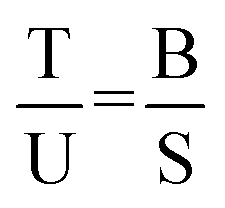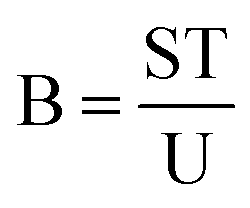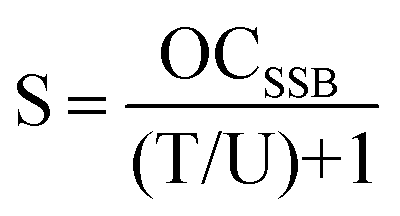Ectoine interaction with DNA: influence on ultraviolet radiation damage†
Received
7th January 2020
, Accepted 27th February 2020
First published on 18th March 2020
Abstract
Ectoine is a small zwitterionic osmolyte and compatible solute, which does not interfere with cell metabolism even at molar concentrations. Plasmid DNA (pUC19) was irradiated with ultraviolet radiation (UV-C at 266 nm) under quasi physiological conditions (PBS) and in pure water in the presence and absence of ectoine (THP(B)) and hydroxyectoine (THP(A)). Different types of UV induced DNA damage were analysed: DNA single-strand breaks (SSBs), abasic sites and cyclobutane pyrimidine dimers (CPDs). A complex interplay between these factors was observed with respect to the nature and occurrence of DNA damage with 266 nm photons. In PBS, the cosolutes showed efficient protection against base damage, whilst in pure water, a dramatic shift from SSB damage to base damage was observed when cosolutes were added. To test whether these effects are caused by ectoine binding to DNA, further experiments were conducted: small-angle X-ray scattering (SAXS), surface-plasmon resonance (SPR) measurements and Raman spectroscopy. The results show, for the first time, a close interaction between ectoine and DNA. This is in stark contrast to the assumption made by preferential exclusion models, which are often used to interpret the behaviour of compatible solutes within cells and with biomolecules. It is tentatively proposed that the alterations of UV damage to DNA are attributed to ectoine influence on nucleobases through the direct interaction between ectoine and DNA.
1 Introduction
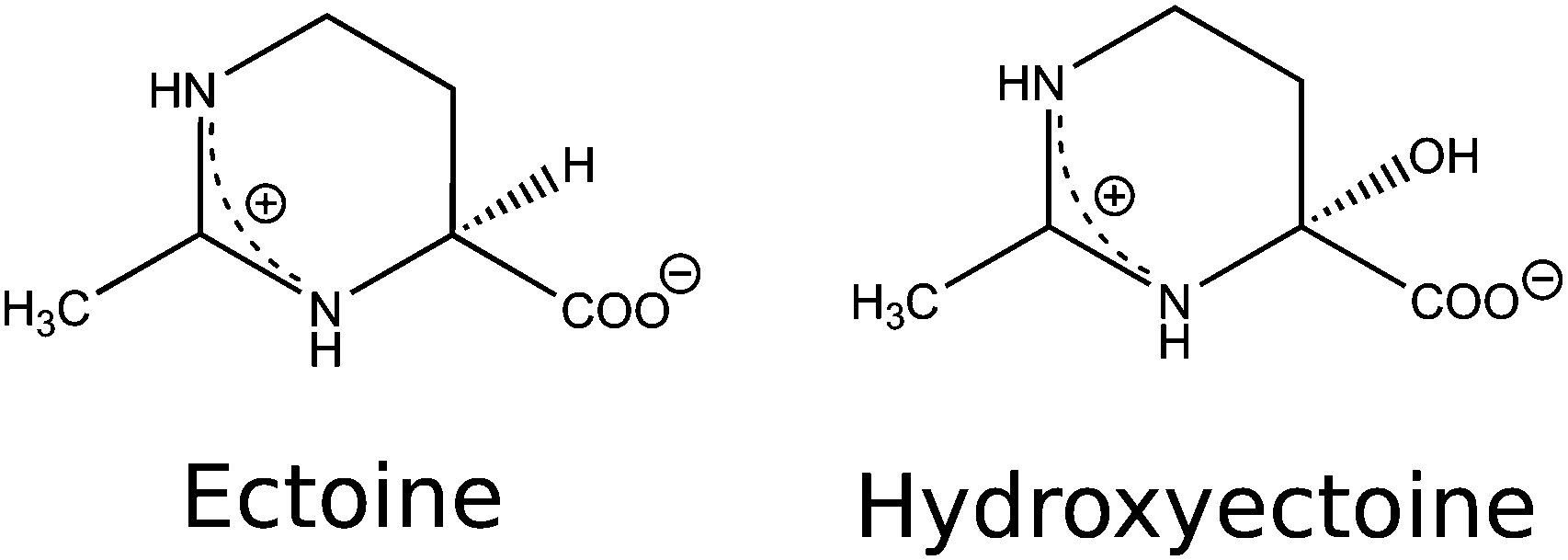
Osmolytes and compatible solutes such as ectoine ((S)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid, THP(B)) and hydroxyectoine ((4S,5S)-5-hydroxy-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid, THP(A)) (Fig. 1) are produced by some halophilic microorganisms to survive in environments with high salinity, such as salt lakes in desert regions.1 These microorganisms achieve osmoadaptation to high external salt concentrations by the regulation of osmotic pressure via the accumulation of molar concentrations of ectoine.2–4 In this context, an interesting property of ectoine is the compensating effect on water dynamics as opposed to that of sodium chloride.5 Ectoine has a zwitterionic structure in the solid state6 and in aqueous solution7,8 at neutral pH. Its water structuring (kosmotropic) effects,5,7–9 role as a hydroxyl radical scavenger,10,11 and influence on protein functions,9,12–16 gene expression,17,18 DNA melting temperature,12,19 DNA structure,20,21 DNA–protein binding,9,12 and protection of biomolecules against ionising radiation10 and oxidative stress22 were investigated over the last decade. Due to its versatile properties, ectoine is currently used in a multitude of biotechnological, cosmetic and medical applications.2,3,22–25 Most prominently, it is used in sunscreens, despite evidence from recent studies concerning its influence on UV irradiation of biological targets, showing Janus-faced behaviour.17,20,26–28 Bünger et al. irradiated human keratinocytes with UV-A (340–400 nm).17 They showed a decrease in mitochondrial DNA mutations for cells pretreated with ectoine and a suppression of radiation induced signaling mechanisms within the cells. UV/Vis (315–800 nm) irradiation of human keratinocytes was performed by Botta et al.27 Cells that were incubated before irradiation within an ectoine solution showed a decrease in DNA single-strand breaks (SSBs) compared to that of the control samples. They hypothesised that the protective effect was due to the ectoine induced expression of the heat shock protein 70 (Hsp70s), which is known to protect cells against heat induced stress and toxic chemicals. In contrast to these positive effects, studies by Beblo-Vranesevics et al.28 and Meyer et al.20 showed contrasting results. Beblo-Vranesevic et al.28 irradiated the thermophilic microorganisms H. marinus and A. fulgidus with UV-C (254 nm) photons with and without external addition of 0.5 mol L−1 ectoine. For both microorganisms, a decrease of the cellular survival rate was found upon addition of ectoine. Meyer et al.20 performed in vitro experiments with plasmid DNA (pUC19) in pure water irradiated by UV-A (365 nm) photons. After irradiation, an increasing amount of SSB with increasing ectoine (0 mol L−1, 0.1 mol L−1 and 0.5 mol L−1) concentrations was detected. To resolve some of the inconsistencies in the aforementioned literature, we aim to investigate the interaction of ectoine with DNA and determine the changes in the amount and type of DNA radiation damage upon the addition of ectoine and hydroxyectoine under in vitro conditions. Irradiation was performed with a pulsed laser at a well defined wavelength (UV-C at 266 nm) in ultra pure water and under quasi physiological conditions (1× phosphate buffered saline, PBS). The damage was analysed in terms of induction of single-strand breaks, abasic sites (base loss) and base damage (cyclobutane pyrimidine dimers (CPDs)) in the form of endonuclease sensitive sites (T4).29 These types of lesions are known to be precursors to mutation and the development of cancer.30,31 To gain insight into the mechanisms that change the DNA damage yields in the presence of ectoine, the interaction between the two molecules was investigated. To our knowledge, only indirect information is available here.9,20,21 Meyer et al. observed structural changes in plasmid DNA after incubation with ectoine, without clarifying whether these effects were based on direct or indirect interactions between both molecules.20 Computational work by Oprzeska-Zingrebe et al. estimated that ectoine accumulates between 0.5–2 nm distance to the sugar–phosphate backbone of dsDNA due to electrostatic and dispersion interactions.21 To test this prediction experimentally, we studied DNA–ectoine interactions utilising three complementary methods, small-angle X-ray scattering (SAXS), surface-plasmon resonance (SPR) and Raman spectroscopy.
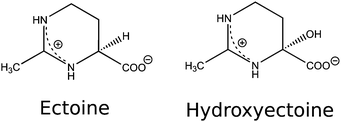 |
| | Fig. 1 The structural formula of ectoine and hydroxyectoine. | |
2 Materials and methods
2.1 Sample preparation
Double-stranded plasmid DNA, pUC19 with 2686 base pairs, at a concentration of 200 ng μL−1 in 1× PBS, was obtained from PlasmidFactory (Bielefeld, Germany). For experiments performed in ultrapure water, dsDNA was dialysed following a standard protocol (Float-a-lyzer G2 with a molecular weight cutoff of 8–10 kDa, Spectrum Labs) in ultrapure water (LiChrosolv, Merck) to remove buffer compounds and organic contaminations. Ectoine and hydroxyectoine with >95% purity were purchased from Sigma Aldrich, and sterile PBS was purchased from Roth Chemie. Solutions (60 μL) with a final plasmid concentration of 17 ng μL−1 in ultra pure water and 1× PBS (pH 7.4), with 0 mmol L−1 and 10 mmol L−1 ectoine and hydroxyectoine were prepared directly before irradiation in quartz micro cuvettes with 10 mm path length and 2 mm width (Suprasil, Hellma Analytics). The cuvettes were extensively cleaned before filling. They were first washed with ethanol (PhEur, ChemSolute) and then washed three times with ultra pure water and dried with high purity Argon (5.0, Linde) before filling.
2.2 UV irradiation
Irradiation with a pulsed laser source at 266 nm (FQSS266-Q2, max. pulse energy 1.12 μJ, beam diameter 777 μm, pulse FWHM 0.88 ns, CryLas GmbH) was performed at a repetition rate of 10 kHz. The laser power during irradiation was adjusted using a mechanical attenuator and determined with a laser power meter (Model 407A, SpectraPhysics). Here, fluence refers to the energy passed through the liquid that was exposed behind the cuvette surface. The liquid in the cuvette was exposed over a surface of (2 × 3) mm2 as defined by the cuvette width and filling height. During irradiation, the plasmid DNA diffuses through the volume. The diffusion behaviour of pUC19 DNA in confined geometries was discussed in detail and simulated in our previous work.32,33 The plasmids reside partly within and out of the direct laser beam. Fluence values have to therefore be interpreted as spatially and time averaged. Irradiation at 266 nm was performed for (120 ± 4) s at a laser power of (10.3 ± 0.5) mW with a fluence of (20 ± 2) J cm−2. After irradiation, the samples were immediately analysed by gel electrophoresis.
2.3 Measurements of DNA single-strand breaks
The samples were analysed by gel electrophoresis within a 1% agarose gel (5 V cm−1, 30 minutes, 1× TAE buffer) and SYBR Gold. The undamaged, supercoiled (SC) form of the plasmid and the plasmid with a SSB in its open circular (OC) form can be distinguished by their different electrophoretic mobilities within the gel. For a detailed description, see the previous work.10 The intensity profiles from the gel image were subjected to linear background subtraction. Afterwards, an integration over the different bands, which correspond to the supercoiled/undamaged (SCSSB) and open circular/SSB (OCSSB) forms of the plasmids, was performed. The difference in the attachment efficiency of SYBR Gold to the supercoiled plasmids in comparison to the open circular plasmids was determined as 1.05. Therefore, no correction is needed. The details on the determination of this value can be found in the ESI.†
2.4 Determination of DNA base damage and base loss
To quantify the induction of abasic sites and cyclobutane pyrimidine dimers, 20 μL of DNA solution of each sample was treated with T4 endonuclease V (T4, New England Biolabs) following the protocol given by the supplier (1 unit of endonuclease in 1× reaction buffer, 2 μg bovine serum albumin per 20 μL of DNA solution at 37 °C for 30 minutes). T4 endonuclease V introduces a strand-break at an abasic site or at a CPD, which will be denoted in the following text as T4.29 Therefore, SC plasmid DNA, which contains such a lesion and does not contain a SSB prior to T4 endonuclease V treatment, relaxes afterwards to the OC form, which can be detected by gel electrophoresis as described above. This results in a decrease of the relative fluorescence intensities of the SC (SCT4) band of the plasmid when compared to the intensity of the untreated sample (SCSSB). Likewise, the relative intensity of the OC band (OCT4) in the T4 treated sample increases when compared to the untreated OC band (OCSSB).
2.5 Calculation of the different types of DNA damage
To properly quantify and compare the effects of the presence of cosolutes on UV induced DNA damage, one has to consider the different types of damage and their combinations: in every sample, up to four different sets of plasmids can exist. They are either undamaged (U), have only SSB (S), have only T4 sensitive sites (T) or both (B), SSB and T4s (base damage: CPD or abasic site29). The measured and normalised (100 = SC + OC) band intensities correspond to the four species such that undamaged plasmids are given by U = SCT4 and all plasmids that have one or both types of damage correspond to OCT4 = S + T + B. All plasmids that have at least one SSB (SSBtotal) are given by:| | | OCSSB = SSBtotal = S + B | (1) |
from what follows, the amount of plasmids with T4 only is:Under the assumption of independent production of SSB and T4s and a random distribution of T4s over all plasmids, the following ratios are equal:| | 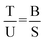 | (3) |
which gives the amount of plasmids with both types of damage as:| | 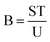 | (4) |
With eqn (1)–(3), the number of plasmids with SSB only is determined as:| | 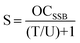 | (5) |
With these relations, the size of the different species (U, S, T, and B) can be determined from the gel electrophoresis data. The total amount of plasmids with T4 can be calculated as:
2.6 Raman spectroscopy measurements
Raman measurements were performed using a confocal Alpha300R instrument (WITec, Germany), equipped with a 20× Zeiss EX Epiplan DIC objective, a 532 nm laser (Excelsior 532-60) and a laser power of 18 mW. The spectrometer has a UHTS-300-VIS (grid of 1800 gratings per mm) and a thermoelectrically cooled CCD-camera Andor DV-401A-BV-532 (at −64 °C). Herring sperm DNA (Sigma) was used to prepare the DNA stock solution with a DNA concentration of 8.4 mg mL−1. The ectoine stock solution was prepared with a concentration of 1 M. Ultra pure water was used as the solvent (Merck, LiChrosolv). Three different samples were analysed (Fig. 5): (DNA): 100 μL of DNA stock solution + 10 μL of water; (Ect.): 100 μL of water and 10 μL of ectoine stock solution; (DNA + Ect.): 100 μL of DNA stock solution + 10 μL of ectoine stock solution. The spectra of the liquids were obtained through high precision Zeiss cover glasses. Eight spectra, each with an accumulation time of 500 s, were averaged. Measurements were performed around 851 rel. cm−1 where the characteristic ectoine mode is located, which is very sensitive to changes in the hydration shell (see below).8 The unprocessed Raman spectra are shown in Fig. 5.
2.7 Surface plasmon resonance measurements
Surface plasmon resonance (SPR) experiments allow monitoring the biomolecular interactions in real time under the conditions of continuous flow over a sensor chip surface. The observed change in resonance angle (expressed as resonance units, RU) is proportional to the surface concentration (mass per unit area) of adsorbed molecules. For details, see our previous work.9 Regarding the binding of ectoine to immobilized DNA strands, an experimental study34 established a conversion factor of 1.3 RU per 10−10 g cm−2 for biomolecules directly attached to a gold surface without a covering dextran layer. HPLC-purified thiol modified oligonucleotides (5′-HS-C6-dT25-3′) were obtained from Thermo Fischer Scientific (Ulm, Germany). Water for chromatography (99.96% for NMR) was obtained from Merck (Darmstadt, Germany). SPR gold-on-glass chips were obtained from GE HealthCare (Munich, Germany). The dT25 DNA adlayer was formed by immersing carefully cleaned (piranha solution, ultra pure water, nitrogen gas) gold chips for 120 min in a 10 μM solution of oligonucleotides in 2× SSC followed by washing with 1× PBS and ultra pure water. The SPR experiments were carried out at a flow rate of 10 μL min−1 using a Biacore T-100 instrument (Uppsala, Sweden).
2.8 Small angle X-ray scattering
Small-angle X-ray scattering (SAXS) measurements were performed on a “Multiscale Analyser for Ultrafine Structures” (MAUS), a customized Xeuss 2.0 system (Xenocs, France). X-rays were generated from a microfocus X-ray tube with a copper target, and a multilayer optic technique was employed to parallelise and monochromatise the beam to the Cu Kα wavelength of 0.1542 nm. The liquid samples were examined in flow by continuous pumping through a cell bearing silicon nitride windows. Data collection was performed using an in-vacuum EIGER 1M detector (Dectris, Switzerland), which was placed at a distance of 557 mm from the sample. The resulting data have been processed, background subtracted and scaled to absolute units using the DAWN software package according to standardised procedures.35,36 To maximize the signal from elastic X-ray scattering, herring sperm DNA in water (Sigma), which comigrates with the 587 and 831 base pair marker, was used to obtain a high concentration solution (35 mg mL−1). Four samples were prepared: 1200 μL of ultrapure water (Merck LiChrosolv), (900 μL of DNA solution + 300 μL of ultrapure water), ectoine in water (900 μL of ultrapure water and 300 μL of 0.24 M ectoine solution), DNA + ectoine in water (900 μL of DNA solution + 300 μL of 0.24 M ectoine solution).
3 Results
3.1 Modification of UV induced DNA damage by ectoines
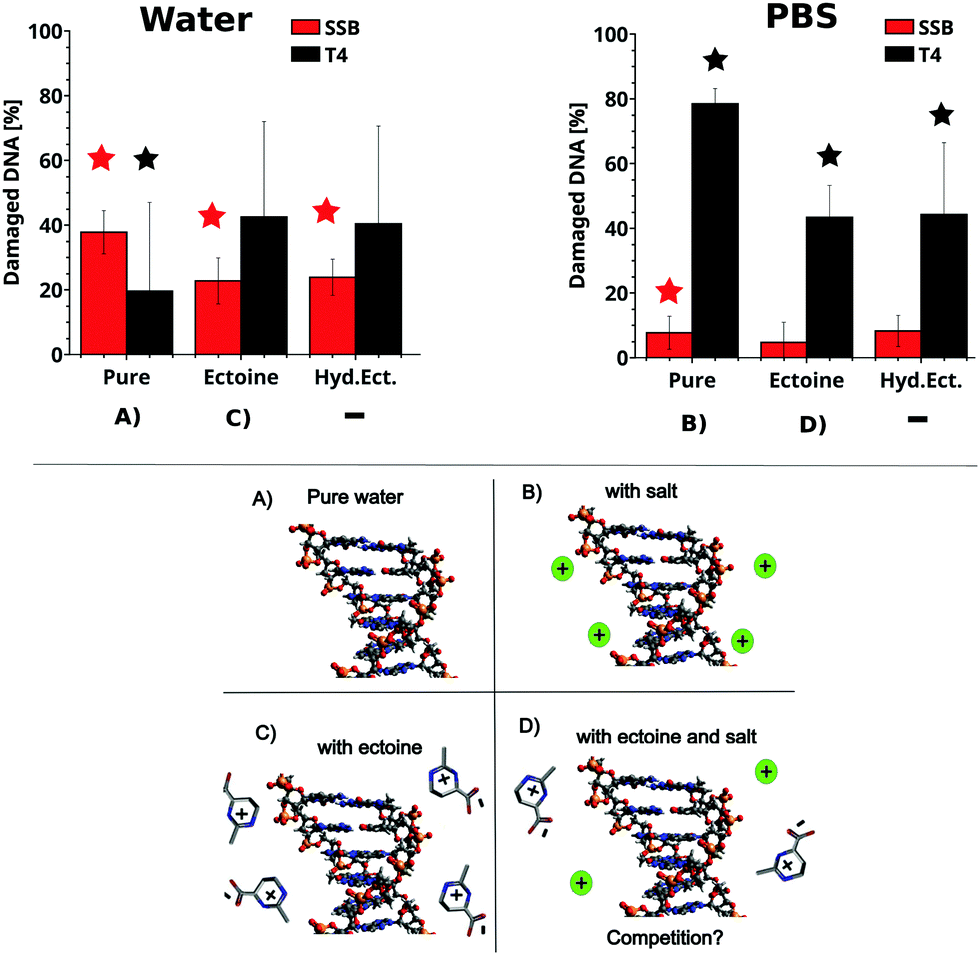
The influence of ectoine and hydroxyectoine on the induction of SSB and base damage (T4 endonuclease sensitive sites (T4s)) by UV-C radiation (266 nm, 4.66 eV) in ultrapure water and PBS buffer is shown in Fig. 2 (top), together with a schematic representation of the different experimental conditions (bottom). Total damage yields were calculated according to eqn (1)–(6), and the detailed experimental data for each data point and statistics are tabulated in the ESI.† In general, SSB induction is much lower than formation of T4s (Fig. 2). After irradiation in PBS, a slight production (below 20%) of SSB is observed, alongside a strong increase in T4s for all samples (over 40%). Hereby, the presence of both cosolutes leads to a significant decrease in the induction of T4s compared to the irradiated sample without cosolute. A significant influence of the cosolutes on SSB induction under these conditions is not observed. The above situation changes dramatically for 266 nm irradiation in ultra pure water, where a significant production of SSB is observed in all samples. However, the presence of both ectoines provides significant protection against SSB induction in ultra pure water. In contrast, the induction of T4s is not prevented by the presence of ectoines, which is contrast to the behaviour observed in PBS.
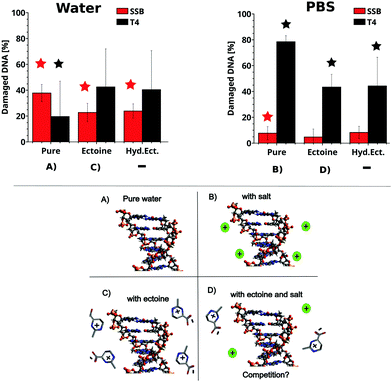 |
| | Fig. 2 Top: The percentage of DNA with SSB and T4 sensitive sites in the presence of the cosolute after irradiation with 266 nm photons in PBS (right) and ultra pure water (left) with respect to unirradiated samples. The irradiation corresponds to a fluence of 20 J cm−2. The relative amounts of SSBtotal (red) and T4total (black) were calculated from the gel electrophoresis data and eqn (1)–(6). The number of SSB and T4 sites of the unirradiated samples was subtracted. For details on the calculation, see the methods section. The depicted error bars represent the standard deviation (n = 4–6, see ESI†). Stars over samples with cosolutes indicate significant differences (P < 0.05; two sample t-test) compared to the irradiated pure samples. Stars over irradiated pure samples indicate significant damage compared to the unirradiated sample. Bottom: Schematic representation of DNA in pure water (A), with salt (B), ectoine (C) and ectoine + salt (D). | |
3.2 Ectoine binding to DNA
The influence of ectoine on UV induced DNA damage can be either mediated by a direct or an indirect influence on DNA. To test the hypothesis of whether ectoine binds to DNA or accumulates in its vicinity, small angle X-ray scattering (SAXS), surface plasmon resonance (SPR) and Raman spectroscopy measurements were performed.
Small angle X-ray scattering of ectoine DNA complexes.
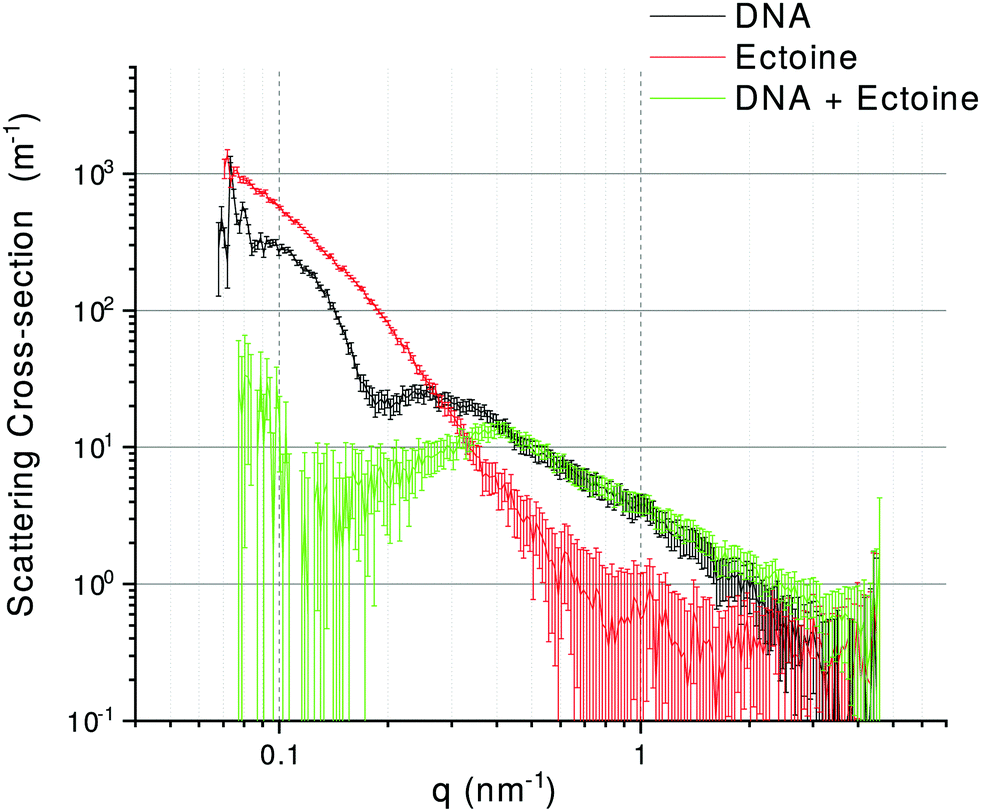
SAXS patterns of DNA, ectoine and DNA + ectoine samples are shown in Fig. 3. As the scattering patterns from DNA and ectoine both have distinct features present within the scattering towards low-q, it is assumed that a non-interacting solution of both DNA and ectoine would result in scattering equal to the sum of these contributions. The scattering from the DNA + ectoine sample does not resemble the combined scattering from the separate DNA and ectoine samples. This gives strong indications that there is a direct interaction between the DNA and ectoine within the DNA + ectoine sample. The evidence of this interaction is most distinct towards lower values of q and could be explained by a change in conformation of the DNA due to direct interactions with ectoine.21
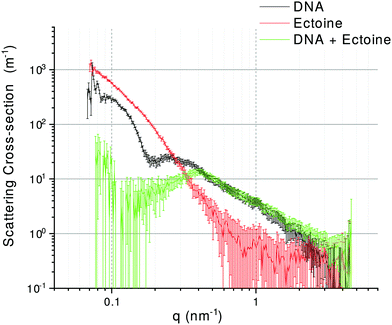 |
| | Fig. 3 Background subtracted SAXS curves for DNA (black), ectoine (red), and DNA + ectoine (green) samples. For details, see the text. | |
Surface plasmon resonance measurements of ectoine binding to DNA.
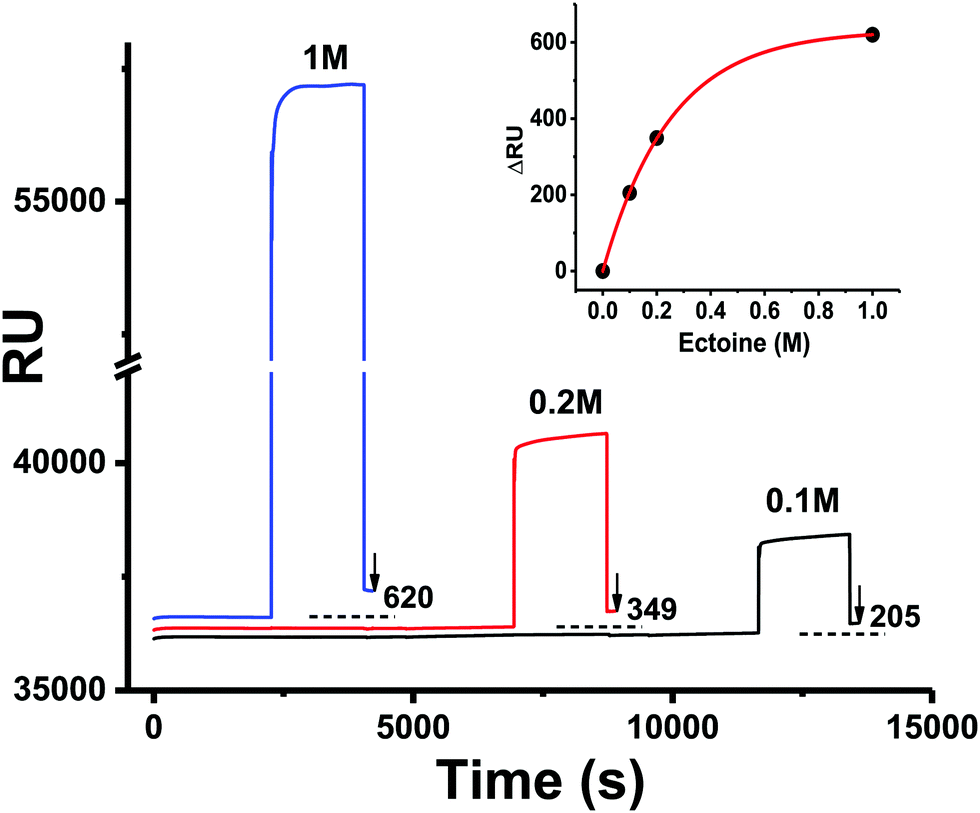
Surface tethered ssDNA was exposed to solutions of different ectoine concentrations and the resulting surface plasmon resonance (SPR) signal was measured. The SPR data from before, during and after exposure of the surface with bound ssDNA to ectoine solutions are shown in Fig. 4. When the resonance signals (RU) from before (black dotted lines) and after exposure (solid lines) are compared, a concentration dependent increase is observed (inset Fig. 4). This increase is directly related to the amount of ectoine remaining bound to DNA after initiation of the surface washing. Based on previous studies,37 the surface coverage of dT25 was estimated to be σ ≈ 5 × 1012 ssDNA molecules per cm2. From the above, it can be calculated that at a 1 M bulk concentration (ΔRU 620), roughly 1.8 × 1014 ectoine molecules per cm2 are bound at the surface. With the present dT25 surface coverage, one has 1.25 × 1014 bases per cm2. Hence, the exposure to 1 M ectoine concentration results in the binding of 1.44 ectoine molecules per nucleotide on average. For the concentration of 0.2 M ectoine, exposure results in ΔRU 349 and 0.81 ectoine molecules per nucleotide, whilst 0.1 M ectoine results in ΔRU 205 and 0.47 ectoine molecules per nucleotide. We note here that the short and therefore conformationally non-flexible DNA (dT25) is accommodated in the SAM on gold in a near vertical geometry. Considering the amount of ectoine bound to dT25 at 1 M ectoine solution concentration (about 1 ectoine molecule per 1 nucleotide, that is, a 25 times higher number of ectoine molecules bound to DNA than there are DNA molecules bound to the surface), there would be at such high DNA coverage no space for ectoine to bind to the surface in any significant amount.
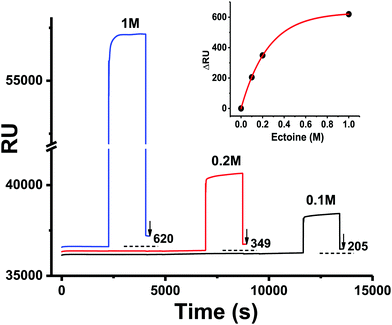 |
| | Fig. 4 Surface plasmon resonance (SPR) data concerning the interaction of ectoine with ssDNA (dT25) immobilized on a gold chip (see details in the Experimental section). The surface was exposed to ectoine at a flow rate of 10 μL min−1 with various ectoine concentrations (1 M, 0.2 M, 0.1 M in 1× PBS) and then washed with 1× PBS solution. The corresponding changes in RU due to the binding of ectoine to DNA are indicated in the figure. The inset shows the amount of bound ectoine in ΔRU units as a function of the molar concentration of ectoine. The ΔRU values are obtained from the baseline before exposure (black dotted line) and the resulting SPR signal after exposure. The red line is a guide for the eye. | |
Raman spectroscopy of characteristic markers of hydrogen bonding.
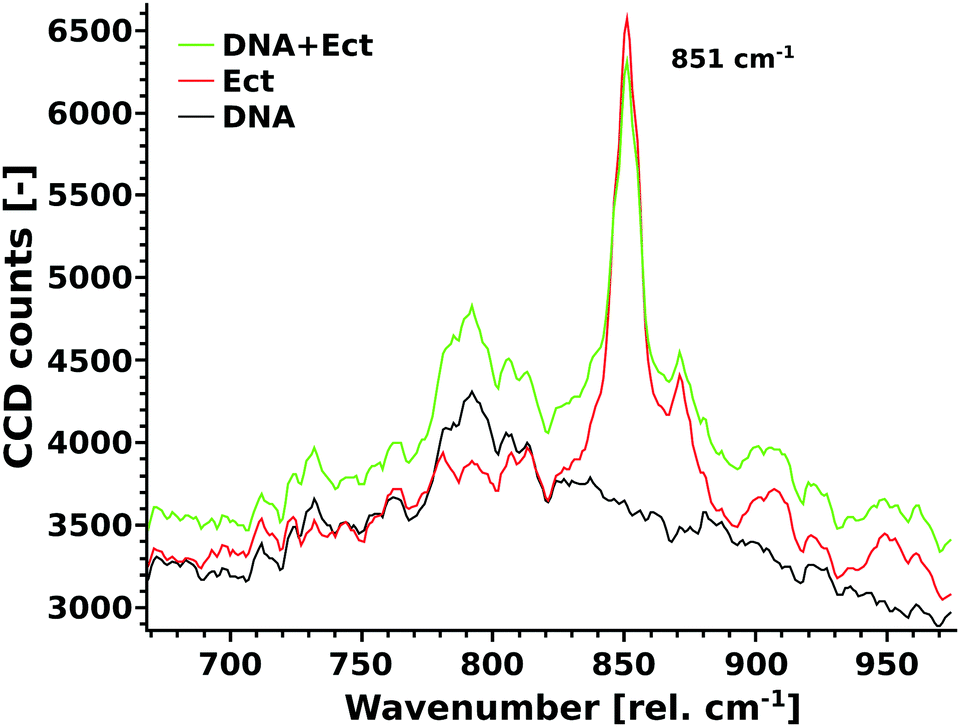
In a different study, we have shown that the ring breathing mode of ectoine at 851 rel. cm−1 (Fig. 5) is a sensitive measure for the solvation process in a crowded molecular environment.8 The Raman scattering intensity of this mode is dependent on the amount of water in its hydration shell and the formation of hydrogen bonds between ectoine and its surroundings. Hence, a change in the number of water molecules found in the vicinity of ectoine in the presence of DNA can be monitored by the intensity of this vibrational mode. The rationale behind this analysis is that the results from the melting temperature measurements (see ESI†) and recent simulation works21 suggest the denaturing effects of ectoine on DNA with an influence on the hydrogen bonding behaviour of DNA. This approach has the advantages of much better signal to noise ratios, as well as less ambiguity in comparison to the analysis of the multitude of vibrational modes of DNA (compare Fig. 5). The results show an absolute decrease of the 851 rel. cm−1 mode of ectoine upon addition of DNA. This behaviour is in striking contrast to the intensity increase of all other modes, especially in the direct vicinity of the 851 rel. cm−1 peak. Thus, it can be concluded that DNA and ectoine mutually influence their hydrogen interactions with the surroundings.
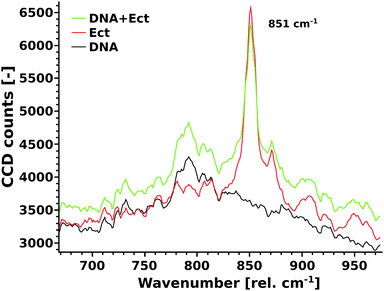 |
| | Fig. 5 Unprocessed Raman spectra of DNA (black), ectoine (red), and DNA + ectoine (green) in water are shown. The ectoine mode at 851 rel. cm−1, which is characteristic of ectoine interaction with its local environment, decreases due to DNA–ectoine interactions. | |
4 Discussion
The UV radiation damage of DNA has shown a complex dependence on the presence of ectoine, hydroxyectoine and salt. To understand the underlying mechanisms, it is first necessary to discuss the results regarding ectoine–DNA interaction. Afterwards, the relevant processes leading to different types of UV radiation damage to DNA and their possible modifications by the presence of cosolutes are analysed. Finally, implications for an in vivo system and the interpretation of results obtained by other authors will be presented.
To our knowledge, previous experimental studies only speculated about the possibility that ectoine binds to DNA without providing experimental evidence.9,20 One computational study predicted that ectoine accumulates between 0.5–2 nm distance to the sugar–phosphate backbone of dsDNA due to electrostatic and dispersion interactions.21 Here, we provide for the first time experimental evidence that ectoine binds directly to DNA. From the SPR resonance measurements, it was revealed that ectoine molecules bind to ssDNA in a concentration dependent manner (Fig. 4). At 0.1 M and 0.2 M ectoine concentrations, 0.47 and 0.81 ectoine molecules bind per nucleotide, respectively. At 1 M ectoine concentration, on average, 1.44 ectoine molecules bind per nucleotide. Further evidence for DNA–ectoine interaction was provided by SAXS measurements. Here, the X-ray scattering patterns (Fig. 3) from DNA (black curve) and ectoine (red curve) solutions revealed both distinct features towards low-q values. It is assumed that a non-interacting solution of both DNA and ectoine would result in scattering equal to the sum of these contributions. However, the scattering from the DNA + ectoine sample (Fig. 3 green curve) does not resemble the combined scattering observed from the separate DNA and ectoine samples. This gives an indication that there is a direct interaction between DNA and ectoine within the DNA + ectoine sample. The evidence of this interaction is most distinct towards lower values of q and could be explained by a change in the conformation of DNA due to direct interactions with ectoine, as was predicted by a computational study by Oprzeska-Zingrebe et al.21 In the same study, it was predicted that ectoine influences intermolecular H-bond formation and replaces other molecules in the first hydration shell of the DNA strand.21 Such an influence on intermolecular H-bond formation is in agreement with the results obtained from Raman spectroscopy, where the characteristic ring breathing mode of ectoine at 851 rel. cm−1 (Fig. 5), a sensitive measure for the amount of water in its hydration shell, along with the formation of hydrogen bonds between ectoine and its surroundings, decreased in the presence of DNA.8 Thus, a change in the number of water molecules in the vicinity of ectoine in the presence of DNA and vice versa can be monitored from the intensity of this vibrational mode. Therefore, it can be concluded that DNA and ectoine interact directly with each other and that ectoine is not preferentially excluded from the DNA surface, which is in line with the SAXS and SPR results. According to Oprzeska-Zingrebe et al., this is caused by electrostatic and dispersion interactions that lead to ectoine accumulation in 0.5–2 nm distance to the sugar–phosphate backbone.21 Such an interaction has potential to influence the precursors of UV induced DNA damage, the excited states of DNA and their decay mechanisms, as previously seen with other organic cosolutes.38
Besides the influence of cosolutes on DNA, through direct interaction, the structural and electronic properties of biomolecules can be modified by cosolute induced changes in the solvent properties.7,9,15,38–40 For example, zwitterionic osmolytes and compatible solutes are known to modify the dielectric constant of the solvents and, in turn, the counterion atmosphere around the DNA.9,40–42 This was studied by dielectric spectroscopy,42 nuclear magnetic resonance spectroscopy,41 and computational studies.9,40 For instance, the presence of ectoine increases the water dielectric constant of the solution (water + cosolute) in comparison to pure water and therefore decreases electrostatic interactions within the solution.9,42 Such an increase in the dielectric constant of the medium can lead to an increase in the net charge at the DNA backbone.41 Furthermore, it is important to note that ectoine has an influence on the hydrogen bonding behaviour of water and forms a tightly bound first hydration shell.5,9,42 Meyer et al. assumed that the formation of such strong ectoine water complexes can lead to conformational changes of DNA and to a decrease in base pairing in pure water.20 These indirect influences could result in additional modifications of electron transfer properties, DNA conformation, hole trapping sites and free electronic states.38,43 The relevance of these direct and indirect mechanisms for DNA excitation and decay mechanisms is discussed below.
The different types of observed DNA damage, SSB and T4 sensitive sites, are located at different DNA building blocks. SSBs occur at the sugar–phosphate backbone. In vivo, they can be caused by the radiation induced formation of sugar radicals, electron induced bond breakage or secondary radiation products, such as reactive oxygen species (ROS), etc.29,32,44,45 Under the presented in vitro conditions, direct damage can be caused by ionisation or excitation of the DNA molecules.30,46,47 The vertical ionisation energies of the DNA constituents in water lie between (7.3–8.1) eV for the base components and (8.9–9.5) eV for the phosphate groups.48,49 Therefore, the direct damage occurring in this study from UV photons with 266 nm (4.66 eV) is not due to the production of free (kinetic) electrons by a vertical ionisation process.
Still, photoionisation of DNA constituents with 266 nm laser pulses can lead to the production of (non-kinetic) hydrated electrons, as observed in other studies by their characteristic absorption at around 700 nm.50,51 It was found that single or two photon processes contribute simultaneously to ionisation of DNA bases at high laser intensities (order of MW cm−2).51 Nevertheless, at 650 kJ (cm2 pulse)−1, the contribution for dsDNA was found to be governed (over 70%) by single photon processes.51 Due to the even lower laser intensities in this study (≪24 kJ (cm2 pulse)−1), single photon processes dominate. After single photon ionisation, the ejected electron is localised in a (partially/pre) hydrated state in water or at the water–DNA interface. The exact nature and interactions of such electrons with DNA are currently under debate and beyond the focus of this work.52–57 Nevertheless, prehydrated electrons are known to effectively cause reductive DNA damage such as SSB.53,55–57 Their damage efficiency is likely to be influenced by the change in the electronic states of the DNA itself and of the DNA–water interface through the presence and interaction of ectoine and the displacement of water in the hydration shell of DNA. Similar mechanisms were observed in our previous work regarding the protection of DNA by ectoine against ionizing radiation (high energy electrons), where, besides the aforementioned prehydrated electrons, many additional damaging species and pathways were also present.10 From the presented irradiation data (Fig. 2), it is evident that in the case of the pure samples, the buffer conditions without additional cosolute already have a huge impact on the amount and type of damage observed, highlighting the importance of the counterion atmosphere around the sugar–phosphate backbone. In the case of irradiation in PBS solution, SSB contributes little to the total damage. Here, no significant differences in SSB induction between pure and cosolute solutions could be detected. In pure water, SSB formation increases compared to PBS, but decreases relatively in the presence of ectoine and hydroxyectoine. There, the DNA backbone is more susceptible to damage due to the absence of stabilising positive ions, particularly Na+, which is a weak scavenger of prehydrated electrons.10,58 In pure water, the stabilising role is partly taken over by the cosolutes as is evident from the direct ectoine–DNA binding interaction reported above. This effect leads to a decrease in SSB induction upon addition of ectoine in pure water (Fig. 2 left, red bars, sample (A) and (C)). At the same time, the amount of base damage (discussed further below) increases in ultrapure water when cosolutes are added. This shift from SSB to T4 damage upon addition of cosolutes might be explained by a change in the probability of competing decay channels of excited electronic states. Thus, leading either to a higher amount of SSB or T4 sensitive sites, as rationalized below.
Under the presented in vitro conditions, the T4 sensitive sites (base damages) are caused by direct absorption of UV photons at the bases.29,30,46,47 CPDs, the most frequent UV induced lesion in dsDNA, are predominantly formed from excited electronic 1ππ* states. The electronic transition from the ground state to the excited ππ* state in single nucleobases gives rise to the characteristic absorption observed at around 260 nm, where contributions from excitations into delocalised states (Frenkel excitons) have to be considered.30,46 The properties of these excitons depend on the dipolar coupling between the ππ* transitions, which is influenced by the relative orientation of the nucleobases and the ground state conformation.46,59 This, in turn, is dependent on the ionic strength of the solvent and the presence of osmolytes.30,38,39 Another type of collective excited states emerges when electronic orbitals between adjacent nucleobases overlap.46 Here, a photon induced charge transfer between the nucleobases and the resulting charge separation makes this type of excited state more sensitive to ions and/or polar cosolutes. Both types of collective excited states can mix or decay into each other, a behaviour that is favoured by base stacking. The charge transfer states in DNA are related to the hypochromism of DNA duplexes, which is exploited in the study of duplex stability and in the determination of DNA melting curves.46 The observed lowering of pUC19 DNA melting temperature (see ESI† and ref. 12, 19) (Tm = 84.2 ± 0.4) indicates a decrease in base interactions in the presence of ectoine (Tm = 82.7 ± 0.4) and hydroxyectoine (Tm = 81.6 ± 0.7), through modification of the hydrogen bonds is also indicated by the Raman measurements. As stated above, such a decrease in base pairing changes the decay channels of the excited states and influences the yields of different types of damage. This might explain the strong decrease in T4 sensitive sites under 266 nm irradiation in pure water (Fig. 2 left, black bars). To provide more detailed information, and to test our model of the cosolute effects on the modification of excited states, different decay and damaging channels, future studies should involve time resolved spectroscopy to monitor them directly. Additionally, a detailed investigation of different types of base damage (e.g. by Endo III and Fpy glycosylase assays), and UV-A and UV-B radiation would be of interest to complete the picture of the influence of ectoine interaction with DNA on UV radiation damage.
The results presented in this in vitro study are representative of UV-C radiation damage caused by direct absorption at the DNA. We note here that for human exposure, the most relevant damage contributions originate from UV-A and UV-B photons via indirect mechanisms. In vivo, indirect DNA damage represents an additional contribution that will be briefly discussed below to put our results into a broader context. In vivo, indirect damage is caused by ROS (e.g. ˙OH, O2˙−, 1O2) that can introduce a broad variety of chemical alterations to all DNA constituents.31 In this case, ROS are produced by UV absorption at intracellular chromophores,31 since UV absorption of water becomes only relevant for photon energies above 6 eV60 and ectoine and hydroxyectoine provide protection against ROS attack due to their radical scavenging capabilities10,45 as demonstrated by the electron paramagnetic resonance (EPR) measurements that can be found in the ESI† (compare as well ref. 10 and 11). These radical scavenging properties are likely to play a part in the in vivo protection by ectoine against UV-A induced SSB27 in human keratinocytes and the decrease in mutation of mitochondrial DNA,17 as observed by other authors. We would like to add here that a protective effect of ectoine by the direct absorption of UV light can be ruled out, since its UV absorption at a wavelength of 266 nm or above is neglectable at the given concentrations compared with DNA. For details on UV absorption measurements of ectoine at different wavelengths, see the ESI.† Furthermore, in the cytoplasm of living cells, the stabilisation of biomolecules, the type of radiation damage, repair efficiency and biological relevance of a lesion depend on a multitude of factors. This is especially relevant in medical or cosmetic applications17,24–26 and when an uptake of ectoine by mammalian cells takes place, which was reported for human keratinocytes.18 On the other hand, Beblo-Vranesevic et al.28 found a decrease in the cellular survival rate in two different microorganisms (H. marinus and A. fulgidus) upon irradiation with UV-C (254 nm) and the external addition of ectoine (0.5 mol L−1). In such a context, when other stressors are present, the combined interaction of radiation and cosolutes within the cell membranes, of the specific organisms, becomes relevant and has to be studied for different types of cells individually.25 Thus, to provide a complete picture of the interplay of direct and indirect damaging effects, further studies are needed combining in vitro and in vivo systems.
5 Summary
Plasmid DNA pUC19 was irradiated with UV-C photons in PBS and in ultra pure water in the presence and absence of ectoine and hydroxyectoine. The yields of SSB, base damage and loss were altered depending on the cosolute and buffer conditions. When either ectoine or hydroxyectoine was present, base damage decreased strongly and SSB was unaltered when irradiation was performed in PBS. Under non-physiological conditions (in ultra pure water), SSB induction decreased when ectoines were present, which is in contrast to the behaviour of the base damage. A direct binding interaction between DNA and ectoine could be shown experimentally by the combination of small angle X-ray scattering, surface plasmon resonance and Raman spectroscopy. It was found that the exposure of ssDNA to 1 M ectoine solute results in the binding of about 1.44 ectoine molecules per nucleotide. Additionally, ectoine and hydroxyectoine were both found to lower the DNA melting temperature and to change the local hydrogen bonding environment.
Based on our findings, we tentatively propose that the change in the type and amount of DNA damage is caused by the alterations of decay channels of the excited states in DNA. These alterations are assumed to have their origin within the combined effects of direct electrostatic interaction and solvent mediated effects by ectoine.
Author contributions
M. B. H. conceived the study, wrote the manuscript, and performed the irradiation, Raman and EPR experiments. H. Se. and T. S. performed and analyzed SPR measurements. G. J. S. performed and analyzed SAXS measurements. All authors contributed to the writing of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Discussions with M. Weller, V. Wachtendorf, L. Cordsmeier, A. Pellegrino, J. Smiatek, T. A. Waniek and R. Wellhausen are gratefully acknowledged. We thank L. Cordsmeier for performing the UV spectroscopy measurements.
References
- E. A. Galinski, H.-P. Pfeiffer and H. G. Trueper, Eur. J. Biochem., 1985, 149, 135–139 CrossRef CAS PubMed
 .
.
- F. Piubeli, M. Salvador, M. Argandoña, J. J. Nieto, V. Bernal, J. M. Pastor, M. Cánovas and C. Vargas, Microb. Cell Fact., 2018, 17, 2 CrossRef PubMed .
- L. Czech, L. Hermann, N. Stöveken, A. A. Richter, A. Höppner, S. H. J. Smits, J. Heider and E. Bremer, Genes, 2018, 9, 177 CrossRef PubMed .
- N. Gunde-Cimerman, A. Plemenitaš and A. Oren, FEMS Microbiol. Rev., 2018, 42, 353–375 CrossRef CAS PubMed .
- M. B. Hahn, F. Uhlig, T. Solomun, J. Smiatek and H. Sturm, Phys. Chem. Chem. Phys., 2016, 18, 28398–28402 RSC .
- W. Schuh, H. Puff, E. A. Galinski and H. G. Trüper, Z. Naturforsch., C: J. Biosci., 2014, 40, 780–784 Search PubMed .
- J. Smiatek, R. K. Harishchandra, O. Rubner, H.-J. Galla and A. Heuer, Biophys. Chem., 2012, 160, 62–68 CrossRef CAS PubMed .
-
M. B. Hahn, T. Solomun and J. Smiatek, 2020, Manuscript in preparation.
- M. B. Hahn, T. Solomun, R. Wellhausen, S. Hermann, H. Seitz, S. Meyer, H.-J. Kunte, J. Zeman, F. Uhlig, J. Smiatek and H. Sturm, J. Phys. Chem. B, 2015, 119, 15212–15220 CrossRef CAS PubMed .
- M. B. Hahn, S. Meyer, M.-A. Schröter, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Chem. Chem. Phys., 2017, 19, 25717–25722 RSC .
- S. Brands, P. Schein, K. F. Castro-Ochoa and E. A. Galinski, Arch. Biochem. Biophys., 2019, 108097 CrossRef PubMed .
- G. Malin, R. Iakobashvili and A. Lapidot, J. Biol. Chem., 1999, 274, 6920–6929 CrossRef CAS PubMed .
- Y. Oberdörfer, S. Schrot, H. Fuchs, E. Galinski and A. Janshoff, Phys. Chem. Chem. Phys., 2003, 5, 1876–1881 RSC .
- A. Narayanan Krishnamoorthy, C. Holm and J. Smiatek, J. Phys. Chem. B, 2014, 118, 11613–11621 CrossRef CAS PubMed .
- G. Zaccai, I. Bagyan, J. Combet, G. J. Cuello, B. Demé, Y. Fichou, F.-X. Gallat, V. M. G. Josa, S. von Gronau, M. Haertlein, A. Martel, M. Moulin, M. Neumann, M. Weik and D. Oesterhelt, Sci. Rep., 2016, 6, 31434 CrossRef CAS PubMed .
- D. V. Yanykin, M. Malferrari, S. Rapino, G. Venturoli, A. Y. Semenov and M. D. Mamedov, Photosynth. Res., 2019, 141, 782–801 CrossRef PubMed .
- J. Buenger and H. Driller, Skin Pharmacol. Physiol., 2004, 17, 232–237 CrossRef CAS PubMed .
- E. Buommino, C. Schiraldi, A. Baroni, I. Paoletti, M. Lamberti, M. De Rosa and M. A. Tufano, Cell Stress Chaperones, 2005, 10, 197–203 CrossRef CAS PubMed .
- M. Schnoor, P. Voß, P. Cullen, T. Böking, H.-J. Galla, E. A. Galinski and S. Lorkowski, Biochem. Biophys. Res. Commun., 2004, 322, 867–872 CrossRef CAS PubMed .
- S. Meyer, M.-A. Schröter, M. B. Hahn, T. Solomun, H. Sturm and H. J. Kunte, Sci. Rep., 2017, 7, 7170 CrossRef CAS PubMed .
- E. A. Oprzeska-Zingrebe, S. Meyer, A. Roloff, H.-J. Kunte and J. Smiatek, Phys. Chem. Chem. Phys., 2018, 20, 25861–25874 RSC .
- W. Sajjad, S. Qadir, M. Ahmad, M. Rafiq, F. Hasan, R. Tehan, K. L. McPhail and A. A. Shah, J. Appl. Microbiol., 2018, 125, 457–467 CrossRef CAS PubMed .
- G. Lentzen and T. Schwarz, Appl. Microbiol. Biotechnol., 2006, 72, 623–634 CrossRef CAS PubMed .
- V. A. Dao, A. Bilstein, S. Overhagen, L. Géczi, Z. Baráth and R. Mösges, Oncology and Therapy, 2018, 1–14 Search PubMed .
- T. Rieckmann, F. Gatzemeier, S. Christiansen, K. Rothkamm and A. Münscher, Sci. Rep., 2019, 9, 6594 CrossRef PubMed .
- J. Buenger, J. Degwert and H. Driller, IFSCC Mag., 2001, 4, 127 Search PubMed .
- C. Botta, C. Di Giorgio, A.-S. Sabatier and M. De Meo, J. Photochem. Photobiol., B, 2008, 91, 24–34 CrossRef CAS PubMed .
- K. Beblo-Vranesevic, E. A. Galinski, R. Rachel, H. Huber and P. Rettberg, Arch. Microbiol., 2017, 199, 17–28 CrossRef CAS PubMed .
- Y. Jiang, M. Rabbi, M. Kim, C. Ke, W. Lee, R. L. Clark, P. A. Mieczkowski and P. E. Marszalek, Biophys. J., 2009, 96, 1151–1158 CrossRef CAS PubMed .
- W. J. Schreier, P. Gilch and W. Zinth, Annu. Rev. Phys. Chem., 2015, 66, 497–519 CrossRef CAS PubMed .
- B. Epe, Photochem. Photobiol. Sci., 2011, 11, 98–106 RSC .
- M. B. Hahn, S. Meyer, M.-A. Schröter, H. Seitz, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Chem. Chem. Phys., 2017, 19, 1798–1805 RSC .
- M. B. Hahn, S. Meyer, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Rev. E, 2017, 95, 052419 CrossRef PubMed .
- R. L. Schoch and R. Y. H. Lim, Langmuir, 2013, 29, 4068–4076 CrossRef CAS PubMed .
- B. R. Pauw, A. J. Smith, T. Snow, N. J. Terrill and A. F. Thünemann, J. Appl. Crystallogr., 2017, 50, 1800–1811 CrossRef CAS PubMed .
- J. Filik, A. W. Ashton, P. C. Y. Chang, P. A. Chater, S. J. Day, M. Drakopoulos, M. W. Gerring, M. L. Hart, O. V. Magdysyuk, S. Michalik, A. Smith, C. C. Tang, N. J. Terrill, M. T. Wharmby and H. Wilhelm, J. Appl. Crystallogr., 2017, 50, 959–966 CrossRef CAS PubMed .
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, Nucleic Acids Res., 2001, 29, 5163–5168 CrossRef CAS PubMed .
- M. Tanaka, T. Matsumoto and H. Iida, Org. Biomol. Chem., 2018, 16, 6695–6702 RSC .
- G. Portella, M. Terrazas, N. Villegas, C. González and M. Orozco, Angew. Chem., Int. Ed., 2015, 54, 10488–10491 CrossRef CAS PubMed .
- E. A. Oprzeska-Zingrebe and J. Smiatek, Biophys. Rev., 2018, 1–16 Search PubMed .
- S. Flock, R. Labarbe and C. Houssier, Biophys. J., 1996, 71, 1519–1529 CrossRef CAS PubMed .
- A. Eiberweiser, A. Nazet, S. E. Kruchinin, M. V. Fedotova and R. Buchner, J. Phys. Chem. B, 2015, 119, 15203–15211 CrossRef CAS PubMed .
- M. Tanaka, H. Iida and T. Matsumoto, Chem. Lett., 2017, 47, 62–64 CrossRef .
- J. Simons, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS PubMed .
-
C. von Sonntag, Free-Radical-Induced DNA Damage and Its Repair, Springer, Berlin, Heidelberg, 2006 Search PubMed .
- D. Markovitsi, Photochem. Photobiol., 2016, 92, 45–51 CrossRef CAS PubMed .
- A. Banyasz, I. Vayá, P. Changenet-Barret, T. Gustavsson, T. Douki and D. Markovitsi, J. Am. Chem. Soc., 2011, 133, 5163–5165 CrossRef CAS PubMed .
- E. Pluhařová, C. Schroeder, R. Seidel, S. E. Bradforth, B. Winter, M. Faubel, P. Slavíček and P. Jungwirth, J. Phys. Chem. Lett., 2013, 4, 3766–3769 CrossRef .
- C. A. Schroeder, E. Pluhařová, R. Seidel, W. P. Schroeder, M. Faubel, P. Slavíček, B. Winter, P. Jungwirth and S. E. Bradforth, J. Am. Chem. Soc., 2015, 137, 201–209 CrossRef CAS PubMed .
- C. E. Crespo-Hernández and R. Arce, J. Phys. Chem. B, 2003, 107, 1062–1070 CrossRef .
- S. Marguet, D. Markovitsi and F. Talbot, J. Phys. Chem. B, 2006, 110, 11037–11039 CrossRef CAS PubMed .
- K. R. Siefermann, Y. Liu, E. Lugovoy, O. Link, M. Faubel, U. Buck, B. Winter and B. Abel, Nat. Chem., 2010, 2, 274–279 CrossRef CAS PubMed .
- K. R. Siefermann and B. Abel, Angew. Chem., Int. Ed., 2011, 50, 5264–5272 CrossRef CAS PubMed .
- B. Abel, U. Buck, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2011, 14, 22–34 RSC .
- B. Abel, Annu. Rev. Phys. Chem., 2013, 64, 533–552 CrossRef CAS PubMed .
- J. Nguyen, Y. Ma, T. Luo, R. G. Bristow, D. A. Jaffray and Q.-B. Lu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11778–11783 CrossRef CAS PubMed .
- J. Ma, F. Wang, S. A. Denisov, A. Adhikary and M. Mostafavi, Sci. Adv., 2017, 3, e1701669 CrossRef PubMed .
- T. W. Kee, D. H. Son, P. Kambhampati and P. F. Barbara, J. Phys. Chem. A, 2001, 105, 8434–8439 CrossRef CAS .
- A. A. Voityuk, Photochem. Photobiol. Sci., 2013, 12, 1303–1309 RSC .
- A. Mozumder, Phys. Chem. Chem. Phys., 2002, 4, 1451–1456 RSC .
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental methods. See DOI: 10.1039/d0cp00092b |
|
| This journal is © the Owner Societies 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Glen J.
Smales
b,
Harald
Seitz
cd,
Tihomir
Solomun
b and
Heinz
Sturm
*ab,
Glen J.
Smales
b,
Harald
Seitz
cd,
Tihomir
Solomun
b and
Heinz
Sturm