
Halogen bonded metal bis(dithiolene) 2D frameworks†
Hadi
Hachem
,
Olivier
Jeannin
 ,
Marc
Fourmigué
*,
Frédéric
Barrière
and
Dominique
Lorcy
*
,
Marc
Fourmigué
*,
Frédéric
Barrière
and
Dominique
Lorcy
*
Univ Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: marc.fourmigue@univ-rennes1.fr; dominique.lorcy@univ-rennes1.fr
First published on 6th May 2020
Abstract
Halogenated metal dithiolene complexes have been designed together with halogen bond acceptor sites in order to favor halogen bonding interactions between these electroactive architectures. For the synthesis, we explored the reactivity of the protected N-tert-butyl-1,3-thiazoline-2-thione-4,5-dithiolate ligand towards DMSO and I2 and successfully prepared the appropriate protected 2-iodo-1,3-thiazole-4,5-dithiolate ligand, (I-tzdt). Nickel and gold monoanionic complexes, [M(I-tzdt)2]−1 (M = Au, Ni), have been synthesized and the Au complexes were isolated in the solid state in their trans and cis isomers. The trans-Ni and trans-Au complexes are isostructural and X-ray diffraction analysis confirmed the occurrence of short and directional I⋯S halogen bonding interactions leading to 2D metal bis(dithiolene) frameworks with channels hosting the counterions. On the other hand, additional chalcogen⋯chalcogen interactions are observed for the cis-Au complex, transforming the 2D into a 3D network.
Introduction
In the broad field of (semi) conducting molecular materials, besides the appropriate intrinsic properties of the precursor molecules, the electronic properties of the materials strongly rely on their solid-state organization and the strength of the intermolecular interactions,1,2 allowing for possible charge carrier delocalization and high conductivities. Among them, π–π interactions and chalcogen⋯chalcogen3 interactions have been thoroughly investigated. In order to modify these organizations, other more directional and predictable intermolecular interactions have been used and particularly hydrogen bonding (HB).4 For example, tetrathiafulvalene derivatives5 or dithiolene complexes6–9 were accordingly functionalized with HB donor groups such as alcohols, amides or imides. More recently, halogen bonding was rediscovered as an efficient and directional tool to control solid state associations.10,11 In conducting materials derived from TTF derivatives, this approach has led to numerous salts of iodinated TTFs acting as XB donors.5,12 Indeed, oxidation of the TTF was shown to activate the σ-hole of the iodine substituent13 and reinforce its interaction with the counterions in cation radicals salts. In conducting materials derived from dithiolene complexes, all examples reported so far consider dithiolene complexes only as XB acceptors. For example, [Ni(mnt)2],14 [Ni(dmit)2]15,16 [Pd(dmit)2]17 (Scheme 1a) have been reported to interact, through the nitrile nitrogen atom or through the thiocarbonyl sulfur atom, with numerous halogenated cations.18 Looking for dithiolene complexes which could act themselves as XB donors reveals a very limited number of halogenated complexes, and most of them were not structurally characterized. One finds for example derivatives of the tetrachlorobenzene-1,2-dithiolate,19 the 4,5-dichlorobenzene-1,2-dithiolate20 or the tetrabromobenzene-1,2-dithiolate (Scheme 1b).21 One single example reports XB interaction in the crystal structure of the neutral nickel complex [Ni(ClPhdt)2] (ClPhdt = 2-(p-chlorophenyl)-1,2-dithiolate).22 The Cl⋯Cl intermolecular contact is described as a weak type-II contact with a Cl⋯Cl distance of 3.275 Å (Reduction Ratio, RR = 0.94).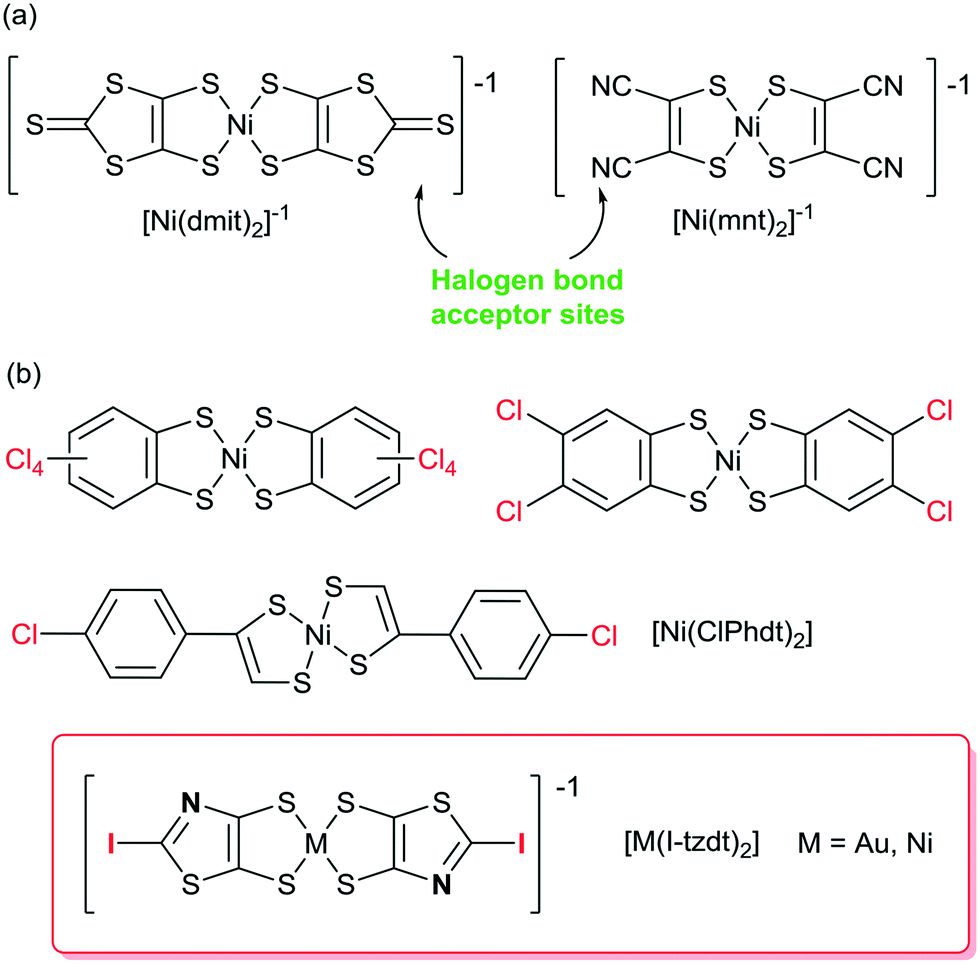 | ||
| Scheme 1 (a) Dithiolene complexes known as XB acceptors. (b) Dithiolene complexes as potential XB donors. | ||
As it is well known that the XB donor ability strength decreases in the order I > Br > Cl > F, it is thus of interest to build metal bis(dithiolene) complexes functionalized by iodine atoms to act as XB donor groups. Furthermore, since the conducting and/or magnetic properties of bis(dithiolene) complexes are essentially due to the intermolecular interactions of their electroactive skeleton, it is also of interest to build such complexes with, at the same time, not only the XB donor moiety (iodine) but also efficient XB acceptor sites (sp2 nitrogen, sp2 sulfur, …) to potentially increase these interactions.
Based on our know-how for the synthesis of dithiolene complexes, we investigated the synthesis of 2-iodo-1,3-thiazole-4,5-dithiolate (I-tzdt) ligand and its ability to generate the corresponding nickel and gold monoanionic complexes. This ligand fulfills both requirements by bearing simultaneously the iodine atom and the sp2 nitrogen atom of the thiazole core. In this study, we report the syntheses of monoanionic and neutral nickel and gold complexes as well as their structural and electronic properties. Electrostatic potential (ESP) calculations carried out on the precursors and the complexes provide a complementary understanding of the organization of the molecules in the solid state.
Results and discussion
The proligands, syntheses and properties
In order to form the iodinated thiazole core, we first considered the strategy developed in the literature for the synthesis of 2-iodo-1,3-thiazole derivatives which can usually be successfully prepared starting from either 2-amino-1,3-thiazole derivatives (nBuONO, KCuI2),23 or 4,5-disubstituted-1,3-thiazole by subsequent lithiation and iodination at the 2 position.24 However, none of these approaches can be easily used to form the targeted 2-iodothiazole dithiolate ligand. We recently reported an original reactivity of N-tert-butyl-1,3-thiazoline-2-thione derivatives such as 1 in the presence of alkyliodides such as MeI to form 2-alkylthiothiazole dithiolate proligands (Scheme 2a).25 The advantage of this transformation is that the proligand of the dithiolate is already formed and the reaction leads to a 2-functionalized thiazole core without modifying the protected dithiolate ligand. Indeed, 1,3-dithiole-2-ones are well known to react in basic medium (MeONa, tBuOK, KOH) to generate the corresponding dithiolate ligand. Therefore, we focused on the reactivity of the proligand 1 in the presence of iodine itself as a possible way to introduce iodine on a thiazole ring. Looking at the literature, we found that the use of iodine in DMSO is a versatile tool which could promote many oxidative reactions26 or aromatizations,27 but also the formation of iodinated alkenes from arylacetylenes.28 Thus we decided to explore the reactivity of 1 in DMSO in the presence of iodine (Scheme 2b).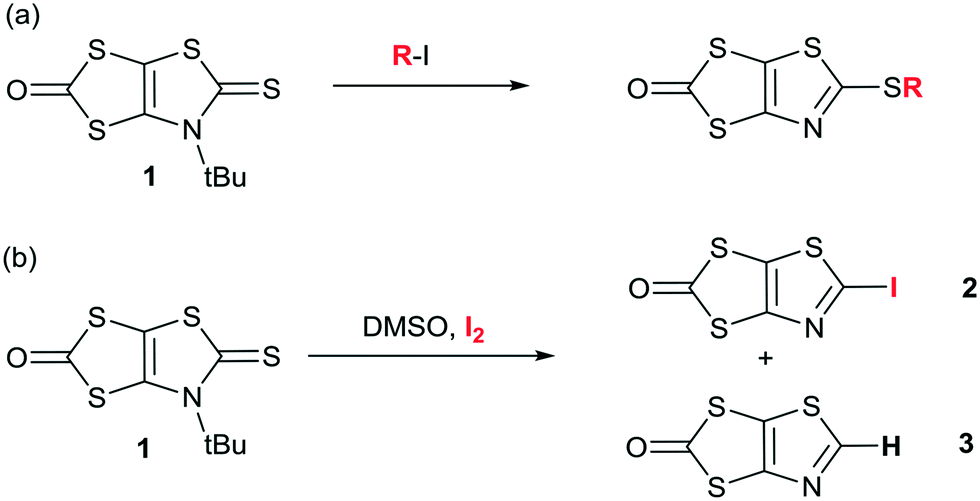 | ||
| Scheme 2 Synthetic path to the proligands 2 and 3. | ||
To our delight, this procedure afforded the desired proligand 2 with a 2-iodothiazole core in 70% yield. The optimized conditions are 1 mmol of 1 in 2 mL of DMSO in the presence of 4 equivalents of I2 at 55 °C for 4 h. We also found that the non-substituted 1,3-thiazole 3 was formed in the medium but in low yields (5–10%) (Scheme 2b). Crystals of sufficient quality for an X-ray diffraction study were obtained by slow concentration of a CH2Cl2 solution of both 2 and 3. Their solid-state organization is reported in Fig. 1 and 2 respectively. In both cases the fused heterocyclic cores are planar. For compound 2, the sulfur, oxygen and nitrogen atoms of the molecule can act as a Lewis base and form intermolecular halogen bonding with the iodine atom of a neighboring molecule. It appears that a strong XB is established with the thiazole nitrogen atom, with a I⋯N interatomic distance at 3.118(17) Å, corresponding to a reduction ratio (RR) of 88.3% relative to the sum of the van der Waals radii (3.53 Å) (Fig. 1), and a marked linearity as the C–I⋯N angle is found at 157.6(3)°. Besides, a closer look at the structure shows also the existence of chalcogen bonding (ChB) interactions in the same plane with a short S⋯O interatomic distance of 2.940(14) Å that corresponds to a reduction ratio of 88.5% relative to the van der Waals contact distance (3.32 Å).
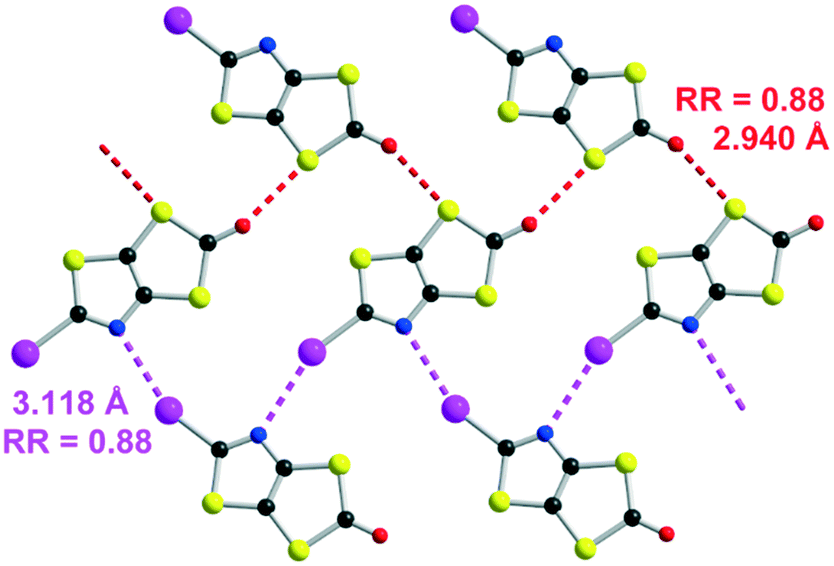 | ||
| Fig. 1 Detail of the I⋯N XB (pink dotted line) and S⋯O ChB (red dotted line) interactions in 2. | ||
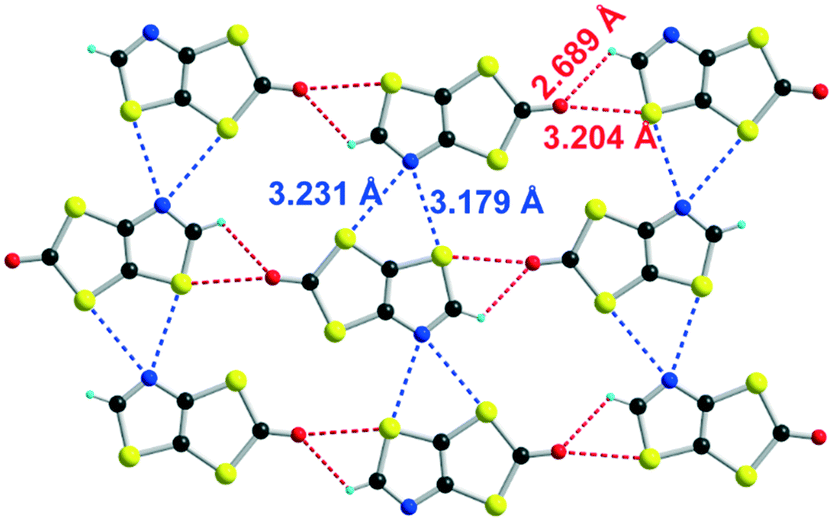 | ||
| Fig. 2 Details of the shortest intermolecular H⋯O, S⋯O and S⋯N contacts in 3. | ||
Unlike the iodo proligand 2, no strong intermolecular interactions are observed in 3. As shown in Fig. 2, the shortest H⋯O, S⋯O and S⋯N contacts correspond respectively to RR values of 0.99, 0.96 and 0.95.
Electrostatic potential (ESP) calculations have been carried out on the optimized geometry of the two molecules that have been crystallographically characterized, namely 2 and 3. These calculations were performed in order to rationalize the interactions taking place in the crystal. In 2 (Fig. 3a), two positive maxima are found, one inbetween the sulfur atoms belonging to the thiazole and the dithiole rings (+36.87 kcal mol−1) and one, as expected, on the iodine atom (+34.73 kcal mol−1). The most negative extrema are found on the carbonyl oxygen atom (−26.56 kcal mol−1) and on the thiazole nitrogen atom (−21.81 kcal mol−1). This calculated charge repartition is in good agreement with the structural organization of molecule 2 (Fig. 1) where predominant ChB (Sδ+⋯Oδ−) and XB (Iδ+⋯Nδ−) interactions are observed. For the dithiol-2-thione 3 with no iodine atom, the situation is somehow comparable, with a larger but more spreaded positive area around the sulfur and hydrogen atoms of the thiazole ring (+37.29 kcal mol−1), and a smaller, diffuse one inbetween the sulfur atoms belonging to the thiazole and the dithiole rings (+33.82 kcal mol−1). We note that albeit weaker in 3 than in 2, the shortest intermolecular interactions are also systematically associated with favorable δ+⋯δ− contacts.
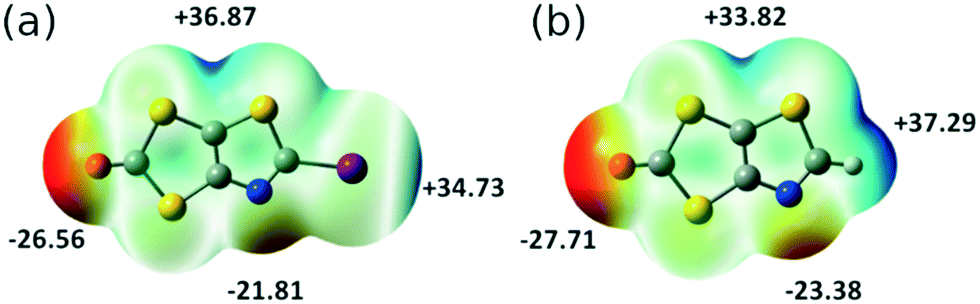 | ||
| Fig. 3 Molecular electrostatic potential surfaces mapped at 0.001 e− au−3 isodensity surface for (a) 2; (b) 3. The common color scale ranges from −28 kcal mol−1 (red) to +40 kcal mol−1 (blue). | ||
The monoanionic complexes [M(I-tzdt)2]−1 (M = Au, Ni)
The monoanionic complexes [M(I-tzdt)2]−1 (M = Au, Ni) were prepared (Scheme 3) as Bu4N+ or Ph4P+ salts by adding sodium methanolate to the proligand 2 to generate the dithiolate ligand, followed by the successive addition of KAuCl4 or NiCl2·H2O, and Bu4NBr (Scheme 3). According to a similar procedure, but using 3 as proligand and Ph4PBr in the last step, we also prepared the monoanionic complex [Ph4P][Au(H-tzdt)2]. Interestingly, the 1H NMR spectrum of this complex reveals clearly two signals for the proton of the thiazole core (Fig. S4†). Due to the dissymmetry of the ligand, all these complexes can exist under two different configurations, the cis and the trans isomers. Therefore, these two singlets of equal intensity are attributed to the presence of the cis and trans isomers in equal proportion in solution. Recrystallizations afforded crystals amenable to single crystal X-ray diffraction only for the iodinated gold and nickel complexes (see below).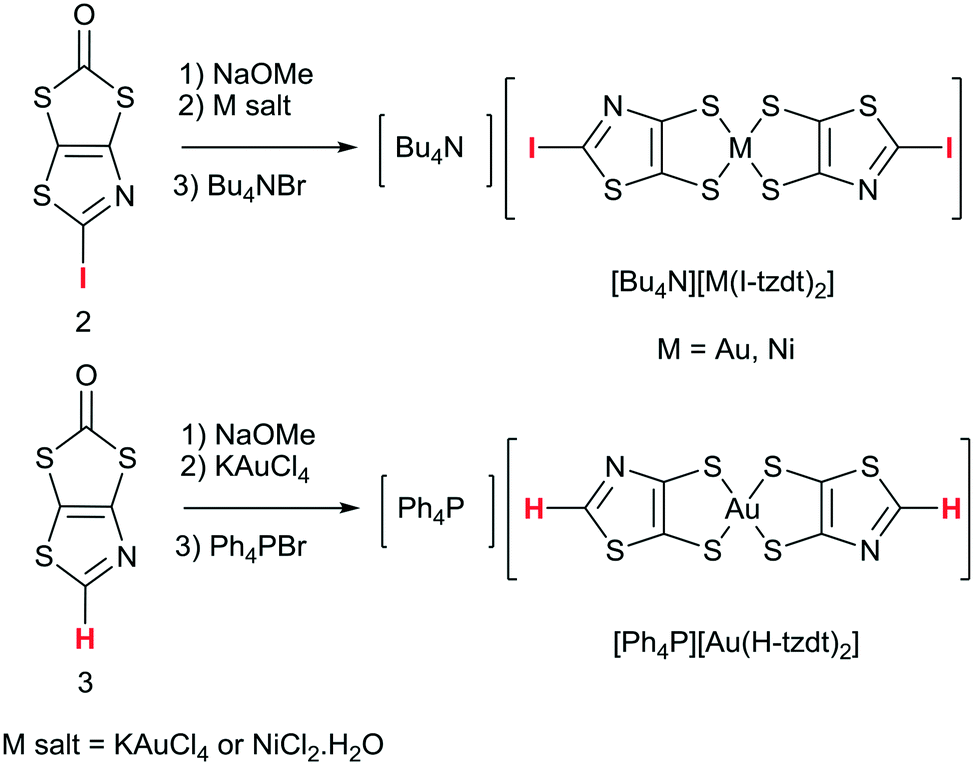 | ||
| Scheme 3 Synthetic path to the anionic [M(I-tzdt)2]−1 (M = Au, Ni) and [Au(H-tzdt)2]−1 complexes. | ||
The redox properties of the four complexes were investigated by cyclic voltammetry performed in CH2Cl2 and relevant data are collected in Table 1. For the gold complexes [Au(R-tzdt)2]−1 (R = I, H), upon oxidation only one pseudo reversible system is observed. As shown in Fig. 4 for [Bu4N][Au(I-tzdt)2], an anodic peak exhibiting an abrupt increase of the intensity at 0.65 V vs. SCE occurs while on the reverse scan, a sharp cathodic peak with a higher intensity than the one observed for the anodic peak is observed at 0.46 V. As often encountered with these metal complexes, the shape of this voltammogram clearly indicates the electrodeposition of an oxidized species at the electrode which is further reduced with the appearance of a sharp desorption reduction peak (Fig. 4). This oxidation process corresponds to the oxidation of the monanion [Au(R-tzdt)2]−1 into the neutral radical species [Au(R-tzdt)2]˙.
| E −2/−1 | E pa/Epc−1/0 | |
|---|---|---|
| a Irreversible process; Epa/Epc: anodic peak potential and cathodic peak potential. | ||
| [Ni(I-tzdt)2] | −0.35 | +0.40/+0.23 |
| [Ni(MeS-tzdt)2] | −0.48 | +0.20/+0.03 |
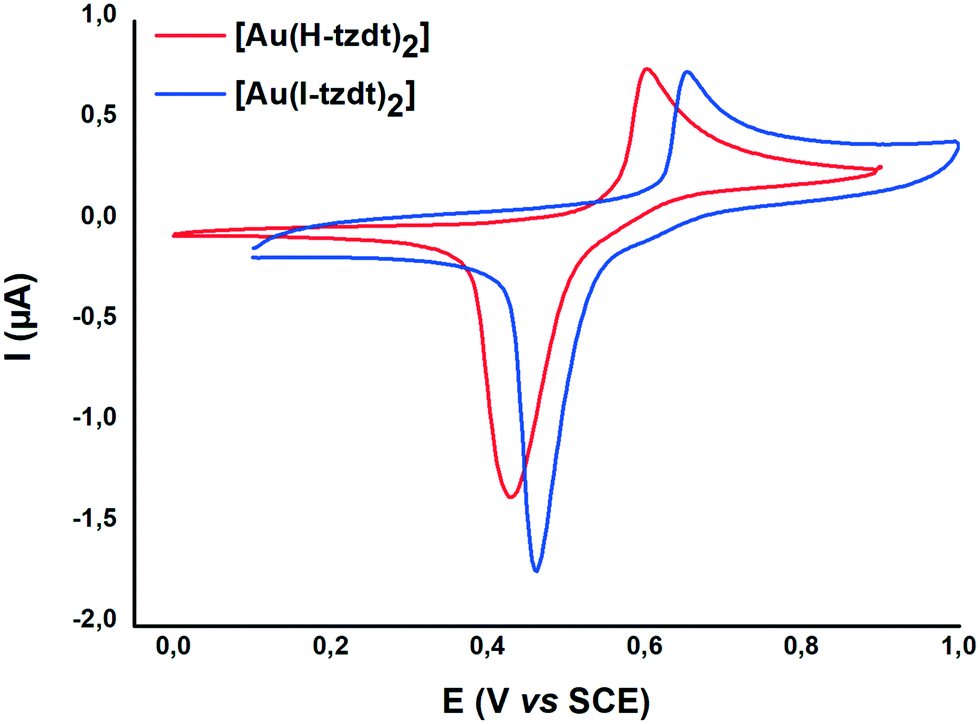 | ||
| Fig. 4 Cyclic voltammograms of Au(R-tzdt) (R= H, I) in CH2Cl2 0.1 M NBu4PF6, at 100 mV s−1. | ||
We note that the replacement of the hydrogen atom by an iodine atom induces an anodic shift of the oxidation potential, E−1/0, due to the electron withdrawing effect of the halogen. Besides, an interesting comparison can be made with the thiomethyl analog (R = SMe), [Au(MeS-tzdt)2]−1 reported earlier25 and analyzed here in the same conditions and reported in Table 1. [Au(MeS-tzdt)2]−1 exhibits indeed a notable cathodic shift for this −1/0 oxidation wave indicating that the thiomethyl substituent has, in these tzdt complexes, an electron donating effect. The Ni complex exhibits two reversible redox processes corresponding respectively to the −2/−1 and −1/0 redox processes (Fig. S6†). The same trends about the influence of the substituents on the redox potentials noticed for the Au complexes are found for the Ni ones, with for the −1/0 redox process potential, ESMe < EI.
Only the monoanionic Au and Ni complexes with the I-tzdt ligand were obtained in the crystalline form after recrystallization. Depending on the solvent used for recrystallization, two types of crystals were obtained for [Bu4N][Au(I-tzdt)2], which proved actually to be the cis and trans isomers. As demonstrated by X-ray diffraction analysis, recrystallization of [Bu4N][Au(I-tzdt)2] in acetone leads to the trans isomer, trans-[Au(I-tzdt)2]−1, while recrystallization of [Bu4N][Au(I-tzdt)2] in acetonitrile affords the cis isomer of the monoanionic gold complex, cis-[Au(I-tzdt)2]−1. Recrystallization of the [Bu4N][Ni(I-tzdt)2] in acetonitrile leads to crystals which are composed of the trans isomer of the monoanionic nickel complex, trans-[Ni(I-tzdt)2]−1. The gold and nickel trans isomers are isostructural, they crystallize in the monoclinic system, space group C2/c, with the complex located on an inversion center and the Bu4N+ cation on a two-fold axis. [Bu4N]cis-[Au(I-tzdt)2] crystallizes in the monoclinic system, space group P21, with both anion and cation in general position.
The molecular structures of the cis and trans isomers of [Au(I-tzdt)2]−1 are depicted in Fig. 5. Intramolecular bond distances are in the expected ranges. For both isomers, the metallacycles are slightly distorted along the S---S axis with angles from 2.4–2.8° for the trans and to 4.6° for the cis isomers. Comparison of the bond distances shows that the bond lengths of the thiazole cores are similar for both isomers while some minor differences are observed on the C–S bond lengths of the metallacycle.
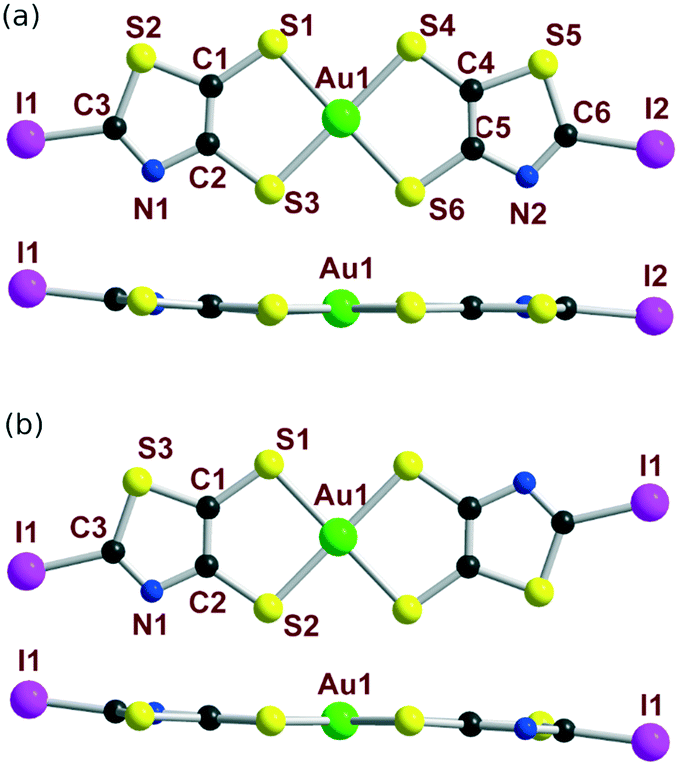 | ||
| Fig. 5 Molecular view (top) and side view (bottom) of the monoanionic (a) cis-[Au(I-tzdt)2] and (b) trans-[Au(I-tzdt)2] complexes. | ||
The solid-state organization of the three anionic complexes reveals the presence of strong halogen bonding interactions involving the iodine atom as XB donor and, surprisingly, one sulfur atom of the dithiolate ligand as XB acceptor (Table 2). In the isostructural trans-[M(I-tzdt)2]1− (M = Au, Ni) complexes, the I⋯S distances amount to 3.335(3) (Fig. 6a) and 3.406(2) Å respectively with C−I⋯S angles around 172°, indicative of a strong XB (RR = 0.88) with pronounced directionality. As depicted in Fig. 7a, these interactions lead to the formation of a two-dimensional anionic molecular framework, delineating rectangular channels running along (a and b), of internal dimensions ≈9.3 × 5.0 Å where the Bu4N+ counterions arms are inserted.
| I⋯S dist. (Å) | RR | ∠C–I⋯S (°) | |
|---|---|---|---|
| cis-[Au(I-tzdt)2]1– | 3.349(4)3.459(5) | 0.890.91 | 172.4(4)163.0(5) |
| trans-[Au(I-tzdt)2]1– | 3.335(3) | 0.88 | 172.3(3) |
| trans-[Ni(I-tzdt)2]1– | 3.406(2) | 0.90 | 171.7(2) |
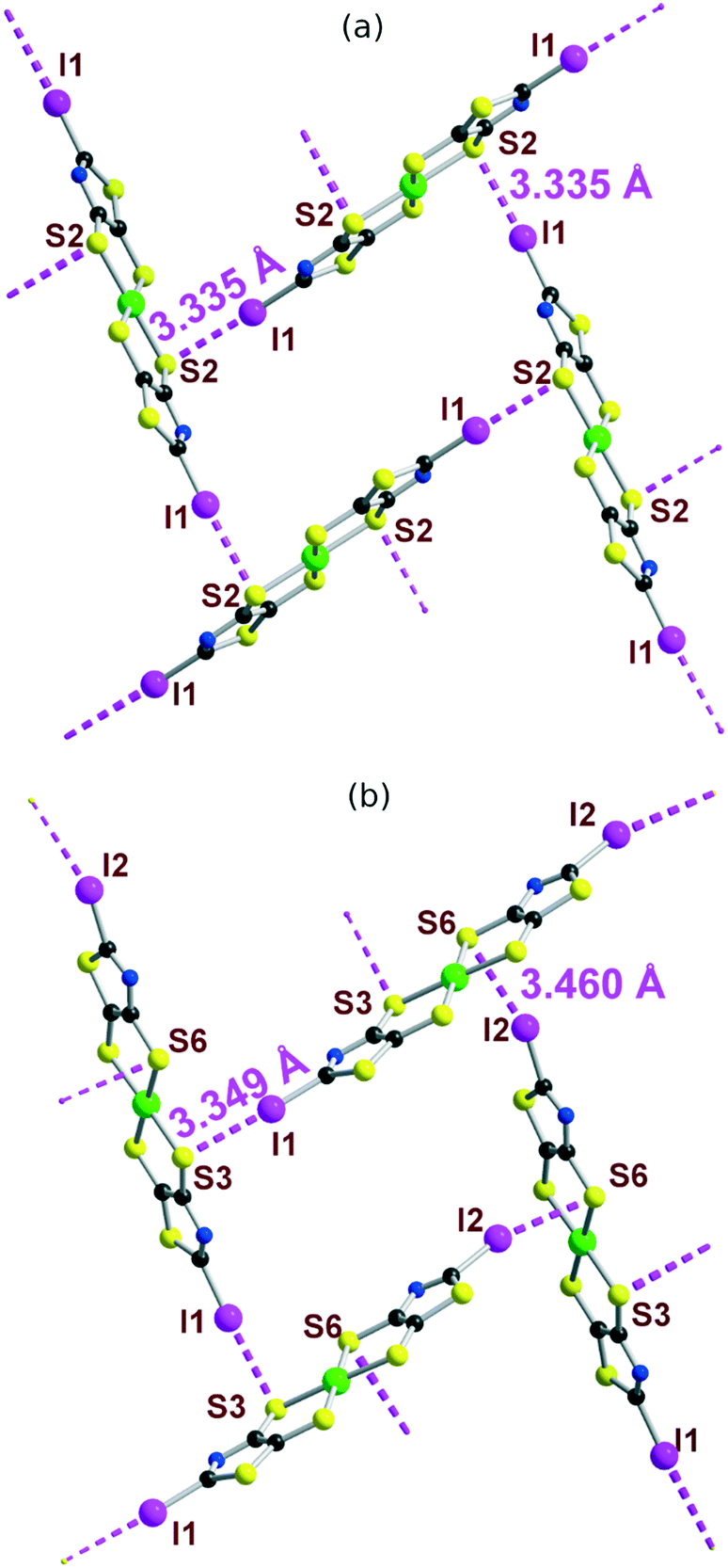 | ||
| Fig. 6 Organization of the monoanionic species, (a) trans-[Au(I-tzdt)2]1– and (b) cis-[Au(I-tzdt)2]1− in the solid state showing the XB interaction network. The counterions have been omitted for clarity. | ||
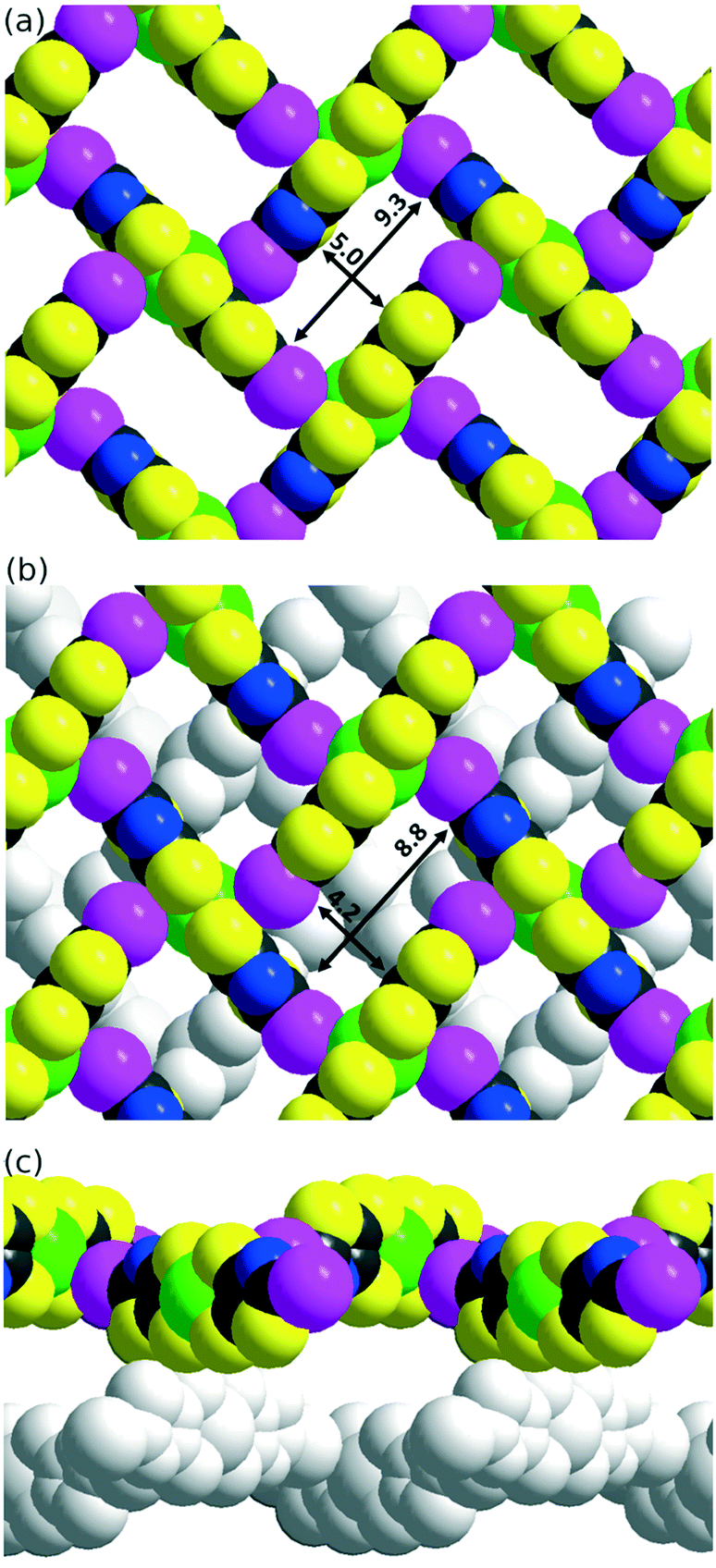 | ||
| Fig. 7 (a) View of the solid state organization of trans-[Au(I-tzdt)2]−1 anions with I⋯S XB interactions between complexes delineating channels. (b and c) Solid state organization of the cis-[Au(I-tzdt)2]−1 anions with two alternating layers. | ||
The situation is more complex in the less symmetric cis-[Au(I-tzdt)2]−1 anionic network (Fig. 6b). Two I⋯S XB interactions, I1⋯S3 3.349(4) and I2⋯S6 3.460(5) Å (RR = 0.89–0.92), are found to generate a similar two-dimensional but more corrugated anionic framework with an added short S⋯S interlayer contact involving the sulfur atom of the thiazole ring of neighboring complexes with S2⋯S5 = 3.451(8) Å (RR = 0.96). The rectangular cavity is then slightly smaller (8.8 × 4.2 Å) but more importantly the alternating layers are rotated relative to each other hindering the formation of channels (Fig. 7b). The recurrent involvement of the metallacycle sulfur atoms as XB acceptor is particularly striking here if we refer to all other examples where dithiolene complexes such as [M(dmit)2] or [M(mnt)2] act as XB acceptor through the thiocarbonyl sulfur atom or the nitrile nitrogen atom respectively. The original feature observed here leads to an orthogonal orientation of the complexes' molecular planes and the stabilization of these peculiar 2D or 3D anionic molecular frameworks.
Electronic properties
While the d8 gold monoanionic complexes are diamagnetic, the nickel monoanions are paramagnetic with S = ½. The temperature dependence of the magnetic susceptibility of a polycrystalline sample of [Bu4N][trans-Ni(I-tzdt)2] show that the complex follows a Curie–Weiss law with an effective moment of 1.83 μB and θ = −0.54 K (Fig. S7†). This behavior is consistent with the overall structural description detailed above which indicates the absence of any overlap interactions between the orthogonal complexes.The oxidation potentials of both the nickel and gold monoanionic complexes (R = H, I) infer that their oxidation could lead to the corresponding neutral complexes. Electrocrystallization, a convenient procedure to generate such neutral species, was used in order to generate the neutral radical species [Au(I-tzdt)2]˙. However, all our attempts to grow monocrystals at the anode failed. The material deposited at the electrode nevertheless gives the expected elemental analysis (see Exp. section). Transport measurements were performed on a compressed pellet of [Au(I-tzdt)2]˙. The room temperature conductivity amounts to 4–5 × 10−3 S cm−1. The magnetic susceptibility was measured with a SQUID magnetometer in the temperature range of 2−300 K. The paramagnetic susceptibility corrected for the Pascal diamagnetic term (Fig. S8†) is essentially temperature independent (4.7 × 10−4 cm3 mol−1), in accordance with a Pauli-type susceptibility of a good conductor. A Curie tail observed at the lower temperatures corresponds to 8% of S = ½ species attributable to paramagnetic defects.
In order to possibly rationalize the structural features of the anionic species and the recurrent stabilization of these rectangular frameworks through directional I⋯S XB interactions, we have also carried out electrostatic potential calculations on the monoanionic trans-[M(I-tzdt)2]−1 (M = Au, Ni). As shown in Fig. 8, because of the anionic character of both complexes, the electrostatic potential surface is negative allover. Charge concentration is observed on the nitrogen atom of the thiazole ring as well as on the sulfur atoms of the metallacycles while the most depleted charged area is associated with the σ-hole on the iodine atoms. This charge distribution is consistent with the obtained solid-state arrangement, and the preferred XB interaction with the metallacycle sulfur atoms, rather than the thiazole nitrogen atom, a probable consequence of the decreased accessibility of the latter.
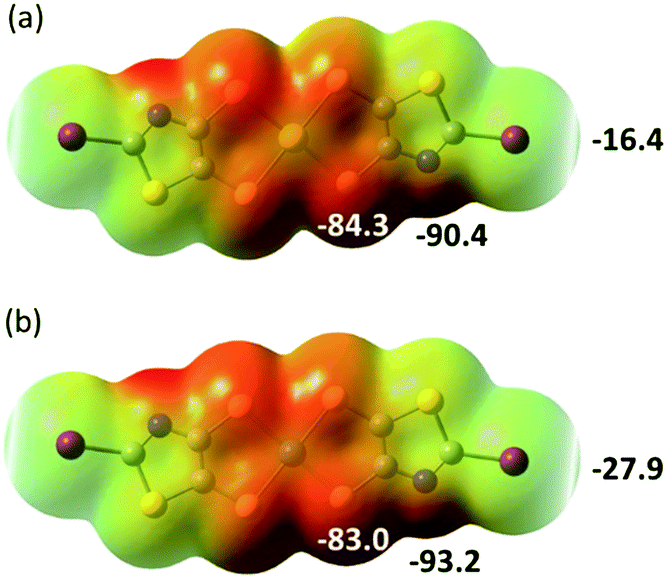 | ||
| Fig. 8 Molecular electrostatic potential surfaces at the 0.001 e− au−3 isodensity level for the monoanionic complexes trans-[M(I-tzdt)2]−1 with (a) M = Au; (b), M = Ni. Colour scale ranges from +36.89 kcal mol−1 (blue) to −93.16 kcal mol−1 (red). | ||
To follow with a same analysis, the electrostatic potential calculations were also performed on the neutral trans-[M(I-tzdt)2] (M = Au, Ni) complexes in order to evaluate the evolution with the charge of the complexes (Fig. 9). In both cases the extrema values are located on the nitrogen (δ−) and the iodine (δ+) of the complexes. They differ in that respect of the proligand 2 described above (Fig. 3a) where the most partial positive site was located inbetween the sulfur atoms belonging to the thiazole and the dithiole rings, and not on the iodine atom.
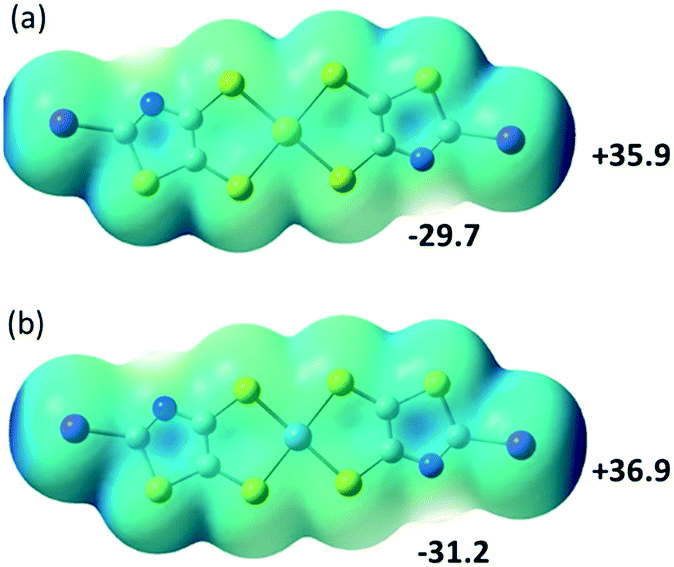 | ||
| Fig. 9 Molecular electrostatic potential surfaces at the 0.001 e− au−3 isodensity level for the neutral complexes trans-[M(I-tzdt)2]−1 with (a) M = Au; (b), M = Ni. Colour scale ranges from +36.89 kcal mol−1 (blue) to −93.16 kcal mol−1 (red). | ||
Conclusion
In summary, we have designed and synthesized a 2-iodo-1,3-thiazole dithiolene proligand with both XB donor and acceptor sites, through an original DMSO/I2 driven transformation of the N-tBu-1,3-thiazoline-2-thione proligand. The corresponding iodo-substituted gold and nickel monoanionic complexes [M(I-tzdt)2]−1 (M = Au and Ni) organize in the crystalline state into layered anionic structures due to dominant I⋯S XB interactions. A noticeable difference is observed between the cis and trans isomers. In the solid state, the trans-[M(I-tzdt)2]−1 (M = Au and Ni) isomers form a 2D network with the formation of sizeable rectangular channels delineated by the skeletons of the complexes. On the other hand, the cis-[Au(I-tzdt)2]−1 isomer exhibits additional S⋯S intermolecular contacts transforming the 2D into a 3D anionic framework. We are currently investigating the formation of single-component29,30 and mixed-valence conducting salts31 from these attractive dithiolene complexes blessed with XB donor ability.Experimental section
General information
All commercial chemicals were used without further purification. The solvents were purified and dried by standard methods. NMR spectra were obtained in CDCl3 unless indicated otherwise. Chemical shifts are reported in ppm, 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm) and 13C NMR spectra were referenced to CHCl3 (77.2 ppm). Melting points were measured on a Kofler hot-stage apparatus and are uncorrected. Mass spectra were recorded by the Centre Régional de Mesures Physiques de l'Ouest, Rennes. Elemental analyses were performed at the Service de Microanalyse, Gif sur Yvette. Cyclic voltammetry were carried out on a 10−3 M solution in CH2Cl2, containing 0.1 M nBu4NPF6 as supporting electrolyte. Voltammograms were recorded at 0.1 V s−1 on a platinum electrode and the potentials were measured versus the saturated calomel electrode (SCE). The starting derivative 1 was prepared according to the procedure previously described.25Synthesis of 5-iodo-[1,3]dithiolo[4,5-d]thiazol-2-one 2
To a solution of 1 (300 mg, 1.14 mmol) in 3 mL of DMSO, under inert atmosphere, iodine crystals (1.15 g, 4.28 mmol) were added. The reaction mixture was stirred at 55 °C for 4 hours. Then, the reaction was quenched with a saturated solution of sodium thiosulfate (200 mL), and the product was extracted with CH2Cl2. The organic phase was washed with water and dried over MgSO4. The solvent was then evaporated under vacuum and the crude product was purified by flash chromatography (eluent: CH2Cl2/petroleum ether; 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85) to give compound 2 as a yellowish white powder in 70% yield together with compound 3 in 5% yield.
:85) to give compound 2 as a yellowish white powder in 70% yield together with compound 3 in 5% yield.
2: Mp: 168 °C. 13C NMR (75 MHz) δ 190.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 144.1 (CC), 122.4 (CC), 99.1 (CN); HRMS (ASAP) calcd for C4HINOS3 [M + H] +: 301.82596. Found: 301.8257; anal calcd for C4INOS3: C, 15.95; N, 4.65. Found: C, 16.17; N, 4.67.
O), 144.1 (CC), 122.4 (CC), 99.1 (CN); HRMS (ASAP) calcd for C4HINOS3 [M + H] +: 301.82596. Found: 301.8257; anal calcd for C4INOS3: C, 15.95; N, 4.65. Found: C, 16.17; N, 4.67.
3: Mp: 120 °C; 1H NMR (300 MHz) δ 8.97 (s, 1H); 13C NMR (75 MHz) δ 190.7 (CO), 153.50 (NC–H), 143.9(CC), 118.1 (CC); HRMS (ESI) calcd for C4H2NOS3 [M + H] +: 175.9293. Found: 175.9294. Anal calcd for C4HNOS3: C, 27.42; H, 0.58; N, 7.99; S: 54.89. Found C, 27.43; H, 0.70; N, 8.02; S, 55.05.
Synthesis of [NBu4][M(I-tzdt)2]
Under inert atmosphere, a solution of Na (23 mg, 1 mmol) in MeOH (20 mL) was added to the dithiole-2-one 2 (100 mg, 0.33 mmol). After complete dissolution, the solution was stirred for 30 min at room temperature. Then a solution of KAuCl4 (63 mg, 0.16 mmol) or NiCl2·6H2O (40 mg, 0.16 mmol) in MeOH (5 mL) was added followed, 6 hours later, by the addition of Bu4NBr (54 mg, 0.16 mmol for the Au complex and 110 mg, 0.33 mmol for the Ni complex). After stirring for 15 h, the formed precipitate was filtered and recrystallized to afford the monoanionic complexes.[NBu4][Au(I-tzdt)2], brown orange crystals; yield 80% (130 mg); Mp: 209 °C; 1H NMR ((CD3)2SO, 300 MHz) δ 3.16 (m, 8H), 1.57 (m, 8H), 1.31 (m, 8H), 0.93 (t, 12H); 13C NMR ((CD3)2SO, 75 MHz) δ 150.7 (CC), 129.1 (CC), 105.4 (NC–I), 57.9 (N–C), 23.5 (NCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 19.6 (H2CH3), 13.9 (CH3); HRMS (ESI) calcd for C6N2I2S6Au [M-Bu4N]−: 742.61464 found: 742.6128; anal. calcd. For C22H36AuI2N2S6: C, 26.81; H, 3.68; N, 4.26; S, 19.52. Found: C, 27.14; H, 3.85; N, 4.32; S, 19.45.
H2), 19.6 (H2CH3), 13.9 (CH3); HRMS (ESI) calcd for C6N2I2S6Au [M-Bu4N]−: 742.61464 found: 742.6128; anal. calcd. For C22H36AuI2N2S6: C, 26.81; H, 3.68; N, 4.26; S, 19.52. Found: C, 27.14; H, 3.85; N, 4.32; S, 19.45.
[NBu4][Ni(I-tzdt)2], dark blue crystals yield 50%(70 mg); Mp > 240 °C; anal calcd for C22H36I2N2NiS6: C, 31.18; H, 4.28; N, 4.96; S, 22.70. Found: C, 31.32; H, 4.06; N, 5.08; S, 22.69.
Synthesis of [PPh4][M(H-tzdt)2]
Under inert atmosphere, a solution of Na (40 mg, 1.74 mmol) in MeOH (20 mL) was added to the dithiole-2-one 3 (100 mg, 0.57 mmol). After complete dissolution, the solution was stirred for 30 min at room temperature. Then a solution of KAuCl4 (110 mg, 0.29 mmol) in MeOH (5 mL) was added followed 6 hours later by the addition of P(Ph)4Br (125 mg, 0.3 mmol). After stirring for 15 h, the formed brown precipitate was filtered and recrystallized from 1,2-dichloroethane to afford the monoanionic complex. [PPh4][Au(H-tzdt)2], brown orange crystals in 40% yield (96 mg). Mp = 222 °C; 1H NMR (CD2Cl2, 300 MHz) of the cis and trans isomers δ 9.03 (s, 2H)/9.05(s, 2H), 7.99–7.58 (m, 20H); 13C NMR (CD2Cl2, 75 MHz) δ 158.3, 135.6, 134.5, 134.4, 130.5, 118.1, 116.9; HRMS (ESI) calcd for C54H42N2P2S6Au [2C+, A]+: 1169.08077 found: 1169.0811; anal. calcd. For C30H22N2AuPS6: C, 43.37; H, 2.67; N, 3.37; S, 23.15. Found: C, 43.06; H, 2.65; N, 3.42; S, 23.83.Electrocrystallizations
[Au(I-tzdt)2] was prepared electrochemically under an argon atmosphere using a standard U-shaped cell with Pt electrodes. A solution of [NBu4][Au(I-tzdt)2] (10 mg) together with nBu4NCl (200 mg) in 12 mL of acetonitrile/benzonitrile solution was placed in the cell. Black powder of [Au(I-tzdt)2] was obtained on the anode upon application of a constant current of 0.5 μA for 10 days. [Au(I-tzdt)2] anal. calcd. For C6AuI2N2S6: C, 9.70; N, 3.77; S, 25.88. Found: C, 10.14; N, 4.05; S, 26.43.Resistivity measurements
The temperature-dependent resistivity for the Au complex was measured using a four-point Van der Pauw geometry on a pressed pellet with a thickness of 0.35(4) mm. Graphitic carbon paste and gold wires (10 μm in diameter) were used to create Ohmic contacts, and a AC current in the range of Idc = 0.1–10 μA was applied.Crystallography
Data were collected on an APEXII Bruker-AXS diffractometer for 2, 3, [NBu4]cis-[Au(I-tzdt)2] and [NBu4]trans-[Ni(I-tzdt)2] and on D8 VENTURE Bruker AXS diffractometer for [NBu4]trans-[Au(I-tzdt)2]. Both diffractometers, equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å), are located at the centre de Diffractométrie (CDFIX), Université de Rennes 1, France. The structures were solved by dual-space algorithm using the SHELXT program,32 and then refined with full-matrix least-square methods based on F2 (SHELXL).33 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions. The higher residual electron density (1–2 e− Å−3) in the structures of [NBu4]cis-[Au(I-tzdt)2] and [NBu4]trans-[Au(I-tzdt)2] gold complexes is located in the neighborhood of the gold atom. Crystallographic data on X-ray data collection and structure refinements are given in Table 3. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC: 1994381–1994385.| Compound | 2 | 3 | [NBu4] cis[Au(I-tzdt)2] | [NBu4] trans[Au(I-tzdt)2] | [NBu4] trans[Ni(I-tzdt)2] |
|---|---|---|---|---|---|
| Formula | C4INOS3 | C4HNOS3 | C22H36AuI2N3S6 | C22H36AuI2N3S6 | C22H36I2N3NiS6 |
| FW (g mol−1) | 301.13 | 175.24 | 985.66 | 977.6 | 847.41 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/a | P21/n | P21 | C2/c | C2/c |
| a (Å) | 9.017(3) | 3.9123(2) | 11.1817(5) | 15.6439(10) | 15.9488(17) |
| b (Å) | 9.118(3) | 16.5922(11) | 13.2215(5) | 12.2704(8) | 12.5276(12) |
| c (Å) | 9.594(3) | 9.7464(5) | 11.5412(4) | 16.6152(11) | 16.2505(18) |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 95.676(13) | 98.329(3) | 91.649(2) | 93.368(3) | 92.452(6) |
| γ (°) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 784.9(4) | 626.00(6) | 1705.53(12) | 3183.9(4) | 3243.9(6) |
| T (K) | 296(2) | 296(2) | 296(2) | 150(2) | 296(2) |
| Z | 4 | 4 | 2 | 4 | 4 |
| D calc (g cm−3) | 2.548 | 1.859 | 1.919 | 2.056 | 1.735 |
| μ (mm−1) | 4.803 | 1.083 | 6.507 | 6.971 | 2.905 |
| Total refls. | 4843 | 4310 | 12067 |
14081 |
10918 |
| Abs. Corr . | Multi-scan | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| Uniq. Refls. (Rint) | 1819(0.0578) | 1415(0.0252) | 7227(0.0283) | 3641(0.0305) | 3668(0.0593) |
| Uniq. Refls. (I > 2σ(I)) | 1278 | 1183 | 5252 | 3197 | 1765 |
| R 1, wR2 | 0.0521, 0.1214 | 0.0377, 0.0763 | 0.0509, 0.0946 | 0.0671, 0.1651 | 0.0562, 0.1314 |
| R 1, wR2 (all data) | 0.0843, 0.1477 | 0.0485, 0.0805 | 0.0831, 0.1078 | 0.0773, 0.1765 | 0.1443, 0.1621 |
| GoF | 1.091 | 1.09 | 1.028 | 1.034 | 0.983 |
Theoretical Modeling
Electrostatic potential calculations were carried out on the optimized geometry of the molecules (with density functional theory using the Gaussian 09 Revision D.01 software, the B3LYP functional and the 6-31+G** basis set for all atoms and the LANLdp basis set for iodine). GaussView 5.0.9 was used to generate the figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by University Rennes 1 through a PhD grant (to H. H.). This work was granted access to the HPC resources of CINES under the allocation 2020-A0080805032 made by GENCI.Notes and references
- S. Sutton, C. Risko and J. L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed.
- G. R. Desiraju, Cryst. Growth Des., 2011, 11, 896–898 CrossRef CAS.
- M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed.
- S. A. Baudron, N. Avarvari and P. Batail, Inorg. Chem., 2005, 44, 3380–3382 CrossRef CAS PubMed.
- Y. Le Gal, T. Roisnel, P. Auban-Senzier, T. Guizouran and D. Lorcy, Inorg. Chem., 2014, 53, 8755–8761 CrossRef CAS PubMed.
- (a) F. Camerel and M. Fourmigué, Eur. J. Inorg. Chem., 2020, 508–522 CrossRef CAS; (b) S. Debnath, H. S. Srour, B. Donnio, M. Fourmigué and F. Camerel, RSC Adv., 2012, 2, 4453–4462 RSC.
- S. Yokomori, A. Ueda, T. Higashino, R. Kumai, Y. Murakami and H. Mori, CrystEngComm, 2019, 21, 2940–2948 RSC.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- M. Fourmigué and J. Lieffrig, Top. Curr. Chem., 2015, 359, 91–114 CrossRef PubMed.
- (a) R. Oliveira, S. Groni, C. Fave, M. Branca, F. Mavré, D. Lorcy, M. Fourmigué and B. Schöllhorn, Phys. Chem. Chem. Phys., 2016, 18, 15867–15873 RSC; (b) R. Oliveira, S. Groni, A. Vacher, F. Barrière, D. Lorcy, M. Fourmigué, E. Maisonhaute, B. Schöllhorn and C. Fave, ChemistrySelect, 2018, 3, 8874–8880 CrossRef CAS; (c) H. Hijazi, A. Vacher, S. Groni, D. Lorcy, E. Levillain, C. Fave and B. Schöllhorn, Chem. Commun., 2019, 55, 1983–1986 RSC.
- T. Devic, B. Domercq, P. Auban-Senzier, P. Molinié and M. Fourmigué, Eur. J. Inorg. Chem., 2002, 2844–2849 CrossRef CAS.
- K. Fukuroi, K. Takahashi, T. Mochida, T. Sakurai, H. Ohta, T. Yamamoto, Y. Einaga and T. Mori, Angew. Chem., Int. Ed., 2014, 53, 1983–1986 CrossRef CAS PubMed.
- T. Kusamoto, H. M. Yamamoto, N. Tajima, Y. Oshima, S. Yamashita and R. Kato, Inorg. Chem., 2012, 51, 11645–11654 CrossRef CAS PubMed.
- T. Imakubo, H. Sawa and R. Kato, J. Chem. Soc., Chem. Commun., 1995, 1097–1098 RSC.
- (a) Y. Kosaka, H. M. Yamamoto, A. Nakao, M. Tamura and R. Kato, J. Am. Chem. Soc., 2007, 129, 3054–3055 CrossRef CAS PubMed; (b) Y. Kosaka, H. M. Yamamoto, A. Tajima, A. Nakao, H. Cui and R. Kato, CrystEngComm, 2013, 15, 3200–3211 RSC; (c) T. Kusamoto, H. M. Yamamoto, N. Tajima, Y. Oshima, S. Yamashita and R. Kato, Inorg. Chem., 2012, 51, 11645–11654 CrossRef CAS PubMed; (d) T. Kusamoto, H. M. Yamamoto and R. Kato, Cryst. Growth Des., 2013, 13, 4533–4541 CrossRef CAS.
- L. L. Gonçalves, V. Gama, R. T. Henriques and M. Almeida, Synth. Met., 1991, 40, 397–401 CrossRef.
- S. Tsukada, M. Kondo, H. Sato and T. Gunji, Polyhedron, 2016, 117, 265–272 CrossRef CAS.
- C. J. Bowlas, A. E. Underhill and D. Thetford, Phosphorus Sulfur Relat. Elem., 1992, 67, 301–304 CrossRef CAS.
- V. Madhu and S. K. Das, Inorg. Chem., 2008, 47, 5055–5070 CrossRef CAS PubMed.
- F. G. Siméon, M. T. Wendahl and V. W. Pike, J. Org. Chem., 2009, 74, 2578–2580 CrossRef PubMed.
- B. G. Van den Hoven and H. Alper, J. Am. Chem. Soc., 2001, 123, 1017–1022 CrossRef CAS PubMed.
- (a) A. Filatre-Furcate, P. Auban-Senzier, M. Fourmigué, T. Roisnel, T. Dorcet and D. Lorcy, Dalton Trans., 2015, 44, 15683–15689 RSC; (b) A. Filatre-Furcate, T. Roisnel and D. Lorcy, J. Organomet. Chem., 2016, 819, 182–188 CrossRef CAS.
- (a) A. Monga, S. Bagchi and A. Sharma, New J. Chem., 2018, 42, 1551–1576 RSC; (b) S. Kolita, P. Borah, P. S. Naidu and P. J. Bhuyan, Tetrahedron, 2016, 72, 532–538 CrossRef CAS; (c) V. Venkateswarlu, K. A. A. Kumar, S. Gupta, D. Singh, R. A. Vishwakarma and S. D. Sawant, Org. Biomol. Chem., 2015, 13, 7973–7978 RSC.
- (a) Y. F. Liang, X. Li, X. Wang, M. Zou, C. Tang, Y. Liang, S. Song and N. Jiao, J. Am. Chem. Soc., 2016, 138, 12271–12272 CrossRef CAS PubMed; (b) Y. F. Liang, S. Song, L. Ai, X. Lia and N. Jiao, Green Chem., 2016, 18, 6462–6467 RSC; (c) N. Mupparapu, S. Khan, S. Battula, M. Kushwaha, A. P. Gupta, Q. N. Ahmed and R. A. Vishwakarma, Org. Lett., 2014, 16, 1152–1155 CrossRef CAS PubMed.
- S. A. Rather, A. Kumar and Q. N. Ahmed, Chem. Commun., 2019, 55, 4511–4514 RSC.
- (a) N. Tenn, B. Bellec, O. Jeannin, L. Piekara-Sady, P. Auban-Senzier, J. Íñiguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2009, 131, 16961–16967 CrossRef CAS PubMed; (b) Y. Le Gal, T. Roisnel, P. Auban-Senzier, N. Bellec, J. Íñiguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2018, 140, 6998–7004 CrossRef CAS PubMed.
- (a) H. B. Cui, T. Tsumuraya, T. Miyazaki, Y. Okano and R. Kato, Eur. J. Inorg. Chem., 2014, 24, 3837–3840 CrossRef; (b) A. Filatre-Furcate, N. Bellec, O. Jeannin, P. Auban-Senzier, M. Fourmigué, A. Vacher and D. Lorcy, Inorg. Chem., 2014, 53, 8681–8690 CrossRef CAS PubMed.
- (a) R. Kato, Chem. Rev., 2004, 104, 5319–5346 CrossRef CAS PubMed; (b) R. Kato, Bull. Chem. Soc. Jpn., 2014, 87, 355–374 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic and crystallographic data CCDC 1994381–1994385. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00512f |
| This journal is © The Royal Society of Chemistry 2020 |