
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesorption products during linear heating of copper zeolites with pre-adsorbed methanol†
Xueting
Wang
 ab,
Adam A.
Arvidsson
bc,
Magnus
Skoglundh
ab,
Anders
Hellman
bc and
Per-Anders
Carlsson
*ab
ab,
Adam A.
Arvidsson
bc,
Magnus
Skoglundh
ab,
Anders
Hellman
bc and
Per-Anders
Carlsson
*ab
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden. E-mail: per-anders.carlsson@chalmers.se
bCompetence Centre for Catalysis, Chalmers University of Technology, 412 96 Gothenburg, Sweden
cDepartment of Physics, Chalmers University of Technology, 412 96 Gothenburg, Sweden
First published on 26th February 2020
Abstract
Desorption products from zeolites with medium (MFI) and small (CHA) pores and with and without ion-exchanged copper were studied during linear heating after the pre-adsorption of methanol using a chemical flow reactor with a gas phase Fourier transform infrared spectrometer. The methanol desorption profiles were deconvoluted and compared with those predicted from first-principles calculations. In situ diffuse reflectance infrared Fourier transform spectroscopy was used to study the samples during methanol desorption following a step-wise increase of the sample temperature. It is shown that well-dispersed copper species in the Cu-zeolite samples interact more strongly with methanol and its derivatives as compared to the bare zeolites, resulting in methanol desorption at higher temperatures. Moreover, the introduction of Cu leads to CO formation and desorption in larger amounts at lower temperatures compared to the bare zeolites. The formation and desorption of dimethyl ether (DME) from pre-adsorbed methanol takes place at different temperatures depending on both the influence of Cu and the zeolite topology. The Cu sites in zeolites lead to higher DME formation/desorption temperatures, while a small shift of DME desorption towards higher temperatures is observed for the CHA framework structure compared to the MFI framework structure.
1 Introduction
Zeolites have been used as catalysts in many applications due to their microporosity and surface acidity that can be utilised for controlling the product selectivity in acid catalysed reactions. Over the years, their hydrothermal stability has been improved and the pores and cages can accommodate various elements that function as active species, such as copper ions, in reactions.1–3 Despite their high framework crystallinity, mechanistic studies and modelling studies are challenging due to their complex framework structures and rich variety of their chemical composition and defect density. The protonic form of zeolites is active for many acid catalysed reactions such as methanol to hydrocarbons or olefins, where the Brønsted acid sites play important roles.4–6 Upon ion-exchange with copper, various Cu species, such as monomers, dimers and small clusters, are formed depending on the parent zeolite composition/structure and the ion-exchange level.7–9 The redox properties of these Cu species can help in the catalysis of selective oxidation reactions such as methane to methanol or acetic acid.10–12 When the Cu/Al ratio is below 0.5, both Brønsted acid sites and Cu sites are present in the Cu-zeolite, which may facilitate the interplay of these sites and contribute to more complex mechanisms for reactions such as NOx reduction13 and methane to methanol.14 Commonly, probe molecules such as NH3 combined with temperature-programmed desorption (TPD) are used for the identification of acid sites in zeolites,15,16 whereas molecules such as H2, N2, CO or suitable hydrocarbons are used to probe Cu sites in Cu-zeolites.17–19In this study we use methanol-TPD as a valuable method for the investigation of the dynamic interaction between methanol and zeolite materials.20–22 This can bring valuable insights into reactions such as methanol-to-hydrocarbons, where the selectivity for different products depends largely on the framework structure and acidity of the zeolites.2,6 Methanol is adsorbed and converted to surface methoxy species on the Brønsted acid sites, which initiate further reactions towards hydrocarbons.23–25 In this respect, the characterization of zeolites using methanol as a probe molecule can reveal the impact of topology and acidity on important elementary steps and chemical reactions.
Zeolites with medium and small pore sizes, such as ZSM-5 with MFI and SSZ-13 with CHA framework structures, are commonly used as catalysts. The MFI framework structure includes channels whereas the CHA framework consists of cages.26 The shape and pore dimensions steer the product selectivity in the methanol-to-hydrocarbon process2,6,27 and can possibly host different copper species active for the conversion of methane to methanol.3,11,28,29 In this study, we focus on two Cu-zeolite samples with the MFI or CHA framework structure (Cu-ZSM-5 and Cu-SSZ-13), respectively. The typical Cu species present in Cu-ZSM-5 and Cu-SSZ-13 samples have been studied in the previous literature7,29–33 and can give guidance on the Cu speciation of our samples. According to these studies the Cu-ZSM-5 sample with a medium Cu/Al ratio (0.38) and a relatively low Si/Al ratio (13.5) should contain a large proportion of Cu-oxo species such as dimers and trimers.7,30,31 However, for the Cu-SSZ-13 sample with both low Cu/Al (0.13) and Si/Al (10) ratios, Cu monomers located in the six-membered rings with two Al are common, while Cu dimers are not commonly observed.29,31–33
This study speciates the desorption products during linear heating of zeolites and Cu-zeolites (with MFI and CHA framework structures) after methanol exposure, with SiO2 and Cu/SiO2 as references. The methanol desorption profiles have been deconvoluted and compared with those predicted from first-principles calculations. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) has been used to study the surface species on the samples during methanol desorption following a step-wise increase of the temperature. It is shown that copper species in the zeolites interacts more strongly with methanol and its derivatives than the zeolitic Brønsted acid sites. Moreover, the formation and desorption of DME from pre-adsorbed methanol occur at higher temperatures for the samples with Cu modification. Furthermore, the zeolite topology has a minor influence on the DME desorption temperature, which is slightly higher for the CHA framework structure than for the MFI framework structure.
2 Methods
2.1 Sample preparation
The preparation of the Cu-ZSM-5, Cu-SSZ-13 and Cu/SiO2 samples has been described in detail previously.22,34 Briefly, the Cu-ZSM-5 and Cu-SSZ-13 samples were prepared using aqueous ion-exchange in which each parent zeolite (H-ZSM-5, Si/Al = 13.5; or H-SSZ-13, Si/Al = 10) was mixed with an aqueous solution of Cu(NO3)2 (Sigma-Aldrich, ACS reagent, 0.1 M, 100 ml g−1 zeolite) for 24 h. The suspension was filtered and dried at 120 °C overnight to give the powder sample. The Cu/SiO2 sample was prepared using incipient wetness impregnation where an aqueous solution of Cu(NO3)2·5H2O (Sigma-Aldrich, ACS reagent, 0.29 M, 1.66 ml g−1 silica) was slowly added to the silica (Akzo Nobel, Kromasil, 200 Å, 5 μm). The mixture was then freeze-dried over night and calcined in air at 350 °C for 3 h.The basic characteristics of the powder samples have been reported in our previous study.34 Shortly, the X-ray diffraction (XRD) results show that the zeolite framework structures of the Cu-zeolite samples are well-preserved during the Cu ion-exchange process. The Cu species in the Cu-ZSM-5 and Cu-SSZ-13 samples are mainly isolated Cu ions and clusters according to ex situ X-ray absorption spectroscopy analysis, whereas the Cu/SiO2 sample (predominantly) consists of CuO nanoparticles with an average size of 28.3 nm as calculated from the XRD pattern. The copper loading measured with an inductively coupled plasma-sector field mass spectrometer (ICP-SFMS) is 2.8 and 1.3 wt% for the Cu-ZSM-5 and Cu-SSZ-13 samples, respectively. The calculated Cu/Al ratio is 0.38 and 0.13 for the Cu-ZSM-5 and Cu-SSZ-13, respectively. The Cu loading for the Cu/silica sample was calculated to be 3.0 wt%.
Monolith samples for the flow reactor study were prepared by wash-coating each powder sample on cordierite monolith substrates (Corning, 400 cpsi, L = 15 mm, and Ø = 13 mm). The wash-coating procedure is described elsewhere.34 Approximately 0.16 g of each powder sample was coated on the monoliths using water dispersible boehmites (Sasol, Disperal P2) as the binder.
2.2 Desorption experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1) with Ar as the balancing gas during the pre-treatment and methanol exposure. Subsequently, the samples were stabilized under a 500 ml min−1 Ar flow (GHSV = 11300 h−1) at 80 °C (10 min). The desorption part of the experiment was then carried out under a 500 ml min−1 flow of Ar with a temperature ramp of 5 °C min−1.
000 cm−1, a Praying Mantis™ diffuse reflectance accessory and a high-temperature stainless steel reaction chamber (Harrick Scientific Products Inc.). The spectra were recorded between 900 and 4000 cm−1 and the spectral resolution is 1 cm−1. The instrumental aperture was 4 mm wide during measurements. A sieved fraction between 38 and 75 μm of the samples was used and about 85 μl of the sample was loaded in the sample cup.
000 h−1) with Ar as the balancing gas during the pre-treatment and methanol exposure. Subsequently, the samples were stabilized under a 500 ml min−1 Ar flow (GHSV = 11300 h−1) at 80 °C (10 min). The desorption part of the experiment was then carried out under a 500 ml min−1 flow of Ar with a temperature ramp of 5 °C min−1.
000 cm−1, a Praying Mantis™ diffuse reflectance accessory and a high-temperature stainless steel reaction chamber (Harrick Scientific Products Inc.). The spectra were recorded between 900 and 4000 cm−1 and the spectral resolution is 1 cm−1. The instrumental aperture was 4 mm wide during measurements. A sieved fraction between 38 and 75 μm of the samples was used and about 85 μl of the sample was loaded in the sample cup.
Methanol desorption experiments were carried out for the Cu-ZSM-5, H-ZSM-5, Cu-SSZ-13, H-SSZ-13, Cu/SiO2 and SiO2 samples. After pre-treatment of the samples with O2 at 550 °C for one hour, a few droplets of methanol (99.8%, Sigma-Aldrich) were added to the samples at 30 °C. The following desorption experiments were carried out under a 100 ml min−1 flow of pure Ar with a stepwise temperature increase from 30 °C to 450 °C. The backgrounds were recorded in pure Ar at 30 °C before methanol adsorption.
2.3 Theoretical methods
Theoretical calculations were performed with the Vienna ab initio simulation package (VASP) software36,37 using the spin-polarized version of the Perdew–Burke–Ernzerhof (PBE)38 exchange–correlation functional together with D3 van der Waals correction.39 Only the Γ-point was used to sample the Brillouin zone. The energy cutoff was 400 eV and Gaussian smearing with a value of 0.05 eV was used. The convergence criterion between two subsequent steps in the self-consistent loop was 10−8 eV.The geometry relaxation was considered to be converged when the maximum force was less than 0.03 eV Å−1. Vibrational frequencies were calculated within the harmonic limit, using central differences to obtain the Hessian matrix with a displacement of 0.01 Å. Structural relaxations and vibrational frequencies were calculated using the Atomic Simulation Environment (ASE)40 and VASP.
Cu monomer and dimer sites were modelled in the MFI (ZSM-5) and CHA (SSZ-13) frameworks (see Fig. S9 and S10, ESI†). In MFI, the Cu monomer sites were modelled over the Al T12 site5 (Fig. S9a and b, ESI†), and the dimer was modelled over the [Cu–O–Cu]2 + site41,42 (Fig. S9d, ESI†). It is a known problem that semilocal exchange–correlation functionals delocalize the open spin states over the copper atoms in the system.43 However, this only has a minor effect on methanol adsorption energies.44 The silanol was modelled over the monomer silicon T-site from a dissociated water molecule (Figs. S9c and S10c, ESI†), forming two hydroxyl species with a calculated vibration frequency of around 3770 cm−1, which agrees well with the experimental results.22
 | (1) |
In the mean-field assumption, the change of coverage of methanol, θ, is
 | (2) |
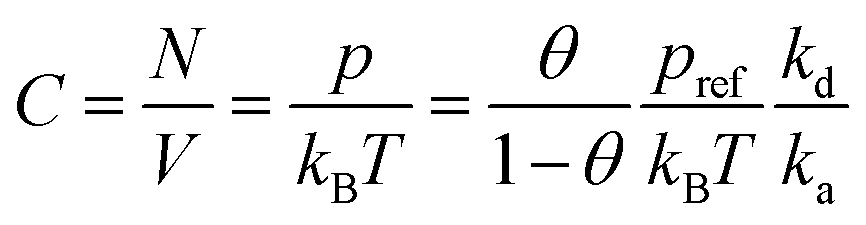 | (3) |
 | (4) |
Using these two expressions for the concentration (3) and (4), the following relationship is obtained:
 | (5) |
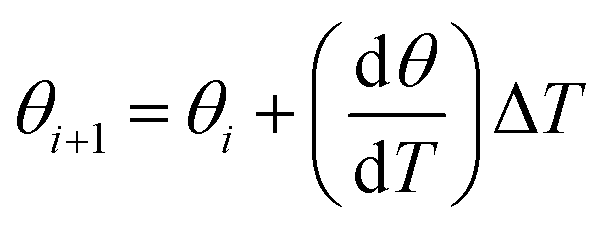 | (6) |
3 Results and discussion
The major gas phase products (methanol, DME and CO) formed during linear heating of all methanol exposed samples are shown in Fig. 1. In all cases, methanol desorbs mainly in the lower temperature range (below 250 °C), followed by DME formation at intermediate temperatures (130 to 350 °C), and CO formation solely at higher temperatures (above 250 °C). As shown in Fig. 1a, the desorption experiment for the Cu-ZSM-5 and Cu-SSZ-13 samples results in a larger amount of released methanol and also a broader desorption profile as compared to the parent H-ZSM-5 and H-SSZ-13 samples, respectively. Both observations indicate that the Cu ions/clusters in the Cu-ZSM-5 and Cu-SSZ-13 samples interact more strongly with methanol as compared to the sites in the parent zeolites. The desorption profiles for the Cu/SiO2 and SiO2 samples, however, exhibit considerably lower intensity with a desorption maximum at around 130 °C. The methanol desorbed from the SiO2 sample is presumably attributed to the methanol pre-adsorbed on the silanol groups.47 The slightly lower intensity and broader shape of the methanol desorption profile of the Cu/SiO2 sample compared to that of the SiO2 sample are likely due to CuO nanoparticles in the Cu/SiO2 sample covering silanol sites responsible for methanol adsorption and creating new adsorption sites on the CuO nanoparticles. The small difference, however, between the methanol desorption profile for the Cu/SiO2 and SiO2 samples indicates that the adsorption of methanol is only affected to a minor extent by the presence of CuO, which is in contrast with the Cu ions/clusters in the Cu-zeolite samples.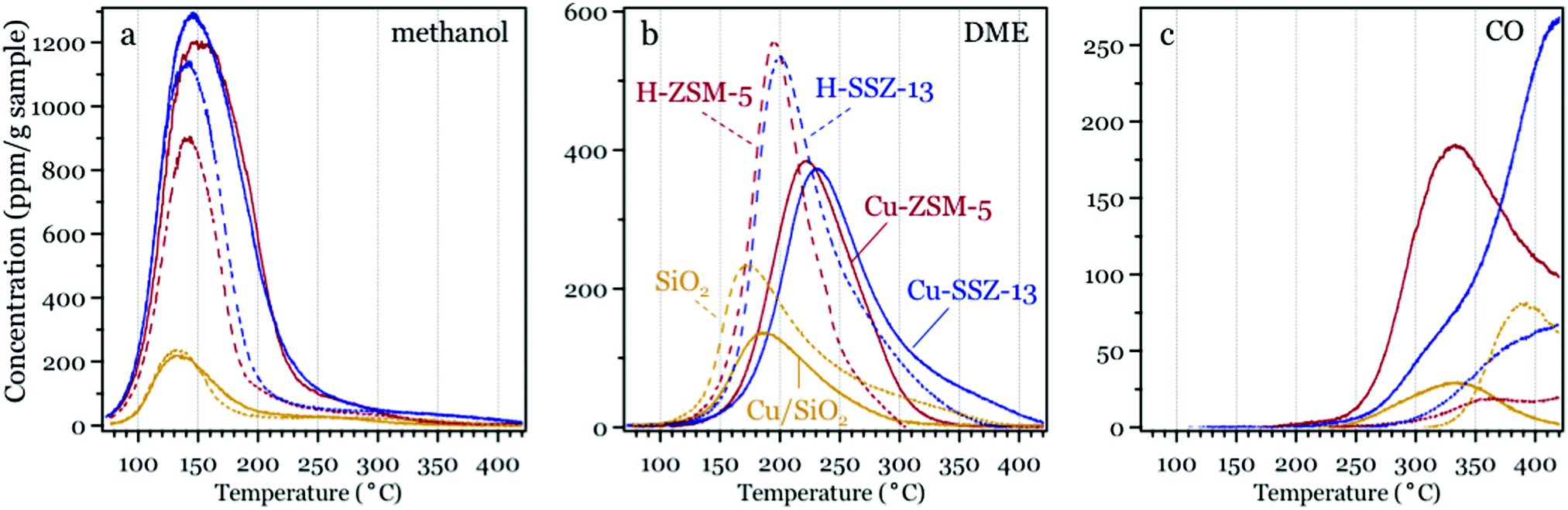 | ||
| Fig. 1 Desorption products during linear heating of oxidized Cu-ZSM-5 (red solid line), H-ZSM-5 (red dashed line), Cu-SSZ-13 (blue solid line), H-SSZ-13 (blue dashed line), Cu/SiO2 (yellow solid line), and SiO2 (yellow dashed line) samples with pre-adsorbed methanol. Panel (a) methanol, (b) DME and (c) CO. | ||
During linear heating of the samples, DME is formed due to methanol dehydration over the acidic sites on the samples.4,48 The DME desorption profiles of all samples are presented in Fig. 1b. It is evident that the Cu-exchanged zeolites release lower amounts of DME at temperatures lower than 220 °C and higher amounts of DME at temperatures exceeding 220 °C compared to the parent zeolites. The lower amount of DME desorption at lower temperatures for the Cu-ZSM-5 and Cu-SSZ-13 samples compared to the H-ZSM-5 and H-SSZ-13 samples might be due to the ion-exchange of Cu ions at the Brønsted acid sites and at framework defects in the Cu-ZSM-5 and Cu-SSZ-13 samples during the ion-exchange, resulting in blocking of acidic sites for low temperature DME formation. The DME desorption at higher temperatures (above 220 °C) for the Cu-zeolites is presumably a result from the stronger interaction between methoxy/DME and the Cu species in these samples compared to the zeolite samples. Moreover, DME desorbs at slightly lower temperatures from the samples with the MFI type of framework structure, i.e. the H-ZSM-5 and Cu-ZSM-5 samples, compared to the corresponding samples with the CHA type of framework structure, i.e. the H-SSZ-13 and Cu-SSZ-13 samples, respectively. This difference is presumably due to the different framework structures. It is anticipated that DME diffusion is faster in the MFI framework structure where the channels are easily accessed through 10-membered rings, while slower in the CHA framework structure where diffusion through 8-membered rings is limiting. Additionally, a considerable amount of DME forms from pre-adsorbed methanol at temperatures above 150 °C over the Cu-ZSM-5 and Cu-SSZ-13 samples, which suggests that low temperatures (<200 °C) are necessary for selectivity towards methanol when Cu-zeolites are used as catalysts for partial oxidation of methane to methanol. For the Cu/SiO2 and SiO2 samples, lower DME desorption with desorption maximum at lower temperatures is observed compared to all zeolite samples. The lower amount of DME formed over the Cu/SiO2 sample compared to that of the SiO2 sample is likely due to the CuO nanoparticles covering some silanol groups on the silica surface that are responsible for DME formation. Moreover, as the Cu/SiO2 sample contains CuO nanoparticles dominantly while the Cu-zeolite samples consist of mostly Cu ions/clusters, the difference in DME desorption temperatures for the Cu/SiO2 and the Cu-zeolite samples indicates that isolated Cu ions/clusters are responsible for the strong interaction with methanol and its derivative species rather than the CuO particles.
At temperatures higher than 250 °C, CO starts to desorb from the Cu containing samples while the CO desorption starts at a higher temperature (around 300 °C) for the zeolite and silica samples. The amount of CO desorbed from the Cu-zeolite samples is higher than that of the zeolite samples. The higher amount of CO formed over the Cu-zeolite samples is likely due to the ability of the Cu ions/clusters to oxidize the adsorbed methanol/methoxy species in the Cu-zeolite samples, which is in agreement with our previous study of methanol desorption using in situ IR spectroscopy.22 Moreover, a large amount of CO desorbs at much lower temperatures for the Cu-ZSM-5 sample than the Cu-SSZ-13 sample with around 100 °C difference for the desorption maximum, while the amount of CO2 formed and desorbed is much higher for the Cu-ZSM-5 sample compared to the Cu-SSZ-13 sample (Fig. S1b and S2b, ESI†). It is anticipated that such difference in the CO/CO2 profile is a result of the difference in proportion of various Cu species in these two samples.
Interestingly, the oxidizing or reducing pre-treatment condition affects the desorption profile of the Cu containing samples (Fig. S1, S2, S3, ESI† panels a and b) but not the Cu free samples (Fig. S1, S2 and S3, ESI† panels c and d), which indicates that some Cu species, depending on the pre-treatment environment, are prone to changes. For both Cu-ZSM-5 and Cu-SSZ-13 samples, a higher amount of methanol, DME and formaldehyde are desorbed when the samples are pre-oxidized. The CO and CO2 desorption profiles, however, are affected by pre-treatment conditions more significantly for the Cu-ZM-5 sample than the Cu-SSZ-13 sample. As discussed previously, the Cu-ZSM-5 sample should contain higher amounts of small Cu clusters such as dimers and trimers while the Cu-SSZ-13 sample contains Cu monomers dominantly. The difference in CO and CO2 desorption profiles for the Cu-ZSM-5 could be due to the changes of these small Cu clusters which are insufficient in the Cu-SSZ-13 sample.
The surface species formed during methanol desorption were followed by DRIFTS and selected regions of the spectra are presented in Fig. 2. The presented spectra were recorded at low temperatures (Fig. 2a) where the methanol desorption profile peaks and medium temperatures (Fig. 2b) where the DME desorption profile peaks and high temperatures (Fig. 2c) where CO desorption profile peaks for each sample (Fig. 1a–c), respectively. As shown in Fig. 2a, a double band at 2957 and 2855 cm−1, originated from adsorbed methoxy groups,22,49–51 is evident for all samples, indicating that methanol adsorbs and dissociates over all samples. Moreover, the negative bands at 3747, 3667 and 3614 cm−1 for the Cu-SSZ-13, H-SSZ-13, Cu-ZSM-5 and H-ZSM-5 samples are associated with the O–H stretching vibrations in isolated silanol, isolated hydroxyl on extra framework Al and Brønsted acid sites,4,22,49–51 which are isolated vibrational structures disappearing due to their interaction with adsorbed methanol. For the Cu/SiO2 and SiO2 samples, however, only a negative band at 3747 cm−1 exists due to methanol interactions with isolated silanol groups. The lack of other negative bands coincides with the absence of Al in these two samples. At medium temperatures (Fig. 2b), absorption features originated from DME, i.e. the bands at 3011 and 2834 cm−1,4 are visible for the zeolite and Cu-zeolite samples. The absorption bands associated with methoxy species, however, are prominent for all six samples. As minor amounts of methanol desorb for each sample in the medium temperature range (Fig. 1a), we speculate that the surface methoxy species on all six samples readily form DME in the medium temperature range that desorb into the gas phase. Moreover, the absorption bands associated with CO on Cu+ (2157 cm−1)52 are evident for the Cu-ZSM-5 and Cu-SSZ-13 samples, suggesting that the methoxy species is further oxidized to CO on the Cu sites at medium temperatures. At high temperatures, as shown in Fig. 2c, absorption features in the C–H stretching vibration range of 2800–3000 cm−1 are evident but lower in intensity for all six samples. This indicates that surface methanol, methoxy and DME are desorbed to a large extent at such high temperatures. The remaining absorption features at this region, however, suggests that some methoxy species are strongly bonded to the surface and are probably the carbon source for CO formation. For the Cu-ZSM-5 and the Cu-SSZ-13 samples, Cu bonded CO is evident at high temperatures, suggesting that Cu species are responsible for the large amount of CO desorbed in Fig. 1c.
 | ||
| Fig. 2 IR spectra of selected wavenumber regions collected during methanol desorption at low temperatures (a), medium temperatures (b) and high temperatures (c) for the pre-oxidized Cu/SiO2 (yellow), SiO2 (light yellow), Cu-ZSM-5 (red), H-ZSM-5 (light red), Cu-SSZ-13 (blue) and H-SSZ-13 (light blue) sample. | ||
3.1 Deconvolution of the methanol desorption profiles
Deconvolution of the methanol profiles from the H-ZSM-5, Cu-ZSM-5, H-SSZ-13 and Cu-SSZ-13 samples is shown in Fig. 3. Four Gaussian functions are fitted to the methanol desorption profiles for both the H-ZSM-5 and H-SSZ-13 samples. For the Cu-ZSM-5 and Cu-SSZ-13 samples, three additional Gaussian functions (blue lines) are fitted to the methanol desorption profiles based on the four Gaussian functions from the H-ZSM-5 and H-SSZ-13 samples. For this, the three Gaussian functions centered at 133, 200 and 300 °C identical to the parent zeolites are used in the Cu-zeolites, respectively, while the Gaussian functions centered at 153 or 163 °C for the H-ZSM-5 or H-SSZ-13 sample are applied to the corresponding Cu-zeolite samples with reduced amplitude according to the ratio of Brønsted acid site concentrations in the Cu-zeolites and their parent zeolites, see the ESI,† Table S1. For the deconvolution, the least number of Gaussian curves are used to achieve a reasonable fitting. The origin of these fitted functions is related to certain adsorption sites or desorption mechanisms which will be explained later in the Discussion section. Fig. 4 presents the theoretical methanol desorption profile from various Cu and zeolite sites in the Cu-ZSM-5 and Cu-SSZ-13 samples. Compared to the experiments, only the Brønsted acid site, the silanol site, and the Cu–OH–Cu desorb at relevant temperatures. The CuOCu, CuO, and CuOH sites bind methanol too weakly and desorb at too low temperatures, while the Cu monomer and Cu–Cu dimer binds methanol too strongly. From the calculations, one could be tempted to draw the conclusion that only the Cu–OH–Cu dimer is the relevant adsorption site with copper, since the bare copper species (Cu and Cu–Cu) bind too strongly and are relevant where DME and CO have formed at high temperatures. Although appealing, one should be careful about drawing such bold conclusions from these results since this is a relatively simple model only including desorption and adsorption. Furthermore, we performed a sensitivity analysis wherein we changed the binding energy of methanol ±0.1 eV (see ESI,† Fig. S9). This energy is much smaller than the uncertainty attributed to e.g. different exchange–correlation functionals.53 This introduced change in binding energy shifts the desorption curves around ±35 °C. This demonstrates that there is some uncertainty as to the exact position of the desorption peaks. Due to the higher Al content in Cu-SSZ-13, there are likely to be Cu monomers with two Al in the same six-membered ring, forming CuII species. This introduces a shift in methanol binding energies and can make these relevant in the temperature window for methanol formation, see Fig. S12 (ESI†). What we can see from the calculations is that there is a trend in bond strength and the bonding configuration between the different sites. The strongest methanol bond is to the bare Cu sites, both monomers and dimers, where methanol binds oxygen to the copper(s) forming a strong Cu–O bond or bonds. The second strongest methanol bond is to the OH sites where methanol binds oxygen forming a hydrogen bond situation with an intermediate water-like structure. This is the case as well for the Cu monomer and dimer as for the Brønsted and silanol sites. The weakest methanol bond is to the CuO sites where methanol binds hydrogen forming a hydrogen bond but without the intermediate water-like structure. This is sketched in Fig. 5. | ||
| Fig. 3 Deconvolution of methanol signals during methanol desorption from the pre-oxidized (a) H-ZSM-5, (b) Cu-ZSM-5, (c) H-SSZ-13 and (d) Cu-SSZ-13 samples. For the Cu-ZSM-5 and Cu-SSZ-13 samples, extra deconvoluted peaks (blue) were added based on the deconvolution of the H-ZSM-5 and H-SSZ-13 samples. The sum of the deconvoluted peaks is shown as red dashed lines and the center of the deconvoluted peaks is marked with numbers. | ||
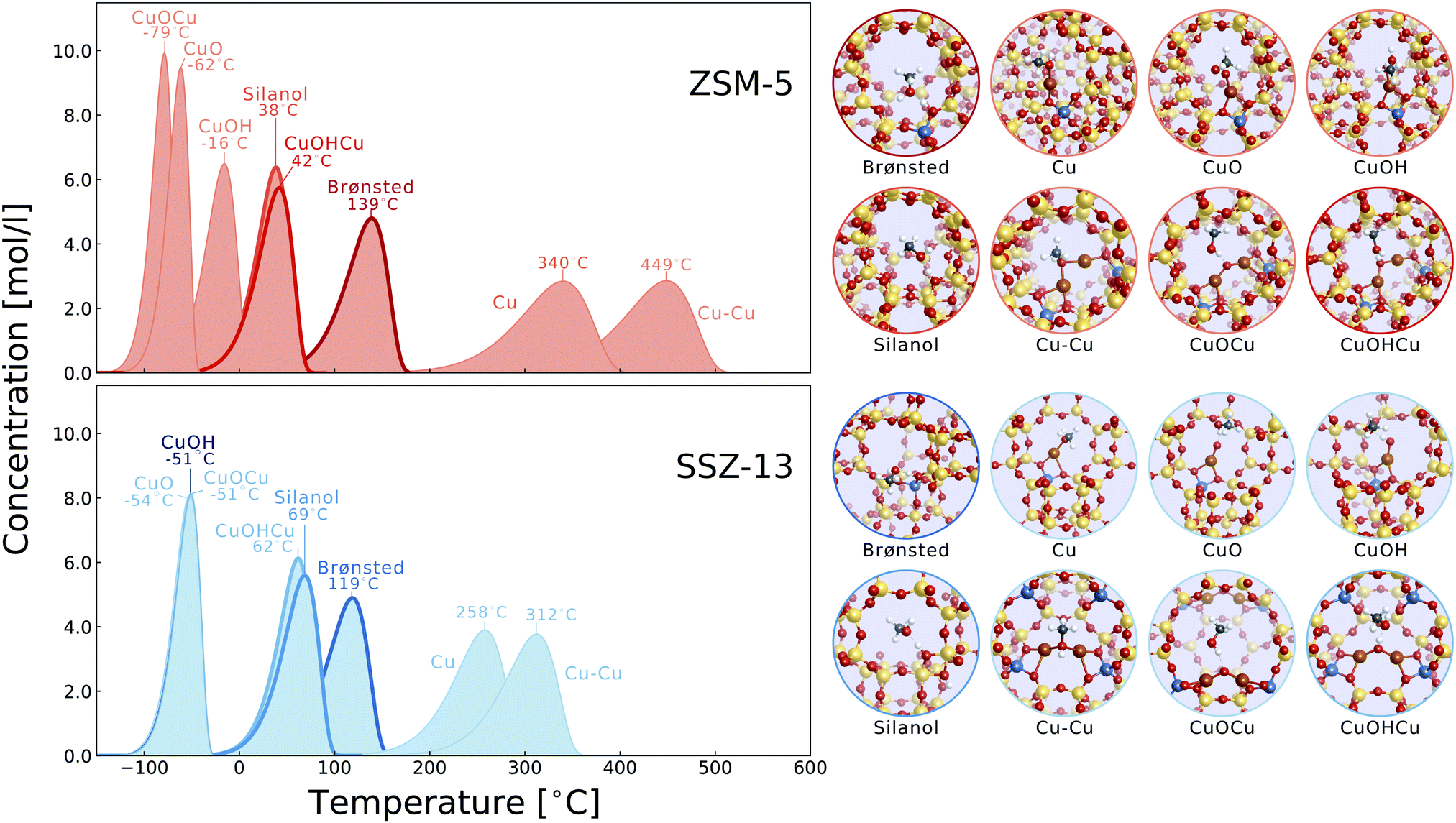 | ||
| Fig. 4 Calculated desorption profiles from first-principles calculations for ZSM-5 in the top panel and SSZ-13 in the lower panel. The peak temperature is shown for each site and an illustration of the site is shown as insets to the right. The relevant curves are highlighted. Atomic colours: silicon: yellow, oxygen: red, aluminum: blue, copper: brown, hydrogen: white, and carbon: gray. | ||
 | ||
| Fig. 5 Sketch of the methanol bonding to the different sites. The strongest bond to methanol is on the Cu sites, where methanol binds oxygen forming a strong Cu–O bond. The second to strongest bond is on the OH-sites (including the Brønsted and silanol sites) where methanol bonds with oxygen forming a hydrogen bond, also forming a water similar structure. Methanol binds the weakest to the CuO site where a hydrogen bond is formed to the methanol hydrogen. | ||
As shown in Fig. 3a and c, for both the H-ZSM-5 and H-SSZ-13 samples, two peaks centered at 133 and 153–163 °C contribute largely to the methanol desorption profiles. The position of the peak centered at around 133 °C coincides with the methanol desorption profile of the SiO2 sample (Fig. 1a). It is anticipated that the origin of this peak is mainly associated with methanol desorption from the silanol groups in the zeolites. While for the peak centered at 153–163 °C, one possible explanation is that it is related to methanol desorption from the Brønsted acid sites in the zeolites. These anticipations are supported by the theoretical calculations shown in Fig. 4, where methanol desorbs at higher temperatures from the Brønsted acid sites than from the silanol groups. Additionally, minor contributions from methanol desorption at temperatures higher than 170 °C are evident for both the H-ZSM-5 and H-SSZ-13 samples. At temperatures higher than 170 °C, DME starts to form from pre-adsorbed methanol over the H-ZSM-5 and H-SSZ-13 samples (as shown in Fig. 1b), indicating that methanol dissociates at such temperatures. Therefore, the two deconvoluted peaks at higher temperatures (centered above 200 °C) for the H-ZSM-5 and H-SSZ-13 samples are presumably associated with methanol adsorbed, dissociated, then recombined and desorbed from the zeolites.
For the Cu-ZSM-5 and Cu-SSZ-13 samples, contributions of methanol desorbed from the Cu sites are clearly indicated by the blue curves shown in Fig. 3b and d. It should be noted that the negative contribution from the Brønsted acid sites covered by Cu is taken into consideration here by reducing the amplitude of the Gaussian functions centred at 153 or 163 °C accordingly for the Cu-ZSM-5 and Cu-SSZ-13 samples, respectively. For both Cu-ZSM-5 and Cu-SSZ-13 samples, contributions of methanol desorption from the Cu sites at low temperatures (peak centered at 133 and 142 °C, respectively), medium temperatures (peak centered around 173 °C) and high temperatures (peak centered above 220 °C) are evident. Though it is clear that different Cu species are involved in methanol adsorption and desorption, the nature of these sites remains unclear. From the theoretical calculations, one may anticipate that hydrated Cu dimer species, i.e. Cu–OH–Cu, contribute largely to the low temperature peaks for both Cu-zeolite samples. Moreover, the formation of hydrated Cu species is likely due to the OH rich environment upon methanol adsorption. The largest contribution for the Cu-ZSM-5 and Cu-SSZ-13 samples, however, is the Gaussian curve centred around 173 °C. At such medium temperatures (170–230 °C), methanol and DME desorb simultaneously from the Cu-ZSM-5 and Cu-SSZ-13 samples. Moreover, methoxy species is evidently observed in the IR spectra of the Cu-ZSM-5 and Cu-SSZ-13 samples at 225 °C (Fig. 2b). Therefore, it is speculated that methanol dissociates over the Cu species at this temperature range, and the formed methoxy species is subsequently converted to either methanol or DME which desorbs from the Cu-zeolite samples. Similar to the high-temperature desorption peaks of the zeolite samples, the high-temperature peaks for the Cu-ZSM-5 and Cu-SSZ-13 samples are presumably associated with the desorption of dissociatively adsorbed methanol on the Cu sites.
4 Conclusions
This study shows that well-dispersed Cu ions/clusters in the zeolites interact strongly with methanol and its derivatives. Formation and desorption of DME are influenced by the ion-exchanged Cu as well as the zeolite framework structure. Effluent DME is observed at higher temperatures for zeolites functionalized with copper and the CHA framework structure shifts the DME desorption to higher temperatures compared to the MFI framework structure. Moreover, the introduction of Cu species increases the amount of CO formed and decreases its formation/desorption temperature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Swedish Research Council through the Röntgen-Ångström Cluster [No. 349-2013-567], the Knut and Alice Wallenberg foundation [No. 2015.0058], and the Competence Centre for Catalysis, which is hosted by Chalmers University of Technology and financially supported by the Swedish Energy Agency and the member companies: AB Volvo, ECAPS AB, Johnson Matthey AB, Preem AB, Scania CV AB, Umicore AG & Co. KG and Volvo Car Corporation AB. The simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX.Notes and references
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- S. M. Campbell, X.-Z. Jiang and R. F. Howe, Microporous Mesoporous Mater., 1999, 29, 91–108 CrossRef CAS.
- R. Y. Brogaard, B. M. Weckhuysen and J. K. Nørskov, J. Catal., 2013, 300, 235–241 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- M. A. C. Markovits, A. Jentys, M. Tromp, M. Sanchez-Sanchez and J. A. Lercher, Top. Catal., 2016, 59, 1554–1563 CrossRef CAS.
- R. Bulánek, B. Wichterlová, Z. Sobalík and J. Tichý, Appl. Catal., B, 2001, 31, 13–25 CrossRef.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- M. H. Mahyuddin, Y. Shiota and K. Yoshizawa, Catal. Sci. Technol., 2019, 9, 1744–1768 RSC.
- K. Narsimhan, V. K. Michaelis, G. Mathies, W. R. Gunther, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2015, 137, 1825–1832 CrossRef CAS PubMed.
- F. Gao, N. M. Washton, Y. Wang, M. Kollár, J. Szanyi and C. H. Peden, J. Catal., 2015, 331, 25–38 CrossRef CAS.
- V. L. Sushkevich and J. A. van Bokhoven, Catal. Sci. Technol., 2018, 8, 4141–4150 RSC.
- K.-H. Steinberg, H. Bremer and P. Falke, Z. Chem., 1974, 14, 110–111 CrossRef CAS.
- V. Kanazirev and N. Borisova, Zeolites, 1982, 2, 23–28 CrossRef CAS.
- V. Kazansky and E. Pidko, Catal. Today, 2005, 110, 281–293 CrossRef CAS.
- G. Spoto, S. Bordiga, A. Zecchina, D. Cocina, E. Gribov, L. Regli, E. Groppo and C. Lamberti, Catal. Today, 2006, 113, 65–80 CrossRef CAS.
- V. Bolis, A. Barbaglia, S. Bordiga, C. Lamberti and A. Zecchina, J. Phys. Chem. B, 2004, 108, 9970–9983 CrossRef CAS.
- S. Ceckiewicz, J. Colloid Interface Sci., 1982, 90, 183–190 CrossRef CAS.
- B. Hunger, S. Matysik, M. Heuchel and W.-D. Einicke, Langmuir, 1997, 13, 6249–6254 CrossRef CAS.
- X. Wang, A. A. Arvidsson, M. O. Cichocka, X. Zou, N. M. Martin, J. Nilsson, S. Carlson, J. Gustafson, M. Skoglundh, A. Hellman and P.-A. Carlsson, J. Phys. Chem. C, 2017, 121, 27389–27398 CrossRef CAS.
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128, 11679–11692 CrossRef CAS PubMed.
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li, B. Yu, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2017, 56, 9039–9043 CrossRef CAS PubMed.
- C.-M. Wang, R. Y. Brogaard, B. M. Weckhuysen, J. K. Nørskov and F. Studt, J. Phys. Chem. Lett., 2014, 5, 1516–1521 CrossRef CAS PubMed.
- International Zeolite Association Webpage, http://europe.iza-structure.org.
- P. N. Plessow and F. Studt, ACS Catal., 2017, 7, 7987–7994 CrossRef CAS.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- W. Gruenert, N. W. Hayes, R. W. Joyner, E. S. Shpiro, M. R. H. Siddiqui and G. N. Baeva, J. Phys. Chem., 1994, 98, 10832–10846 CrossRef.
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC.
- F. Göltl, A. M. Love and I. Hermans, J. Phys. Chem. C, 2017, 121, 6160–6169 CrossRef.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- X. Wang, N. M. Martin, J. Nilsson, S. Carlson, J. Gustafson, M. Skoglundh and P.-A. Carlsson, Catalysts, 2018, 8, 545 CrossRef.
- M. Männikkö, X. Wang, M. Skoglundh and H. Härelind, Appl. Catal., B, 2016, 180, 291–300 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- M.-L. Tsai, R. G. Hadt, P. Vanelderen, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 3522–3529 CrossRef CAS PubMed.
- A. A. Arvidsson, V. P. Zhdanov, P.-A. Carlsson, H. Grönbeck and A. Hellman, Catal. Sci. Technol., 2017, 7, 1470–1477 RSC.
- L. Chen, T. V. W. Janssens and H. Grönbeck, Phys. Chem. Chem. Phys., 2019, 21, 10923–10930 RSC.
- U. Engedahl, H. Grönbeck and A. Hellman, J. Phys. Chem. C, 2019, 123, 26145–26150 CrossRef CAS.
- M. Niwa, N. Katada, M. Sawa and Y. Murakami, J. Phys. Chem., 1995, 99, 8812–8816 CrossRef CAS.
- L. Chen, T. V. W. Janssens, M. Skoglundh and H. Grönbeck, Top. Catal., 2018, 62, 93–99 CrossRef.
- A. G. Pelmenschikov, G. Morosi, A. Gamba, A. Zecchina, S. Bordiga and E. A. Paukshtis, J. Phys. Chem., 1993, 97, 11979–11986 CrossRef CAS.
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed.
- M. C. Kung, S. S. Y. Lin and H. H. Kung, Top. Catal., 2012, 55, 108–115 CrossRef CAS.
- T. Nobukawa, M. Yoshida, S. Kameoka, S.-I. Ito, K. Tomishige and K. Kunimori, J. Phys. Chem. B, 2004, 108, 4071–4079 CrossRef CAS.
- B. R. Wood, J. A. Reimer, A. T. Bell, M. T. Janicke and K. C. Ott, J. Catal., 2004, 225, 300–306 CrossRef CAS.
- K. I. Hadjiivanov, M. M. Kantcheva and D. G. Klissurski, J. Chem. Soc., Faraday Trans., 1996, 92, 4595–4600 RSC.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation of the amount of adsorption sites; desorption profile of main gas phase species; DME desorption and NH3 desorption profiles of the Cu-ZSM-5, H-ZSM-5, Cu-SSZ-13, H-SSZ-13, Cu/SiO2 and SiO2 samples; IR spectra recorded during methanol desorption; a table of binding energies and desorption peak maxima for all calculated adsorption sites; sensitivity analysis on the calculated desorption spectra. See DOI: 10.1039/c9cp05479k |
| This journal is © the Owner Societies 2020 |