
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
F-doping of nanostructured ZnO: a way to modify structural, electronic, and surface properties†
Elisabeth Hannah
Wolf
 a,
Marie-Mathilde
Millet
a,
Friedrich
Seitz
a,
Frenio A.
Redeker
b,
Wiebke
Riedel
b,
Gudrun
Scholz
c,
Walid
Hetaba
ad,
Detre
Teschner
ad,
Sabine
Wrabetz
a,
Frank
Girgsdies
a,
Alexander
Klyushin
a,
Thomas
Risse
b,
Sebastian
Riedel
b and
Elias
Frei
*a
a,
Marie-Mathilde
Millet
a,
Friedrich
Seitz
a,
Frenio A.
Redeker
b,
Wiebke
Riedel
b,
Gudrun
Scholz
c,
Walid
Hetaba
ad,
Detre
Teschner
ad,
Sabine
Wrabetz
a,
Frank
Girgsdies
a,
Alexander
Klyushin
a,
Thomas
Risse
b,
Sebastian
Riedel
b and
Elias
Frei
*a
aFritz-Haber-Institute of the Max-Planck-Society, Department of Inorganic Chemistry, 14195 Berlin, Germany. E-mail: efrei@fhi-berlin.mpg.de
bFreie Universität Berlin, Institut für Chemie u. Biochemie, 14195 Berlin, Germany
cHumboldt – Universität Berlin, Institut für Chemie, 12489 Berlin, Germany
dMax Planck Institute for Chemical Energy Conversion, Department of Heterogeneous Reactions, 45470 Mülheim a.d.R., Germany
First published on 6th April 2020
Abstract
Polycrystalline ZnO is a material often used in heterogeneous catalysis. Its properties can be altered by the addition of dopants. We used gaseous fluorine (F2(g)) as direct way to incorporate fluoride in ZnO as anionic dopants. Here, the consequences of this treatment on the structural and electronic properties, as well as on the acidic/basic sites of the surface, are investigated. It is shown that the amount of F incorporation into the structure can be controlled by the synthesis parameters (t, T, p). While the surface of ZnO was altered as shown by, e.g., IR spectroscopy, XPS, and STEM/EDX measurements, the F2 treatment also influenced the electronic properties (optical band gap, conductivity) of ZnO. Furthermore, the Lewis acidity/basicity of the surface was affected which is evidenced by using, e.g., different probe molecules (CO2, NH3). In situ investigations of the fluorination process offer valuable insights on the fluorination process itself.
1 Introduction
ZnO is an extensively studied metal oxide with multiple applications in catalysis or within optoelectronic devices.1–6 It is commonly employed in heterogeneous catalysis as a support or co-catalyst, for example in Cu/ZnO based systems applied in methanol synthesis7 or the reverse water–gas shift reaction.8 ZnO is an intrinsic n-type semiconductor, containing a high concentration of defects, including oxygen vacancies.9,10 Doping provides a versatile route to engineer the properties of the material. Doping of the cation lattice was shown to affect the structural, electronic, and catalytic properties of ZnO as part of catalytic systems.11–13 Anionic N-doping of ZnO is applied to create a p-type semiconductor.14 Here, we present the effects of using fluoride as an anionic dopant for ZnO to increase its n-type character.In the literature, fluorine doping of oxides has been employed for the manipulation of electronic and optical properties, for example in case of fluorine modified TiO2.15,16
Fluorine doped ZnO has mainly been studied for the application as a transparent conducting oxide and synthesised by methods such as reactive magnetron sputtering17,18 or chemical vapour deposition.19 Previous studies have thus focused on the investigation of the F-doped ZnO materials with respect to their optical and electronic properties.20,21 Further applications of F-doped ZnO thin films include the use as a gas sensing material.22,23 While ZnO for the application in optoelectronics is required to form thin solid layers, in heterogeneous catalysis a high surface area and thus the generation of large amounts of polycrystalline powder is required. Such polycrystalline F-doped ZnO materials have been previously studied with applications in, e.g., photocatalysis.24 The influence of fluorine treatment on the catalytic properties of the Cu/ZnO system has been subject of a recent study,25 however, the role of fluorine remains elusive.
In this contribution, we show the impact of direct F2(g) treatment on ZnO with respect to structural and electronic properties. To control the amount of fluoride in ZnO, different F2(g) partial pressures were applied with the aim of providing a synthetic pathway to a F-modified nanostructured ZnO. We employ various techniques (XRD, XPS, NEXAFS, SEM-EDX, STEM-EDX, 19F NMR, EPR and in situ FT-IR) to probe the effect of fluorine doping on the structural and electronic properties of the material. In light of the applicability of this material as a support in heterogeneous catalysis, the surface properties are thoroughly analysed as well (FT-IR, micro-calorimetry and TDS).
2 Experimental
2.1 Sample preparation
Zinc oxide was synthesised by precipitation from the nitrate as follows: 357 g of zinc nitrate hexahydrate were dissolved in 500 mL deionised water with 15 mL nitric acid and filled up to give 600 mL solution. A second solution of 339.16 g sodium carbonate in deionised water to give 2 L was prepared. In an automated reactor (LabMax, Mettler-Toledo), 400 mL deionised water were placed, and 20 g min−1 of the nitrate solution were added for 30 min while the carbonate solution was added to stabilise the pH at 6.5. The mixture was aged for 60 min before the product was filtered and washed twice with 600 mL water until the conductivity of the filtrate was below 0.5 mS cm−1. The resulting white powder (zinc hydroxycarbonate) was then dried at 353 K over night before calcining it in a rotating furnace at 603 K (ramp rate 2 K min−1, dwell time 3 h) to yield the white zinc oxide powder (called “ZnO” hereafter).Two samples were fluorinated by application of fluorine gas of differing partial pressures. The needed amount of F2 was calculated based on the stoichiometry of Zn in ZnO for nominal 1 at% and 10 at% of fluorination. The partial pressure of F2(g) was adjusted with a calibrated volume. The fluorination was conducted at RT, 1 bar (mixed with N2) and for one hour time of reaction. The sample treated with the lower F2 partial pressure is termed “ZnO_F1” in the following, and the sample resulting from the treatment using a higher partial pressure “ZnO_F10”, respectively.
Commercial zinc fluoride (anhydrous 99 min%, alfa aesar ZnF2 11521, Lot U01C031) was used as a reference.
2.2 X-ray diffraction
The samples were analysed using X-ray diffraction (XRD) on a Bruker AXS D8 Advance II theta/theta diffractometer in Bragg–Brentano geometry using Ni filtered Cuα1+2 radiation and a position sensitive energy dispersive LynxEye silicon strip detector. Subsequently, the diffractograms were analysed by whole powder pattern fitting, employing the Rietvelt method as implemented in the TOPAS software (Bruker AXS).2.3 UV-vis spectroscopy
Diffuse Reflectance UV-vis spectroscopy was performed on a Cary5000 photospectrometer (Agilent), employing a Praying Mantis Diffuse Reflection Accessory and Reaction Chamber (Harrick) using Spectralon as a white standard. The optical band gap of each sample was determined from the appropriate Tauc plots26 by linear extrapolation of the absorption edge to zero intensity.2.4 Electron microscopy
Scanning Electron Microscopy (SEM) was performed using a Hitachi S 4800 SEM equipped with a Genesis 4000 Energy Dispersive X-ray (EDX) detector. The microscope was operated at an acceleration voltage of 1.5 kV. EDX spectra were obtained using an acceleration voltage of 5 kV.For the TEM measurements, a Thermo Fisher Scientific Talos F200X S/TEM was used with an acceleration voltage of 200 kV. STEM measurements were performed using an HAADF detector, and the ChemiSTEM super-X EDX system using four SDD detectors was employed to generate energy dispersive X-ray spectroscopy (EDX) maps.
2.5 Electron paramagnetic resonance
Electron paramagnetic resonance (EPR) measurements of the fluorinated ZnO samples were conducted in quartz tubes (2.9 mm outer diameter). Continuous wave (CW) EPR spectra at X-band frequencies were obtained at room temperature using a Bruker B-ER420 spectrometer upgraded with a Bruker ECS 041XG microwave bridge and a lock-in amplifier (Bruker ER023M). The samples were measured in a Bruker SHQ or TM110 resonator applying a modulation amplitude of 5 G and a modulation frequency of 100 kHz. All spectra were background corrected by subtracting a spectrum of an empty quartz tube and the signals were normalised to the sample mass.2.6 Solid state nuclear magnetic resonance
19F MAS NMR spectra were recorded on a Bruker AVANCE 400 spectrometer (Larmor frequency 376.4 MHz) using a 2.5 mm magic angle spinning (MAS) probe (Bruker Biospin) and applying a spinning speed of 20 kHz if not otherwise indicated. 19F MAS NMR (I = 1/2) spectra were recorded with a π/2 pulse duration of 4.4 μm, a spectrum width of 400 kHz, and a recycle delay of 5 s. Spectral changes for longer recycle delays were checked. Isotropic chemical shifts of 19F are given with respect to the CFCl3 standard. Background signals of 19F were completely suppressed with the application of a phase-cycled depth pulse sequence according to Cory and Ritchey.272.7 X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy was performed on pressed pellets of the samples using non-monochromatised Al Kα excitation (1486.6 eV) and a hemispherical analyser (Phoibos 150, SPECS). The standard Au 4f(7/2) and Cu 2p(3/2) procedure was employed to calibrate the binding energy scale, and no correction for charging effects was applied. Elemental compositions were calculated using theoretical cross sections from Yeh and Lindau.282.8 Conductivity measurements
The contact-free and in situ Microwave Cavity Perturbation Technique (MCPT)29 was employed to measure the conductivity of the (fluorinated) zinc oxide samples at 503 K in inert gas (N2) as well as hydrogen- (5% H2/N2) and oxygen-containing (5% O2/N2) conditions.The gas flow was held constant at 10 mL min−1. The resulting data were fitted and corrected to account for the powder samples’ filling fractions using an established formalism.30,31
2.9 NEXAFS
NEXAFS measurements were performed at the UE56-2 PGM-2 beamline at BESSY II/HZB (Berlin, Germany). All measurements were carried out in a stainless steel NAP-XPS chamber described in detail elsewhere.32 The samples were pelletised and mounted on a sapphire sample holder between a stainless steel back-plate and a stainless steel lid with a 6 mm hole.The NEXAFS spectra of these samples were obtained in the total electron yield mode by recording a sample current. The photon-energy resolution was set to 700 meV at the Zn L3-edge (≈1020 eV), 400 meV at the F K-edge (≈680 eV) and 250 meV at the O K-edge (≈520 eV) The photon energy was calibrated using the Au 4f line. All NEXAFS spectra were normalised to the incident photon flux.
2.10 IR spectroscopy
The fluorine treated ZnO samples were characterised by FTIR and compared to ZnO and ZnF2. The measurements were carried out using a Varian 670 FTIR spectrometer equipped with a MCT detector. The spectra were recorded at a resolution of 2 cm−1 accumulating 128 scans. Self-supported wafers (area weight of 20–30 mg cm−2) were transferred into an IR cell that is connected to a vacuum line and a gas delivery system. Before the spectroscopic measurements, surface impurities on the samples were removed by heating in the IR cell to 240 °C (200 °C in the case of ZnO_F10) in 50 mbar O2 until the IR spectra did not show any further changes. The presence of oxygen is necessary in order to avoid the formation of donor centres which may obscure changes of the vibrational bands as a result of their intense and broad absorption in the IR range.After cooling to 25 °C the cell was evacuated and 67 mbar of NH3 were added. All spectra shown are recorded after evacuation (to remove gas phase NH3) and are referenced against a spectrum taken immediately before NH3 addition. For the desorption experiments the cell was heated at a rate of 1 K min−1, with spectra taken every 10 K.
In situ spectroscopy was performed by a Vertex 70 FT-IR spectrometer from Bruker® equipped with a MCT detector. The spectra were recorded at a resolution of 0.5 cm−1 accumulating 100 scans. Self-supporting wafers of 13 mm diameter were placed in an IR cell.33 The sample was heated to 240 °C in vacuum for 10 h in order to remove surface impurities and subsequently treated with O2 (120 mbar) for 1 h. 43 mbar 10% F2/Ar mixture was added to the cell, while simultaneously taking IR spectra in order to monitor the changes to the sample.
2.11 Microcalorimetry
The microcalorimetric experiments were performed in a Calvet calorimeter (SETARAM MS70) equipped with a custom-designed high vacuum and gas dosing apparatus, which allows for the dosage of probe molecules within a range of 0.02 mmol and the calculation of the amount of adsorbed molecules. The samples were heated to 240 °C at a rate of 5 K min−1 in UHV for 1 h dwell time. After degassing, oxidation was performed in 5% O2 for 30 min two times. Thereafter, the cell was cooled to room temperature and pumped to UHV (10−7 mbar final pressure), placed inside the calorimeter without contact to air, and connected to the microcalorimetric gas adsorption system. CO2 was then dosed stepwise at 30 °C, while recording pressure, adsorption temperature, and heat signals during all dosing steps.2.12 Thermal desorption spectroscopy
Thermal desorption spectroscopy (TDS) was applied for the temperature programmed desorption of oxygen. For this, a self-constructed setup which enables the testing of powder samples was used. The setup is equipped with mass flow controllers, an IR-light furnace (Behr IRF 10) and a mass spectrometer (Pfeiffer Vacuum QME 200). The powder sample was placed on a small quartz-glass boat which is placed in a quartz tube (inner diameter of 14 mm, outer diameter of 20 mm, length of 450 mm) located inside the furnace and connected to the system using Ultra Torr vacuum fittings. Afterwards the system was stepwise brought to 9 × 10−7 mbar and directly connected to the mass spectrometer. The surface was treated at 503 K for 2.5 h under reductive atmospheres (5% H2 in Ar). Subsequently, at 348 K the surface was saturated with double labelled C18O2 at 1 bar. The desorption experiment was conducted at 10−6 mbar with a heating rate of 25 K min−1.3 Results and discussion
3.1 Sample designation
The co-precipitated and calcined polycrystalline ZnO powder was directly treated with F2(g). By employing two different p(F2), fluoride concentrations within the ZnO sample of 1 and 10 at%, respectively, were targeted. Accordingly, the samples are named as ZnO_F1 and ZnO_F10. For details about the preparation method, please see the experimental section. As reference samples, the pristine ZnO and commercial ZnF2 were investigated.3.2 Structural characterisation of fluorine treated ZnO
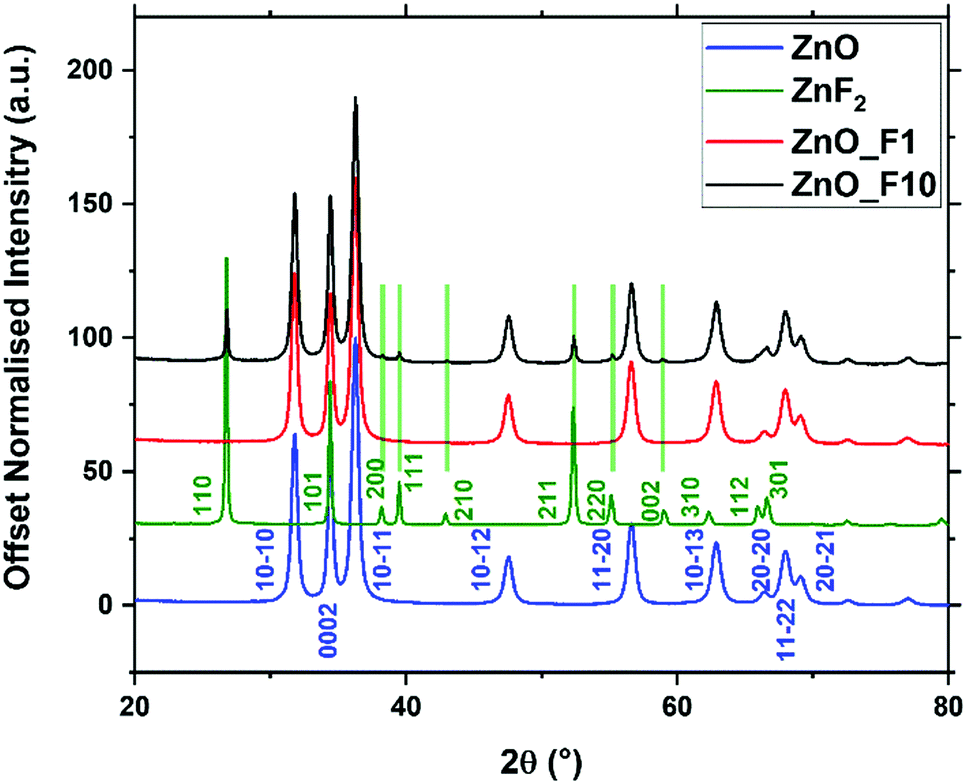 | ||
| Fig. 1 X-ray diffraction patterns of fluorine treated and untreated ZnO, and commercial ZnF2 as a reference. Indices for ZnO and ZnF2 are given from literature ref. 34 and 35. | ||
The whole powder pattern of the fluorinated ZnO samples were fitted (Table 1) to reveal possible changes of the lattice parameters and for the quantification of the ZnF2-phase. The fluorination had no impact on the lattice parameters a and c, whereas the oxygen position z(O) increased with increasing fluorine doping. From the Rietveld refinement, the crystalline ZnF2 by-phase observed for the ZnO_F10 sample amounted to 8 wt% (or ∼6 at%). The slightly increasing domain size of ZnO, in particular for Zn_F10, is interpreted as preferential reactivity of F2(g) with small crystallites to form, e.g., ZnF2 moieties.
| a (Å) | c (Å) | z (O) | Size a/c (nm) | |
|---|---|---|---|---|
| ZnO | 3.2507(1) | 5.2089(2) | 0.3808(6) | 17.8 (6)/23.9 (6) |
| ZnO_F1 | 3.2508(1) | 5.2090(2) | 0.3810(6) | 18.4 (6)/24.0 (6) |
| ZnO_F10 | 3.2508(2) | 5.2089(3) | 0.3815(8) | 19.7 (8)/24.4 (8) |
Additionally, the surface-near molar element ratios of the samples were investigated using XPS (Table 2). While the total oxygen content measured through the O/Zn ratio remains constant upon fluorine treatment, fitting of the O 1s line shapes reveal a decrease of lattice oxygen content with increasing F-content in the sample. The latter can be interpreted as a substitutional fluorine doping of ZnO upon treatment with gaseous F2. Fitting the O1s spectra (Fig. SI1, ESI†) reveals an increase of spectral components with higher binding energy, which points to the fact that the sample with higher F-content contains more absorbed oxygen in the form of OH groups or adsorbed water. This might be a sign for an increased surface Lewis acidity. However, estimating the OH-concentration on the ZnO surface, based on the information depth and calculated quantities (Table 2), the coverage is <1 mono layer (ML) without significant influence on the wurtzite lattice. The F 1s spectra show, as expected, a larger F/Zn ratio for ZnO_F10 than for ZnO_F1 (Fig. SI4, ESI†). The slight increase for ZnO in the F 1s region is explained by minor Zn Auger contributions. The stoichiometric disbalance of the O/Zn ratio (<1) is a typical sign for nanostructured ZnO with an oxygen deficient character, originated in the synthesis history of the precursor.
| O/Zn | O(lattice)/Zn | F/Zn | |
|---|---|---|---|
| ZnO | 0.86 | 0.67 | — |
| ZnO_F1 | 0.85 | 0.655 | 0.023 |
| ZnO_F10 | 0.86 | 0.58 | 0.13 |
| ZnF2 | 0.12 | — | 1.67 |
Interestingly, for the ZnO_F1 sample the F/Zn ratio near the surface (XPS information depth, here: ≈2 nm) is larger than the bulk F/Zn ratio (determined from EDX/SEM, here: ≈100 nm). This finding indicates that the concentration of F might be larger near the sample surface.
 | ||
| Fig. 2 SEM images of ZnO (a), ZnO_F1 (b) and ZnO_F10 (c). | ||
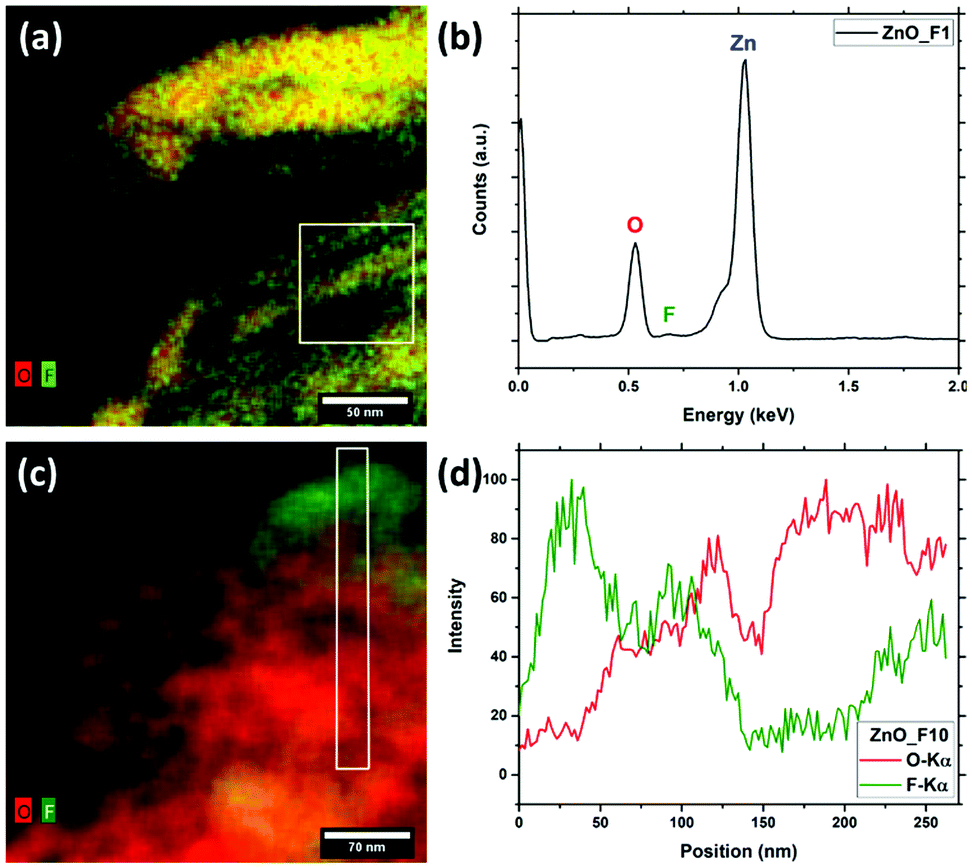 | ||
| Fig. 3 STEM-EDX of the fluorinated ZnO samples – combined O (red) and F(green) EDX-maps of ZnO_F1 (a) and ZnO_F10 (c) as well as EDX spectrum of the marked region of ZnO_F1 (b) and EDX-line scan of marked region of ZnO_F10 (d). | ||
In Fig. 4, a high resolution TEM (HRTEM) image of the same region as Fig. 3(c) is presented along with the fast Fourier transform (FFT) patterns of the indicated regions. The FFTs can be used to identify different crystalline phases present in the sample by referencing the reflections to simulated patterns. These patterns were generated from structures taken from the ICS database (ZnO:36 671![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 330, ZnF2:35 9169) using the JEMS software.37 The indexed reflections are given in Fig. SI5 (ESI†). However, as the reflections in (c) are less defined, the radii of the rings on which the reflections are located were used to determine the corresponding lattice plane distances and possible reflections of the two materials. The ring corresponding to a distance of ca. 0.26 nm, can be attributed to ZnO 002 and/or ZnF2 101.
330, ZnF2:35 9169) using the JEMS software.37 The indexed reflections are given in Fig. SI5 (ESI†). However, as the reflections in (c) are less defined, the radii of the rings on which the reflections are located were used to determine the corresponding lattice plane distances and possible reflections of the two materials. The ring corresponding to a distance of ca. 0.26 nm, can be attributed to ZnO 002 and/or ZnF2 101.
 | ||
| Fig. 4 HR-TEM image of ZnO_F10 (a) with FFTs of the indicated regions (b) and (c). Selected reflections are indexed in the figure for ZnO (blue) and ZnF2 (green). | ||
In both regions indicated in Fig. 4(b) and (c), ZnO was identified as the main structure, but ZnF2 was found as a minor phase, which agrees well with the results obtained from EDX (Fig. 3(c and d)). Since not all reflections present in Fig. 4(c) correspond to only one of the two phases ZnO and ZnF2, along with the results from EDX mapping it can be concluded that this region contains a higher amount of ZnF2 than region (b).
The results of the electron microscopy investigations are in agreement with the elemental quantifications and structural analysis already discussed. High p(F2) was shown to form a heterogeneous system as it leads to the formation of a ZnF2 by-phase, while the low p(F2) results in a phase pure system. The spatially heterogeneous distribution observed for the system exposed to high fluorine pressure is readily explained by a ZnF2 particle adjacent to an F-doped ZnO one.
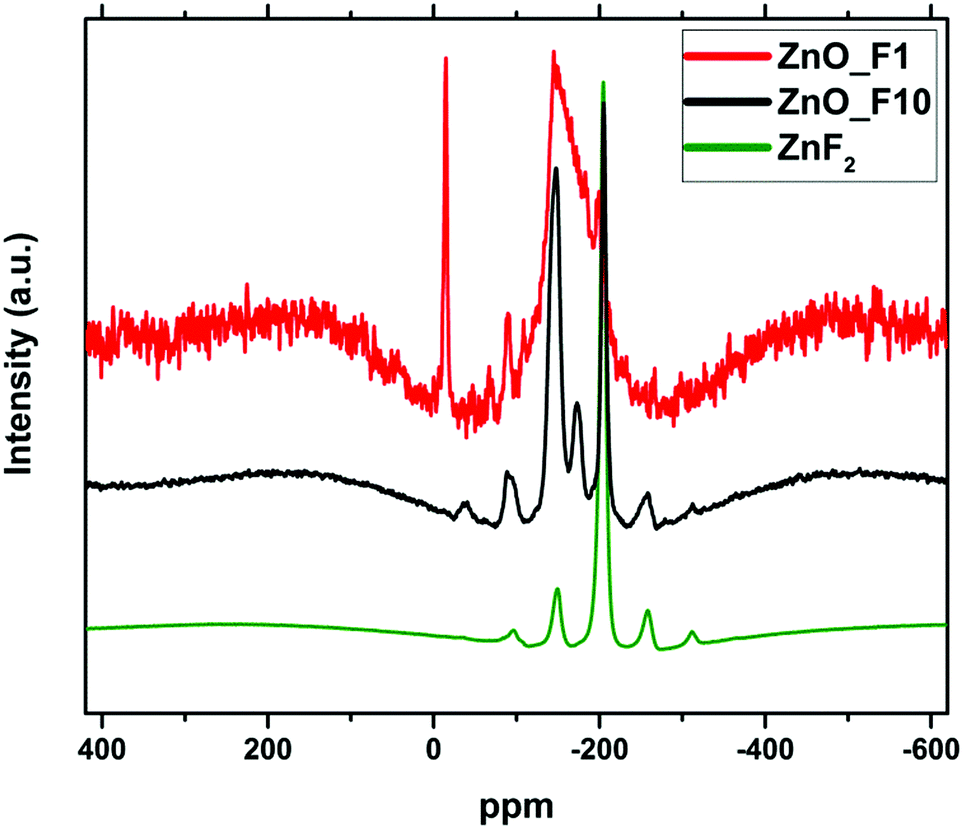 | ||
| Fig. 5 19F MAS NMR spectra of the fluorinated samples and ZnF2 as a reference. The spectra were recorded with different accumulation numbers (ZnO_F1; 256, ZnO_F10: 64, ZnF2: 32) and have been offset and rescaled for clarity. | ||
The spectrum of the ZnO_F10 sample shows a signal at 205 ppm indicating the presence of ZnF2 in line with the X-ray diffraction results. The variation of the spinning frequency reveals that the signals at −36 ppm, −90 ppm, −255 ppm and −310 ppm are due to spinning side bands. In contrast, the well-resolved signals at −148 ppm and −173 ppm indicating a rather uniform coordination environment of the F-nuclei are not found for crystalline ZnF2. The signals are in the typical region for a fluorine doped oxide (or “fluoro-oxide”), as could be expected for ZnO:F.38
The spectrum of ZnO_F1 exhibits a much reduced signal intensity in line with the lower F content, nevertheless the presence of fluorine-related centres is still detectable. The spectrum is characterized by a broad asymmetric line ranging from −150 to −220 ppm. The large width together with the asymmetric line shape gives evidence for a broad distribution of local environments for the fluorine species in this sample. This means, despite a homogeneous spatial distribution of the fluorine within the sample (confirmed by STEM-EDX-maps in Fig. 3(a)), their local arrangements is rather non-uniform (different, non-preferential coordination environments). The spectrum shows narrow signal at −14.8 ppm, which does not disappear in a spin-echo test and is neither found for ZnF2 nor for the ZnO_F10 sample. It is a typical solid state signal (line width ≈1.5 kHz) and it appears at the position found for fluorine in BaF2, however, the elemental analysis did not show any trace of Ba2+. Therefore, we cannot assign this signal unambiguously.
For all three samples, the spin lattice relaxation behaviour of 19F was tested by varying the recycle delay time D1. The intensity of the 19F signals is almost unchanged with longer D1 times for the low fluorine doped sample ZnO_F1, which is typical for a high degree of disorder of 19F species in the sample. Instead, the 19F signal intensity of prominent signals (−148, −173, and −205 ppm; Fig. SI1, ESI†) of the ZnO_F10 sample shows distinct changes upon varying the D1 time, which can be considered an additional indication for a homogeneous environment of the fluorine anions. Finally, the amount of fluorine incorporation (1–2 at% for ZnO_F1 and ∼6 at% for ZnO_F10) might serve as an orientation while interpreting the process of gaseous fluorination as a sequence of doping (without/with preferential coordination environment), saturation and recrystallization (ZnF2).
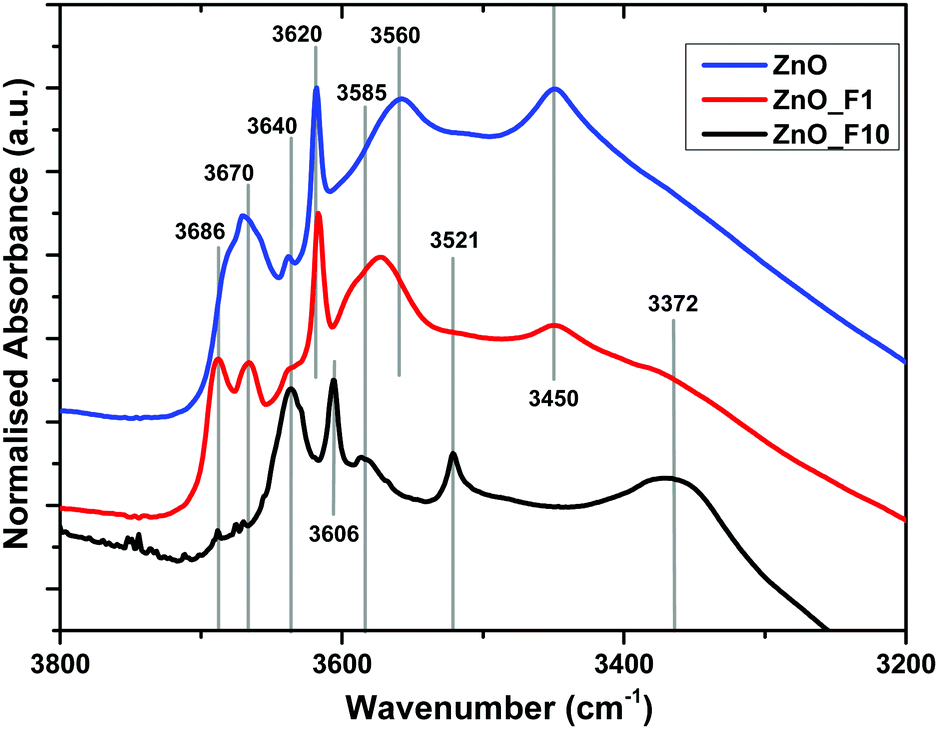 | ||
| Fig. 6 Normalized IR spectra of ZnO, ZnO_F1 and ZnO_F10. The samples were pre-treated in situ at 200 °C in synair to remove moisture. | ||
The bands above 3670 cm−1 are assigned to OH stretching modes associated with adsorbed water on the ZnO 1010 facet. These bands are absent for the highly fluorinated sample ZnO_F10. For ZnO_F1, the complex line shape becomes more resolved with two dedicated signals at 3686 cm−1 and 3670 cm−1, respectively. In addition, the relative intensity of the band assigned to OH groups on the ZnO surface not coupled to co-adsorbed water on this facet (3640 cm−1) increases significantly with the degree of fluorination. Therefore, it becomes apparent that the fluorine treatment reduces and finally removes adsorption sites of H2O on these facets. Furthermore, broad defect bands at 3450 cm−1 and 3560 cm−1 are affected. The intensity at 3450 cm−1 is reduced while the band at 3560 cm−1 is shifted to higher wavenumbers (from 3560 cm−1 to 3570 cm−1) upon low-pressure fluorination. Upon increasing the degree of fluorination, the band at 3570 cm−1 shifts to 3585 cm−1 and loses intensity, whereas the signal at 3450 cm−1 disappears. Concomitantly, two new features at 3521 cm−1 and 3372 cm−1 grow, which can be assigned to OH groups on ZnF240 as also evidenced by the XRD and NMR results. The relative ratio between the defect related bands in, e.g., ZnO and ZnO_F1 changes and becomes for ZnO_F1 at 3570 cm−1 and 3585 cm−1 more intense (or relatively weaker at 3450 cm−1). It seems that fluorine occupies parts of the existing intrinsic defect sites like, e.g., vacancies, on the expanse of new ones (extrinsic, fluorine induced). Thus, it appears plausible that fluorine reacts preferably with surface sites coupled to H2O or existing defects and subsequently exchanges with oxygen (with the consequence of new F-related defect sites). In addition, our IR spectroscopic insights are in agreement with the XPS results discussed above (increase in surface OH-groups on the expense of lattice oxygen for ZnO_F10).
To create a deeper understanding on the ongoing processes of fluorination and fluoride incorporation, in situ reference experiments were conducted. To this end, nanostructured ZnO is treated with F2(g) inside the spectrometer cell, and changes relative to the pristine ZnO were monitored as a function of time. Fig. 7 and 8 show the changes for the OH- and the carbonate-region regions of the IR spectrum. The reaction starts instantaneously after the addition of fluorine gas, and the spectrum continuously evolves for several hours. The most significant changes occur in the OH stretching region as already described above (Fig. 6). Upon exposure of the sample to F2, the bands at 3690 cm−1 and 3670 cm−1, which are assigned to H2O and OH with co-adsorbed water, respectively, both on the ZnO 1010 surface, decline. In contrast, the band at ∼3640 cm−1 increases as a result of the removal of the co-adsorbed water and the possible formation of new OH-groups on former defect sites. This band shifts to 3634 cm−1 at longer reaction times and with higher degrees of fluorination. Analogous changes are observed for the bands due to OH groups on the O-terminated 0001 surface of ZnO. The expected increase at 3616 cm−1 for ZnO with a low degree of fluorination is masked by the strong decrease of the ZnO related band at 3620 cm−1. Instead, an increase at around 3610 cm−1, an OH on the 0001 surface of ZnO, is found (shifts to lower wavenumbers, increased acidity). At longer reaction times, the OH vibration band at 3585 cm−1 which can be assigned to defects becomes visible, in line with a decline of the ZnO defects at 3560 cm−1 and 3450 cm−1. While a quantitative evaluation is not possible due to the strong overlap of bands, a comparison of the water related bands (at 3686 cm−1, 3670 cm−1) indicate that the former reacts somewhat faster with the provided F2. Furthermore, the OH band related to (extrinsic) defects upon F incorporation (or O substitution) at 3585 cm−1 is visibly delayed, interpreted as a consecutive reaction. Little difference is observed in how quickly F2 reacts with the mixed terminated 1010 and the polar terminated 0001 surfaces of ZnO. Concurrent with the changes of the OH stretching vibrations, the formation of gas phase CO2 as well as an overall decline of the intensity of the carbonate bands is observed (Fig. 8). Carbonates might be present as trace residuals of the synthesis process or formed on the surface by contact to the ambient.41 The formation of CO2 by the replacement of carbonate groups at the ZnO surface by fluoride can be expected to occur according to the following equation:
| 2F2 + 2CO32− → 4F− + 2CO2 + O2 |
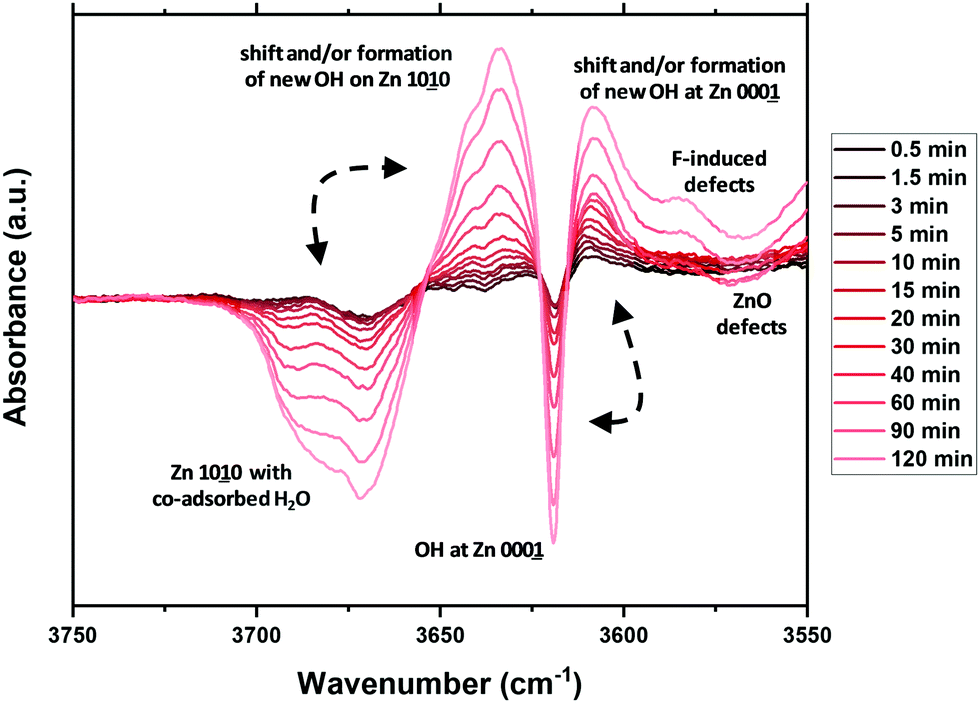 | ||
| Fig. 7 In situ difference infrared spectra following the fluorine gas treatment of ZnO, relative to the untreated sample (0 min). | ||
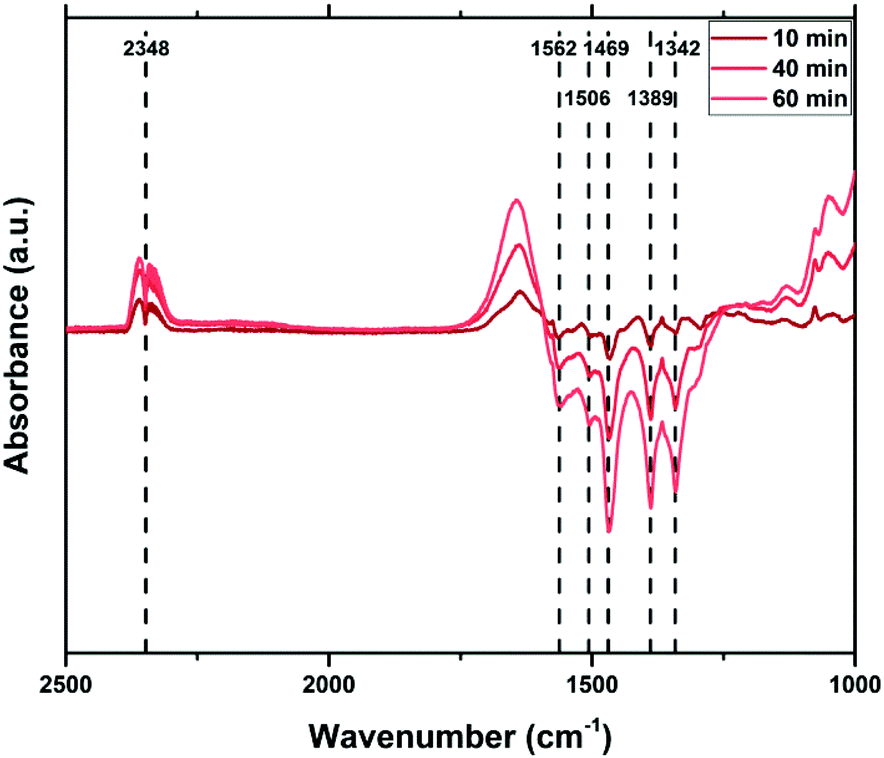 | ||
| Fig. 8 In situ IR spectra during F2 treatment of ZnO, relative to the untreated sample (0 min). | ||
In summary, the IR spectroscopic investigations unravel a couple of changes on the surface of ZnO upon fluorine treatment. The quality and quantity of the OH-groups is significantly altered. OH groups related to co-adsorbed water are gradually decreasing and new OH groups are formed. Besides, some existing OH-groups are shifted to lower energy as a sign for an increased Lewis acidity. No preferential surface termination for fluorination is identified, which might be explained by the overall high reactivity of F2(g). However, the hydroxyl- and carbonate-related functions seem to react faster than the defect related sites, in particular upon the formation of new (extrinsic or F-induced) ones.
3.3 Investigation of the electronic properties of fluorine treated ZnO
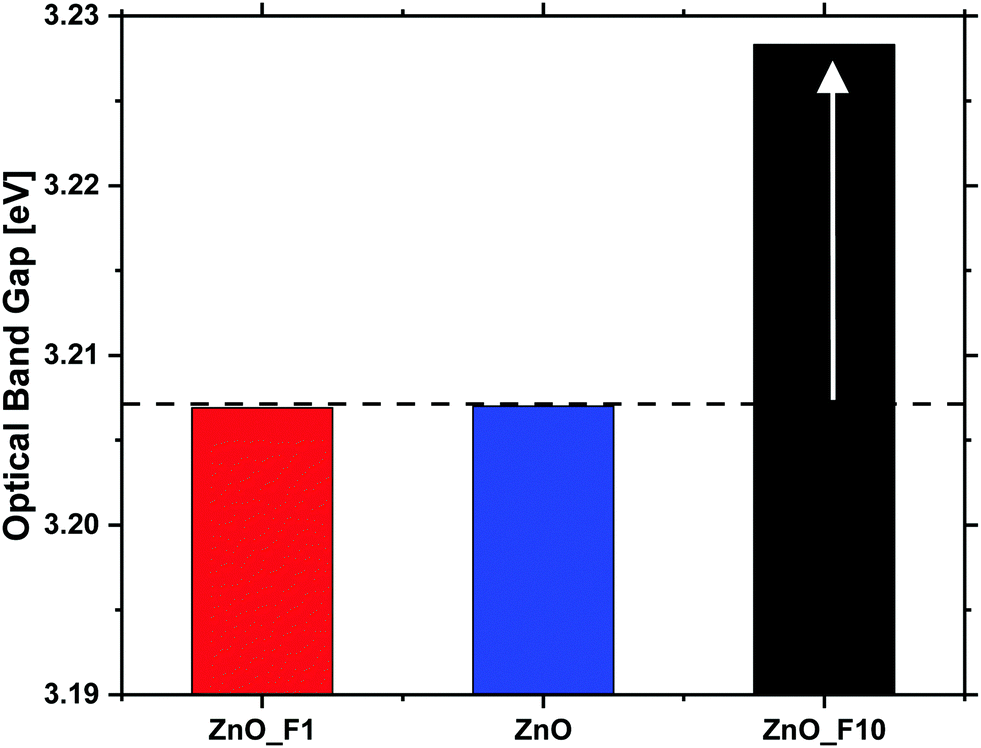 | ||
| Fig. 9 Optical band gaps of the untreated and fluorine treated ZnO samples as determined by UV-vis spectroscopy. | ||
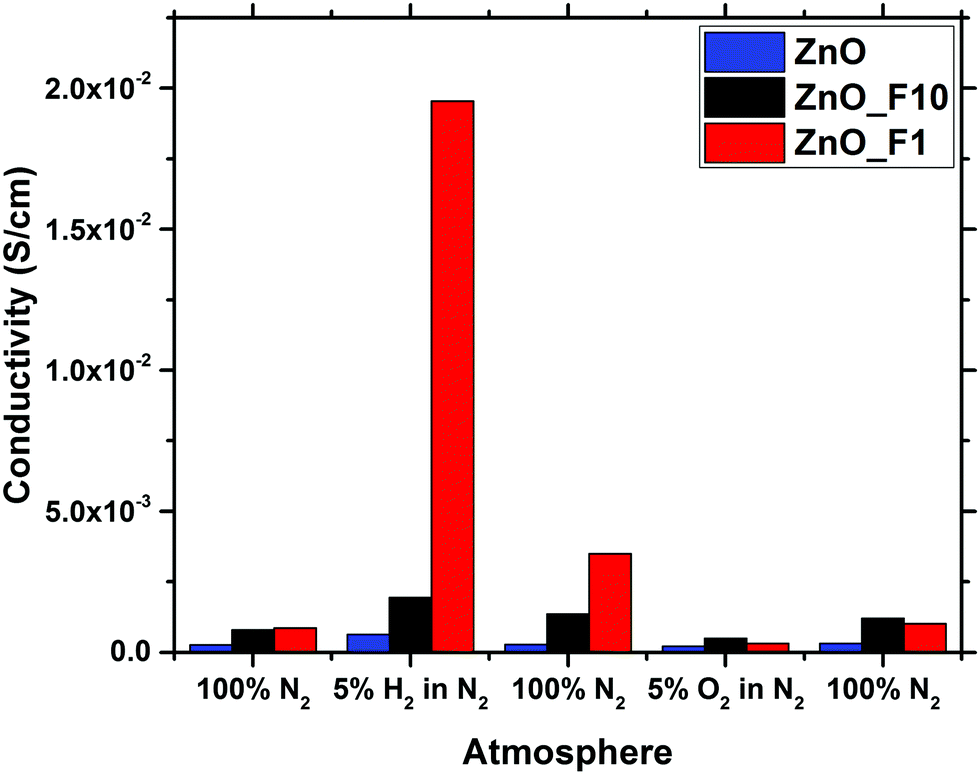 | ||
| Fig. 10 Conductivity of ZnO, ZnO_F1 and ZnO_F10 in different gas feeds determined by MCPT. | ||
In contrast to that, the increase of the conductivity upon reductive treatment is significantly larger for both of the fluorinated samples, indicating that the fluorination has altered the electronic properties of the materials. Since the optical band gap of ZnO_F1 was not affected by the fluorination, this increase in conductivity is rather explained by surface effects. This means that the activation and chemisorption of H2, providing/donating the electrons, is significantly enhanced. The changes induced by reduction are only partially reversible under inert conditions in contrast to the pristine ZnO. This indicates that the reduced state of the material is stabilized in case of the fluorinated systems, and that the activated H2 is bound rather weakly. Furthermore, it is interesting to note that an oxidative treatment, which has little effect on the pristine ZnO, reduced the conductivity of the fluorinated systems. This is, however, in line with the weak bonding of hydrogen on ZnO and thus already removed under inert conditions.
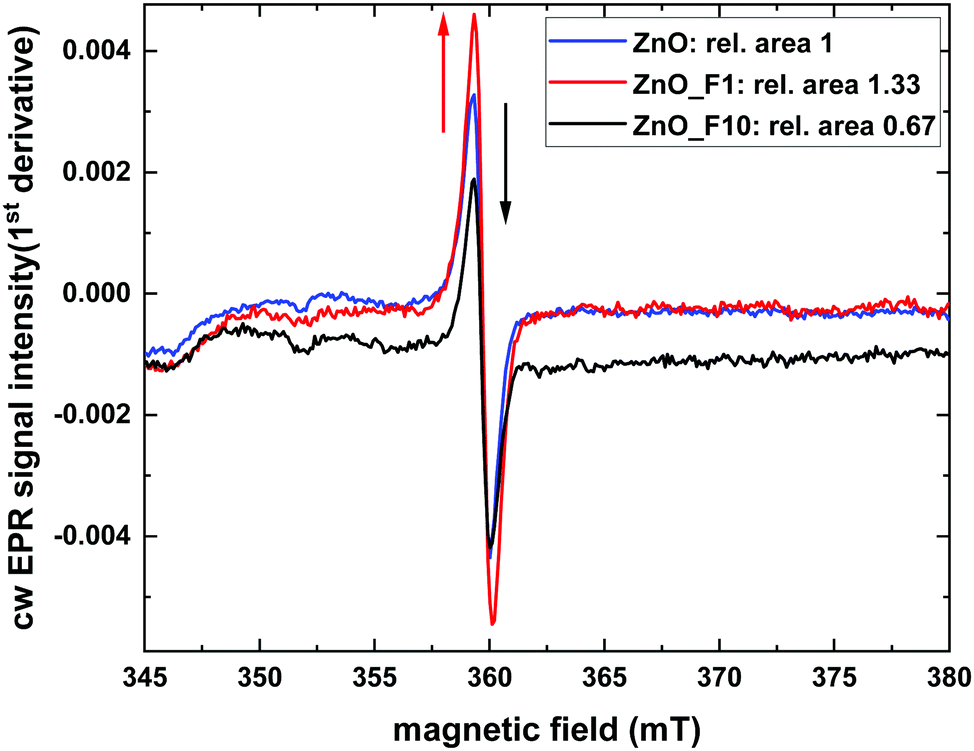 | ||
| Fig. 11 9.8 GHz cw EPR spectra at room temperature, normalised to catalyst mass. | ||
It is interesting to look for a spatial proximity of the defect centers and the F-anions in the lattice. The latter is possible by scrutinizing the coupling between the paramagnetic states to nuclear spins in its surrounding. To this end pulsed EPR measurements of ZnO_F1 were performed, which showed no direct coupling of the ZnO defect at g = 1.958 to fluorine nuclei (see Fig. SI15, ESI†). This excludes a simple substitutional F-doping of the oxygen sublattice and an associated trapping of the hole center close by.
While the XPS data provide evidence for a significant degree of hydroxylation of the ZnO surfaces the pulsed EPR spectra show no evidence for a coupling of the paramagnetic states to hydrogen atoms, which renders the corresponding species not to be located in the bulk of the sample. However, due to the limited impact of bulk F-doping (indicated by optical BG measurements), the ZnO-defect signal at g = 1.958 might rather be surface related and further explain the conductivity measurements, which significantly increased for ZnO_F1. Besides the higher quantity of defect sites for ZnO_F1, another criterion like surface acidity might influence the H2 activation (and conductivity) and is probed later by NH3-IR investigations.
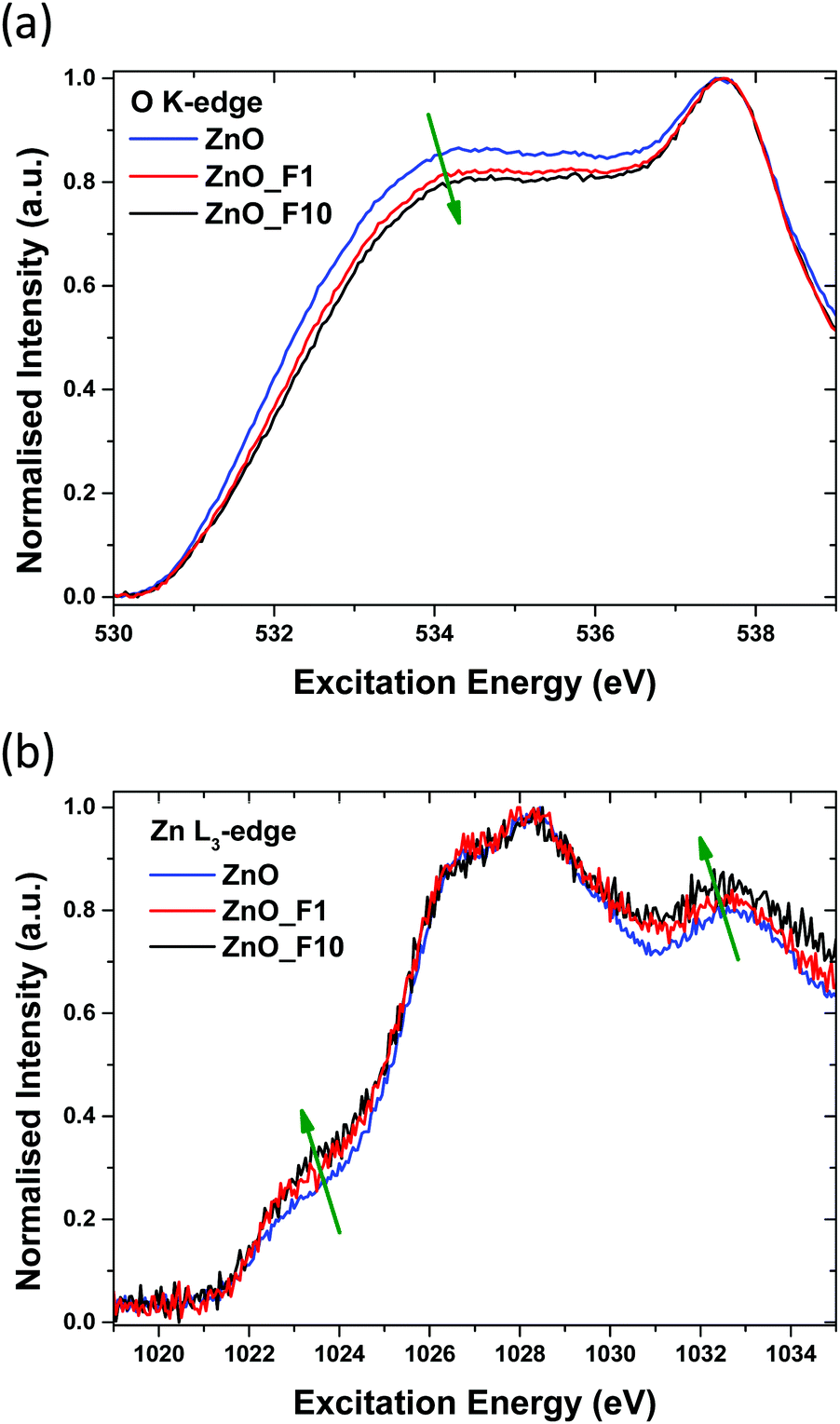 | ||
| Fig. 12 (a) O K-edge and (b) Zn L-edge of unfluorinated and fluorinated ZnO samples. | ||
3.4 Surface acid–base properties of fluorine treated ZnO
49,51 and MoO3 at 1260 cm−149). From a comparison with the IR spectrum of ZnF2 after NH3 adsorption (Fig. SI17, ESI†), the band cannot be assigned to adsorption sites on ZnF2 particles. Thus, it is likely that this band is also associated with an electron poor adsorption site, which is already present on ZnO in low concentrations. The concentration of such adsorption sites (defects) in fluorinated samples increases and possibly represents a Zn cation in a particularly fluoride enriched (likely ordered) environment.
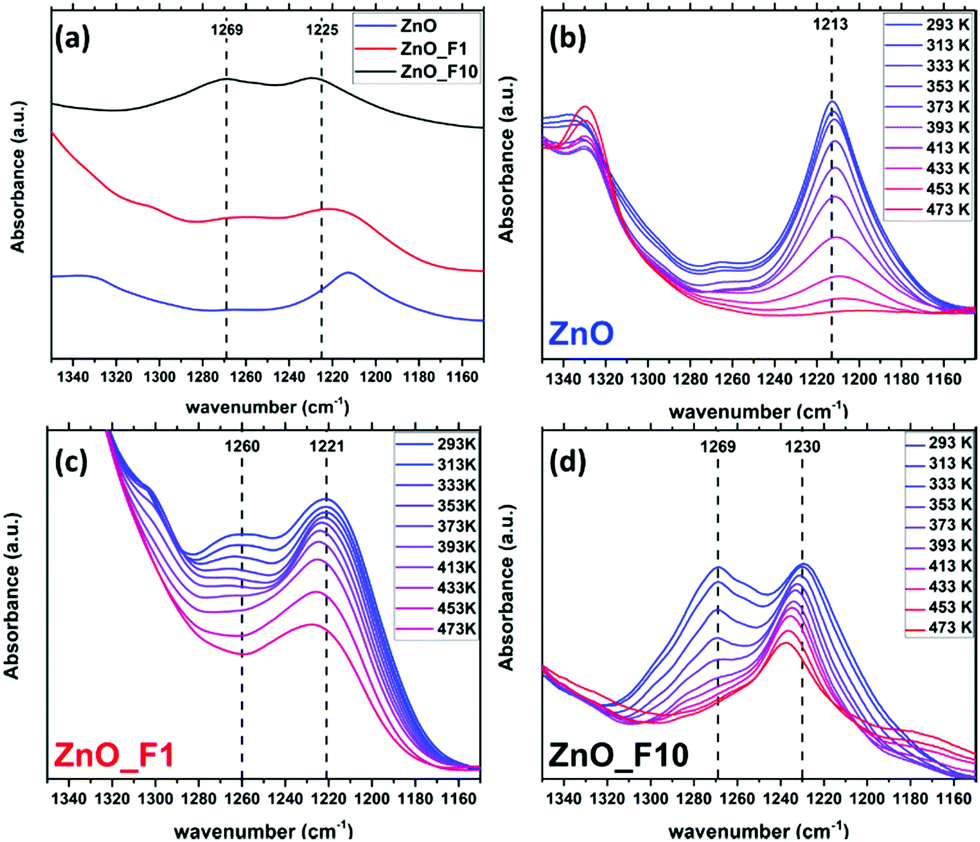 | ||
| Fig. 13 IR spectra after NH3 adsorption relative to pristine samples (a), and spectra taken during NH3 temperature programmed desorption of ZnO (b), ZnO_F1 (c) and ZnO_F10 (d). For reference, dashed lines in (a) are given at 1225 cm−1 and 1269 cm−1. | ||
Temperature programmed IR spectra (Fig. 13(b)–(d)) were measured to elucidate the adsorption strength of NH3 on the different adsorption sites. The stability of the weakly perturbed sites in ZnO_F1 and ZnO_F10 (1221 cm−1 and 1230 cm−1) is significantly enhanced (compared to ZnO at 1213 cm−1). The bands at 1260–1269 cm−1, related to Zn cations likely in a fluoride rich and ordered environment, decline more rapidly than the bands at 1221 cm−1 (ZnO_F1) and 1230 cm−1 (ZnO_F10), respectively. This might be an indication that an additional stabilisation (e.g., by neighbouring OH groups) of the adsorbed molecule on the highly Lewis acidic and fluorine enriched sites is absent (or present for the sites at 1209–1230 cm−1). However, the latter bands have a gradually increased stability with the degree of fluorination and Lewis acidity, also visible at the residual intensity at 473 K for, e.g., ZnO_F10. This might further explain the higher conductivity values under hydrogen atmosphere, since Lewis acid sites are known to be relevant for H2 activation (and here electron donation).52
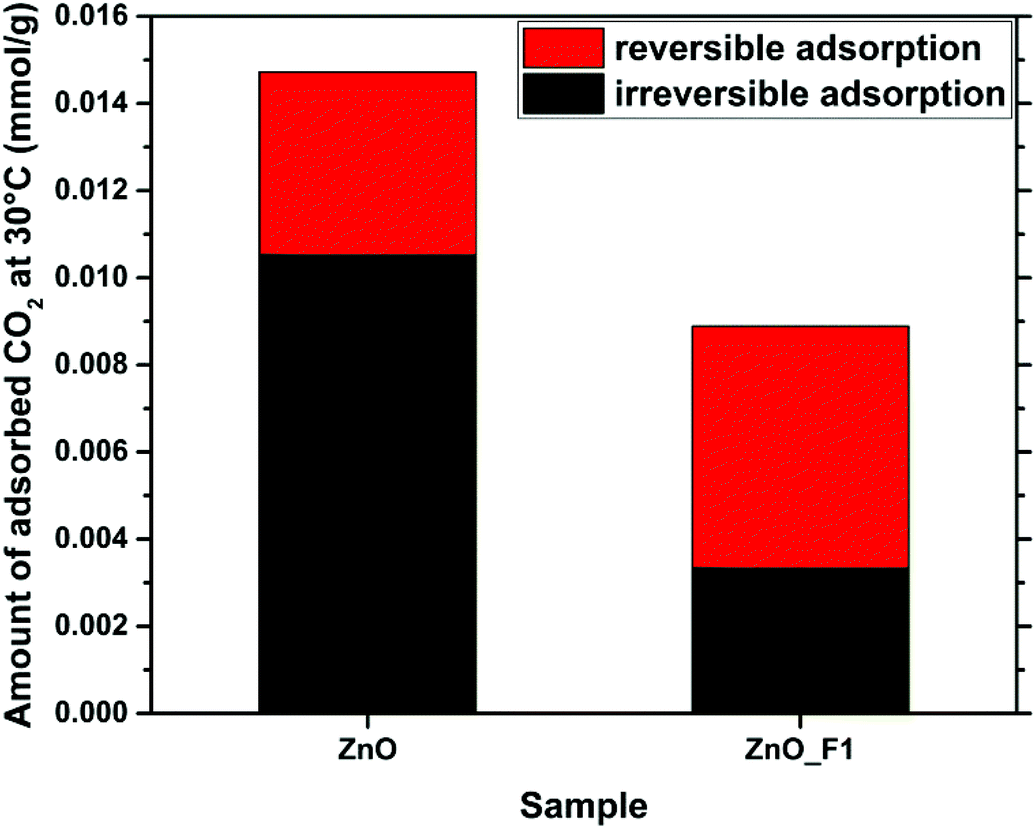 | ||
| Fig. 14 Adsorption of CO2 on the surfaces of ZnO and ZnO_F1. The reversible and irreversible contributions to the total adsorption are indicated. | ||
4 Conclusions and final discussion
In this contribution, we have presented the direct treatment of ZnO with F2(g) as a convenient way to modify both the electronic properties and the surface structure of ZnO. Hereby, the degree of fluorine incorporation into the sample can be controlled by variation of the synthesis parameters (partial pressure of F2, residence time of the fluorine gas on the sample). The time resolved fluorination indicates a kinetically controlled reaction of F2(g) with ZnO. In situ IR spectroscopy obtained during this reaction showed that fluorine removes carbonates on the ZnO surface and displaces adsorbed water, possibly by changing the interaction strength of the surface with H2O through replacement of O by F. No preferred ZnO surface termination with respect to their fluorine interaction was identified. The OH groups, also related to defect sites, were found to be altered in the spectrum as well. The treatment resulted in two types of fluorine modified sample: ZnO_F1 shows a spatially homogeneous distribution of fluorine throughout the sample, as evidenced by STEM/EDX mapping and its crystal structure shows only peaks that can be assigned to ZnO (in line with a possible F-doping of this material). In contrast, ZnO_F10, resulting from treatment of ZnO with a higher partial pressure of F2, resulted in “over-fluorination” as indicated by the appearance of a ZnF2 by-phase. This is demonstrated by NMR and X-ray diffraction and underlined by the agglomeration of F near the particle surface in the EDX map presented. In addition, the 19F-NMR experiments reveal as function of the fluorine content two different kinds of ZnO:F doping: In the low F-doping regime (1–2 at%) a rather heterogeneous coordination environment (see broad 19F-NMR signals) and a well ordered coordination with higher degree of doping (until 6 at%). A combination of XRD, NMR, EPR and in situ IR studies identifies the fluorination as intriguing process where F is very likely locates on lattice and interstitial sites. The substitution of lattice oxygen seems to be the primary process, since F-induced defect sites are detectable only delayed, as possible consecutive adaption of the structure. Comparing the F/Zn ratios of surface-near and bulk sensitive methods, a homogeneous surface enrichment of fluorine for ZnO_F1 and a heterogeneous for ZnO_F10 is indicated. This behaviour is to be expected for samples resulting from gas–solid-reactions and is currently under investigation for ZnO single crystals as part of a separate study.In both samples, the F2 treatment affected the electronic properties and the interaction of the sample with various gaseous probe molecules. Adsorption of NH3 and CO2 as Lewis-acidic and -basic probes, respectively, revealed, in the IR spectra of the samples and in the microcalorimetry investigation, that the Lewis basic sites in ZnO were quenched, while the Lewis acidity of the surface increased incrementally with the F-content. As a consequence, the interaction of the samples with CO2 was greatly reduced, as confirmed by microcalorimetry and TDS. Hereby, the fluorinated samples differed significantly in their behaviour, with ZnO_F10 showing an interaction with CO2 close to ZnF2 (and close to zero). Furthermore, using conductivity as an indicator, we showed that the less fluorinated ZnO_F1 showed a drastic increase in conductivity (and consequently in H2 activation) as compared to untreated ZnO and the sample with a higher F-content. Generally, with the degree of fluorination the surface Lewis acidity (on the expense of basicity) is gradually increased as monitored by IR and various probe molecules. The electronic properties, however, are mainly affected by the F-doping, evidenced by DRUVS, NEXAFS and conductivity measurements. This is explained by a combination of more defect sites near the surface (EPR) and a higher Lewis acidity (NH3-IR). The increase of the Lewis acidity alone seems not to be sufficient to increase the conductivity significantly (see ZnO_F10). Within an attempt to understand the complex processes upon fluorination, the 19F MAS NMR experiments serve as important bridge between these parts, identifying that nature and consequence of the occurring doping might also be different (at different levels of F-incorporation). Based on the numerous applied methods, the complexity of the nanostructured ZnO and relevant processes upon fluorination are identified and in parts unravelled. The impact of different local arrangements on, e.g., the surface properties has to be investigated within a separated DFT study.
Since ZnO often finds application as part of a CO2 reduction catalyst, these findings suggest that the catalytic performance of catalysts derived from fluorine treated ZnO may be influenced significantly.25 Hereby, an “over-fluorination” might result in a rather inactive catalyst, while a sample with a lower F-content (i.e. moderately doped ZnO:F) may improve the catalytic performance due to the interaction of this material with H2 and its dual site character of the surface (acid/basic surface sites). Further work on this sample system will be conducted in order to investigate the limits of F-doping without the formation of the ZnF2 by-phase, as well as the catalytic performance in CO2 reduction of ZnO:F adding a metal component.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank W. Frandsen and A. S. Tragl for SEM measurements and T. Drews for the treatment of ZnO with elemental fluorine. Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projekt-ID 387284271 – SFB 1349. (Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – Projektnummer 387284271 – SFB 1349) E. H. W. thanks the International Max Planck Research School “Functional Interfaces in Physics and Chemistry” for funding and support. Open Access funding provided by the Max Planck Society.Notes and references
- G. Wang, X. Yang, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2010, 10, 1088–1092 CrossRef CAS PubMed.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2015, 4, 4596 CrossRef PubMed.
- R. Schölin, M. Quintana, E. M. J. Johansson, M. Hahlin, T. Marinado, A. Hagfeldt and H. Rensmo, J. Phys. Chem. C, 2011, 115, 19274–19279 CrossRef.
- L. E. Greene, M. Law, J. Goldberger, F. Kim, J. C. Johnson, Y. Zhang, R. J. Saykally and P. Yang, Angew. Chem., Int. Ed., 2003, 42, 3031–3034 CrossRef CAS PubMed.
- Z. R. Tian, J. A. Voigt, J. Liu, B. Mckenzie, M. J. Mcdermott, M. A. Rodriguez, H. Konishi and H. Xu, Nat. Mater., 2003, 2, 821–826 CrossRef CAS PubMed.
- Y. J. Xing, Z. H. Xi, Z. Q. Xue, X. D. Zhang, J. H. Song, R. M. Wang, J. Xu, Y. Song, S. L. Zhang and D. P. Yu, Appl. Phys. Lett., 2003, 83, 1689–1691 CrossRef CAS.
- T. Lunkenbein, F. Girgsdies, T. Kandemir, N. Thomas, M. Behrens, R. Schlögl and E. Frei, Angew. Chem., Int. Ed., 2016, 55, 12708–12712 CrossRef CAS PubMed.
- C. Álvarez Galván, J. Schumann, M. Behrens, J. L. G. Fierro, R. Schlögl and E. Frei, Appl. Catal., B, 2016, 195, 104–111 CrossRef.
- M. B. Fichtl, J. Schumann, I. Kasatkin, N. Jacobsen, M. Behrens, R. Schlögl, M. Muhler and O. Hinrichsen, Angew. Chem., Int. Ed., 2014, 53, 7043–7047 CrossRef CAS PubMed.
- I. Beinik, M. Hellström, T. N. Jensen, P. Broqvist and J. V. Lauritsen, Nat. Commun., 2015, 8845 CrossRef CAS PubMed.
- J. Schumann, M. Eichelbaum, T. Lunkenbein, N. Thomas, M. C. Álvarez Galván, R. Schlögl and M. Behrens, ACS Catal., 2015, 3260–3270 CrossRef CAS.
- J. Schumann, T. Lunkenbein, A. Tarasov, N. Thomas, R. Schlögl and M. Behrens, ChemCatChem, 2014, 6, 2889–2897 CrossRef CAS.
- M. Behrens, G. Lolli, N. Muratova, I. Kasatkin, M. Hävecker, R. Naumann D’Alnoncourt, O. Storcheva, K. Köhler, M. Muhler and R. Schlögl, Phys. Chem. Chem. Phys., 2013, 15, 1374–1381 RSC.
- M. Wang, F. Ren, J. Zhou, G. Cai, L. Cai, Y. Hu, D. Wang, Y. Liu, L. Guo and S. Shen, Sci. Rep., 2015, 5, 12925 CrossRef CAS PubMed.
- A. M. Czoska, S. Livraghi, M. Chiesa, E. Giamello, S. Agnoli, G. Granozzi, E. Finazzi, C. Di Valentin and G. Pacchioni, J. Phys. Chem. C, 2008, 112, 8951–8956 CrossRef CAS.
- E. M. Samsudin and S. B. Abd Hamid, Appl. Surf. Sci., 2017, 391, 326–336 CrossRef CAS.
- X. Noirfalise, T. Godfroid, G. Guisbiers and R. Snyders, Acta Mater., 2011, 59, 7521–7529 CrossRef CAS.
- R. E. Treharne and K. Durose, Thin Solid Films, 2011, 519, 7579–7582 CrossRef CAS.
- H. Liang and R. G. Gordon, J. Mater. Sci., 2007, 42, 6388–6399 CrossRef CAS.
- P. M. Ratheesh Kumar, C. Sudha Kartha, K. P. Vijayakumar, F. Singh and D. K. Avasthi, Mater. Sci. Eng., B, 2005, 117, 307–312 CrossRef.
- R. Gonzalez-Hernandez, A. I. Martinez, C. Falcony, A. A. Lopez, M. I. Pech-Canul and H. M. Hdz-Garcia, Mater. Lett., 2010, 64, 1493–1495 CrossRef CAS.
- E. Şennik, S. Kerli, Ü. Alver and Z. Z. Öztürk, Sens. Actuators, B, 2015, 216, 49–56 CrossRef.
- E. Gunasekaran, M. Ezhilan, G. K. Mani, P. Shankar, A. J. Kulandaisamy, J. B. B. Rayappan and K. J. Babu, Semicond. Sci. Technol., 2018, 33, 095005 CrossRef.
- M. W. Kadi, D. McKinney, R. M. Mohamed, I. A. Mkhalid and W. Sigmund, Ceram. Int., 2016, 42, 4672–4678 CrossRef CAS.
- V. Dybbert, S. M. Fehr, F. Klein, A. Schaadt, A. Hoffmann, E. Frei, E. Erdem, T. Ludwig, H. Hillebrecht and I. Krossing, Angew. Chem., Int. Ed., 2019, 1–6 Search PubMed.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- D. G. Cory and W. M. Ritchey, J. Magn. Reson., 1988, 80, 128–132 Search PubMed.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- M. Eichelbaum, R. Stösser, A. Karpov, C.-K. Dobner, F. Rosowski, A. Trunschke and R. Schlögl, Phys. Chem. Chem. Phys., 2012, 14, 1302–1312 RSC.
- D. C. Dube, J. Phys. D: Appl. Phys., 1970, 3, 1648–1652 CrossRef CAS.
- H. Looyenga, Physica, 1965, 31, 401–406 CrossRef CAS.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, Adv. Catal., 2009, 52, 213–272 CAS.
- R. Farra, M. García-Melchor, M. Eichelbaum, M. Hashagen, W. Frandsen, J. Allan, F. Girgsdies, L. Szentmiklósi, N. López and D. Teschner, ACS Catal., 2013, 3, 2256–2268 CrossRef CAS.
- Y.-N. Xu and W. Y. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4335–4351 CrossRef CAS PubMed.
- W. H. Baur and A. A. Khan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 2133–2139 CrossRef CAS.
- S. C. Abrahams and J. L. Bernstein, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1233–1236 CrossRef CAS.
- P. A. Stadelmann, Ultramicroscopy, 1987, 21, 131–146 CrossRef CAS.
- G. Scholz, C. Stosiek, J. Noack and E. Kemnitz, J. Fluorine Chem., 2011, 132, 1079–1085 CrossRef CAS.
- H. Noei, H. Qiu, Y. Wang, E. Löffler, C. Wöll and M. Muhler, Phys. Chem. Chem. Phys., 2008, 7092–7097 RSC.
- Y. Guo, S. Wuttke, A. Vimont, M. Daturi, J. C. Lavalley, K. Teinz and E. Kemnitz, J. Mater. Chem., 2012, 22, 14587–14593 RSC.
- J. Strunk, K. Kähler, X. Xia and M. Muhler, Surf. Sci., 2009, 603, 1776–1783 CrossRef CAS.
- G. P. Papari, B. Silvestri, G. Vitiello, L. De Stefano, I. Rea, G. Luciani, A. Aronne and A. Andreone, J. Phys. Chem. C, 2017, 121, 16012–16020 CrossRef CAS.
- J. W. Chiou, J. C. Jan, H. M. Tsai, C. W. Bao, W. F. Pong, M.-H. Tsai, I.-H. Hong, R. Klauser, J. F. Lee, J. J. Wu and S. C. Liu, Appl. Phys. Lett., 2004, 84, 3462–3464 CrossRef CAS.
- P. J. Møller, S. A. Komolov and E. F. Lazneva, J. Phys.: Condens. Matter, 1999, 11, 9581–9588 CrossRef.
- J. G. Chen, Surf. Sci. Rep., 1997, 30, 1–152 CrossRef CAS.
- T. Yamamoto, T. Mizoguchi, K. Tatsumi, I. Tanaka, H. Adachi, Y. Muramatsu, E. M. Gullikson and R. C. C. Perera, Mater. Trans., 2004, 45, 1991–1993 CrossRef CAS.
- J. K. Wilmshurst, Can. J. Chem., 1960, 38, 467–472 CrossRef CAS.
- G. F. Svatos, D. M. Sweeny, S.-I. Mizushima, C. Curran and J. V. Quagliano, Infrared Spectrosc. Adsorbed Species Surf. Transit. Met. Oxides, 1957, 3313–3315 CAS.
- A. A. Davydov, Infrared Spectroscopy of Adsorbed Species on the Surface of Transition Metal Oxides, John Wiley & Sons, Chichester, 1990 Search PubMed.
- F. Yin, A. L. Blumenfeld, V. Gruver and J. J. Fripiat, J. Phys. Chem. B, 1997, 101, 1824–1830 CrossRef CAS.
- A. A. Davydov, Kinet. Catal., 1993, 34, 803–808 Search PubMed.
- J. Nováková, L. Kubelková and P. Jírů, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 1331–1339 RSC.
- M. M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlögl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected XPS, electron microscopy, UV-vis, microcalorimetry, EPR, TDS and IR data are listed. See DOI: 10.1039/d0cp00545b |
| This journal is © the Owner Societies 2020 |