
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethods for easy recognition of isostructurality – lab jack-like crystal structures of halogenated 2-phenylbenzimidazoles†
Petra Bombicz *a,
Nóra V. Maya,
Dániel Fegyvernekib,
Avirmed Saranchimega and
Laura Bereczki*a
*a,
Nóra V. Maya,
Dániel Fegyvernekib,
Avirmed Saranchimega and
Laura Bereczki*a
aChemical Crystallography Research Laboratory, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2, 1117 Budapest, Hungary. E-mail: bombicz.petra@ttk.hu; nagyne.bereczki.laura@ttk.hu
bInstitute of Organic Chemistry, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2, 1117 Budapest, Hungary
First published on 18th May 2020
Abstract
Tools to describe isostructurality are important in the understanding of close packing principles and in the fine-tuning of crystal properties. In order to present how different methods work in practice, a series of 2-phenylbenzimidazole derivatives substituted on the phenyl ring in the ortho, meta and para positions or simultaneously in two different positions by F, Cl and Br were selected. The flexibility of the phenylbenzimidazole frame permits a gradual isostructural change of the structures with step-by-step alteration of the internal arrangement as well as of the lengths of the unit cells perpendicular to the determining N–H⋯N hydrogen bonded chains. The exchange of the different halogen substituents alters the angle between the neighbouring benzimidazole moieties and the system of the secondary interactions, and finally the isostructurality is terminated. The series of isostructural crystals look like a lab jack lifted at different heights. Although the neighbouring members of the series are highly similar, the extremes of the list vary deliberately keeping the space group and Z. This raises the question about the extents of structural differences what we still consider isostructural. The preference of certain intermolecular interactions divides the investigated isostructural Pbca crystals into two subgroups like a switch. The definition of isostructurality does not consider supramolecular similarity, although it may have a determining role as shown. It is presented how isostructurality can be described by numerical descriptors. Cell similarity (π), isostructurality (Is), and molecular isometricity indices are calculated. Correlations of the molecular conformation, secondary interactions and the crystallographic parameters are revealed by statistical methods. With the use of these methods, we provide an easy way to recognise and to characterize isostructurality. We show that the prerequisites of isostructurality are the similar composition and conformation of the compounds, with their analogous molecular and supramolecular arrangement in the crystals having the same space group and Z. Exploitation of the Cambridge Structural Database for systematical investigations to complete the isostructural series is essential.
Introduction
More and more new substances with tailor-made properties are produced by crystal engineering.1–3 This can be achieved based on the knowledge of the molecular and supramolecular properties4,5 and crystallographic and non-crystallographic symmetries.6 Mastering the supramolecular packing architecture, in effect synthon engineering,7,8 can be achieved through fine chemical changes. Firm transformation of crystal packing arrangements can be accomplished by influencing the electrostatic and steric properties by application of substituents, by changing their placement and/or chemical composition or by the use of compounds with a similar chemical composition in multi-component structures. Molecular placement and molecular conformation of flexible molecules may adjust to the chemical and supramolecular features, thus isostructural crystals can be achieved.9,10 This way we may succeed to produce a series of crystal structures with a gradual transition in crystal packing arrangements through chemical change. Isostructural crystals are highly similar in their packing arrangement, but may differ to a lesser or greater extent in chemical properties, like seeding and crystal growth, recognition processes, stability, and biological activity. The balance of spatial requirements and electrostatic effects ultimately determines the crystal packing arrangement.There is a slight structural difference between the neighbouring members in the series of fine-tuned isostructural crystals, but structural variations can already be severe among the different members of the series. The question is how large the extent of difference can be among crystal structures that we may still consider as isostructural crystals.6
Investigation of isostructurality is a tool for understanding close packing principles. Recognition of isostructurality is not necessarily straightforward. Isostructurality calculations and statistical analyses are efficient tools for the discovery of isostructural crystals as presented in this paper.
Crystal structures of halogenated 2-phenylbenzimidazole (PBI) derivatives were analysed to investigate the effect of substitution on isostructurality. The molecules are substituted on the phenyl ring in the ortho, meta and para positions or simultaneously in two different positions with fluorine, chlorine and bromine. Eight structures were retrieved from the Cambridge Structural Database11 for comparison. To complete the series, four new compounds were synthesised, and their single crystal structures were determined. Exploitation of the Cambridge Structural Database is essential in order to have a complete isostructural series for the systematic investigations.
The moderate rotational freedom around the single bond between the aromatic rings of the substituted PBI chain-forming molecules generates flexible structures with variable unit cell dimensions depending on derivatisation. Owing to the presence of the determinative N–H⋯N hydrogen bonded chains in all crystal structures, the halogenated PBI structures will be similar to different extents. Halogen interactions are more diversified than hydrogen bonding regarding both interaction strength and directionality and are therefore applicable for more sophisticated crystal engineering purposes.12–16 With the presence of the different halogen substituents at different positions, we have obtained a series of structures to test the limits of isostructurality.
Correlations of the halogen atomic positions, secondary interactions and crystallographic parameters have been revealed by statistical methods. The likeness of the structures is quantified by different kinds of similarity and isostructurality calculations. The arrangement of the molecules resembles a laboratory jack of which the height is determined by the halogen substitution.
Experimental
Synthesis and single crystal growth of OFPBI, OBRPBI, MCLPBI and MBRPBI
Synthesis of the new substituted benzimidazole compounds was performed by a slightly modified process described by Secci.17 The corresponding halogenated benzaldehyde or benzoic acid (1 mmol), 1,2-diaminobenzene (1 equiv.) and sodium metabisulfite (Na2S2O5, 1 equiv.) were dissolved in 4 ml DMF in a 10 ml vial suitable for an automatic single-mode microwave reactor (2.45 GHz high-frequency microwaves, power range 0–300 W). The mixture was stirred for 30 s and then heated by microwave irradiation for 40 min at 100 °C. The internal vial temperature was controlled by an IR sensor. After cooling with pressurized air, the reaction mixture was poured onto ice, filtered, and dried in air. For further purification, products were recrystallized from diethyl ether (ortho-substituted compounds) or ethanol (meta-substituted compounds).Single crystal X-ray diffraction data of OFPBI, OBRPBI, MCLPBI and MBRPBI
X-ray diffraction data were collected on a Rigaku RAXIS-RAPID II diffractometer using CuKα (OBRPBI, OFPBI) or MoKα (MCLPBI, MBRPBI) radiation with a graphite monochromator. Numerical absorption correction was applied to the data. The structures were solved by direct methods (and subsequent difference syntheses). Sir2014 (ref. 21) and SHELXT22 under WinGX23 software were used for structure solution and refinement, respectively. Anisotropic full-matrix least-squares refinements on F2 for all non-hydrogen atoms were performed.Hydrogen atomic positions were calculated from assumed geometries except for the H2N amine hydrogens that were located in difference maps. Hydrogen atoms were included in structure factor calculations, but they were not refined. The isotropic displacement parameters of the hydrogen atoms were approximated from the U(eq.) value of the atom they were bonded to.
Statistical analysis
Multivariate data analysis was performed on the crystallographic data of the twelve investigated substituted benzimidazoles to reveal correlations and similarities between crystal parameters, molecular structures and supramolecular interactions.24,25 Cluster analysis24 was performed on the standardized dataset (collected in Tables 1 and 2), where the samples are grouped based on similarities without taking into account the information about the group membership, e.g. substitution and space groups. This technique is based on the idea that the similarity is inversely related to the distance (differences between parameters) between samples. Cluster analysis calculates the distances (or correlation) between all samples using a defined metric which is Euclidean distance in our case. Grouping of the samples was performed by the Ward's method clustering algorithm. Principal component analysis (or factor analysis, PCA) was applied as well on the data in order to reduce the data dimensionality.25 PCA transforms the original measured variables into new uncorrelated variables called principal components which are the linear combination of the original measured variables. All statistical evaluations were accomplished with the software Statistica.26| Code | Substituent | SG | V | a | b | c | β | Z | KPI | Density | Packing |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MCLPBI | mCl | P21/c | 1082.2(2) | 12.589(1) | 9.5051(8) | 9.7495(7) | 111.933(8) | 4 | 68.0 | 1.400 | H–T |
| MBRPBI | mBr | P21/c | 1110.9(3) | 12.676(2) | 9.656(2) | 9.765(2) | 111.638(9) | 4 | 58.0 | 1.625 | H–T |
| GOLNOM | oClmCl | Pbca | 2301.73(6) | 5.6259(8) | 9.739(1) | 42.007(6) | 90 | 8 | 69.7 | 1.519 | H–H |
| OBRPBI | oBr | Pbca | 2279.8(2) | 7.007(3) | 9.936(3) | 32.745(3) | 90 | 8 | 66.1 | 1.592 | H–H |
| LUJWIW | oCl | Pbca | 2220.55(5) | 7.0197(1) | 9.9261(1) | 31.8686(4) | 90 | 8 | 66.9 | 1.368 | H–H |
| OFPBI | oF | Pbca | 2117.9(2) | 7.1246(3) | 10.0327(4) | 29.629(1) | 90 | 8 | 66.1 | 1.331 | H–H |
| DETKIX | oClpCl | Pbca | 2435.0(9) | 8.563(2) | 9.910(2) | 28.694(6) | 90 | 8 | 65.6 | 1.436 | H–H |
| JEYTUD | pF | Pbca | 2080(1) | 8.574(2) | 9.830(3) | 24.680(7) | 90 | 8 | 67.4 | 1.355 | H–H |
| MINHOI | mFpF | Pbca | 2028.3(7) | 8.7195(17) | 9.9454(19) | 23.389(4) | 90 | 8 | 71.9 | 1.508 | H–H |
| QERTIR | pCl | Pbca | 2157.2(8) | 9.1284(18) | 9.783(2) | 24.156(5) | 90 | 8 | 68.3 | 1.408 | H–H |
| TUDXIB | mFpCl | Pbca | 2103.35(15) | 9.2302(4) | 9.8500(4) | 23.1247(9) | 90 | 8 | 72.5 | 1.558 | H–H |
| SEZNUH | pBr | Pbca | 2205.7(7) | 9.3969(17) | 9.7876(18) | 23.982(4) | 90 | 8 | 68.5 | 1.645 | H–H |
| Code | Angle BI–BI | Dist. N–H⋯N | Angle N–H⋯N | Angle BI–Ph | oH(BI)⋯oH(BI) | oH(BI)⋯oH(Ph) | Dist.b X⋯π | Dist. C–H⋯X | Dist.b N–H⋯X | Dist.b X⋯X | Dist.b π⋯π |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Angle BI–BI: angle between the planes of the neighbouring benzimidazole moieties [°], dist. N–H⋯N: distance between the hydrogen bonded nitrogens [Å], angle N–H⋯N: N–H⋯N angle of the hydrogen bond [°], angle BI–Ph: angle between the planes of the benzimidazole and the phenyl moieties of the same molecule [°], oH(BI)⋯oH(BI): distance of the ortho hydrogens of the hydrogen bonded benzimidazole moieties [Å], oH(BI)⋯oH(Ph): distance of the ortho hydrogens of the phenyl and benzimidazole moieties of the hydrogen bonded molecules [Å], X⋯π: intra-chain halogen⋯π interaction length of the hydrogen bonded molecules [Å], dist. C–H⋯X: hydrogen bonds to halogen atoms [Å], dist. N–H⋯X: distance of the amine hydrogen and the halogen [Å], dist. X⋯X: inter-chain X⋯X halogen bond lengths [Å], dist. π⋯π: presence of π⋯π stacked aromatic interactions in the structures [Å].b Missing interactions are indicated by —. | |||||||||||
| MCLPBI | 67.83° | 2.868(3) | 164(3)° | 26.8(1)° | 2.854 | 3.921 | 4.058 | 3.132 | — | — | 3.799(2) |
| 3.912(2) | |||||||||||
| MBRPBI | 68.53° | 2.867(7) | 154(3)° | 28.4(3)° | 2.900 | 3.931 | 4.073 | 3.130 | — | — | 3.872(4) |
| 3.972(4) | |||||||||||
| GOLNOM | 74.49° | 2.824 | 148° | 27.70° | 2.934 | 3.710 | 3.3077 | 3.020 | 2.64 | — | — |
| 3.5551 | 3.082 | ||||||||||
| 3.6125 | 3.139 | ||||||||||
| OBRPBI | 92.88° | 2.801(6) | 162(4)° | 42.4(2)° | 4.033 | 2.641 | 4.186 | 3.060 | 2.91(4) | — | — |
| 3.221 | |||||||||||
| LUJWIW | 94.72° | 2.801 | 163° | 39.60° | 4.023 | 2.696 | 4.341 | 2.901 | 2.77 | — | — |
| 3.095 | |||||||||||
| OFPBI | 99.21° | 2.863(4) | 159(3)° | 35.2(2)° | 4.048 | 2.868 | 4.738 | 2.498 | 2.46(3) | — | — |
| DETKIX | 112.33° | 2.866 | 155° | 42.00° | 4.911 | 2.429 | 4.295 | 2.945 | 2.79 | 3.506 | — |
| 4.698 | |||||||||||
| JEYTUD | 117.76° | 2.875 | 163° | 29.23° | 5.066 | 2.391 | — | 2.703 | — | 3.45 | 3.7341 |
| 2.844 | |||||||||||
| MINHOI | 119.17° | 2.874 | 158° | 30.00° | 5.030 | 2.308 | — | 2.508 | 3.04 | 2.849 | 3.5892 |
| 2.746 | 3.387 | ||||||||||
| QERTIR | 123.57° | 2.905 | 167° | 27.00° | 5.365 | 2.322 | — | 3.096 | — | 3.346 | 3.8442 |
| 3.135 | |||||||||||
| TUDXIB | 124.40° | 2.924 | 166° | 26.90° | 5.416 | 2.286 | — | 2.896 | — | 2.691 | 3.6839 |
| 2.899 | 3.017 | ||||||||||
| 3.069 | 3.370 | ||||||||||
| SEZNUH | 126.66° | 2.925 | 168° | 26.77° | 5.476 | 2.307 | — | 3.212 | — | 3.433 | 3.9360 |
| 3.217 | |||||||||||
Cell similarity, isostructurality and molecular isometricity calculations27–30
The prerequisite of isostructurality is the similarity of the unit cells. The cell similarity index (π) describes the dissimilarity of the unit cell dimensions of the compared crystals:where a, b, and c and a′, b′, and c′ are the orthogonalized lattice parameters of the two related crystals. In the event of a great similarity of two unit cells, π is close to zero.
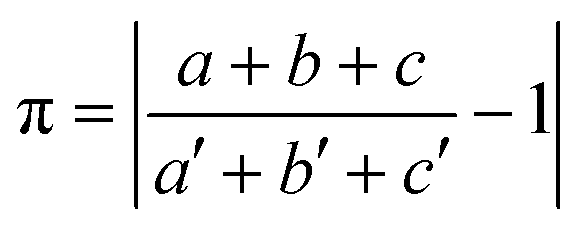
Isostructurality implies equal Z′ and a similar internal arrangement in the related crystal lattices. In contrast to polymorphism, isostructurality can be characterized by a numerical descriptor.
The isostructurality index (Is) of two related crystal structures can be calculated as follows:
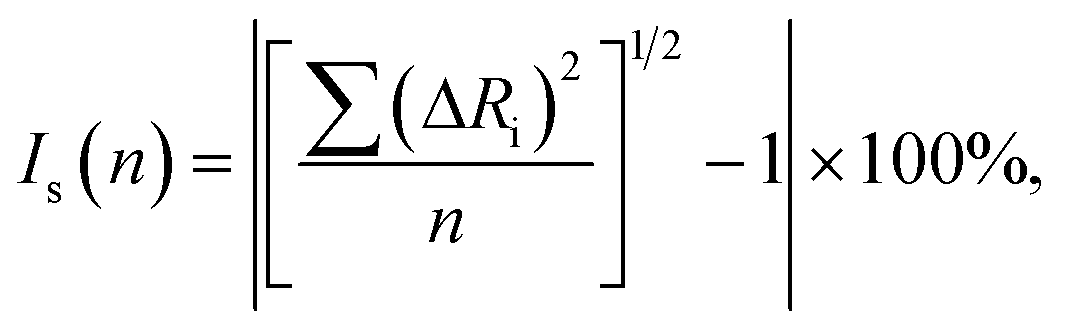
The cell similarity and isostructurality calculations for the pairs of similar crystal structures were performed using software ISOS.30
The isometricities of the related molecular moieties in different crystal structures were determined using the software Mercury31 of the CSD11 superimposing them and calculating the root mean square of the distance differences of the related atoms (rmsD), giving also the largest distance difference (maxD) between them.
The Kitaigorodskii packing index (KPI) was calculated using software Platon.32
Discussion
Structural analysis of the lab jack crystals
Eight halogenated 2-phenylbenzimidazole crystal structures from the CSD11 (JEYTUD,33 MINHOI,34 TUDXIB,35 LUJWIW01,36 DETKIX,37 GOLNOM,38 QERTIR,39 SEZNUH40) and 4 new systematically synthesised halogenated 2-phenylbenzimidazole structures (ortho-fluoro- (OFPBI), meta-chloro- (MCLPBI), ortho-bromo- (OBRPBI) and meta-bromo- (MBRPBI)phenylbenzimidazole (PBI)) (Table S1†) have been analysed and compared to reveal the structural similarities and differences in their fine-tuned series (Fig. 1). The studied PBIs have been substituted on the phenyl ring in the ortho, meta and para positions or they are substituted simultaneously at two different positions with F, Cl and Br. Our aim is to understand the close packing principles and the role of molecular conformation, supramolecular interactions and symmetries in order to be able to perform directed manipulation of molecular packing arrangements as well as the investigation of the possible application of halogen interactions for the fine-tuning of hydrogen bonded structures, while maintaining isostructurality.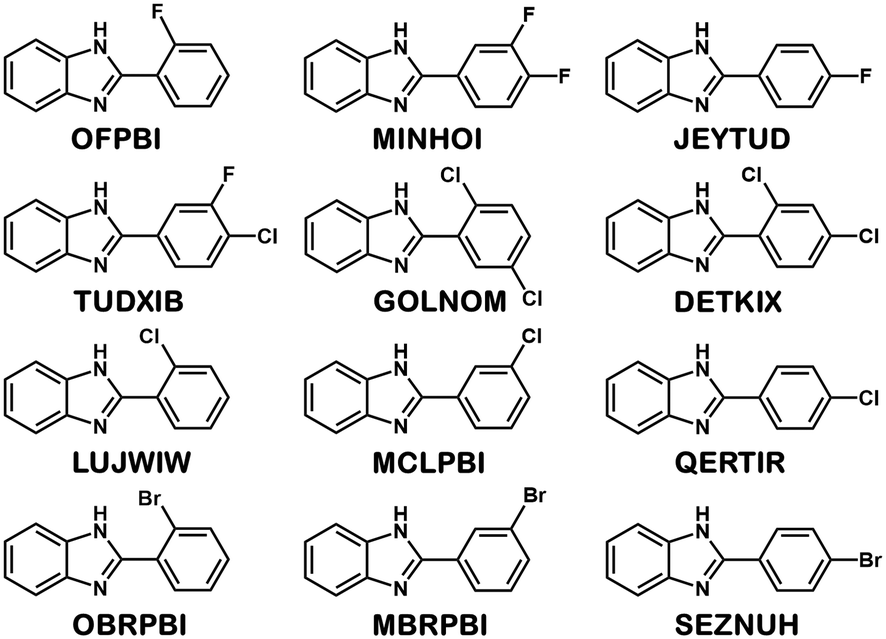 | ||
| Fig. 1 Formula diagrams of the studied halogenated 2-phenylbenzimidazoles. | ||
PBI has a rather rigid molecular structure that can be twisted around the benzimidazole-phenyl axis. The arrangements of the molecules in the crystal lattices of the halogenated PBI derivatives are determined by only one intermolecular interaction, that is significantly stronger than any other secondary interaction in the crystals. This most characteristic secondary interaction is the N–H⋯N type hydrogen bond in the crystals, which incorporates the amino groups of the imidazole moieties (Fig. 2). It threads the halogenated PBIs into chains, which organize primarily the packing arrangements, and only a few different supramolecular interactions are possible among the hydrogen bonded molecular chains.
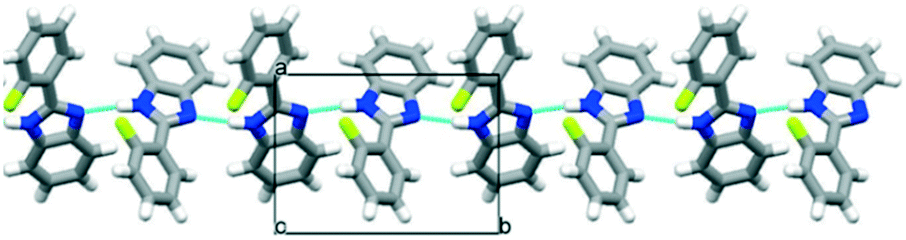 | ||
| Fig. 2 The N–H⋯N hydrogen bonds of the substituted phenylbenzimidazoles (PBIs) in the crystal structure of OFPBI as a typical example. | ||
However, the phenylbenzimidazole derivative structures are flexible to some extent, and this flexibility permits the considerable alteration of the lattice parameters. The relative tilt of the hydrogen bonded molecules within the chain is determined by the substituents of the 2-phenylbenzimidazole skeleton. The investigated question is how and to what extent the type and the position of a halogen substituent(s) may affect and fine-tune the crystal structures, while the other influencing factors are reduced as much as possible.
Ten of the twelve 2-phenylbenzimidazole molecules crystallize in the Pbca space group (Z = 8) and the packing arrangements of the Pbca structures are all similar. The single meta-substituted compounds (namely MCLPBI and MBRPBI) crystallize in the P21/c space group (Z = 4) and are on their part isostructural (Fig. S1†).
All PBI derivatives are organized into hydrogen bonded chains by the determining N–H⋯N type intermolecular interactions between the imidazole moieties. In the Pbca structures these molecular chains are arranged in the crystallographic b direction (Fig. 2). The length of the b axes is basically governed by these hydrogen bonded chains and therefore are similar for all compounds within 0.5 Å (Table 1). The molecules can tilt around the hydrogen bonds and the degree of the tilt determines the lengths of the crystallographic a and c axes. The length of the a unit cell axes is increased by 67% and the length of the c unit cell axis is decreased by 43% within the series (Fig. S1†). There is a linear correlation between the opening angle of the benzimidazole moieties (Fig. 3 and Table 2) and the lengths of the a and c unit cell axes. In the P21/c structures, the N–H⋯N hydrogen bonded chains are found in the crystallographic c direction, and their lengths are in the range of the length of the b axes of the Pbca structures.
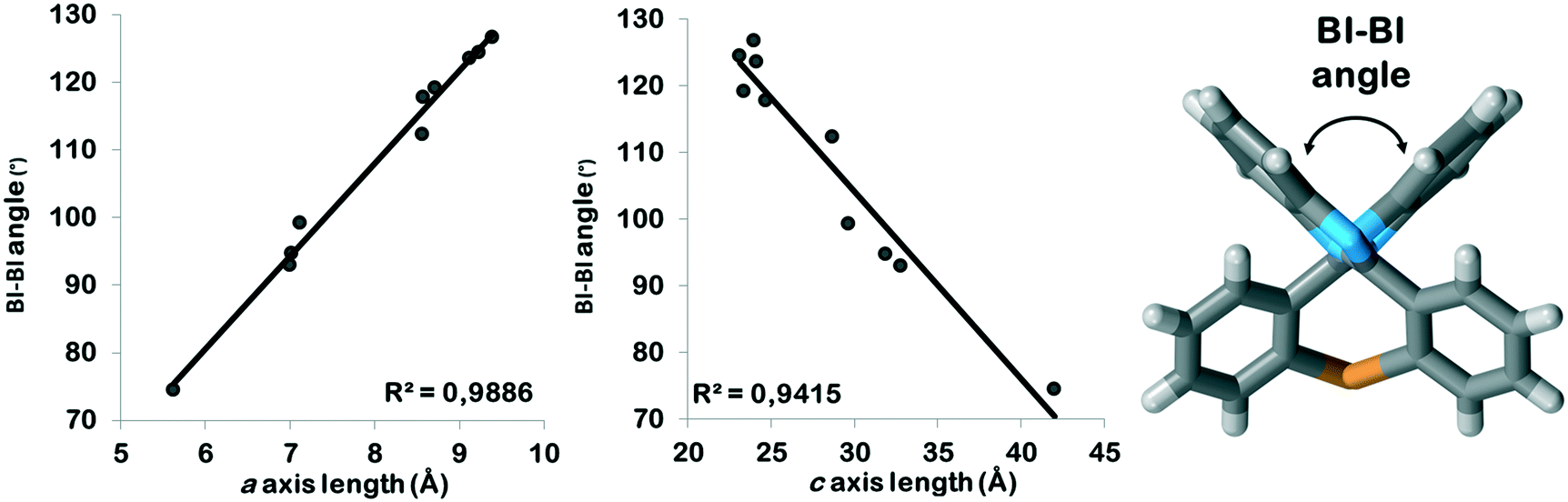 | ||
| Fig. 3 Correlation between the BI–BI tilt angles and the a as well as the c axis lengths in the Pbca structures. | ||
The halogenated non-single-meta substituted 2-phenylbenzimidazole molecules crystallize in the Pbca space group, and are oriented in a head-to-head (H–H) arrangement (Fig. 4). With this sort of organisation, the similar molecular moieties become close to each other and the formation of halogen–halogen interactions is promoted. The halogenated phenyl rings form either X⋯X, X⋯H or X⋯π weak interactions and the benzimidazole moieties form aromatic H⋯π or π⋯π interactions. In contrast, the meta-halogenated compounds, crystallized in the P21/c space group, are oriented in a head-to-tail (H–T) arrangement and the meta-halogens form intra-chain X⋯π interactions.
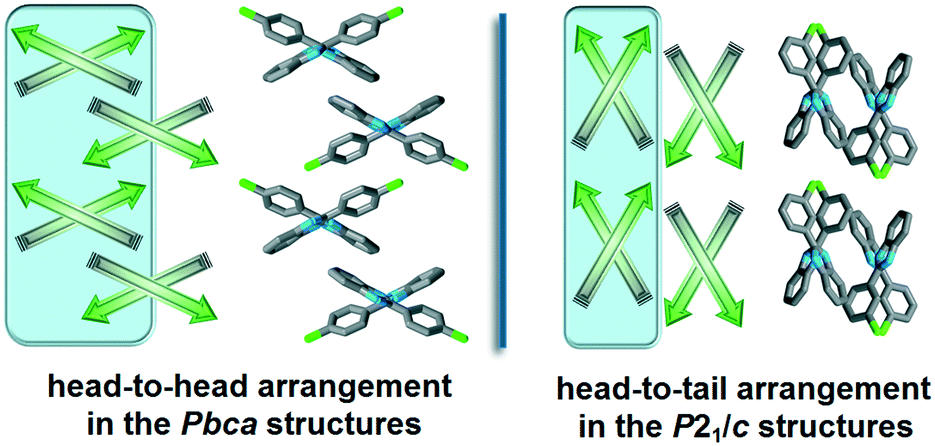 | ||
| Fig. 4 Schematic structural patterns in the halogenated 2-phenylbenzimidazole structures. Comparison of the packing arrangement in the Pbca (QERTIR) and P21/c (MCLPBI) crystals. | ||
In the presence of an ortho halogen substituent, the a unit cell axis is elongated (5.63–8.56 Å) and the c axis is shortened (28.69–42.01 Å); the tilt angle between the benzimidazole moieties (BI–BI angle) is in a smaller range (75–112°) compared to the other Pbca isomers – “the lab jack is pulled to reach the high shelves” (Fig. 5, Table 2). The gradual changes can be observed from GOLNOM (oClmCl) via OBRPBI (oBr), LUJWIW (oCl) and OFBPI (oF) to DETKIX (oClpCl). In the ortho derivatives, an intramolecular N–H⋯X hydrogen bond is formed, which hampers the free rotation of the halogenated phenyl ring on the benzimidazole moiety.
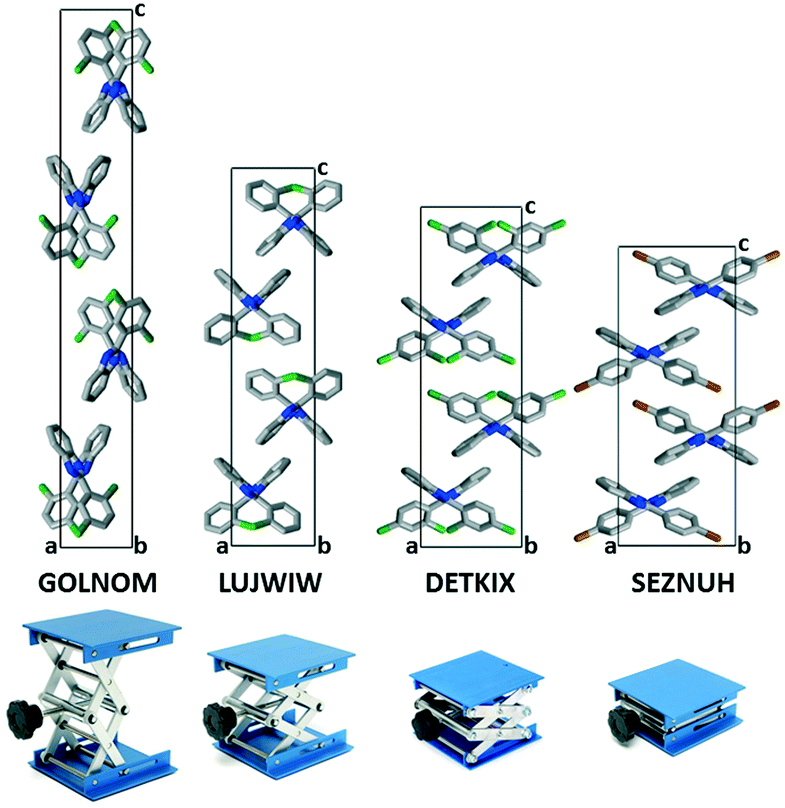 | ||
| Fig. 5 The lab jack in pulled (GOLNOM), medium (LUJWIW and DETKIX) and pushed (SEZNUH) positions as an effect of the placement of the halogen substituents. Crystallographic c and a axes are proportional to the real values. | ||
A further gradual increase of the length of the a axis (8.57–9.40 Å), decrease of the c axis (23.98–24.68 Å), and the opening of the benzimidazoles' tilt angle (118–127°) can be observed within the group of the para-halogen substituted derivatives – “the lab jack is now pushed in lower positions” (Fig. 5). The BI–BI angle increases together with the order of the polarizability of the halogen substituents JEYTUD (pF), MINHOI (mFpF), QERTIR (pCl), TUDXIB (mFpCl) and SEZNUH (pBr).
In order to reveal what is behind the isostructural gradual change in the supramolecular interactions, the synthons were analysed. C–H⋯X interactions are common in all studied structures. The detected BI–BI angle is mainly affected by the intermolecular X⋯π and X⋯X interactions (Fig. 6) depending on the placement, type and number of substitutions. They make the fine-tuned alteration of the crystal properties possible (Fig. S2†).
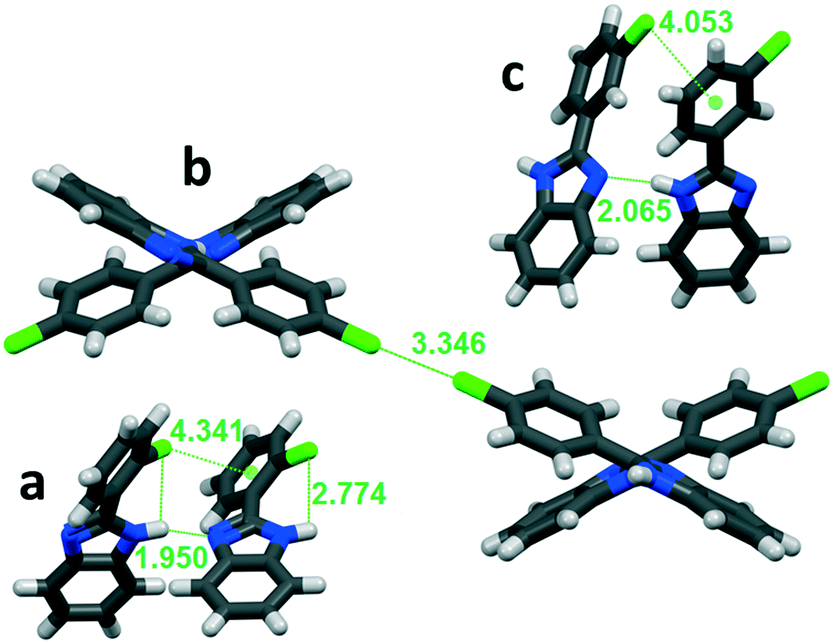 | ||
| Fig. 6 Secondary interactions in a: ortho-substituted LUJWIW, b: para-substituted QERTIR, and c: meta-substituted MCLPBI. | ||
The ortho position of the halogen atoms inhibit the formation of X⋯X interactions, because the halogens cannot be in close proximity to each other. The most favourable intermolecular interactions in the ortho halogen substituent containing structures are the intra-chain X⋯π interactions with the phenyl ring of a neighbouring molecule (Fig. 6a). The tilt angle decreases with increasing polarizability of the halogens in the F, Cl, and Br order. This interaction fixes the PBI molecules in a relatively small tilt angle.
In the case of the para-halogen substitution of the BPI, X⋯X halogen bonds can be formed between the hydrogen bonded chains. It results in an open position: the angles between the benzimidazole moieties become higher compared to those of the ortho isomers. The stronger interchain X⋯X interaction results in a higher BI–BI angle, the angle increases in the F, Cl, and Br order among the para-halogen derivatives (Table 2 and Fig. 6b), and the BI–BI angle is the largest in the case of para-bromo-phenylbenzimidazole (SEZNUH). The halogen–halogen interactions can only be formed if the phenyl-benzimidazole compounds contain a para halogenato substituent on the phenyl ring, otherwise it will not be present because of steric reasons. An additional ortho substituent lowers the BI–BI angle, while the additional meta substituent slightly increases the BI–BI angle.
In the isostructural series, the presence of the X⋯π (low BI–BI angle) or the X⋯X (high BI–BI angle) interaction divides the crystals into two distinct subgroups (Table 2). Both X⋯π and X⋯X interactions are present in the crystal structure of the simultaneously ortho and para chlorinated DETKIX molecule.
In the case of ortho substitution, the BI–BI angle is in the range of 92–112°. An additional meta substituent lowers the BI–BI angle, and it is 75° in GOLNOM. Single meta substitution (MCLPBI and MBRPBI) further decreases the BI–BI angle to 68°, where the benzimidazole rings of the neighbouring hydrogen bonded chains have not enough space to be in a parallel position to form π⋯π interactions with each other. Therefore, the packing changes from H–H to H–T and thus, as the space group transforms from Pbca into P21/c, the isostructurality is terminated. The space group change means the loss of a two-fold screw axis symmetry, as P21/c is a maximal non-isomorphic subgroup of Pbca.
In summary, in the fine-tuned isostructural Pbca series of the halogenated phenylbenzimidazole molecules, the supramolecular interactions and the tilt angle of the neighbouring BPIs depend highly on the placement of the substituents: the meta substitution resulted in the lowest, the ortho substitution in medium, and the para substitution in the highest values of BI–BI angles. The length of the most dependent crystallographic c axes changes just the opposite way. The presence of two different kinds of substituents in different positions results in an average of the structural parameters as a consequence of different cumulative effects. In the simultaneously ortho and meta chloro substituted GOLNOM, the BI–BI angle is lowered, the c axis is elongated to an extreme extent, and the Pbca space group is retained.
Multivariate data analysis of the structural data
Statistical tools were used for the analysis of the crystal structures in order to apply them in the recognition of isostructurality. Multivariate data analysis was performed to reveal correlations and similarities among the members of the series of structures of systematically halogenated 2-phenylbenzimidazole derivatives. The data of crystal parameters, molecular conformations and secondary interactions involved in the analysis are summarized in Tables 1 and 2 (all data except for β, Z, space group and H–H/H–T packing arrangement). The missing interactions indicated by ‘—’ in the tables were taken into account with a value of 7 in the statistical calculations (Table S2†). Significant correlations among the values of the lattice parameters, the molecular conformations and the secondary interactions of the investigated crystals were found using the software Statistica25 (Fig. S3 and Table S3†). Cluster analysis was performed on the standardized dataset, where samples are grouped based on similarities without considering the information about the class membership.It has to be highlighted that substitution and space group information were not taken into consideration in the cluster analysis merely based on the crystal data (a, b, c, V, KPI, and density), the molecular conformation (BI–BI angle and BI–Ph angle) and the secondary interaction data (N–H⋯N length and angle, oH(Bi)⋯oH(BI), oH(Bi)⋯oH(Ph), X⋯π and X⋯X distances).
The tree diagram obtained by cluster analysis indicates that the structures can be divided into three distinct similarity groups (Fig. 7) taking into account the linkage distances between their parameters. Group 1 contains the single-meta substituted MBRPBI and MCLPBI. They are the P21/c structures. GOLNOM is a sort of outlier, and the crystal of the ortho- and meta-substituted molecule is rather different from all other structures but more similar to the group 2 and 3 structures, which belong to the Pbca structures. OBRPBI, LUJWIW, OFPBI and DETKIX belong to group 2, and they are the ortho-substituted derivatives. Finally, JEYTUD, MINHOI, QERTIR, TUDXIB and SEZNUH belong to group 3, and they are the PBIs with para-substituents (Fig. 8). Principal component analysis has been performed for the same datasets and the same grouping of the elements could be identified as with cluster analysis (Fig. S4 and Table S4 and S5†). This grouping is in thorough agreement with our previous crystallographic analyses (check the very same order of structures in Tables 1 and 2) based only on mathematical data analysis without any prior knowledge of substitutions, space groups and structural analysis.
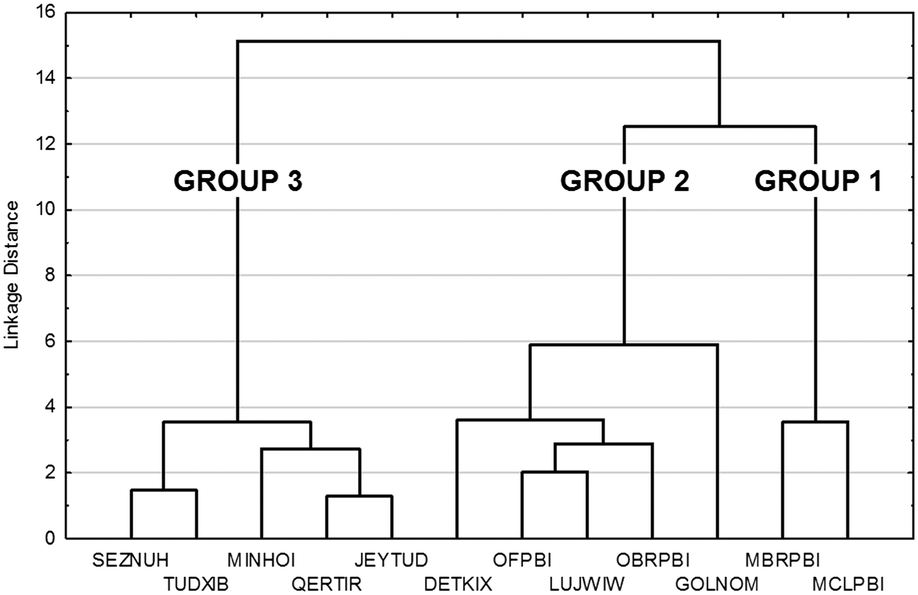 | ||
| Fig. 7 Cluster analysis of the crystal data, molecular conformation data and supramolecular interaction data (Tables 1 and 2 and S2–S4) in the fine-tuned series of halogen substituted PBI. | ||
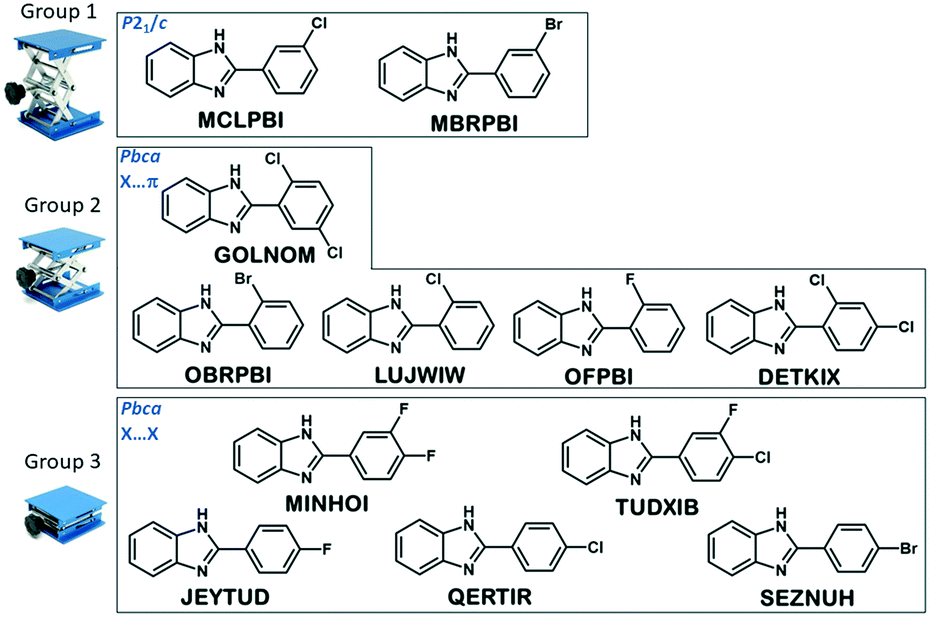 | ||
| Fig. 8 Grouping of the structures in the series of halogen substituted phenylbenzimidazoles based on the result of the cluster analysis. | ||
The multivariate data analysis was repeated with a reduced number of parameters choosing 5 instead of 14 to investigate the chance of simplification and the limits of the methods in the isostructurality studies. The parameters were selected to represent crystallographic data (a, b and c cell lengths), molecular conformation (BI–Ph angle) and supramolecular interaction (BI–BI angle) which is indirect information about the placement of the molecules in the unit cell. The same grouping was obtained by the data analysis presented on a tree diagram (Fig. S5†), while orders within the groups are occasionally interchanged.
The presented multivariate data analysis proves that the cluster analysis is a quick and easy-to-use tool to discover isostructurality before performing a thorough structure analysis to filter isostructural crystals out from the abundance of structures with similar cell parameters or even similar internal arrangements.
Cell similarity, isostructurality and molecular isometricity indices
In view of the similar cell dimensions, identical space groups and analogous molecular arrangements, the Pbca crystals of the halogenated 2-phenylbenzimidazoles are considered isostructural. However, the tilt between the molecules increases by 70% and the length of the a and c unit cell axes varies by ca. 70% within this group (Fig. 9). | ||
| Fig. 9 Superimposed Pbca unit cells of the halogenated 2-PBI structures indicating the arrangement of the molecules in their asymmetric unit. GOLNOM is coloured by elements, group 2 molecules are coloured green, and group 3 molecules are yellow. | ||
Cell similarity (π) and isostructurality (Is) indices were calculated (Fig. 10a) for the halogenated PBI derivatives in the Pbca space group. In the case of high similarity, π is close to zero. For more than half of the pairs of the structures (23 out of 45), π is less than 0.1. For the 84.4% of the structures, π is less than 0.2. Higher π values (between 0.2 and 0.4) are calculated only in the case of GOLNOM (GOLNOM vs. DETKIX, OFPBI, MINHOI, SEZNUH, JEYTUD, QERTIR, TUDXIB in ascending order) which is in accordance with the observation that this is the most outlier structure from the other Pbca structures. This result was also received from the multivariate data analysis on the cell parameters.
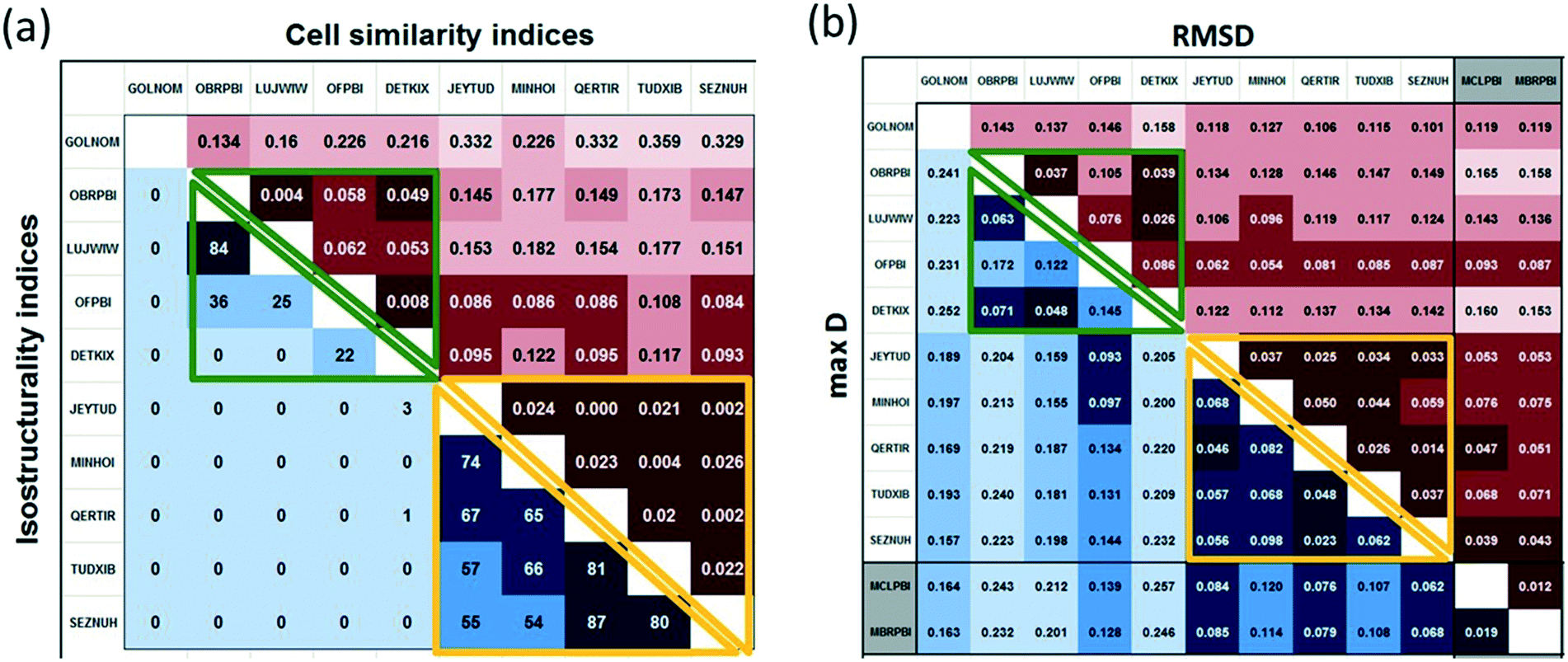 | ||
| Fig. 10 a. Cell similarity (π) and isostructurality (Is) indices calculated for the halogenated 2-phenylbenzimidazole structures crystallized in the Pbca space group. b. Molecular isometricity calculations:31 rmsD and maxD values for all the halogenated 2-phenylbenzimidazole structures. Header of the P21/c structures are highlighted in grey. Calculated values belong to group 2 structures are framed with green lines, and values belong to group 3 structures are framed with yellow lines. | ||
The isostructurality index (Is) takes into account both the differences in the geometry of the molecules and the positional differences caused by rotation and translation. As the similarity between two structures becomes higher, the value of Is becomes closer to 100%. The cell similarity and isostructurality indices form a well-structured pattern as a function of the BI–BI angle which is in good agreement with the above detailed structure analysis and the cluster analysis as well (Fig. 10a).
Both π and Is indices show high similarity in the group of JEYTUD, MINHOI, QERTIR, TUDXIB and SEZNUH compounds which corresponds to the cluster analysis of group 3 (Fig. 10a, yellow highlight). These are the para substituted derivatives eventually with an additional meta-fluoro substituent. Based on the pattern of the cell similarity indices considering the entries with π < 0.08, another group of compounds can be selected at lower BI–BI angles including OBRPBI, LUJWIW, OFPBI and DETKIX (top left) corresponding to the cluster analysis of group 2 (Fig. 10a, green highlight). The variation of the a unit cell axis lengths is 18–22% within this group.
It can be deduced from the data indicated in Fig. 10a that the isostructurality index gives a strict criterion for the structural similarity and decreases rapidly to zero. While the two isostructural subgroups (group 2 having X⋯π interactions and group 3 containing X⋯X interactions) are seemingly separated by Is calculation, the cell similarity change within the series is rather continuous.
The molecular conformation of flexible molecules may adjust to the supramolecular features. Molecular isometricity is a direct measure of the degree of approximate isomorphism of the compared molecules. It gives information about the molecular conformation only, and it is space group independent. The superimposed molecules are characterized by the root mean square of the distance differences of the corresponding atoms (rmsD) and by the largest distance difference of the compared atoms (maxD).31 The molecular conformations of the PBI frames irrespective of their substitution are compared (Fig. 10b). Because of the presence of the aromatic rings in the PBI molecule, the flexibility of the molecules is provided by the single bond. The groups 2 and 3 of the Pbca compounds can be recognised based on their small calculated rmsD and maxD values. The BI–Ph angles vary in rather narrow ranges in the two groups, and they are between 35.2 and 42.1° in group 2 compounds (except GOLNOM), while these angles are between 26.7 and 30.0° in group 3. Group 1 compounds can also be included in the molecular isometricity calculations and it seems that their molecular geometry is closer to group 3 compounds than group 2 compounds in the different P21/c space group (BI–Ph angles are 26.8 and 28.2°).
Packing coefficient, density, powder diffraction and isostructurality
Kitaigorodskii packing coefficients (KPI)32 were calculated for the crystals of the 2-phenylbenzimidazoles derivatives. The KPI values correspond with the supramolecular interaction patterns, e.g. group 2 and 3 structures separate well (Table 1).The KPI is also sensitive to the number of substituents within the group. Calculating the density of the crystals, it can be seen that the atomic weight of the substituents has higher influence on density than close packing. Notwithstanding, tendencies in the correspondence of the KPI, type and number of the substituents and the density can be observed.
The powder diffractograms show unique fingerprints of the solid compounds. PXRD is suitable for identification of individual crystals within the isostructural series (Fig. S6†). Comparing the calculated powder patterns of the isostructural materials, they are distinctive, although sometimes some similarities can be observed. Research on the match between the PXRD patterns of isomorphic crystals has just commenced.42
Isostructurality in the PBI series
The placement of the molecules in all halogen substituted 2-phenylbenzimidazole Pbca crystals is similar and is systematically changing in the presented fine-tuned series, indicated by the increasing angle of the neighbouring benzimidazole molecules. In general, para substitution (group 3) increases the tilt angle of the neighbouring benzimidazole molecules, and we may say that the scissors are more open, or the lab jack is in its lower positions. In the case of ortho substituents (group 2), the tilt angle of the neighbouring benzimidazole molecules is shrinking, the scissors are less open, or the lab jack is in its higher positions. A further decrease in the tilt angle of the neighbouring benzimidazole molecules achieved by meta substitution (group 1) terminates isostructurality and results in a change of the space group to P21/c.The flexibility of the structures permits a gradual change of the lengths of the unit cells. In the presented examples, it is the most pronounced (nearly two-fold) in the crystallographic c direction, with unchanged space group and Z, perpendicular to the common, determining N–H⋯N hydrogen bonded chains.
There is a switch in the intra- and intermolecular interactions (DETKIX) which divides the investigated isostructural Pbca structures into two subgroups: there are X⋯π intermolecular interactions in group 2 (lab jack upper positions) and there are X⋯X interactions in the group 3 subgroup (lab jack lower positions).
It is presented in the exemplary series of the substituted 2-phenylbenzimidazole structures how the isostructural similarities of Pbca crystals can be described by numerical descriptors and how the relationship between the molecular and supramolecular properties with the structural features can be revealed and characterized. Cell similarity, isostructurality, molecular isometricity calculations are congruent with multivariate data analysis and made the identification and description of the structural similarities of the PBI compounds possible. The types of structural information used in the different kinds of structural comparisons are summarized in Table 3.
| F | S | SG | Z | UC | V | Co | Pl | SI | |
|---|---|---|---|---|---|---|---|---|---|
| a CA1: cluster analysis on 14 parameters, CA2: cluster analysis on 5 parameters, π: cell similarity index, Is: isostructurality index, Im: molecular isometricity, KPI: Kitaigorodskii packing coefficient and calculated density, F: formula, S: substitution, SG: space group, Z: Z value, UC: unit cell lengths, V: unit cell volume, Co: molecular conformation, Pl: placement of the molecule in the cell, and SI: secondary interactions. | |||||||||
| CA1 | — | — | — | — | ✓ | ✓ | ✓2 | — | ✓6 |
| CA2 | — | — | — | — | ✓ | — | ✓1 | — | ✓1 |
| π | — | — | — | — | ✓ | — | — | — | — |
| I s | ✓ | ✓ | ✓ | ✓ | — | — | ✓ | ✓ | — |
| I m | ✓ | ✓ | — | — | — | — | ✓ | — | — |
| KPI | ✓ | — | — | — | — | ✓ | — | — | — |
Conclusions
Property engineering, the fine tuning of structural properties, can be achieved by application of substituents, changing their placement and/or chemical composition influencing electrostatic and sterical properties. Both placement of the molecules in the crystal lattice and molecular conformation of flexible molecules may adjust to the supramolecular features. A given packing motif may tolerate small molecular changes, and the structures remain isostructural despite the chemical changes within a limit.The packing arrangements of the neighbouring structures in the fine-tuned series – like during the up or down movement of the lab jack – are highly similar. The structures from the two ends of the ordered list – like the open and the closed lab jack positions – have low similarity. It is a question whether we recognise and consider the two opposite terminal members of the structure series being isostructural. It is necessary to work out a method in order to determine and define the criteria for the limits of isostructurality: what the extent of differences do we still consider similar.
There are crystals whose cell parameters are similar, space groups are the same, arrangements of the molecules are analogous in all structures, but the only difference is in the preference of intermolecular interactions – like groups 2 and 3 in the example series above. According to the IUCr definition,41 “two crystals are said to be isostructural if they have the same structure, but not necessarily the same cell dimensions nor the same chemical composition, and with a ‘comparable’ variability in the atomic coordinates to that of the cell dimensions and chemical composition”. This definition does indicate the need to “have the same structure”, but it does not say anything about the probable necessity for the similarity of the supramolecular interactions as a criterion of the isostructurality of crystals.
The unit cells of different compounds with different internal arrangements may be occasionally similar. Notwithstanding, the cells of chemically similar compounds with analogous internal arrangements are necessarily similar. Anyhow, recognition of isostructurality is not necessarily straightforward, see the case of the two termini of the presented series of 2-phenylbenzimidazoles in the Pbca space group.
Here is a recommendation for a technique on how to recognise and numerically describe isostructurality step by step:
1. Isostructurality analysis of crystals of similar composition should start with the calculation of the cell similarity index (π).
2. Conformations of molecules in the isostructural crystals are alike. Their similarity can be compared by molecular isometricity calculations.
3. Multivariate data analysis (cluster analysis or principal component analysis) of the structural data, especially in the case of a higher number of crystals, can assist grouping of the structures into subclasses.
4. The prerequisite of isostructurality is the similar molecular geometry and the analogous placement of molecules in the crystal lattice; it is described numerically by the isostructurality index (Is).
5. Isostructurality investigations have to be completed with the similarity check of the synthons and the supramolecular interactions among the compared crystals.
The calculation of cell similarity (π) and isostructurality (Is), as well as molecular isometricity indices, completed with a prior multivariate data analysis of the structural data contributes to the easy recognition and characterization of isostructural crystals. Tools to describe isostructurality are important in fine-tuning of crystal properties with the final aim to prepare new materials with desired properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gergely O. Szabó is acknowledged for his contribution to the synthesis, and Zita Makó participated in the crystallisation and structure determination of MCLPBI. The authors are grateful to Gyula T. Gál for the solution of the MBRPBI structure. Tamás Holczbauer is acknowledged for his advices about the diffraction measurements and his continuous support for the crystallography group members. This work was supported by the National Research, Development and Innovation Office-NKFIH through OTKA K124544 and KH129588 and the J. Bolyai Research Scholarship BO/00109/17 of the Hungarian Academy of Sciences (N. V. M.).Notes and references
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
- G. R. Desiraju, Nature, 2001, 412, 397–400 CrossRef CAS PubMed.
- V. A. Russell and M. D. Ward, Chem. Mater., 1996, 8, 1654–1666 CrossRef CAS.
- G. Resnati, E. Boldyreva, P. Bombicz and M. Kawano, IUCrJ, 2015, 2, 675–690 CrossRef CAS PubMed.
- R. Taylor, J. C. Cole and C. R. Groom, Cryst. Growth Des., 2016, 16, 2988–3001 CrossRef CAS.
- P. Bombicz, Crystallogr. Rev., 2017, 23, 118–151 CrossRef.
- P. Bombicz, T. Gruber and C. Fischer, CrystEngComm, 2014, 16, 3646–3654 RSC.
- E. J. C. de Vries, S. Kantengwa, A. Ayamin and N. Báthori, CrystEngComm, 2016, 18, 7573–7579 RSC.
- C. Fischer, P. Bombicz and G. Lin, Cryst. Growth Des., 2012, 12, 2445–2454 CrossRef CAS.
- S. Ranjan, R. Devarapalli, S. Kundu, S. Saha, S. Deolka, V. R. Vangalac and C. M. Reddy, IUCrJ, 2020, 7, 173–183 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno and M. P. Lightfoot, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- C. B. Aakeröy, P. D. Chopade and J. Desper, Cryst. Growth Des., 2011, 11, 5333–5336 CrossRef.
- P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12, 5835–5838 CrossRef CAS.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS.
- D. Secci, A. Bolasco, M. D'Ascenzio, F. Sala, M. Yánez and S. Carradori, J. Heterocyclic Chem., 2012, 49, 1187–1195 CrossRef CAS.
- N. A. Weires, J. Boster and J. Magolan, Eur. J. Org. Chem., 2012, 33, 6508–6512 Search PubMed.
- P. Sang, Y. Xie, J. Zou and Y. Zhang, Org. Lett., 2012, 15, 3894–3897 CrossRef PubMed.
- J. Feng, S. Handa, F. Gallou and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 8979–8983 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarno, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- T. Hastie, R. Tibshirani and J. Friedman, The Elements of Statistical Learning, Data Mining, Inference, and Prediction, Springer, 2008 Search PubMed.
- S. Wold, K. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37–52 CrossRef CAS.
- Dell Inc., Dell Statistica (data analysis software system), version 13, software.dell.com, 2016.
- A. Kálmán, L. Párkányi and Gy. Argay, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1039–1049 CrossRef.
- A. Kálmán and L. Párkányi, Isostructurality of organic crystals in Advances in molecular structure research 3 ed. M. Hargittai and I. Hargittai, Greenwich JAI Press, 1997, pp. 189–226 Search PubMed.
- A. Kálmán In, Fundamental principles of molecular modeling, ed. W. Gans, New York Plenum Press, 1996 Search PubMed.
- L. Párkányi, ISOS, Software for isostructurality calculation, http://www.chemcryst.hu/isos/.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76, 1–11 CrossRef CAS PubMed.
- N. Rashid, M. K. Tahir, S. Kanwal, N. M. Yusof and B. M. Yamin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1402 CrossRef CAS.
- M. S. Krishnamurthy, N. Fathima, H. Nagarajaiah and N. S. Begum, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o1689 CrossRef CAS PubMed.
- M. S. Krishnamurthy and N. S. Begum, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o387 CrossRef CAS PubMed.
- M. Azam, A. A. Khan, S. I. Al-Resayes, M. S. Islam, A. K. Saxena, S. Dwivedi, J. Musarrat, A. Trzesowska-Kruszynska and R. Kruszynski, Spectrochim. Acta, Part A, 2015, 142, 286 CrossRef CAS PubMed.
- F.-F. Jian, H.-Q. Yu, Y.-B. Qiao, T.-L. Liang and P.-S. Zhao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o321 CrossRef CAS.
- R. T. Stibrany and J. A. Potenza, CSD Communication (Private Communication), 2010.
- F.-F. Jian, H.-Q. Yu, Y.-B. Qiao, P.-S. Zhao and H.-L. Xiao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o5194 CrossRef CAS.
- N. Rashid, M. K. Tahir, N. M. Yusof and B. M. Yamin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1260 CrossRef CAS.
- IUCr, Definition of isostructurality, https://dictionary.iucr.org/Isostructural_crystals.
- S. Ranjan, R. Devarapalli, S. Kundu, S. Saha, S. Deolka, V. R. Vangalac and C. M. Reddy, IUCrJ, 2020, 7, 173–183 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1990350–1990353 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00410c |
| This journal is © The Royal Society of Chemistry 2020 |