
Surface grafted agents with various molecular lengths and photochemically active benzophenone moieties†
Gaby
Nordendorf
 a,
Samuel L.
Schafforz
a,
Eireen B.
Käkel
b,
Shunyi
Miao
a and
Alexander
Lorenz
*a
a,
Samuel L.
Schafforz
a,
Eireen B.
Käkel
b,
Shunyi
Miao
a and
Alexander
Lorenz
*a
aDepartment of Chemistry, Paderborn University, Warburger Str. 100, 33098 Paderborn, Germany. E-mail: alexander.lorenz@uni-paderborn.de
bInstitute of Chemistry, University of Kassel, Heinrich-Plett-Str. 40, 34132 Kassel, Germany
First published on 11th December 2019
Abstract
Homologues of benzophenone silane, a covalently graftable, photochemically active surface functionalizing agent, are investigated as surface functionalization agents for both small particles and planar substrates. In these homologues, a chlorosilane functional group and a photochemically active benzophenone oxo moiety are separated with an aliphatic spacer of varying length. The species obtained are first investigated by surface grafting on substrates (Si wafers, glass plates, and indium tin oxide coated glass plates). Si wafer samples are investigated with ellipsometry clearly indicating monolayer formation. The monolayer thickness can be controlled by the size of the aliphatic spacer group and also by the doping concentration of the solution used in the spin-casting step. The functionalized surfaces are further investigated by measuring the contact angle of a suitable organic fluid, a nematic liquid crystal. Photo exposure of these samples results in a drastically varied contact angle: The surface grafted species are still photochemically active and photo exposure leads to the addition of a nearby organic molecule (from the liquid crystalline phase) to each activated surface agent molecule. The synthesized species are then investigated as (covalently binding) surfactants in the wet planetary ball milling process aimed to fabricate solid–liquid dispersions (of Fe doped lithium niobate particles). It was found that the use of species with higher molecular length results in dispersions of small particles, functionalized with photochemically active surface agents. Indeed, they show better performance than conventional surfactants.
1. Introduction
Benzophenone silane, BPS, is a surface graftable crosslinker with one chlorosilane anchoring group and one photochemically active benzophenone oxo moiety. In BPS, these two functional moieties are separated by an aliphatic spacer (oxoalkyl-spacer) with a length of 3 carbon atoms. Via the chlorosilane anchoring group, BPS can easily attach to a hydroxyl-group-bearing surface in a condensation reaction. Benzophenone silane has been widely used to photochemically immobilize thin films of functional materials,1–21 in biologically relevant research,22–28 or to obtain fast switching polymer liquid crystal (LC) composites.29,30 Various types of planar substrates can be coated in a straight-forward spin casting approach by using mixtures of BPS and toluene. At the low concentrations usually applied for spin casting, one can probably assume that these mixtures are perfect solutions at least at the time when they are dropped on the spinning substrates. However, the solvent evaporates during spin casting and thermal annealing.Benzophenone is a well-known type II photoinitiator31–33 and frequently used with a tertiary amine as co-initiator. It can be applied as a photoinitiator even without the use of tertiary amines (added co-initiators), efficiently. The process of forming an active radical is a two-step process. First, benzophenone is excited. At a wavelength of ≈250 nm the excitation initially results in an exited singlet state followed by intersystem crossing to a triplet state (triplet benzophenone). At a slightly higher wavelength (≈350 nm), an nπ* transition (with lower absorption coefficients) occurs and triplet benzophenone can be obtained, directly. In the second step, triplet benzophenone undergoes hydrogen abstraction from an aliphatic chain of a nearby molecule, for example a macromolecule,34 which results in the formation of a BP-H˙ radical, which is usually not active towards carbon–carbon double bonds, and a second, active radical. However, the BP-H˙ radical is also well-known to initiate the formation of a new carbon–carbon covalent bond with nearby aliphatic moieties of neighboring molecules, where the carboxylic oxygen is simultaneously converted to an OH group. Thereby, neighboring molecules can be covalently attached to the former benzophenone group. The photochemical properties of surface grafted benzophenone silanes are very much comparable to those of benzophenone. BPS can be used as a photo crosslinker in experiments where surface hydroxyl groups1,13,24,30,35,36 or surfaces with surface-near hydroxyl groups16 (such as polymer films) are first grafted with BPS and then macromolecules get photochemically attached to these benzophenone silane bearing surfaces. Alternatively, benzophenone silanes can be used as surface grafted photoinitiators30,37 to initiate radical polymerization in reactive mixtures containing suitable monomers, such as acrylates. In both cases, the activated and/or growing macroradicals can recombine with the surface bound BP-H˙ moieties and form surface grafted macromolecules.
In the ball-milling based fabrication process of dispersions of inorganic particles such as BaTiO3 or Fe doped LiNbO3, surfactants play an important role. In order to obtain small (even in the nano-range) particles, these inorganic substances have been milled in the presence of heptane as an organic solvent and oleic acid as a surfactant.38–44 Using an organic solvent is desirable to make use of the dispersions obtained and dope target samples such as LCs, polymers or other samples with small, surface functionalized particles. An organic solvent can be removed from these samples easily (without varying the chemical composition of the doped substances). It has become clear that oleic acid can in some cases bind covalently42 to the particle surfaces during the milling process and result in small particles functionalized with a stable organic monolayer, which might be very important to obtain stable dispersions or nano-dispersions with the desired stability. The ball milling process is delicate and depends for example on the milling time, the type of balls used, and the milling energy. Anyway, alternative surface functionalizing agents should be investigated.
In this work, homologues (with an alkyloxo spacer length of n = 4–12 carbon atoms) of BPS were synthesized (the synthesis route is summarized in Scheme 1) by following the two-stepped procedure published for BPS24 and replacing 3-bromopropen with higher homologues of terminal bromoalkenes. In the first step, the intermediate products (n-ENE intermediates) were obtained and then converted to the final BPS homologues (n-BPS).
 | ||
| Scheme 1 Reactions for the two-step synthesis of the intermediate products n-ENE and the final products n-BPS (with n = m + 2 carbon atoms). | ||
For the surface functionalization of substrates, the BPS homologues obtained were dissolved in toluene. These solutions were spin cast on silicon wafers. The spin coated samples were thermally annealed and subsequently cleansed with an organic solvent. The surface functionalized, cleansed samples were investigated with ellipsometry to reveal thickness variations of the monolayers of BPS homologues. The results were very interesting and partially unexpected. The variations were caused by the varying spacer length of the BPS homologues and also by concentration variations of the solution used in the spin casting step. Contact angle variations were observed upon photo exposure: The species were indeed still photochemically active. The species were added as surface agents in the wet planetary ball milling process aimed to fabricate solid–liquid dispersions (of iron doped lithium niobate, Fe:LiNbO3) in heptane. It was found that species with higher molecular length were indeed suitable to yield the desired dispersions of particles bearing a photochemically active surface functionalization.
2. Results and discussion
The experimental results are presented in the following subsections along with a brief discussion of the experimental outcome.2.1 Synthesis results
BPS and its homologues (with spacer length between 4 and 12 carbon atoms) were successfully synthesized. The synthesis procedure turned out to be feasible for all investigated homologues, as expected. Anyway, the yields obtained varied. The yield for the intermediate n-ENE products (Table 1) was high (78%) for 3-ENE, and low for 4-ENE (34%) and 5-ENE (25%). The synthesis of 6- and 7-ENE led to yields higher than 65% and the yield then steadily decreased with the increasing length of the aliphatic chain of the terminal bromoalkene used.| Compound | Molar mass [g mol−1] | Melting point [°C] | Color | Mass [g] | Conversion [%] |
|---|---|---|---|---|---|
| 3-ENE | 238 | 76.9 | White | 3.65 | 78 |
| 4-ENE | 252 | Liquid | Clear | 1.70 | 34 |
| 5-ENE | 266 | 37.3 | White | 0.66 | 25 |
| 6-ENE | 280 | Liquid | Clear | 3.78 | 65 |
| 7-ENE | 294 | Liquid | Clear | 4.78 | 81 |
| 8-ENE | 308 | 34.4 | White | 3.15 | 51 |
| 9-ENE | 322 | 35.3 | White | 2.63 | 41 |
| 10-ENE | 336 | 44.7 | White | 2.81 | 42 |
| 11-ENE | 350 | 47.8 | White | 2.57 | 37 |
| 12-ENE | 364 | 53.5 | White | 0.35 | 25 |
In contrast, the hydrosilation step always resulted in high yields (Table 2): the average yield (not considering the outlier value of 10-BPS) was 76% with a standard deviation of 11%. The chain length of the aliphatic spacer had no influence on the conversion seen.
| Compound | Molar mass [g mol−1] | Melting point [°C] | Color | Mass [g] | Conversion [%] |
|---|---|---|---|---|---|
| 3-BPS | 332.6 | ≈33 | White | 3.31 | 67 |
| 4-BPS | 346.6 | Liquid | White | 0.88 | 76 |
| 5-BPS | 360.6 | 53 | White | 0.45 | 80 |
| 6-BPS | 374.6 | Liquid | Brown | 3.49 | 79 |
| 7-BPS | 388.6 | 42 | Brown | 1.29 | 65 |
| 8-BPS | 402.6 | Liquid | Brown | 3.35 | 93 |
| 9-BPS | 416.6 | 57.5 | White | 2.73 | 87 |
| 10-BPS | 430.6 | 42 | White | 0.70 | 20 |
| 11-BPS | 444.6 | 53 | White | 2.00 | 61 |
| 12-BPS | 458.6 | 46 | White | 0.31 | 74 |
2.2 Monolayer thickness
Initially, the length of n-BPS-O-SiH3 molecules in their all-trans conformation was measured in a ball-and-stick model in an optimized molecular geometry (UFF force field) in Avogadro.45 In this straight-forward approach, the Cl-atom of n-BPS was replaced by a O-SiH3 moiety, which probably was a preferable approximation for a surface-bound molecule (preferable to a Cl-ligand).The furthest distance between the Si-atom (silane group) and one selected hydrogen atom of the benzophenone group was measured as shown in Fig. 1 for 11-BPS. The obtained values for all investigated BPS homologues are listed in Table 3. The length obtained represents the maximum possible thickness of a n-BPS monolayer on a substrate surface in the case of perpendicular (brush-like) molecular alignment (when neglecting the covalent silicon–oxygen bonds connecting the Si-atom covalently to the substrate). In the approach presented, the part of the molecules actually contributing to the measurable layer thickness is accounted for. This thickness obtained is a feasible value, because the covalently bound surface-oxygen is part of a native SiO2 layer present at the substrate surfaces.
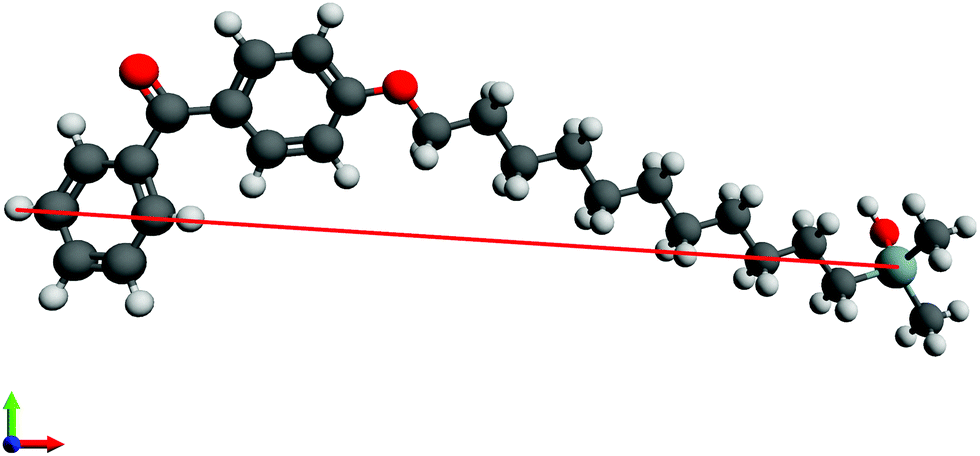 | ||
| Fig. 1 The ball-and-stick model of 11-BPS in all-trans conformation in an optimized geometry (Avogadro, UFF force field) is shown exemplarily. The red colored line indicates the molecular length measured (23.75 Å). Here, showing the crystal structure of the SiO2 surface (to which the BPS molecule is attached in a real sample) would reduce the clarity, and hence a covalently bound BPS-O-SiH3 is shown instead. | ||
| Compound | Molecular length [Å] |
|---|---|
| 3-BPS | 14.48 |
| 4-BPS | 15.06 |
| 5-BPS | 16.73 |
| 6-BPS | 17.40 |
| 7-BPS | 19.02 |
| 8-BPS | 19.81 |
| 9-BPS | 21.14 |
| 10-BPS | 22.23 |
| 11-BPS | 23.75 |
| 12-BPS | 24.66 |
The film thickness derived from ellipsometric measurements (colored symbols) along with the estimated maximum values for monolayers of surface grafted molecules (gray shaded squares) is shown in Fig. 2. A linear trend line can be used to describe the estimated molecular length in the all-trans conformation, as indicated in the figure. The slope of this trend line was ≈0.12 nm (slightly lower than a typical C–C bond length of 0.154 nm) per carbon atom n. For all samples, the experimentally measured film thickness was below the estimated maximal monolayer thickness, which clearly indicated the formation of monolayers, where the molecular conformation and/or orientation differed (with a few exceptions) from the all-trans conformation with perpendicular molecular alignment. In the samples obtained from solutions with an intermediate concentration, the film thickness initially (for a spacer length of 3, 5 or 6 carbon atoms) decreased with increasing spacer length. Looking at the higher (5 to 11 carbon atoms) spacer length, one can also see that the concentration now increased linearly with increasing spacer length. Again, the data were fitted with a linear trend line. The slope obtained was ≈0.11 nm per carbon atom n, and thus comparable to the slope seen in the estimated molecular length in the all-trans confirmation. However, the layer thickness was much (0.8 nm) smaller than would be expected for monolayers with all-trans conformation and perpendicular anchoring. Using a low concentration (Fig. 2, yellow diamonds) led to even smaller layer thickness and a smaller slope (0.055 nm per carbon atom) of the trend line. The layer thickness measured for the high concentration (taupe colored triangles) resulted in an almost constant value of ≈0.7 nm. A monolayer with perpendicular orientation of the surface grafted molecules was selectively found in 3-BPS (independent of the concentration) and 12-BPS (at a medium concentration). Monolayers were successfully formed in all samples prepared. The impact of the doping concentration on the monolayer thickness was, however, surprising. The samples were spin coated from a liquid phase, which possessed various n-BPS concentrations. In general, a high initial concentration led to a constant monolayer thickness. In the samples made with lower concentrations of the liquid phase, the monolayer thickness (determined by the molecular conformation of the grafted molecules and the grafting density) was varied. An alternative approach to coat surfaces with BPS is to immerse samples in a BPS solution,1,6,13 which can be heated or slowly evaporated during the grafting step without creating convection fluxes as during spin casting. If so, the grafting density can be even higher than in spin cast samples and the resulting layer thickness of monolayers can be affected. In the spin casting approach, two opposing processes took place simultaneously: The n-BPS concentration in the bulk of the liquid phase was lowered by the condensation reaction with surface OH-groups (reaction at the solid–liquid interface), while it was increased by evaporation of the solvent. In the spin casting approach, the concentration of n-BPS in the bulk of the liquid phase was simultaneously increased, since the solvent evaporated.
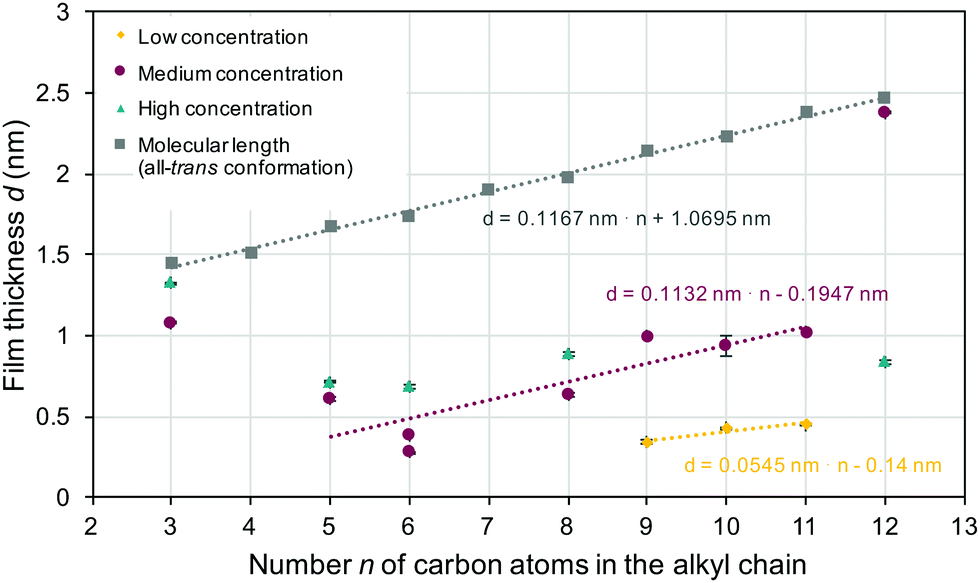 | ||
| Fig. 2 Film thickness derived by spectroscopic ellipsometry for surface grafted n-BPS monolayers obtained from coating solutions with various n-BPS concentrations. In addition, the estimated molecular length of the homologues in the all-trans conformation as the limiting thickness of a monolayer is shown. | ||
A non-polar solvent was used. Therefore, one can speculate that the dissolved n-BPS molecules (with polar end-groups) should possess a surface excess and were thus pushed to the surfaces upon increasing the overall concentration in the liquid phase. Maybe even lyotropic phases can be formed at higher (increasing with time) concentrations of n-BPS. Here, the grafting from such a lyotropic phase can be described with two processes: One possibility, which can lead to characteristic grafting kinetics, is that the concentration of the freely dissolved species is kept constant in the liquid phase, if a lyotropic phase (such as a micellar phase) was formed. Should the concentration decrease due to the grafting process, species will re-dissolve from the micelles present. Alternatively, the monolayer could be grafted through a process, where first micelles were adsorbed on the surfaces. It would be desirable to study the phase behavior of n-BPS species at various concentrations in scattering experiments. Such experiments turned out to be challenging in a preliminary approach, which might be caused by the non-typical structure of n-BPS with two (polar) functional groups, separated by an aliphatic spacer. Anyway, the monolayer thickness seen in the higher homologues (n > 9) (where formation of lyotropic phases seems more likely) was less concentration dependent, than in the smaller molecules. Understanding the phase behavior of n-BPS in more detail can potentially help to develop a detailed understanding of the grafting process of these versatile, surface graftable, photochemically active molecules.
2.3 Contact angle of E7
The contact angle of a suitable organic fluid, the nematic liquid crystal E7, on the treated substrates (references and spin coated samples) was investigated. The results obtained in the reference samples are summarized in Table 4. The cleansed surfaces were activated first by plasma treatment and then by UV exposure. In the non-treated substrates, small contact angles of 6.5° (Si-wafers), 8.9° (glass plates), and 10.4° (ITO glass plates) were found. After plasma treatment, these angles were varied to smaller values of 2.1° (Si-wafers), 5° (glass plates), and 2.7° (ITO glass plates), which can be explained by the increased surface polarity (activated surfaces, presence of surface OH groups). UV treatment of cleaned samples resulted in contact angles of 10.3° (Si-wafers), 11° (glass plates), and 11.2° (ITO glass plates). These changes were small. Anyway, UV exposure of previously plasma treated samples led to a slight increase to 5.8° (Si-wafers), 3.5° (glass plates), and 4.6° (ITO glass plates), which were intermediate values smaller than in the bare samples but higher than in the plasma treated samples. UV exposure in the bare samples or in the presence of E7 led to a slight deactivation of the surfaces as compared to the plasma treated samples.| Substrate | Plasma treatment | UV exposure | Contact angle [°] |
|---|---|---|---|
| Si wafer | — | — | 6.5 ± 1.3 |
| Post | — | 2.1 ± 0.3 | |
| — | Post | 10.3 ± 0.5 | |
| Post | Post | 5.8 ± 0.2 | |
| Glass | — | — | 8.9 ± 0.9 |
| Post | — | 5.0 ± 0.0 | |
| — | Post | 11.0 ± 0.0 | |
| Post | Post | 3.5 ± 0.4 | |
| ITO coated glass | — | — | 10.4 ± 1.3 |
| Post | — | 2.7 ± 0.6 | |
| — | Post | 11.2 ± 1.0 | |
| Post | Post | 4.6 ± 1.7 | |
In surface grafted samples (9-BPS) (Table 5), which were plasma treated prior to the grafting step, the contact angles were drastically increased to ≈20° (Si-wafers) and >13° (glass plates). For ITO coated glasses, such an increase was selectively seen, if methyl cyclohexane was used as a solvent. If the surface grafted samples were exposed to UV light, the contact angle decreased, as expected, since the surfaces were now functionalized with photochemically attached LC molecules. This decrease (from 22.6° to 14°; Δ = 8.6°) was most pronounced in Si-wafer samples grafted by using 2-butanone 9-BPS solutions and ITO glasses treated with methyl cyclohexane (from 15.6° to 10.6°; Δ = 5°). Example images recorded with a suitable camera attached to the contact angle meter are shown (Fig. 3). The effect was less pronounced (i.e. <2°) in the alternative samples. One can speculate that the UV intensity at the surfaces was maybe too small (exposure through the LC layer) in the latter experiments.
| Substrate | Solvent | UV exposure | Contact angle [°] |
|---|---|---|---|
| Si wafer | 2-Butanone | — | 22.6 ± 2.5 |
| Post | 14.0 ± 0.8 | ||
| Methyl cyclohexane | — | 19.9 ± 0.8 | |
| Post | 19.5 ± 0.4 | ||
| Glass | 2-Butanone | — | 13.0 ± 2.7 |
| Post | 12.1 ± 0.9 | ||
| Methyl cyclohexane | — | 19.1 ± 2.7 | |
| Post | 20.4 ± 0.8 | ||
| ITO coated glass | 2-Butanone | — | 5.0 ± 0.0 |
| Post | 5.7 ± 1.1 | ||
| Methyl cyclohexane | — | 15.6 ± 1.7 | |
| Post | 10.6 ± 1.7 | ||
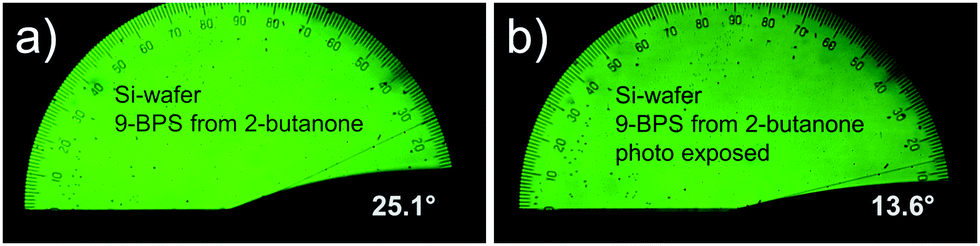 | ||
| Fig. 3 Contact angle of E7 investigated on Si wafer samples. (a) Coated with 9-BPS and (b) after UV exposure. | ||
2.4 DLS on functionalized particles
The light scattering data obtained in samples milled with added 7-BPS are shown in Fig. 4. Useful data could already be obtained after 16 h of grinding time. The curves shown had a polydispersity index <0.3 and showed a Gaussian-like distribution of particle sizes. At a grinding time of 18 h, the average particle diameter was ≈500 nm. The particles obtained were comparably well dispersed. For reference, also lecithin and oleic acid were used as surfactants. Milling in the presence of 7-BPS resulted in samples which could be easily investigated with DLS, whereas the reference samples were still contaminated with larger particles which corrupted the measurements. No data could be extracted from these measurements. It is probably possible to find small particles in such samples after elimination of large particles by filtration or centrifugation. However, the use of n-BPS homologues was preferable, since the well-known alternative surfactants cannot yield photochemically active particles.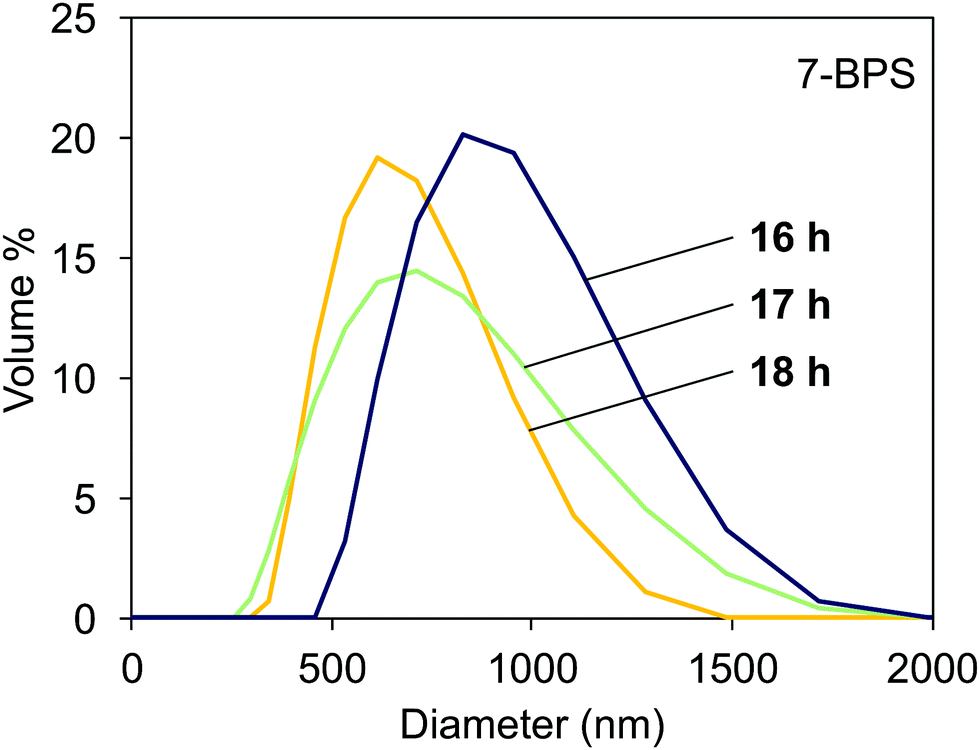 | ||
| Fig. 4 Particle distribution measured with DLS in dispersions of milled Fe:LiNbO3 particles, milled in the presence of 7-BPS for 16, 17, and 18 h. | ||
The DLS data measured in samples milled in the presence of 11-BPS after 20, 21, and 22 h of milling time had passed are shown in Fig. 5. The polydispersity index measured again was <0.3, which showed that samples were not monodisperse but DLS was still a suitable method to study their properties. After 20 h of milling time had passed, the particle size distribution curve was relatively narrow and had a maximum at ≈1000 nm. The particle size distribution became wider at higher milling times. High milling times were required and the samples initially contained two types of particles, smaller and larger particles. The contribution of large particles is not seen in the size distribution at 20 h, because these particles were still large enough to segregate at the bottom plane of the DLS cuvettes, where they could not contribute to the scattering. Due to further progress of the milling process, the particle size distribution became wider as seen in the curves recorded at 21 h and 22 h milling time. Now, also smaller particles were dispersed than in the samples taken at a milling time of 20 h. In addition, these dispersions also contained even larger particles than were seen at 20 h milling time. These particles could be formed by milling progress of larger particles still present in the grinding jars, but (as explained before) not contributing to the scattering in the DLS experiments conducted in samples taken at a milling time of 20 h. It is also possible that the large particle sizes seen in the scattering curves in samples at 21 h and 22 h were due to aggregation of smaller particles. The particle size distributions shown are promising, since they show how the milling progress resulted in dispersions of small, surface functionalized inorganic particles, which may find applications in doped polymer films and as dopants for soft phases, such as liquid crystals and lyotropic phases.
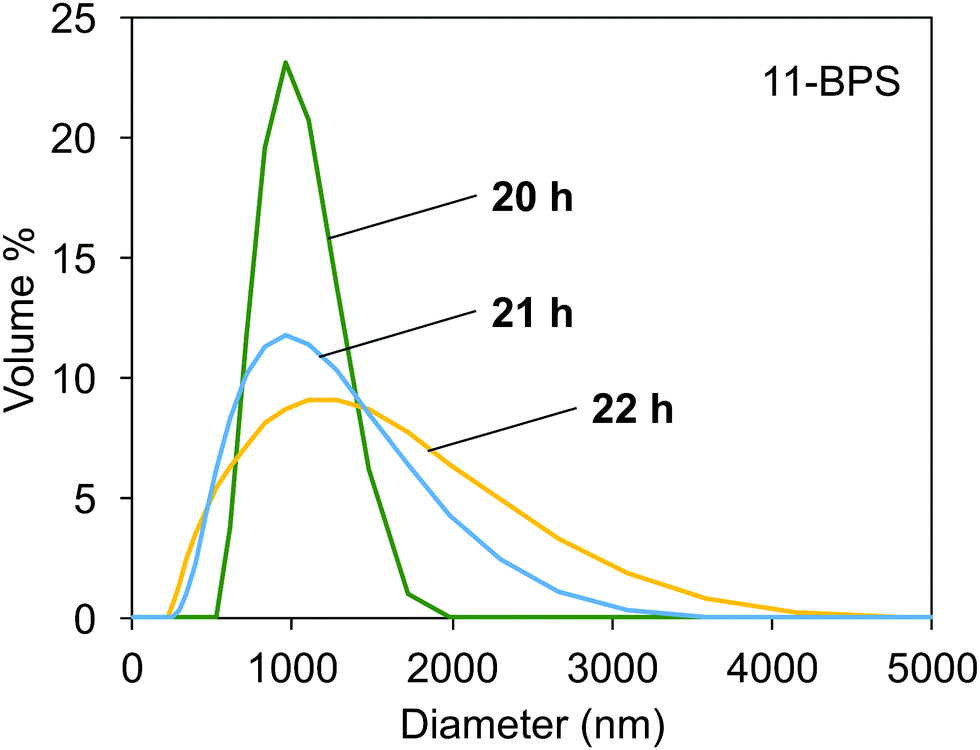 | ||
| Fig. 5 Particle distribution measured with DLS in dispersions of milled Fe:LiNbO3 particles, milled in the presence of 11-BPS for 20, 21, and 22 h. | ||
The milling experiments clearly showed that the presence of 7-BPS and 11-BPS led to the successful generation of dispersions of relatively small, milled particles. The milling process even worked more efficiently than in reference experiments with oleic acid and lecithin. The particles obtained in the presence of n-BPS can be expected to possess a shell, probably even a monolayer shell, of n-BPS attached to their polar Fe:LiNbO3 surface.
The surface free energy of particles can be reduced by the presence of a solvent (through wetting of the particle's surfaces) and (more efficiently) by the presence of a surfactant. The use of surfactants in the wet high energy planetary ball milling process (surfactant assisted high energy ball milling) has been comprehensively reviewed.46 A surfactant (for example the polar head group) can be physically adsorbed on the particle's surfaces and stabilize the internal interface of the solid–liquid dispersion through London-dispersion forces, residing between the aliphatic tail of the surfactant molecule and solvent molecules. The presence of a surface-active substance can prevent particle agglomeration or cold welding processes. Moreover, high energy ball milling is sometimes discussed as a means of mechanochemical synthesis:42,46,47 Oleic acid can in general be used as a surfactant. However, in milled dispersions of barium titanate particles it was found that oleic acid was transformed to a metal oleate compound (with ionic bonds between the particle surface and the surface covering layer). The presence of a surface grafted layer can stabilize a dispersion more efficiently than a physically adsorbed interfacial agent. The role of n-BPS is schematically shown and compared to milling with just a solvent in Fig. 6.
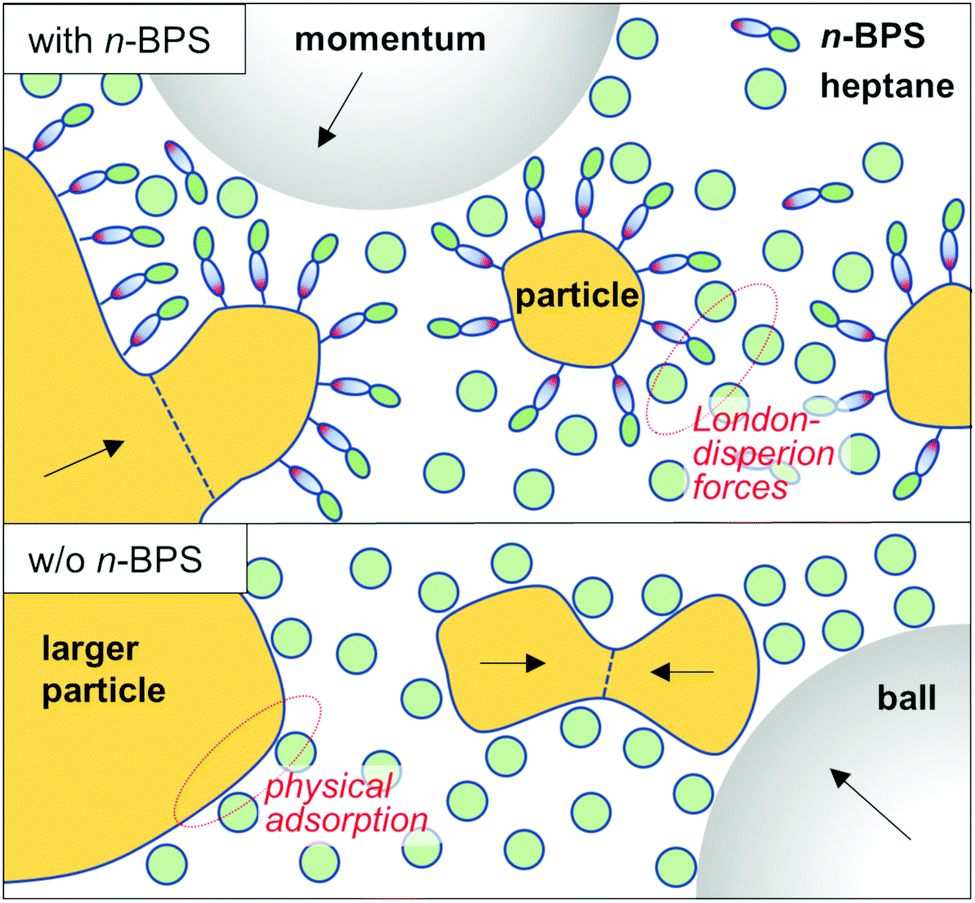 | ||
| Fig. 6 Schematic of the milling process in the presence of n-BPS and without n-BPS. In the first case, the surfaces of oxide particles are grafted with a monolayer of n-BPS. The surface free energy is thereby reduced. The solid–liquid dispersion is stabilized through London-dispersion forces (between the grafted molecules and the solvent). Without n-BPS, the surface free energy can also be lowered but now, selectively, by physisorption of solvent molecules. If so, agglomeration and even fusion of smaller particles cannot be efficiently prevented and the particles can easily segregate: The solid–liquid dispersion is not well stabilized. | ||
The n-BPS coated particles obtained are very promising to be incorporated in bulk materials such as polymer films (where the benzophenone group can attach to the polymer via the photo-induced formation of C–C-bonds, or particles with an attached photoinitiator). Such particles with an attached photoinitiator can be applied in grafting from experiments, where surface attached oligomers or polymers can be grown in from the particle surfaces. Here, the particles should be dispersed in a solution containing suitable monomers (such as acrylate monomers). Exposure to light will initiate a radical polymerization reaction where the growing chains are attached to the particle surfaces.
3. Experimental section
3.1 Synthesis
The two-stepped synthesis route for BPS has been well described previously.6,13 The description of the synthesis of one higher homologue (6-BPS) is given in the following for completeness. Comments on the appearance, melting point data (as seen in melting point tubes), the obtained masses, and conversion for both synthesis steps are summarized in Table 1 (intermediate products) and Table 2 (final products). The homologous series was synthesized by replacing allylbromide in the first synthesis step of BPS6,13 with linear terminal bromo-alkenes (homologues of allylbromide). Therefore, a higher homologue of the intermediate product was obtained in the Williamson ether synthesis step, which was subsequently hydrosilated (second step) with chlorodimethylsilane, to finally obtain BPS-homologues with oxoalkyl spacer length in the range of 4–12 carbon atoms (n-BPS). As an example, the synthesis of (4-(hex-5-en-1-yloxy)phenyl)(phenyl)methanone (6-ENE) and (4-((6-(chlorodimethylsilyl)hexyl)oxy)phenyl)(phenyl)methanone (6-BPS) is described below. The hydrosilation step was conducted in a dry argon atmosphere. All synthesis- and post-synthesis purification steps were carried out under yellow light conditions (in a yellow-lit lab and fume hood) since the benzophenone moiety was sensitive to UV-C radiation.1H NMR (500 MHz, CDCl3, δ in ppm): 1.60 (m, 2H, CH2–CH2), 1.92 (m, 2H, CH2–CH2), 2.15 (m, 2H, CH2–CH2), 4.05 (m, 2H, O–CH2), 4.98 (m, 1H, CH2 = CH2), 5.05 (m, 1H, CH2 = CH2), 5.83 (m, 1H, CH2 = CH2), 6.95–7.82 (various m, 9H, C–Harom.).
The NMR data of the other homologues are included in the NMR section of the ESI.†
1H NMR (500 MHz, CDCl3, δ in ppm): 0.01–0.09 and 0.41 (m, 6H, Si–CH3), 0.53 (m, 2H, Si-CH2), 1.30–1.52 (m, 6H, CH2–CH2–CH2), 1.82 (m, 2H, CH2–CH2–CH2), 4.04 (m, 2H, O–CH2), 6.94–7.81 (various m, 9H, C–Harom.).
The measured NMR data of the other homologues are included in the ESI.† These data showed that the final product was (at least in the CDCl3 solutions contained in the NMR tubes) partially converted: the chlorosilane functional group was converted to a silanol functional group (in the presence of residual water). Anyway, the results obtained in the following investigation steps clearly showed that the surface grafting step (condensation reaction with surface OH-groups) worked well and surface grafted monolayers were obtained from all homologues investigated.
3.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 min at 50 rps. Subsequently, the spin cast substrates were placed on a temperature controlled hot plate (55 °C) for 40 min in a fume hood to promote occurrence of the condensation reaction (surface attachment) and to remove the solvent. The substrates were allowed to cool down to room temperature again and any excess (non-grafted) BPS homologue was removed by rinsing with 350 μL of toluene four times. After every second rinse, the sample was dried (blowing ball) with air.
:15 min at 50 rps. Subsequently, the spin cast substrates were placed on a temperature controlled hot plate (55 °C) for 40 min in a fume hood to promote occurrence of the condensation reaction (surface attachment) and to remove the solvent. The substrates were allowed to cool down to room temperature again and any excess (non-grafted) BPS homologue was removed by rinsing with 350 μL of toluene four times. After every second rinse, the sample was dried (blowing ball) with air.
The concentration of the coating solutions was adjusted and three series of concentrations were used, as shown in Table 6.
| Compound | Low concentration [mol L−1] | Medium concentration [mol L−1] | High concentration [mol L−1] |
|---|---|---|---|
| 3-BPS | 0.00120 | 0.00240 | |
| 5-BPS | 0.00129 | 0.00258 | |
| 6-BPS | 0.00125 | 0.00250 | |
| 8-BPS | 0.00166 | 0.00332 | |
| 9-BPS | 0.00064 | 0.00128 | |
| 10-BPS | 0.00062 | 0.00124 | |
| 11-BPS | 0.00075 | 0.00150 | |
| 12-BPS | 0.00116 | 0.00232 |
Preparation of milled particle dispersions
Dispersions (in heptane) of iron doped lithium niobate particles were fabricated in a planetary ball mill (Retsch PM 200). First, pieces of an Fe:LiNbO3 crystal were manually crushed using an agate mortar and pestle until a powder was obtained. This powder (500 mg) was transferred to a ZrO2 grinding jar and 7-BPS (300 mg) or 11-BPS (300 mg) was added, selectively. Subsequently, each grinding jar was partially filled (1/3rd of the jar height) with small (1.5 mm) ZrO2 balls. These balls were preconditioned; they were previously milled in the presence of added heptane and the added heptane was regularly exchanged, and the balls were washed with fresh heptane. During the first repetitions, the fluid phase in the grinding jars had become turbid and was contaminated with abrasion of the balls. The milling process (the preconditioning) was continued until the heptane remained clear. The balls and the milling jars were then washed with filtered heptane. Once the preconditioned balls were added to the grinding jars, heptane (5 mL) was added through a syringe filter (PTFE 0.2 μm). The filter was used to avoid the presence of dust in the solvent. The grinding jars were placed in the planetary ball mill, fixed and the milling was conducted at 100 rpm. The grinding process was divided in 15 minute intervals, to avoid damage of the mill and continued for ≈20 h. Samples were taken during the milling process and stored in Eppendorf tubes.Conclusions
Homologues of n-BPS with n = 4–12 were investigated. The yield of the Williamson ether synthesis step was higher for intermediate spacer length (65%) and smaller for both small and larger spacer length. The yield of the hydrosilation step (76%) was relatively high and independent of the spacer length. Spin casting from solution followed by thermal annealing and rinsing was found to result in monolayer formation in all homologues as seen in the ellipsometric investigations of surface functionalized Si wafers.The monolayer thickness was found to depend on both the spacer length and the concentration of the solution used in the spin casting step. These results were instructive to learn about the typical thickness of the obtained monolayers. The use of diluted solutions of species with a short aliphatic spacer group will lead to small monolayer thickness (d < 0.7 nm). One can speculate whether the formation (at least during the annealing process on the spin cast substrates) of lyotropic phases in concentrated solutions of higher homologues can control the kinetics of the grafting process and thereby lead to constant monolayer thickness (apparently with a tilted molecular alignment on the surfaces).
The photochemistry of the benzophenone oxo functional group was tested by exposing surface functionalized samples covered with the LC E7 to UV light. These investigations revealed that the surface energy (contact angle of E7) was indeed varied by photo exposure. These results are promising also for liquid crystal related research because the pretilt angle49 of the LC director (which is important for the electro-optic performance) on a surface depends on the (now photo controllable) contact angle. Moreover, n-BPS functionalized surfaces can potentially be applied straight-forwardly (for example by spin casting of high molecular weight solvents followed by photo exposure, for example through a photo mask) to fabricate surface coatings with controllable wetting properties.
The synthesized species were used as surfactants in the wet ball milling process of dispersions of small inorganic particles. It turned out that higher homologues are capable of yielding dispersions of functionalized particles and they can outperform well-known surfactants already during the milling process. The particle dispersions obtained are highly useful, because their photochemically active surface functionalization can be addressed by photo-exposure in order to incorporate them in polymer films or in general in soft matter samples.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their thanks to Prof. Dr Klaus Huber and Ms Mahnaz Doostdar Kejdehi for fruitful discussions on the properties of solutions and their potential role in the grafting process of n-BPS homologues. In addition, the authors would also like to express their thanks to Pd. Dr Hans Egold and Ms Karin Stolte for the measurement of the NMR spectra and assistance with the analysis of the recorded spectra. Funding by the German Research Council (DFG LO 1922/4-1) is gratefully acknowledged.Notes and references
- G. K. Raghuraman, R. Dhamodharan, O. Prucker and J. Rühe, A Robust Method for the Immobilization of Polymer Molecules on SiO2 Surfaces, Macromolecules, 2008, 41, 873–878 CrossRef CAS.
- S. Hattori, K. Asakawa, S. Mikoshiba, H. Nakamura, A. Hieno, Y. Seino, M. Kanno and T. Azuma, Blockcopolymer Self-Assembly Directed by Photochemically Attached Polymer Surface Layer, J. Photopolym. Sci. Technol., 2011, 24, 581–585 CrossRef CAS.
- Y. Kim, A. Bulusu, A. J. Giordano, S. R. Marder, R. Dauskardt and S. Graham, Experimental Study of Interfacial Fracture Toughness in a SiNx/PMMA Barrier Film, ACS Appl. Mater. Interfaces, 2012, 4, 6711–6719 CrossRef CAS PubMed.
- K. M. Carroll, A. J. Giordano, D. Wang, V. K. Kodali, J. Scrimgeour, W. P. King, S. R. Marder, E. Riedo and J. E. Curtis, Fabricating Nanoscale Chemical Gradients with ThermoChemical NanoLithography, Langmuir, 2013, 29, 8675–8682 CrossRef CAS PubMed.
- Y. Kado, M. Mitsuishi and T. Miyashita, Fabrication of Three-Dimensional Nanostructures Using Reactive Polymer Nanosheets, Adv. Mater., 2005, 17, 1857–1861 CrossRef CAS.
- P. Aliuos, A. Sen, U. Reich, W. Dempwolf, A. Warnecke, C. Hadler, T. Lenarz, H. Menzel and G. Reuter, Inhibition of fibroblast adhesion by covalently immobilized protein repellent polymer coatings studied by single cell force spectroscopy, J. Biomed. Mater. Res., Part A, 2014, 102, 117–127 CrossRef PubMed.
- M. Gianneli, P. W. Beines, R. F. Roskamp, K. Koynov, G. Fytas and W. Knoll, Local and Global Dynamics of Transient Polymer Networks and Swollen Gels Anchored on Solid Surfaces, J. Phys. Chem. C, 2007, 111, 13205–13211 CrossRef CAS.
- C. Dorrer and J. Rühe, Micro to nano: surface size scale and superhydrophobicity, Beilstein J. Nanotechnol., 2011, 2, 327–332 CrossRef CAS PubMed.
- J. D. Jeyaprakash, S. Samuel and J. Ruehe, Microstructured fluorinated nanofilms, PMSE Prepr., 2004, 90, 290–291 CAS.
- M. Muramatsu, M. Iwashita, T. Kitano, T. Toshima, M. H. Somervell, Y. Seino, D. Kawamura, M. Kanno, K. Kobayashi and T. Azuma, Nanopatterning of diblock copolymer directed self-assembly lithography with wet development, J. Micro/Nanolithogr., MEMS, MOEMS, 2012, 11, 031305 CrossRef.
- A.-K. Schuler, O. Prucker and J. Rühe, On the Generation of Polyether-Based Coatings through Photoinduced C, H Insertion Crosslinking, Macromol. Chem. Phys., 2016, 217, 1457–1466 CrossRef CAS.
- K. Li, C. K. Pandiyarajan, O. Prucker and J. Rühe, On the Lubrication Mechanism of Surfaces Covered with Surface-Attached Hydrogels, Macromol. Chem. Phys., 2016, 217, 526–536 CrossRef CAS.
- O. Prucker, C. A. Naumann, J. Rühe, W. Knoll and C. W. Frank, Photochemical Attachment of Polymer Films to Solid Surfaces via Monolayers of Benzophenone Derivatives, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS.
- Y. Shiraishi and T. Hirai, Photochemical desulfurization and denitrogenation of light oil by using benzophenone-modified silica gel as a heterogeneous triplet photosensitizer, Solvent Extr. Res. Dev., Jpn., 2003, 10, 79–85 CAS.
- F. Hoffmann, T. Wolff, S. Minko and M. Stamm, Photochemical structuring and fixing of structures in binary polymer brush layers via 2π + 2π photodimerization, J. Colloid Interface Sci., 2005, 282, 349–358 CrossRef CAS PubMed.
- K. A. Ibrahim, A. H. Al-Muhtaseb, O. Prucker and J. Rühe, Preparation of hydrophilic polymeric nanolayers attached to solid surfaces via photochemical and ATRP techniques, J. Polym. Res., 2013, 20, 124 CrossRef.
- A. Hieno, S. Hattori, H. Nakamura, K. Asakawa, Y. Seino, M. Kanno and T. Azuma, Quick Formation of DSA Neutralization Polymer Layer Attached by Reactive Self-Assembled Monolayer, J. Photopolym. Sci. Technol., 2012, 25, 73–76 CrossRef CAS.
- R. Toomey and Juergen. Ruehe, Surface-attached polymer networks: synthesis and swelling behavior, PMSE Prepr., 2003, 89, 157–158 CAS.
- S. Lenz, S. K. Nett, M. Memesa, R. F. Roskamp, A. Timmann, S. V. Roth, R. Berger and J. S. Gutmann, Thermal Response of Surface Grafted Two-Dimensional Polystyrene (PS)/Polyvinylmethylether (PVME) Blend Films, Macromolecules, 2010, 43, 1108–1116 CrossRef CAS.
- J. Jarvholm, M. Srinivasarao and L. M. Tolbert, Traversing the “Top-Down/Bottom-Up” Divide: Molecular-Scale Lithography of Self-Assembled Ribbons, J. Am. Chem. Soc., 2009, 131, 398–399 CrossRef CAS PubMed.
- A. Alkan, C. Steinmetz, K. Landfester and F. R. Wurm, Triple-Stimuli-Responsive Ferrocene-Containing PEGs in Water and on the Surface, ACS Appl. Mater. Interfaces, 2015, 7, 26137–26144 CrossRef CAS PubMed.
- S. V. Orski, S. Kundu, R. Gross and K. L. Beers, Design and Implementation of Two-Dimensional Polymer Adsorption Models: Evaluating the Stability of Candida antarctica Lipase B/Solid-Support Interfaces by QCM-D, Biomacromolecules, 2013, 14, 377–386 CrossRef CAS PubMed.
- L. Y. Hwang, H. Götz, C. J. Hawker and C. W. Frank, Glyco-acrylate copolymers for bilayer tethering on benzophenone-modified substrates, Colloids Surf., B, 2007, 54, 127–135 CrossRef CAS PubMed.
- W. W. Shen, S. G. Boxer, W. Knoll and C. W. Frank, Polymer-Supported Lipid Bilayers on Benzophenone-Modified Substrates, Biomacromolecules, 2001, 2, 70–79 CrossRef CAS PubMed.
- L. Y. Hwang, H. Götz, W. Knoll, C. J. Hawker and C. W. Frank, Preparation and Characterization of Glycoacrylate-Based Polymer-Tethered Lipid Bilayers on Benzophenone-Modified Substrates, Langmuir, 2008, 24, 14088–14098 CrossRef CAS PubMed.
- S. Loschonsky, K. Shroff, A. Wörz, O. Prucker, J. Rühe and M. Biesalski, Surface-Attached PDMAA-GRGDSP Hybrid Polymer Monolayers that Promote the Adhesion of Living Cells, Biomacromolecules, 2008, 9, 543–552 CrossRef CAS PubMed.
- W. W. Shen, W. Knoll and C. W. Frank, Toward biomimetic lipid bilayers on solid substrates: design and characterization of lipopolymer support, Polym. Mater.: Sci. Eng., 2001, 84, 400–401 CAS.
- A. Woerz, B.-J. Chang, O. Prucker, M. Biesalski and J. Ruehe, Ultrathin polymer layers for the control of cell adhesion – from biocompatible surfaces to living cell chips, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2005, 46, 114–115 CAS.
- L. Braun, S. L. Schafforz and A. Lorenz, Surface grafted crosslinker in polymer network liquid crystals, J. Mol. Liq., 2018, 267, 109–114 CrossRef CAS.
- L. Braun, Z. Gong, A. Habibpourmoghadam, S. L. Schafforz, L. Wolfram and A. Lorenz, in Emerging Liquid Crystal Technologies XIII, International Society for Optics and Photonics, 2018, vol. 10555, p. 105550N Search PubMed.
- W. A. Green, Industrial photoinitiators: a technical guide, CRC Press, Boca Raton, 2010 Search PubMed.
- Radiation curing: coatings and printing inks; technical basics, applications and trouble shooting, ed. P. Glöckner, T. Jung, S. Struck and K. Studer, Vincentz Network, Hannover, 2008 Search PubMed.
- Radiation curing in polymer science and technology: photoinitiating systems, ed. J. P. Fouassier and J. F. Rabek, Elsevier Applied Science, London, 1993, vol. 2 Search PubMed.
- N. D. Carbone, M. Ene, J. R. Lancaster and J. T. Koberstein, Kinetics and Mechanisms of Radical-Based Branching/Cross-Linking Reactions in Preformed Polymers Induced by Benzophenone and Bis-Benzophenone Photoinitiators, Macromolecules, 2013, 46, 5434–5444 CrossRef CAS.
- R. Toomey, D. Freidank and J. Rühe, Swelling Behavior of Thin, Surface-Attached Polymer Networks, Macromolecules, 2004, 37, 882–887 CrossRef CAS.
- G. Morgese and E. M. Benetti, Polyoxazoline biointerfaces by surface grafting, Eur. Polym. J., 2017, 88, 470–485 CrossRef CAS.
- A. Lorenz, L. Braun, V. Kolosova, R. Hyman and T. D. Wilkinson, Continuous phase modulation in polymer-stabilized liquid crystals, in Emerging Liquid Crystal Technologies XI, ed. L.-C. Chien, D. J. Broer, H. Kikuchi and N. V. Tabiryan, Proc. SPIE 9769, 2016, p. 976912 Search PubMed.
- S. A. Basun, G. Cook, V. Yu. Reshetnyak, A. V. Glushchenko and D. R. Evans, Dipole moment and spontaneous polarization of ferroelectric nanoparticles in a nonpolar fluid suspension, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 024105 CrossRef.
- A. Lorenz, N. Zimmermann, S. Kumar, D. R. Evans, G. Cook, M. Fernández Martínez and H.-S. Kitzerow, Doping a Mixture of Two Smectogenic Liquid Crystals with Barium Titanate Nanoparticles, J. Phys. Chem. B, 2013, 117, 937–941 CrossRef CAS.
- A. Lorenz, N. Zimmermann, S. Kumar, D. R. Evans, G. Cook and H.-S. Kitzerow, Doping the nematic liquid crystal 5CB with milled BaTiO3 nanoparticles, Phys. Rev. E, 2012, 86, 051704 CrossRef PubMed.
- G. Cook, J. L. Barnes, S. A. Basun, D. R. Evans, R. F. Ziolo, A. Ponce, V. Yu. Reshetnyak, A. Glushchenko and P. P. Banerjee, Harvesting single ferroelectric domain stressed nanoparticles for optical and ferroic applications, J. Appl. Phys., 2010, 108, 064309 CrossRef.
- I. U. Idehenre, Y. A. Barnakov, S. A. Basun and D. R. Evans, Spectroscopic studies of the effects of mechanochemical synthesis on BaTiO3 nanocolloids prepared using high-energy ball-milling, J. Appl. Phys., 2018, 124, 165501 CrossRef.
- Y. A. Barnakov, I. U. Idehenre, S. A. Basun, T. A. Tyson and D. R. Evans, Uncovering the mystery of ferroelectricity in zero dimensional nanoparticles, Nanoscale Adv., 2019, 1, 664–670 RSC.
- A. Lorenz, N. Zimmermann, S. Kumar, D. R. Evans, G. Cook, M. F. Martínez and H.-S. Kitzerow, X-ray scattering of nematic liquid crystal nanodispersion with negative dielectric anisotropy [Invited], Appl. Opt., 2013, 52, E1–E5 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- M. Ullah, E. Ali and S. B. A. Hamid, Surfactant-Assisted Ball Milling: A Novel Route to Novel Materials with Controlled Nanostructure – A Review, Rev. Adv. Mater. Sci., 2014, 37, 1–14 CAS.
- C. Dhand, N. Dwivedi, X. Jun Loh, A. N. J. Ying, N. Kumar Verma, R. W. Beuerman, R. Lakshminarayanan and S. Ramakrishna, Methods and strategies for the synthesis of diverse nanoparticles and their applications: a comprehensive overview, RSC Adv., 2015, 5, 105003–105037 RSC.
- C. M. Herzinger, B. Johs, W. A. McGahan, J. A. Woollam and W. Paulson, Ellipsometric determination of optical constants for silicon and thermally grown silicon dioxide via a multi-sample, multi-wavelength, multi-angle investigation, J. Appl. Phys., 1998, 83, 3323–3336 CrossRef CAS.
- B.-Y. Liu, C.-H. Meng and L.-J. Chen, Role of Monomer Alkyl Chain Length in Pretilt Angle Control of Polymer-Stabilized Liquid Crystal Alignment System, J. Phys. Chem. C, 2017, 121, 21037–21044 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data for synthesized n-ENE and n-BPS compounds. See DOI: 10.1039/c9cp05722f |
| This journal is © the Owner Societies 2020 |