
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrophosphination using [GeCl{N(SiMe3)2}3] as a pre-catalyst†
A. N.
Barrett
 ,
H. J.
Sanderson
,
M. F.
Mahon
and
R. L.
Webster
*
,
H. J.
Sanderson
,
M. F.
Mahon
and
R. L.
Webster
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: r.l.webster@bath.ac.uk
First published on 9th October 2020
Abstract
Transformations catalyzed by germanium are scarce, with examples mainly limited to widely catalyzed processes such as polymerisation of lactide and hydroboration of carbonyls. Reported is the first example of hydrophosphination using a germanium pre-catalyst, yielding anti-Markovnikov products when diphenylphosphine is reacted with styrenes or internal alkynes at room temperature.
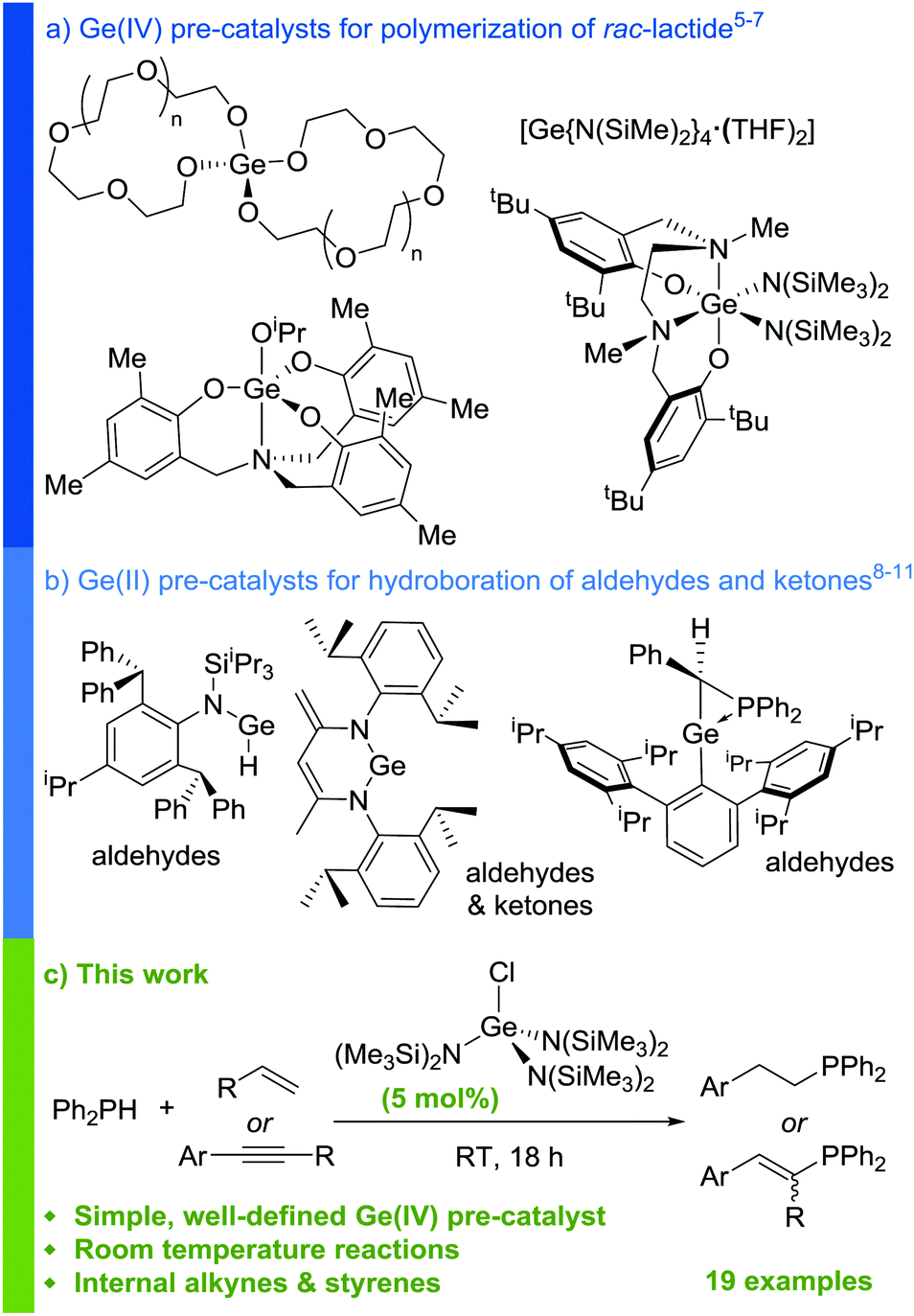
Over the last two decades, the use of main group compounds as catalysts for organic reactions has seen a vast increase in focus.1–3 Amongst this work, significant progress has been made in the field of germanium catalysis. A fundamental challenge lies in unlocking the catalytic potential of this group 14 element, which is far behind tin in terms of its exploitation in catalysis, and such work may prove instrumental to furthering understanding and harbouring new discoveries in main group catalysis. It is not surprising, based on the ability of Sn(IV) complexes to act as polymerization catalysts,4 that initial disclosures in the literature demonstrated the ability of Ge(IV) complexes to act as initiators for the ring-opening polymerization of lactide (Scheme 1a).5–7 Subsequently, there have been numerous reports on low-coordinate Ge(II) species acting as efficient pre-catalysts for the hydroboration of carbonyls (Scheme 1b).8–11 This reactivity has also been extended to the catalytic cyanosilylation of aldehydes.12,13
 | ||
| Scheme 1 Examples of germanium catalysis reported in the literature to-date. | ||
Despite this progress, lactide polymerization and carbonyl hydrofunctionalization are commonly reported catalytic transformations,14,15 therefore a desire remains to uncover use for germanium (both Ge(II) and Ge(IV)) in the catalysis of a wider range of transformations.16 It is clear from the current literature that, in the presence of at least one labile co-ligand, Ge(IV) complexes can catalyze non-redox, σ-bond metathesis type transformations such as lactide polymerization. This indicates that opportunities exist for Ge(IV) catalysts, particularly in hydrofunctionalization reactions with pertinent substrates.
The hydrophosphination (HP) of unsaturated hydrocarbons is an atom economical process of forming P–C bonds.17 Using a main group element to transform another is an area of growing interest, amongst which examples of main group catalyzed HP have emerged.18 Reaction mechanisms for these transformations involving main group, and lanthanide,19–23 complexes are often postulated to proceed via σ-bond metathesis type steps at the catalyst center,24 leading us form the hypothesis that a Ge catalyst may have success in this area. To the best of our knowledge, no examples of Ge-catalyzed HP have been reported. However, the heavier group 14 element, Sn, has been shown to be active catalytically in the HP of alkenes and alkynes by Waterman and co-workers, who, in 2014, reported a modest scope of HP reactions using PhPH2.25 Subsequent studies on the same Sn(IV) system using Ph2PH in 2016 expanded the scope to further substrates.26 However, these transformations required an atmosphere of H2 due to the catalyst's propensity to undergo homodehydrocoupling of the phosphine substrate over the desired HP reaction. This, along with the elevated temperatures necessary (65 °C) and high catalyst loading (10 mol%), makes the reaction only marginally more competitive than the analogous catalyst-free, thermal process.27,28 We were therefore keen to investigate if a more active catalyst could be found within group 14 in the form of a Ge(IV) species. Rather than start our studies with an over-engineered catalyst structure, we looked to initiate our studies with an operationally simple motif; understand the catalytic process and thus be in a strong position to build in ligand design at a later stage, based on understanding. We herein present the first HP of alkenes using a structurally simple Ge(IV) pre-catalyst under mild conditions (Scheme 1c).
Reaction of GeCl4 with 10 equiv. of Na(HMDS) yields [GeCl{N(SiMe3)2}3], 1, in good yield (63%, Scheme 2), the structure of which was confirmed by X-ray diffraction.29 In an analogous reaction, Thomas and co-workers propose the formation of tetra(amido) THF adduct [Ge(HMDS)4(THF)2]:7 in our synthesis 1H DOSY analysis indicates lack of persistent THF coordination in solution.29
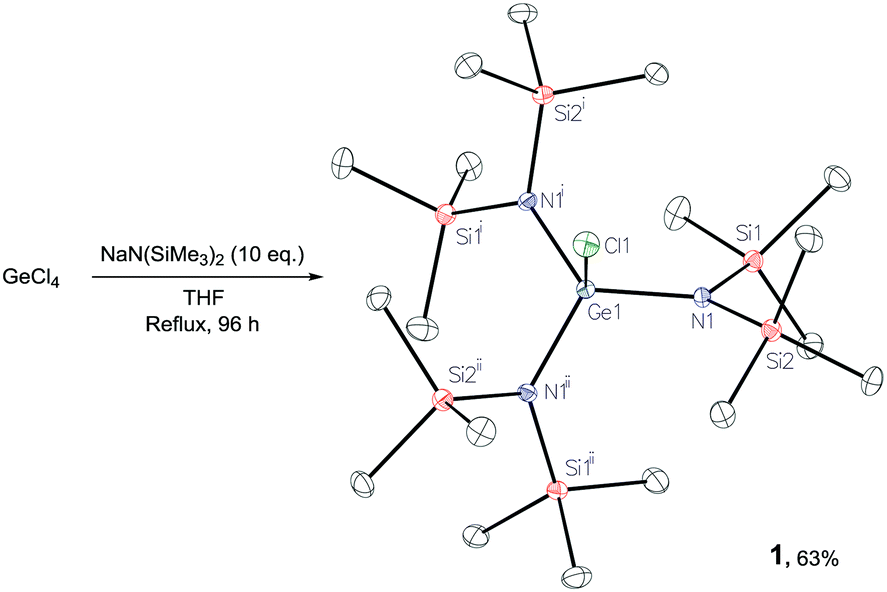 | ||
Scheme 2 Synthesis and single crystal X-ray structure of 1 displaying the inherent C3v symmetry of the molecule. Ellipsoids are represented at 30% probability and hydrogen atoms have been omitted for clarity. Symmetry operations: i![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 + y − x, 1 − x, z; ii−1 − y, x − y, z. Bond distance (Å) and angles (°): Ge1–Cl1, 2.2154(11); Ge1–N1, 1.8542(16); Cl–Ge–N1 102.12(6); N1–Ge–N1′ 115.71(4). 1 + y − x, 1 − x, z; ii−1 − y, x − y, z. Bond distance (Å) and angles (°): Ge1–Cl1, 2.2154(11); Ge1–N1, 1.8542(16); Cl–Ge–N1 102.12(6); N1–Ge–N1′ 115.71(4). | ||
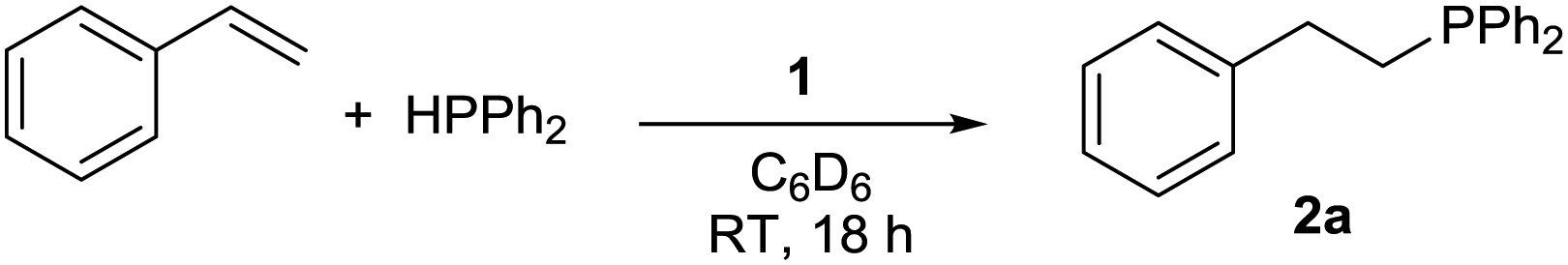
Pleasingly, 1 is an efficient pre-catalyst for the HP of styrene with diphenylphosphine (Ph2PH) and provides 91% conversion to the anti-Markovnikov HP product 2a after 18 hours at RT with a catalyst loading of 5 mol%, using C6D6 as solvent (Table 1, entry 1). Only trace quantities of homodehydrocoupling by-product P2Ph4 is formed, therefore any concerns over a competing reaction of this type prove insignificant. Markovnikov product is never observed. Catalyst loadings of 2.5 mol% and 1 mol% result in the reduction of spectroscopic yield to 81% and 38% respectively (entries 2 and 3). Notably, no reaction is observed when GeCl4 is used as the catalyst. Change of solvent to THF gives similar conversion compared to benzene (entry 4). Other solvents (MeCN (vide infra), DCM, CDCl3) show reactivity with 1 and are therefore not suitable for the HP reaction.29 A modest excess of styrene (1.2 equiv.) gives 94% conversion in an inexpensive NMR solvent (entry 5).
|
|
|||
|---|---|---|---|
| Entry | Catalyst loading/mol% | Solvent | Spectroscopic yieldb/% |
| a Conditions: styrene (0.5 mmol, 57 μL), Ph2PH (0.5 mmol, 87 μL), 1 (5 mol%, 0.025 mmol, 14.7 mg), C6D6 (0.6 mL). b Spectroscopic yield calculated via integration against 1,3,5-trimethoxybenzene internal standard. c 0.6 mmol styrene. | |||
| 1 | 5 | C6D6 | 91 |
| 2 | 2.5 | C6D6 | 81 |
| 3 | 1 | C6D6 | 38 |
| 4c | 5 | THF | 99 |
| 5c | 5 | C6D6 | 94 |
With the optimized conditions in hand (Table 1, entry 5) we then set out to investigate the substrate scope for the reaction. The reaction proceeds with a wide range of styrenes (Table 2). Biphenyl-substituted phosphine 2b is formed in excellent yield (entry 2). p-Me-, p-MeO- and p-CF3-substituted phosphines (2c–2e) are formed in reduced conversions (entries 3 to 5). Halide substituents are well tolerated, with phosphine products 2f to 2h formed in good to moderate conversion (entries 6 to 8). m- and o-substituted phosphines (2i to 2m) are also formed in good yield (entries 9 to 13).
| Entry | Alkene | Product | Spec. yieldb (%) | |
|---|---|---|---|---|
|
a Conditions: styrene (0.6 mmol), Ph2PH (0.5 mmol, 87 μL), 1 (5 mol%, 0.025 mmol, 14.7 mg), C6D6 (0.6 mL), 18 h, RT.
b Spectroscopic yield calculated via integration of product peak against a trimethoxybenzene internal standard.
c 50 °C, 24 h.
d RT, 96 h.
e RT, 48 h. Spec. yield and E:Z calculated via31P inverse gated NMR spectroscopy against a P(OEt)3 internal standard.
f RT, 72 h. Spec. yield and E:Z ratio calculated via31P inverse gated NMR spectroscopy against a P(OEt)3 internal standard.
|
||||
|
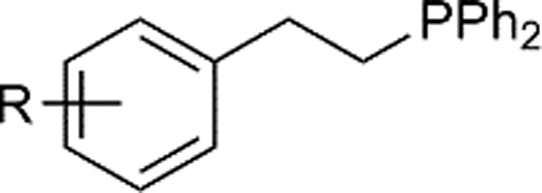
|
|||
| 1 | R = H | 2a | 94 | |
| 2 | 4-Ph | 2b | 98 | |
| 3 | 4-Me | 2c | 94c | |
| 4 | 4-OMe | 2d | 56c | |
| 5 | 4-CF3 | 2e | 83c | |
| 6 | 4-F | 2f | 76c | |
| 7 | 4-Cl | 2g | 94 | |
| 8 | 4-Br | 2h | 64 | |
| 9 | 3-Me | 2i | 92 | |
| 10 | 3-Br | 2j | 82 | |
| 11 | 2-Me | 2k | 89 | |
| 12 | 2-OMe | 2l | 89 | |
| 13 | 2-Br | 2m | 84 | |
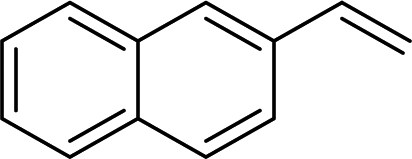
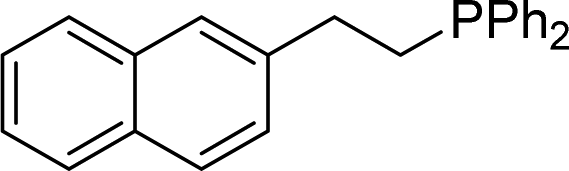
| 14 |
|
|
2n | 90 |
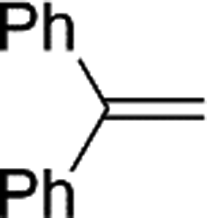
| 15 |
|

|
2o | 70c |
| 16 |

|
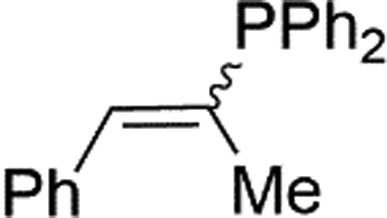
|
2p | 84d |
| 3.9:1, E:Z |
||||
| 17 |

|
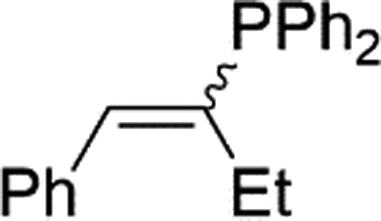
|
2q | 99c |
| 1:1, E:Z |
||||
| 18 |

|
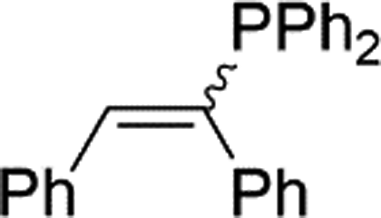
|
2r | 81e |
| 1.3:1, E:Z |
||||
| 19 |
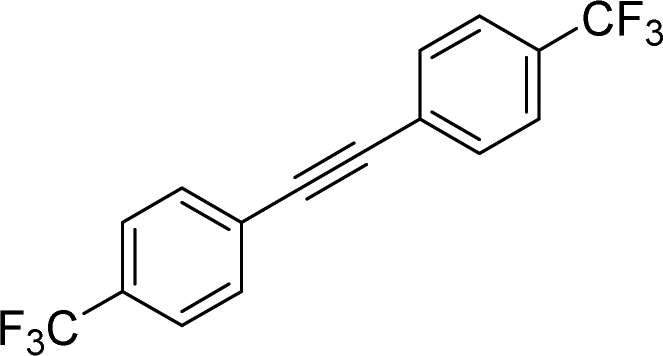
|
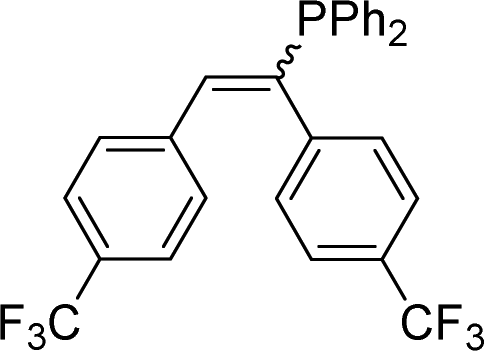
|
2s | 41f |
| 2:1, E:Z |
||||
Naphthyl substituted phosphine 2n is formed in excellent yield (entry 14) and 1,1-diphenylethylene performs comparably well, albeit under slightly more forcing conditions (50 °C, 24 h, entry 15). Phenyl acetylene does not react, but internal alkynes (entries 16 to 19) react at RT: conditions that are highly competitive compared to the leading literature, for example Schmidt's RT, La-catalyzed process in pyridine solvent30 and Cui's Ca- and Y-mediated chemistry that uses temperatures ranging from RT-75 °C.31 Unfortunately, reactions of 2- and 4-vinylpyridine and acrylonitrile with 1 all result in the formation of unidentified polymers irrespective of order of addition of Ph2PH. Methyl acrylate, butyl acrylate, cis- and trans-stilbene, along with β-methyl styrene do not react. HP reactions using unactivated alkenes (e.g. cyclohexene, allyl benzene) result in no conversion while no stoichiometric reaction of 1 and HPCy2, or catalytic HP of styrene using 1 and HPCy2, is observed.29 However, scale-up synthesis of 2a is successful: using 5 mmol Ph2PH (0.93 g/0.87 mL), 6 mmol styrene, 2.5 mol% Ge, at RT gives 94% after 24 h. Catalyst robustness was tested through an iterative addition protocol: 0.5 mmol Ph2PH, 0.6 mmol styrene, 5 mol% 1, were reacted at RT in C6D6 until complete conversion of Ph2PH was observed. Further equivalents of Ph2PH and styrene were added sequentially until six additions had been achieved (3 mmol Ph2PH added) giving 87% spectroscopic yield.29
Focus was then turned to investigating the mechanism of the reaction. A kinetic study of the reaction to form 2a was undertaken via variable time normalization analysis,32 implicates the reaction is approximately first order with respect to 1 and Ph2PH and a more complex relationship with styrene (≤1, ≥0.5), in-line with a complex reaction where multiple species can propagate from one Ge-center.29 There is no reaction when a stoichiometric quantity of styrene is added to 1. Conversely, reaction of Ph2PH with 1 results in the formation of a bright yellow color, which is also observed during catalysis. 1H DOSY analysis of this species shows fast diffusing HN(SiMe3)2 peaks and slower diffusing aryl resonances, consistent with protonation of Ge-bound N(SiMe3)2 by HPPh2.29 Monitoring catalysis (15 mol% 1) as a function of time by 31P NMR spectroscopy shows the Ph2PH doublet decreasing in intensity as product 2a, and a singlet at −36 ppm, increase. The signal at −36 ppm reaches a maximum when all Ph2PH is consumed (∼60% 2a formed), and then shows a slow decrease of this signal into 2a for the remainder of the reaction. There is also a very small singlet at −27 ppm that decreases in intensity slowly over the course of the reaction. Our data is consistent with bis- and tris-phosphido complexes (−27 and −36 ppm respectively) due to the loss of P–H, the fact that integration of the −36 ppm signal is consistent with 3 equiv. Ph2PH being sequestered from solution, alongside 31P NMR signals being more shielded than [GeCl{N(SiMe3)2}2(PPh2)] (not observed).33 Noteworthy is that a stabilized radical, (•Ge{N(SiMe3)2}3), from photochemical disproportionation of [Ge{N(SiMe3)2}2] has been reported. However, we have conclusively ruled-out radicals from this reaction.29
Combining these experimental observations, we propose that the tris-phosphido species (3tris) is the principal active catalyst for this transformation, and therefore postulate a mechanism based upon this (Scheme 3). After the formation of on-cycle 3tris, styrene may then insert into the Ge–P bond in a regioselective manner to form an alkyl intermediate (4mono/bis/tris). We have discounted this step occurring via attack of the β-position of styrene by the P-atom as no telomerization side products are observed.24 We propose 1,2-insertion to take place in a non-stepwise manner in accordance with the reaction being non-integer order with respect to styrene. From short-lived alkyl intermediates 4, stepwise protonolysis accounts for the approximate integer relationship in Ph2PH. This step liberates the product and reforms active species 3.
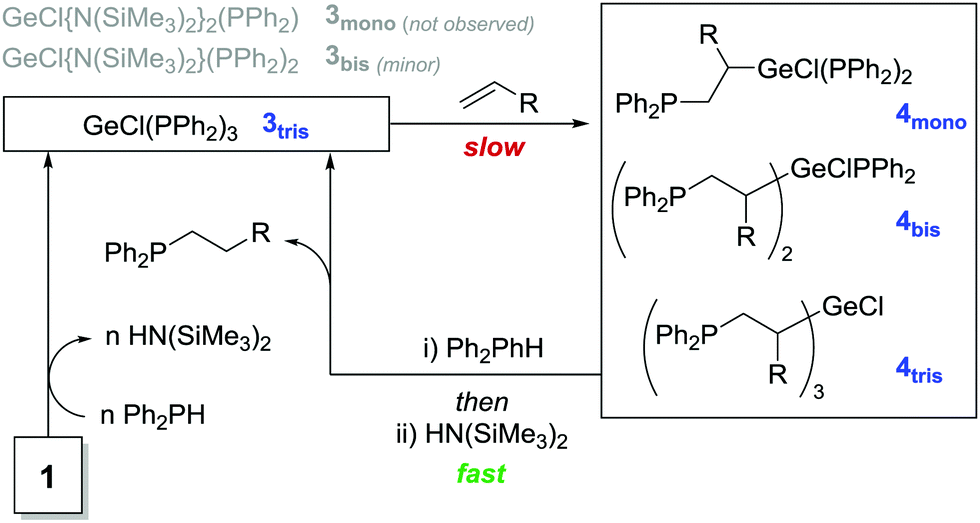 | ||
| Scheme 3 Proposed method by which 1 catalyzes the HP of styrenes. 3bis is only observed in trace quantities. | ||
During solvent screening, we also observed that 1 could catalytically induce the trimerization of MeCN to 2,6-dimethylpyrimidin-4-amine (Scheme 4). Base-mediated cascade reactions34,35 and an iridium hydride catalyst36 have previously been used to obtain the same product under more forcing conditions (e.g. Murahashi's Ir-catalyzed method uses IrH5(PiPr3)2 at 140 °C for 12 h giving TON = 12). This reactivity represents a new avenue of reactions this germanium species is capable of catalyzing and will be explored in due course.
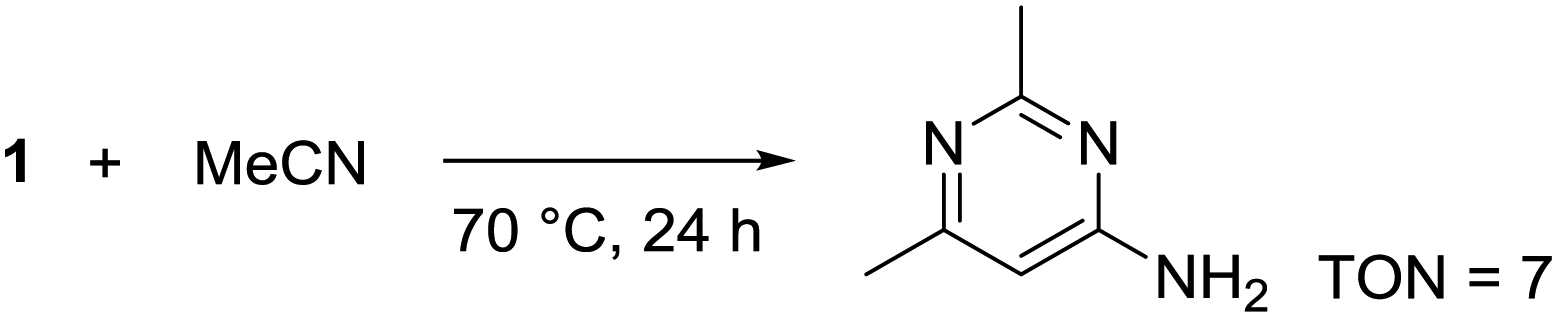 | ||
| Scheme 4 Trimerization of acetonitrile catalyzed by 1. | ||
In summary, we have disclosed a rare example of a Ge(IV)-catalyzed (non-polymerization) reaction. An operationally simple Ge(IV) tris(amido) complex has shown excellent ability as a pre-catalyst for the hydrophosphination of a range of styrenes and internal alkynes. The transformations proceed at room temperature over a time period competitive with transition metal catalyzed analogues. Preliminary mechanistic studies implicate the formation of a Ge-(tris)phosphido complex as the active catalyst and a redox-neutral mechanistic pathway was postulated. We hope that this work, using such a simple pre-catalyst, serves as a platform for future research in germanium-catalyzed hydrofunctionalization reactions.
The EPSRC and the University of Bath are thanked for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS.
- T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176–4197 RSC.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS.
- A. Finne, Reema and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3074–3082 CrossRef CAS.
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem. Int. Ed., 2007, 46, 2280–2283 CrossRef CAS.
- J. Guo, P. Haquette, J. Martin, K. Salim and C. M. Thomas, Angew. Chem. Int. Ed., 2013, 52, 13584–13587 CrossRef CAS.
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS.
- Y. Wu, C. Shan, Y. Sun, P. Chen, J. Ying, J. Zhu, L. Liu and Y. Zhao, Chem. Commun., 2016, 52, 13799–13802 RSC.
- J. Schneider, C. P. Sindlinger, S. M. Freitag, H. Schubert and L. Wesemann, Angew. Chem. Int. Ed., 2017, 56, 333–337 CrossRef CAS.
- S. Sinhababu, D. Singh, M. K. Sharma, R. K. Siwatch, P. Mahawar and S. Nagendran, Dalton Trans., 2019, 48, 4094–4100 RSC.
- R. K. Siwatch and S. Nagendran, Chem. – Eur. J., 2014, 20, 13551–13556 CrossRef CAS.
- R. Dasgupta, S. Das, S. Hiwase, S. K. Pati and S. Khan, Organometallics, 2019, 38, 1429–1435 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- T. Sugahara, J. D. Guo, T. Sasamori, S. Nagase and N. Tokitoh, Angew. Chem. Int. Ed., 2018, 57, 3499–3503 CrossRef CAS.
- I. Wauters, W. Debrouwer and C. V. Stevens, Beilstein J. Org. Chem., 2014, 10, 1064–1096 CrossRef.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- M. R. Douglass, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 2001, 123, 10221–10238 CrossRef CAS.
- A. M. Kawaoka, M. R. Douglass and T. J. Marks, Organometallics, 2003, 22, 4630–4632 CrossRef CAS.
- I. V. Basalov, S. C. Roşca, D. M. Lyubov, A. N. Selikhov, G. K. Fukin, Y. Sarazin, J.-F. Carpentier and A. A. Trifonov, Inorg. Chem., 2014, 53, 1654–1661 CrossRef CAS.
- I. V. Basalov, V. Dorcet, G. K. Fukin, J.-F. Carpentier, Y. Sarazin and A. A. Trifonov, Chem. – Eur. J., 2015, 21, 6033–6036 CrossRef CAS.
- J. Yuan, H. Hu and C. Cui, Chem. – Eur. J., 2016, 22, 5778–5785 CrossRef CAS.
- L. Rosenberg, ACS Catal., 2013, 3, 2845–2855 CrossRef CAS.
- K. A. Erickson, L. S. H. Dixon, D. S. Wright and R. Waterman, Inorg. Chim. Acta, 2014, 422, 141–145 CrossRef CAS.
- J. P. W. Stelmach, C. A. Bange and R. Waterman, Dalton Trans., 2016, 45, 6204–6209 RSC.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Green Chem., 2012, 14, 2699–2702 RSC.
- Y. Moglie, M. J. Gonzalez-Soria, I. Martin-Garcia, G. Radivoy and F. Alonso, Green Chem., 2016, 18, 4896–4907 RSC.
- See ESI†.
- M. M. I. Basiouny, D. A. Dollard and J. A. R. Schmidt, ACS Catal., 2019, 9, 7143–7153 CrossRef CAS.
- H. Hu and C. Cui, Organometallics, 2012, 31, 1208–1211 CrossRef CAS.
- J. Burés, Angew. Chem. Int. Ed., 2016, 55, 16084–16087 CrossRef.
- J. K. West and L. Stahl, Organometallics, 2012, 31, 2042–2052 CrossRef CAS.
- R. Bossio, S. Marcaccini, V. Parrini and R. Pepino, J. Heterocycl. Chem., 1986, 23, 889–891 CrossRef CAS.
- A. Verma, S. Jana, C. Durga Prasad, A. Yadav and S. Kumar, Chem. Commun., 2016, 52, 4179–4182 RSC.
- H. Takaya, T. Naota and S.-I. Murahashi, J. Am. Chem. Soc., 1998, 120, 4244–4245 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analysis data. CCDC 2023897 for the crystal structure of 1. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05792d |
| This journal is © The Royal Society of Chemistry 2020 |