DOI:
10.1039/D0CE01137A
(Paper)
CrystEngComm, 2020,
22, 6783-6795
Modelling of an aza-Michael reaction from crystalline naphthalene derivatives containing peri–peri interactions: very long N–C bonds?†
Received
4th August 2020
, Accepted 16th September 2020
First published on 17th September 2020
Abstract
The separation between a pair of peri-located dimethylamino and ethene-2,2-dinitrile groups in a naphthalene molecule, which models the progress of a Michael reaction, can be controlled by the installation of a short ethylene bridge or the introduction of repulsive interactions at the opposite set of peri positions. Introduction of a dimethylammonium substituent produced a hydrated chloride salt in which the Me2N⋯C(H)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CN)2 separation between reactive groups decreases, reversibly, from 2.167 Å at 200 K to 1.749 Å at 100 K, with the maximum rate of change in the range 128–140 K, which was studied by variable temperature X-ray crystallography and solid state NMR. From these and other crystallographic data a correlation between Me2N⋯C bond formation and alkene bond breaking was constructed for the first step of an aza-Michael reaction.
C(CN)2 separation between reactive groups decreases, reversibly, from 2.167 Å at 200 K to 1.749 Å at 100 K, with the maximum rate of change in the range 128–140 K, which was studied by variable temperature X-ray crystallography and solid state NMR. From these and other crystallographic data a correlation between Me2N⋯C bond formation and alkene bond breaking was constructed for the first step of an aza-Michael reaction.
Introduction
Bond formation and breaking is a core theme in chemical reactivity but it is very difficult to monitor the whole process, including the correlation between bond formation and breaking, due to the high energy of the transition state which involves partially broken and formed bonds. Indeed transition states have only been accessed experimentally by femtosecond spectroscopy as demonstrated by Zewail,1 and calculations offer a more pragmatic approach to exploring transition state structures.2 The Michael reaction is a very well-known reaction, involving the addition of a nucleophile such as an amine, thiolate or enolate, to an electron deficient alkene or alkyne. It is used to prepare a wide variety of substances including enantiopure ones, and may be promoted, for example, by organocatalysis.3 The aza-Michael reaction, in particular, is used in the construction of heterocycles, alkaloids and molecules of pharmaceutical relevance.4 Some calculations of the transition states structures and/or energies for such reactions have been reported, in some cases aimed at predicting mutagenicity of the Michael acceptor.5,6
The idea of modelling reaction pathways based on structural data from crystals is well established for both organic and some inorganic systems though the experimental data generally lies closer to the start or end of the reaction process.7 The naphthalene framework has been used for observing interactions and reactions between pairs of groups placed at the peri-positions by crystallographic and solid-state NMR methods.8 Apart from nucleophile/electrophile interactions, peri-substituted naphthalenes have provided chiral hydrocarbons with very large steric interactions,9 frustrated lone pairs systems10 and proton sponges,11 with experimental charge density studies carried out in selected cases.12 Attractive interactions between functional groups were first reported in the pioneering work from Dunitz et al. on the interactions of nucleophiles with carbonyl groups as in naphthalene 1.13 Such n–π* interactions affect the chemistry of the electrophile, for example the aldehyde 2 both protonates and acylates on the O atom with formation of a N–C bond between the two groups,14 as do several closely related ketones.15 We have reported the structure of a series of compounds where a dimethylamino group lies next to a polarised alkene, such as 3 and 4.16 In the first of these the nucleophile impinges on an alkene terminated by two nitriles with a Me2N⋯C separation of 2.413(2) Å while in the latter case, where the alkene is activated by two in-plane lactone carbonyls, the groups reacts to form a zwitterion with a long Me2N–C bond of 1.651(3) Å. Although a range of such systems has been studied,17 no such structures with Me2N⋯C separations in the range 1.7–2.4 Å have been found. Indeed, the only molecular structure with such N⋯C separations are for transannular interactions across the ring systems of particular alkaloids, such as clivorine 5 (1.93 Å) and senkirkine 6 (2.25 Å),18 as highlighted by Bürgi, Dunitz and Schefter.19 n–π* interactions between peptide carbonyls have been particularly highlighted recently for their role in determining the conformation of proteins.20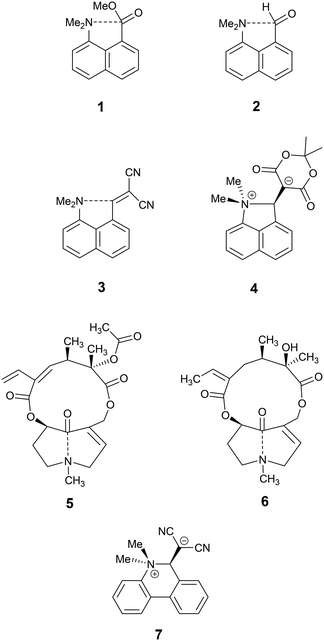
To modify peri-interactions and find such intermediate separations between the peri-groups, we decided to explore whether a pair of substituents at the second set of peri-positions of the naphthalene skeleton could control the electrophile/nucleophile separation at the first pair. In particular, would repulsion between a second pair of substituents lead to the first pair being pushed closer together, and conversely would contraction at the second pair lead to an increased electrophile/nucleophile separation? We describe here results for compounds containing a dimethylamino group and a dicyanoethenyl group as the nucleophile/electrophile pair. The results of a reaction between this pair of groups to form a zwitterion has been observed in crystals of the corresponding biphenyl system 7, with new Me2N–C bonds of 1.586(3) and 1.604(3) Å in the two independent molecules.21 In this case the reaction involves the formation of a less strained ring than in the naphthalene series.
Discussion
Structural effects of a second set of peri-substituents in a naphthalene derivative
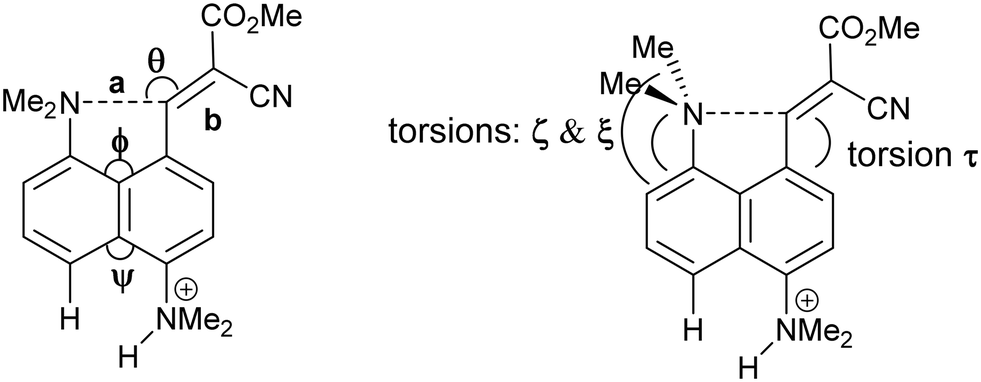
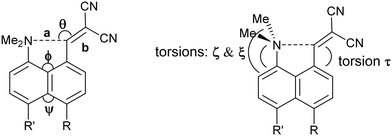
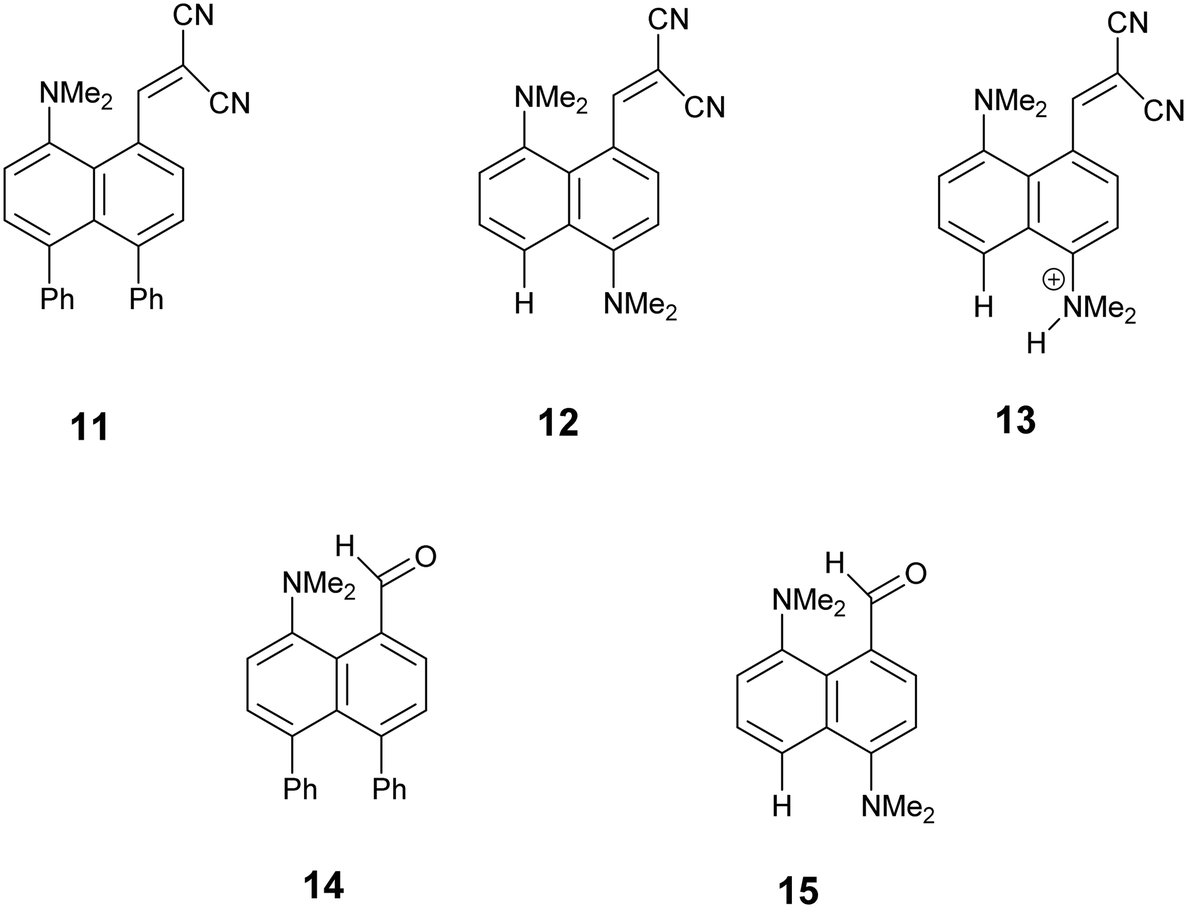
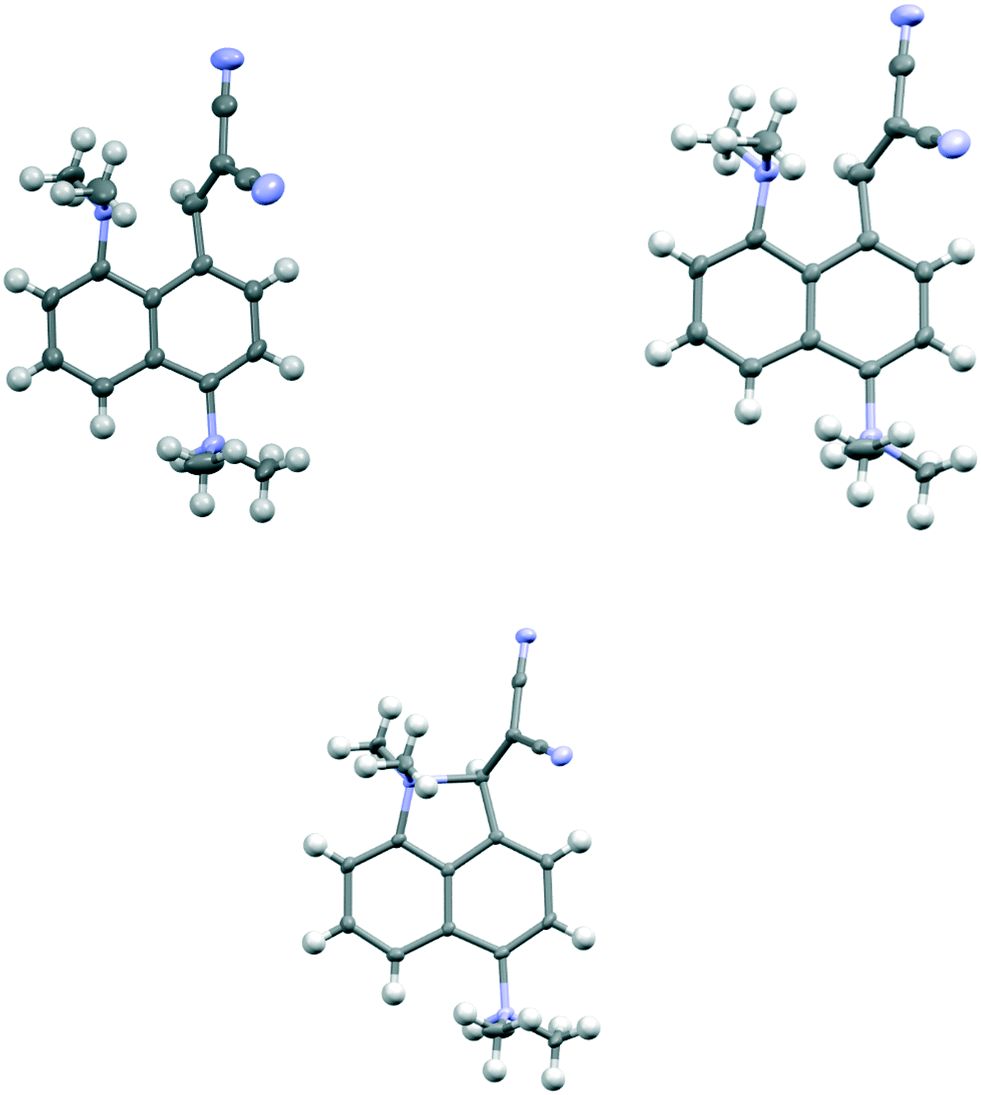
We started by exploring the effect of installing an ethylene bridge between the second set of peri-positions on the Me2N⋯C(H)C(CN)2 separation by studying acenaphthene derivative 8. The molecule was prepared from acenaphthene in six steps (ESI†). Its X-ray crystal structure, determined at 150 K, showed that the installation of the ethylene bridge had led to a significant increase in the distance between the functional groups: the Me2N⋯CHC(CN)2 distances in the two crystallographically unique molecules are 2.755(2) and 2.846(2) Å compared to 2.413(2) Å in the unsubstituted analogue 3 (Fig. 1). Furthermore, the difference of 16–17° in the exo angles at the fusion the benzene rings between the two peri positions (ϕ and ψ in Table 1) shows how the widening of the ϕ angle between the amino and alkene groups has been caused by the compression of the ψ angle which was imposed by the short ethylene bridge: i.e. ϕ: 127.30(18)/128.54(17)° vs. ψ: 111.73(17)/111.25(18)°. In 3, without the ethylene bridge, these two angles differ only by ca. 2° (122.6(2) and 120.42(12)°). The increased separation, and reduced n–π* interaction between the –NMe2 and –C(H)C(CN)2 groups, allows the electrophilic group in one of the two crystallographically independent molecules to rotate closer to the acenaphthene plane and to increase the conjugation between the alkene and the aromatic system. Indeed, this general strategy of a short peri-bridge was also implemented in the design of acenaphthylene derivative 9 by Ishigaki and Suzuki which shows an exceptionally long C–C bond between peri-substituents22 and also in materials with frustrated lone pairs such as 10.10
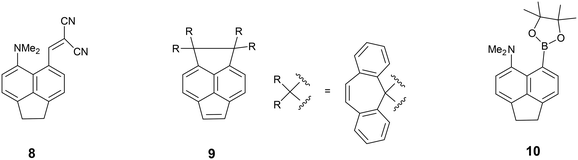
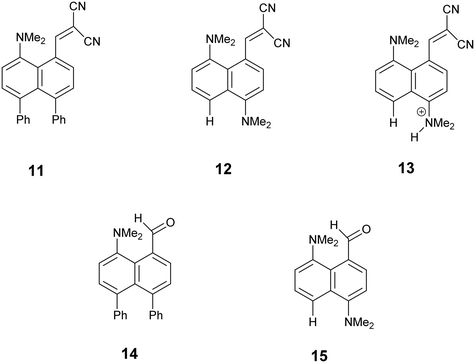
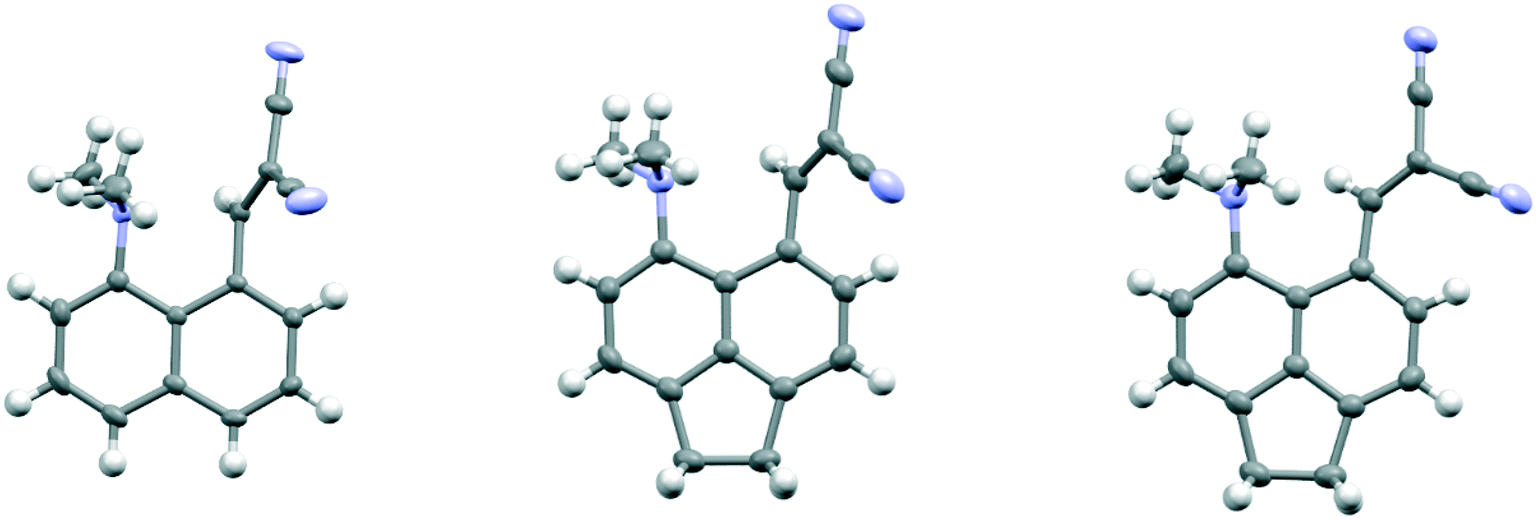
To explore the opposite effect, the ethylene bridge was replaced by two phenyl rings which should repel each other since the peri-naphthalene ring carbon atoms are only ca. 2.5 Å apart. Thus, the peri-diphenyl derivative 11 with a Me2N⋯C(H)C(CN)2 interaction at the opposite peri-positions was prepared from 1,8-diphenylnaphthalene in five steps via aldehyde 14, and the dinitrile 11 was crystallised as a toluene solvate. The X-ray crystal structure was determined and the molecular structure is shown in Fig. 2. The two phenyl groups are tilted at 62.2 and 63.2° to the naphthalene, and they are repelled apart with an angle of 20.9° between them and a widening of the nearby exo-angle (ψ) at the fusion of the naphthalene rings to 126.24(13)°. The opposite exo-angle (ϕ) is compressed to 117.74(12)° leading to the other two peri groups being oriented closer together so that the Me2N⋯C(H) C(CN)2 separation is reduced to 2.3603(19) Å, 0.053 Å shorter than in the dinitrile 3 without peri phenyl groups.
 |
| | Fig. 1 Molecular structures of 316 (left) and the two independent molecules of 8 (middle and right) in the second of which (right) the alkene has rotated to be closer to the naphthalene plane. | |
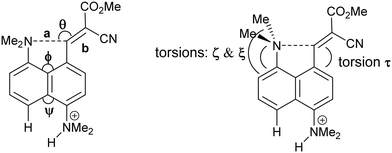
Table 1 Selected molecular geometry for naphthalenes 3, 8, 11 and 12 and cation 13
|
|
| Compound |
T/K |
a/Å |
b/Å |
θ/° |
Me2NH⋯H/Å |
|
3, R = R′ = H |
150 |
2.413(2) |
1.354(2) |
112.5(2) |
— |
| |
|
8, R,R′ = CH2CH2 |
150 |
2.755(2) |
1.343(3) |
122.71(14) |
— |
| 2.846(2) |
1.348(3) |
133.00(15) |
| |
|
11, R = R′ = Ph |
150 |
2.3603(19) |
1.356(2) |
114.21(11) |
— |
| |
|
12, R = NMe2, R′ = H |
150 |
2.455(2) |
1.356(2) |
116.29(12) |
— |
| 2.512(2) |
1.350(2) |
117.19(12) |
| |
|
13·Cl, |
200 |
2.167(4) |
1.369(4) |
113.2(2) |
2.07 |
| R = +NHMe2, |
150 |
2.098(4) |
1.378(4) |
113.4(2) |
2.09 |
| R′ = H |
134 |
1.932(4) |
1.401(4) |
115.0(2) |
2.11 |
|
|
100 |
1.749(3) |
1.431(3) |
115.9(2) |
2.14 |
|
|
T/K |
φ/° |
ψ/° |
τ/° |
ζ/° |
ξ/° |
|
3, R = R′ = H |
150 |
120.42(14) |
122.6(2) |
56.5(2) |
49.7(2) |
−81.5(2) |
| |
|
8, R,R′ = CH2CH2 |
150 |
127.30(18) |
111.73(17) |
51.9(3) |
45.8(2) |
−82.1(2) |
| 128.54(17) |
111.25(18) |
35.9(3) |
27.5(3) |
−100.8(2) |
| |
|
11, R = R′ = Ph |
150 |
126.24(13) |
117.74(12) |
51.0(2) |
94.95(18) |
−36.9(2) |
| |
|
12, R = NMe2, R′ = H |
150 |
120.27(15) |
122.74(15) |
53.1(2) |
77.8(2) |
−54.9(3) |
| 120.81(15) |
122.75(15) |
49.9(2) |
90.5(2) |
−43.0(3) |
| |
|
13·Cl
|
200 |
117.8(2) |
126.5(3) |
53.0(4) |
48.9(5) |
−82.3(4) |
| R = +NHMe2, |
150 |
117.1(2) |
127.1(2) |
51.4(4) |
49.1(4) |
−81.0(3) |
| R′ = H |
134 |
116.0(2) |
128.1(2) |
46.4(4) |
49.6(4) |
−78.6(3) |
|
|
100 |
114.3(2) |
129.3(2) |
42.0(4) |
48.0(3) |
−76.4(2) |
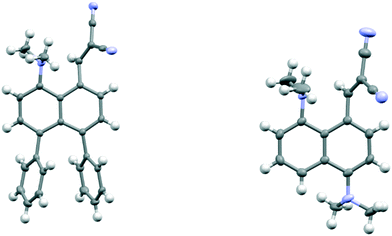 |
| | Fig. 2 Molecular structures of naphthalene 11 with two peri phenyl groups (left) and one of the two crystallographically unique molecules of naphthalene 12 with a dimethylamino group and hydrogen atom at the second set of peri-positions (right). | |
Continuing the idea of introducing a repulsive peri-interaction, the corresponding naphthalenes with just one –NMe2 group, unprotonated or protonated, in the opposite peri-position, 12 and 13·Cl respectively, were prepared. Protonation was expected to increase the effective size of the dimethylamino group. Compound 12 was prepared by peri-lithiation of 1,5-bis(dimethylamino)naphthalene and treatment with DMF to yield aldehyde 15 followed by Knoevenagel condensation with malonitrile. The X-ray structure of the unprotonated material 12 (Fig. 2) shows that the peri-repulsion between the H and –Me2N groups in the two crystallographically unique molecules is not very effective, even though there is van der Waals contact between the peri H atom and a N-methyl H atom. The exo angles at the fusion of the benzene rings differ only by ca. 2°, and are similar to those in naphthalene 3 which has no second dimethylamino group. The Me2N⋯C interactions are just slightly longer than in 3 (Table 1). The results from the X-ray crystal structure of the HCl salt, however, showed a much more significant repulsion between the dimethylammonium group and the peri H atom.
Variable temperature peri-interactions in 13·Cl
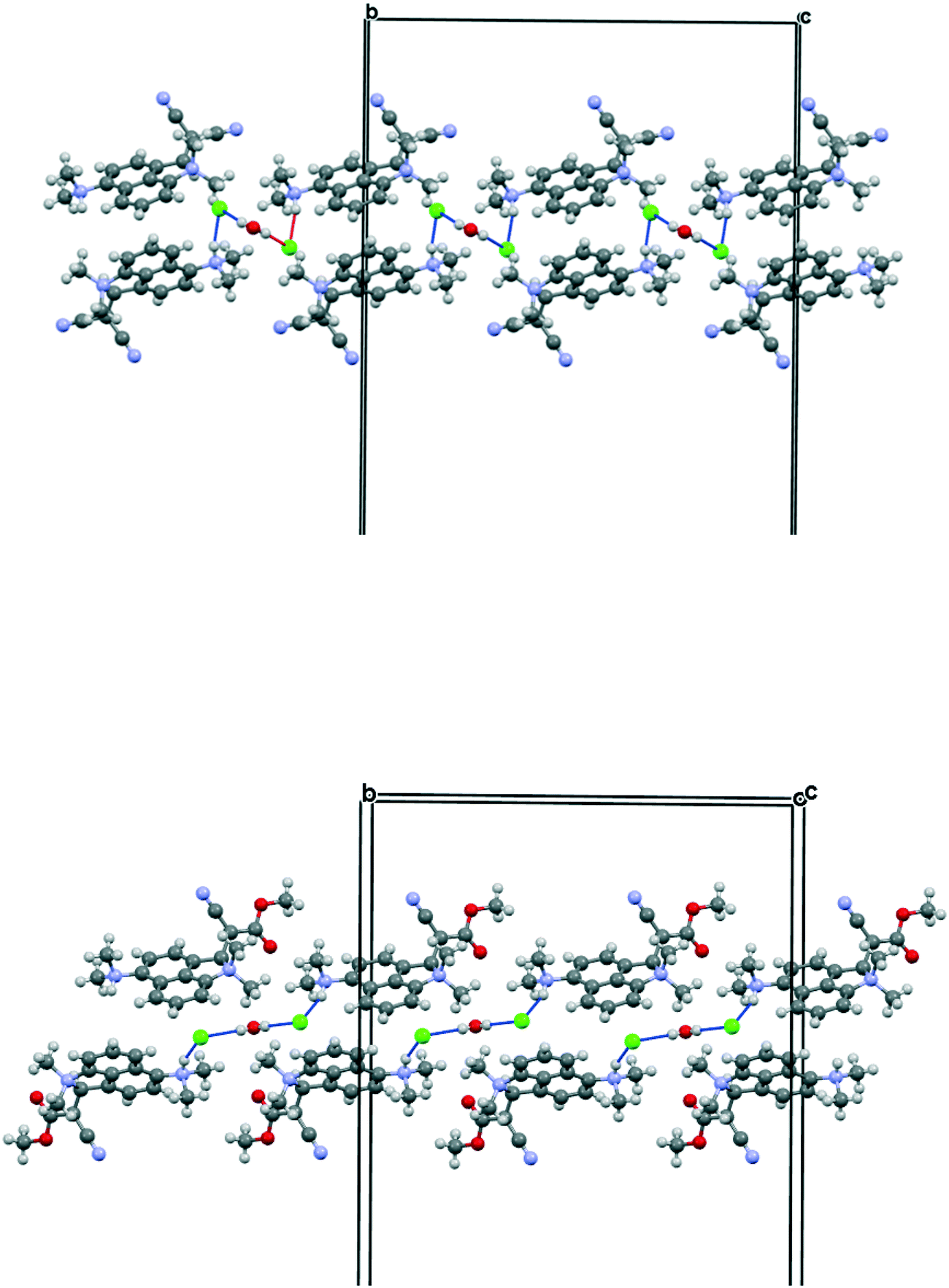
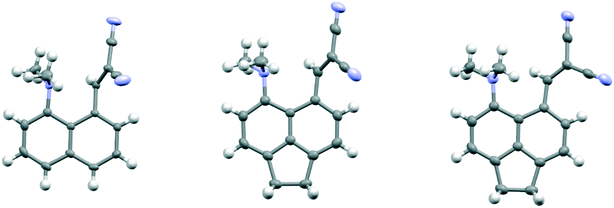
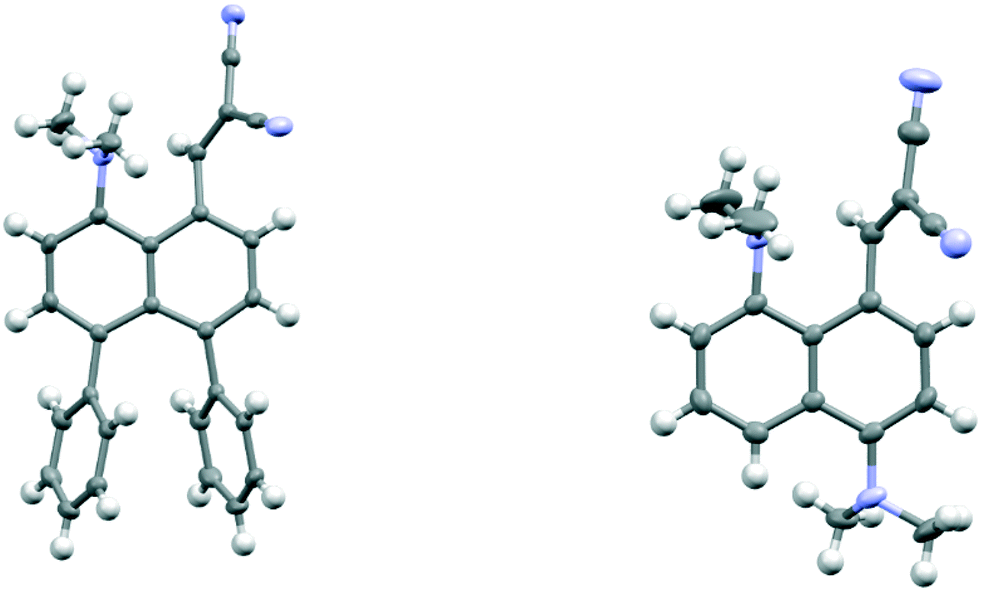
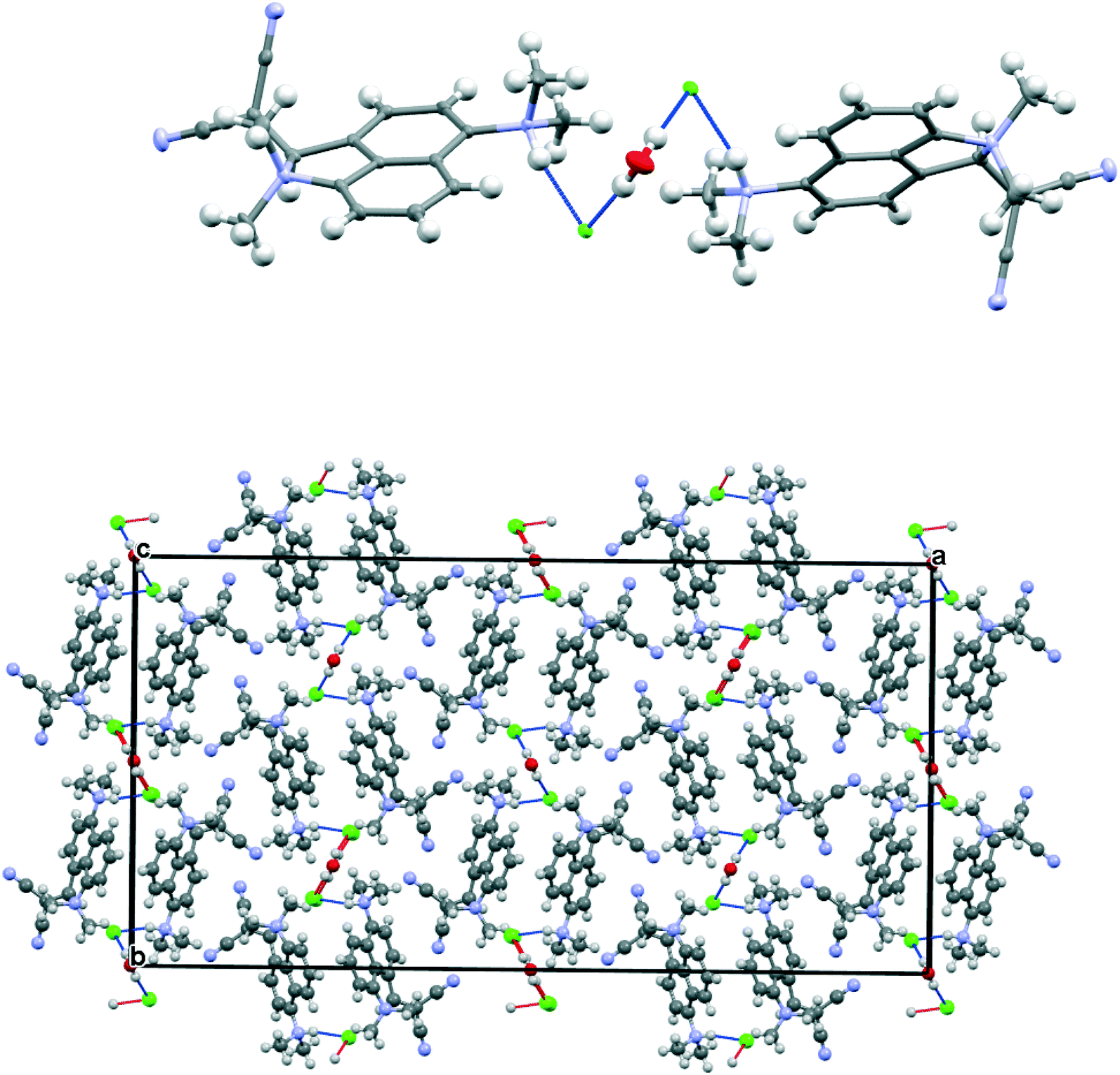
The salt 13·Cl, readily prepared from 12, crystallised in the orthorhombic space group Fdd2 with one molecule of water per two cations of 13 and two chloride anions. In the crystal structure of this salt at 200 K the cation shows a remarkably short Me2N⋯C(H)C(CN)2 interaction of just 2.167(4) Å, which is ca. 1 Å within the separation predicted by the sum of van der Waals radii (Fig. 3). This is also ca. 0.25 Å shorter than is observed in the corresponding naphthalene derivative 3 with just a pair of H atoms at the second set of peri-positions. The dimethylamino group is well aligned with the alkene-dinitrile group for a reaction to take place, with a Me2N⋯CC angle of 113.2(2)°. Furthermore, the alkene bond is lengthened by 0.015 Å compared to the unsubstituted molecule 3, suggesting that there has been a small degree of progress along the reaction coordinate for the addition of a tertiary amine the alkene. At the other side of the molecule the dimethylammonium substituent and peri-H atom are repelled apart, so there is van der Waals contact (2.07 Å) between the N–H and the peri-H atom. The N-methyl groups lie to either side of the naphthalene plane to minimise steric interaction with the ortho H atom. This peri-repulsion leads to an exo angle ψ of 126.5(3)° and consequentially to a much smaller exo ϕ angle of 117.8(2)°, and the closer approach between the dimethylamino group and alkene. In the crystal structure the dimethylammonium group forms a hydrogen bond to the chloride anion, and a water molecule bridges two chloride anions (Fig. 4). Pairs of cations of 13, linked by hydrogen bonding through a Cl⋯HOH⋯Cl unit, lie in columns along the b axis, with four parallel columns lying side by side along the a axis (44.4094 Å at 200 K). The two cations are related by the two fold axis along the short c axis which cuts the water's oxygen atom. The column is completed by a second such pair, related to the first by a two-fold screw axis along the c axis (Fig. 4, vide infra).
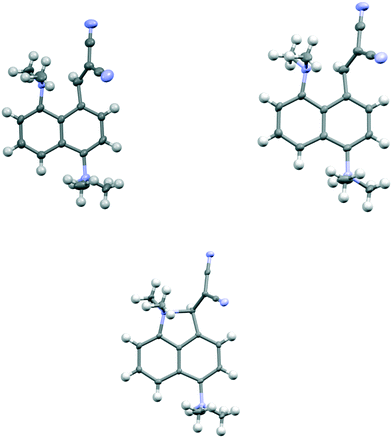 |
| | Fig. 3 The molecular structure of the cation 13 at 200 K (top left), at 150 K (top right) and 100 K (below), with Me2N⋯C separations of 2.167(4), 2.098(4) and 1.749(3) Å respectively. A bond between the functional groups is drawn in the latter case, to indicate the closer distance. | |
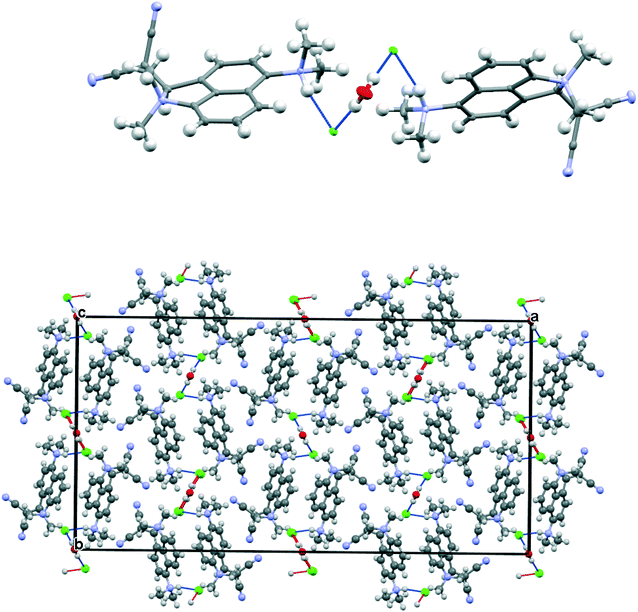 |
| | Fig. 4 Crystal packing for 13·Cl·0.5H2O at 100 K viewed down the c axis: a pair of chloride anions bridged by a water molecule each form a hydrogen bond to the N–H of the dimethylammmonio group of cation 13 (above) and the full crystal packing arrangement (below). | |
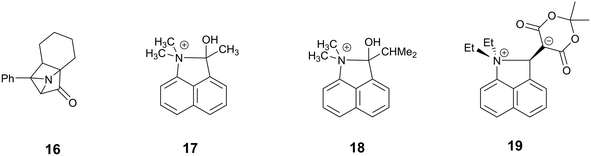
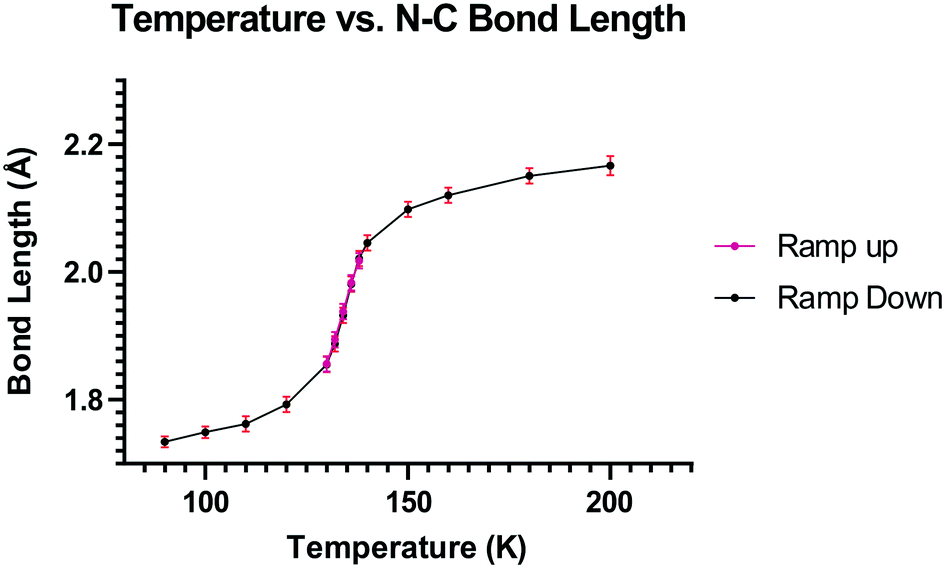
On cooling the crystal of this salt to 150 K the molecular structures changes by a small amount, with the Me2N⋯CC distance contracting by 0.069 Å to 2.098(4) Å, and the alkene lengthening slightly by 0.009 Å. However, on cooling further to 100 K the structure of the cation underwent a more significant change. The contraction of the crystal lattice applies pressure to the cation, and the dimethylamino and dicyanoethenyl groups are pushed closer together to give a Me2N⋯C separation of 1.749(3) Å, though this is still not as short as in the biphenyl derivative 7 (1.586/1.604 Å). The alkene bond has also partly broken and its length (1.431(3) Å) lies intermediate between that for this species at 200 K (1.369 Å), and that in the biphenyl case 7 (1.487/1.493 Å). Furthermore, the molecular geometry at the attacked alkene carbon has changed from planar to partially tetrahedral. The coordinates of the hydrogen atom at this centre were found in a difference Fourier map and refined in the X-ray analysis. Thus the angles between the two C–C bonds and the C–H bond at this centre change from 122.2, 116.2, 117.9° at 200 K to 118.6, 112.7 and 109.6° at 100 K. As this addition reaction proceeds partial charges, positive and negative, should develop respectively on the dimethylamino nitrogen atom and on the carbon atom between the nitrile groups. The N-methyl bonds have lengthened in response, from 1.462(5) and 1.476(4) Å at 200 K to 1.486(3) and 1.490(3) Å at 100 K. These four values lie in-between those for naphthalenes 3 and 4, 1.459 and 1.502 Å respectively, which have longer and shorter Me2N⋯C separations (2.413 and 1.651 Å).16 There is also a tightening of the bond angles at the N atom in the dimethylamino group on going from 200 K to 100 K. The bonds from the developing carbanion to the nitrile groups have shortened from 1.430(4) and 1.434(4) Å at 200 K to 1.416(3) and 1.423(3) Å at 100 K, with slight increases in the nitriles' bond lengths: from 1.135(4)/1.142(4) Å to 1.149(3)/1.149(3) Å, as the nitriles play a greater role in stabilising the negative charge. All these changes are consistent with progress along the reaction coordinate on cooling the crystal. Overall, this suggests that there is a partially formed bond of length 1.749 Å between the two functional groups. The Me2N⋯C interaction is aligned approximately along the c axis of the cell, and this axis contracts by ca. 2.8% on cooling from 200 K to 100 K, while there are much smaller changes on the a axis (+0.6%) and the b (−0.7%). The overall packing arrangement observed at 200 K is maintained (Fig. 4). The longest C–N bonds in the Cambridge Structural Database, neglecting artefacts due to disordered side chains or inaccurate structures and excluding peri-naphthalenes, are ca. 1.61 Å e.g. in the highly strained system 16 where the nitrogen atom is involved in the fusion of two four-membered rings and a three-membered ring.23 Among the peri-naphthalenes the longest N–C bonds between peri-substituents are in cations 17 and 18 and zwitterion 19 and are in the range 1.66–1.68 Å.15,17,24
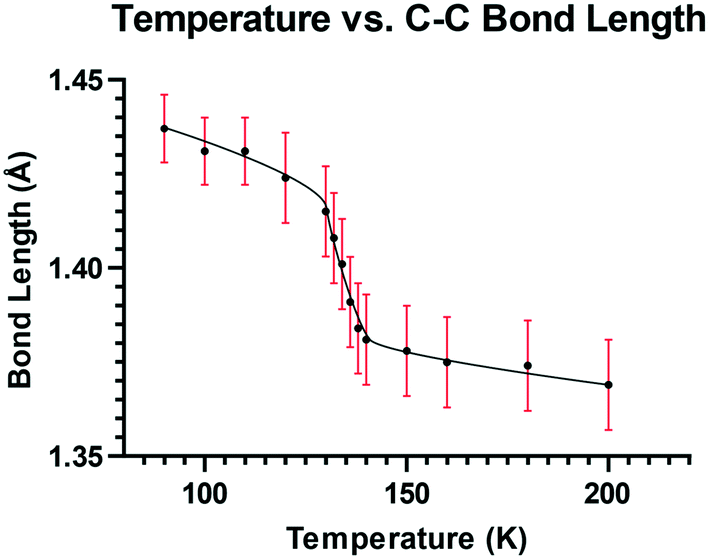
To explore this temperature dependent change of the Me2N⋯C interaction, the crystal structure of 13·Cl·0.5H2O was determined at a set of intermediate temperatures, 10 K apart, and since the most notable structural changes were found to take place between 140 and 128 K, the crystal structure was carefully measured at intervals of 2 K within this range. The crystal structure was redetermined at 132, 134, 136 and 138 K as the crystal was warmed back up to 150 K, and the results indicated that the process is reversible. The changes in the Me2N⋯C separation and bond length of the C(H)C(CN)2 alkene with temperature are shown in Fig. 5 and 6. The region of fastest change with temperature is centred on ca. 134 K.
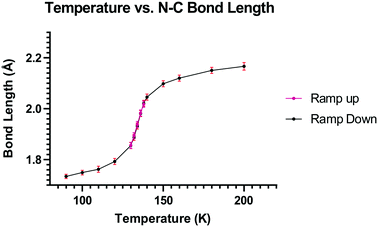 |
| | Fig. 5 Graph of the Me2N⋯C distance (Å) versus temperature (K) for 13·Cl·0.5H2O. | |
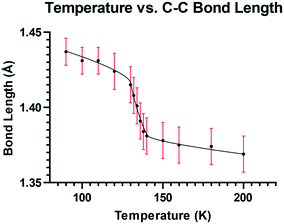 |
| | Fig. 6 Graph of the C(H)C(CN)2 distance (Å) versus temperature (K) for 13·Cl·0.5H2O. | |
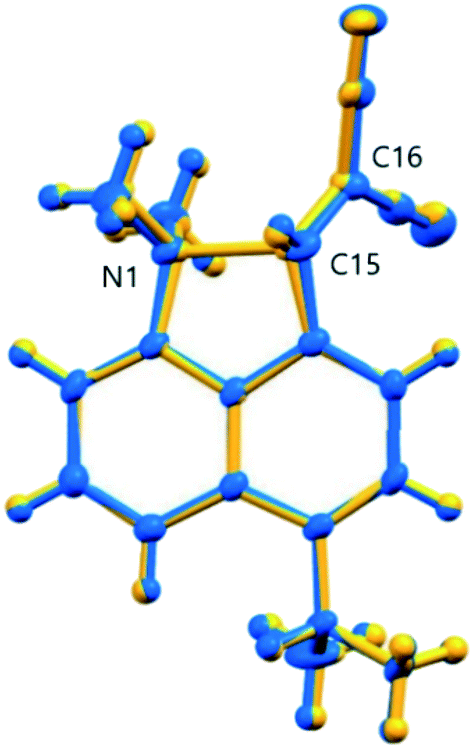
The overall structural change in cation 13 on cooling from 150 to 100 K is small as illustrated by the overlaid structures (Fig. 7), though significant for the two reacting groups. Two possible interpretations of the observed changes could be considered. Either there is just a steady reduction in the Me2N⋯C distance on cooling with a more rapid change in the 140 to 128 K range as the structure passes through a higher energy state, or there are two structures represented by those at ca. 140 and 128 K, and within this temperature range an increasing percentage flip over to the “128 K structure” as the temperature falls. The former interpretation would require that the structure was progressing through a transition state. The latter interpretation should lead to increased atomic displacement parameters for the interacting atoms in the temperature range where a mixture of two structures was present.
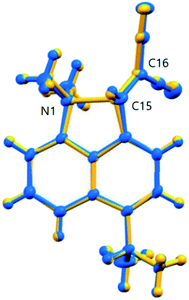 |
| | Fig. 7 An overlay of the two structures of cation 13 at 150 K (blue) and 100 K (yellow) demonstrating the changes in the overall skeleton for 13·Cl·0.5H2O, and selected atomic labels for those atoms most directly involved in the interaction. | |
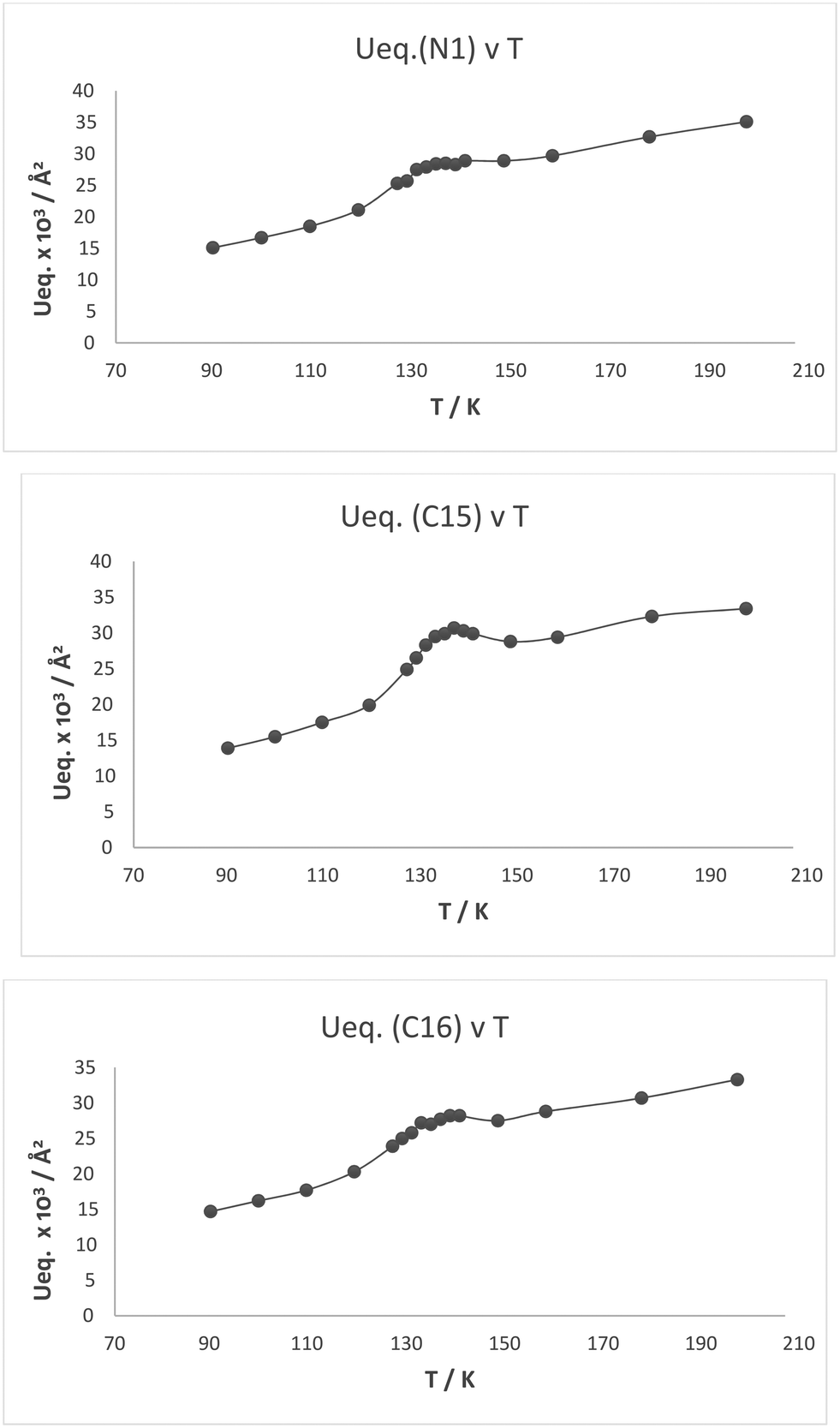
Plots of the atomic Ueq. value versus temperature where Ueq. is an isotropic parameter derived from the six atomic anisotropic displacement parameters to represents the uncertainty in the atomic position due to thermal motion and/or disorder,25 for the three atoms primarily involved in the interaction process are shown in Fig. 8. Superimposed on the expected steady decrease in the Ueq. value with falling temperature, there is an increase which is centred at ca. 134 K: by ca. 25% for the two directly interacting atoms, the amino nitrogen N1 and the alkene carbon C15, and by ca. 10% for the remote alkene carbon C16 (Fig. 7). This would be consistent with the presence of two similar structural forms in this temperature range.
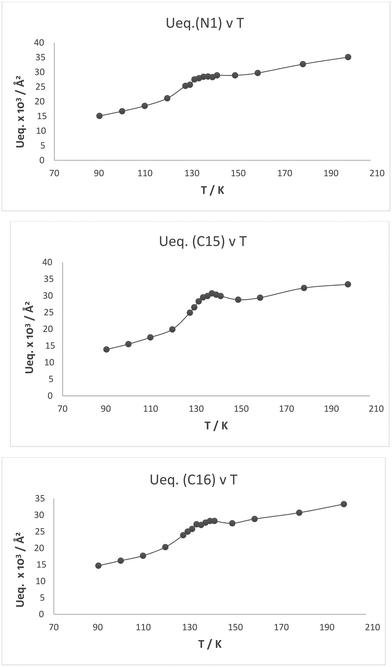 |
| | Fig. 8 Plot of Ueq. (Å2) against temperature (K) for the three atoms involved in the Me2N⋯C(H)C(CN)2 interaction/bond formation process in cation 13: N1, C15 and C16. | |
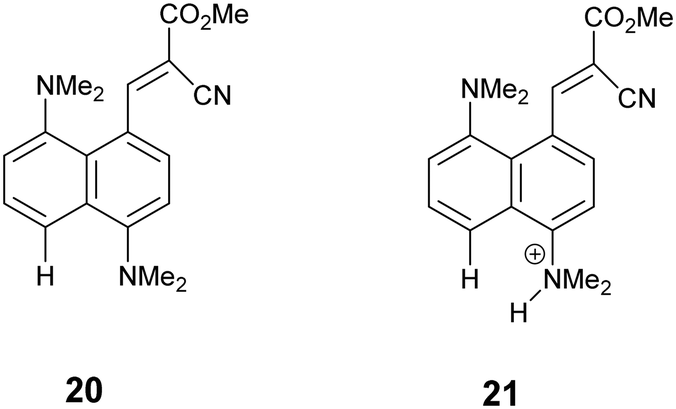
Structure of the cyano ester analogue, 21·Cl
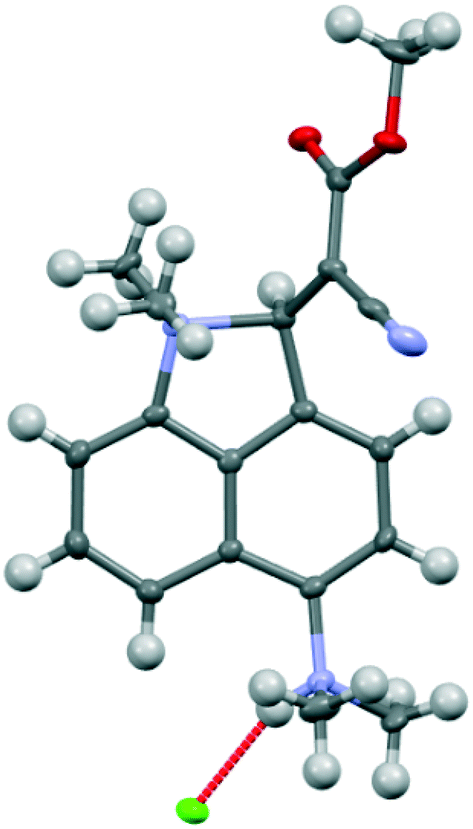
 For comparative purposes, the corresponding methyl cyanoester 20 was prepared and converted to its chloride salt 21·Cl. Its crystal structure was measured at 260 and 150 K. The crystal structure of this salt resembles that of the dinitrile chloride salt 13·Cl·0.5H2O in a number of respects. It includes a molecule of water per two chloride anions, and has similar cell parameters and the same space group (Fig. 9 and 10). However, the Me2N⋯C separation in 21·Cl·0.5H2O at 260 K is 1.754(6) Å, significantly shorter than for the dinitrile salt 13·Cl·0.5H2O at 200 K (2.167(4) Å), and the partially broken alkene bond has a length of 1.442(5) cf. 1.369(3) Å (Table 2). Thus this structure at 260 K most closely resembles that of the dinitrile salt at 100 K. On cooling to 150 K the Me2N⋯C separation contracts by 0.049 Å to 1.705(3) Å, and the former alkene bond extends slightly to 1.452(3) Å. So in this case, there is a substantial degree of N–C bond formation and CC bond breaking at 260 K.
For comparative purposes, the corresponding methyl cyanoester 20 was prepared and converted to its chloride salt 21·Cl. Its crystal structure was measured at 260 and 150 K. The crystal structure of this salt resembles that of the dinitrile chloride salt 13·Cl·0.5H2O in a number of respects. It includes a molecule of water per two chloride anions, and has similar cell parameters and the same space group (Fig. 9 and 10). However, the Me2N⋯C separation in 21·Cl·0.5H2O at 260 K is 1.754(6) Å, significantly shorter than for the dinitrile salt 13·Cl·0.5H2O at 200 K (2.167(4) Å), and the partially broken alkene bond has a length of 1.442(5) cf. 1.369(3) Å (Table 2). Thus this structure at 260 K most closely resembles that of the dinitrile salt at 100 K. On cooling to 150 K the Me2N⋯C separation contracts by 0.049 Å to 1.705(3) Å, and the former alkene bond extends slightly to 1.452(3) Å. So in this case, there is a substantial degree of N–C bond formation and CC bond breaking at 260 K.
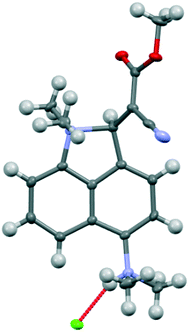 |
| | Fig. 9 Molecular structure of the cation 21 and associated chloride anion in the salt 21·Cl·0.5H2O at 150 K. | |
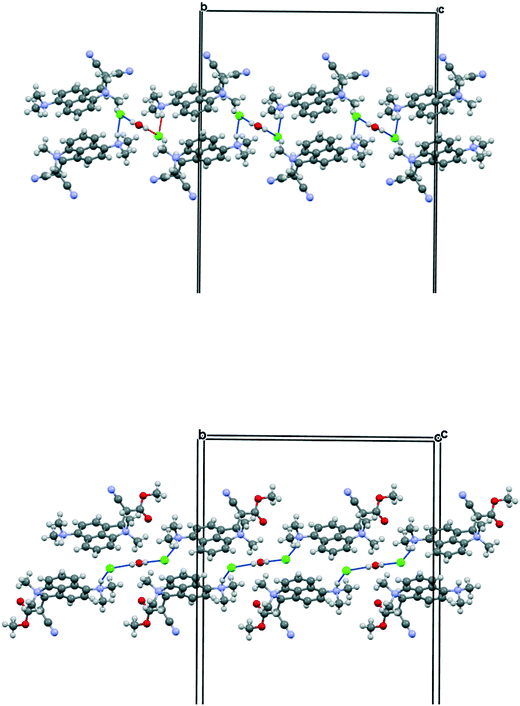 |
| | Fig. 10 Linking of pairs of cations by a Cl⋯HOH⋯Cl unit along the b axis for dinitrile salt 13·Cl·0.5H2O (top), and for cyanoester salt 21·Cl·0.5H2O (bottom), viewed down the c axis, with the b axis horizontal. | |
Table 2 Selected molecular geometry for naphthalene cation 21 in 21·Cl·0.5H2O
|
|
| Compound |
T/K |
a/Å |
b/Å |
θ/° |
Me2NH⋯H/Å |
|
21·Cl
|
260 |
1.754(6) |
1.442(5) |
115.4(3) |
2.18 |
|
|
150 |
1.705(3) |
1.452(3) |
115.23(18) |
2.24 |
|
|
T/K |
φ/° |
ψ/° |
τ/° |
ζ/° |
ξ/° |
|
21·Cl
|
260 |
114.2(4) |
128.9(4) |
46.4(6) |
56.6(5) |
−66.9(5) |
|
|
150 |
113.7(2) |
129.0(2) |
45.7(3) |
55.7(3) |
−66.7(3) |
The crystal structures of the dinitrile and cyanoester salts 13·Cl·0.5H2O and 21·Cl·0.5H2O have similar orthorhombic unit cells, and the space group is Fdd2 in both. For the cyano ester salt, the cations are organised into columns along the b axis. To accommodate the ester group in place of the cyano group the a axis is expanded by ca. 9%. The c axis is contracted by 5%. Cations are connected in pairs by the Cl⋯HOH⋯Cl unit and are related to each other by the two fold axis through the water molecule. A second pair of cations related to the first by a 21 axis provides the second component of the column. However, there is a large difference in the relative orientation of the cations and the Cl⋯HOH⋯Cl unit when compared to the dinitrile salt (Fig. 10). First of all, the Cl⋯O⋯Cl angle is much wider (108.7 cf. 82.9°) which places the chloride ions further apart (5.310 cf. 4.220 Å), and the Cl⋯Cl vector is more closely aligned along the b axis lying at 9.2° cf. 29.2°. This pushes the two symmetry related cations further away from each other, with their protonated nitrogen atoms now 8.994 Å apart cf. 5.522 Å in 13·Cl·0.5H2O. This is further illustrated by the much larger Cl⋯O⋯Cl⋯(H)N torsion angle of 141.7 cf. 59.5°, though the Me2N(H)⋯Cl⋯O angle remains almost constant. (91.9 cf. 89.1°).
Solid state NMR studies on 13·Cl·0.5H2O
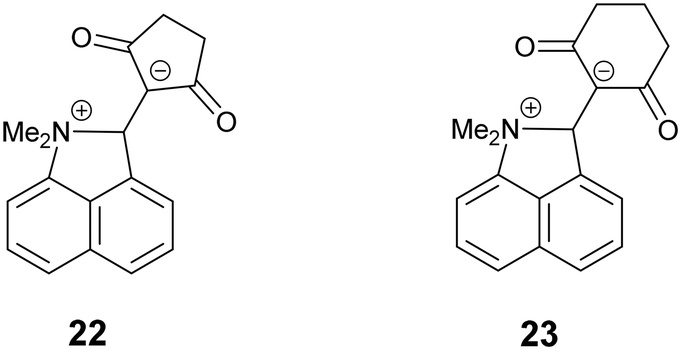
To gain some further insight into the increasing progress of the reaction between the functional groups in the crystalline salt 13·Cl·0.5H2O on decreasing the temperature, the analogue which carried a 13C label at the attacked alkene carbon was prepared and crystallised and then converted to a powder and studied by variable temperature solid state 13C NMR. The expected unit cell dimensions were confirmed by powder X-ray diffraction The “open” analogue 3 (Me2N⋯C: 2.413 Å) and the “closed” analogues 4 and 22–23 (Me2N⋯C: 1.612(2)–1.651(3) Å) provide references for the chemical shift of this labelled carbon atom at its possible extreme positions: 165.6 and 88.9–99.1 ppm respectively.16,17 In solution both the salt 13·Cl and free base 12 show that they are in the “open” structure with the alkene carbon atoms at 163.1 and 73.4 ppm for 13·Cl and 165.8 and 73.0 ppm for 12 in CD2Cl2.
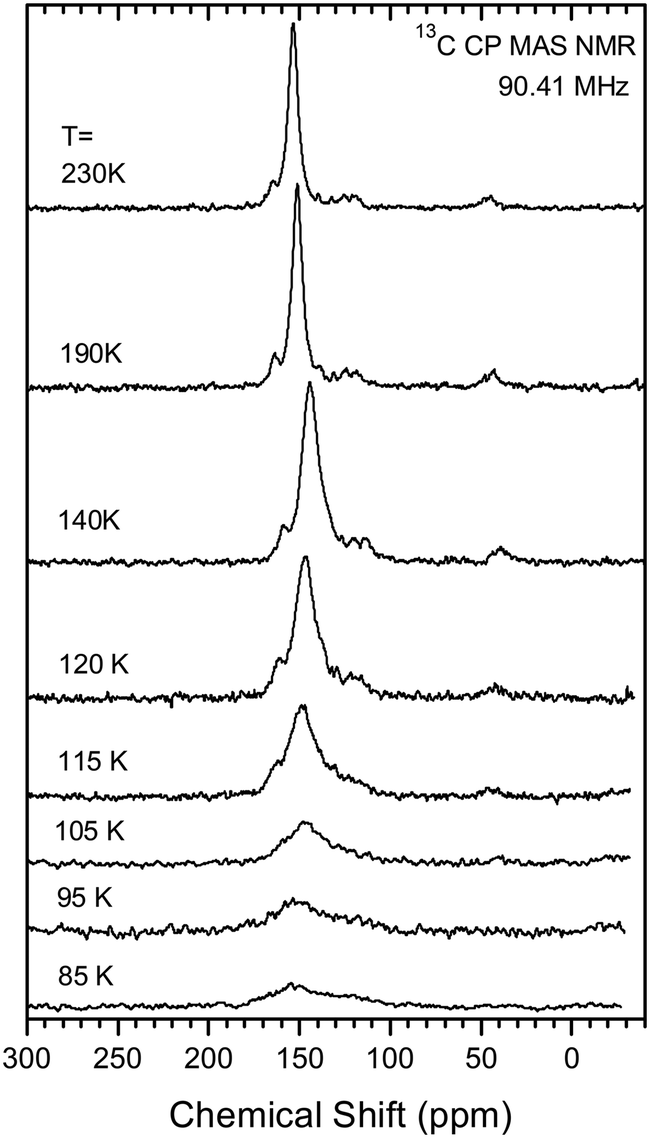
As pointed out above, the two possible scenarios of the structural changes on cooling should show up in the NMR spectrum in a different way. If there is just a steady reduction in the Me2N⋯C distance on cooling, one would expect the continuous change in the chemical shift of a single line of the labelled carbon from 156 ppm to ∼90 ppm. In the other scenario one would expect two lines in the spectrum corresponding to the high- and low-temperature phases, with the intensity ratio depending on the temperature. The change with temperature in the NMR spectrum of the labelled carbon is given in Fig. 11. At high temperature the spectrum shows only one signal at a chemical shift of ca. 153 ppm at 230 K falling to ca. 144 ppm at 120 K. On decreasing the temperature further, the signal amplitude decreases drastically, it becomes broader and the maximum moves back towards 150 ppm at 85 K. From this behaviour one can rule out the first scenario with a steady change in the Me2N⋯C bond/interaction distance. Assuming coexistence of the two phases as proposed in the second scenario seems not to be fully adequate as well. The most probable explanation for such temperature dependence of the spectrum is assuming a slow exchange between the two configurations of the molecule. According to the general understanding of the NMR line shape of molecular switching between two states26 the broadening of the line occurs when the switching rate is close to the difference of the resonances in these states. In the present case, we expect severe broadening of the line if the exchange rate between the two configurations of the molecule is of the order of few kHz.
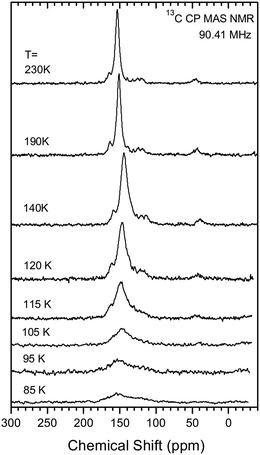 |
| | Fig. 11 Temperature dependence of 13C CP MAS NMR spectrum of 13·Cl·0.5H2O sample. The details of the measurement are given in ESI.† | |
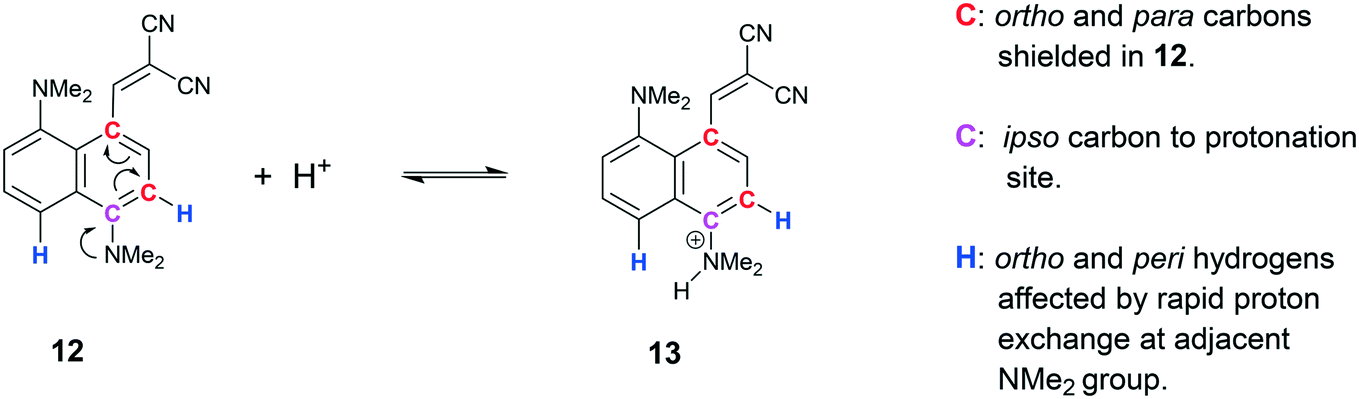
Solution NMR studies on 13·Cl
The solution state 1H and 13C NMR spectra of 13·Cl have been studied in both CD2Cl2 and in DMSO-d6. In CD2Cl2 there appears to be a fast equilibrium between the cation 13 and the corresponding free base 12. This leads to broadening of the 13C signals ipso, ortho and para to the protonated NMe2 group (Scheme 1). When not protonated electron donation into the naphthalene ring leads to upfield shifts of the ortho and para carbons and a downfield shift of the ipso carbon.27 Furthermore, the two neighbouring H signals (peri and ortho) are also broadened. In DMSO-d6 the equilibrium is more strongly towards the free base. Of further note is the upfield movement of the –C(CN)2 carbon from 73.4 to 66.2 ppm on changing from CD2Cl2 to DMSO-d6, consistent with an increase in electron density at this carbon. The corresponding carbanion would be expected to resonate at ca. 20 ppm.28 The observed change in shift may be due to the more polar solvent stabilising a more advanced interaction between the peri groups. We have indeed observed a shift for the corresponding carbon at 65.5 ppm for 11 in CDCl3. Interestingly the SSNMR spectrum shows this carbon at 56.5 ppm at 260 K.
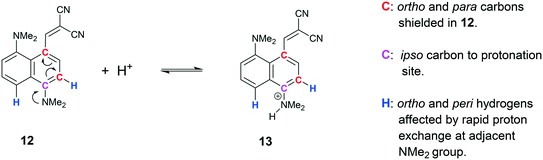 |
| | Scheme 1 Equilibrium of cation 13 with free base 12 observed in solution by NMR. | |
Mapping of the reaction coordinate
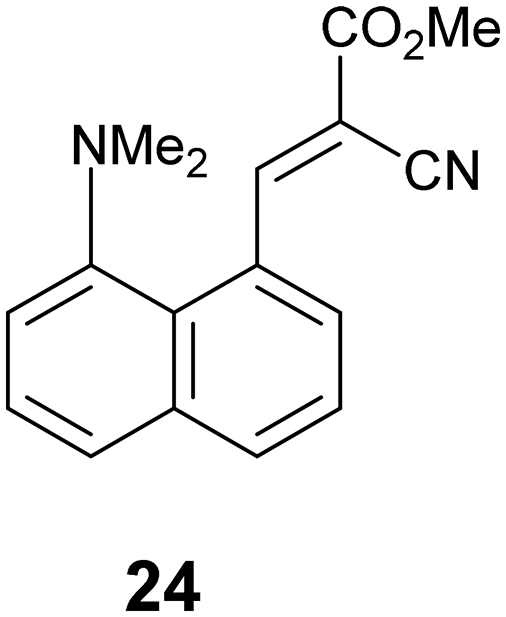
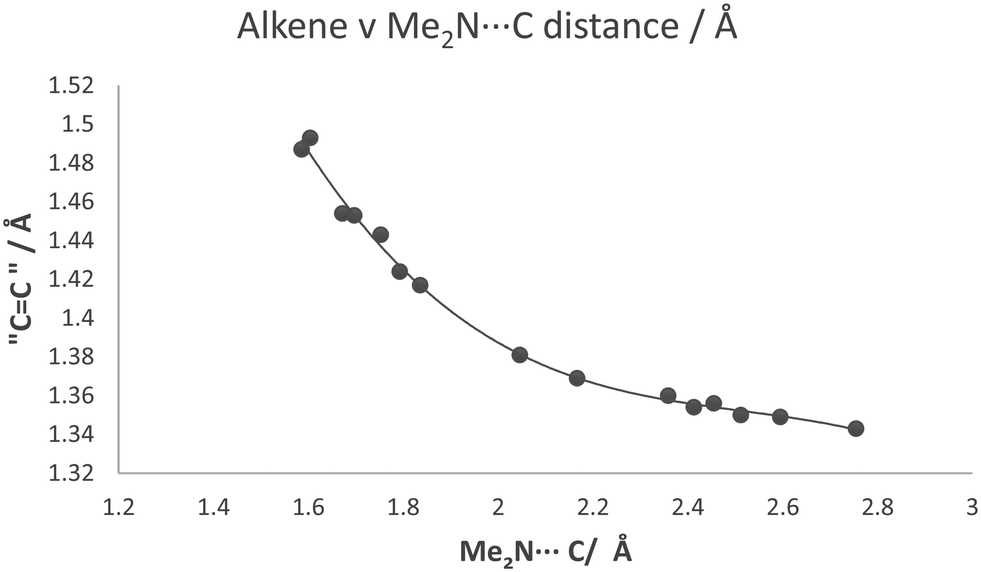
The partial breaking of the alkene bond in cation 13, in which it changes from a bond length of 1.369 Å at 200 K to 1.431 Å at 100 K, takes place while the Me2N⋯C distance changes from just 2.167 Å to 1.749 Å. In the cyclised biphenyl derivative 7, the Me2N–C bonds are shorter (1.604(3) and 1.586(3) Å), and the “broken” alkene bonds are longer (1.487 and 1.493 Å), while in several compounds containing the C–C(−)(CN)2 fragment the single C–C(CN)2 bond has a length in the range 1.514–1.536 Å.29 In contrast, for the unsubstituted naphthalene 3 with a Me2N⋯C separation of 2.413 Å, the alkene bond is 1.354 Å long. These data give insight into the bond formation and breaking in an aza-Michael reaction. For 13·Cl at 150 K the nucleophile approaches close to the alkene (Me2N⋯C: 2.098 Å) with only a small increase of 0.024 Å in the length of double bond, but then on cooling to 100 K the alkene bond is partially broken while the Me2N⋯C distance contracts by just 0.349 Å. To get a fuller picture of this correlation the corresponding distances from a range of structures containing an interaction or bond between a dimethylamino group and an ethene-2,2-dinitrile (3, 7, 8, 11, 12, 13@200 K, 13@150 K, 13@140 K, 13@128 K, 13@120 K, 13@100 K) are plotted in Fig. 12, from a Me2N⋯C separation of ca. 2.8 Å in acenaphthene 8 to ca. 1.6 Å in the biphenyl 7. We also included three data points where one of the nitriles is replaced by a methyl ester, two from 18·Cl (@260 K and 150 K) and also the corresponding peri-naphthalene with no extra peri substituents 24 (ESI†). The graph shows a curved correlation, though of course it is short of data in the range 1.75–2.01 Å.
Goodman's calculations on the transition state for attack by methylamine on acrolein and acrylamide where the double bond is activated by an aldehyde or a primary amide group show MeN(H2)⋯C contacts of 2.04 and 1.84 Å, and alkene bond lengths of 1.393 and 1.412 Å respectively.5 From our crystallographic data, where the reaction is intramolecular and thus the product has the additional consideration of strain energy due to formation of a fused ring, a very approximate estimate for the position of the transition state from the crystallographic data could be the mid-point of the zone in which the Me2N⋯C distance changes quickly, i.e. with a Me2N⋯C separation of ca. 1.94 Å and the former CC bond at ca. 1.40 Å.
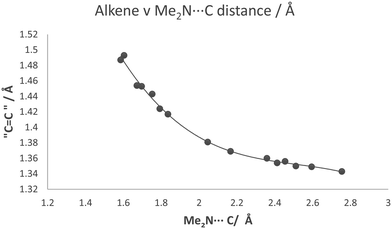 |
| | Fig. 12 For a series of compounds containing the Me2N⋯C(H)C(CN)2 grouping, a plot of the length of approached alkene bond against Me2N⋯C separation. | |
Conclusion
This study provided insight into the correlation between the bond breaking and bond formation processes in an aza-Michael reaction using groups positioned at the peri-positions of a naphthalene ring, with their separation modified by steric interactions at the opposite peri-positions. An ethylene bridge leads to increased separation while repulsion between two phenyl groups leads to decreased separation. Having established the principle, then further compression between the reactive groups can be attempted using larger substituents. Of particular interest is the reversible behaviour with temperature of the naphthalene based salt 13·Cl·0.5H2O in which the Me2N⋯C separation closes from 2.167 to 1.749 Å on cooling from 200 to 100 K. The balance of evidence currently suggests that there is a dynamic equilibrium between two close structural forms in the range 140–128 K. Using the Pauling expression for bond order, at 200 K the peri N⋯C interaction corresponds to a bond order of ca. 0.15, while at 100 K the bond order is ca. 0.47,30 though these may be underestimates since the formula applies to bond between neutral atoms. Further investigations are warranted, including charge density measurements by accurate X-ray diffraction at different temperatures on this material and closely related salts, a structural study of the effect of externally applied pressure on the separation between the reactive groups as well as appropriate calculations and spectroscopic investigations. Single crystal to single crystal phase transitions are well known in molecular materials, though usually involve the reorientation of the molecules relative to each other and reorganisation of the attractive interactions between them.31 In contrast, the case discussed here involves just a step change in the close intramolecular interaction/partial bond formation between two groups, and is very rare. Crystallographic studies suggest that at the lower limit (1.75 Å) there is some form of bond between the reactive groups, given the strong induction of pyramidality at the carbon atom and extension of the alkene bond, though it may be more appropriate to consider this a partially formed bond. Charge density studies may provide further insight into this. Only a few N⋯C separations in this range are known, in particular in systems with a transannular N⋯CO interaction: clivorine 5 with a N⋯C separation of 1.993(3) Å and a somewhat lengthened CO bond (1.258(3) Å), and senkirkine 6 where the N⋯C separation is longer (2.293 Å) but the carbonyl bond in the usual range (1.213 Å).18 Just why the cation 13 can tolerate the higher energy interactions between functional groups in the 2.2–1.7 Å region may be due to the role of strong hydrogen bonding stabilising this particular type of crystal structure.
Experimental
Full details of the synthesis and characterisation of new substances, the determination of crystal structures by X-ray diffraction, solid-state NMR studies and calculations are provided in the ESI.† Crystal structures have been deposited at the Cambridge Structural Database with numbers: CCDC 2016747–2016754 and 2019629.
Conflicts of interest
There are no conflicts of interest to report.
Acknowledgements
We thank Nottingham Trent University for a studentship (JCB) and for financial support for materials. We wish to thank Dr Tõnis Pehk for additional assignment of the solution NMR resonances. I. H. and R. S. were supported by the European Regional Development Fund (Grant No. TK134), and by the Estonian Research Council (PRG4, IUT23-7).
References
- J. C. Polanyi and A. H. Zewail, Acc. Chem. Res., 1995, 28, 119–122 Search PubMed; D. Zhong and A. R. Zewail, J. Phys. Chem. A, 1998, 102, 4031–4058 Search PubMed; A. R. Attar, A. Bhattacherjee and S. R. Leone, J. Phys. Chem. Lett., 2015, 8, 5072–5077 Search PubMed; A. A. Ischenko, P. M. Weber and R. J. D. Miller, Russ. Chem. Rev., 2017, 86, 1173–1253 Search PubMed.
- L. D. Jacobson, A. D. Bochevarov, M. A. Watson, T. F. Hughes, D. Rinaldo, S. Ehrlich, T. B. Steinbrecher, S. Vaitheeswaran, D. M. Philipp, M. D. Halls and R. A. Friesner, J. Chem. Theory Comput., 2017, 13, 5780–5797 Search PubMed; A. B. Birkholz and H. B. Schlegel, J. Comput. Chem., 2015, 36, 1157–1166 Search PubMed; L. K. Beland, P. Brommer, F. El-Mellouhi, J.-F. Joly and N. Mousseau, Phys. Rev. E, 2011, 84, 046704 Search PubMed.
-
M. B. Smith, March's Advanced Organic Chemistry, John Wiley and Sons Inc., New Jersey, 8th edn, 2020 Search PubMed; P. Wadhu, A. Kharbanda and A. Sharma, Asian J. Org. Chem., 2018, 7, 634–661 Search PubMed; C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988–999 Search PubMed; C. F. Nising and S. Bräse, Chem. Soc. Rev., 2008, 37, 1218–1228 Search PubMed; R. D. Little, M. R. Masjedizadeh, O. Wallquist and J. I. McLoughlin, Org. React., 1995, 47, 315–552 Search PubMed.
- R. W. Bates, W. Ko and V. Barát, Org. Biomol. Chem., 2020, 18, 810–829 Search PubMed; M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2019, 17, 3670–3708 Search PubMed; M. Sánchez-Roselló, J. L. Aceňa, A. Simón-Fuentes and C. Del Pozo, Chem. Soc. Rev., 2014, 43, 7430–7453 Search PubMed; Z. Amara, J. Caron and D. Joseph, Nat. Prod. Rep., 2013, 30, 1211–1225 Search PubMed; A. Y. Rulev, Russ. Chem. Rev., 2011, 8, 197–218 Search PubMed.
- T. E. H. Allen, M. N. Grayson, J. M. Goodman, S. Gutsell and P. J. Russell, J. Chem. Inf. Model., 2018, 58, 1266–1271 Search PubMed.
- J. A. Izzo, Y. Myshchuk, J. S. Hirschi and M. J. Vetticatt, Org. Biomol. Chem., 2019, 17, 3934–3939 Search PubMed; S. Romanini, E. Galletti, L. Caruana, A. Mazzanti, F. Himo, S. Santoro, M. Fochi and L. Bernardi, Chem. – Eur. J., 2015, 21, 17578–17582 Search PubMed; E. H. Krenske, R. C. Petter, Z. Zhu and K. N. Houk, J. Org. Chem., 2011, 76, 5074–5081 Search PubMed; D. Mulliner, D. Woudrousch and G. Schüürmann, Org. Biomol. Chem., 2011, 9, 8400–8412 Search PubMed; J. A. H. Schwöbel, J. C. Madden and M. T. D. Cronin, SAR QSAR Environ. Res., 2010, 21, 693–710 Search PubMed; E. E. Kwan and D. A. Evans, Org. Lett., 2010, 12, 5124–5127 Search PubMed.
-
Structure Correlation, ed. H.-B. Bürgi and J. D. Dunitz, VCH, Weinheim, 1994, vol. 1 Search PubMed; E. Hupf, M. Olaru, C. I. Rat, M. Fugel, C. B. Hübschle, E. Lork, S. Grabowsky, S. Mebs and J. Beckmann, Chem. – Eur. J., 2017, 23, 10568–10579 Search PubMed.
- J. C. Bristow, M. A. Addicoat and J. D. Wallis, CrystEngComm, 2019, 21, 1009–1018 Search PubMed; F. R. Knight, R. A. M. Randall, K. S. Athukorala Arachchige, L. Wakefield, J. M. Griffin, S. E. Ashbrook, M. Bühl, A. M. Z. Slavin and J. D. Woolins, Inorg. Chem., 2012, 51, 11087–11097 Search PubMed; K. S. M. Mickoleit, R. Kempe and H. Oehme, Angew. Chem., Int. Ed., 2000, 39, 1610–1612 Search PubMed; G. P. Schiemenz, Z. Naturforsch., B: J. Chem. Sci., 2006, 61, 535–554 Search PubMed.
- K. Yamamoto, N. Oyamada, S. Xia, Y. Kobayashi, M. Yamaguchi, H. Maeda, H. Nishihara, T. Uchimaru and E. Kwon, J. Am. Chem. Soc., 2013, 135, 16526–16532 Search PubMed.
- D. Pla, O. Sadek, S. Cadet, B. Mestre-Voegtlé and E. Gras, Dalton Trans., 2015, 44, 18340–18346 Search PubMed.
- H. A. Staab and T. Saupe, Angew. Chem., Int. Ed. Engl., 1988, 27, 865–879 Search PubMed.
- S. Sarkar and T. N. Guru Row, IUCrJ, 2017, 4, 37–49 Search PubMed; P. R. Mallinson, G. T. Smith, C. C. Wilson, E. Grech and K. Wozniak, J. Am. Chem. Soc., 2003, 125, 4259–4270 Search PubMed.
- W. B. Schweizer, G. Procter, M. Kaftory and J. D. Dunitz, Helv. Chim. Acta, 1978, 61, 2783–2808 Search PubMed.
- A. Wannebroucq, A. P. Jarmyn, M. B. Pitak, S. J. Coles and J. D. Wallis, Pure Appl. Chem., 2016, 88, 317–331 Search PubMed.
- N. Mercadal, S. P. Day, A. Jarmyn, M. B. Pitak, S. J. Coles, C. Wilson, G. J. Rees, J. V. Hanna and J. D. Wallis, CrystEngComm, 2014, 16, 8363–8374 Search PubMed.
- P. C. Bell and J. D. Wallis, Chem. Commun., 1999, 257–258 Search PubMed.
- A. Lari, M. B. Pitak, S. J. Coles, G. J. Rees, S. P. Day, M. E. Smith, J. V. Hanna and J. D. Wallis, Org. Biomol. Chem., 2012, 10, 7763–7779 Search PubMed; J. O'Leary, W. Skranc, X. Formosa and J. D. Wallis, Org. Biomol. Chem., 2005, 3, 3273–3283 Search PubMed.
- K. B. Birnbaum, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 2825–2833 Search PubMed; G. I. Birnbaum, J. Am. Chem. Soc., 1974, 96, 6165–6168 Search PubMed.
- H.-B. Bürgi, J. D. Dunitz and E. Schefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 Search PubMed.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 Search PubMed; R. W. Newberry, G. J. Bartlett, B. VanVeller, D. N. Woolfson and R. T. Raines, Protein Sci., 2014, 23, 284–288 Search PubMed.
- J. O'Leary and J. D. Wallis, Org. Biomol. Chem., 2009, 7, 225–228 Search PubMed.
- Y. Ishigaki, T. Shimajiri, T. Takeda, R. Takoono and T. Suzuki, Chem, 2018, 4, 795–806 Search PubMed.
-
S. Pusch, D. Schollmeyer and T. Opatz, Exp. Cryst. Struct. Determ., 2016, REFCODE: UTOBUC Search PubMed.
- I. I. Schuster, A. J. Freyer and A. L. Rheingold, J. Org. Chem., 2000, 65, 5752–5759 Search PubMed.
- R. X. Fischer and E. Tillmans, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 775–776 Search PubMed.
-
C. P. Slichter, Principles of Magnetic Resonance, Springer-Verlag, 3rd edn, 1992, p. 596 Search PubMed.
- D. Škalamera, L. Cao, L. Isaacs, R. Glaser and K. Mlinarić-Majerski, Tetrahedron, 2016, 72, 1541–1546 Search PubMed.
- W. F. Richter, K. Hartke, W. Massa and G. Münninghof, Chem. Ber., 1989, 122, 1133–1137 Search PubMed.
- H. Song, Y. Kim, J. Park, Y. H. Ko and E. Lee, Eur. J. Org. Chem., 2017, 1231–1235 Search PubMed; L. Carlucci, G. Ciani, D. M. Proserpio and A. Sironi, Angew. Chem., Int. Ed. Engl., 1996, 35, 1088–1090 Search PubMed REFCODES: KANZUX, NACVOC, NACVUI, ULAVAG; M. Y. Belikov, M. Y. Ievlev, I. V. Belikova, O. V. Ershov, V. A. Tafeenko and M. D. Surazhskaya, Chem. Heterocycl. Compd., 2015, 51, 518–525 Search PubMed.
- N. E. Brese and M. O'Keefe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 Search PubMed;
I. D. Brown, 2016, https://www.iucr.org/resources/data/datasets/bond-valence-parameters, (accessed July 2020);
L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, New York, USA, 1960 Search PubMed.
- V. K. Srirambhatla, R. Guo, D. M. Dawson, S. L. Price and A. Florence, Cryst. Growth Des., 2020, 20, 1800–1810 Search PubMed; M. M. H. Smets, E. Kalkman, A. Krieger, P. Tinnemans, H. Meeks, E. Vlieg and H. M. Cuppen, IUCrJ, 2020, 7, 331–341 Search PubMed; C. Ge, J. Liu, X. Ye, Q. Han, L Zhang, S. Cui, Q. Guo, G. Liu, Y. Liu and X. Tao, J. Phys. Chem. C, 2018, 122, 15744–15752 Search PubMed; R. Enjalbert and J. Galy, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 1005–1010 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Ivo
Heinmaa
c,
Raivo
Stern
b,
Ivo
Heinmaa
c,
Raivo
Stern
 For comparative purposes, the corresponding methyl cyanoester 20 was prepared and converted to its chloride salt 21·Cl. Its crystal structure was measured at 260 and 150 K. The crystal structure of this salt resembles that of the dinitrile chloride salt 13·Cl·0.5H2O in a number of respects. It includes a molecule of water per two chloride anions, and has similar cell parameters and the same space group (Fig. 9 and 10). However, the Me2N⋯C separation in 21·Cl·0.5H2O at 260 K is 1.754(6) Å, significantly shorter than for the dinitrile salt 13·Cl·0.5H2O at 200 K (2.167(4) Å), and the partially broken alkene bond has a length of 1.442(5) cf. 1.369(3) Å (Table 2). Thus this structure at 260 K most closely resembles that of the dinitrile salt at 100 K. On cooling to 150 K the Me2N⋯C separation contracts by 0.049 Å to 1.705(3) Å, and the former alkene bond extends slightly to 1.452(3) Å. So in this case, there is a substantial degree of N–C bond formation and C
For comparative purposes, the corresponding methyl cyanoester 20 was prepared and converted to its chloride salt 21·Cl. Its crystal structure was measured at 260 and 150 K. The crystal structure of this salt resembles that of the dinitrile chloride salt 13·Cl·0.5H2O in a number of respects. It includes a molecule of water per two chloride anions, and has similar cell parameters and the same space group (Fig. 9 and 10). However, the Me2N⋯C separation in 21·Cl·0.5H2O at 260 K is 1.754(6) Å, significantly shorter than for the dinitrile salt 13·Cl·0.5H2O at 200 K (2.167(4) Å), and the partially broken alkene bond has a length of 1.442(5) cf. 1.369(3) Å (Table 2). Thus this structure at 260 K most closely resembles that of the dinitrile salt at 100 K. On cooling to 150 K the Me2N⋯C separation contracts by 0.049 Å to 1.705(3) Å, and the former alkene bond extends slightly to 1.452(3) Å. So in this case, there is a substantial degree of N–C bond formation and C