
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidation of the role of guanidinium incorporation in single-crystalline MAPbI3 perovskite on ion migration and activation energy†
Apurba
Mahapatra
 a,
Rashmi
Runjhun
b,
Jan
Nawrocki
b,
Janusz
Lewiński
bc,
Abul
Kalam
d,
Pawan
Kumar
a,
Suverna
Trivedi
e,
Mohammad Mahdi
Tavakoli
f,
Daniel
Prochowicz
*b and
Pankaj
Yadav
*g
a,
Rashmi
Runjhun
b,
Jan
Nawrocki
b,
Janusz
Lewiński
bc,
Abul
Kalam
d,
Pawan
Kumar
a,
Suverna
Trivedi
e,
Mohammad Mahdi
Tavakoli
f,
Daniel
Prochowicz
*b and
Pankaj
Yadav
*g
aDepartment of Physics & Astronomy, National Institute of Technology, Rourkela, 769008, India
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dprochowicz@ichf.edu.pl
cFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
dDepartment of Chemistry, Faculty of Science, King Khalid University, P.O. Box 9004, Abha 61413, Saudi Arabia
eDepartment of Chemical Engineering, National Institute of Technology, Rourkela, 769008, India
fDepartment of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
gDepartment of Solar Energy, School of Technology, Pandit Deendayal Petroleum University, Gandhinagar-382 007, Gujarat, India. E-mail: Pankajphd11@gmail.com
First published on 15th April 2020
Abstract
Ion migration plays a significant role in the overall stability and power conversion efficiency of perovskite solar cells (PSCs). This process was found to be influenced by the compositional engineering of the A-site cation in the perovskite crystal structure. However, the effect of partial A-site cation substitution in a methylammonium lead iodide (MAPbI3) perovskite on the ion migration process and its activation energy is not fully understood. Here we study the effect of a guanidinium (GUA) cation on the ion transport dynamics in the single crystalline GUAxMA1−xPbI3 perovskite composition using temperature-dependent electrochemical impedance spectroscopy (EIS). We find that the small substitution of MA with GUA decreases the activation energy for iodide ion migration in comparison to pristine MAPbI3. The presence of a large GUA cation in the 3D perovskite structure induces lattice enlargement, which perturbs the atomic interactions within the perovskite lattice. Consequently, the GUAxMA1−xPbI3 crystal exhibits a higher degree of hysteresis during current–voltage (J–V) measurements than the single-crystalline MAPbI3 counterpart. Our results provide the fundamental understanding of hysteresis, which is commonly observed in GUA-based PSCs and a general protocol for in-depth electrical characterization of perovskite single crystals.
Introduction
Hybrid organic–inorganic lead halide perovskites have inspired researchers in the field of optoelectronic devices over the last years due to their excellent light absorption, long diffusion length, great photovoltaic (PV) properties, and low-cost processing.1 These properties extend the scope of application of perovskites in tandem solar cells,2 photodetectors3–6 and light-emitting diodes.7–9 Apart from these encouraging factors, many important challenges in the perovskite semiconductors such as low operational stability,10 polarization at low frequencies/long time scales,11,12 and instability to humidity, light exposure and high temperature restrict the commercialization of the PSCs.13 In addition, ion migration is regarded as the main issue that increases the current–voltage (J–V) hysteresis14,15 and reduces the stability of PV devices.15–17The migration process of ions in perovskite films has been intensively studied using photo-thermal induced resonance (PTIR) microscopy,18 temperature-dependent impedance spectroscopy,19,20 Muon spin relaxation (μSR),19 defect spectroscopy,21 current–potential curves22,23 and Kelvin probe force microscopy (c-KFM).24 There are several ions that act as migrating ions in the prototypical MAPbI3 perovskite i.e., (i) methylammonium (MA), (ii) iodide and (iii) proton or hydrogen ions (H+).15,25,26 The H+ ions play a considerably minor role in comparison with other ions due to their low concentration in PSCs.25,27 Moreover, Pb2+ migration within the perovskite structure is hindered due to its high activation energy.28,29 Thus, MA and iodide ions are primarily migrating ions that affects the stability and hysteresis of PV devices due to their low activation energy to move through the perovskite structure (0.5–0.8 eV for MA and 0.2–0.7 eV for iodide ions).20,23,28,30–33 Moreover, the observed hysteresis in perovskite solar cells (PSCs) is ascribed to the iodide ion because it has a diffusion coefficient of 10−12 cm2 s−1, which is 5 times larger than that of MA cations (10−16 cm2 s−1).14,32 By controlling the chemical composition and surface passivation of the perovskite film, the ion motion through the perovskite lattice can be retarded, leading to improved stability and suppressed J–V hysteresis.31,34 For example, the performance, and stability of PSCs are improved, when a part of iodide ions is replaced by smaller bromide ions, which show stronger hydrogen-bonding interactions with the organic ammonium cations35,36 due to higher electronegativity (Mulliken, bromide: 6.76, iodide: 4.24) or hardness (bromide: 4.24, iodide: 3.70).35,37 On the other hand, the stability, and efficiency of PSCs can be enhanced by substitution of MA cations with formamidinium (FA), azetidinium (Az) or guanidinium (GUA) cations.19,38–40 As theoretically studied, GUA cations show potential to solve the hysteresis issue in the PSCs due to its zero dipole moment and larger size of 278 pm as compared to the MA cation (217 pm).41,42 Surprisingly, it was found that the incorporation of GUA in the MAPbI3 perovskite structure enhances the hysteresis effect in comparison to the pristine MAPbI3 based PSCs.38,43 Initially, Yang et al. assumed that GUA is not confined within the perovskite lattice but rather is weakly hydrogen-bonded to the under-coordinated ionic species mainly at grain boundaries.38 They proposed that mobile GUA cations can migrate to the interfaces and screen the applied electric field leading to enhanced hysteresis in the device. However, more recent studies revealed that GUA is able to replace MA cations forming a pure phase of the GUAxMA1−xPbI3-type perovskite.40,44 Under these circumstances, the determination of the precise effect of GUA incorporation into the MAPbI3 perovskite lattice on the ionic migration needs further investigations. Notably, the mentioned studies concerned the investigation of completed devices, where the ion motions can be influenced by the grain boundaries in the perovskite active layer, interfaces of the absorber layer with the electron and hole transporting layers and aging of the solar cells. The presence of grain boundaries provides fast paths for the migration of ions leading to a higher hysteresis and lower stability. Thus, the elucidation of the exact role of the cations and halides on the ion migration process by investigating the polycrystalline perovskite films has not been achieved.
In this work, we study the effect of GUA on ion transport in mixed-cation GUAxMA1−xPbI3 perovskite single crystals using temperature-dependent electrochemical impedance spectroscopy (EIS). The investigation of single crystals allows elimination of the effect of grain boundaries and interfaces on the kinetics of ion migration, and it is crucial to study the intrinsic behavior of perovskite semiconductors for a better understanding of the charge transport and recombination phenomena. We show that the GUAxMA1−xPbI3 perovskite single crystals exhibit lower activation energy for the iodide ion migration than MAPbI3 single crystals. The GUAxMA1−xPbI3 single crystals also exhibit a higher degree of hysteresis during current–voltage (J–V) measurements than the single-crystalline MAPbI3 counterparts, demonstrating higher ionic motion in the GUAxMA1−xPbI3 perovskite system. These results provide a basic insight into the precise effect of the GUA incorporation into the MAPbI3 perovskite lattice on the ion migration process.
Results and discussion
MAPbI3 and GUAxMA1−xPbI3 perovskite single crystals were synthesized using the inverse temperature crystallization method in γ-butyrolactone.45 In the case of GUAxMA1−xPbI3 single crystals, a 1 M precursor solution containing PbI2, MAI and GUAI with the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8:0.2 were employed (for details see the Experimental section). The ratio of the organic cations incorporated into the perovskite structure was estimated using liquid-state 1H NMR analysis by dissolving one separate crystal in 500 μL of DMSO-d6. By comparing the integrals of the MA and GUA peaks, we find that the actual MA/GUA ratio is 0.985:0.015, which remarkably differs from the nominal ratio in the solution (Fig. S1, ESI†). Three crystals from the same batch were measured and the results show the same ratio with a low concentration of GUA in the perovskite crystals. The high discrepancy in the GUA content between the actual and nominal GUA/MA ratio in crystal and solution is likely due to the very low yield of the crystallization process (∼10%), which can be attributed to the high solubility of the perovskite precursors. We also attempted the synthesis of GUAxMA1−xPbI3 single crystals containing a higher GUA content by mixing PbI2, MAI and GUAI with the molar ratios of 1:0.7:0.3 and 1:0.6:0.4, but using the above-mentioned procedure, we failed to crystallize the materials as single crystals. Thus, the crystals with the formula GUA0.015MA0.985PbI3 (GUAMA) were used to further study the properties of GUA-based perovskite crystals. Powder X-ray diffraction (pXRD) measurements were performed on the resulting MAPbI3 and GUAMA crystals to check the structure and phase purity. As shown in Fig. S2 (ESI†), both samples display a similar perovskite crystal structure with diffraction peaks centered at 14.07°, 28.1°, and 31.8°.46 We observed a small shift of the diffraction peaks to lower angles upon introduction of GUA cations, which is in accordance with the literature.40,44 Next, thin layers of silver (Ag) were deposited on both sides of MAPbI3 and GUAMA crystals with dimensions of 4 × 4 × 1.92 mm and 1.7 × 1.7 × 1.2 mm (Fig. S3, ESI†), respectively.
:0.8:0.2 were employed (for details see the Experimental section). The ratio of the organic cations incorporated into the perovskite structure was estimated using liquid-state 1H NMR analysis by dissolving one separate crystal in 500 μL of DMSO-d6. By comparing the integrals of the MA and GUA peaks, we find that the actual MA/GUA ratio is 0.985:0.015, which remarkably differs from the nominal ratio in the solution (Fig. S1, ESI†). Three crystals from the same batch were measured and the results show the same ratio with a low concentration of GUA in the perovskite crystals. The high discrepancy in the GUA content between the actual and nominal GUA/MA ratio in crystal and solution is likely due to the very low yield of the crystallization process (∼10%), which can be attributed to the high solubility of the perovskite precursors. We also attempted the synthesis of GUAxMA1−xPbI3 single crystals containing a higher GUA content by mixing PbI2, MAI and GUAI with the molar ratios of 1:0.7:0.3 and 1:0.6:0.4, but using the above-mentioned procedure, we failed to crystallize the materials as single crystals. Thus, the crystals with the formula GUA0.015MA0.985PbI3 (GUAMA) were used to further study the properties of GUA-based perovskite crystals. Powder X-ray diffraction (pXRD) measurements were performed on the resulting MAPbI3 and GUAMA crystals to check the structure and phase purity. As shown in Fig. S2 (ESI†), both samples display a similar perovskite crystal structure with diffraction peaks centered at 14.07°, 28.1°, and 31.8°.46 We observed a small shift of the diffraction peaks to lower angles upon introduction of GUA cations, which is in accordance with the literature.40,44 Next, thin layers of silver (Ag) were deposited on both sides of MAPbI3 and GUAMA crystals with dimensions of 4 × 4 × 1.92 mm and 1.7 × 1.7 × 1.2 mm (Fig. S3, ESI†), respectively.
In order to calculate the activation energy of ions, we performed electrochemical impedance spectroscopy (EIS) measurements as a function of temperature in the frequency range from 1 MHz to 1 Hz at an AC perturbation voltage of 20 mV under dark conditions. Fig. 1a and b show the EIS Nyquist spectra of both MAPbI3 and GUAMA single crystals at different temperatures. To check the thermal stability of the investigated crystals, the EIS measurements as a function of decreasing temperature were also measured (Fig. S4, ESI†). We found that the EIS spectra recorded during both measurement cycles exhibit a similar behavior, which signifies that the perovskite crystals are thermally stable in the temperature range of 303–363 K. Notably, the obtained Nyquist spectra of both crystals show a single semicircle, which is consistent with our recent report.47 However, two or three semicircles corresponding to the high, mid and low-frequency regions are observed for PSCs. The interpretation of these semicircles is usually explained with the help of an electrical equivalent circuit, which is still controversial in the literature. Nevertheless, it is generally acceptable to ascribe the mid or low frequency (from 1 to 103 Hz) response to ion motion. In the present work, Nyquist spectra of both crystals decrease with increasing temperature due to the increase in the conductivity. The rate of change in the real part of Nyquist spectra is found to be higher for MAPbI3 than that of GUAxMA1−xPbI3 crystals (Fig. S5, ESI†). The higher rate of change in real part of Nyquist spectra for MAPbI3 crystal signifies its lower temperature stability.
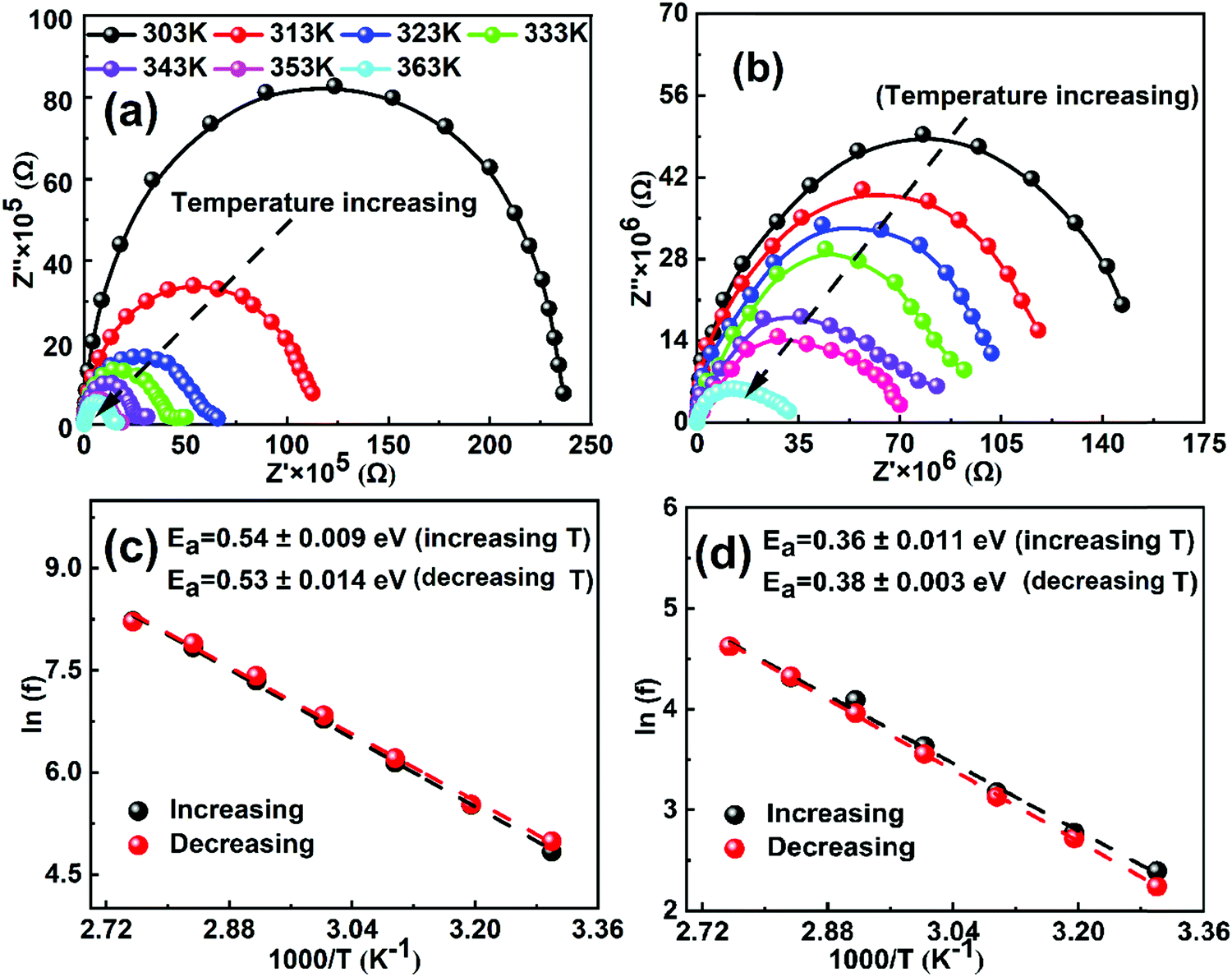 | ||
| Fig. 1 Nyquist plots of the (a) MAPbI3 and (b) GUA0.015MA0.985PbI3 single crystals at 0 V DC bias in the frequencies ranging from 1 MHz to 1 Hz as a function of temperature (313–363 K). Arrhenius plots of the inflection frequencies vs. 1000/T (ln(f0) vs. 1000/T) during increasing and decreasing temperature of (c) MAPbI3 and (d) GUA0.015MA0.985PbI3 crystals. Ea is the activation energy for the traps. | ||
Fig. S6 (ESI†) shows the plots of the complex part of impedance as functions of frequency and temperature. The peaks in the complex impedance spectra at frequencies ranging from 101 to 103 Hz and 101 to 102 Hz are clearly visible for MAPbI3 and GUAxMA1−xPbI3 crystals, respectively. The observed frequency ranges and corresponding time constant is on the same order as the low frequency response in PSCs, which suggests that the net response of perovskite crystals is due to the ion migration. In our previous reports on GUAxMA1−xPbI3 PSCs, we observed a complex impedance peak between 105 and 106 Hz and the corresponding time constant depicts the recombination kinetics in the cell, which is significantly suppressed upon GUA incorporation.43,48 However, such high-frequency response is not observed in the present study. In turn, we observed that with incorporation of a small amount of the GUA cation in the MAPbI3 crystal structure, the rate of change in the apparent time constant is significantly suppressed with a change in temperature. The higher apparent time constant is found to be associated with the hysteresis phenomenon in PSCs.49 The activation energies were extracted from the apparent time constant for both crystals as a function of increasing and decreasing temperature and plotted in Fig. 1c and d. The activation energy of 0.53 and 0.36 eV is found for MAPbI3 and GUAMA, respectively, which is consistent with our47 and other group's results.38 The lower activation energy of the GUAMA crystal signifies that ion migration occurred more easily in this crystal in comparison with the pristine MAPbI3. The activation energy of ions calculated for both MAPbI3 and GUAxMA1−xPbI3 thin films using EIS and temperature-dependent ion conductivity are summarized in Table 1.
| Activation energy of ions for MAPbI3 and GUAxMA1−xPbI3 | |||
|---|---|---|---|
| Activation Energy | Measurements | Ref. | |
| MAPbI3 | 0.624 eV (I−) (single crystal) | Temperature-dependent ion conductivity | 50 |
| 0.82 eV (thin film) | Temperature-dependent ion conductivity | 51 | |
| 0.53 eV (I−) | Temperature-dependent electrochemical impedance spectroscopy (EIS) | 30 | |
| 0.51 eV (I−) | Temperature-dependent ion conductivity from Warburg impedance | 30 | |
| 1.05 eV (single crystal) | Temperature-dependent ion conductivity | 31 | |
| 0.5 eV (thin film) | |||
| 0.43 eV (I−) (pellets) | Temperature-dependent ion conductivity | 52 | |
| MAPbI3 | 0.0211 eV (thin film) | AdmittanceSpectroscopy | 38 |
| MA0.95GA0.05PbI3 | 0.0148 eV | ||
| MAPbI3 | 0.29 eV (thin film) | Temperature-dependent electrochemical impedance spectroscopy (EIS) | 19 |
| MA0.95GA0.05PbI3 | 0.38 eV | ||
| Mid frequency (1 Hz–105 Hz) | |||
| MAPbI3 | 0.4 eV | ||
| MA0.95GA0.05PbI3 | N/A | ||
| Low frequency (>1 Hz) | |||
Most of the values for MAPbI3 and GUAxMA1−xPbI3 thin films are found in the range of 0.3–0.6 eV and 0.1 to 0.7 eV, respectively. For example, Yang et al., have calculated an activation energy of 21.11 meV for MAPbI3 PSC and a significantly lower activation energy of 14.86 meV for GUAxMA1−xPbI3 devices.38 This indicates that the recombination within the bulk of the perovskite film has been successfully suppressed. In turn, Ferdani et al., calculated an activation energy of 0.44 eV for the MAPbI3 PSC by plotting the EIS low-frequency time constant as a function of temperature.19 The obtained activation energy is close to our calculation and is mainly found to be associated with the diffusion of iodide ions. The activation energy for the GUA0.05MA0.95PbI3 PSC has not been derived from EIS, however, the value of 0.78 eV was obtained from Ab initio simulation, which is comparatively higher with respect to our values and the reported values by Yang et al.38
To further confirm the values of activation energies derived from EIS measurements, the conductivity of perovskite crystals was measured under dark conditions (Fig. S7, ESI†) and plotted in the Nerst–Einstien formalism (Fig. 2). It is well-established for mixed conductors that ionic conduction is dominant in the high-temperature region, while electronic conduction is dominant in the low-temperature region.53 From the obtained plot, the activation energy is derived using the expression of 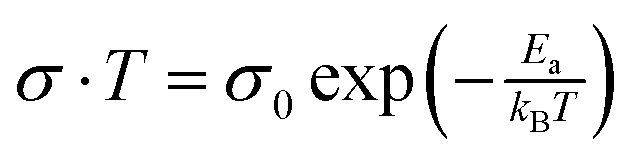 , where kB is the Boltzman constant, T is the absolute temperature and σ0 is a constant.54 By using the Arrhenius plot, the activation energies of 0.52 and 0.36 eV are obtained for MAPbI3 and GUAMA crystals, respectively. The obtained activation energies are in good agreement with our EIS findings. For metal halide perovskites, iodide vacancies have been found to have a more detrimental influence on the charge trapping and non-radiative recombination because of their deep energy levels.
, where kB is the Boltzman constant, T is the absolute temperature and σ0 is a constant.54 By using the Arrhenius plot, the activation energies of 0.52 and 0.36 eV are obtained for MAPbI3 and GUAMA crystals, respectively. The obtained activation energies are in good agreement with our EIS findings. For metal halide perovskites, iodide vacancies have been found to have a more detrimental influence on the charge trapping and non-radiative recombination because of their deep energy levels.
 | ||
| Fig. 2 Temperature-dependent conductivity of (a) MAPbI3 and (b) GUA0.015MA0.985PbI3 single crystals (Arrhenius plot). | ||
The ion activation energy is also defined as a result of the interaction between the ion and the perovskite lattice. This interaction can be easily tuned by the substitution of the halide or A-site cations in the perovskite structure. In our previous works, using solid-state NMR, we showed that when MA with a radius of 2.17 Å is substituted by a cation with larger ionic radius, i.e., FA (2.53 Å)55 or GUA (2.78 Å),40 these organic groups exhibit a higher degree of rotation. This behavior can cause an additional strain and weaken the atomic interaction inside the lattice and consequently decrease the ion activation energy (see Fig. 3).56 However, the substitution of the A-site cation with a lower radius cation, e.g. Rb (1.52 Å) or Cs (1.67 Å), compresses the perovskite lattice and suppresses the ion motion.57 For instance, Zhou et al., have shown that Cs-based perovskites possess higher ion activation energy as compared to MA-based ones, which led to better light stability.58
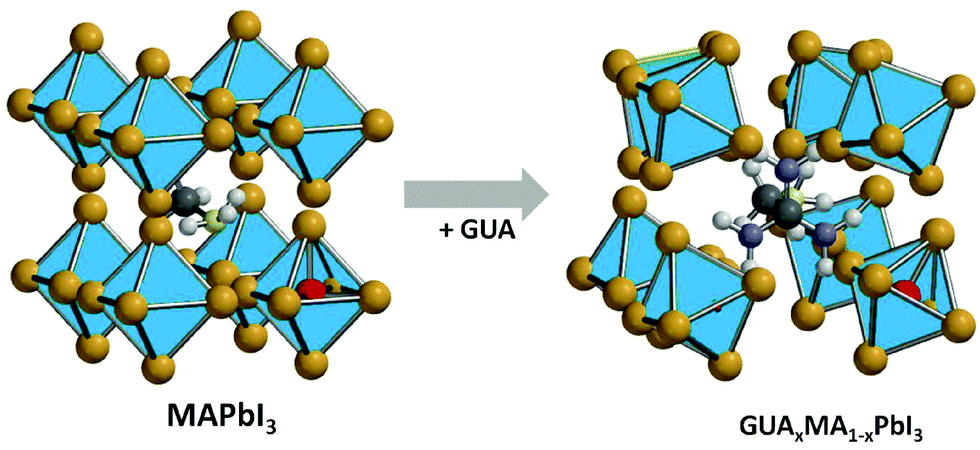 | ||
| Fig. 3 Schematic representation of the introduction of the GUA cation into the MAPbI3 crystal lattice. | ||
The suppression of ion migration with increasing ion activation energy can be easily observed during current–voltage (I–V) measurement at different scan rates. Fig. S8 (ESI†) shows the hysteresis behavior of both crystals at different scan rates. A higher hysteresis effect is observed for the GUAMA crystal, which confirms the higher ionic motion in the GUAxMA1−xPbI3 perovskite systems.19,42,44
Conclusions
The effect of GUA incorporation into the MAPbI3 perovskite lattice on the ion transport properties has been investigated using a combination of electrochemical impedance spectroscopy (EIS) and temperature-dependent ion conductivity techniques. This study provides the first insights into the effect on iodide ion transport in the GUAxMA1−xPbI3 single crystal. The calculated average activation energy corresponding to the ion migration for MAPbI3 and GUAxMA1−xPbI3 crystals are 0.52 and 0.36 eV, respectively. The substitution of MA with GUA cations in the MAPbI3 crystal lattice is found to cause an additional strain and weaken the atomic interaction inside the lattice. This consequently decreases the ion activation energy of MAPbI3 crystals. The present study enhances our fundamental understanding of ion migration and provides a basic insight into the mixed-cation perovskite single crystals.Experimental section
Synthesis of MAPbI3 and GUAxMA1−xPbI3 single crystals
1.0 M MAPbI3 solution was prepared by dissolving equimolar amounts of PbI2 and MAI in γ-butyrolactone at 60 °C overnight. Similarly, perovskite solution containing PbI2, MAI and GUAI in a molar ratio of 1:0.8:0.2 was prepared in γ-butyrolactone and kept at 60 °C overnight. Before crystallization, the solution was filtered using 0.2 μm pore size PTFE filter. Next, 2 mL filtered precursor solution was kept at 160 °C for 30 min in an oil bath, which resulted in the growth of very small seed crystals. The seed crystal solution was further heated at 120 °C for 3 h. Finally, the remaining solution was discarded, and crystals were washed with acetone 2–3 times and then dried.
Characterization of perovskite single crystals
The 1H NMR spectra were recorded at 298 K on a Bruker 600 AVANAC III spectrometer and the spectra were analyzed using Nova software. Powder X-ray diffraction patterns were recorded with a Empyrean diffractometer (PANalytical) equipped with a copper lamp (40 kV, 40 mA). The pXRD patterns were recorded for finely ground crystals over a 2θ range of 10° to 40° without rotating the sample using a low background Si sample holder. EIS and temperature-dependent ion conductivity measurements were performed using a potentiostat (Autolab) equipped with a frequency response analyzer. EIS measurements were performed as a function of temperature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. is thankful to the Dean of Scientific Research, King Khalid University for financial support by grant number G.R.P. 42–40. R. R. acknowledge the funding from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska Curie grant agreement No. 711859 and Polish Ministry of Science and Higher Education from the co-funded project, grant agreement no. 3549/H2020/COFUND2016/2. D. P. acknowledges the financial support from the HOMING programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (POIR.04.04.00-00-5EE7/18-00). P. Y. acknowledges the ORSP of Pandit Deendayal Petroleum University and the DST SERB (CRG/2018/000714) and DST Nano Mission (DST/NM/NT/2018/174) for financial support.References
- A. Mahapatra, D. Prochowicz, M. M. Tavakoli, S. Trivedi, P. Kumar and P. Yadav, J. Mater. Chem. A, 2019, 8, 27–54 RSC.
- G. E. Eperon, T. Leijtens, K. A. Bush, R. Prasanna, T. Green, J. T.-W. W. Wang, D. P. McMeekin, G. Volonakis, R. L. Milot, R. May, A. Palmstrom, D. J. Slotcavage, R. A. Belisle, J. B. Patel, E. S. Parrott, R. J. Sutton, W. Ma, F. Moghadam, B. Conings, A. Babayigit, H.-G. G. Boyen, S. Bent, F. Giustino, L. M. Herz, M. B. Johnston, M. D. McGehee and H. J. Snaith, Science, 2016, 354, 861–865 CrossRef CAS PubMed.
- Y. Guo, C. Liu, H. Tanaka and E. Nakamura, J. Phys. Chem. Lett., 2015, 6, 535–539 CrossRef CAS PubMed.
- H. Deng, X. Yang, D. Dong, B. Li, D. Yang, S. Yuan, K. Qiao, Y.-B. Cheng, J. Tang and H. Song, Nano Lett., 2015, 15, 7963–7969 CrossRef CAS PubMed.
- L. Dou, Y. Yang, J. You, Z. Hong, W.-H. Chang, G. Li and Y. Yang, Nat. Commun., 2014, 5, 5404 CrossRef CAS PubMed.
- K. Pandey, M. Chauhan, V. Bhatt, B. Tripathi, P. Yadav and M. Kumar, RSC Adv., 2016, 6, 105076–105080 RSC.
- O. A. Jaramillo-Quintero, R. S. Sanchez, M. Rincon and I. Mora-Sero, J. Phys. Chem. Lett., 2015, 6, 1883–1890 CrossRef CAS PubMed.
- N. K. Kumawat, D. Gupta and D. Kabra, Energy Technol., 2017, 5, 1734–1749 CrossRef CAS.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 RSC.
- E. J. Juarez-Perez, R. S. Sanchez, L. Badia, G. Garcia-Belmonte, Y. S. Kang, I. Mora-Sero and J. Bisquert, J. Phys. Chem. Lett., 2014, 5, 2390–2394 CrossRef CAS PubMed.
- Z. Xiao, Y. Yuan, Y. Shao, Q. Wang, Q. Dong, C. Bi, P. Sharma, A. Gruverman and J. Huang, Nat. Mater., 2015, 14, 193–198 CrossRef CAS PubMed.
- P. Zhao, B. J. Kim and H. S. Jung, Mater. Today Energy, 2018, 7, 267–286 CrossRef.
- M. De Bastiani, G. Dell’Erba, M. Gandini, V. D’Innocenzo, S. Neutzner, A. R. S. Kandada, G. Grancini, M. Binda, M. Prato, J. M. Ball, M. Caironi and A. Petrozza, Adv. Energy Mater., 2016, 6, 1501453 CrossRef.
- M. L. Petrus, J. Schlipf, C. Li, T. P. Gujar, N. Giesbrecht, P. Müller-Buschbaum, M. Thelakkat, T. Bein, S. Hüttner and P. Docampo, Adv. Energy Mater., 2017, 7, 1700264 CrossRef.
- J. Carrillo, A. Guerrero, S. Rahimnejad, O. Almora, I. Zarazua, E. Mas-Marza, J. Bisquert and G. Garcia-Belmonte, Adv. Energy Mater., 2016, 6, 1502246 CrossRef.
- M. Bag, L. A. Renna, R. Y. Adhikari, S. Karak, F. Liu, P. M. Lahti, T. P. Russell, M. T. Tuominen and D. Venkataraman, J. Am. Chem. Soc., 2015, 137, 13130–13137 CrossRef CAS PubMed.
- Y. Yuan, J. Chae, Y. Shao, Q. Wang, Z. Xiao, A. Centrone and J. Huang, Adv. Energy Mater., 2015, 5, 1500615 CrossRef.
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak, A. B. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, M. S. Islam and P. J. Cameron, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- A. Pockett, G. E. Eperon, N. Sakai, H. J. Snaith, L. M. Peter and P. J. Cameron, Phys. Chem. Chem. Phys., 2017, 19, 5959–5970 RSC.
- Y. Hu, E. M. Hutter, P. Rieder, I. Grill, J. Hanisch, M. F. Aygüler, A. G. Hufnagel, M. Handloser, T. Bein, A. Hartschuh, K. Tvingstedt, V. Dyakonov, A. Baumann, T. J. Savenije, M. L. Petrus and P. Docampo, Adv. Energy Mater., 2018, 8, 1703057 CrossRef.
- S. Meloni, T. Moehl, W. Tress, M. Franckeviius, M. Saliba, Y. H. Lee, P. Gao, M. K. Nazeeruddin, S. M. Zakeeruddin, U. Rothlisberger and M. Graetzel, Nat. Commun., 2016, 7, 10334 CrossRef CAS PubMed.
- I. Levine, P. K. Nayak, J. T.-W. Wang, N. Sakai, S. Van Reenen, T. M. Brenner, S. Mukhopadhyay, H. J. Snaith, G. Hodes and D. Cahen, J. Phys. Chem. C, 2016, 120, 16399–16411 CrossRef CAS.
- J. S. Yun, J. Seidel, J. Kim, A. M. Soufiani, S. Huang, J. Lau, N. J. Jeon, S. Il Seok, M. A. Green and A. Ho-Baillie, Adv. Energy Mater., 2016, 6, 1600330 CrossRef.
- J. M. Frost and A. Walsh, Acc. Chem. Res., 2016, 49, 528–535 CrossRef CAS PubMed.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS PubMed.
- D. A. Egger, L. Kronik and A. M. Rappe, Angew. Chem., Int. Ed., 2015, 54, 12437–12441 CrossRef CAS PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- N. K. Elumalai, M. A. Mahmud, D. Wang and A. Uddin, Energies, 2016, 9, 861 CrossRef.
- M. N. F. Hoque, N. Islam, Z. Li, G. Ren, K. Zhu and Z. Fan, ChemSusChem, 2016, 9, 2692–2698 CrossRef CAS PubMed.
- J.-W. W. Lee, S.-G. G. Kim, J.-M. M. Yang, Y. Yang and N.-G. G. Park, APL Mater., 2019, 7, 041111 CrossRef.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- Y. Yuan, Q. Wang, Y. Shao, H. Lu, T. Li, A. Gruverman and J. Huang, Adv. Energy Mater., 2016, 6, 1501803 CrossRef.
- A. Mahapatra, D. Prochowicz, M. M. Tavakoli, S. Trivedi, P. Kumar and P. Yadav, J. Mater. Chem. A, 2020, 8, 27–54 RSC.
- R. Ruess, F. Benfer, F. Böcher, M. Stumpp and D. Schlettwein, ChemPhysChem, 2016, 17, 1505–1511 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- N. De Marco, H. Zhou, Q. Chen, P. Sun, Z. Liu, L. Meng, E. P. Yao, Y. Liu, A. Schiffer and Y. Yang, Nano Lett., 2016, 16, 1009–1016 CrossRef PubMed.
- S. R. Pering, W. Deng, J. R. Troughton, P. S. Kubiak, D. Ghosh, R. G. Niemann, F. Brivio, F. E. Jeffrey, A. B. Walker, M. S. Islam, T. M. Watson, P. R. Raithby, A. L. Johnson, S. E. Lewis and P. J. Cameron, J. Mater. Chem. A, 2017, 5, 20658–20665 RSC.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, M. Saski, P. Yadav, D. Bi, N. Pellet, J. Lewiński, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 3345–3351 CrossRef CAS PubMed.
- G. Giorgi, J.-I. I. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. C, 2015, 119, 4694–4701 CrossRef CAS.
- G. Giorgi and K. Yamashita, Nanotechnology, 2015, 26, 442001 CrossRef PubMed.
- D. Prochowicz, M. M. Tavakoli, A. Q. Alanazi, S. Trivedi, H. Tavakoli Dastjerdi, S. M. Zakeeruddin, M. Grätzel and P. Yadav, ACS Omega, 2019, 4, 16840–16846 CrossRef CAS PubMed.
- A. D. Jodlowski, C. Roldán-Carmona, G. Grancini, M. Salado, M. Ralaiarisoa, S. Ahmad, N. Koch, L. Camacho, G. D. Miguel and M. K. Nazeeruddin, Nat. Energy, 2017, 2, 972–979 CrossRef CAS.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- Z. Lian, Q. Yan, Q. Lv, Y. Wang, L. Liu, L. Zhang, S. Pan, Q. Li, L. Wang and J. L. Sun, Sci. Rep., 2015, 5, 16563 CrossRef PubMed.
- A. Kalam, R. Rashmi, M. Apurba, M. M. Tavakoli, T. Suverna, H. T. Dastjerdi, K. Pawan, J. Lewiński, M. Pandey, D. Prochowicz and P. Yadav, J. Phys. Chem. C, 2020, 124, 3496–3502 CrossRef CAS.
- D. Prochowicz, M. M. Tavakoli, A. Kalam, R. D. Chavan, S. Trivedi, M. Kumar and P. Yadav, J. Mater. Chem. A, 2019, 7, 8218–8225 RSC.
- F. Ebadi, N. Taghavinia, R. Mohammadpour, A. Hagfeldt and W. Tress, Nat. Commun., 2019, 10, 1574 CrossRef PubMed.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365, 473–478 CrossRef CAS PubMed.
- Y. C. Zhao, W. K. Zhou, X. Zhou, K. H. Liu, D. P. Yu and Q. Zhao, Light: Sci. Appl., 2017, 6, e16243 CrossRef CAS PubMed.
- T.-Y. Yang, G. Gregori, N. Pellet, M. Grätzel and J. Maier, Angew. Chem., Int. Ed., 2015, 54, 7905–7910 CrossRef CAS PubMed.
- H. Lee, S. Gaiaschi, P. Chapon, D. Tondelier, J. E. Bourée, Y. Bonnassieux, V. Derycke and B. Geffroy, J. Phys. Chem. C, 2019, 123, 17728–17734 CrossRef CAS.
- R. A. McKee, Solid State Ionics, 1981, 5, 133–136 CrossRef CAS.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. Péchy, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 10055–10061 CrossRef CAS PubMed.
- J. Shi, Y. Li, Y. Li, D. Li, Y. Luo, H. Wu and Q. Meng, Joule, 2018, 2, 879–901 CrossRef CAS.
- M. Saliba, T. Matsui, K. Domanski, J. Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J. P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- W. Zhou, Y. Zhao, X. Zhou, R. Fu, Q. Li, Y. Zhao, K. Liu, D. Yu and Q. Zhao, J. Phys. Chem. Lett., 2017, 8, 4122–4128 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01119c |
| This journal is © the Owner Societies 2020 |