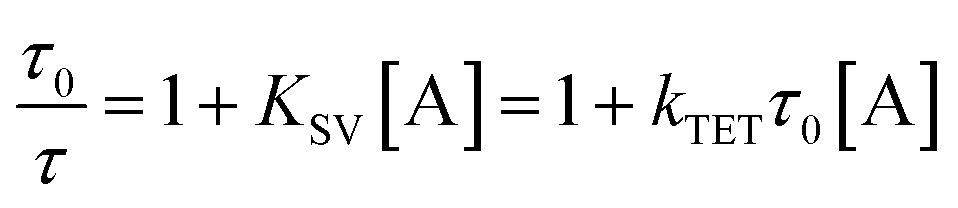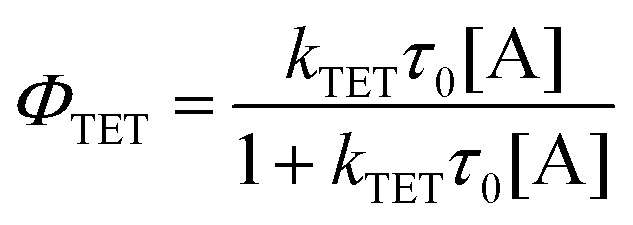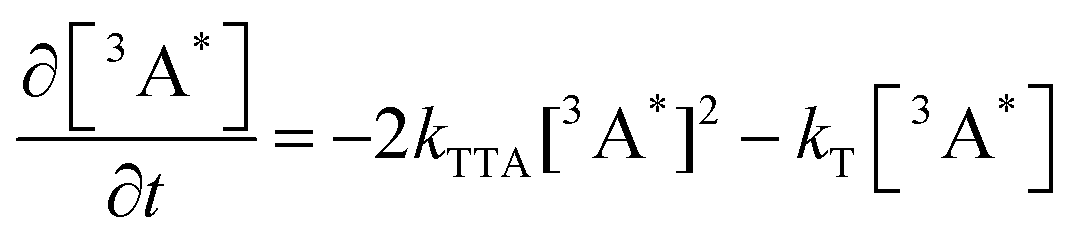Optimizing photon upconversion by decoupling excimer formation and triplet triplet annihilation†
Received
4th December 2019
, Accepted 18th December 2019
First published on 18th December 2019
Abstract
Perylene is a promising annihilator candidate for triplet–triplet annihilation photon upconversion, which has been successfully used in solar cells and in photocatalysis. Perylene can, however, form excimers, reducing the energy conversion efficiency and hindering further development of TTA-UC systems. Alkyl substitution of perylene can suppress excimer formation, but decelerate triplet energy transfer and triplet–triplet annihilation at the same time. Our results show that mono-substitution with small alkyl groups selectively blocks excimer formation without severly compromising the TTA-UC efficiency. The experimental results are complemented by DFT calculations, which demonstrate that excimer formation is suppressed by steric repulsion. The results demonstrate how the chemical structure can be modified to block unwanted intermolecular excited state relaxation pathways with minimal effect on the preferred ones.
Introduction
Triplet–triplet annihilation photon upconversion (TTA-UC) can help light harvesting materials to overcome their bandgap limitation and thus achieve higher solar energy conversion efficiencies beyond the Shockley–Queisser limit.1–7 The mechanism has recently been successfully incorporated in solar cells,8–14 photo-catalysts,15–18 optical devices,19–24 and light imaging.25–31 It requires two functional species, a sensitizer and an annihilator. The sensitizer absorbs a photon and transfers the energy to an annihilator through triplet energy transfer.32 Two annihilator molecules in their triplet excited states undergo triplet–triplet annihilation29 upon close contact, promoting one molecule to its excited singlet state from where emission of a high energy photon occurs. Perylene is among the most promising annihilator candidates due to its high stability, commercial availability, and excellent photo-physical properties.34 TTA-UC systems based on perylene as the annihilator hold the photon upconversion quantum yield record and have now been used in solar cells, photo-redox catalysis and bio-imaging.18,35–37
The drawback of perylene is its tendency to form excited dimers (excimers). Excimer formation in TTA-UC reduces the overall energy conversion efficiency, both due to the lower energy of the emitted photons and due to the significant rate constant of non-radiative decay from excimers.38 Dover et al. have shown the conversion between monomers and excimers through triplet pair and free diffusion during both singlet fission and TTA-UC.39 We have further suggested that another main reason for the surprisingly large amount of excimer emission in perylene based photon upconversion is due to a preassembly step on the triplet energy surface, already before the upconversion event.40 Alkyl substitution offers the opportunity to manipulate the stacking interactions of perylene with minimal impact on the excited levels of the individual molecules. Indeed, perylene derivatives like tetra-tert-butyl-perylene show low excimer formation in TTA-UC, since the molecular aggregation is sterically hindered.41,42 However, the steric bulk also reduces mobility and molecular contact, and thus can inhibit triplet energy transfer32,43 and TTA. It is therefore of great importance to control the pi–pi interactions in perylene in order to limit excimer formation without compromising photon upconversion performance.
Here, a series of alkyl substituted perylene derivatives are used to investigate the interplay between upconversion performance and excimer formation blockade. Static and dynamic upconversion experiments show that an ethyl group is sufficient to block excimer formation. This result is corroborated by calculations, which show that steric hindrance prevents coupling on the excimer landscape. The results presented here shed light on molecular design of TTA-UC annihilators and help to optimize annihilator molecules for TTA-UC.
Results
Spectroscopic characterization of annihilators
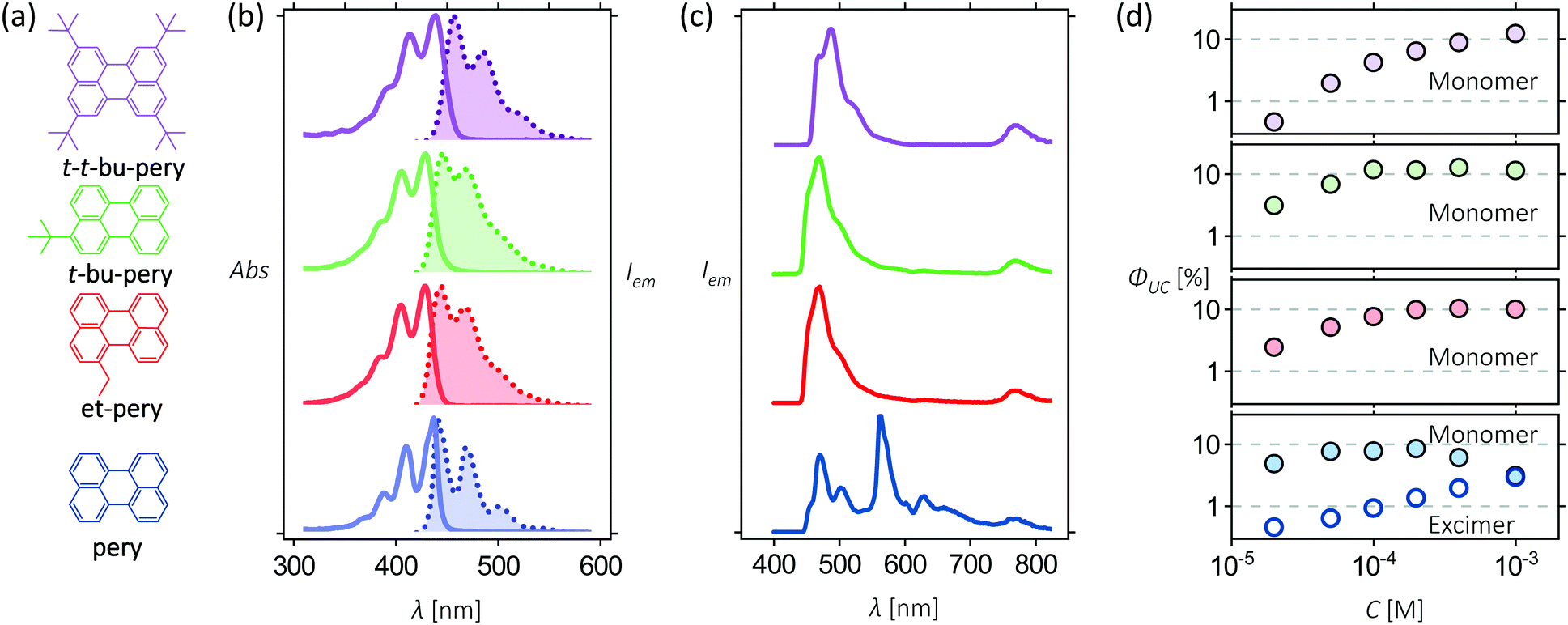
To assess the effect of increasing steric hindrance on excimer formation and upconversion efficiency, four perylene derivatives were used: perylene (pery), 1-ethyl-perylene (et-pery), 3-tert-butyl-perylene (t-bu-pery) and 2,5,8,11-tetra-tert-butylperylene (t-t-bu-pery), which are available from our previous work (Fig. 1a).44 The absorption and emission spectra of these molecules in tetrahydrofuran (THF) are shown in Fig. 1b revealing only minor changes to the energy of the singlet excited state. The Stokes shifts of pery, et-pery, t-bu-pery and t-t-bu-pery are 103 cm−1, 736 cm−1, 938 cm−1 and 849 cm−1, respectively. The increased Stokes shift after alkyl substitution indicates that the difference in polarizability between the ground and excited states increases when alkylated.45 All four annihilators demonstrate fluorescence quantum yields near unity, and a fluorescence lifetime of 3.8 ns, indicating that the excited states configuration is not affected much by the substitution (Fig. S1, ESI†).
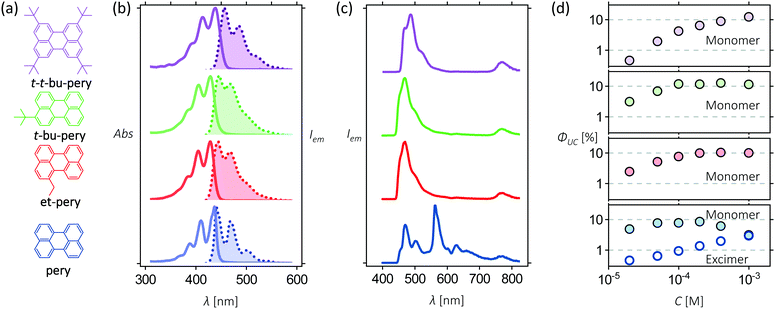 |
| | Fig. 1 (a) Molecular structure of pery, et-pery, t-bu-pery, and t-t-bu-pery; (b) absorption/emission spectra of 10 μM pery, et-pery, t-bu-pery, and t-t-bu-pery in THF when excited at 400 nm; (c) upconverted emission spectra from solutions of 10 μM PtTBTP and 1 mM perylene derivatives upon excitation at 617 nm in THF.33 The absolute photon upconversion quantum yields at different concentrations of annihilators. | |
Photon upconversion with different annihilators
In order to construct TTA-UC systems with these perylene derivatives, we used Platinum tetra-benzo-tetra-phenyl-porphyrin (PtTBTP) as a triplet sensitizer. PtTBTP has a high phosphorescence quantum yield, long triplet lifetime, and a triplet energy that matches the one of perylene.46 The potential photon energy increase of the PtTBTP/perylene couple is 0.75 to 0.83 eV, as determined by the difference of the E00 energies (Fig. S2, ESI†).47 Photon upconversion using PtTBTP in combination with the presented perylene derivatives was performed using a light-emitting diode (LED) as excitation source (617 nm; Fig. 1c). For all perylene derivatives, strong emission can be observed in the range 450 nm to 540 nm, which is assigned to perylene fluorescence due to TTA-UC. Excimer emission at 565 nm from perylene is also present.48 The quantum yield of excimer emission increases with perylene concentration, indicating a process for perylene excimer formation that involves diffusion. No excimer emission was, however, observed for the three perylene derivatives with steric side groups over a wide range of annihilator concentrations (Fig. 1d). We can thus conclude that even such small substituents as an ethyl group are enough to block excimer formation in perylene derivatives. Photon-upconversion emission of the four annihilators show linear and quadratic dependence on the excitation intensity (Fig. S3, ESI†), as a typical feature of TTA-UC. The light intensity threshold of quadratic to linear dependence increases after alkyl substitution, reducing the TTA-UC efficiency at low excitation intensity.
Energetics of annihilator dimers
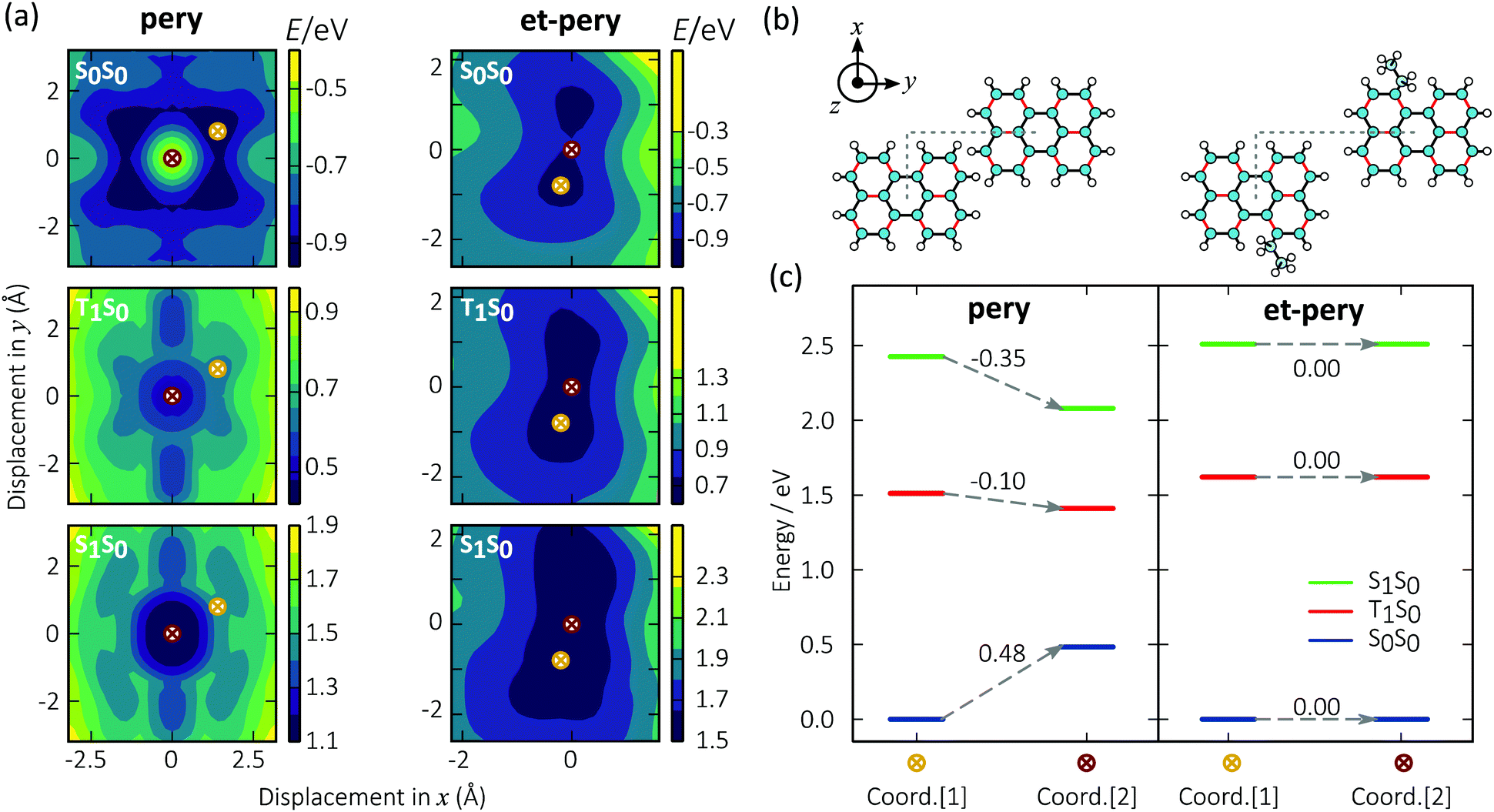
It has recently been suggested that a reason for strong excimer emission in perylene based TTA-UC is the formation of excimers on the triplet surfaces before the annihilation event.40 The relationship between excimer formation and molecular structure can be explored from an energetic point of view (Note S1.6, ESI†). To this end, we analysed the potential energy surface for the interaction between a pair of perylene molecules within the framework of time-dependent density functional theory (TD-DFT).49–52 We relaxed the structure of the molecular dimers on the ground state (S0) potential energy surface (PES). Subsequently, we mapped out the PES as a function of the lateral displacement of the two monomers (Fig. 2a and b). The z offset distances for pery and et-pery are about 3.32 and 3.48 Å, respectively. The first excited singlet (S1) and triplet (T1) energy surfaces of the dimer were then created by adding the respective excitation energies to the ground state energy.
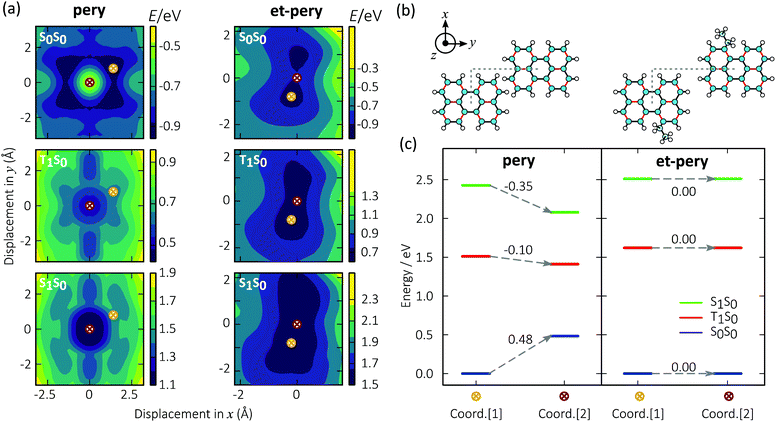 |
| | Fig. 2 (a) Calculated energy surfaces of the confined systems of pery and et-pery dimers. The energy minimum geometry on the S0S0 energy surface is labelled with a yellow cross, and the energy minimum geometry on the T1S0 and S1S0 energy surfaces is labelled with a red cross; (b) bi-molecular coordination of pery and et-pery dimer on xy plane; (c) energy diagram of pery and et-pery dimer at relaxation. | |
In the ground state of the pery dimer, the two monomers are shifted relative to each other, similar to the situation in a graphene bilayer.53 The singlet and triplet excimers on the other hand, are characterized by perfect lateral alignment of the two monomers. The energies of the two configurations differ by 0.48 eV, −0.10 eV, and −0.35 eV on the S0, T1, and S1 surfaces, respectively. These energy differences correlates with low energy excimer emission (Fig. 2c).
The PES of et-pery is less symmetric than in the case of pery and the lowest energy configurations on the S0, T1, and S1 landscape are almost identical to each other. The distortion of the molecular geometry caused by the et groups impacts the ability of the two monomers to move relative to each other (note that the core of et-pery is slightly twisted). This prevents them from achieving an alignment that, as in the case of pery, enables efficient coupling on the excited landscape, and thus excimer formation. We can conclude that alkyl substitution is an effective means to disrupt the favourable excimer geometry of perylene on both singlet and triplet energy surfaces.
Kinetics of photon upconversion
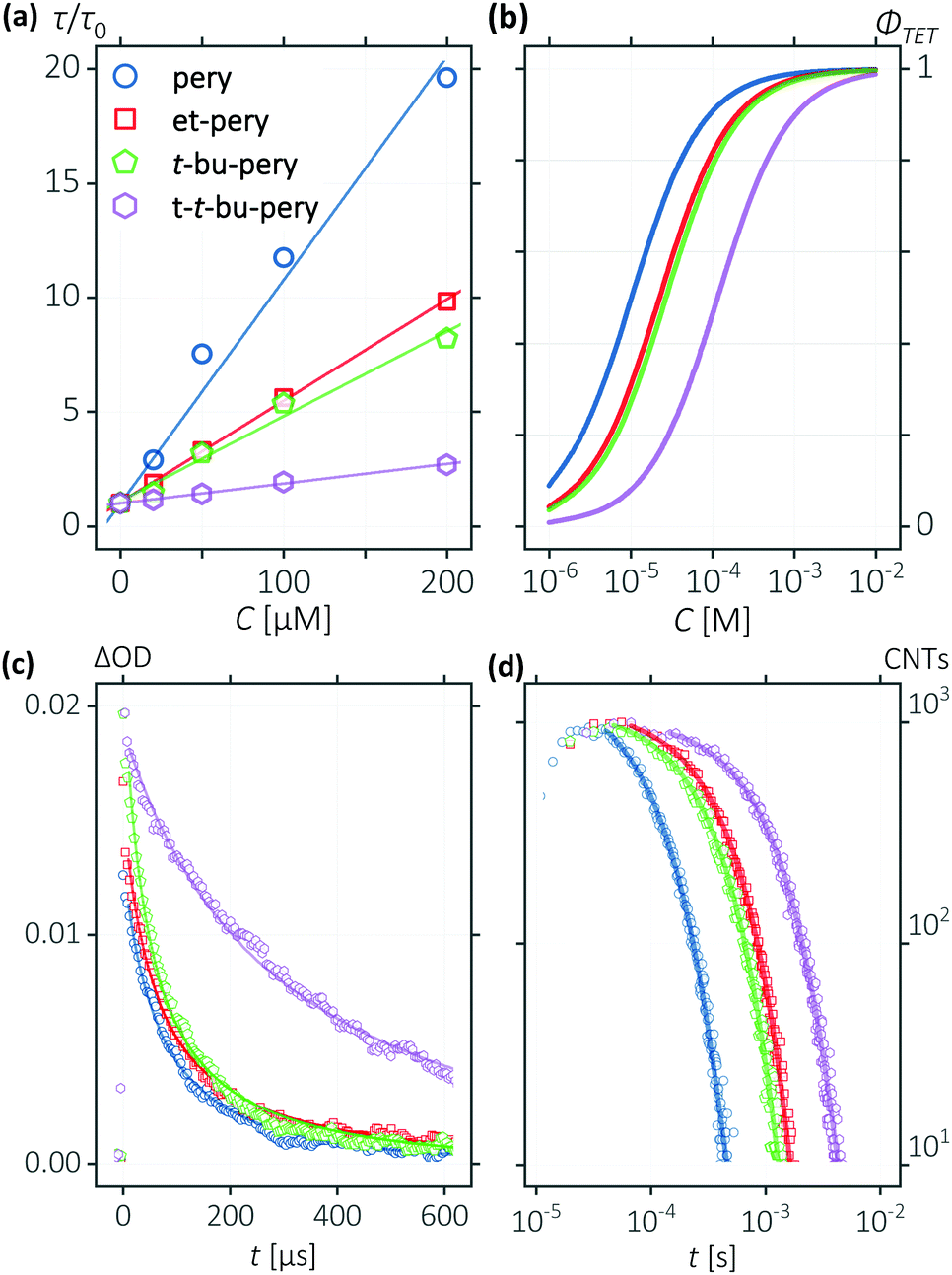
While alkyl substitution prevents excimer formation, it also has drawbacks. Increasing the molecular size by alkylation reduces the molecular mobility and thus the bimolecular reaction rates of triplet energy transfer32 and TTA. The TET rate constant can be determined by examining the quenching efficiency of the sensitizer phosphorescence by the annihilator. The relationship between the quenching effect and quencher concentration follows the Stern–Volmer equation (eqn (1) and (2)).54| | 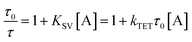 | (1) |
| | 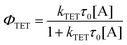 | (2) |
In eqn (1) and (2), τ0 is the phosphorescence lifetime of the sensitizer in absence of an annihilator, τ is the lifetime of the sensitizer in presence of an annihilator, KSV is the Stern–Volmer constant, kTET is the TET rate constant, and [A] is the concentration of the annihilator. Fig. 3a displays the sensitizer quenching as the concentration of annihilator is increased. With increasing alkyl substitution, the rate of TET is decreasing. It is instructive to compare the TET quantum yield (ΦTET) as a function of annihilator concentration calculated by eqn (2) (Fig. 3b). To reach a TET quantum efficiency of 0.99, the concentration of pery, et-pery, t-bu-pery, and t-t-bu-pery should be 1.0, 2.2, 2.6 and 11.5 mM, respectively. The relatively low TET efficiency of t-t-bu-pery thus requires highly concentrated conditions to quench the triplet sensitizer effectively. This effect is directly related to the bigger molecular size and the steric hindrance caused by the substitution. Therefore, as small substitution as possible is preferable in order to maintain a high TET efficiency.
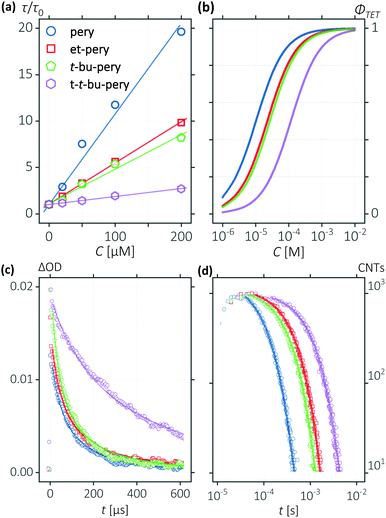 |
| | Fig. 3 (a) Stern–Volmer quenching relationship of PtTBTP sensitizer and perylene derivatives; (b) simulated triplet energy transfer efficiencies; (c) time resolved transient absorption decay at the of annihilator T1 → Tn transition peaks of 10 μM PtTBTP and 1 mM perylene/perylene derivatives in THF when excited at 617 nm with pulse energies between 0.30 and 0.37 mJ; (d) time-resolved delayed fluorescence decay at 470 nm of 10 μM PtTBTP and 1 mM perylene/perylene derivatives in THF when excited at 617 nm with pulse energies of 4 μJ. | |
Alkyl substitution will not only affect the rate of TET, also the rate of TTA will be affected. The generation of annihilators in the excited triplet state is on the timescale of a few hundereds of nanoseconds to a few microseconds, whereas consumption is on the microsecond timescale. We can then describe the kinetics of TTA-UC by the following equations (eqn (3) and (4)), which assume that the annihilators are in the triplet excited state to start with:
| | 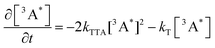 | (3) |
| | 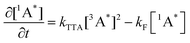 | (4) |
where [
3A*] and [
1A*] are the concentrations of annihilator in the excited triplet and singlet states, respectively,
t is the time after excitation,
kTTA is the TTA rate constant,
kT is the intrinsic decay rate constant of the excited triplet annihilator, and
kF is the fluorescence decay rate constant of the excited singlet annihilator.
55,56
The kinetics of the triplet excited annihilator was measured by transient absorption. The triplet states were monitored by their T1 to Tn transitions (Fig. S4, ESI†). Fig. 3c displays the time resolved transient absorption decays of the T1 to Tn transition of the annihilators, and Fig. 3d displays the time resolved upconverted emission of the annihilators. The bimolecular TTA rate constants were obtained by fitting these decay curves using eqn (3) and (4).55,57 TTA can only occur within the closely bound triplet pair, which is formed when two annihilators in their excited triplet state meet.58–61 The effective interaction radius follows the Einstein–Smoluchowski relation (Note S1.7, ESI†), and can be calculated from eqn (5).62,63
where
D is the molecular diffusion coefficient (eqn (S3), ESI
†), and
N is the Avogadro constant,
RTTA is the effective TTA reaction radius. A large molecular size due to alkyl substitution reduces the mobility of annihilator molecules and also restricts the formation of an effective triplet pair by steric hindrance.
Table 1 lists the calculated TTA-UC parameters of all four annihilators. The TTA rate constants and effective radii decrease with size of the alkyl substituents. Substitution with big alkyl groups is then unfavourable for TTA kinetics for the above two reasons.
Table 1 Dynamic parameters of perylene and perylene derivatives for TTA-UC, observed and calculated from Fig. 3 and Fig. S4 (ESI)
| Molecule |
K
SV/M−1 |
k
TET/M−1 s−1 |
λ (T1–Tn)/nm |
ε (T1–Tn)/M−1 cm−1 |
k
TTA/M−1 s−1 |
k
T/s−1 |
D/m2 s−1 |
R
TTA/Å |
|
pery
|
9.06 × 104 |
2.77 × 109 |
485 |
13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
8.40 × 109 |
3.55 × 103 |
1.72 × 10−9 |
6.62 |
|
et-pery
|
4.43 × 104 |
1.26 × 109 |
497 |
11369 |
5.58 × 109 |
1.35 × 103 |
1.56 × 10−9 |
4.57 |
|
t-bu-pery
|
3.70 × 104 |
1.05 × 109 |
501 |
11995 |
5.82 × 109 |
1.79 × 103 |
1.44 × 10−9 |
5.17 |
|
t-t-bu-pery
|
8.61 × 103 |
2.44 × 108 |
486 |
14939 |
1.15 × 109 |
5.42 × 102 |
1.02 × 10−9 |
1.52 |
Given the negative effect of large steric substituents on the rate of TTA, the TTA efficiency can be compensated by the reduced intrinsic decay of the triplet annihilator. In a typical TTA-UC process, TTA is competing with the intrinsic decay of triplet annihilators, and the TTA quantum yield (ΦTTA) follows eqn (6)
| | 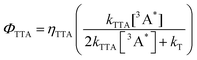 | (6) |
where
ηTTA is the possibility of TTA forming a singlet excited state according to spin statistics. Since the maximum upconversion quantum yields are at the same level, we assume the same spin statistical parameters for the four annihilators. The quantum yield of TTA is then determined by the competition between TTA and the intrinsic decay of excited triplet annihilators. Previous reports have pointed out that the radiative decay rate constant of perylene is less than 100 s
−1.
64 However, perylene has a high inclination to aggregate and decay non-radiatively, which increases the effective
kT.
65 We noticed that the intrinsic rate of decay decreases in the alkylated derivatives as compared to perylene, leading to an improved performance of alkylated perylene as TTA-UC annihilators.
Conclusions
Here, we have examined the TTA-UC performance of perylene and alkyl-perylene derivatives. The comparison revealed that mono-substitution with small alkyl group is sufficient in order to prevent excimer emission, while maintaining high performance. Furthermore, we demonstrate computationally how alkyl substitution prevents excimer formation on both singlet and triplet surfaces. Considering that the excess steric effect reduces the TTA-UC efficiency by restricting triplet–triplet energy transfer, a proper level of steric hindrance benefits TTA-UC systems. Furthermore, from the efficiency of annihilation it is also clear that triplet pair formation is much less sensitive to geometrical constraints than excimer formation. This comparative study thus leads to design rules for TTA-UC systems with high efficiency and contributes to the development of solar energy conversion.
Author contributions
C. Y. and K. B. conceived the project and wrote the manuscript. C. Y. carried out all the photon upconversion experiments and spectroscopy measurements. K. K. synthesized the four annihilator molecules. C. Y. and V. G. carried out all the analysis for spectroscopy. S. K. S. and P. E. carried out the quantum chemistry calculation of the energetics of annihilator dimers. All authors joined discussion and contributed to improvement of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge financial support from the Swedish Research council (2016-03354), the European Research council (ERC-2017-StG-757733), and the Knut and Alice Wallenberg Foundation. VG acknowledges funding from the Swedish research council, Vetenskapsrådet 2018-00238. C. Y. and K. B. gratefully acknowledge Dr Christoph Kerzig for proof reading and useful discussion.
References
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC
 .
.
- M. J. Tayebjee, D. R. McCamey and T. W. Schmidt, J. Phys. Chem. Lett., 2015, 6, 2367–2378 CrossRef CAS PubMed .
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC .
- L. Frazer, J. K. Gallaher and T. W. Schmidt, ACS Energy Lett., 2017, 2, 1346–1354 CrossRef CAS .
- Y. Zeng, J. Chen, T. Yu, G. Yang and Y. Li, ACS Energy Lett., 2017, 2, 357–363 CrossRef CAS .
- T. Dilbeck and K. Hanson, J. Phys. Chem. Lett., 2018, 9, 5810–5821 CrossRef CAS PubMed .
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS .
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953–6959 RSC .
- V. Jankus, E. W. Snedden, D. W. Bright, V. L. Whittle, J. A. G. Williams and A. Monkman, Adv. Funct. Mater., 2013, 23, 384–393 CrossRef CAS .
- A. Monguzzi, D. Braga, M. Gandini, V. C. Holmberg, D. K. Kim, A. Sahu, D. J. Norris and F. Meinardi, Nano Lett., 2014, 14, 6644–6650 CrossRef CAS PubMed .
- A. Monguzzi, S. M. Borisov, J. Pedrini, I. Klimant, M. Salvalaggio, P. Biagini, F. Melchiorre, C. Lelii and F. Meinardi, Adv. Funct. Mater., 2015, 25, 5617–5624 CrossRef CAS .
- S. P. Hill, T. Dilbeck, E. Baduell and K. Hanson, ACS Energy Lett., 2016, 1, 3–8 CrossRef CAS .
- S. P. Hill and K. Hanson, J. Am. Chem. Soc., 2017, 139, 10988–10991 CrossRef CAS PubMed .
- B. Shan, T.-T. Li, M. K. Brennaman, A. Nayak, L. Wu and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 463–471 CrossRef CAS PubMed .
- K. Börjesson, D. Dzebo, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. A, 2013, 1, 8521–8524 RSC .
- C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC .
- D. Choi, S. K. Nam, K. Kim and J. H. Moon, Angew. Chem., Int. Ed., 2019, 58, 6891–6895 CrossRef CAS PubMed .
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed .
- Y.-H. Chen, C.-C. Lin, M.-J. Huang, K. Hung, Y.-C. Wu, W.-C. Lin, R.-W. Chen-Cheng, H.-W. Lin and C.-H. Cheng, Chem. Sci., 2016, 7, 4044–4051 RSC .
- K. Börjesson, P. Rudquist, V. Gray and K. Moth-Poulsen, Nat. Commun., 2016, 7, 12689 CrossRef PubMed .
- J. Han, P. Duan, X. Li and M. Liu, J. Am. Chem. Soc., 2017, 139, 9783–9786 CrossRef CAS PubMed .
- R. Vadrucci, A. Monguzzi, F. Saenz, B. D. Wilts, Y. C. Simon and C. Weder, Adv. Mater., 2017, 29, 1702992 CrossRef PubMed .
- D. Yang, J. Han, M. Liu and P. Duan, Adv. Mater., 2018, 0, 1805683 Search PubMed .
- X. Yang, J. Han, Y. Wang and P. Duan, Chem. Sci., 2019, 10, 172–178 RSC .
- Q. Liu, T. Yang, W. Feng and F. Li, J. Am. Chem. Soc., 2012, 134, 5390–5397 CrossRef CAS PubMed .
- Q. Liu, W. Feng, T. Yang, T. Yi and F. Li, Nat. Protoc., 2013, 8, 2033 CrossRef CAS PubMed .
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 135, 5029–5037 CrossRef CAS PubMed .
- O. S. Kwon, H. S. Song, J. Conde, H.-i. Kim, N. Artzi and J.-H. Kim, ACS Nano, 2016, 10, 1512–1521 CrossRef CAS PubMed .
- S. Mattiello, A. Monguzzi, J. Pedrini, M. Sassi, C. Villa, Y. Torrente, R. Marotta, F. Meinardi and L. Beverina, Adv. Funct. Mater., 2016, 26, 8447–8454 CrossRef CAS .
- J. Park, M. Xu, F. Li and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 5493–5499 CrossRef CAS PubMed .
- D. Yildiz, C. Baumann, A. Mikosch, A. J. C. Kuehne, A. Herrmann and R. Gostl, Angew. Chem., Int. Ed., 2019, 58, 12919–12923 CrossRef CAS PubMed .
- M. Kitazawa, T. Yabe, Y. Hirata and T. Okada, J. Mol. Liq., 1995, 65–66, 321–324 CrossRef .
- R. Casillas, M. Adam, P. B. Coto, A. R. Waterloo, J. Zirzlmeier, S. R. Reddy, F. Hampel, R. McDonald, R. R. Tykwinski, M. Thoss and D. M. Guldi, Adv. Energy Mater., 2019, 9, 1802221 CrossRef .
- J. H. Schön, C. Kloc and B. Batlogg, Appl. Phys. Lett., 2000, 77, 3776–3778 CrossRef .
- J.-H. Kim, F. Deng, F. N. Castellano and J.-H. Kim, ACS Photonics, 2014, 1, 382–388 CrossRef CAS .
- C. Li, C. Koenigsmann, F. Deng, A. Hagstrom, C. A. Schmuttenmaer and J.-H. Kim, ACS Photonics, 2016, 3, 784–790 CrossRef CAS .
- S. Hoseinkhani, R. Tubino, F. Meinardi and A. Monguzzi, Phys. Chem. Chem. Phys., 2015, 17, 4020–4024 RSC .
- A. J. Musser, S. K. Rajendran, K. Georgiou, L. Gai, R. T. Grant, Z. Shen, M. Cavazzini, A. Ruseckas, G. A. Turnbull, I. D. W. Samuel, J. Clark and D. G. Lidzey, J. Mater. Chem. C, 2017, 5, 8380–8389 RSC .
- C. B. Dover, J. K. Gallaher, L. Frazer, P. C. Tapping, A. J. Petty, M. J. Crossley, J. E. Anthony, T. W. Kee and T. W. Schmidt, Nat. Chem., 2018, 10, 305–310 CrossRef CAS PubMed .
- C. Ye, V. Gray, J. Mårtensson and K. Börjesson, J. Am. Chem. Soc., 2019, 141, 9578–9584 CrossRef CAS PubMed .
- J. Shi and C. W. Tang, Appl. Phys. Lett., 2002, 80, 3201–3203 CrossRef CAS .
- B. K. Shah, D. C. Neckers, J. Shi, E. W. Forsythe and D. Morton, J. Phys. Chem. A, 2005, 109, 7677–7681 CrossRef CAS PubMed .
- V. Gray, A. Dreos, P. Erhart, B. Albinsson, K. Moth-Poulsen and M. Abrahamsson, Phys. Chem. Chem. Phys., 2017, 19, 10931–10939 RSC .
- K. Kushwaha, L. Yu, K. Stranius, S. K. Singh, S. Hultmark, M. N. Iqbal, L. Eriksson, E. Johnston, P. Erhart, C. Muller and K. Börjesson, Adv. Sci., 2019, 6, 1801650 CrossRef .
- J. Yang, A. Dass, A.-M. M. Rawashdeh, C. Sotiriou-Leventis, M. J. Panzner, D. S. Tyson, J. D. Kinder and N. Leventis, Chem. Mater., 2004, 16, 3457–3468 CrossRef CAS .
- A. Monguzzi and J. Pedrini, J. Photonics Energy, 2017, 8, 022005 Search PubMed .
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC .
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. Lett., 2010, 1, 195–200 CrossRef CAS .
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS .
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2005, 123, 124107 CrossRef .
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed .
- B. Karpichev, L. Koziol, K. Diri, H. Reisler and A. I. Krylov, J. Chem. Phys., 2010, 132, 114308 CrossRef CAS PubMed .
- O. Bludský, M. Rubeš, P. Soldán and P. Nachtigall, J. Chem. Phys., 2008, 128, 114102 CrossRef PubMed .
- X. Liu and J. Qiu, Chem. Soc. Rev., 2015, 44, 8714–8746 RSC .
- C. Bohne, E. B. Abuin and J. C. Scaiano, J. Am. Chem. Soc., 1990, 112, 4226–4231 CrossRef CAS .
- T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062–4072 CrossRef CAS PubMed .
- J. Saltiel, G. R. March, W. K. Smothers, S. A. Stout and J. L. Charlton, J. Am. Chem. Soc., 1981, 103, 7159–7164 CrossRef CAS .
- S. Baluschev, V. Yakutkin, G. Wegner, B. Minch, T. Miteva, G. Nelles and A. Yasuda, J. Appl. Phys., 2007, 101, 023101 CrossRef .
- T. L. Keevers and D. R. McCamey, Phys. Rev. B, 2016, 93, 045210 CrossRef .
- B. S. Basel, J. Zirzlmeier, C. Hetzer, S. R. Reddy, B. T. Phelan, M. D. Krzyaniak, M. K. Volland, P. B. Coto, R. M. Young, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Chem, 2018, 4, 1092–1111 CAS .
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X. Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed .
- J. Keizer, Chem. Rev., 1987, 87, 167–180 CrossRef CAS .
- B. Nickel, P. Borowicz, A. A. Ruth and J. Troe, Phys. Chem. Chem. Phys., 2004, 006, 3350–3363 RSC .
- P. J. Walla, F. Jelezko, P. Tamarat, B. Lounis and M. Orrit, Chem. Phys., 1998, 233, 117–125 CrossRef CAS .
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp06561j |
|
| This journal is © the Owner Societies 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  bc,
Khushbu
Kushwaha
a,
Sandeep
Kumar Singh
d,
Paul
Erhart
d and
Karl
Börjesson
bc,
Khushbu
Kushwaha
a,
Sandeep
Kumar Singh
d,
Paul
Erhart
d and
Karl
Börjesson