
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trends in stabilisation of Criegee intermediates from alkene ozonolysis†
Mike J.
Newland
 *a,
Beth S.
Nelson
a,
Amalia
Muñoz
b,
Milagros
Ródenas
b,
Teresa
Vera
b,
Joan
Tárrega
b and
Andrew R.
Rickard
ac
*a,
Beth S.
Nelson
a,
Amalia
Muñoz
b,
Milagros
Ródenas
b,
Teresa
Vera
b,
Joan
Tárrega
b and
Andrew R.
Rickard
ac
aWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, UK. E-mail: mike.newland@gmail.com
bFundación CEAM, EUPHORE Laboratories, Avda Charles R. Darwin 14. Parque Tecnológico, Valencia, Spain
cNational Centre for Atmospheric Science, Wolfson Atmospheric Chemistry Laboratories, University of York, UK
First published on 4th June 2020
Abstract
Criegee Intermediates (CI), formed in the ozonolysis of alkenes, play a central role in tropospheric chemistry as an important source of radicals, with stabilised CI (SCI) able to participate in bimolecular reactions, affecting climate through the formation of inorganic and organic aerosol. However, total SCI yields have only been determined for a few alkene systems, while speciated SCI yields from asymmetrical alkenes are almost entirely unknown. Here we report for the first time a systematic experimental exploration of the stabilisation of CH2OO and (CH3)2COO CI, formed from ten alkene–ozone systems with a range of different sizes and structures, under atmospherically relevant conditions in the EUPHORE chamber. Experiments in the presence of excess SO2 (an SCI scavenger) determined total SCI yields from each alkene–ozone system. Comparison of primary carbonyl yields in the presence/absence of SO2 determined the stabilisation fraction of a given CI. The results show that the stabilisation of a given CI increases as the size of the carbonyl co-product increases. This is interpreted in terms of the nascent population of CI formed following decomposition of the primary ozonide (POZ) having a lower mean energy distribution when formed with a larger carbonyl co-product, as more of the energy from the POZ is taken by the carbonyl. These findings have significant implications for atmospheric modelling of alkene ozonolysis. Higher stabilisation of small CI formed from large alkenes is expected to lead to lower radical yields from CI decomposition, and higher SCI concentrations, increasing the importance of SCI bimolecular reactions.
Introduction
The formation of Criegee intermediates (CI) from gas phase alkene ozonolysis has received attention over the past five decades owing to their role as important non-photolytic sources of radicals (OH, HO2 and RO2) to the troposphere.1–5 More recently, the potential importance of bimolecular reactions of stabilised CI (SCI) has been the subject of much research (see Vereecken et al.6 and references within). These reactions contribute significantly to the sulfuric acid budget in certain environments through oxidation of SO2,7 and the acidity of the atmosphere through removal of organic and inorganic acids.8 Bimolecular reactions of SCI have also been implicated in the formation of aerosol from monoterpene ozonolysis9 through dimerization,10,11 oligomerization12 and reaction with peroxy radicals,13 contributing to new particle formation14 and so directly affecting cloud condensation nuclei,15,16 rainfall, and climate.The Criegee ozonolysis reaction mechanism17 proceeds via concerted cycloaddition of the ozone molecule across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to form a chemically activated primary ozonide (POZ), followed by cleavage of the C–C bond and one of the O–O bonds forming a carbonyl molecule and a carbonyl oxide, or ‘Criegee intermediate’,18 see Scheme 1. A population of ozonolysis derived CIs in the gas-phase is formed with a broad internal energy distribution.19 A fraction of CI may be formed ‘cold’ (although this is not the case for all alkenes20,21), that is, without enough internal energy to undergo prompt decomposition, these are termed stabilised Criegee intermediates (SCI). The remainder are formed chemically activated (CI*). These CI* tend to undergo prompt decomposition on a timescale of nanoseconds.22 However, they can also be collisionally deactivated to add to the SCI population. The distinction between these two routes to SCI formation has been demonstrated in laboratory experiments performed as a function of pressure.20,21,23–25 The fraction of each type of CI that is formed will depend on the initial energy distribution of the CI population.24
C double bond to form a chemically activated primary ozonide (POZ), followed by cleavage of the C–C bond and one of the O–O bonds forming a carbonyl molecule and a carbonyl oxide, or ‘Criegee intermediate’,18 see Scheme 1. A population of ozonolysis derived CIs in the gas-phase is formed with a broad internal energy distribution.19 A fraction of CI may be formed ‘cold’ (although this is not the case for all alkenes20,21), that is, without enough internal energy to undergo prompt decomposition, these are termed stabilised Criegee intermediates (SCI). The remainder are formed chemically activated (CI*). These CI* tend to undergo prompt decomposition on a timescale of nanoseconds.22 However, they can also be collisionally deactivated to add to the SCI population. The distinction between these two routes to SCI formation has been demonstrated in laboratory experiments performed as a function of pressure.20,21,23–25 The fraction of each type of CI that is formed will depend on the initial energy distribution of the CI population.24
 | ||
| Scheme 1 Simplified mechanism for the production of CI and primary and secondary carbonyl products in the ozonolysis of 2-methyl propene. | ||
Unimolecular decomposition of CI yields a range of radical products, with decomposition of syn-CI (i.e. CI with an alkyl group on the same side of the terminal O atom of the carbonyl oxide moiety), via a vinyl hydroperoxide, producing OH with (near) unit yield. SCI can have sufficiently long lifetimes to undergo bimolecular reactions with H2O, RCOOH, and SO2, amongst other species, in the atmosphere. Hence the relative yields of CI/SCI from a particular alkene–ozone system determine the effect of that system on atmospheric composition.
Inclusion of alkene ozonolysis chemistry in regional and global chemical transport models is essential to correctly estimate radical concentrations, and the product distribution from the removal of alkenes by reaction with ozone. However, such mechanisms must firstly parameterize SCI yields for a structurally diverse range of alkenes for which no measurements exist, lumping together SCI of different structures and hence reactivities, and secondly lumping together stabilisation of different SCI. This leads to large uncertainties on the SCI yields and hence on the effect of alkene–ozone reactions on atmospheric composition.
Many laboratory studies probing the chemistry of SCI have utilised the facile photolysis of alkyl iodides to yield SCI in the presence of oxygen.26 This has proved an invaluable resource for the study of SCI chemistry. However, alkene ozonolysis is expected to be the dominant source of CI to the atmosphere, and certain atmospherically relevant questions, such as the fractional CI and SCI yields from alkene ozonolysis, can only be answered by probing the alkene–ozone system.
Total SCI yields have previously been measured from a number of atmospherically relevant alkene–ozone systems including short chain alkenes,27–31 isoprene,32–34 and monoterpenes.7,32 These yields are generally measured indirectly, either by the removal of an SCI scavenger (e.g. SO2) or formation of a product from an SCI scavenging reaction (e.g. production of H2SO4, via the SO2 + SCI reaction). However, very little experimental information exists on the relative amounts of different SCI formed in non-symmetrical systems. For non-symmetrical, non-cyclic, alkenes, a pair of CI and carbonyl co-products are formed (Scheme 1). The yield of each SCI depends on: (i) the yield of each CI – determined by the fragmentation of the POZ (α); (ii) the fraction of each CI that is stabilised (S). The stabilisation of a given CI produced from different alkene–ozone systems might be expected to differ. For example, while the fraction of CH2OO stabilised in ethene ozonolysis has been determined to be 0.35–0.54 based on a wide range of experimental studies, the chamber studies of Nguyen et al.34 suggest that the majority of CH2OO formed in isoprene ozonolysis is stabilised.
Here, we present results of a series of alkene ozonolysis experiments carried out at the European PhotoReactor facility (EUPHORE), Valencia, Spain, in which yields of chemically activated and stabilised CH2OO and (CH3)2COO are determined, for the first time, for a systematic range of alkene–ozone systems under atmospherically relevant conditions. An empirical structure–activity relationship for the stabilisation of these types of Criegee intermediates, based on the size of the carbonyl co-product formed in the decomposition of the primary ozonide is also presented, with the atmospheric implications of the results discussed.
Experimental
EUPHORE
EUPHORE is a 200 m3 simulation chamber used for studying reaction mechanisms under atmospheric boundary layer conditions. The chamber is fitted with large horizontal and vertical fans to ensure rapid mixing (three minutes). Further details of the chamber setup and instrumentation are available elsewhere.35–37 Experiments comprised time-resolved measurement of the formation of carbonyl products and the loss of alkene and ozone (and in some experiments SO2). SO2 and O3 abundance were measured using conventional fluorescence and UV absorption monitors, respectively; alkene and oxygenated volatile organic compound abundance was determined via FTIR spectroscopy and PTR-MS. The precision of the SO2 and O3 monitors were 0.25 and 0.47 ppbv respectively (evaluated as 2 standard deviations of the measured value prior to SO2 or O3 addition). Experiments were performed in the dark (i.e., with the chamber housing closed; j(NO2) ≤ 10−6 s−1), at atmospheric pressure (ca. 1000 mbar) and temperatures between 297 and 305 K, on timescales of ca. 30–90 minutes. Chamber dilution was monitored via the first order decay of an aliquot of SF6, added prior to each experiment. Cyclohexane (ca. 75 ppmv) was added at the beginning of each experiment to act as an OH scavenger, such that SO2 reaction with OH was calculated to be ≤1% of the total chemical SO2 removal in all experiments.Experimental approach
Experimental procedure comprised addition of SF6 and cyclohexane, followed by O3 (ca. 1000 ppbv for the experiments with alkenes producing CH2OO, which generally have reaction rates with ozone ∼1 × 10−17 cm3 s−1; and ca. 500 ppbv for the experiments with alkenes producing (CH3)2COO, which generally have reaction rates with ozone ∼1 × 10−16 cm3 s−1) and SO2 if used (ca. 2000 ppbv). A gap of five minutes was left prior to addition of the alkene, to allow complete mixing. The reaction was then initiated by addition of the alkene (ca. 800 ppbv for the systems producing CH2OO, and 400 ppbv for those producing (CH3)2COO). The chamber was monitored for 30–90 minutes subsequent to the addition of the alkene depending on the rate of reaction with ozone, the rate of alkene/ozone consumption being dependent on k(alkene + ozone). Roughly 50% of the CH2OO producing alkenes were consumed after 60 minutes, while 90% of the faster reacting alkenes were consumed within roughly 25 minutes. The experiments were performed under dry conditions (RH < 1%). A full experiment list is given in Table S1 (ESI†).Results and discussion
Total SCI yields
Alkene ozonolysis experiments were performed in the presence of ca. 2000 ppbv SO2, such that the overwhelming majority (≥94%) of the SCI was scavenged and converted to a carbonyl. The total SCI yield, ϕ, was calculated by regressing the loss of ozone (dO3) against the loss of SO2 (dSO2) (eqn (E1)), both corrected for chamber dilution. In reality the experimentally determined value of ϕ is a minimum value, ϕmin since other loss channels for the SCI (e.g. decomposition) may still be having a small but non-negligible effect (Newland et al. (2015a)), accounted for in eqn (E1) by f. | (E1) |
Total SCI yields were calculated for ten alkene–ozone systems in this work (C2–C10). Fig. 1 shows an example plot of ΔSO2vs. ΔO3 for an ozonolysis experiment with 2,4-dimethyl-pent-2-ene. Uncertainties are ±2σ and represent the combined systematic (estimated measurement uncertainty) and precision components. Fig. S1 (ESI†) shows total SCI plots for each of the ten alkenes studied. Table 1 gives the total SCI yields corrected for additional SCI losses, i.e. f, (see Tables S2 and S3, ESI† this correction generally increased the yields by ∼5%) calculated for each alkene, and values previously reported in the literature where available.
 | ||
| Fig. 1 Total SCI yield (ϕSCI-min) in 2,4-dimethyl pent-2-ene ozonolysis, derived from the removal of SO2 (ΔSO2) relative to the removal of ozone (ΔO3) (eqn (E1)). Dashed line: linear regression of measurements. Data is not corrected for additional loss processes – see text for details. Vertical and horizontal error bars represent precision uncertainties. | ||
| Alkene | SCI yielda | Literature values | Method |
|---|---|---|---|
| a Uncertainties are ±2σ and represent the combined systematic (estimated measurement uncertainty) and precision components. | |||
| Ethene (C2) | 0.43 (±0.02) | 0.35–0.54 (see Newland et al.;31 Alam et al.36 for refs) | |
| Propene (C3) | 0.34 (±0.01) | 0.25 (±0.02)28 | ΔH2SO4/ΔO3 |
| 0.4441 | Carbonyl product yields | ||
| 2-Methylpropene (C4) | 0.24 (±0.03) | 0.17 (±0.03)28 | ΔH2SO4/ΔO3 |
| 2-Methyl-but-1-ene (C5) | 0.33 (±0.01) | ||
| 1-Heptene (C7) | 0.61 (±0.03) | ||
| 2-Methyl-but-2-ene (C5) | 0.28 (±0.01) | ||
| 2,3-Dimethyl-but-2-ene (C6) | 0.31 (±0.04) | 0.10–0.65 (see Newland et al.31 for refs) | |
| 2,4-Dimethyl-pent-2-ene (C7) | 0.41 (±0.01) | ||
| 2,3,4-Trimethyl-pent-2-ene (C8) | 0.49 (±0.01) | ||
| Myrcene (C10) | 0.46 (±0.03) | 0.3542 | Theoretical calculations |
For alkenes for which total SCI yields have been measured previously, there is good agreement between the values measured here and those reported in the literature. The ethene–ozone system is the most studied, with reported SCI yields ranging from 0.35–0.54, from a range of different techniques – see Newland et al.;31 Alam et al.36 for further references. The value of 0.43 (±0.02) derived here lies towards the lower end of this range. For propene only two values exist in the literature. The value of 0.28 (±0.02) derived here is in good agreement with the value of 0.25 (±0.02) from Hatakeyama et al.28 from determination of H2SO4 production relative to ozone loss in chamber experiments, and considerably lower than the value of 0.44 from Horie and Moortgat,41 derived from analysis of carbonyl products from the propene–ozone reaction. For 2-methylpropene, Hatakeyama et al.28 derived a value of 0.17 (±0.03), using the method described above, compared to our value of 0.21 (±0.04). Deng et al.42 recently reported a total SCI yield of 0.35 for the monoterpene myrcene based on theoretical calculations (82% (CH3)2COO, 15% large anti-SCI, 3% large syn-SCI), compared to our measured value of 0.46. It may be expected that our experimental yield is an underestimation due to the very fast unimolecular reaction of the large anti-SCI formed in this system (calculated by Deng et al.42 to be 7600 s−1) which means that probably <50% would be scavenged by 2000 ppbv SO2. Based on the yield of anti-SCI predicted by Deng et al.42 (15%), this may lead to an underestimation of the total SCI yield of ∼8%. None of the other alkenes studied here have had total SCI yields reported previously to the authors’ knowledge.
Fig. 2 shows the total SCI yields measured in this study plotted against carbon number of the parent alkene. Previous studies43 have noted that total SCI yields do not appear to display a strong dependence on alkene size. The systems studied here suggest a weak dependence on the size of the parent alkene, with total SCI yield increasing with alkene size. Propene and 2-methylpropene have total SCI yields of 0.24–0.34, while the larger trisubstituted alkenes, 2,3,4 trimethyl-pent-2-ene and myrcene have yields of 0.41–0.49, and the largest terminal alkene, 1-heptene has a yield of 0.61 (±0.03). However, as shown later the total SCI yield is the product of a number of effects and hence any simple relation to alkene size must be treated with caution.
 | ||
| Fig. 2 Total SCI yields derived from ten alkene–ozone systems in this work. | ||
Primary carbonyl yields
For non-symmetrical, non-cyclic, alkenes, a pair of CI and carbonyl co-products are formed in the decomposition of the POZ (Scheme 1). The sum yield of pathways α1 + α2 (i.e. the sum yield of the two possible primary carbonyls) should be equal to one. This has been confirmed to be the case in the extensive experimental dataset of Grosjean and Grosjean44 for systems in which the smaller carbonyl is not also formed from decomposition of the larger CI. Therefore, by determining the yield of just one of the carbonyls (ideally the larger one, as it cannot be formed in CI decomposition) it is possible to determine both the primary yield of the other carbonyl, and the yield of both CI. For most CI, syn and anti conformers can also be formed because of the negligible rotation about the CO bond of the CI moiety (Vereecken and Francisco, 2012). However, the two CI of focus in this study, CH2OO and (CH3)2COO, are both symmetrical.
The relative magnitude of pathways α1 and α2 is determined by the fragmentation of the POZ. This appears to be determined predominantly by the structure around the double bond at which ozonolysis occurs.3,45 A fraction, S, of each of the two CI formed is stabilised, either due to being formed below the energy threshold for being chemically activated, or via collisional stabilisation. The yield of each SCI is thus given by the product of αi and Si (eqn (E2); Scheme 1). The total yield of SCI from a given alkene is then the sum of the yields of the two specific SCI (eqn (E3)).
| ϕSCI-min = α × S | (E2) |
| ϕSCI-tot = α1 × S1 + α2 × S2 | (E3) |
 | (E4) |
| CH2OO + SO2 → HCHO + SO3 | (R1) |
 | ||
| Fig. 3 Measured ΔHCHO vs. ΔO3 (both corrected for dilution) for a hept-1-ene ozonolysis experiment with and without 2000 ppbv of the SCI scavenger SO2 present. Precision uncertainties are smaller than the presented data points. | ||
| Alkene | Zero SO2 | High SO2 | α 1 | α 2 | ||
|---|---|---|---|---|---|---|
| Carb1 yield | Carb2a yield | Carb1 yield | Carb2 yield | |||
| a Carb2 = the carbonyl formed from POZ decomposition that is not HCHO/acetone. b Assumed to be 1 by definition. | ||||||
| Carb1 = HCHO | ||||||
| Ethene | — | — | 1.31 (±0.07) | N.A. | 1.00b | — |
| Propene | 0.61 (±0.04) | 0.38 (±0.06) | 0.84 (±0.02) | 0.62 (±0.02) | 0.62 | 0.38 |
| 2-Methyl-but-1-ene | 0.93 (±0.01) | 0.40 (±0.01) | 1.05 (±0.03) | 0.50 (±0.01) | 0.60 | 0.40 |
| 1-Heptene | 0.53 (±0.01) | 0.42 (±0.01) | 0.84 (±0.03) | 0.80 (±0.12) | 0.53 | 0.47 |
| Carb1 = Acetone | ||||||
| 2-Methylpropene | 0.28 (±0.01) | 1.19 (±0.04) | 0.35 (±0.00) | 1.26 (±0.02) | 0.28 | 0.72 |
| 2-Methyl-but-2-ene | 0.34 (±0.01) | 0.80 (±0.07) | 0.44 (±0.02) | 0.69 (±0.12) | 0.34 | 0.66 |
| 2,3-Dimethyl-but-2-ene | 1.05 (±0.01) | — | 1.38 (±0.05) | N.A. | 1.00b | — |
| 2,4-Dimethylpent-2-ene | 0.22 (±0.01) | 1.04 (±0.03) | 0.50 (±0.02) | 0.94 (±0.05) | 0.22 | 0.78 |
| 2,3,4-Trimethylpent-2-ene | 0.44 (±0.01) | 0.78 (±0.01) | 0.59 (±0.07) | 0.72 (±0.08) | 0.22 | 0.78 |
| Myrcene | 0.22 (±0.01) | — | 0.61 (±0.03) | — | 0.22 | 0.78 |
For alkenes in which HCHO or acetone are expected to be formed in the decomposition of the larger CI, α1 was determined based on the measured yield of the larger carbonyl. HCHO is expected to be a decomposition product of any CI with a methyl group syn or anti to the carbonyl oxide moiety.46 For syn-CI this comes via decomposition of a β-oxo-alkoxy radical formed via the vinyl-hydroperoxide mechanism; for anti-CI the methyl radical is formed in decomposition of a bis-oxy radical, formed via 1,3 ring closure of the CI, leading to CH3O2 and ultimately to HCHO. For longer alkyl chains, HCHO is not expected to be formed. For short chain terminal alkenes this is consistent with the extensive database of ozonolysis experiments by Grosjean and Grosjean44 (and references therein). These show total primary carbonyl yields of unity for straight chain terminal alkenes with the exception of propene (that forms CH3CHOO), for which the sum carbonyl yield is well in excess of 1 (1.30). Any alkene that produces (CH3)2COO will also have a large secondary HCHO yield from decomposition of this CI.
CI stabilisation
The stabilisation, S, of a given CI is calculated as the ratio of the yield, ϕ, of the SCI to the yield of the CI (eqn (E5)). ϕCI is calculated as described above, based on the primary carbonyl yields in experiments with no SCI scavenger. ϕSCI for a specific CI is determined from the difference between the carbonyl yields in experiments with and without an SCI scavenger. The calculated stabilisation of CH2OO and (CH3)2COO in the alkene systems studied here are given in Table 3. | (E5) |
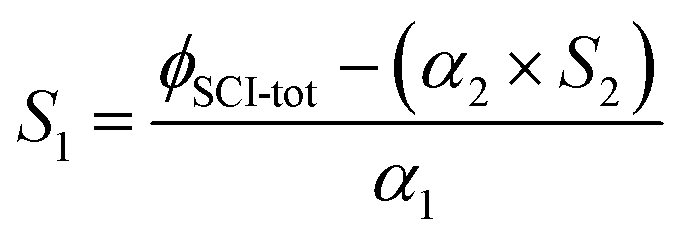 | (E6) |
| Alkene | Stab. (S) | Method |
|---|---|---|
| a Symmetrical, therefore total SCI yield (Table 2). b From model fit to data (Fig. S2, ESI). | ||
| CH2OO | ||
| Ethene | 0.43 (±0.02) | |
| Propene | 0.60 (±0.10) | |
| 2-Methylpropene | 0.61 (±0.12) | (E6) |
| 2-Methyl-but-1-ene | 0.56 (±0.10) | (E6) |
| Hept-1-ene | 0.73 (±0.08) | (E5) |
| (CH3)2COO | ||
| 2-Methylpropene | 0.10 (±0.04) | (E5) |
| 2-Methyl-but-2-ene | 0.16 (±0.04) | (E5) |
| 2,3-Dimethyl-but-2-ene | 0.31 (±0.03) | |
| 2,4-Dimethylpent-2-ene | 0.38 (±0.04) | (E5) |
| 2,3,4-Trimethylpent-2-ene | 0.53 (±0.04) | |
| Myrcene | 0.53 (±0.04) | (E5) |
Discussion
Fig. 4 shows the observed relationship between the stabilisation of the CI and the carbon number of the carbonyl co-product formed in POZ decomposition. There is a clear increase in stabilisation of both CH2OO and (CH3)2COO with increasing carbon number of the carbonyl co-product. This can be considered in terms of the distribution of the total energy available from decomposition of the POZ. If it is assumed that the total energy liberated in decomposition of the POZ is independent of the size of the alkene,19 and that the available energy has time to become distributed equally between the non-hydrogen atoms (i.e. C and O), then as the size of the carbonyl co-product increases relative to the CI, so the proportion of the available energy that is taken by the CI will decrease. This would be expected to yield a CI population with a lower mean energy distribution, both increasing the yield of CI that are ‘born cold’, and also increasing the amount of CI that will be collisionally stabilised (Fig. 5). While theoretical work has shown that the energy distribution between CI and carbonyl fragments in POZ decomposition may be non-statistical,47,48 it might still be expected that the general relationship will hold. | ||
| Fig. 4 Black triangles: variation of the stabilisation (S), of CH2OO and (CH3)2COO with size of carbonyl co-product determined from experiment. Red dashed line: modelled stabilisation using eqn (E7), assumes SCI yields of 0.43 (±0.04) and 0.31 (±0.04) for ethene (CH2OO) and 2,3-dimethyl-but-2-ene ((CH3)2COO) respectively. | ||
 | ||
| Fig. 5 Schematic showing the effect of increasing the size of the carbonyl co-product on the nascent mean energy distribution of a population of CI formed following POZ decomposition. As the mean energy decreases, the fraction of SCI increases. | ||
Based on the discussion above, the data presented in Fig. 4 can be fitted by the general relationship given in eqn (E7).
| S = 1 − (ACI/Atot) × F | (E7) |
The relationship is plotted in Fig. 4 (red dashed line) for a SCI yield from ethene of 0.43 (lower limit of 0.39, upper limit of 0.43), and from 2,3-dimethyl-but-2-ene of 0.31 (0.27–0.35). It is seen that the prescribed relationship fits the observed data well. While there are certainly likely to be additional factors influencing the stabilisation of CI, such as specific substituents and more complex structures, this work shows that the number of carbons in the system is a strong determinant of CI stabilisation, particularly for systems with similar structures.
For CI stabilisation from a given alkene, there is expected to be three important effects: (i) collisional stabilisation of the POZ, which will then decompose to yield exclusively stabilised CI – this has been predicted to be significant (65%) for the C15 sesquiterpene β-caryophyllene49 but to be insignificant for smaller (<C15) alkenes;50 (ii) an increased stabilisation of a given CI with increasing parent alkene size, as shown herein; and (iii) an increased stabilisation of larger CI, which have longer lifetimes (due to distributing the initial energy from the POZ decomposition among a greater number of degrees of freedom) allowing greater collisional stabilisation.20 This effect is particularly noticeable for 1-heptene in this dataset, with stabilisation of the larger CI, hexanal oxide being high (∼50%).
Using the relationships shown here it is possible to calculate the stabilisation of a given CI produced from an alkene that also produces either CH2OO or (CH3)2COO if the total SCI yield and the POZ decomposition branching ratio (α) are known. However, there are still relatively few alkenes for which total SCI yields have been measured. Further useful information to inform structure activity relationships used in atmospheric models would cover: (i) the dependence of α on alkene structure; (ii) trends in the total SCI yields from symmetrical alkenes of increasing size and complexity, to provide values on which to pin the relationships observed herein; (iii) the effect of the size of the CI itself on stabilisation. In addition, information on the relative yields and stabilisation of (E)/(Z) CI is required to fully represent the impact of alkene ozonolysis on atmospheric composition. In addition to further experimental studies, a detailed theoretical study would provide strong corroborating evidence both for the relationship derived in this work, and the further work suggested here.
Atmospheric implications
This work suggests that small CI produced from large alkenes found in the atmosphere, e.g. isoprene, monoterpenes, sesquiterpenes, etc., will predominantly be formed stabilised. For CH2OO, the SCI of which reacts almost entirely with water vapour in the boundary layer, this will result in a high yield of the products of the CH2OO + H2O/(H2O)2 reaction, and low radical (OH and HO2) yields (although it has been suggested that radical yields from CH2OO decomposition are low anyway51,52). This finding is particularly important for isoprene, the most abundantly emitted alkene to the atmosphere.53 Nguyen et al.34 suggested that the total SCI yield (∼0.6)32–34 measured from isoprene ozonolysis is almost entirely stabilised CH2OO (rather than the C4-CI). The main products of the reaction of CH2OO with water vapour are thought to be hydroxy-methylhydroperoxide (HMHP) and formic acid (HCOOH),54 but recent flow tube experiments55 suggest a roughly equal split between HMHP and formaldehyde (HCHO) from the CH2OO + (H2O)2 reaction, with direct HCOOH formation <5%. The dominant fate of HMHP in the atmosphere is unclear, with a relatively long lifetime against reaction with OH of about 1 day.56 OH reaction was shown to yield HCOOH and HCHO in a ratio of 0.88.56 For (CH3)2COO, and other small syn-CI, for which reaction with water vapour is a negligible sink under boundary layer conditions,55 a higher stabilisation will lead to an increased atmospheric concentration of these SCI. Under typical boundary layer conditions these SCI will participate in bimolecular reactions (e.g. with SO2 and organic acids), contribute to aerosol formation, or undergo unimolecular decomposition.Conclusions
Ozonolysis experiments were performed at the EUPHORE atmospheric simulation chamber on a range of alkenes that produce either the CH2OO or (CH3)2COO Criegee intermediates. Total stabilised Criegee intermediate (SCI) yields were determined from the temporal decay of the SCI scavenger SO2. Speciated CI yields were determined based on FTIR measurements of primary carbonyl products. Speciated SCI yields were determined from comparison of carbonyl yields in the presence/absence of the SCI scavenger SO2. From this information, the stabilisation of CH2OO and (CH3)2COO from each alkene was determined. The stabilisation is shown to increase with increasing carbon number of the carbonyl co-product formed in the decomposition of the primary ozonide. Stabilisation of CH2OO increases from a minimum of ∼0.4 from ethene, to 0.71 from 1-heptene. Stabilisation of (CH3)2COO increases from <0.1 from isobutene, to 0.50 from the monoterpene myrcene. An empirical relationship based on the energy distribution through the molecule on dissociation of the POZ fits the observed data well. This trend has implications for predicted tropospheric concentrations of SCI, with current models generally using SCI yields based on the total yield from the relevant symmetrical alkene–ozone system. From this work it is shown that stabilisation of small CI from many atmospherically relevant alkenes, such as isoprene and monoterpenes, is likely to be considerably higher than currently predicted. This would increase the importance of bimolecular reactions, and reduce radical yields from CI decomposition.Data availability
Experimental data will be available in the Eurochamp database, http://www.eurochamp.org, from the H2020 EUROCHAMP2020 project, GA no. 730997.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Luc Vereecken for very helpful discussions. The assistance of the EUPHORE staff is gratefully acknowledged. This work has received funding from the European Union's Horizon 2020 research and innovation programme through the EUROCHAMP-2020 Infrastructure Activity under grant agreement No. 730997, by means of its Transnational Activities, and Fundacion CEAM and the Generalitat Valenciana for the IMAGINA-Prometeo project (PROMETEO2019/110). Fundación CEAM is partly supported by Generalitat Valenciana. Mike Newland and Andrew Rickard also acknowledge support from the Mechanisms for Atmospheric chemistry: GeneratioN, Interpretation and FidelitY – MAGNIFY project, funded by the UK Natural Environment Research Council (NERC, via grant NE/M013448/1). Beth Nelson acknowledges the NERC SPHERES Doctoral Training Partnership (DTP) for her studentship.Notes and references
- H. Niki, P. D. Maker, C. M. Savage, L. P. Breitenbach and M. D. Hurley, J. Phys. Chem., 1987, 91, 941–946 CrossRef CAS.
- S. E. Paulson and J. J. Orlando, Geophys. Res. Lett., 1996, 23, 3727–3730 CrossRef CAS.
- A. R. Rickard, D. Johnson, C. D. McGill and G. Marston, J. Phys. Chem. A, 1999, 103, 7656–7664 CrossRef CAS.
- G. Salisbury, A. R. Rickard, P. S. Monks, B. J. Allan, S. J. B. Bauguitte, S. A. Penkett, N. Carslaw, A. C. Lewis, D. J. Creasey, D. E. Heard, P. J. Jacobs and J. D. Lee, J. Geophys. Res.: Atmos., 2001, 106, 12669–12687 CrossRef CAS.
- D. E. Heard, L. J. Carpenter, D. J. Creasey, J. R. Hopkins, J. D. Lee, A. C. Lewis, M. J. Pilling, P. W. Seakins, N. Carslaw and K. M. Emmerson, Geophys. Res. Lett., 2004, 31, L18112 CrossRef.
- L. Vereecken, A. Novelli and D. Taraborrelli, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- M. J. Newland, A. R. Rickard, T. Sherwen, M. J. Evans, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Atmos. Chem. Phys., 2018, 18, 6095–6120 CrossRef CAS.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, M. Anwar, H. Kahn, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Angew. Chem., Int. Ed., 2014, 18, 4547–4550 CrossRef PubMed.
- F. A. Mackenzie-Rae, H. J. Wallis, A. R. Rickard, K. L. Pereira, S. M. Saunders, X. Wang and J. F. Hamilton, Atmos. Chem. Phys., 2018, 18, 4673–4693 CrossRef CAS.
- K. Kristensen, T. Cui, H. Zhang, A. Gold, M. Glasius and J. D. Surratt, Atmos. Chem. Phys., 2014, 14, 4201–4218 CrossRef.
- X. Zhang, R. C. McVay, D. D. Huang, N. F. Dalleska, B. Aumont, R. C. Flagan and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2015, 46, 14168–14173 CrossRef PubMed.
- Y. Sakamoto, S. Inomata and J. Hirokawa, J. Phys. Chem. A, 2013, 117, 12912–12921 CrossRef CAS PubMed.
- Y. Zhao, L. M. Wingen, V. Perraud, J. Greaves and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2015, 17, 12500–12514 RSC.
- M. Ehn, E. Kleist, H. Junninen, T. Petaja, G. Lonn, S. Schobesberger, M. Dal Maso, A. Trimborn, M. Kulmala, D. R. Worsnop, A. Wahner, J. Wildt and T. F. Mentel, Atmos. Chem. Phys., 2012, 12, 5113–5127 CrossRef CAS.
- J. R. Pierce, M. J. Evans, C. E. Scott, S. D. D’Andrea, D. K. Farmer, E. Swietlicki and D. V. Spracklen, Atmos. Chem. Phys., 2013, 13, 3163–3176 CrossRef.
- T. Jokinen, T. Berndt, R. Makkonen, V. M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther, D. R. Worsnop, M. Kulmala, M. Ehn and M. Sipila, Proc. Natl. Acad. Sci. U. S. A., 2015, 23, 7123–7128 CrossRef PubMed.
- R. Criegee and G. Wenner, Liebigs Ann. Chem., 1949, 564, 9–15 CrossRef CAS.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- G. T. Drozd and N. M. Donahue, J. Phys. Chem. A, 2011, 115, 4381–4387 CrossRef CAS PubMed.
- M. Campos-Pineda and J. Zhang, Chem. Phys. Lett., 2017, 683, 647–652 CrossRef CAS.
- G. T. Drozd, T. Kurtén, N. M. Donahue and M. I. Lester, J. Phys. Chem. A, 2017, 121, 6036–6045 CrossRef CAS PubMed.
- S. Hatakeyama and H. Akimoto, Res. Chem. Intermed., 1994, 20, 503–524 CrossRef CAS.
- G. T. Drozd, J. Kroll and N. M. Donahue, J. Phys. Chem. A, 2011, 115, 161–166 CrossRef CAS PubMed.
- J. P. Hakala and N. M. Donahue, J. Phys. Chem. A, 2016, 14, 2173–2178 CrossRef PubMed.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204–207 CrossRef CAS PubMed.
- H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, Chem. Phys. Lett., 1977, 46, 327–330 CrossRef CAS.
- S. Hatakeyama, H. Kobayashi and H. Akimoto, J. Phys. Chem., 1984, 88, 4736–4739 CrossRef CAS.
- T. Berndt, T. Jokinen, R. L. Mauldin III, T. Petaja, H. Herrmann, H. Junninen, P. Paasonen, D. R. Worsnop and M. Sipila, J. Phys. Chem. Lett., 2012, 3, 2892 CrossRef CAS.
- T. Berndt, T. Jokinen, M. Sipilä, R. L. Mauldin, H. Herrmann, F. Stratmann, H. Junninen and M. Kulmala, Atoms. Environ., 2014, 89, 603 CrossRef CAS.
- M. J. Newland, A. R. Rickard, M. S. Alam, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Phys. Chem. Chem. Phys., 2015, 17, 4076–4088 RSC.
- M. Sipila, T. Jokinen, T. Berndt, S. Richters, R. Makkonen, N. M. Donahue, R. L. Mauldin III, T. Kurtén, P. Paasonen, N. Sarnela, M. Ehn, H. Junninen, M. P. Rissanen, J. Thornton, F. Stratmann, H. Herrmann, D. R. Worsnop, M. Kulmala, V. M. Kerminen and T. Petäjä, Atmos. Chem. Phys., 2014, 14, 12143–12153 CrossRef.
- M. J. Newland, A. R. Rickard, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Atmos. Chem. Phys., 2015, 15, 9521–9536 CrossRef CAS.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Milller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. B. Wild, S. Stephen, A. H. Goldstein, J. A. de Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Phys. Chem. Chem. Phys., 2016, 18, 10241–10254 RSC.
- K. H. Becker, EUPHORE: Final Report to the European Commission, Contract EV5V-CT92-0059, Bergische Universität Wuppertal, Germany, 1996.
- M. S. Alam, M. Camredon, A. R. Rickard, T. Carr, K. P. Wyche, K. E. Hornsby, P. S. Monks and W. J. Bloss, Phys. Chem. Chem. Phys., 2011, 13, 11002–11015 RSC.
- A. Muñoz, E. Borrás, M. Ródenas, T. Vera and H. A. Pedersen, Environ. Sci. Technol., 2018, 52, 9136–9144 CrossRef PubMed.
- IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation, http://iupac.pole-ether.fr.
- R. M. I. Elsamra, A. Jalan, Z. J. Buras, J. E. Middaugh and W. H. Green, Int. J. Chem. Kinet., 2016, 48, 474–488 CrossRef CAS.
- A. Jalan, J. W. Allen and W. H. Green, Phys. Chem. Chem. Phys., 2013, 15, 16841–16852 RSC.
- O. Horie and G. K. Moortgat, Atmos. Environ., Part A, 1991, 24, 1881–1896 CrossRef.
- P. Deng, L. Wang and L. Wang, J. Phys. Chem. A, 2018, 22, 3013–3020 CrossRef PubMed.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- E. Grosjean and D. Grosjean, Environ. Sci. Technol., 1997, 31, 2421–2427 CrossRef CAS.
- M. E. Jenkin, R. Valorso, B. Aumont, M. J. Newland and A. R. Rickard, Atmos. Chem. Phys., 2020 Search PubMed , in preparation.
- E. C. Tuazon, S. M. Aschmann, J. Arey and R. Atkinson, Environ. Sci. Technol., 1997, 31, 3004–3009 CrossRef CAS.
- G. Vayner, S. V. Addepalli, K. Song and W. L. Hase, J. Chem. Phys., 2006, 125, 014317 CrossRef PubMed.
- M. Pfeifle, Y.-T. Ma, A. W. Jasper, L. B. Harding, W. L. Hase and S. J. Klippenstein, J. Chem. Phys., 2018, 148, 174306 CrossRef PubMed.
- T. L. Nguyen, R. Winterhalter, G. Moortgat, B. Kanawati, J. Peeters and L. Vereecken, Phys. Chem. Chem. Phys., 2009, 11, 4173–4183 RSC.
- N. M. Donahue, G. T. Drozd, S. A. Epstein, A. A. Presto and J. H. Kroll, Phys. Chem. Chem. Phys., 2011, 13, 10848–10857 RSC.
- T. L. Nguyen, H. Lee, D. A. Matthews, M. C. McCarthy and J. F. Stanton, J. Phys. Chem. A, 2015, 119, 5524–5533 CrossRef CAS PubMed.
- D. Stone, K. Au, S. Sime, D. J. Medeiros, M. Blitz, P. W. Seakins, Z. Decker and L. Sheps, Phys. Chem. Chem. Phys., 2018, 20, 24940–24954 RSC.
- K. Sindelarova, C. Granier, I. Bouarar, A. Guenther, S. Tilmes, T. Stavrakou, J.-F. Müller, U. Kuhn, P. Stefani and W. Knorr, Atmos. Chem. Phys., 2014, 14, 9317–9341 CrossRef.
- P. Neeb, F. Sauer, O. Horie and G. K. Moortgat, Atmos. Environ., 1997, 31, 1417–1423 CrossRef.
- L. Sheps, B. Rotavera, A. J. Eskola, D. L. Osborn, C. A. Taatjes, K. Au, D. E. Shallcross, M. A. H. Khan and C. J. Percival, Phys. Chem. Chem. Phys., 2017, 19, 21970–21979 RSC.
- H. M. Allen, J. D. Crounse, K. H. Bates, A. P. Teng, M. P. Krawiec-Thayer, J. C. Rivera-Rios, F. N. Keutsch, J. M. St. Clair, T. F. Hanisco, K. H. Møller, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. A, 2018, 122, 6292–6302 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00897d |
| This journal is © the Owner Societies 2020 |