2CO3. However, the conclusion that K2CO3 is simply dispersed on the Al2O3 surface is complicated by the clear evidence of crystalline KHCO3 and K2CO3·xH2O in fresh K/α-Al2O3 from XRD. Specifically, this shows that a significant proportion of potassium species are in crystalline bulk state. Comparison between the XANES of K/α-Al2O3 and KHCO3 provides better agreement regarding the position of multiple scattering features, although the band is broader and at slightly lower energy in K/α-Al2O3. Therefore, it is suggested that a mixture of KHCO3 and domains and K2CO3·xH2O account for the perceived lack of long-range order without it being highly dispersed over the α-Al2O3 surface. Changes in the local RuO2 rutile structure on hydration have previously been seen in Ru K-edge XANES with a similar splitting of a broad feature into 2 clear peaks on dehydration.45 The observation is of clear importance when considering future potassium XANES experiments. The hygroscopic and reactive nature of potassium compounds under atmospheric conditions can result in the formation of alternative structures and can be confused or conflated with particle dispersion, if not combined with other characterisation techniques.
| Sample | Band A (1s →3d) | Band B (1s → 4p) | Band C (1s → CR) |
|---|---|---|---|
| a Feature possibly two overlapping bands as seen in KHCO3. However, accurate position of each feature could not be determined and was treated as a single feature. | |||
| K2CO3 | 3612.4 (shoulder) | 3614.8 | 3622.0 |
| KHCO3 | 3612.1(shoulder) | 3615.2 | 3619.8 + 3621.2 |
| K/α-Al2O3 | 3612.4 (shoulder) | 3615.0 | 3619.9a |
In the first operando experiment K/α-Al2O3 was heated in synthetic air (20% O2 in He) in the absence of soot as a control experiment, that showed observable changes in the position of XANES features on heating to 600 °C (Fig. 3). The relatively poor signal to noise observed in these operando experiments demonstrates the challenge of the low energy X-rays measurements through the reactor windows. Yet, while an improvement in signal to noise would be desirable, the peak positions and XANES features are clearly discernible. No new features were observed in the XANES but splitting of the B and C bands was observed. Changes in peak positions were confirmed from the 2nd derivatives of the data, which show that band A remained constant throughout experiments, while B and C shifted (Fig. 3b). The position of band B was found to shift from 3614.9 eV to 3614.1 eV on heating from room temperature to 250 °C and thereafter decreased marginally by a total of 0.2 eV between 250 and 600 °C (Fig. 3c). Band C position was found to initially increase from 3619.9 eV to a maximum of 3621.5 eV, on heating to 160 °C. Further heating from 160 °C to 600 °C resulted in the multiple scattering peak slowly reducing in energy to 3620.4 eV.
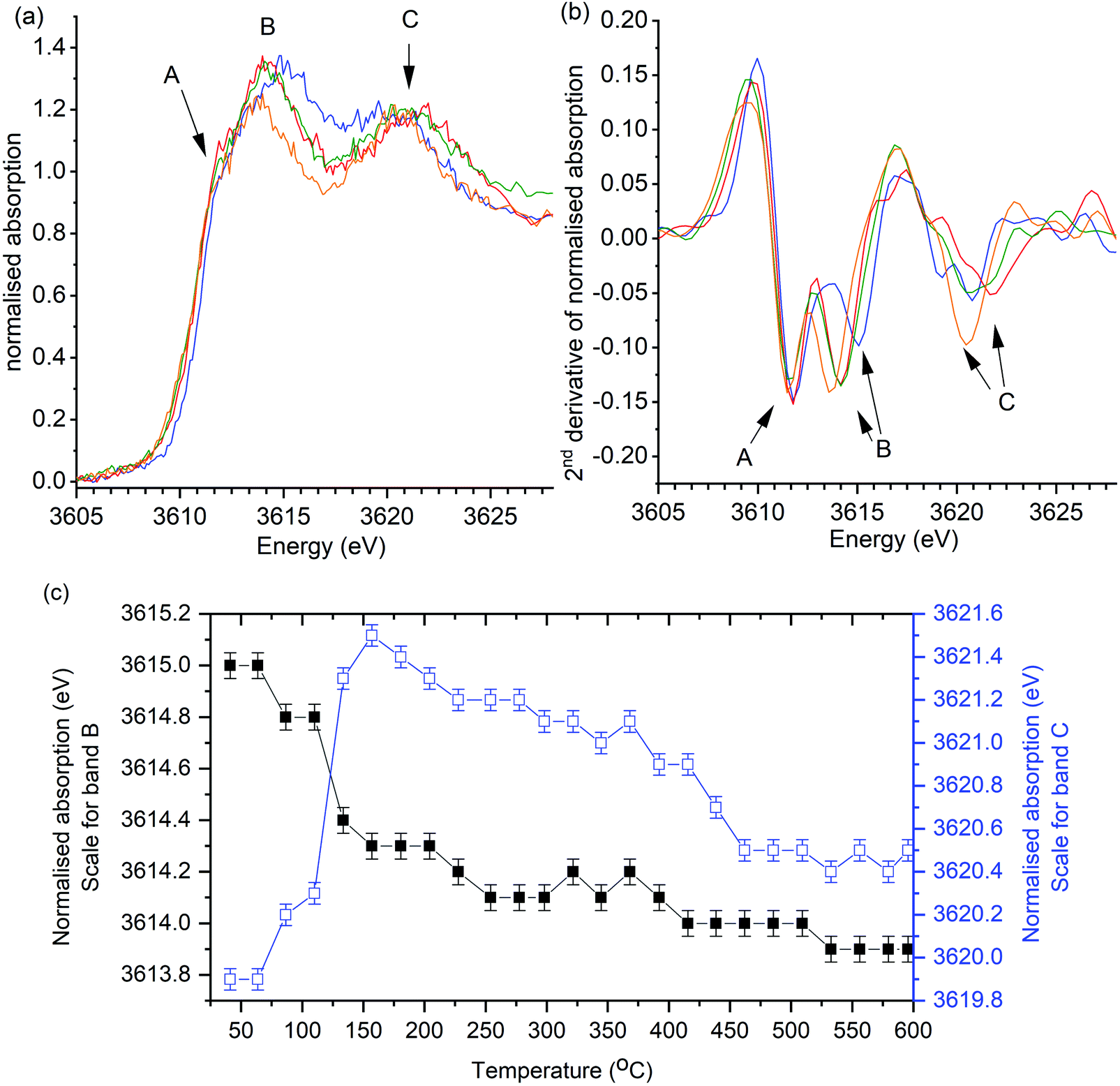 | ||
| Fig. 3 Control operando potassium K-edge XANES of K/α-Al2O3 during heating to 600 °C in the absence of soot. Heating rate of 2 °C min−1 (a) Selection of normalised XANES data showing key changes in catalyst structure during reaction. (b) 2nd derivative of data processed XANES spectra shown in (a). XANES scan at; 40 °C (blue), 160 °C (red), 300 °C (green) and 600 °C (orange). Fig. 6c shows recorded energy of features B (1s → 4d) and C (multiple scattering feature) at all recorded temperatures. Error of ±0.1 eV determined as confidence in measuring peak maximum. | ||
The same general trend in the progression of band positions was observed in the operando XANES analysis of K/α-Al2O3 during soot combustion (Fig. 4). In the presence of soot, the shifting of band B was found to be comparable to heating in the absence of soot, with a significant decrease from 3615.0 eV to 3614.2 eV on heating to 150 °C, followed by a relatively stable energy position thereafter. The progression of band C also followed the same trend as in the control experiment, although the decrease in band position on heating above 220 °C in the presence of soot was more rapid (Fig. 4c), with a sudden 0.5 eV drop relative to the slow decline in energy seen in the control experiment.
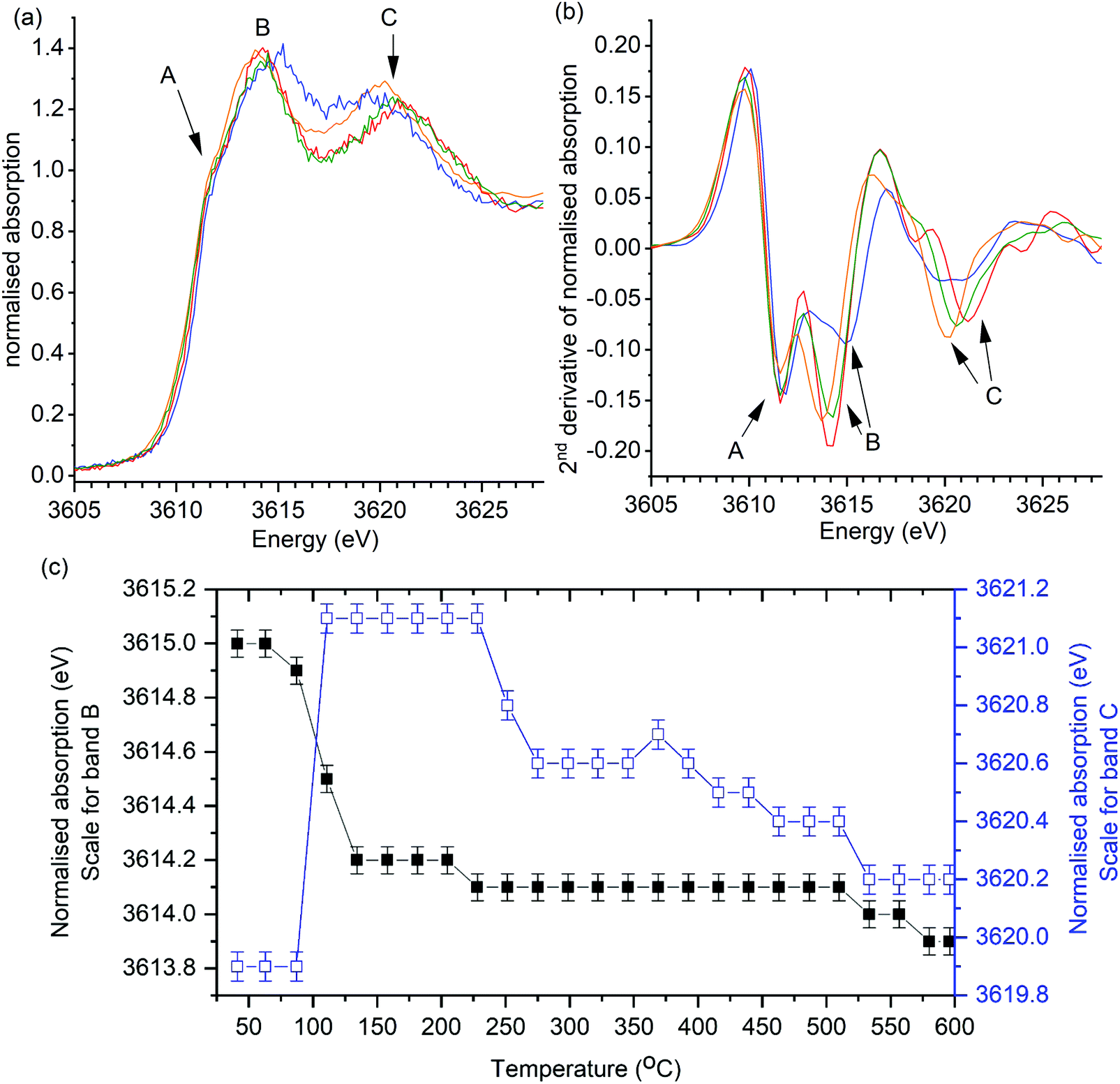 | ||
| Fig. 4 Operando potassium K-edge XANES of K/α-Al2O3 during temperature programmed soot oxidation. Catalyst: soot ratio of 10:1. Heating rate of 2 °C min−1. (a) Selection of normalised XANES data showing key changes in catalyst structure during reaction. (b) 2nd derivative of data processed XANES spectra shown in (a). XANES scan at; 40 °C (blue), 160 °C (red), 300 °C (green) and 600 °C (orange). Fig. 7c shows recorded energy of features B (1s → 4d) and C (multiple scattering feature) at all recorded temperatures. Error of ±0.1 eV determined as confidence in measuring peak maximum. | ||
Regarding correlation between potassium XANES features and structural changes of a sample during a catalytic reaction, there are only three published papers to aid our interpretation of the data. Jacobs et al. studied potassium promoted Fischer–Tropsch catalysts ex situ, while Huffman et al. reported an in situ study of potassium XANES at 200 °C under different H2/CO/CO2 gas compositions and the decomposition of potassium containing lignite chars.20,24,25 A further complication is that these studies provide alternative hypothesis for observed changes in XANES features. As discussed above, changes in band C position have been attributed by Jacobs et al. as being due to changes in the long-range order of a compound (K2CO3), due to their dispersion on support structures.20 Alternatively, Huffman/Huggins et al. associate changes in band C position as being due to evolution of different potassium compounds.24 Variation in band B, associated with the 1s → 4p, is not discussed, with Jacobs et al. stating it remains fixed due to the simple nature of the electronic transition. However, the variations in position of features associated with the 1s → 4p transition, seen by Hung and Muramatsu's studies of potassium compounds and the standards reported in this study, show that changes in the local environment of K+ do influence band B energy in the range of 2 eV.
Both effects discussed in the literature can be considered during the reaction of K/α-Al2O3 with soot and the control experiment; i.e., a change in the potassium compound during reaction, or a change in local structure of a given compound through dispersion/aggregation or crystallisation. Given there are no clear additional features associated with splitting of the B band or pre-edge 1s → 3d features, the shifting of features B and C are difficult to assign to definitive changes in ligand environment around K+. Specifically, the absence of the distinctive 1s → 3d seen in KO2 (band A1 in Fig. 2) suggest this proposed reactive species was not present to a significant extent during soot oxidation. Given the high loading of potassium within the catalyst and the transient nature of reactive species like KO2 the lack of characteristic XANES features for this species does not exclude its existence entirely but shows that it is not present as a stable resting state under reaction conditions. It can be noted that final positions of band B and C in K/α-Al2O3, on heating to 600 °C (3613.9 and 3620.2 eV) are close to that recorded for KOH·xH2O at 3613.9 eV and 3619.7 eV. However, assignment to KOH would not be made with confidence. Firstly, K+ environment in KOH is highly distorted with a significant range of K–O distances between 2.69 and 3.15 Å.46 Additionally, it has a propensity to hydrate or react with carbon dioxide. KOH·xH2O is therefore less dependable as a descriptor of K–OH interactions and perhaps more of an indicator of a disordered K+ environment. Further, post reaction XPS showed that carbonate was present within the K/α-Al2O3 sample, suggesting literal assignment of the final XANES species to KOH is dubious. Clearly, the difficulty in assigning the observed XANES changes show the need for detailed XANES modelling to provide a more definitive understanding and assignment of specific structures. Therefore, results of the current study will initially be interpreted more generally as being due to changes in the local structure of K+; predominantly in the form of K2CO3 and KHCO3 as indicated by the similarity of the initial XANES spectra to potassium carbonates and the evidence from post reaction XPS. To measure this structural change, a simplistic indicator parameter, based on the difference in energy between bands B and C (ΔCB) was determined. XANES modelling of KHCO3 and K2CO3 will then be considered to support these initial hypothesised structural changes.
Changes in C–B energy with respect to reaction temperature, for reactions with and without soot, are shown in Fig. 5 alongside the simultaneously recorded mass spectrometry of mass 44 (CO2) and mass 18 (H2O). In the absence of soot, a notable increase in ΔCB from 4.9(2) eV to 7.2(2) eV was seen between 60 and 160 °C, which coincided with evolution of both CO2 and H2O. In the presence of soot, a similar trend was observed but with a slightly smaller increase in ΔCB to 6.9(2) eV. These spectroscopic observations are in agreement with those seen by Huggins et al. which was attributed as the conversion of a dispersed carboxyl-bond potassium species to K2CO3 on heating.25 The evolution of H2O and CO2 gases, monitored in the experiments reported in our paper are strongly suggestive of dehydration of K2CO3·xH2O and KHCO3 decomposition to K2CO3 being reasonable for the notable increase in ΔCB. At 160 °C the ΔCB of 7.2(2) eV and 6.9(2) eV is comparable to the K2CO3 standard (7.1 eV), showing that that an ideal bulk K2CO3 is formed in both cases.
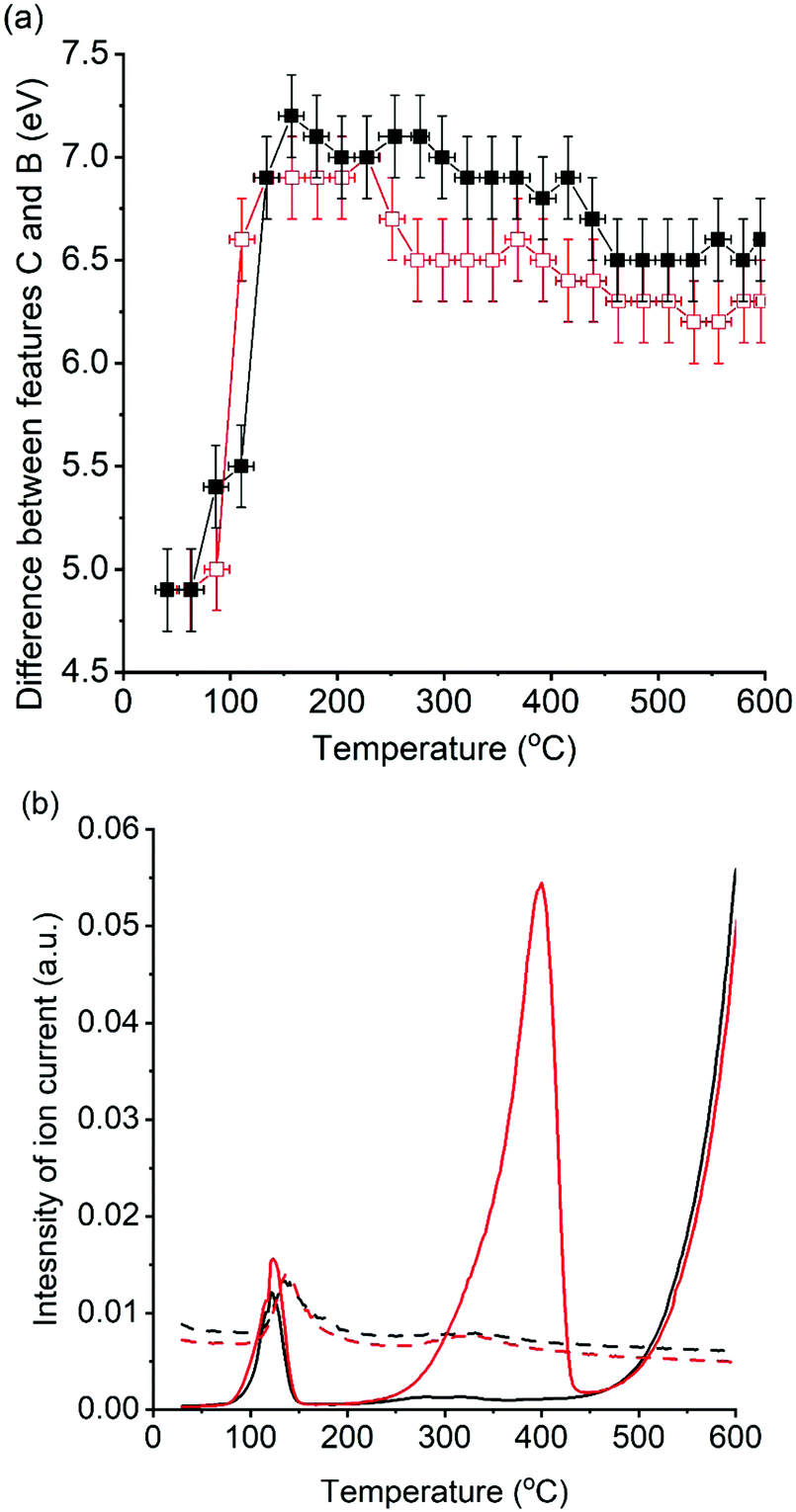 | ||
| Fig. 5 Comparison of changes in potassium XANES features with recorded gas evolution for K/α-Al2O3 during temperature programmed soot oxidation. Control experiment in the absence of soot (black) and during soot oxidation (red). (a) Difference between XANES features C and B with respect to temperature. Estimated confidence error of ±0.2 eV determined from sum of measurement errors for energies of features B and C. (b) Evolved gas analysis of CO2 (solid line) and H2O (dotted line). | ||
In the absence of soot, further heating to 415 °C resulted in a slight decline in ΔCB from 7.2(2) eV to 6.9(2) eV, with low levels of CO2 or H2O being detected by the mass spectrometer. The evolved gas observed during this blank was attributed to the oxidation of surface carbon species, as seen by XPS. This reduction in ΔCB was dominated by a decrease in energy of band C (Fig. 3) and following Jacobs et al. hypothesis is attributed to the dispersion of K2CO3 over the alumina surface, as the potassium species become mobile. In the presence of soot, onset of CO2 and H2O formation began at 250 °C and was attributed to the oxidation of specific soot functional groups in close contact with the catalyst. Interestingly, a decrease in ΔCB, from 7.0(2) eV to 6.5(2) eV, was observed at comparable temperatures (230–270 °C) in the soot containing experiment. While relatively subtle, this change is notably different to the gradual decrease in ΔCB seen in the absence of soot. The significant drop in ΔCB in the presence of soot was repeatable in a duplicate experiment (Fig. S4, ESI†). Between 225 °C and 450 °C ΔCB values were markedly lower in the soot containing experiment and coincided with the temperature range that CO2 evolution was observed (250–440 °C). These findings show that the local structure of potassium changes during the catalytic oxidation of soot. The cause of this structural change with the onset of soot oxidation can be speculatively attributed to; (1) enhanced mobility of K2CO3 facilitated by the local exotherm generated by soot oxidation, i.e. observing the effect of soot combustion on the average structure of K2CO3 in the catalyst. (2) The formation of defects in the K2CO3 lattice caused by the formation of “active” potassium oxide centres, or an increased proportion of disordered active species in interaction with the soot. In either case, there is a small but discernible structure–reactivity relationship observed for the first-time by using operando potassium K-edge XANES.
Heating from 415 °C to 600 °C resulted in a further decrease ΔCB in the control experiment from 6.9(2) eV to a stable value of 6.5(2) eV by 460 °C, indicating further potassium mobility and loss of long-range order through dispersion. In the presence of soot ΔCB remained relatively constant with a slight 0.2 eV drop to a final value of 6.3(2) eV, suggesting that soot combustion had assisted in lower temperature dispersion of the K2CO3, which was then in a pre-dispersed state during further heating. In both experiments CO2 formation was observed from 450 °C. The production of CO2 at these temperatures, in the absence of soot, can be attributed to the initial decomposition of the graphitic windows of the reaction cell. Although there is no direct contact between the K/α-Al2O3 catalyst and the window, potassium species are clearly highly mobile and have facilitated graphite combustion. Given the unstable nature of the reactor cell windows above 600 °C it was not possible to study the high temperature changes seen by TGA at ca. 750 °C. Although, given that soot oxidation was complete by 600 °C, the use of XANES to study the influence of potassium local structure was not compromised. The final ΔCB values are considerably greater than seen in the fresh catalyst, but lower than that of bulk K2CO3 with a value of 7.1 eV, showing that the local structure of potassium species is highly dynamic during heating.
Potassium K-edge XANES simulation: influence of KHCO3 and K2CO3 local structure
XANES spectra of KHCO3 and K2CO3 were simulated using FEFF9 with cluster sizes varying from 6–233 atoms or 6–50 atoms, for K2CO3 and KHCO3 respectively. The potassium environment (Fig. S5, ESI†) for KHCO3 is 7 coordinate to oxygen, while two equally weighted environments of 9 (K1) and 6 (K2) coordinate are seen in K2CO3. Each potassium environment in K2CO3 was studied separately, with the influence of cluster size of the K1 component on the XANES shown in Fig. S6 and of the K2 component in Fig. S7 (ESI†). For both environments at 6 atom cluster sizes the XANES comprised of two broad features at 3615 and 3621–3623 eV, with a third scattering feature observable at 3642 eV. As the cluster sizes were increased the broad feature at 3621–3623 eV split into two features, identified as being the B and C bands discussed in the literature (vide supra). The presence of the A feature also becomes clear as the cluster size is increased. Fig. 6 shows the average XANES spectra of the 1:1 K1:K2 environments found in K2CO3 at cluster sizes representative of these changes, namely 6, 22, 121 and 223 atoms (full cluster size data set shown in Fig. S8, ESI†), alongside the experimental XANES of K2CO3 and the K/α-Al2O3 catalyst. Excellent agreement between the energies of the simulated larger clusters (121 and 223 atoms) and experimentally obtained XANES of K2CO3 was observed (Table S1, ESI†), intensities of features were not well reproduced as commonly seen for XANES modelling, that also does not include structural and thermal disorder effects.
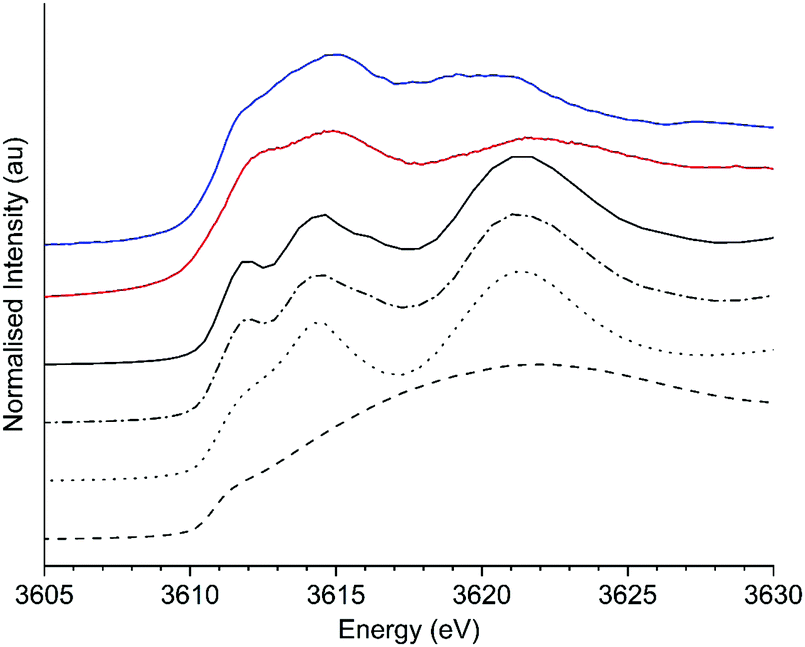 | ||
| Fig. 6 Comparison of simulated potassium edge XANES of K2CO3 (both environments) at different cluster sizes with experimental K2CO3 and K/α-Al2O3. Black lines: 6 atoms (dashed), 22 atoms (dotted), 121 atoms (dashed–dotted) and 223 atoms (solid). Red line: experimental K2CO3 and blue line K/α-Al2O3. | ||
The lack of identifiable B and C features at 6 atom cluster size is not observed experimentally in K/α-Al2O3 both ex situ or at any point during the operando experiments, suggesting such isolated species are not present. In simulation cluster sizes of 22 and >121 atoms the features B and C are clearly present, but with feature B being at lower energy in the 22 atom simulation, at 3614.3 eV vs. 3614.5 eV in larger clusters, and feature C also shifted 0.2 eV higher than seen in the larger cluster. The shift in B feature position seen experimentally in K/α-Al2O from 3614.9 eV to 3614.0 eV could therefore reflect the formation of smaller clusters. The ΔCB for the 22 atom cluster was 7.1 eV, 0.4 eV higher than in the larger cluster. Although a change in ΔCB was seen by simulation it was found to be opposite, i.e. greater ΔCB for small particles, to that seen experimentally and proposed by Jacobs. Therefore, it was concluded that simply altering cluster size did not sufficiently replicate experimental findings.
As discussed vide supra, K/α-Al2O3 at room temperature contained significant amounts of KHCO3. Therefore, the XANES spectra of this compound were simulated for clusters between 6–50 atoms in size (Fig. 7). Features simulated for clusters of 50 atoms broadly correlated with experimentation, but with significant discrepancy in the position of bands C and Ci which was at significantly higher energy in simulation. The 22 atom cluster reproduced the experimental energy of band C better with a ΔCB value of 6.1 eV, which is notably smaller than the 6.8 eV seen by simulation and experiment for K2CO3. At <22 atom cluster simulations were comparable with those for K2CO3; while not seen during the currently reported operando study, similar spectra were observed by Huggins et al. for potassium within a lignate structure.25 The simulations suggest that the species observed could be isolated small potassium compound clusters. Further simulation studies across a range of potassium compounds are required to verify how well the 6 atom cluster simulations for KHCO3/K2CO3 translate to other compounds. In summary simulation of KHCO3 reproduced the reduced ΔCB experimentally only in the 22 atom cluster, with the divergence from experiment at larger cluster size raising concern over the validity of the simulation.
 | ||
| Fig. 7 Comparison of simulated potassium edge XANES of KHCO3 at different cluster sizes with experimental KHCO3 and K/α-Al2O3. Black lines: 6 atoms (dashed), 22 atoms (dotted), 50 atoms (dashed–dotted) Red line: experimental KHCO3 and blue line K/α-Al2O3. | ||
Two further strategies were employed in the simulation of K2CO3 XANES to support the operando study. Firstly, the expansion or contraction of the K2CO3 lattice by 5% was simulated, to reflect interaction with the α-Al2O3 support or the reported expansion with high θ coverage of water. Models for each K site at the full range of cluster sizes are shown in Fig. S7–S11 (ESI†). As seen in Fig. S11, expansion of the lattice resulted in a lowering of the energy position of the simulated XANES, contraction conversely shifted energies higher. The ΔCB values show (Table S1, ESI†) a change on expansion or contraction, with expansion giving a ΔCB 6.3 eV for the 223 atom cluster and a ΔCB of 7.4 eV on contraction of the same cluster. As clearly shown in Fig. 5a, during operando soot oxidation the shift in features B and C in K/α-Al2O3 were between 7.0 and 6.5 eV, which is within a similar range to that seen in these simulations. Importantly, while in simulation both bands B and C shifted, experimentally ΔCB was influenced above 200 °C predominantly by shifts in band C. Hence, while simulation did replicate ΔCB changes, shifts were more symmetric in nature than observed experimentally (Fig. 8).
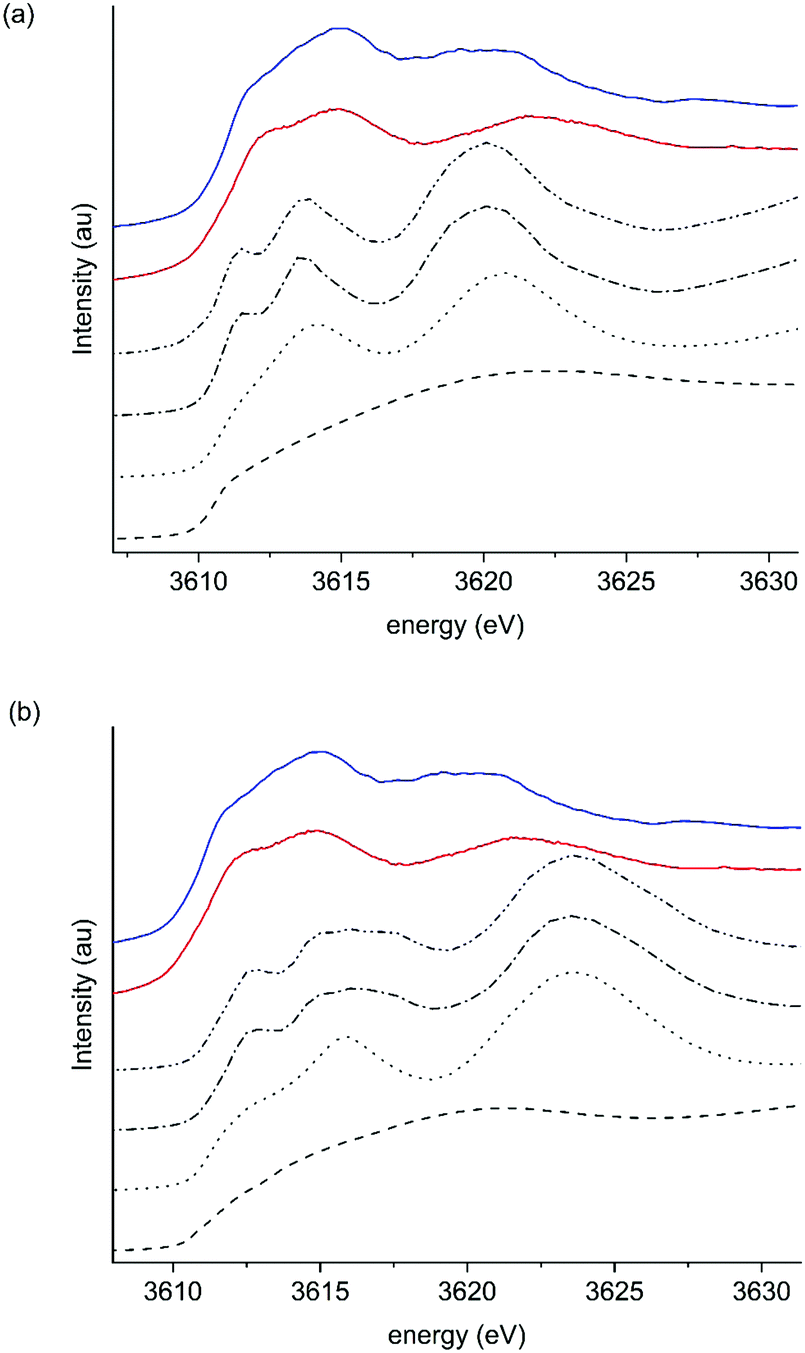 | ||
| Fig. 8 Comparison of simulated potassium edge XANES of K2CO3 at different cluster sizes with 5% expansion/contraction of the lattice. (a) 5% lattice expansion. (b) 5% lattice contraction. Black lines: 6 atoms (dashed), 22 atoms (dotted), 50 atoms (dashed–dotted). Red line: experimental K2CO3 and blue line K/α-Al2O3. | ||
The second simulation strategy considered the potential for the K1:K2 environment ratio to alter during reaction, due to particle size effects or the formation of structural defects. Systematic variation of the K1:K2 ratio, shown in Fig. 9, demonstrates that such a variation can alter the relative positions of bands B and C. For K1:K2 extremes of 10:90 and 90:10 the ΔCB values were calculated to be 4.9 and 7.5 eV. These differences are comparable to the observed changes in experimental ΔCB values from 7.0 and 6.5 eV Although, experimentally band C was observed to drop at high temperatures while B remained constant, which is not reflected in simultaneous reduction of B and increase in C band energy in simulation.
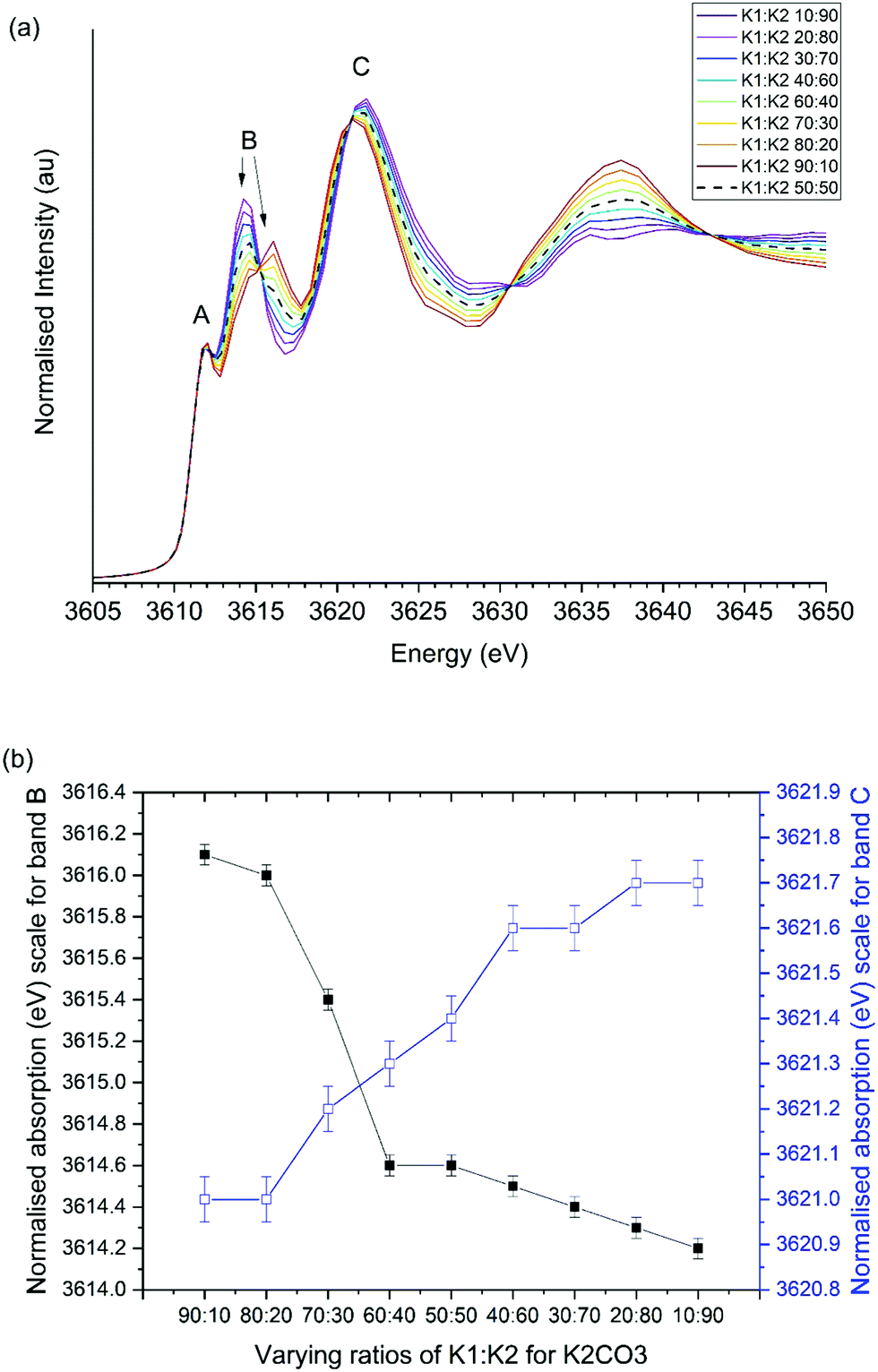 | ||
| Fig. 9 Influence of K1:K2 ratio on simulated XANES spectra of K2CO3. (a) Simulated XANES, (b) position of observed bands B and C. | ||
Therefore, it can be concluded that current simulations provide a rationale for the changes in XANES features observed experimentally. However, no single set of experiments precisely replicated experimental observation. Generally, the simulations support the hypothesis that the fresh catalyst comprises of a potassium environment inconsistent with bulk K2CO3. Practically, this is most likely due to the presence of KHCO3 that is only partially substantiated in the simulations at a specific cluster size. Further, heating above the decomposition of the KHCO3 resulted in shifts in band C which couldn’t be directly replicated by simulation. Yet, simulation clearly demonstrated that structural changes in K2CO3 can influence the positions of features B and C.
Conclusion
Operando potassium K-edge XANES has been used to study the evolution of potassium species in the catalytic oxidation of soot. The experiment is, to our knowledge, the first reported XANES operando study at a potassium K-edge and demonstrates the potential for further XANES analysis of alkali and alkaline earth catalysts, promotors and poisons, during reaction. Two identifiable phenomena were observed during the temperature programmed oxidation experiments. Firstly, significant changes in the potassium XANES occurred during the dehydration and decomposition of bicarbonate, between 100–160 °C, to produce bulk K2CO3. The result provides a note of caution when interpreting potassium XANES; namely that the spectra are highly sensitive to local structure, which is itself sensitive to hydration and adsorption of CO2. This sensitivity is further confirmed by XANES simulations which showed changes in cluster size, expansion or contraction of the lattice, and changes in the different abundance of potassium crystallographic environment significantly changed simulated XANES spectra of K2CO3. At higher temperatures of 200–600 °C K2CO3 becomes mobile and disperses over the Al2O3 support, as evidenced by progressive shifting of a feature in the XANES associated with multiple scattering resonance to lower energy. This finding is supported by XPS and XRD analysis of the catalyst post reaction. In the presence of soot, this change in XANES feature is significantly more pronounced than in a control experiment without soot and demonstrates interaction between potassium species and soot during its combustion. Simultaneously recorded mass spectroscopy showed that this change in XANES was co-current with the onset of soot oxidation. Current simulations could not model shifts in the purported multiple scattering feature to changes in lattice size or cluster size. However, changes in the ratio of the two potassium sites in K2CO3 did result in such a change. Although the explicit nature of this interaction is unresolved, it appears to exclude the possibility of the stoichiometric reaction of potassium carbonate with elemental carbon in the soot as a key step in the reaction mechanism. Instead, the alkali metal appears to have the classic catalytic role of activating O2 into reactive oxygen species, with the mobility of K2CO3 facilitating the transport of these species to immobilised soot particles. This represents a significant finding that can be investigated further, both for this specific example of soot oxidation, and in the broader context of other alkali promotors/poisons in a range of catalytic processes. Finally, the important role of simulation in ongoing studies is clearly highlighted.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the EPSRC-funded XMaS Mid-Range facility at BM28 in the European Synchrotron Radiation Facility (ESRF), Grenoble, France where the XAFS measurements were performed (28-01-1223). We thank the XMaS beamline team for support of BM28 experiments. We acknowledge the X-ray absorption measurement time provided at the Diamond Light Source on B18 (SP19850). We also acknowledge the Environment theme of the UK Catalysis Hub (EPSRC grant EP/K014854/1). Alex Mayer thanks Diamond Lightsource for PhD funding. Dr Laurence Bouchenoire and Prof. K.G. Upul Wijaynatha are thanked for their time and support.References
- Z. Kowalczyk, S. Jodzis, W. Rarog, J. Zielinski and J. Pielaszek, Effect of potassium and barium on the stability of a carbon-supported ruthenium catalyst for the synthesis of ammonia, Appl. Catal., A, 1998, 173, 153–160 CrossRef CAS.
- D. R. Strongin and G. A. Somorjai, Effects of potassium on ammonia synthesis over iron single-crystal surfaces, J. Catal., 1988, 109, 51–60 CrossRef CAS.
- K. Aika, H. Hori and A. Ozaki, Activation of nitrogen by alkali metal promoted transition metal. I. Ammonia synthesis over ruthenium promoted by alkali metal, J. Catal., 1972, 27, 424–431 CrossRef CAS.
- D. B. Bukur, D. Mukesh and S. A. Patel, Promoter effects on precipitated iron catalysts for Fischer-Tropsch synthesis, Ind. Eng. Chem. Res., 1990, 29, 194–204 CrossRef CAS.
- V. R. R. Pendyala, U. M. Graham, G. Jacobs, H. H. Hamdeh and B. H. Davis, Fischer-Tropsch Synthesis: Deactivation as a Function of Potassium Promoter Loading for Precipitated Iron Catalyst, Catal. Lett., 2014, 144, 1704–1716 CrossRef CAS.
- M. Akimoto and E. Echigoya, Participation of double bond type lattice oxygen in vapor-phase catalytic oxidation of olefins, J. Catal., 1974, 35, 278–288 CrossRef CAS.
- L. Chen, J. Li and M. Ge, The poisoning effect of alkali metals doping over nano V2O5-WO3/TiO2 catalysts on selective catalytic reduction of NOx by NH3, Chem. Eng. J., 2011, 170, 531–537 CrossRef CAS.
- S. A. Kondrat, P. J. Smith, J. H. Carter, J. S. Hayward, G. J. Pudge, G. Shaw, M. S. Spencer, J. K. Bartley, S. H. Taylor and G. J. Hutchings, The effect of sodium species on methanol synthesis and water-gas shift Cu/ZnO catalysts: Utilising high purity zincian georgeite, Faraday Discuss., 2017, 197, 287–307 RSC.
- S. A. Kondrat, P. J. Smith, P. P. Wells, P. A. Chater, J. H. Carter, D. J. Morgan, E. M. Fiordaliso, J. B. Wagner, T. E. Davies, L. Lu, J. K. Bartley, S. H. Taylor, M. S. Spencer, C. J. Kiely, G. J. Kelly, C. W. Park, M. J. Rosseinsky and G. J. Hutchings, Stable amorphous georgeite as a precursor to a high-activity catalyst, Nature, 2016, 531, 83–87 CrossRef CAS PubMed.
- S. Wilke and M. H. Cohen, Influence of potassium adatoms on the dissociative adsorption of hydrogen on Pd(100), Surf. Sci., 1997, 380, L446–L454 CrossRef CAS.
- N. D. Lang, S. Holloway and J. K. Noerskov, Electrostatic adsorbate-adsorbate interactions: the poisoning and promotion of the molecular adsorption reaction, Surf. Sci., 1985, 150, 24–38 CrossRef CAS.
- P. J. Feibelman and D. R. Hamann, Electronic structure of a ‘poisoned’ transition-metal surface, Phys. Rev. Lett., 1984, 52, 61–64 CrossRef CAS.
- G. Pirug and H. P. Bonzel, Short range interaction of potassium and carbon monoxide coadsorbed on platinum(111), Surf. Sci., 1988, 199, 371–390 CrossRef CAS.
- J. E. Crowell and G. A. Somorjai, The effect of potassium on the chemisorption of carbon monoxide on the rhodium(111) crystal face, Appl. Surf. Sci., 1984, 19, 73–91 CrossRef CAS.
- Z.-P. Liu and P. Hu, An Insight into Alkali Promotion: A Density Functional Theory Study of CO Dissociation on K/Rh(111), J. Am. Chem. Soc., 2001, 123, 12596–12604 CrossRef CAS PubMed.
- K. Nagase, T. Shimodaira, M. Itoh and Y. Zheng, Kinetics and mechanisms of the reverse Boudouard reaction over metal carbonates in connection with the reactions of solid carbon with the metal carbonates, Phys. Chem. Chem. Phys., 1999, 1, 5659–5664 RSC.
- F. Kapteijn, O. Peer and J. A. Moulijn, Kinetics of the alkali carbonate catalyzed gasification of carbon. 1. Carbon dioxide gasification, Fuel, 1986, 65, 1371–1376 CrossRef CAS.
- D. W. McKee, Mechanisms of the alkali metal catalyzed gasification of coal, Fuel, 1983, 62, 170–175 CrossRef CAS.
- B. Lamontagne, F. Semond and D. Roy, K overlayer oxidation studied by XPS: the effects of the adsorption and oxidation conditions, Surf. Sci., 1995, 327, 371–378 CrossRef CAS.
- G. Jacobs, V. R. R. Pendyala, M. Martinelli, W. D. Shafer, M. K. Gnanamani, S. Khalid, A. MacLennan, Y. Hu and B. H. Davis, Fischer-Tropsch Synthesis: XANES Spectra of Potassium in Promoted Precipitated Iron Catalysts as a Function of Time On-stream, Catal. Lett., 2017, 147, 1861–1870 CrossRef CAS.
- F. Xu, Z. Huang, P. Hu, Y. Chen, L. Zheng, J. Gao and X. Tang, The promotion effect of isolated potassium atoms with hybridized orbitals in catalytic oxidation, Chem. Commun., 2015, 51, 9888–9891 RSC.
- M. Ding, Y. Yang, J. Xu, Z. Tao, H. Wang, H. Wang, H. Xiang and Y. Li, Effect of reduction pressure on precipitated potassium promoted iron-manganese catalyst for Fischer-Tropsch synthesis, Appl. Catal., A, 2008, 345, 176–184 CrossRef CAS.
- J. Iranmahboob, D. O. Hill and H. Toghiani, Characterization of K2CO3/Co-MoS2 catalyst by XRD, XPS, SEM, and EDS, Appl. Surf. Sci., 2001, 185, 72–78 CrossRef CAS.
- G. P. Huffman, N. Shah, J. Zhao, F. E. Huggins, T. E. Hoost, S. Halvorsen and J. G. Goodwin Jr, In situ XAFS investigation of K-promoted Co catalysts, J. Catal., 1995, 151, 17–25 CrossRef CAS.
- F. E. Huggins, N. Shah, G. P. Huffman, F. W. Lytle, R. B. Greegor and R. G. Jenkins, In situ XAFS investigation of Ca and K catalytic species during pyrolysis and gasification of lignite chars, Fuel, 1988, 67, 1662–1667 CrossRef CAS.
- R. Ramdas, E. Nowicka, R. Jenkins, D. Sellick, C. Davies and S. Golunski, Using real particulate matter to evaluate combustion catalysts for direct regeneration of diesel soot filters, Appl. Catal., B, 2015, 176–177, 436–443 CrossRef CAS.
- J. P. A. Neeft, M. Makkee and J. A. Moulijn, Metal oxides as catalysts for the oxidation of soot, Chem. Eng. J., 1996, 64, 295–302 CAS.
- D. Fino, N. Russo, G. Saracco and V. Specchia, Removal of NOx and diesel soot over catalytic traps based on spinel-type oxides, Powder Technol., 2008, 180, 74–78 CrossRef CAS.
- Y. Wei, Z. Zhao, J. Liu, S. Liu, C. Xu, A. Duan and G. Jiang, Multifunctional catalysts of three-dimensionally ordered macroporous oxide-supported Au@Pt core-shell nanoparticles with high catalytic activity and stability for soot oxidation, J. Catal., 2014, 317, 62–74 CrossRef CAS.
- W. F. Shangguan, Y. Teraoka and S. Kagawa, Promotion effect of potassium on the catalytic property of CuFe2O4 for the simultaneous removal of NOx and diesel soot particulate, Appl. Catal., B, 1998, 16, 149–154 CrossRef CAS.
- H. An, C. Kilroy and P. J. McGinn, Combinatorial synthesis and characterization of alkali metal doped oxides for diesel soot combustion, Catal. Today, 2004, 98, 423–429 CrossRef CAS.
- G. Corro, U. Pal, E. Ayala and E. Vidal, Diesel soot oxidation over silver-loaded SiO2 catalysts, Catal. Today, 2013, 212, 63–69 CrossRef CAS.
- R. Matarrese, L. Castoldi, L. Lietti and P. Forzatti, Soot combustion: Reactivity of alkaline and alkaline earth metal oxides in full contact with soot, Catal. Today, 2008, 136, 11–17 CrossRef CAS.
- M. S. Gross, M. A. Ulla and C. A. Querini, Catalytic oxidation of diesel soot: New characterization and kinetic evidence related to the reaction mechanism on K/CeO2 catalyst, Appl. Catal., A, 2009, 360, 81–88 CrossRef CAS.
- L. Castoldi, R. Matarrese, L. Lietti and P. Forzatti, Intrinsic reactivity of alkaline and alkaline-earth metal oxide catalysts for oxidation of soot, Appl. Catal., B, 2009, 90, 278–285 CrossRef CAS.
- A. Nzihou, B. Stanmore and P. Sharrock, A review of catalysts for the gasification of biomass char, with some reference to coal, Energy, 2013, 58, 305–317 CrossRef CAS.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Parameter-free calculations of X-ray spectra with FEFF9, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513 RSC.
- J. J. Rehr, J. J. Kas, M. P. Prange, A. P. Sorini, Y. Takimoto and F. Vila, Ab initio theory and calculations of X-ray spectra, C. R. Phys., 2009, 10, 548–559 CrossRef CAS.
- R. L. Lehman, N. G. Glumac and J. S. Gentry, Thermal stability of potassium carbonate near its melting point, Thermochim. Acta, 1998, 316, 1–9 CrossRef CAS.
- A. P. de Kroon, G. W. Schaefer and F. Aldinger, Direct Synthesis of Binary K-β- and K-β’’-Alumina. 1. Phase Relations and Influence of Precursor Chemistry, Chem. Mater., 1995, 7, 878–887 CrossRef CAS.
- J. Sokolowski and A. Kotarba, The structure of potassium aluminium oxide KAlO2, Mater. Sci. Forum, 2000, 321–324, 954–959 CAS.
- M. Kantschewa, E. V. Albano, G. Ertl and H. Knoezinger, Infrared and X-ray photoelectron spectroscopy study of potassium carbonate/γ-alumina, Appl. Catal., 1983, 8, 71–84 CrossRef CAS.
- C. Sugiura and S. Muramatsu, K-edge X-ray absorption spectra from potassium compounds, Phys. Status Solidi B, 1985, 132, K111–K115 CrossRef CAS.
- C. M. Dutta and H. W. Huang, K-edge absorption spectra of ionic potassium and its Z + 1 analogy, Phys. Rev. Lett., 1980, 44, 643–646 CrossRef CAS.
- S. Iqbal, S. A. Kondrat, D. R. Jones, D. C. Schoenmakers, J. K. Edwards, L. Lu, B. R. Yeo, P. P. Wells, E. K. Gibson, D. J. Morgan, C. J. Kiely and G. J. Hutchings, Ruthenium Nanoparticles Supported on Carbon: An Active Catalyst for the Hydrogenation of Lactic Acid to 1,2-Propanediol, ACS Catal., 2015, 5, 5047–5059 CrossRef CAS.
- J. A. Ibers, J. Kumamoto and R. G. Snyder, Structure of potassium hydroxide: an X-ray and infrared study, J. Chem. Phys., 1960, 33, 1164–1170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01227k |
| This journal is © the Owner Societies 2020 |