
Real-time degradation dynamics of hydrated per- and polyfluoroalkyl substances (PFASs) in the presence of excess electrons†

Abstract
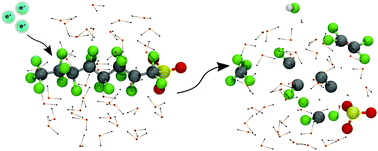
Per- and polyfluoroalkyl substances (PFASs) are synthetic chemicals that are harmful to both the environment and human health. Using self-interaction-corrected Born–Oppenheimer molecular dynamics simulations, we provide the first real-time assessment of PFAS degradation in the presence of excess electrons. In particular, we show that the initial phase of the degradation involves the transformation of an alkane-type C–C bond into an alkene-type C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the PFAS molecule, which is initiated by the trans elimination of fluorine atoms bonded to these adjacent carbon atoms.
C bond in the PFAS molecule, which is initiated by the trans elimination of fluorine atoms bonded to these adjacent carbon atoms.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

