
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inter-protein interactions govern protein loading into porous vaterite CaCO3 crystals
Natalia A.
Feoktistova
ab,
Nadezhda G.
Balabushevich
a,
Andre G.
Skirtach
 c,
Dmitry
Volodkin
ad and
Anna S.
Vikulina
*b
c,
Dmitry
Volodkin
ad and
Anna S.
Vikulina
*b
aDepartment of Chemistry, Lomonosov Moscow State University, Leninskiye gory 1-3, 119991 Moscow, Russia
bFraunhofer Institute for Cell Therapy and Immunology, Branch Bioanalytics and Bioprocesses, Am Mühlenberg 13, 14476 Potsdam-Golm, Germany. E-mail: anna.vikulina@izi-bb.fraunhofer.de; Tel: +49-331 58187-122
cDepartment of Biotechnology & NB-Photonics, University of Ghent, Coupure Links 653, 9000 Gent, Belgium
dSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK
First published on 24th April 2020
Abstract
The fast development of protein therapeutics has resulted in a high demand for advanced delivery carriers that can effectively host therapeutic proteins, preserve their bioactivity and release them on demand. Accordingly, vaterite CaCO3 crystals have attracted special attention as sacrificial templates for protein encapsulation in micro- and nanoparticles (capsules and beads, respectively) under mild biofriendly conditions. This study aimed to better understand the mechanism of protein loading into crystals as a primary step for protein encapsulation. The loading of three therapeutic proteins (250 kDa catalase, 5.8 kDa insulin, and 6.5 kDa aprotinin) was investigated for crystals with different porosities. However, unexpectedly, the protein loading capacity was not consistent with the protein molecular weight. It solely depends on the inter-protein interactions in the bulk solution in the presence of crystals and that inside the crystals. The smallest protein aprotinin aggregates in the bulk (its aggregate size is about 100 nm), which prohibits its loading into the crystals. Insulin forms hexamers in the bulk, which can diffuse into the crystal pores but tend to aggregate inside the pores, suppressing protein diffusion inward. Catalase, the largest protein tested, does not form any aggregates in the bulk and diffuses freely into the crystals; however, its diffusion into small pores is sterically restricted. These findings are essential for the encapsulation of protein therapeutics by means of templating based on CaCO3 crystals and for the engineering of protein-containing microparticles having desired architectures.
1. Introduction
Since the commercialisation of recombinant human insulin as the first therapeutic protein in 1982, the market of protein-based drugs has become one of the most rapidly growing areas in the pharmaceutical industry.1 Nowadays, several therapeutic proteins belonging to different classes (monoclonal antibodies, hormones, enzymes, growth factors, antimicrobial peptides, etc.) are indicated for the treatment of multiple disorders, including various cancers, diabetes, haemophilia, asthma, and plaque psoriasis.2 As natural biological products, proteins are selective, non-toxic and generally possess much lower side effects than that usually caused by small-molecular-weight drugs. However, to date, the therapeutic potential and wide clinical use of protein-based therapeutics are largely limited by their (i) high fragility resulting from their labile and microenvironment-sensitive 3D structure and (ii) short half-life in the blood stream, which is mostly caused by their fast degradation by proteases.3,4 These obstacles can be circumvented via the encapsulation of therapeutic proteins into delivery carriers designed to preserve the structure and bioactivity of protein molecules, protect them against enzymatic degradation, and optionally achieve controlled and targeted protein delivery.5Despite the existence of numerous drug delivery carriers, the majority of them have been designed for the delivery of small-molecular-weight drugs and the encapsulation conditions are inappropriate for protein loading. This is due to the harsh formulation conditions, which are harmful for protein bioactivity (e.g., exposure to organic solvents), surface modification and low encapsulation capacities.6,7 This has stimulated a high demand for the development of sustainable nano- and micro-particulate containers suitable for the mild encapsulation of proteins.6,8,9
Accordingly, inorganic crystals of vaterite calcium carbonate have attracted special attention from researchers.10,11 CaCO3 is a low-soluble mineral that is abundant in nature12 and can be easily synthesized by mixing precursor salts containing Ca2+ and CO32+ ions.13 Structurally, vaterite crystals are comprised of a multitude of spherical nanocrystallines,13 which aggregate and form a highly developed mesoporous structure with an average pore size in the range of 10–60 nm.14,15 This pore size is ideal for the entrapment of large macromolecules with similar dimensions. These crystals can be formulated in a wide range of sizes starting from tens of nm16,17 up to tens of μm.18 The shape of the particles also can be tuned.19 Importantly, although the synthesis of CaCO3 is commonly performed in the presence of organic solvents20 and polymer additives21–23 and various stimuli24 can be used to control its pore size,9 recently the mild way to produce vaterite CaCO3 crystals in aqueous media at a pH close to near neutral (7–8) and ionic strength close to physiological values has been proposed.13 Additionally, CaCO3 crystals are fully degradable and can be easily hydrolysed under slightly acidic pH and in the presence of Ca2+-binding agents, or recrystallised into more stable calcite polymorphs, changing their porous structure to a non-porous one and releasing encapsulated drugs.25,26 Recent studies also demonstrated the high cellular uptake of CaCO3.27,28 In addition, CaCO3 crystals can be used as sacrificial templates for the formulation of porous protein29 and polymer beads,30,31 and layer-by-layer assemblies to produce multilayer capsules.32–35 All these findings indicate that CaCO3 crystals are promising carriers for the encapsulation of protein-based therapeutics for oral,36–38 intravenous,17 intratumoral,39 intranasal,40 and mucosal drug delivery;41,42 tissue engineering43,44 and regeneration;45,46 and diagnostics.47,48 This diversity of administration routes enabled by CaCO3 for protein delivery is especially important because in current medical practice, protein drugs are predominantly available in injectable formulations, but the demand for non-invasive drug delivery systems for therapeutic proteins is high.49
Despite the apparent potential of CaCO3 crystals for the delivery of protein-based drugs, the inclusion of proteins inside crystals largely remains unexplored. The two main approaches for the inclusion of the proteins in the solid matrix of CaCO3 are co-synthesis (protein entrapment in CaCO3 during the formation of the crystals) and adsorption (physisorption of proteins onto pre-formed crystals).50 It is encouraging that regardless of the loading approach, CaCO3 crystals have shown enormous loading capacities for many therapeutic proteins, and furthermore the bioactivity of the proteins after their release from CaCO3 crystals remains high.51,52 Notably, co-synthesis often results in higher loading efficiencies but lower residual protein activities.51,53 Nowadays it is widely believed that the adsorption of macromolecules onto the porous surface of CaCO3 crystals is presumably governed by electrostatic protein–CaCO3 interactions, which has been proposed for polyelectrolytes,54,55 DNA56 and globular proteins.57,58 Modulation of the strength of electrostatic interactions via the doping of CaCO3 crystals with highly charged polymer matrices has been reported to enhance protein entrapment.59 Additionally, the impact of intermolecular interactions on the adsorption and retention of macromolecules inside the crystal pores has been clearly identified, but to date this remains poorly understood.60 This is indicated by the number of contradictory findings. For instance, the co-synthesis of catalase into vaterite crystals resulted in the entrapment of a large amount of this protein, which greatly exceeded the maximum possible values calculated theoretically. This has been explained by protein aggregation in the presence of Ca2+.53 Another demonstrative example is the adsorption of the linear polymer poly(acrylic acid) on the CaCO3 surface, which has been characterized by drastically different Gibbs energies ranging from −45 kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 61 up to +395 kJ mol−162 in the corresponding studies. Importantly, some studies report a direct correlation57 between the protein net charge and adsorption capacities, while others report no evidence of the correlations.50 Thus, the overall mechanism of macromolecular loading in crystals remains unclear.
61 up to +395 kJ mol−162 in the corresponding studies. Importantly, some studies report a direct correlation57 between the protein net charge and adsorption capacities, while others report no evidence of the correlations.50 Thus, the overall mechanism of macromolecular loading in crystals remains unclear.
This study aimed to reveal the main factors responsible for the protein loading in vaterite crystals, and therefore decipher the mechanism of this type of protein loading. The role of the inter-molecular interaction between three model globular proteins, namely the enzyme catalase (CAT), the hormone insulin (INS), and the proteinase inhibitor aprotinin (APR), was evaluated for protein adsorption on vaterite crystals of different porosity, i.e. average pore sizes of 10 and 15 nm. This was done by analysing the protein diffusion in the crystals, protein distribution inside the crystals, and protein aggregation state in solution. The chosen proteins have different molecular masses and isoelectric points (pI), allowing the impact of the physical–chemical characteristics of the proteins on their loading into the crystals to be assessed as a key to understand the loading mechanism. Besides, nowadays, these proteins are widely used in medicine. Specifically, CAT is an active component of gels applied for the healing of skin ulcers and burns, INS is used to control blood sugar in diabetic patients, and APR is used to decrease bleeding during surgeries and treat some disorders. This makes them favorable candidates for studying the protein interactions with vaterite carriers.
2. Experimental
2.1. Materials
Calcium chloride dihydrate CaCl2·2H2O, sodium carbonate (Na2CO3, anhydrous), and fluorescein isothiocyanate isomer I (FITC) were purchased from Sigma-Aldrich (Germany); aprotinin from bovine lung: preparation “Ingiprol” (60% active centre) from RUE Belmedpreparaty (Belarus); insulin, zinc salt from Human Biosynthetic, IBCh RAS, Russia; and catalase from bovine liver (C-1345) from Sigma, Germany. The water used in all experiments was prepared using a Millipore Milli-Q purification system and had a resistivity higher than 18.2 MΩ cm.2.2. Synthesis of CaCO3 microcrystals
Porous vaterite CaCO3 microcrystals were prepared by rapidly mixing an equal volume of aqueous solutions of CaCl2 and Na2CO3 at two different temperatures according to the protocols described elsewhere.13 Briefly, 3 mL of 1 M CaCl2 was added to 9 mL of water with constant stirring at a temperature of 22 °C or 45 °C. Then 3 mL of 1 M Na2CO3 (1 M) was rapidly added to this solution at 22 °C or 45 °C, respectively. After vigorous agitation with a magnetic stirrer (30 s, 160 rpm) and incubation for 15 min at the corresponding temperature, the precipitate was filtered, washed with water and dried at 80 °C.2.3. FITC-conjugation of proteins
0.1 mg mL−1 FITC solution in 0.5 M carbonate buffer (pH 9.0) was added dropwise to 2 mg mL−1 protein solution in the same buffer under stirring to obtain the FITC:protein molar ratio of 1:5. The prepared solutions were incubated for 4 h in the dark and placed in dialysis bags with a cutoff 8–10 kDa. Then dialysis against water was conducted for all the protein solutions for 2 days. The obtained proteins solutions were stored at −20 °C.
2.4. Confocal laser scanning microscopy (CLSM)
CLSM analysis was performed using a Zeiss LSM 510 Meta (Zeiss, Germany) equipped with an oil-immersion objective with 40× magnification and numerical aperture of 1.3. Standard filter settings for excitation and emission of FITC were used for a laser source with a wavelength of 488 nm. To study the protein interaction with CaCO3 microcrystals, 0.2 mL of 1 mg mL−1 FITC-labelled protein solution (insulin-FITC, aprotinin-FITC, or catalase-FITC) was added to 1 mg of microcrystals followed by short agitation. Kinetic study of the protein accumulation was performed by monitoring the fluorescence signal inside the crystals. Images were processed using Image PRO (Adobe Systems Inc) to optimise brightness and colour.2.5. Dynamic light scattering (DLS)
1 mg mL−1 protein solution in water (pH 10.5) or in the supernatant of the CaCO3 microcrystals was used for DLS measurements using a Zeta-sizer Nano ZS (Malvern, UK). The supernatant was prepared by dispersing 15 mg of CaCO3 microcrystals in 1 mL of water followed by 15 min incubation and centrifugation to completely precipitate the crystals. The protein solutions were filtered through a 0.22 μm filter before the measurements.2.6. Scanning electron microscopy (SEM)
SEM images were recorded using a Gemini LEO 1550 electron microscope at an accelerating voltage of 3 kV. Samples were prepared by dropping the particle suspension on a glass slide followed by 1 h drying at 90 °C and then sputtering with gold.2.7. Brunauer, Emmett and Teller (BET) analysis
N2 adsorption–desorption analysis was performed using a QUADRASORB SI-MP (Quantachrome Instruments, USA) at 77.3 K. Prior to measurement, the samples were degassed at 150 °C for 20 h. The Brunauer–Emmett–Teller (BET) theory and the Barret–Joyner–Halenda model were used for the surface area and pore-size distribution analysis, respectively.2.8. Raman microscopy
The Raman spectra of the prepared CaCO3 crystals was measured using a Raman microscope (CRM200, WITec, Ulm, Germany) equipped with a piezo-scanner (P-500, Physik Insrtumente, Karlsruhe, Germany) and a diode-pumped 785 nm NIR laser excitation (Toptica Photonics AG, Graefelfing, Germany). The laser beam was focused through a 100× (NA 0.95, Olympus) objective. The spectra were acquired with a thermoelectrically cooled CCD detector (DU401ABV, Andor, UK) behind a grating (300 g mm−1) spectrograph (Action, Princeton Instruments Inc., Trenton, Nj, USA) with a spectral resolution of 6 cm−1.2.9. Mathematical fitting
The experimental points for the kinetics of protein adsorption were fitted with the models described below.Pseudo-first order (PFO) kinetics:
| Q = Qmax·(1 − exp(−kt)) | (1) |
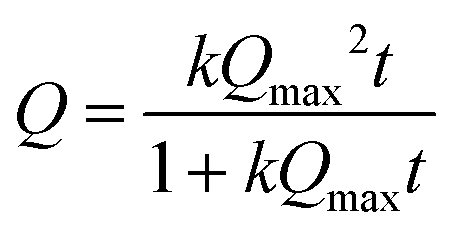 | (2) |
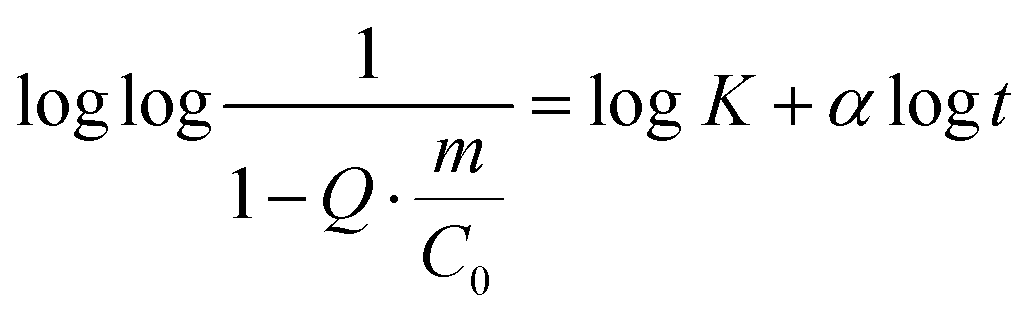 | (3) |
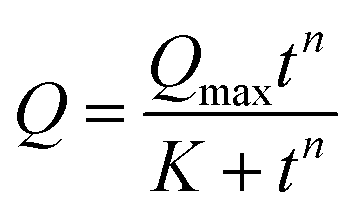 | (4) |
The goodness of the fit was assessed based on adjusted R-squared coefficients.
3. Results and discussion
3.1. Protein state in solution
The role of the protein nature in protein loading/diffusion in vaterite microcrystals was investigated using three model therapeutic proteins with various sizes and different pI (CAT, INS, and APR). CAT and INS have similar pI (about 5.4) but have different molecular weights, where CAT (250 kDa) is much heavier than INS (6 kDa). Additionally, the hydrodynamic diameter of CAT (dh ∼ 10.4 nm)63 exceeds that of INS (dh ∼ 2.7 nm)64 by about 3–4 times. INS and APR are very similar in terms of molecular size (dh ∼ 2.9 nm for APR);65 however, APR has much higher pI (10.5).Since vaterite crystals are partially soluble in water (solubility constant of 1.3 × 10−5 M)26 and prone to hydrolysis, the aqueous suspension of the crystals has a pH of about 10.5. Therefore, in the presence of the crystals, CAT and INS are positively charged and APR possesses no charge.
Besides, the presence of Ca2+ and HCO3−/CO32− ions liberated from the CaCO3 crystals may cause conformational changes in the protein structure. accordingly, the potential role of these ions in protein aggregation was probed by comparing the DLS volume-weighted size distribution profiles of the aqueous protein solutions with the pH adjusted to 10.5 and protein solution in the supernatant of the crystal suspension having the same pH (Fig. 1). The average hydrodynamic diameters (dh) of CAT, APR, and INS in water with pH 10.5 were found to be 7.7 ± 1.5, 1.8 ± 0.9, and 4.5 ± 0.3 nm, respectively, which correlate well with the literature data. Notably, INS was present in the form of a hexamer, which is typical for this protein (dh ∼ 5.1 nm).66
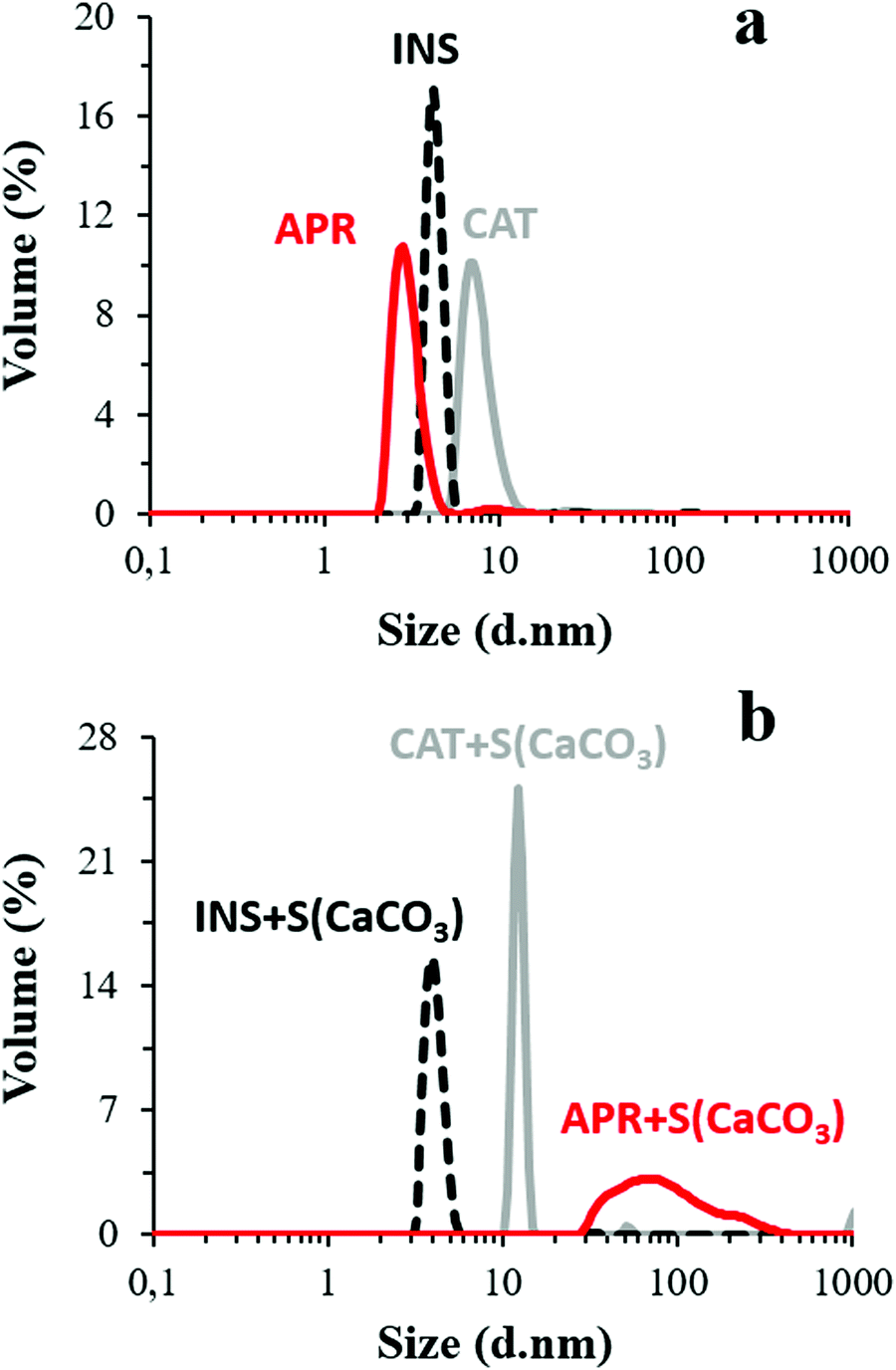 | ||
| Fig. 1 Size distribution by volume of the proteins in aqueous solution with the pH adjusted to 10.5 (a) and protein–FITC dissolved in the supernatant of CaCO3 microcrystals at pH 10.5 (b) at 22 °C. All solutions had a concentration of 0.1 mg mL−1 and were filtrated through a 0.22 μm filter. | ||
The dh of the proteins in the presence of the crystal supernatant is shown in Fig. 1b. CAT and INS remained stable and had the same dh as that in water with pH 10.5. However, APR was prone to strong aggregation, where the average dh of the self-associated APR assemblies increased significantly to ca. 70–80 nm. The rate of the aggregation of APR was very high, as concluded from the analysis of the change in count rate as a function of time (Fig. 2). The aggregation was followed by sedimentation of the aggregates, and consequently, a reduction in the intensity of the scattered light. The count rate drastically decreased after 5 min of replacement of APR in the supernatant solution of the crystals. In contrast, no significant changes were observed for CAT and INS in the supernatant compared to the water solution with pH 10.5. The increase in the average dh of CAT by a factor of almost two indicates the tendency of CAT to aggregate in the presence of the ions in the supernatant due to the hydrolysis of the crystals.
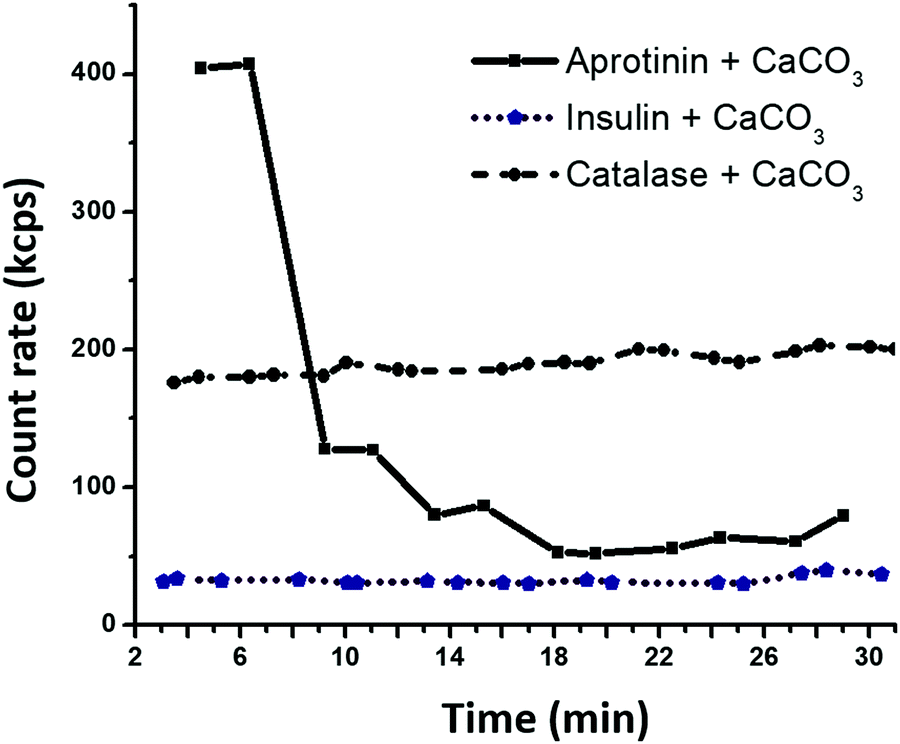 | ||
| Fig. 2 Time dependence of count rate for protein–FITC exposed to the supernatant of CaCO3 microcrystals at 22 °C. | ||
To investigate the role of the pore dimensions on the protein loading into the crystals, as described in the next section, two batches of crystals possessing different porosity were used, where the dimensions of the pore sizes:
(i) exceeded the average size of INS and CAT but remained below the diameter of the APR aggregates and (ii) exceeded the average size of INS but closely corresponded to the diameter of CAT.
3.2. CaCO3 microcrystals of different porosity
Vaterite CaCO3 microcrystals having different porosity were prepared as described in our previous study13 by varying the preparation temperature (22 °C and 45 °C). Both types of crystals have a similar diameter of 10 ± 2 μm, but differ in their porosity. The BET analysis revealed that the total pore volume of the crystals (Vp) is 9.7 × 10−2 mL g−1 and 20.5 × 10−2 mL g−1 for the crystals prepared at 22 °C and 45 °C, respectively. Considering that the apparent (bulk) density of the crystals (ρ) equals 2.54 g mL−1 (Mineralogy Database), the relative porosity of the crystals can be calculated using an equation:| ε = ρ·Vp | (5) |
Fig. 3a and b present the SEM images of the microcrystals prepared at 22 °C and 45 °C, respectively. The shape of both types of crystals is not ideally spherical, and their morphology is rather flocky-like. These morphological features are typical for large CaCO3 crystals (size exceeding 10 μm).13,19 The crystals were the vaterite polymorph of CaCO3, as proven by the Raman microscopy analysis (Fig. 3c). The characteristic peaks of the vaterite polymorph (double peaks at 265 and 302 cm−1 and at 1075–1090 cm−1) are marked by arrows, and no characteristic peaks of calcite were observed.67
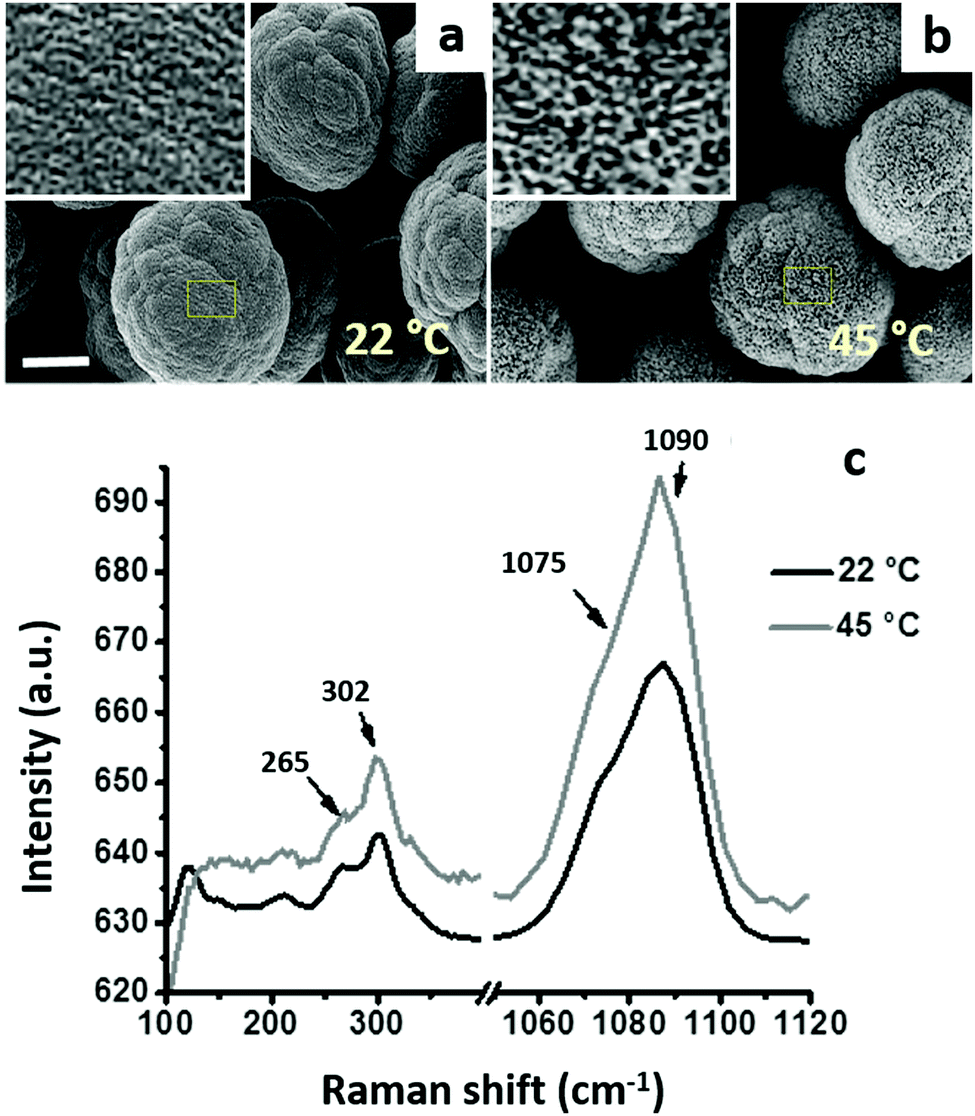 | ||
| Fig. 3 SEM images of CaCO3 microcrystals prepared at 22 °C (a) and 45 °C (b) in water. Scale bar is 4 μm. Insets show 8× zoomed surfaces of the crystals within yellow rectangular areas. (c) Raman spectra of the microcrystals prepared at 22 °C and 45 °C (the arrows point at characteristic picks of vaterite). | ||
3.3. Protein distribution inside the crystals of different porosity
The equilibrium distribution of the proteins on the surface and inside the CaCO3 microcrystals was probed by CLSM for the crystals exposed to an aqueous solution of FITC-labelled proteins. Notably, the significant difference in the refractive indices of water and the crystals (1.55–1.65 and 1.33, respectively) prevented quantitative conclusions from being drawn regarding the protein localization. In addition, the structure of the crystals is mesoporous, which may induce undesirable optical effects. However, a fluorescence signal from the FITC-labelled proteins was detected for the crystals, meaning that the excitation light (wavelength 488 nm) could penetrate the porous crystals, which allowed us to perform a semi-qualitative analysis.Fig. 4 illustrates the CLSM images of the crystals after 1 h of incubation in the protein solutions. CAT was evenly distributed in the whole internal volume of the crystals with large pores (15 nm) exceeding its dh value (Fig. 4b and d). In contrast, the incubation of ∼10 nm CAT into the crystals with a smaller pore sizes (∼10 nm) resulted in the nonhomogeneous distribution of CAT, which only partially penetrated the internal volume of the crystals and largely remained on the crystal surface (Fig. 4a and c). The obvious explanation for this is that CAT fills the larger pores and has diffusion limitations to enter the pores that are smaller or comparable to its dh.
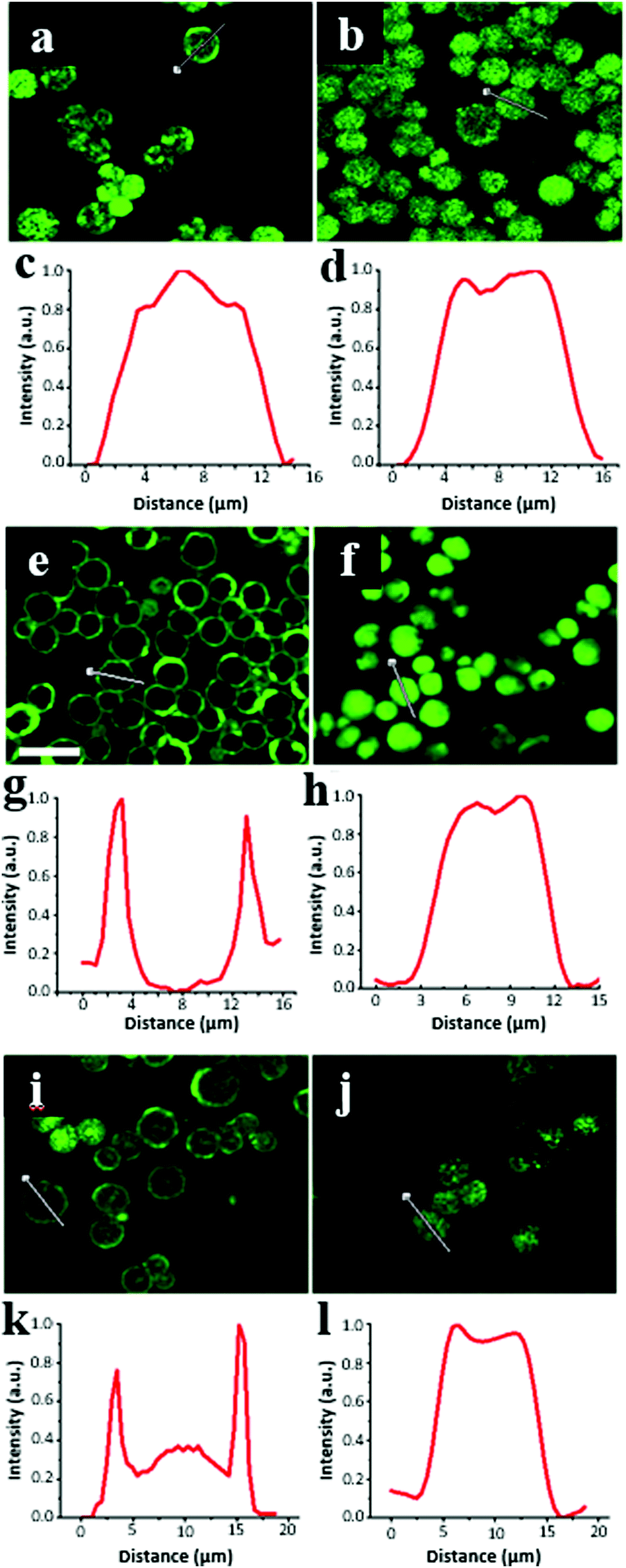 | ||
| Fig. 4 Confocal images and fluorescence profiles taken across crystals incubated for 60 min with proteins labelled by FITC. The crystals prepared at 22 °C (left: a, c, e, g, i, and k) and 45 °C (right: b, d, f, h, j, and l) and have average pore sizes of 10 and 15 nm, respectively. The proteins are catalase (a)–(d), insulin (e)–(h), and aprotinin (i)–(l). Scale bar is 20 μm. | ||
The signal originating from CAT loaded in the crystals also varied in each crystal. As can visually be concluded from the images in Fig. 4a and b, the signal intensity of CAT varied more between the crystals prepared at 22 °C than that at 45 °C, respectively. This indicates that a fraction of large crystal pores is distributed less uniformly among these crystals. It should be noted that the florescence signal from the FITC-labelled CAT in the crystals was much higher than that outside the crystals (in bulk solution), indicating that the equilibrium shifted towards CAT adsorption in the crystals. This is consistent with the high free Gibbs energy (−36 kJ mol−1) reported for CAT loading into crystals via adsorption.51
Surprisingly, INS was homogeneously distributed inside the crystals with large 15 nm-sized pores (Fig. 4f and h), but the protein did not penetrate inside the crystals having smaller 10 nm sized pores (Fig. 4e and g). Notably, although the dimensions of a single INS hexamer are smaller than that of CAT, the distribution of INS on the crystal surface/inside the crystals greatly shifted towards the surface. This indicates a different mechanism for the diffusion of CAT and INS into the crystal pores, as described below.
It can be assumed that the diffusion of CAT is not limited by the crystal pores being blocked by molecules of CAT, which is expected due to very close values of the CAT diameter and the average pore sizes of the crystals. CAT diffusion into the pores did not induce any aggregation of the protein molecules in the pores due to their proximity and potential unfolding, which can enhance inter-protein aggregation. In the case of INS, the mechanism differs. The INS molecules that enter the crystal pores tended to aggregate with each other in the pores due to the hydrophobic inter-protein attractions, which drive the formation of INS hexamers. For the CAT molecules, this hydrophobic effect may not be so prominent and can be compensated by electrostatic repulsion. It is known that INS is prone to the formation of elongated amyloid-like aggregates upon contact with hydrophobic surfaces. The diameter of these amyloid aggregates is typically in the range of 2–5 nm with a length of tens of nm. Thus, we assume that the formation of similar or other types of INS aggregates in the pores or on the vaterite surface may lead to the complete clogging of the smaller 10 nm pores, but only partial blockage of the larger 15 nm pores. This hypothesis is collaborated by the high Gibbs energy for amyloid INS aggregation (−26 ± 8 kJ mol−1),68 which is close to or exceeds that for INS adsorption on the vaterite surface (−27 ± 1 kJ mol−1).51 However, more evidence is required to explain the mechanism of INS adsorption on CaCO3 crystals, which was not the main focus of the present study.
Compared to the other tested proteins, APR had the lowest affinity for the vaterite surface. That was confirmed by the lowest ratio of the average fluorescence signal obtained from the crystals to background fluorescence in solution. Thus, for the crystals with 15 nm sized pores this ratio was found to be 31 ± 7, 6 ± 1 and 2 ± 1 for INS, CAT and APR, respectively. The aggregation of APR in the presence of crystals (Fig. 1) seriously suppressed its diffusion into the crystals with a much smaller size (Fig. 4i–l). Some of the signals detected inside the crystals may be related to the presence of single molecules and APR aggregates of smaller sizes.
Thus, the results above illustrate that protein diffusion cannot be predicted based on the dh and pI of proteins. Hence, for all three tested proteins, their adsorption on vaterite crystals is thermodynamically favourable, but to varying degrees.51 Thus, to gain more insight into the protein interaction with the crystals, the kinetics of protein loading into the crystals was further investigated.
3.4. Kinetics of protein adsorption
Herein, the kinetics of protein loading into the crystals was assessed by means of the relative fluorescence signal accumulated in the vaterite crystals during the loading of FITC-labelled proteins (Fig. 5). Notably, the absolute values of the fluorescence signals cannot be compared because of the various and hardly evaluated effects of the crystal internal microenvironment on the fluorescence of the loaded proteins due to quenching. However, the shapes of the normalized loading profiles can be compared to draw a conclusion about the kinetics of the protein loading.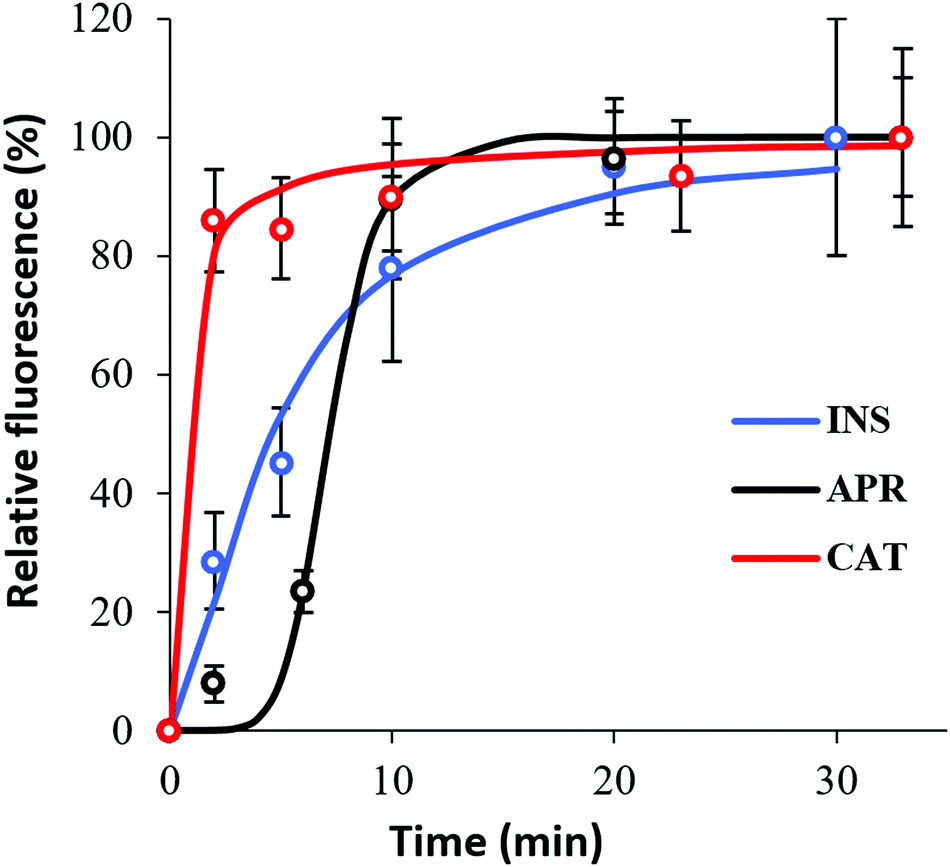 | ||
| Fig. 5 Kinetics of the protein penetration in the particles for 30 min under normal conditions by means of total normalized protein–FITC fluorescence intensity. | ||
The different adsorption kinetics observed for the tested therapeutic proteins indicate the existence of different adsorption mechanisms. The results presented in Fig. 5 show that the time when the protein signal reaches a plateau (equilibrium saturation) was a couple of minutes for CAT, whereas saturation was not reached for INS even after 30 min of loading. In the case of APR, saturation was achieved after ca. 10 min of incubation.
Protein adsorption into porous matrices is usually limited either by surface adsorption or molecular diffusion inside the crowded porous matrix. Thus, the possible adsorption mechanism and corresponding rate-controlling steps may be established by applying kinetics models. The following models for the kinetics of protein adsorption were applied in this study: (i) pseudo-first and (ii) pseudo-second order adsorption models and (iii) pore diffusion model. The adjusted R2 coefficient that considers the number of parameters and the number of experimental points was used for the validation of the models. Table 1 lists the fitting parameters obtained for CAT, INS and APR. The best fitting of the CAT adsorption kinetics to the PSO equation indicates that CAT adsorbs on the vaterite surface via chemisorption. The adsorption of INS is significantly slower, which may be related to the lower affinity of INS to the crystal surface in comparison with that of CAT (ΔG of −27 and −36 kJ mol−1, respectively).51 This obeys the PFO equation, and therefore is driven by physisorption.71
| Protein | Model | Parameter | Adj. R2 | Insight to mechanism |
|---|---|---|---|---|
| CAT | PFO | k 0.90 min−1 | 0.984 | PSO chemisorption |
| PSO | k 3.5 × 10 −4 % −1 s −1 | 0.994 | ||
| Pore diffusion | α 0.06 | 0.891 | ||
| K 80.1 | ||||
| m/C0 0.011 | ||||
| Hill | K 0.13; n 0.5 | 0.992 | ||
| INS | PFO | k 0.14 min −1 | 0.994 | PFO physisorption |
| PSO | k 4.3 × 10−5%−1 s−1 | 0.953 | ||
| Pore diffusion | α 0.4 | 0.507 | ||
| K 25.6 | ||||
| m/C0 0.014 | ||||
| Hill | K 10.5; n 1.5 | 0.980 | ||
| APR | PFO | k 0.11 min−1 | 0.881 | Hill, S-shaped protein clustering |
| PSO | k 3.0 × 10−5%−1 s−1 | 0.794 | ||
| Pore diffusion | α 0.6 | 0.301 | ||
| K 16.5 | ||||
| m/C0 0.016 | ||||
| Hill | K 3.7 × 10 5 ; n 6.5 | 0.994 | ||
The low R2 coefficients obtained for both CAT and INS indicate that pore diffusion is not the limiting step and the proteins diffuse sufficiently fast inside the pores of the vaterite crystals, causing the adsorption of the crystals to be the slowest and rate-limiting step.
Interestingly, recently it has been shown that in contrast to these proteins, the adsorption of mucin is controlled by its retarded diffusion inside the pores of vaterite crystals.42 This comparison indicates that the spreading dynamics of compact globular proteins is less affected by their interaction with the walls of the CaCO3 pores when compared to the large and non-globular mucin, which is known for its strong adhesiveness. Although accurate evaluation of the impact of the protein–wall interactions on the actual law of diffusion of these proteins is challengeable and has not been performed thus far (as has been shown for the diffusion of other molecules in crowded and non-inert matrices72,73), it can be assumed that these specific interactions are not strong enough to be determinative for the overall adsorption kinetics.
Moreover, considering the very similar pI of CAT and INS, it may be assumed that the binding of CAT to the vaterite surface is not electrostatic nature and is driven more by hydrophobic and/or other specific interaction with CaCO3. Interestingly, it has recently been demonstrated that the impact of van der Waals forces is prevalent over electrostatic binding of CAT to CaCO3 crystals.74 Here, we demonstrate that the adsorption of INS, which has a similar surface charge to that of CAT, is non-specific, indicating that the electrostatic attraction of INS to vaterite may not play a significant role.
Finally, the adsorption of APR corresponds to an S-shaped curve, which indicates a much more complex mechanism of adsorption involving surface clustering.69 Since the peculiar S-shape of the APR loading kinetics could not be easily fitted to any known kinetic models that have a solid mechanistic basis (some of these models exist but require complex numerical solutions),69 APR adsorption was fitted to the well-known Hill equation.70 This equation is commonly applied for dose–response curves, and therefore allows only some parameters to be extracted, but not the rate constants (Table 1).
Remarkably, the inflection point for the APR adsorption kinetics (8 min) corresponds well to the time needed for the formation of APR aggregates (also about 8 min, Fig. 2). Specifically, while APR is non-aggregated, it can diffuse inside the crystals. Afterwards, APR heavily aggregates in the bulk solution and cannot enter the pores of CaCO3 due to steric limitations. Meanwhile, the large APR aggregates are not anchored to the external surface of the crystals, and therefore do not block the pores (as confirmed by the homogeneous APR distribution, as shown in Fig. 4). At this stage (after inflection point in the kinetic curve), the remaining single APR molecules continue to diffuse inside the crystals until equilibrium between the single molecules, and small and large aggregates of APR is reached in both the solution and the crystal interior.
3.5. Mechanism of protein diffusion
Together with the state of the proteins and protein distribution in the crystals assessed above, the characteristic kinetics for the saturation of the crystal internal surface with the tested proteins strongly supports the mechanism of the adsorption of the three therapeutic proteins, as illustrated in Fig. 6.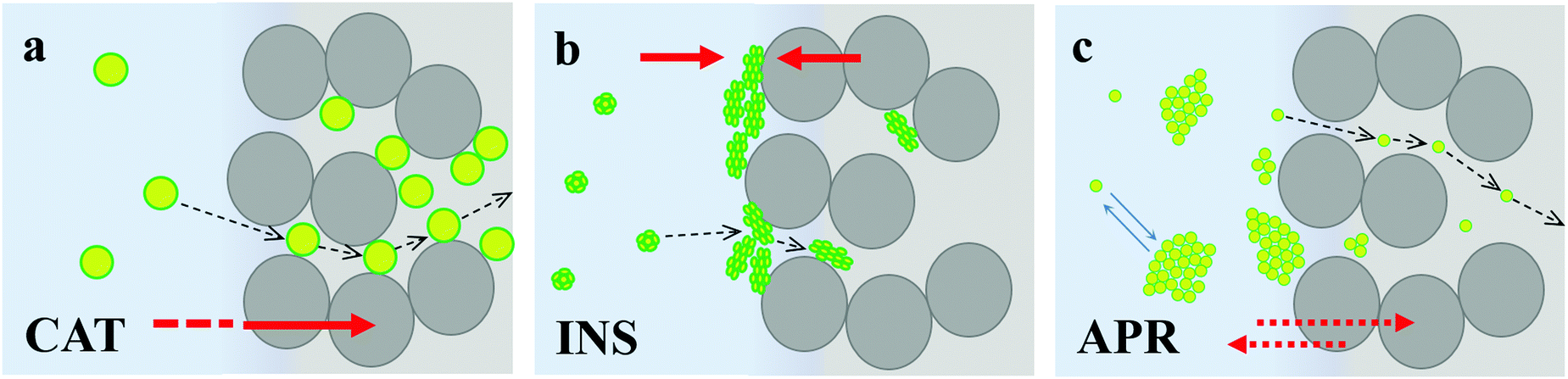 | ||
| Fig. 6 Schematic illustration of protein adsorption in CaCO3 microcrystals prepared at 22 °C: catalase (a), insulin – hexamer (b), and aprotinin – aggregate (c). Red arrows indicate that (a) adsorption equilibrium is shifted towards crystal interior for CAT; (b) crystal external surface for INS, wherein INS aggregates and its further diffusion into the crystal pores is blocked and (c) single APR molecules are equilibrated between the solution and crystal interior (adsorption constant is low, which is shown by the double red arrow); and single APR molecules are in equilibrium with the APR aggregates (blue arrows). | ||
CAT diffuses in the pores of the crystals without significant steric limitations and strongly binds to the entire surface of CaCO3via chemisorption. The equilibrium is strongly shifted towards the crystal volume (Fig. 6a).
INS does not aggregate in solution but tends to cluster at the surface of the crystals, where it predominantly adsorbs on the external surface of the CaCO3 crystals. The equilibrium is shifted towards the formation of surface aggregates (Fig. 6b), and thus its affinity to CaCO3 is much lower in comparison to that of CAT.
In contrast, APR forms aggregates immediately in solution, but these aggregates have a lower affinity to the crystal surface, do not anchor the surface of the CaCO3 crystals, and do not block the crystal pores, and therefore do not hinder the protein diffusion inside the crystals. APR diffusion into the crystals strongly depends on the aggregation kinetics, where the major portion of APR fills the crystal pores when APR is not yet heavily aggregated. Further adsorption results in more single APR molecules and the formation of small APR aggregates (with dh below the pore diameter) until equilibrium is reached between the APR aggregates and single molecules in solution and inside the crystals. Although the negative Gibbs free energy value indicates favourable adsorption, its equilibrium constant is much lower than that for CAT and INS adsorption, indicating that most of the APR remains in the bulk solution (schematically shown with a double arrow in Fig. 6c).
4. Conclusions
Herein, the main factors responsible for protein loading into pre-formed vaterite CaCO3 crystals were revealed. The mechanism of the adsorption of three model therapeutic proteins of various molecular masses and pI, i.e. CAT, INS, and APR, was proposed. The protein loading does not directly correlate with the size and the net charge of the proteins. For instance, the largest protein CAT (250 kDa) showed better diffusion into the crystal pores; however, the diffusion of the much smaller INS and APR (6 kDa) was restricted by steric limitations. These limitations are caused by Ca2+-mediated inter-protein aggregation in the presence of the crystals, either in solution (APR) or on crystal surface (INS). For the proteins that are prone to aggregation, molecular diffusion into the pores is governed by the equilibrium constant of the protein aggregation. In case of strong aggregation (INS) that occurs only at the crystal surface, protein diffusion through the crystal pores is sterically hindered. APR forms large aggregates of ca. 80 nm initially in solution, but these aggregates do not anchor to the crystal surface, and therefore do not block the pores and hinder further diffusion. Single molecules of APR that are present in equilibrium with the APR aggregates can freely diffuse into the pores. Once the protein molecules penetrate the pores of the vaterite crystals, they diffuse through the porous matrix quite fast; however, the methods were used in this study did not allow the estimation of the effective diffusion coefficients of the proteins. Nevertheless, it was concluded that self-diffusion does not determine the overall rate of protein adsorption. Instead, protein adsorption is limited by surface processes. This study strongly supports the pivotal role of inter-protein interaction in solution and on the crystal surface in protein adsorption in vaterite CaCO3. Thus, aggregation should be carefully considered when encapsulating therapeutic proteins inside vaterite crystals. Specifically, it may have a significant impact not only on the loading but also on the release and preservation of the activity of the therapeutics, which play a crucial role in drug delivery applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was performed within the framework of the M. V. Lomonosov Moscow State University state task (government grant AAAA-A16-116052010081-5). This work was supported by the Alexander von Humboldt Foundation in the framework of the Sofja Kovalevskaja program. AGS thanks the special research fund BOF UGent (BOF14/IOP/003, BAS094-18, 01IO3618) and FWO (G043219) for support. D. V. acknowledges QR Fund 2018–2019 and SPF 2019-2020 from Nottingham Trent University. A. V. thanks the Europeans Union's Horizon 2020 research and innovation programme for funding (the Marie-Curie Individual Fellowship LIGHTOPLEX-747245). The authors thank Rona Pitschke and Heike Runge for SEM imaging.References
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7(1), 21 CrossRef CAS PubMed.
- H. A. D. Lagassé, A. Alexaki, V. L. Simhadri, N. H. Katagiri, W. Jankowski, Z. E. Sauna and C. Kimchi-Sarfaty, F1000Research, 2017, 6, 113 Search PubMed.
- M. L. Tan, P. F. M. Choong and C. R. Dass, Peptides, 2010, 31(1), 184 CrossRef CAS PubMed.
- A. Mandal, D. Pal, V. Agrahari, H. M. Trinh, M. Joseph and A. K. Mitra, Adv. Drug Delivery Rev., 2018, 126, 67 CrossRef CAS PubMed.
- F. Moncalvo, M. I. Martinez Espinoza and F. Cellesi, Front. Bioeng. Biotechnol., 2020, 8, 89 CrossRef PubMed.
- D. S. Pisal, M. P. Kosloski and S. V. Balu-Iyer, J. Pharm. Sci., 2010, 99(6), 2557 CrossRef CAS PubMed.
- S. Gupta, A. Jain, M. Chakraborty, J. K. Sahni, J. Ali and S. Dang, Drug Delivery, 2013, 20(6), 237 CrossRef CAS PubMed.
- A. Jain, A. Jain, A. Gulbake, S. Shilpi, P. Hurkat and S. K. Jain, Crit. Rev. Ther. Drug Carrier Syst., 2013, 30(4), 293 CrossRef CAS PubMed.
- M. R. Villegas, A. Baeza and M. Vallet-Regí, Molecules, 2018, 23(5), 1008 CrossRef PubMed.
- N. G. Balabushevich, V. A. Izumrudov and N. I. Larionova, Polym. Sci., Ser. A, 2012, 54(7), 540 CrossRef CAS.
- M. S. Saveleva, K. Eftekhari, A. Abalymov, T. E. L. Douglas, D. Volodkin, B. V. Parakhonskiy and A. G. Skirtach, Front. Chem., 2019, 7, 179 CrossRef CAS PubMed.
- J. H. E. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Díaz, Angew. Chem., Int. Ed., 2012, 51(48), 11960 CrossRef CAS PubMed.
- N. Feoktistova, J. Rose, V. Z. Prokopović, A. S. Vikulina, A. Skirtach and D. Volodkin, Langmuir, 2016, 32(17), 4229 CrossRef CAS PubMed.
- B. V. Parakhonskiy, A. M. Yashchenok, M. Konrad and A. G. Skirtach, Adv. Colloid Interface Sci., 2014, 207, 253 CrossRef CAS PubMed.
- D. Volodkin, Adv. Colloid Interface Sci., 2014, 207, 306 CrossRef CAS PubMed.
- B. V. Parakhonskiy, C. Foss, E. Carletti, M. Fedel, A. Haase, A. Motta, C. Migliaresi and R. Antolini, Biomater. Sci., 2013, 1(12), 1273 RSC.
- A. Som, R. Raliya, L. Tian, W. Akers, J. E. Ippolito, S. Singamaneni, P. Biswas and S. Achilefu, Nanoscale, 2016, 8(25), 12639 RSC.
- R. Salomão, L. M. M. Costa and G. M. d Olyveira, Adv. Tissue Eng. Regen. Med. Open Access, 2017, 3(2), 00059 Search PubMed.
- B. V. Parakhonskiy, A. M. Yashchenok, S. Donatan, D. V. Volodkin, F. Tessarolo, R. Antolini, H. Möhwald and A. G. Skirtach, ChemPhysChem, 2014, 15(13), 2817 CrossRef CAS PubMed.
- K. K. Sand, J. D. Rodriguez-Blanco, E. Makovicky, L. G. Benning and S. L. S. Stipp, Cryst. Growth Des., 2012, 12(2), 842 CrossRef CAS.
- N. A. N. Hanafy, M. El-Kemary and S. Leporatti, J. Nanomed. Res., 2018, 7(4), 235 Search PubMed.
- D. B. Trushina, T. V. Bukreeva and M. N. Antipina, Cryst. Growth Des., 2016, 16(3), 1311 CrossRef CAS.
- D. B. Trushina, T. N. Borodina, S. N. Sulyanov, J. V. Moiseeva, N. V. Gulyaeva and T. V. Bukreeva, Crystallogr. Rep., 2018, 63(6), 998 CrossRef CAS.
- M. Delcea, H. Möhwald and A. G. Skirtach, Adv. Drug Delivery Rev., 2011, 63(9), 730 CrossRef CAS PubMed.
- A. Sergeeva, R. Sergeev, E. Lengert, A. Zakharevich, B. Parakhonskiy, D. Gorin, S. Sergeev and D. Volodkin, ACS Appl. Mater. Interfaces, 2015, 7(38), 21315 CrossRef CAS PubMed.
- P. V. Binevski, N. G. Balabushevich, V. I. Uvarova, A. S. Vikulina and D. Volodkin, Colloids Surf., B, 2019, 181, 437 CrossRef CAS PubMed.
- B. Parakhonskiy, M. V. Zyuzin, A. Yashchenok, S. Carregal-Romero, J. Rejman, H. Möhwald, W. J. Parak and A. G. Skirtach, J. Nanobiotechnol., 2015, 13, 53 CrossRef PubMed.
- V. Vergaro, I. Pisano, R. Grisorio, F. Baldassarre, R. Mallamaci, A. Santoro, G. P. Suranna, P. Papadia, F. P. Fanizzi and G. Ciccarella, Materials, 2019, 12(9), E1481 CrossRef PubMed.
- D. Volodkin, Colloid Polym. Sci., 2014, 292(6), 1249 CrossRef CAS.
- M. Behra, S. Schmidt, J. Hartmann, D. V. Volodkin and L. Hartmann, Macromol. Rapid Commun., 2012, 33(12), 1049 CrossRef CAS PubMed.
- N. Feoktistova, G. Stoychev, N. Puretskiy, L. Ionov and D. Volodkin, Eur. Polym. J., 2015, 68, 650 CrossRef.
- Y. D. Chen, J. J. Xu, Y. Wang, H. Chen, Q. J. Luo, X. D. Li and W. P. Zhu, RSC Adv., 2017, 7(3), 1260 RSC.
- J. M. Silva, S. G. Caridade, R. R. Costa, N. M. Alves, T. Groth, C. Picart, R. L. Reis and J. F. Mano, Langmuir, 2015, 31(41), 11318 CrossRef CAS PubMed.
- C. Vilela, A. R. P. Figueiredo, A. J. D. Silvestre and C. S. R. Freire, Expert Opin. Drug Delivery, 2017, 14(2), 189 CrossRef CAS PubMed.
- T. G. Shutava, P. P. Pattekari, K. A. Arapov, V. P. Torchilin and Y. M. Lvov, Soft Matter, 2012, 8(36), 9418 RSC.
- S. Schmidt, K. Uhlig, C. Duschl and D. Volodkin, Acta Biomater., 2014, 10(3), 1423 CrossRef CAS PubMed.
- M.-K. Kim, J.-A. Lee, M.-R. Jo, M.-K. Kim, H.-M. Kim, J.-M. Oh, N. W. Song and S.-J. Choi, Nanomaterials, 2015, 5(4), 1938 CrossRef CAS PubMed.
- N. Sudareva, O. Suvorova, N. Saprykina, N. Smirnova, P. Bel’tyukov, S. Petunov, A. Radilov and A. Vilesov, J. Microencapsulation, 2018, 35(7–8), 619 CrossRef CAS PubMed.
- X.-W. He, T. Liu, Y.-X. Chen, D.-J. Cheng, X.-R. Li, Y. Xiao and Y.-L. Feng, Cancer Gene Ther., 2008, 15(3), 193 CrossRef CAS PubMed.
- I. Marchenko, T. Borodina, D. Trushina, I. Rassokhina, Y. Volkova, V. Shirinian, I. Zavarzin, A. Gogin and T. Bukreeva, J. Microencapsulation, 2018, 35(7–8), 657 CrossRef CAS PubMed.
- N. G. Balabushevich, E. A. Sholina, E. V. Mikhalchik, L. Y. Filatova, A. S. Vikulina and D. Volodkin, Micromachines, 2018, 9(6), E307 CrossRef PubMed.
- N. G. Balabushevich, E. A. Kovalenko, E. V. Mikhalchik, L. Y. Filatova, D. Volodkin and A. S. Vikulina, J. Colloid Interface Sci., 2019, 545, 330 CrossRef CAS PubMed.
- T. Paulraj, N. Feoktistova, N. Velk, K. Uhlig, C. Duschl and D. Volodkin, Macromol. Rapid Commun., 2014, 35(16), 1408 CrossRef CAS PubMed.
- A. Sergeeva, N. Feoktistova, V. Prokopovic, D. Gorin and D. Volodkin, Adv. Mater. Interfaces, 2015, 2(18), 1500386 CrossRef.
- M. S. Saveleva, A. N. Ivanov, M. O. Kurtukova, V. S. Atkin, A. G. Ivanova, G. P. Lyubun, A. V. Martyukova, E. I. Cherevko, A. K. Sargsyan, A. S. Fedonnikov, I. A. Norkin, A. G. Skirtach, D. A. Gorin and B. V. Parakhonskiy, Mater. Sci. Eng., C, 2018, 85, 57 CrossRef CAS PubMed.
- T. E. L. Douglas, K. Sobczyk, A. Łapa, K. Włodarczyk, G. Brackman, I. Vidiasheva, K. Reczyńska, K. Pietryga, D. Schaubroeck, V. Bliznuk, P. van der Voort, H. A. Declercq, J.d. van Bulcke, S. K. Samal, D. Khalenkow, B. V. Parakhonskiy, J. van Acker, T. Coenye, M. Lewandowska-Szumieł, E. Pamuła and A. G. Skirtach, Biomed. Mater., 2017, 12(2), 25015 CrossRef PubMed.
- A. M. Yashchenok, D. Borisova, B. V. Parakhonskiy, A. Masic, B. Pinchasik, H. Möhwald and A. G. Skirtach, Ann. Phys., 2012, 524(11), 723 CrossRef CAS.
- I. Y. Stetciura, A. V. Markin, A. N. Ponomarev, A. V. Yakimansky, T. S. Demina, C. Grandfils, D. V. Volodkin and D. A. Gorin, Langmuir, 2013, 29(12), 4140 CrossRef CAS PubMed.
- R. Bajracharya, J. G. Song, S. Y. Back and H.-K. Han, Comput. Struct. Biotechnol. J., 2019, 17, 1290 CrossRef CAS PubMed.
- A. I. Petrov, D. V. Volodkin and G. B. Sukhorukov, Biotechnol. Prog., 2005, 21(3), 918 CrossRef CAS PubMed.
- N. G. Balabushevich, A. V. Lopez de Guerenu, N. A. Feoktistova and D. Volodkin, Phys. Chem. Chem. Phys., 2015, 17(4), 2523 RSC.
- S. Donatan, A. Yashchenok, N. Khan, B. Parakhonskiy, M. Cocquyt, B.-E. Pinchasik, D. Khalenkow, H. Möhwald, M. Konrad and A. Skirtach, ACS Appl. Mater. Interfaces, 2016, 8(22), 14284 CrossRef CAS PubMed.
- A. S. Vikulina, N. A. Feoktistova, N. G. Balabushevich, A. G. Skirtach and D. Volodkin, Phys. Chem. Chem. Phys., 2018, 20(13), 8822 RSC.
- C. Wang, C. He, Z. Tong, X. Liu, B. Ren and F. Zeng, Int. J. Pharm., 2006, 308(1–2), 160 CrossRef CAS PubMed.
- G. B. Sukhorukov, D. V. Volodkin, A. M. Günther, A. I. Petrov, D. B. Shenoy and H. Möhwald, J. Mater. Chem., 2004, 14(14), 2073 RSC.
- A. Fujii, T. Maruyama, Y. Ohmukai, E. Kamio, T. Sotani and H. Matsuyama, Colloids Surf., A, 2010, 356(1–3), 126 CrossRef CAS.
- A. Hernández-Hernández, A. B. Rodríguez-Navarro, J. Gómez-Morales, C. Jiménez-Lopez, Y. Nys and J. M. García-Ruiz, Cryst. Growth Des., 2008, 8(5), 1495 CrossRef.
- I. Polowczyk, A. Bastrzyk and M. Fiedot, Materials, 2016, 9(11), E944 CrossRef PubMed.
- P. Shi, J. Qin, J. Hu, Y. Bai and X. Zan, Colloids Surf., B, 2019, 175, 184 CrossRef CAS PubMed.
- A. D. Trofimov, A. A. Ivanova, M. V. Zyuzin and A. S. Timin, Pharmaceutics, 2018, 10(4), E167 CrossRef PubMed.
- B. Zhu, X. Xu and R. Tang, J. Chem. Phys., 2013, 139(23), 234705 CrossRef PubMed.
- U. Aschauer, D. Spagnoli, P. Bowen and S. C. Parker, J. Colloid Interface Sci., 2010, 346(1), 226 CrossRef CAS PubMed.
- V. La Verde, P. Dominici and A. Astegno, Bio-Protoc., 2017, 7(8), 19265 Search PubMed.
- N. G. Balabushevich, M. A. Pechenkin, I. N. Zorov, E. D. Shibanova and N. I. Larionova, Biochemistry, 2011, 76(3), 327 CAS.
- H. Fritz and G. Wunderer, Arzneimittelforschung, 1983, 33(4), 479 CAS.
- J. Brange, D. R. Owens, S. Kang and A. Vølund, Diabetes Care, 1990, 13(9), 923 CrossRef CAS PubMed.
- F. C. Donnelly, F. Purcell-Milton, V. Framont, O. Cleary, P. W. Dunne and Y. K. Gun’ko, Chem. Commun., 2017, 53(49), 6657 RSC.
- T. P. J. Knowles, W. Shu, G. L. Devlin, S. Meehan, S. Auer, C. M. Dobson and M. E. Welland, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(24), 10016 CrossRef CAS PubMed.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162(1–2), 87 CrossRef CAS PubMed.
- S. Goutelle, M. Maurin, F. Rougier, X. Barbaut, L. Bourguignon, M. Ducher and P. Maire, Fundam. Clin. Pharmacol., 2008, 22(6), 633 CrossRef CAS PubMed.
- J.-P. Simonin, Chem. Eng. J., 2016, 300, 254 CrossRef CAS.
- S. K. Ghosh, A. G. Cherstvy, D. S. Grebenkov and R. Metzler, New J. Phys., 2016, 18(1), 13027 CrossRef.
- S. K. Ghosh, A. G. Cherstvy and R. Metzler, Phys. Chem. Chem. Phys., 2015, 17(3), 1847 RSC.
- N. A. Feoktistova, A. S. Vikulina, N. G. Balabushevich, A. G. Skirtach and D. Volodkin, Mater. Des., 2020, 185, 108223 CrossRef CAS.
| This journal is © the Owner Societies 2020 |