
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemote functional group directed C–H activation by an Ir(III) phenanthroline complex†
Rozalie Sharon Genevieve
Corea
 a and
Scott
Gronert
*ab
a and
Scott
Gronert
*ab
aDepartment of Chemistry, Virginia Commonwealth University, 1001 W Main Street, Richmond, VA 23284, USA
bDepartment of Chemistry and Biochemistry, University of Wisconsin – Milwaukee, 3210 N. Cramer St., Milwaukee, WI 53211, USA. E-mail: sgronert@uwm.edu
First published on 20th November 2020
Abstract
The regioselectivity of the C–H activation of 1-butanol and 1-methoxybutane by an iridium(III) phenanthroline complex was studied in the gas phase and revealed activation at gamma and delta carbons. In the ether, nearly exclusive gamma activation was observed. DFT calculations were used to explore the origin of this substrate-driven selectivity. The data show that the iridium(III) complex is a potent and potentially highly selective remote C–H activation agent.
C–H activation has been referred to as the “holy grail” of chemistry1 because the ability to functionalize at C–H bonds in the absence of activating groups has great potential in many synthetic pathways.2–4 For this reason, there is much interest in agents that can selectively activate C–H bonds.5–7 Alcohols and ethers are two of the most commonly-used and readily-available precursors in synthesis,8 and C–H activation of them would offer novel synthetic strategies for adding complexity to these substrates.9 The C–H activation of these molecules was not routinely implemented until the 1990s.10 This is partly due to the inherently low reactivity imparted by these functional groups. Functional group directed C–H activation by organometallic complexes aids in achieving selective activation based on feasible geometry of the final product.11,12 More recently, iridium complexes have proven to be viable catalysts for C–H activation of various alcohol, ether and other substrates.11,13–17 In 2015, our group reported the ability of an iridium(III)-1,10-phenanthroline catalyst to activate secondary C–H bonds in cyclohexane.18 Here, we use gas-phase studies to explore whether the high reactivity of this complex can be harnessed in the selective, remote activation of C–H bonds in a model alcohol and ether, 1-butanol and 1-methoxybutane. Gas-phase methods have proven to be powerful tools for elucidating the mechanistic characteristics of organometallic complexes and their associated reactive intermediates as well as identifying potential reagents for synthetic applications.19,20 The experiments were carried out on a modified Thermo Finnigan LCQ ion trap mass spectrometer with an ESI source using established methodologies.21 Calculations were completed with Gaussian1622 using the M0623 DFT methodology with a mixed basis set for optimizations and frequency calculations (LANL2DZ24 on iridium and 6-311+G**25 on all other atoms) and an all-electron basis set (QZVP) for final electronic energies.
With complex I, C–H activation leads to the loss of H–Cl and the formation of a new Ir–C bond at the site of the hydrogen loss (Scheme 1). Given that I reacts with cyclohexane, it is not surprising that a similar process is seen when simple ethers and alcohols are treated with I. Spectra for the reactions of 1-butanol and 1-methoxybutane with I are given in Fig. 1. Along with C–H activation, the reactions also lead to adducts with the alcohol/ether and adventitious water in the ion trap. The conditions in these gas-phase studies make adducts common (i.e., relatively high pressures, ∼1 mTorr He). With the alcohols and ethers, the site of activation is not clear in mass spectrometric studies because the only evidence of reaction is HCl loss and we do not have effective ways to probe the structure of the product ion – collision-induced dissociation of the product does not reveal useful data about the site of activation. However, selective deuteration of the substrates can be used to identify the sites of activation by monitoring for HCl vs. DCl loss (e.g., Scheme 2).
 | ||
| Scheme 1 C–H activation reaction of complex I. | ||
 | ||
| Fig. 1 Reaction of I with 1-butanol (top) and 1-methoxybutane (bottom). In all experiments, the low-mass isotopomer of I (m/z 441) was used to avoid mixtures of isotopes in the parent ion (e.g., 191Ir/35Cl vs.193Ir/37Cl). Top: m/z 478 is C–H activation (loss of HCl), m/z 515 is adduct of one 1-butanol molecule and m/z 588 is adduct of two 1-butanol molecules. Bottom: m/z 458 is adduct of water molecule, m/z 476 is adduct of two water molecules, m/z 492 is C–H activation (loss of HCl) and m/z 528 is adduct of a 1-methoxybutane molecule. | ||
 | ||
| Scheme 2 Loss of HCl vs. loss of DCl indicates the site of activation. | ||
A series of selectively deuterated 1-butanol and 1-methoxybutane were used for this study (Scheme 3). The deuterated methyl ethers were synthesized from the corresponding, commercially-available alcohols (see ESI†).
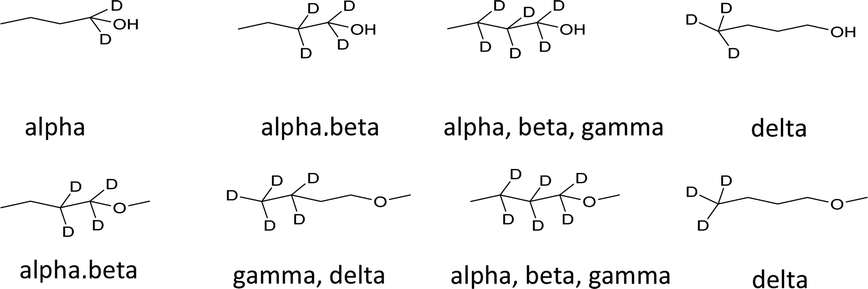 | ||
| Scheme 3 Labeled substrates used in this study. | ||
Each substrate was allowed to react with complex I and product distributions were determined. For the reactions with 1-butanol, the distributions of C–H vs. C–D activation (i.e., H–Cl vs. D–Cl loss) are shown in Fig. 2. C–D activation is only seen when a deuterium is present at the gamma or delta carbon (3- or 4-position) of the butanol. C–H activation is significantly preferred in each case, suggesting important kinetic deuterium isotope effects. To determine the primary deuterium isotope effects in these C–H activations, as well as more accurately assess the regioselectivity, we carried out an experiment with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of the d3 and d6 alcohols. In this experiment, we can differentiate the products of C–H and C–D activation in each alcohol. The relative rates suggest that delta activation is preferred by 55%/45% over gamma activation. If we ignore any secondary isotope effects from vicinal deuteria, the data suggest a kH/kD value of 2.3 for the gamma position and 3.3 for the delta position (ESI,† Fig. S14). These are very reasonable primary deuterium isotope effects for a process with C–D cleavage in the rate-determining step.
:1 mix of the d3 and d6 alcohols. In this experiment, we can differentiate the products of C–H and C–D activation in each alcohol. The relative rates suggest that delta activation is preferred by 55%/45% over gamma activation. If we ignore any secondary isotope effects from vicinal deuteria, the data suggest a kH/kD value of 2.3 for the gamma position and 3.3 for the delta position (ESI,† Fig. S14). These are very reasonable primary deuterium isotope effects for a process with C–D cleavage in the rate-determining step.
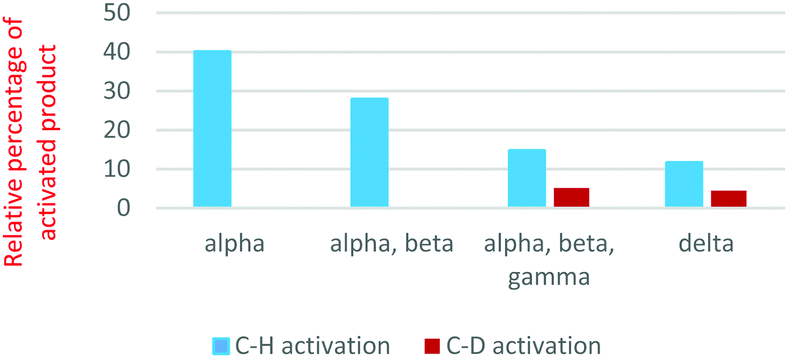 | ||
| Fig. 2 C–H vs. C–D activation of 1-butanol by complex I. The x-axis designates location of deuterium in substrate while the y-axis shows the percentage of the activated product (C–H or C–D) relative to the other products formed with adventitious water, methanol and the substrate. | ||
DFT calculations at the M06 level were carried out on the reaction of 1-butanol with I to further understand the mechanism. Calculation of the reaction enthalpies indicate that both gamma and delta activation are very exothermic processes (>60 kcal mol−1, see ESI†). This implies that the driving force of the selectivity has to be found in the transition state. In these reactions, coordination of the alcohol oxygen to the iridium center is favorable and locks the transition state into a cyclic structure. This clearly is the origin of the preference for the unactivated gamma and delta carbons over the activated alpha carbon of the alcohol. Gamma activation leads to a 5-membered ring while delta activation forms a 6-membered ring (Fig. 3). As we have seen previously in studies of the reaction of I with alkanes, the process is concerted with the iridium guiding the hydrogen from the carbon to the chlorine, without an Ir–H intermediate. Structurally the two transition states are very similar. In each the Ir–O distance is 2.14 Å and the Ir–C distances are between 2.16 and 2.19 Å. The minor difference is that the H–Cl bond formation is more advanced in the delta transition state (H–Cl = 1.45 Å vs. 1.54 Å in gamma), but the surface is relatively flat so this is not particularly consequential. The computed enthalpies of activation favor the delta position by 3.1 kcal mol−1 (−6.0 vs. −2.9 kcal mol−1), but both processes would be viable under the gas-phase conditions and with barriers well below the entrance channel energies, the observed competition is not surprising. The delta transition state benefits from the six-membered ring as well as the absence of some steric interactions that are seen in the gamma transition state between the delta methyl group and a hydrogen on the phenanthrene (H–H distance = 1.85 Å).
 | ||
| Fig. 3 Transition states for C–H activation of 1-butanol by I. Computed at the M06 level with a mixed basis set (LANL2DZ on iridium and 6-311+G** on all other atoms). Gamma activation is the top image and delta activation is the bottom image. Color coding: carbon, grey; hydrogen, light grey; nitrogen, blue; chlorine, green; oxygen, red; and iridium, teal. Structures generated in Avogadro.26 | ||
With this promising evidence of remote functional group driven regioselectivity in the C–H activation process, and the clear indication that coordination to the oxygen was a driver in the selectivity, we decided to shift from the alcohol to a methyl ether. The reasoning was that the added methyl group would not significantly interfere with the oxygen coordination to the iridium, but would put greater conformational constraints on the cyclic transition states, potentially conveying greater regioselectivity. To test this hypothesis, studies were carried out with 1-methoxybutane and isotopomers similar to those used in the 1-butanol study (Scheme 3). As shown in Fig. 1, the iridium(III) complex readily activates a C–H bond in 1-methoxybutane. In this case, there is a much sharper distinction between the sites of activation (Fig. 4). As with the alcohol, there is no evidence of reaction at the alpha or beta carbons. When a deuterium is present at the gamma carbon, there is a high yield of C–D activation (3,3,4,4,4-d5 and 1,1,2,2,3,3-d6), whereas there is almost no C–D activation when deuterium is only present on the delta carbon (4,4,4-d3). This would suggest nearly exclusive reactivity at the gamma carbon, but in the 1,1,2,2,3,3-d6 isotopomer, there is a small yield of what appears to be C–H activation, which suggests reaction at the delta carbon. Again we turned to using a mixture of isotopmers (d3 and d6) to give more accurate relative rate values and kinetic isotope effects. The results indicate that gamma C–H activation is favored 24-fold (96%/4%) over delta activation (Fig. S9, ESI†). The data also suggest a kH/kD value of 2.5 for the gamma position. It was not possible to compute a kH/kD value for the delta position with reasonable certainty given the very low signal intensity for C–D loss at the delta position. The need to correct for 13C isotopomers in the ether also adds to the uncertainty in determining the delta isotope effect (the 13C component of the gamma C–D loss has a significant contribution at the same m/z as the delta C–H loss). In any case, it is clear that the addition of the methyl group has a sharp effect on the regioselectivity and tilts the preference to gamma and its 5-membered ring transition state.
 | ||
| Fig. 4 C–H vs. C–D activation of 1-methoxybutane by complex I. The x-axis designates location of deuterium in substrate while the y-axis shows the percentage of the activated product (C–H or C–D) relative to the other products formed with adventitious water, methanol and the substrate. | ||
DFT calculations identified transition states for 1-methoxybutane that are similar to those found for butanol (Fig. 5). Unlike the 1-butanol system, here the gamma transition is computed to be favored by 1.1 kcal mol−1. Although the 4.2 kcal mol−1 shift in the preference towards gamma is consistent with the experiments, the absolute values suggest that the greatest selectivity should be for delta in 1-butanol. Test calculations with a double hybrid functional did not materially change this result (Table S2, ESI†) nor did free energy corrections. It is not clear whether the discrepancy is the result of the challenges of computationally modeling steric effects or if dynamics effects in the experiments are playing a role given that the barriers are well below the entrance channel (>20 kcal mol−1). The preference for gamma over delta in 1-methoxybutane is due to the steric interactions with the ligand and the added methyl group of the ether. In examining the structures, it is clear that in the delta TS, there is crowding between the phenanthroline ligand and the methoxy's methyl group, which is manifested in a highly distorted C–O–C–C dihedral angle. In the delta TS, it is twisted from the preferred anti orientation (180°) to give a dihedral angle of only 148°. In contrast, the gamma TS can accommodate the methoxy's methyl group and a C–O–C–C dihedral angle of 175° is observed. It should also be noted that in a relative rate experiment, 1-butanol produces the C–H activation product at a rate 30% higher than 1-methoxybutane (ESI,† Fig. S15), which is consistent with the added methyl group suppressing delta C–H activation in the ether.
 | ||
| Fig. 5 Transition states for C–H activation of 1-methoxybutane by I. Computed at the M06 level with a mixed basis set (LANL2DZ on iridium and 6-311+G** on all other atoms). Gamma activation is the top image and delta activation is the bottom image. Color coding: carbon, grey; hydrogen, light grey; nitrogen, blue; chlorine, green; oxygen, red; and iridium, teal. Structures generated in Avogadro.26 | ||
Complex I is a potent C–H activation agent and can lead to selective activation of sites remote from functional groups. In this case, an oxygen can be used to anchor the substrate on the iridium center, driving activity to the gamma or delta carbon. Further tuning is possible with subtle variations near the coordination point, as demonstrated by the ether. These data highlight the potential of complex I and related species to be used as selective agents for C–H activation.
Conflicts of interest
The authors report no conflict of interest.Notes and references
- R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS.
- H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391–3393 CrossRef CAS.
- W. J. Kerr, D. M. Lindsay, P. K. Owens, M. Reid, T. Tuttle and S. Campos, ACS Catal., 2017, 7, 7182–7186 CrossRef CAS.
- L. Niu, J. Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019, 10, 467 CrossRef.
- W. D. Jones, Advances in Carbon–Hydrogen Activation, 2007, vol. 1 Search PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS.
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS.
- Y. Zhang and N. D. Schley, Chem. Commun., 2017, 53, 2130–2133 RSC.
- F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275–296 CrossRef CAS.
- S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937 RSC.
- J. F. Hartwig and E. A. Romero, Tetrahedron, 2019, 75, 4059–4070 CrossRef CAS.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS.
- C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592–595 CrossRef CAS.
- S. Conejero, M. Paneque, M. L. Poveda, L. L. Santos and E. Carmona, Acc. Chem. Res., 2010, 43, 572–580 CrossRef CAS.
- K. I. Goldberg and A. S. Goldman, Acc. Chem. Res., 2017, 50, 620–626 CrossRef CAS.
- K. E. Allen, D. M. Heinekey, A. S. Goldman and K. I. Goldberg, Organometallics, 2013, 32, 1579–1582 CrossRef CAS.
- J. Choi and A. S. Goldman, in Iridium Catalysis, ed. P. G. Andersson, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, vol. 34, pp. 139–167 Search PubMed.
- C. A. Swift and S. Gronert, Angew. Chem., 2015, 127, 6575–6578 CrossRef.
- K. Eller and H. Schwarz, Chem. Rev., 1991, 91, 1121–1177 CrossRef CAS.
- J. C. Traeger, Int. J. Mass Spectrom., 2000, 200, 387–401 CrossRef CAS.
- S. Gronert, J. Am. Soc. Mass Spectrom., 1998, 9, 845–848 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 119, 525 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06298g |
| This journal is © The Royal Society of Chemistry 2020 |