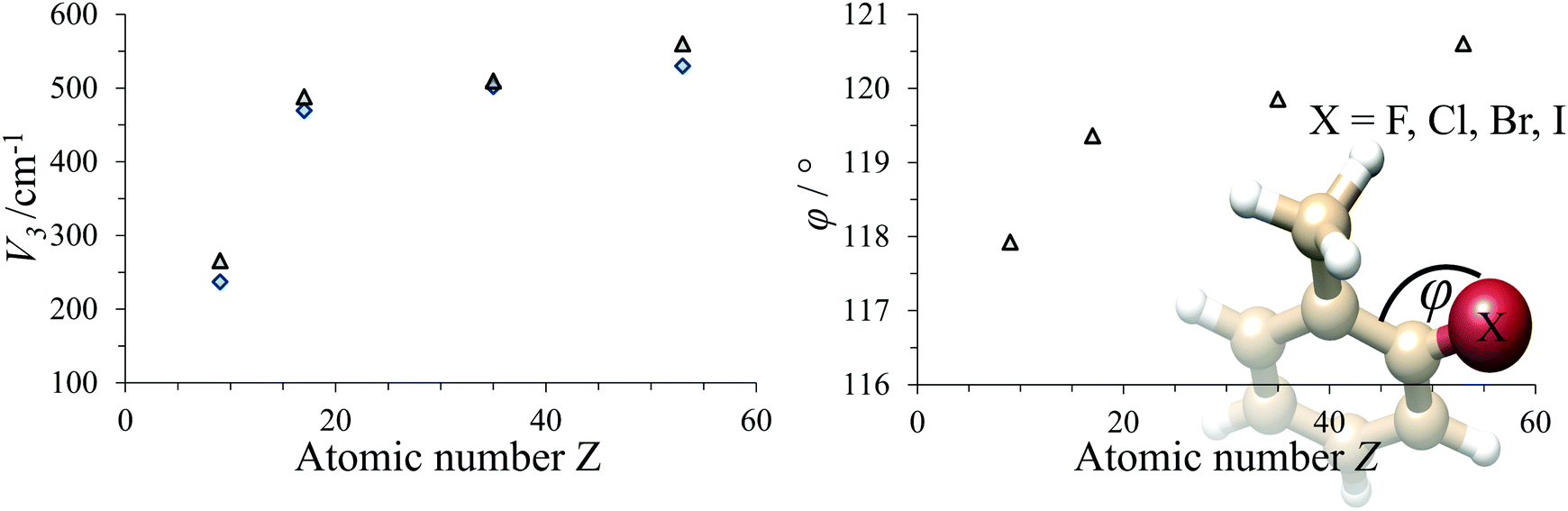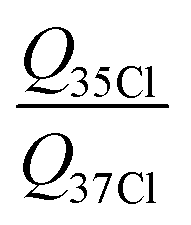Reactivity and rotational spectra: the old concept of substitution effects†
Received
28th February 2020
, Accepted 25th April 2020
First published on 11th May 2020
Abstract
The internal rotation of methyl groups and nuclear quadrupole moments of the halogens Cl, Br, I in o-halotoluenes cause complex spectral fine and hyperfine structures in rotational spectra arising from angular momentum coupling. Building on the existing data regarding o-fluorotoluene and o-chlorotoluene, the investigations of o-bromotoluene and o-iodotoluene allow for a complete analysis of the homologous series of o-halogenated toluenes. The trend in the methyl barriers to internal rotation rising with the size of the halogen can be rationalised by repulsion effects as predicted by MP2 calculations. Furthermore, the analysis of the observed quadrupole coupling serves as a quantitative intra-molecular probe, e.g. for the explanation of the relative reaction yields in the nitration of halotoluenes, related to the different π-bond character of the C–X bond depending on the position of substitution.
1 Introduction
Even in the rising era of computational chemistry, the intuitive understanding of product formation in organic reactions, including the classic concept of substitution effects, is of considerable importance. In the field of synthesising highly functionalised aromatic systems, that are of pharmaceutical importance, a precise idea about the electronic influences of the substituents already introduced into the molecule guides the selection of a suitable synthesis route. For these systems, rules were empirically established from analysing yields of various electrophilic substitutions of substituted benzenes. These concepts were mainly introduced by Robinson and Ingold and applied in countless publications and are still part of the basic course in organic chemistry for undergraduate students.1 One common example is the nitration of aromatic molecules like toluene, where the formation of o- an p-nitrotoluene is preferred over m-nitrotoluene caused by the directing inductive influence of the methyl group. The preferred products are usually explained with the possible resonance structures of the intermediate cation formed after the electrophilic attack. Only for o- and p-attack a cation with the positive charge next to the electron donating methyl group can be formed as illustrated in Fig. 1(a). The electron pulling or electron pushing effect of substituents through the σ bond system is commonly referred to as inductive (−/+I) effect. For halogenated benzenes it might be expected that electrophilic substitution is preferred on meta-positions over ortho- and para-positions, since the halogen is pulling electrons out of the ring (−I), and a positive charge next to the halogen would not be preferred. However, the halogen substituents are ortho- and para-position directing, whereby their −I effect simultaneously reduces the reaction rate. This, in the Lewis structure model, is explained by the lone pairs of the halogen, that can form a double bond to the ring carbon atom corresponding to a resonance structure, which leaves a positive charge at the halogen, illustrated in Fig. 1(b). This is commonly referred to as mesomeric effect (−/+M).1,2
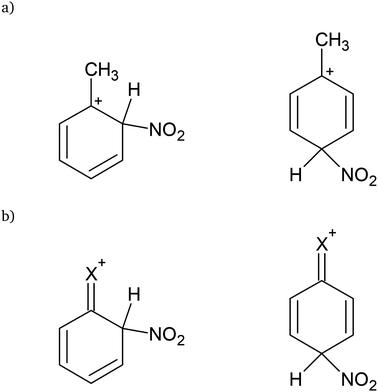 |
| | Fig. 1 Stabilised intermediate cations after electrophilic attack of NO2+ on o- and p-position for (a) toluene (b) halobenzene. | |
To study the influence of inductive and mesomeric effects in the absence of any electrophile experimentally, supersonically cooled jet-expansion techniques combined with high-resolution microwave spectroscopy are excellently suited to test and quantify these concepts: for the halogens chlorine, bromine and iodine, the ranking in −I as well as +M effects is Cl > Br > I. This is rationalised by the decreasing electronegativity as well as orbital overlap regarding the carbon they bind to. Since these nuclei do have an electric quadrupole moment, information on the electronic environment, i.e. their bonding situation, can be obtained from their quadrupole coupling tensor, derived from the hyperfine structure in rotational spectra. From the symmetry of the quadrupole coupling tensor as well as the magnitude of its elements, the population of the valence p-orbitals can be derived and deviations directly reflect the strength of a possible +M and −I effect. In previous investigations of halobenzenes3 and the homologues of the p-halotoluenes4 the trend Cl > Br > I for both, +M and −I effects was verified. This study extends and completes the series of o-halotoluenes by measurements on o-bromotoluene and o-iodotoluene. In addition, the rather large barrier to internal rotation of the methyl group in o-halotoluenes will be discussed in this study.
2 Model Hamiltonian
To derive precise molecular parameters from spectra, an effective model Hamiltonian has to be employed. In many cases a satisfying description of the rotational motion of a molecule can be achieved by using a semi rigid model (Watson's S reduction in Ir representation5):| | | Hrr = A0Pa2 + B0Pb2 + C0Pc2 | (2) |
| | | Hcd = −DJP4 − DJKP2Pa2 − DKPa4 + d1P2(P+2 + P−2) + d2(P+4 + P−4) | (3) |
In this semi rigid model (sr), the rotation is treated separately from vibrational motions as a rigid rotor (rr), corrected for centrifugal distortion (cd). A0, B0, C0 are the ground vibrational state rotational constant, Pa, Pb, Pc the components of the angular momentum operator. DJ, DJK, DK, d1, and d2 are the quartic centrifugal distortion coefficients, P± = Pb ± iPc are commonly known as ladder or step operators. However, in the presence of methyl groups the coupling between the internal rotation along a threefold potential energy path and the overall rotation of the molecule often becomes significant. The semi rigid model is then expanded by an internal rotation Hi term, which includes the coupling:| | 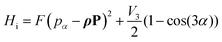 | (5) |
Here, the constant| | 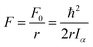 | (6) |
is the reduced constant of internal rotation. V3 is the barrier for the threefold internal rotation and α is the torsional angle. In the limiting case of vanishing coupling F can be derived from the structural moment of inertia of the methyl top Iα but in general the effect of coupling is taken into account by the reducing factor 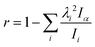 with λi being the direction cosines for the internal rotation axis with respect to the principal axes i. F0, which is the structural constant to internal rotation directly derived from Iα, can easily be estimated from quantum chemically optimised molecular geometries to be about 161.6 GHz for all o-halogenated toluenes. The vector ρ with
with λi being the direction cosines for the internal rotation axis with respect to the principal axes i. F0, which is the structural constant to internal rotation directly derived from Iα, can easily be estimated from quantum chemically optimised molecular geometries to be about 161.6 GHz for all o-halogenated toluenes. The vector ρ with 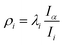 in eqn (5) gives rise to cross-terms originating from the coupling between internal and overall rotation. Its absolute value ranges between zero and one. The zero-value of |ρ| = 0 marks the limiting case of vanished coupling. For |ρ| = 0 the internal rotation part would simply lead to a set of energy levels on the potential energy curve along the torsional degree of freedom. These can be labelled either by ±m quantum numbers, commonly used for small barriers to internal rotation, where the eigenstates are close to free rotor states, or at larger barriers by their torsional vibration v quantum numbers. In this article we will follow the latter labelling scheme. With a finite barrier that allows for tunnelling, the v levels will be split into two symmetry species, labelled with their symmetry quantum number σ = 0, ±1. Levels with σ = 0 are referred to as A species and levels with σ = ±1 are referred to as E species. Transitions from A to E species are forbidden, so that for |ρ| = 0 no splittings would arise in the rotational spectrum, since there is no coupling between rotation and internal rotation. For non vanishing |ρ| values the spectrum will be split and each transition can be assigned to a symmetry species.
in eqn (5) gives rise to cross-terms originating from the coupling between internal and overall rotation. Its absolute value ranges between zero and one. The zero-value of |ρ| = 0 marks the limiting case of vanished coupling. For |ρ| = 0 the internal rotation part would simply lead to a set of energy levels on the potential energy curve along the torsional degree of freedom. These can be labelled either by ±m quantum numbers, commonly used for small barriers to internal rotation, where the eigenstates are close to free rotor states, or at larger barriers by their torsional vibration v quantum numbers. In this article we will follow the latter labelling scheme. With a finite barrier that allows for tunnelling, the v levels will be split into two symmetry species, labelled with their symmetry quantum number σ = 0, ±1. Levels with σ = 0 are referred to as A species and levels with σ = ±1 are referred to as E species. Transitions from A to E species are forbidden, so that for |ρ| = 0 no splittings would arise in the rotational spectrum, since there is no coupling between rotation and internal rotation. For non vanishing |ρ| values the spectrum will be split and each transition can be assigned to a symmetry species.
Treatment of the internal rotation part of the Hamiltonian is usually carried out using one of three methods: principal axis method PAM, ρ-axis method RAM, or the combined axis method CAM, which combines PAM and RAM. A more thorough description of PAM and RAM can be found in ref. 6 and 7. Detailed information on state of the art programs to treat internal rotation problems is given in the review written by Kleiner.8
Further complications arise if the molecule contains a nucleus with electric quadrupole moment. A non-zero electric field gradient at the nucleus together with a significant nuclear quadrupole moment causes strong coupling between nuclear spin and overall rotation of the molecule. The Hamiltonian is then expanded by another term (q):
Matrix elements of
Hq are provided in
ref. 9. The elements of
Hq are parametrised by the matrix elements of the quadrupole coupling tensor
χ, which can be derived from the hyperfine structure of a rotational spectrum. In its diagonalised form, the tensor elements are proportional to the quadrupole moment
Q of the nucleus:
Here
e is the proton charge and
qii is the electric field gradient at the nucleus. Although exact treatment is possible, elements off-diagonal in
J are often omitted for simplicity. Omitting the off-diagonal elements does cause errors, that, in the case of rather small quadrupole moments (like nitrogen or deuterium nuclei) in absence of near-degeneracies, are usually small compared to experimental accuracy. However, chlorine, bromine, and iodine containing compounds can not be treated properly by this approach and are strongly affected by coupling of quadrupolar nuclei. The popular program XIAM,
10–13 which uses the CAM method, does not include elements off-diagonal in
J. Consequently, quadrupolar coupling can not be treated with experimental accuracy in XIAM for the case of the
o-halotoluenes investigated in this study. Instead we will utilise a two step procedure as proposed in the study of (CH
3)SiI.
14 In a first step the internal rotation symmetry species
A and
E are treated separately in terms of the PAM utilizing the program SPFIT, which treats quadrupolar coupling exactly. In the high barrier regime, this yields fits with experimental accuracy. In a second step the effects of quadrupolar coupling are removed and the hyperfine structure free line list of hypothetically unsplit centre frequencies is used in a fit with XIAM to derive the internal rotation parameters and structural rotational constants. While experimental accuracy fits were possible with SPFIT using effective parameters, these lack easy interpretation. Therefore, XIAM was preferred and used for the derivation of internal rotation parameters.
3 Computational
All calculations were carried out with Gaussian16 (Revision A.03).15 All o-halotoluenes optimisations with subsequent frequency calculations were performed using the B3LYP16–19 and the MP220 method with a 6-311++G(2d,2p) basis set as implemented in Gaussian. For iodine a 6-311G** basis set21 taken from the EMSL basis set exchange library22 (https://bse.pnl.gov/bse/) was used. In addition MP2 relaxed potential energy surface scans were performed to predict the internal rotation barrier. Additionally, MP2 calculations on halobenzenes and p-halotoluenes were carried out to consistently predict the π-bond character of the halogen–carbon bonds and compare the results with those obtained for the o-halotoluenes. For o-iodotoluene the influence of relativistic effects on the C–I bond length was estimated by comparing an MP2(full) optimisation with an MP2(full) optimisation using the Douglas–Kroll–Hess second-order scalar relativistic (dkh) Hamiltonian.23–26
4 Experimental
A commercial sample of o-bromotoluene (Aldrich, 99%) was used without further purification. Gas mixtures of ≈5 mbar in 4 bar of helium were prepared and measurements were carried out with the coaxially oriented beam resonator arrangement Fourier transform microwave (COBRA-FTMW) spectrometer in Hannover.27 Segments of the spectrum were recorded between 5 and 21 GHz. From the recorded signals, 190 were identified to belong to the 79Br isotopologue and 240 belong to the 81Br isotopologue. For the investigations of o-iodotoluene, a commercial sample (Alfa Aesar, 98%) was placed into a custom made reservoir nozzle on the broad band inphase/quadrature-phase modulation passage-acquired-coherence technique (IMPACT-FTMW) spectrometer.28 The spectrum from 8.5–18.5 GHz was recorded with neon as carrier gas. The sample was heated up to 70 °C by a resistive coil, wrapped around the nozzle head. 1093 signals were identified from the spectrum and assigned to o-iodotoluene. In both experiments the backing pressure was 1 bar. Fits of molecular parameters to experimental frequencies were carried out using SPFIT29 and XIAM.10–13 In the XIAM fits the values of F0 were fixed on values of the B3LYP optimised equilibrium geometries. An example for a well resolved quadrupole and internal rotation split transition is given in Fig. 2 for o-bromotoluene. All transitions are Doppler-split since the coherently rotating ensemble of molecules emits parallel and anti-parallel to the propagation direction of the molecular jet with both components detected.
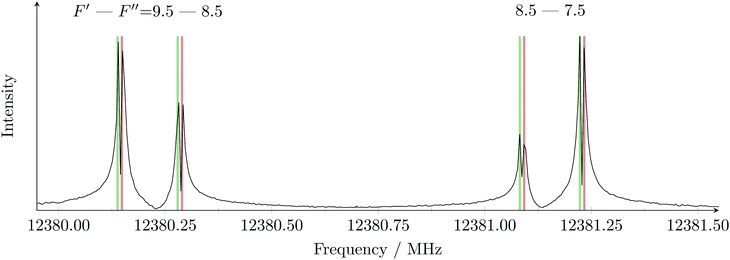 |
| | Fig. 2 Two of the hyperfine components of the 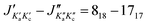 transition of o-bromotoluene. Doppler pairs predicted from the parameters of the fit are coloured red for the A symmetry species and green for the E symmetry species. The separation between the two hyperfine components is 944 kHz, A/E splitting is 12 kHz while the Doppler splitting amounts to 141 kHz with helium as carrier gas. transition of o-bromotoluene. Doppler pairs predicted from the parameters of the fit are coloured red for the A symmetry species and green for the E symmetry species. The separation between the two hyperfine components is 944 kHz, A/E splitting is 12 kHz while the Doppler splitting amounts to 141 kHz with helium as carrier gas. | |
5 Results and discussion
5.1
o-Fluorotoluene
o-Fluorotoluene was already investigated in ref. 30, but the presented fits are rather problematic. Instead of fitting A and E species with the same weight, primarily the deviation of the A species was minimised, which is reflected in the rather high deviations for E species lines up to 545 kHz. Also F0 was fixed to a value, which is inconsistent with our values derived from B3LYP optimised geometries. Since V3 and F0 are strongly correlated, this would lead to barriers inconsistent with the barriers of the other o-halotoluenes investigated in this study. Therefore, we reanalysed the o-fluorotoluene measurements based on the line list presented in ref. 30. An overall standard deviation of σ = 3.0 kHz was achieved with a maximum deviation of 15 kHz and a barrier of 236.841(10) cm−1 was obtained.
5.2
o-Chlorotoluene
o-Chlorotoluene was subject of several spectroscopic investigations.31–35 A reasonable barrier height based on microwave investigations with a COBRA-type spectrometer was determined in ref. 34 in terms of the principal axis method, providing a value of V3,35Cl = 466.44(43) cm−1 and V3,37Cl = 466.2(15) cm−1. The list of microwave transitions used in ref. 34 was expanded by millimetre wave transitions and a global fit was carried out yielding a barrier of about 513 cm−1.35 In this global fit the parameter F0 was floated and took a value of 178.4 GHz for the 35Cl isotopologue, corresponding to Iα = 2.833 amu Å2, which is too small for a methyl group's moment of inertia. Taking the microwave data provided in ref. 34 we reanalysed the spectrum with the SPFIT+XIAM two step fitting procedure. In the XIAM fit we added the millimetre wave lines from ref. 35, for which no splitting due to quadrupole interaction was resolved. To facilitate the determination of accurate internal rotation barriers, only lines for which A as well as E species were provided are used. The fit shows excellent agreement between microwave (σmw,35Cl = 1.3 kHz, σmw,37Cl = 1.4 kHz) and millimetre wave (σmmw,35Cl = 30.3 kHz, σmmw,37Cl = 40.6 kHz) data. The deviations are consistent with the expected experimental accuracy of the microwave transitions of a few kHz and the millimetre wave transitions of about 100 kHz. The determined barriers V3,35Cl = 469.211(90) cm−1 and V3,37Cl = 469.863(204) cm−1 are in satisfactory agreement with each other. Besides the internal rotation, also the quadrupolar coupling can straightforwardly be analysed, since the χyy = χcc = eqccQ component of the quadrupole coupling tensor will not change with rotation of the coordinate system around the c-axis due to the molecule's a,b-plane of symmetry. This invariance upon isotopic substitution allows a precise ratio determination of the quadrupole moments of both nuclei 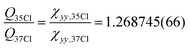 , without having to rely on the off-diagonal element of the tensor, which would otherwise be the largest source of uncertainty. This precise determination re-derived from the lines provided in ref. 34, which agrees with the recent values of Q35 = −0.08165(80) × 10−24 cm2 and Q37 = −0.06435(64) × 10−24 cm2 tabulated in ref. 36, resulting in a ratio of 1.2688(177).
, without having to rely on the off-diagonal element of the tensor, which would otherwise be the largest source of uncertainty. This precise determination re-derived from the lines provided in ref. 34, which agrees with the recent values of Q35 = −0.08165(80) × 10−24 cm2 and Q37 = −0.06435(64) × 10−24 cm2 tabulated in ref. 36, resulting in a ratio of 1.2688(177).
5.3
o-Bromotoluene
o-Bromotoluene was not investigated by means of microwave spectroscopy before. However, internal rotation and bromine nuclear quadrupole coupling were dealt with in other molecules already, e.g. bromopropane37 or, more closely related to this study, p-bromotoluene.4 Since internal rotation splittings are small due to the high barrier in o-halotoluenes, the SPFIT-XIAM two step procedure achieves experimental accuracy. The barriers to internal rotation V3,79Br = 502.333(614) cm−1 and V3,81Br = 503.221(522) cm−1 are in good agreement. Again the ratio of the quadrupole moments 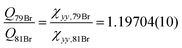 can be derived at high precision, and agrees with the ratio of the recommended quadrupole moments36 of 1.1969(162).
can be derived at high precision, and agrees with the ratio of the recommended quadrupole moments36 of 1.1969(162).
5.4
o-Iodotoluene
Of the investigated halotoluenes, o-iodotoluene has the most pronounced quadrupole splitting. The barrier to internal rotation is V3,I = 530.004(136) cm−1, which is in agreement with the trend to higher barriers with larger atom radii throughout the halogens. Since iodine has only one stable isotope, there is no ratio of quadrupole moments to be derived. Iodine has a very heavy nucleus, so that relativistic effects are expected to have a noticeable influence on the structure of the molecule. The relativistic effect on the C–I bond length was estimated using MP2(full)/6-311++G(2d,2p) calculations with and without DKH Hamiltonian. The inclusion of relativistic effects in the optimisation leads to a decrease in the C–I bond length of 6 mÅ.
5.5 Trend in the potential barrier
The trend in the potential barrier mainly reflects the exchange-repulsion of electrons between halogen and methyl rotor. The internal rotation parameters of all o-halotoluenes are summarised in Table 1. The barrier is monotonically increasing with the atomic number. The exchange-repulsion rising with the halogen's atomic size also affects the structure of the molecule in that the halogen enlarges its distance from the methyl rotor. This is best reflected in the angle φ between the two ring carbons and the halogen. In Fig. 3 the experimental results are plotted together with MP2 predictions, which reproduce the experimental values very well.
Table 1 Vibrational ground state structural rotational constants A0, B0, C0 as well as barrier to internal rotation V3 and angle δ between principal A axis and internal rotation axis α for all o-halotoluenes. The constant to internal rotation F0 was fixed to values based on B3LYP equilibrium geometries, which is 161.6 GHz in all cases. Watson's S reduction in Ir representation was applied
|
|
F |
35Cl |
37Cl |
79Br |
81Br |
I |
|
Number of lines used in the fit.
Standard deviation of the fit.
cm wave and mm wave transitions, respectively.
|
|
A
0/MHz |
3243.09623(55) |
2851.345869(119) |
2842.55382(33) |
2760.01069(162) |
2758.95253(141) |
2732.9609(24) |
|
B
0/MHz |
2180.49303(29) |
1538.315441(41) |
1500.290538(111) |
989.236603(117) |
978.929394(104) |
738.409742(76) |
|
C
0/MHz |
1314.36349(14) |
1005.370796(21) |
987.925653(34) |
731.461419(74) |
725.736795(63) |
583.394506(60) |
|
F
0/GHz |
161.6 (fixed) |
161.6 (fixed) |
161.6 (fixed) |
161.6 (fixed) |
161.6 (fixed) |
161.6 (fixed) |
|
δ/degree |
29.307(14) |
56.612(38) |
57.159(80) |
65.284(179) |
65.146(152) |
68.46(33) |
|
V
3/cm−1 |
236.841(10) |
469.211(90) |
469.86(21) |
502.33(62) |
503.22(53) |
530.004(136) |
|
N
lines
|
86 |
524/430c |
296/452c |
190 |
240 |
1093 |
|
σ/kHzb |
3.0 |
1.3/30.3c |
1.4/40.6c |
2.6 |
2.5 |
4.9 |
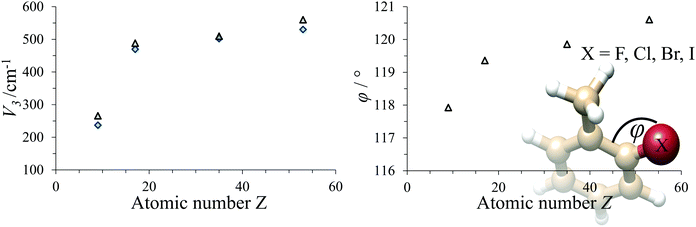 |
| | Fig. 3 Left: Dependence of V3 on the atomic number Z of the halogen. Right: Dependence of the C–C–X angle φ on the atomic number of the halogen. Gray triangles: MP2 prediction, blue diamonds: experimental data. | |
5.6 Quadrupole coupling tensor – the character of the halogen bond
Townes and Dailey provided an intuitive and strikingly successful model to interpret the quadrupolar coupling tensor in terms of π-character (πc) and ionic character (ic) of chemical bonding.38 For halogen substituents, that show approximately unaltered atomic p-orbitals, the relation| | 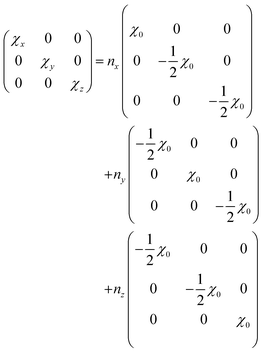 | (9) |
can be used to determine the populations ng of the different pg-orbitals with g = x, y, z.39 The χg coefficients are elements of the diagonalised quadrupole coupling tensor and χ0 is the quadrupole coupling constant due to a single valence p-electron, which can be found tabulated in ref. 6 (Table 14.2, for iodine see ref. 40). By convention, the z-axis points along the carbon–halogen σ-bond and we define, as being customary, the y-axis to be perpendicular to the ring plane. With the tensors being traceless only one additional assumption is necessary to solve eqn (9) for all ng. Since any mesomeric effect caused by the halogen lone pairs will be reflected in a decrease of ny, while nx will remain rather unaffected, we assume its full occupation nx = 2.0. The derived equations| | 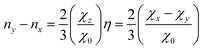 | (10) |
and| | 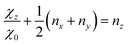 | (11) |
are then used to determine ny and nz. η = (χx − χy)/χz is the asymmetry parameter for the quadrupole coupling tensor. The π-character is then simply the number of electrons removed from the py orbitaland the ionic character is the excess of p-electrons| | | ic = nx + ny + nz − 5. | (13) |
The ionic character can be related to the electronegativity x of the bonding atoms by the rough approximation6,41| | 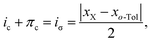 | (14) |
for X = Cl, Br, I with respect to the group electro-negativity of the o-toluene fragment xo-Tol. Here iσ = nz − 1 is the ionic character of the Sigma bond only. The results of the quadrupole coupling tensor analysis can thus be used to determine the group electro-negativity of the o-toluene fragment as presented in Table 4. In all cases πc is rather small and about 4%. That πc remains nearly constant under substitution is somewhat surprising, since for the halobenzenes as well as p-halotoluenes a decreasing trend with the size of the halogen is observed. The πc values of the compounds are summarised in Table 2. The ionic character on the other hand changes from 30% for chlorine to 23% for bromine and 13% for iodine. This is similar to the trend in halobenzenes and p-halotoluenes which is reflected in constant values of group electronegativities for the aromatic ring fragments. A summary of the ionic character of different compounds and the derived fragment electronegativity is given in Table 3.
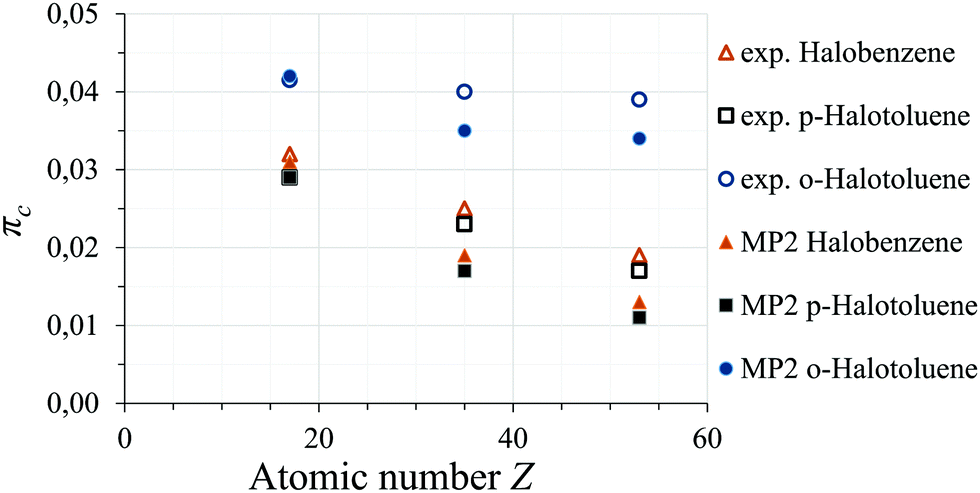
Table 2 C–X bond π-character πc for selected holgenated aromatic molecules
| X |
o-X-Toluene |
m-X-Toluene42 |
p-X-Toluene4 |
X-Benzene3 |
| Cl |
0.0415 |
0.031 |
0.029 |
0.032 |
| Br |
0.040 |
|
0.023 |
0.025 |
| I |
0.039 |
|
0.017 |
0.019 |
Table 3 C–X bond ionic character ic and derived average electronegativity of the ring fragment ![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ring for selected holgenated aromatic molecules
ring for selected holgenated aromatic molecules
| X |
o-X-Toluene |
m-X-Toluene42 |
p-X-Toluene4 |
X-Benzene3 |
|
For chlorine substitution only. The value differs from the value of 2.39 presented in Table 5 of ref. 42, which we corrected for the π contribution to the ionic character.
|
| Cl |
0.30 |
0.30 |
0.31 |
0.30 |
| Br |
0.23 |
|
0.24 |
0.24 |
| I |
0.13 |
|
0.15 |
0.15 |
|
ring
|
2.27 |
2.32a |
2.27 |
2.27 |
Table 4 Quadrupole coupling tensor elements χ obtained from PAM fits using SPFIT. The symmetric quadrupole coupling tensor with corresponding elements χaa, χbb, χcc, and χab was diagonalised to yield χx,y,z. The angle θ between the two axis systems (between a- and z-axis) as well as orbital populations nx,y,z, π-bond character πc, ionic character ic as well as the group electronegativity of the o-toluene fragment xo-Tol are derived from the tensor elements
|
|
35Cl |
37Cl |
79Br |
81Br |
I |
![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) χaa/MHz χaa/MHz |
−101.9576(14) |
−80.8330(14) |
822.148(14) |
686.869(11) |
−2796.915(16) |
| ¼(χbb − χcc)/MHz |
1.21326(43) |
1.03494(46) |
−11.4749(67) |
−9.5947(62) |
33.4838(71) |
|
χ
ab
/MHz |
14.98(28) |
10.20(29) |
12.33(49) |
7.41(14) |
−27.259(26) |
|
χ
z
/MHz |
−70.079 |
−55.125 |
548.279 |
457.990 |
−1864.869 |
|
χ
x
/MHz |
38.520 |
30.251 |
−297.179 |
−248.223 |
999.532 |
|
χ
y
/MHz |
31.559 |
24.874 |
−251.100 |
−209.767 |
865.337 |
|
n
z
|
1.340 |
1.342 |
1.268 |
1.268 |
1.167 |
|
n
x
|
2.000 |
2.000 |
2.000 |
2.000 |
2.000 |
|
n
y
|
1.958 |
1.959 |
1.960 |
1.960 |
1.961 |
|
π
c
|
0.042 |
0.041 |
0.040 |
0.040 |
0.039 |
|
i
c
|
0.298 |
0.301 |
0.228 |
0.228 |
0.128 |
|
x
o-Tol
|
2.320 |
2.316 |
2.264 |
2.264 |
2.216 |
|
θ/degree |
8.007 |
6.912 |
0.836 |
0.601 |
0.545 |
In the o-halotoluenes πc increases with the introduction of the methyl group compared to the halobenzenes, while the p-halotoluenes show a decrease in πc. While we can not give a simple explanation for this trend, MP2 calculations also predict a much higher πc for o-halotoluenes. The MP2 predictions do not reproduce the constant behaviour within in o-halotoluenes, however, also in the bromine and iodine species of p-halotoluenes and halobenzenes πc was underestimated compared to the chlorine values. If this systematic underestimation is taken into account the constant behaviour of the o-halotoluenes can be reproduced. The MP2 predictions of πc are shown together with the experimental data in Fig. 4.
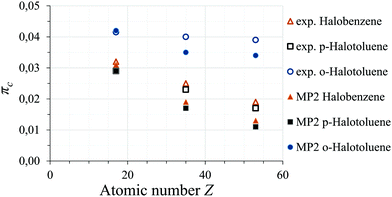 |
| | Fig. 4
π-Character πc for halobenzenes (triangles), p-halotoluenes (squares), and o-halotoluenes (circles): MP2-predicted (filled shapes) and experimental (empty shapes) results. | |
The trend in the ionic characters agrees with the tabulated electro-negativities6 of 3.0 for chlorine, 2.8 for bromine and 2.55 for iodine, which were also used to derive the xo-Tol values of 2.32, 2.26 and 2.22. This slight variance of group electro-negativities upon substitution was already observed in the case of halomethanes with values for chlorine, bromine and iodine containing species of 2.38, 2.30 and 2.29 for the methane fragment41 and is a result of the rough approximation in eqn (14) and errors in the values of electronegativity for the halogens. The χcc ratios for 35Cl/37Cl and 79Br/81Br equal the ratios of quadrupole moments. The experimental ratios found for the o-toluenes are compared to values determined from spectra of halobenzenes3 and p-halotoluenes4 in Table 5. All values agree within 2σ and are consistent with the ratios determined from the recommended quadrupole moments in ref. 36.
Table 5 Ratios of chlorine and bromine quadrupole moments derived in different studies
|
|
o-X-Tol. |
m-X-Tol.42 |
p-X-Tol.4 |
X-Benz.3 |
Tabulated36 |
|
Derived from line list in ref. 34.
This work.
|
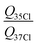
|
1.268745(66)a |
1.271(5) |
1.2691(2) |
1.26880(4) |
1.2688(177) |
| 1.26872(13)34 |
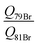
|
1.19704(10)b |
|
1.19697(3) |
1.197036(6) |
1.1969(162) |
6 Conclusions
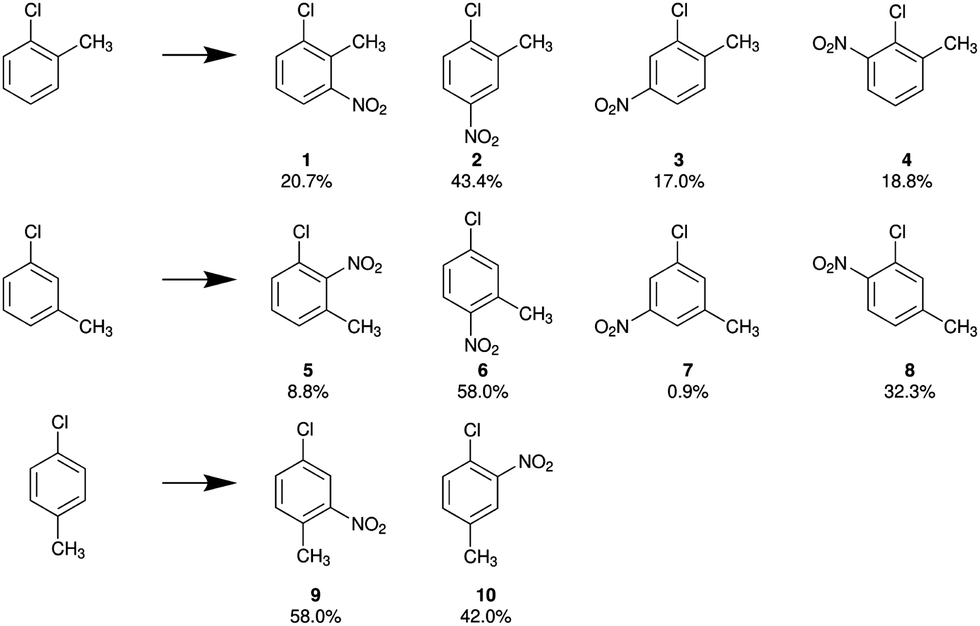
Halogen nuclei are able to serve as direct probe of the arrangement of valence electrons, allowing for conclusions on the reactivity of substituted toluenes to be drawn. Since a chlorine substituent exhibits the most effective interaction due to the largest orbital overlap with the aromatic systems, we focus on the chlorinated aromatic rings in Table 2. While the quite invariant values of group electronegativities reveal no significant change in the σ bonds for all chlorinated species, a comparison of the π-character πc of the C–Cl bond shows that the strongest interaction with the π-system, typically represented by the mesomeric boundary structures, occurs in o-chlorotoluene. Within the concept of substitution effects this indicates a comparably stronger influence of the chlorine substituent on the course of an electrophilic aromatic substitution reaction. In Fig. 5 the product yields of the nitration of all the different monochlorotoluenes are exemplified.
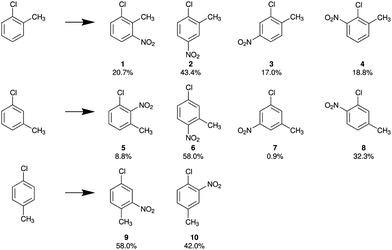 |
| | Fig. 5 Observed product yields for the nitration of o-, m- and p-chlorotoluene.43 | |
Both substituents, chlorine and methyl, are classified as electron pushing relative to the phenyl ring with their +M and +I effects, respectively. In the case of m-chlorotoluene both substituents direct the electrophile towards the same positions. The main product 6 satisfies both, chlorine and methyl directing effect, and the nitro group is introduced in para- and ortho-positions of the chlorine and methyl substituents, respectively.
For p-chlorotoluene the two substituents do not direct towards the same positions anymore. The main product 9, shows that a large portion of reactants follow the directing effect of the methyl substituent rather than the chlorine (ratio 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :42). Surprisingly this changes in o-chlorotoluene. Here more product forms that follows the chlorine directing effect (38:62).
:42). Surprisingly this changes in o-chlorotoluene. Here more product forms that follows the chlorine directing effect (38:62).
Interestingly the π-bond character πc of the C–Cl bond seems to reflect this, by showing a larger πc for o-chlorotoluene than for p-chlorotoluene. From this observation it seems plausible that πc is correlated to a more pronounced mesomeric effect and thus to the observed reaction yields.
We also made an attempt to explain the reaction yields in a more detailed qualitative way, provided in the ESI.† Quantitative predictions of reaction yields might be carried out by use of computational methods, which also allow a more detailed insight into the electronic structure of the molecules involved. An example of a study reproducing accurate quantitative yields is the computational investigation of the nitration of toluene.44
The well determined barriers to internal rotation of the methyl group in o-halotoluenes follow an increasing trend with the size of the halogen nucleus. This is expected to be caused by the increasing exchange-repulsion. However, a quantitative calculation of exchange-repulsion, electronic, polarisation and dispersion contributions was not carried out and could add some additional insight. Such calculations could be provided by effective fragmented potentials.45–47 The trend in potential barriers is well reproduced quantum-chemically utilizing the MP2 method. Benchmark setting are the extremely precise ratios of quadrupolar moments for chlorine and bromine isotopes. These can be used to determine the precise quadrupole moment of both isotopes even if the quadrupole moment of only one of them has been determined accurately. Since the series of o-halotoluenes as well as p-halotoluenes are now complete, future investigations might include the series of m-halotoluenes. These will prove to be an experimentally as well as theoretically challenging problem since strong quadrupolar coupling in the presence of very small barrier to internal rotation with significant V3 and V6 contributions are apparent. An initial microwave study on m-fluorotoluene can already be found in ref. 48 and will be extended to a complete FTMW study on the parent and all singly substituted 13C isotopes.49 A study of m-chlorotoluene was recently published.42
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
J. W. wants to thank the China Scholarships Council (CSC) for financial support. K. G. L. wants to thank the Fonds der Chemischen Industrie (FCI) for his fellowship. We want to acknowledge the financial support of the Land Niedersachsen, the Deutsche Forschungsgemeinschaft (DFG) and the support of the cluster system team at the Leibniz University IT services (LUIS) Hannover, Germany.
Notes and references
- M. D. Saltzman, J. Chem. Educ., 1980, 57, 484 CrossRef CAS.
- G. A. Olah, Q. Wang, X. Li and I. Bucsi, Synthesis, 1992, 1085–1086 CrossRef CAS.
- O. Dorosh, E. Bialkowska-Jaworska, Z. Kisiel and L. Pszczólkowski, J. Mol. Spectrosc., 2007, 2, 228–232 CrossRef.
- V. A. Shubert, D. Schmitz and M. Schnell, Mol. Phys., 2013, 111, 2189–2197 CrossRef.
- J. K. G. Watson, J. Mol. Spectrosc., 1977, 65, 123–133 CrossRef CAS.
-
W. Gordy and R. L. Cook, Microwave Molecular Spectra, Wiley, 3rd edn, 1984 Search PubMed.
- J. D. Swalen, Rev. Mod. Phys., 1959, 31, 841–892 CrossRef.
- I. Kleiner, J. Mol. Spectrosc., 2010, 260, 1–18 CrossRef CAS.
- H. P. Benz, A. Bauder and H. Günthard, J. Mol. Spectrosc., 1966, 21, 156–164 CrossRef CAS.
- R. Woods, J. Mol. Spectrosc., 1966, 21, 4–24 CrossRef CAS.
- R. Woods, J. Mol. Spectrosc., 1967, 22, 49–59 CrossRef CAS.
-
H. Hartwig, Dr. rer. nat., dissertation, Christian-Albrechts-Universität Kiel, 1995.
- H. Hartwig and H. Dreizler, Z. Naturforsch., A: Phys. Sci., 1996, 51a, 923–932 Search PubMed.
- I. Merke, A. Lüchow and W. Stahl, J. Mol. Struct., 2006, 780–781, 295–299 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- F. J. Devlin, J. W. Finley, P. J. Stephens and M. J. Frisch, J. Phys. Chem., 1995, 99, 16883–16902 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- M. N. Glukhovtsev, A. Pross, M. P. McGrath and L. Radom, J. Chem. Phys., 1995, 103, 1878 CrossRef CAS.
- K. Schuchardt, B. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CAS.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 32, 756–763 CrossRef CAS PubMed.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742–3748 CrossRef CAS PubMed.
- G. Jansen and B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1989, 39, 6016–6017 CrossRef PubMed.
- J.-U. Grabow, W. Stahl and H. Dreizler, Rev. Sci. Instrum., 1996, 67, 4072–4084 CrossRef CAS.
- M. K. Jahn, D. A. Dewald, D. Wachsmuth, J.-U. Grabow and S. C. Mehrotra, J. Mol. Spectrosc., 2012, 280, 54–60 CrossRef CAS.
- H. M. Pickett, J. Mol. Spectrosc., 1991, 148, 371–377 CrossRef CAS.
- S. Jacobsen, U. Andresen and H. Mäder, Struct. Chem., 2003, 14, 217–225 CrossRef CAS.
- K. P. R. Nair and K. Epple, Chem. Phys. Lett., 1990, 166, 146–152 CrossRef CAS.
- E. C. Richard, R. A. Walker and J. C. Weisshaar, J. Chem. Phys., 1996, 104, 4451–4469 CrossRef CAS.
- K. P. R. Nair, J. Mol. Struct., 1999, 477, 251–254 CrossRef CAS.
- D. Gerhard, A. Hellweg, I. Merke, W. Stahl, M. Baudelet, D. Petitprez and G. Wlodarczak, J. Mol. Spectrosc., 2003, 220, 234–241 CrossRef CAS.
- K. P. R. Nair, J. Demaison, G. Wlodarczak and I. Merke, J. Mol. Spectrosc., 2006, 237, 137–142 CrossRef CAS.
- P. Pyykkö, Mol. Phys., 2001, 99, 1617–1629 CrossRef.
- M. Meyer, W. Stahl and H. Dreizler, J. Mol. Spectrosc., 1992, 151, 243–259 CrossRef CAS.
- C. H. Townes and B. P. Dailey, J. Chem. Phys., 1949, 17, 782–796 CrossRef CAS.
- S. E. Novick, J. Mol. Spectrosc., 2011, 267, 13–18 CrossRef CAS.
- V. Jaccarino, J. G. King, R. A. Satten and H. H. Stroke, Phys. Rev., 1954, 94, 1798–1799 CrossRef CAS.
- W. Gordy, J. Chem. Phys., 1954, 22, 1470–1471 CrossRef CAS.
- K. P. R. Nair, S. Herbers, A. Lesarri and J.-U. Grabow, J. Mol. Spectrosc., 2019, 361, 1–7 CrossRef CAS.
- H. S. Fry, J. Am. Chem. Soc., 1916, 38, 1327–1333 CrossRef CAS.
- Y. Nieves-Quinones and D. A. Singleton, J. Am. Chem. Soc., 2016, 138, 15167–15176 CrossRef CAS PubMed.
- M. S. Gordon, Q. A. Smith, P. Xu and L. V. Slipchenko, Annu. Rev. Phys. Chem., 2013, 64, 553–578 CrossRef CAS PubMed.
- M. S. Gordon, D. G. Fedorov, S. R. Pruitt and L. V. Slipchenko, Chem. Rev., 2012, 112, 632–672 CrossRef CAS PubMed.
- D. Ghosh, D. Kosenkov, V. Vanovschi, C. F. Williams, J. M. Herbert, M. S. Gordon, M. W. Schmidt, L. V. Slipchenko and A. I. Krylov, J. Phys. Chem. A, 2010, 114, 12739–12754 CrossRef CAS PubMed.
- H. D. Rudolph and A. Trinkaus, Z. Naturforsch., 1968, 23a, 68–76 Search PubMed.
-
K. P. R. Nair, et al., unpublished.
Footnote |
| † Electronic supplementary information (ESI) available: All SPFIT and XIAM fit output files and all quantum chemical frequency calculation outputs that are referred to in the article, as well as a detailed discussion of product yields. See DOI: 10.1039/d0cp01145b |
|
| This journal is © the Owner Societies 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Philipp
Buschmann
*a,
Philipp
Buschmann

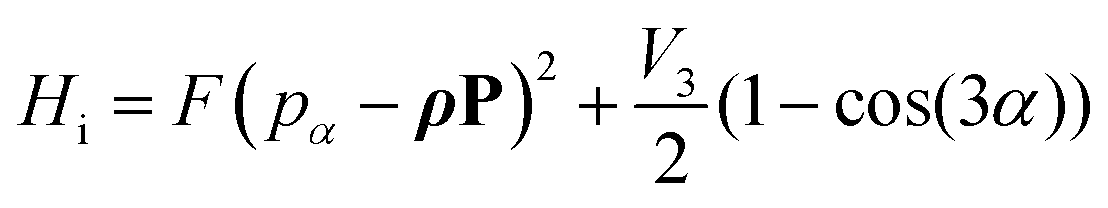

 with λi being the direction cosines for the internal rotation axis with respect to the principal axes i. F0, which is the structural constant to internal rotation directly derived from Iα, can easily be estimated from quantum chemically optimised molecular geometries to be about 161.6 GHz for all o-halogenated toluenes. The vector ρ with
with λi being the direction cosines for the internal rotation axis with respect to the principal axes i. F0, which is the structural constant to internal rotation directly derived from Iα, can easily be estimated from quantum chemically optimised molecular geometries to be about 161.6 GHz for all o-halogenated toluenes. The vector ρ with  in eqn (5) gives rise to cross-terms originating from the coupling between internal and overall rotation. Its absolute value ranges between zero and one. The zero-value of |ρ| = 0 marks the limiting case of vanished coupling. For |ρ| = 0 the internal rotation part would simply lead to a set of energy levels on the potential energy curve along the torsional degree of freedom. These can be labelled either by ±m quantum numbers, commonly used for small barriers to internal rotation, where the eigenstates are close to free rotor states, or at larger barriers by their torsional vibration v quantum numbers. In this article we will follow the latter labelling scheme. With a finite barrier that allows for tunnelling, the v levels will be split into two symmetry species, labelled with their symmetry quantum number σ = 0, ±1. Levels with σ = 0 are referred to as A species and levels with σ = ±1 are referred to as E species. Transitions from A to E species are forbidden, so that for |ρ| = 0 no splittings would arise in the rotational spectrum, since there is no coupling between rotation and internal rotation. For non vanishing |ρ| values the spectrum will be split and each transition can be assigned to a symmetry species.
in eqn (5) gives rise to cross-terms originating from the coupling between internal and overall rotation. Its absolute value ranges between zero and one. The zero-value of |ρ| = 0 marks the limiting case of vanished coupling. For |ρ| = 0 the internal rotation part would simply lead to a set of energy levels on the potential energy curve along the torsional degree of freedom. These can be labelled either by ±m quantum numbers, commonly used for small barriers to internal rotation, where the eigenstates are close to free rotor states, or at larger barriers by their torsional vibration v quantum numbers. In this article we will follow the latter labelling scheme. With a finite barrier that allows for tunnelling, the v levels will be split into two symmetry species, labelled with their symmetry quantum number σ = 0, ±1. Levels with σ = 0 are referred to as A species and levels with σ = ±1 are referred to as E species. Transitions from A to E species are forbidden, so that for |ρ| = 0 no splittings would arise in the rotational spectrum, since there is no coupling between rotation and internal rotation. For non vanishing |ρ| values the spectrum will be split and each transition can be assigned to a symmetry species.

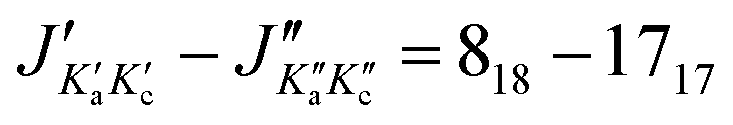 transition of o-bromotoluene. Doppler pairs predicted from the parameters of the fit are coloured red for the A symmetry species and green for the E symmetry species. The separation between the two hyperfine components is 944 kHz, A/E splitting is 12 kHz while the Doppler splitting amounts to 141 kHz with helium as carrier gas.
transition of o-bromotoluene. Doppler pairs predicted from the parameters of the fit are coloured red for the A symmetry species and green for the E symmetry species. The separation between the two hyperfine components is 944 kHz, A/E splitting is 12 kHz while the Doppler splitting amounts to 141 kHz with helium as carrier gas. , without having to rely on the off-diagonal element of the tensor, which would otherwise be the largest source of uncertainty. This precise determination re-derived from the lines provided in ref. 34, which agrees with the recent values of Q35 = −0.08165(80) × 10−24 cm2 and Q37 = −0.06435(64) × 10−24 cm2 tabulated in ref. 36, resulting in a ratio of 1.2688(177).
, without having to rely on the off-diagonal element of the tensor, which would otherwise be the largest source of uncertainty. This precise determination re-derived from the lines provided in ref. 34, which agrees with the recent values of Q35 = −0.08165(80) × 10−24 cm2 and Q37 = −0.06435(64) × 10−24 cm2 tabulated in ref. 36, resulting in a ratio of 1.2688(177).
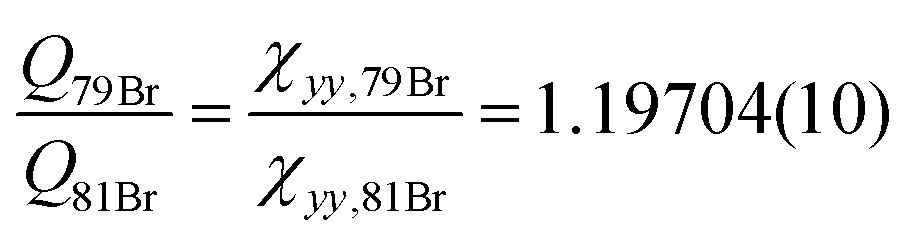 can be derived at high precision, and agrees with the ratio of the recommended quadrupole moments36 of 1.1969(162).
can be derived at high precision, and agrees with the ratio of the recommended quadrupole moments36 of 1.1969(162).