
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudying the cellular distribution of highly phototoxic platinated metalloporphyrins using isotope labelling†
Riccardo
Rubbiani‡
a,
Wenyu
Wu‡
 a,
Anu
Naik‡
a,
Michele
Larocca
a,
Lukas
Schneider
a,
Roxane
Padrutt
a,
Vipin
Babu
a,
Christiane
König
b,
Doris
Hinger
c,
Caroline
Maake
c,
Stefano
Ferrari
bd,
Gilles
Gasser
e and
Bernhard
Spingler
*a
a,
Anu
Naik‡
a,
Michele
Larocca
a,
Lukas
Schneider
a,
Roxane
Padrutt
a,
Vipin
Babu
a,
Christiane
König
b,
Doris
Hinger
c,
Caroline
Maake
c,
Stefano
Ferrari
bd,
Gilles
Gasser
e and
Bernhard
Spingler
*a
aDepartment of Chemistry, University of Zurich, Zurich CH 8057, Switzerland. E-mail: spingler@chem.uzh.ch
bInstitute of Molecular Cancer Research, University of Zurich, Winterthurerstrasse 190, Zurich CH 8057, Switzerland
cInstitute of Anatomy, University of Zurich, Winterthurerstrasse 190, Zurich CH 8057, Switzerland
dInstitute of Molecular Genetics, Academy of Sciences of the Czech Republic, Videnska 1083, Prague, 143 00, Czech Republic
eChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, Paris F-75005, France
First published on 20th October 2020
Abstract
Novel tetraplatinated metalloporphyrin-based photosensitizers (PSs) are reported, which show excellent phototoxic indexes (PIs) up to 5800 against HeLa cells, which is, to the best of our knowledge, the highest value reported for any porphyrin so far. Furthermore, 67Zn isotope labelling allowed the determination of the ratio of zinc to platinum inside the cells using ICP-MS.
PDT is a clinically approved and minimally invasive therapeutic treatment for dermatological, ophthalmic, and infectious diseases, as well as cancer, which involves the application of a photosensitizer (PS) that ideally localizes disease-specifically. The application is followed by low energy irradiation to activate the PS leading to the generation of reactive oxygen species (ROS) which in turn induce cell death.1 A number of publications have reported about PSs conjugated with a chemotoxic unit, e.g., a cisplatin analogue.2 While this approach normally renders the conjugate more toxic,3 it might diminish one of the main advantages of PDT, namely the special selectivity by the applied light source.
Previously, we have reported a series of easily accessible tetrapyridyl-porphyrins (tPt-H24PyP, cPt-H24PyP, dPt-H24PyP) that were coordinated by four platinum(II) complexes to yield highly phototoxic agents (Fig. 1). These compounds displayed a light toxicity down to 19 nM and at the same time low dark toxicities of around 45 μM in human cancer cell lines.4 The ratio of dark to light toxicity is referred to as the phototoxic index (PI), and it should be as high as possible. Our systems displayed PI values higher than 1000 in HeLa cells after being excited with light of a wavelength of 420 nm. These PI values are better than currently clinically used second generation PSs that display a phototoxicity of >260.5
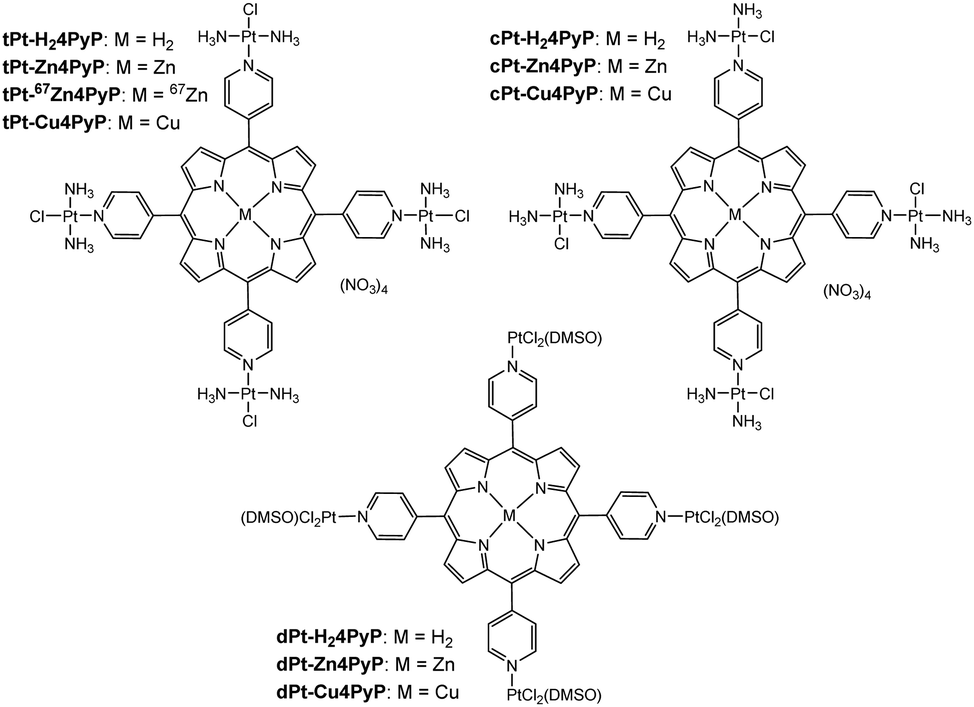 | ||
| Fig. 1 Studied tetraplatinated porphyrins. | ||
Based on our previous studies,1c,4 we investigated a set of important follow-up questions:
(1) How does the insertion of either zinc or copper1c modulate the phototoxicity of tPt-H24PyP, cPt-H24PyP and dPt-H24PyP? The singlet oxygen quantum yield of porphyrins was reported to increase6 or not to change at all7 after insertion of zinc into the macrocycle. In another case, the metal-free porphyrin generated mainly free hydroxy radicals, which were in vitro more cytotoxic than the singlet oxygen produced by the zinc porphyrin.8
(2) What is the fate of the complex tPt-H24PyP in the cell?
(3) What is the reason for the low dark toxicities of our platinum–porphyrin conjugates?
(4) What is the origin of the slow reaction of tPt-H24PyP with guanosine?
To study the effect of a metal inside the center of the porphyrins tPt-H24PyP, cPt-H24PyP and dPt-H24PyP, the novel complexes tPt-Zn4PyP, tPt-Cu4PyP, cPt-Zn4PyP, cPt-Cu4PyP, dPt-Zn4PyP and dPt-Cu4PyP were synthesized following our previously published procedures.4 Subsequently, these complexes were characterized using 1H- and 195Pt-NMR, IR, UV-Vis, MS and elemental analysis (ESI†). Additionally, the cationic unit of tPt-Zn4PyP could be crystallized as the tetraphenylborate salt using the under-oil technique (ESI†) and was studied using X-ray analysis (Fig. 2). We were particularly pleased to obtain this crystal structure, as it is the first example of expanding our robotic, aqueous crystallization trials9 towards organic solvents. The zinc metal center is in a square pyramidal geometry with one DMF molecule as the fifth ligand. This is only the third crystallographic report about a porphyrin with an exocyclic, non-organometallically bound platinum within the porphyrin plane.10
 | ||
| Fig. 2 Ellipsoidal plot of the crystal structure of [trans-PtCl(NH3)2]4-5,10,15,20-tetra(4′-pyridyl)-zinc(II)porphyrin tetraphenylborate·(DMF)7. Hydrogen atoms attached to a carbon atom, non-coordinated anions and DMF molecules not coordinated to the zinc metal center were omitted for clarity. | ||
The compounds were then tested in the cancerous HeLa cell line and the non-cancerous MRC-5 cell line composed of fibroblasts derived from lung tissue to assess their anti-proliferative properties. The results of the cytotoxicity studies of the compounds are presented in Table 1. The insertion of zinc into the complex tPt-H24PyP to yield the zinc complex tPt-Zn4PyP improves the phototoxicity and lowers the dark toxicity at the same time. Together these two effects improve the PI by almost a factor of five. These results can be attributed to the higher singlet oxygen quantum yield (determined as described in ref. 11) of tPt-Zn4PyP compared to tPt-H24PyP (Table S3, ESI†) and are in line with previous reports about other (metallo)porphyrin containing photosensitizers.6a,b,d
| Compound | IC50 MRC5 dark (μM) | IC50 HeLa dark (μM) | IC50 HeLa 420 nm (μM) | PI (HeLa) | Ref. |
|---|---|---|---|---|---|
| PI = phototoxic index, n. d. = not determined, t. w. = this work, n. a. = not applicable. | |||||
| Photofrin | n. d. | >41 | 4.3±0.2 | >9.5 | t. w. |
| Transplatin (tPt) | 86±8 | ∼130 | n. d. | n. d. | 12/13 |
| tPt-H24PyP | 93.4±6.5 | 44.9±8.0 | 0.037±0.02 | 1210 | 4 |
| tPt-Zn4PyP | >100 | >100 | 0.017±0.004 | >5882 | t. w. |
| tPt-Cu4PyP | 82.5±17.5 | 56.4±6.5 | 14.1±6.3 | 4.0 | t. w. |
| Cisplatin (cPt) | 7.9±1.2 | 11.5±2.9 | 22.3±5.7 | 0.52 | 4 |
cPt + H24PyP (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
>100 | >100 | 5.78±1.9 | >17 | 4 |
| cPt-H24PyP | 50.2±0.6 | 35.4±4.4 | 0.054±0.01 | 655 | 4 |
| cPt-Zn4PyP | 65.0±0.6 | 17.1±5.1 | 0.25±0.02 | 68.4 | t. w. |
| cPt-Cu4PyP | >100 | 13.9±1.6 | >100 | <0.14 | t. w. |
| cisPtDMSO (dPt) | >100 | >100 | >100 | n. a. | t. w. |
| dPt-H24PyP | >100 | >100 | 0.15±0.02 | >680 | 4 |
| dPt-Zn4PyP | >100 | >100 | 1.28±0.4 | >78 | t. w. |
| dPt-Cu4PyP | >100 | 61.4±1.5 | >100 | <0.61 | t. w. |
However, the zinc complexes cPt-Zn4PyP and dPt-Zn4PyP are less phototoxic than their corresponding metal-free porphyrins cPt-H24PyP and dPt-H24PyP. This difference in behaviour can be explained for cPt-Zn4PyP. After the insertion of zinc, the complex becomes rather photosensitive, which leads to photo-bleaching under irradiation and therefore to a lower singlet oxygen quantum yield (Table S3, ESI†). The higher phototoxic indexes of complexes tPt-H24PyP, tPt-Zn4PyP and tPt-Cu4PyP compared with cPt-H24PyP, cPt-Zn4PyP and cPt-Cu4PyP, respectively, are further explained below. Furthermore, tPt-Cu-4PyP is, to the best of our knowledge, only the second ever reported copper containing PS,1c which is phototoxic, as normally copper porphyrinoids are non-phototoxic. The DMSO containing complexes dPt-H24PyP and dPt-Zn4PyP have comparable PIs to the cisplatin derived complexes cPt-H24PyP and cPt-Zn4PyP. For a careful discussion on the influence of DMSO upon any platinum containing drug, the reader is directed to the excellent study by Gottesman.14 The singlet oxygen quantum yields (ΦΔ) of tPt-H24PyP, cPt-H24PyP, tPt-Zn4PyP and cPt-Zn4PyP are shown in Table S3 (ESI†).
As previously reported by us, tPt-H24PyP reacts extremely slowly with guanosine, as the platination of N7 progressed to less than 50% completion within 10 days.4 We speculate that the high charge (4+) of the cation of tPt-H24PyP might be responsible for this slow reaction, since the platinum chloride bond must be hydrolysed before the platinum can react with N7 of guanosine. The hydrolysis yields an intermediate that possesses an even higher charge of +5 (Fig. S1, ESI†). In order to test this hypothesis, we synthesized trans-[Pt(NH3)2(pyridine) Cl]NO3 (tPt-Py)15 and repeated the earlier mentioned reaction with guanosine (Fig. S2, ESI†). The reaction of tPt-Py and one equivalent of guanosine was monitored using 1H-NMR spectroscopy in a 1:1 DMF-d7:D2O mixture (see ESI†), showing that 50% of the binding occurred after nearly three days. Compared to previous results of tPt-H24PyP, the reaction of tPt-Py is at least three times faster. This supports the assumption that the rather slow reaction rate of tPt-H24PyP with guanosine is caused by unfavourable electrostatic interactions. Since guanosine and tPt-Py react with a 1:1 stoichiometry, the assumption was made that the reaction can be treated as a second order reaction (see ESI†). The determined slope S is equal to a second order reaction constant k of 0.0034 ± 0.0001 M−1 s−1. This value compares well with the pseudo-first order reaction rate (k1 of 0.00036 ± 0.00002 s−1) for the reaction of the mono aquated phenanthriplatin with 9-methylguanine.16 If one takes the concentration of the excess 9-methylguanine into account, one can convert the pseudo-first order reaction rate into a second order rate (k2 of 0.012 ± 0.007 M−1 s−1). This rate is about 3.5 times faster than the determined rate of tPt-Py with guanosine, which is reasonable since tPt-Py must first hydrolyse before it can react with guanosine.
Next, we investigated the uptake and distribution of the Zn–Pt complex in HeLa cells using ICP-MS. The expected ratio of zinc to platinum is 1:4 in tPt-Zn4PyP, provided the compound does not disintegrate. To study the Zn and Pt uptake, we first synthesized a highly enriched isotope 67Zn complex tPt-67Zn4PyP.17 Using the highly enriched zinc isotope makes it possible to differentiate between naturally occurring zinc and 67Zn18 that was introduced into the cells with compound tPt-67Zn4PyP. As 67Zn is expensive, initially, a careful and lengthy series of optimizations of the synthesis of zinc-5,10,15,20-tetra-(4′-pyridyl)-porphyrin (Zn4PyP) starting from elemental zinc with a natural distribution of isotopes was performed (Table S2, ESI†). Using these optimized conditions, the synthesis of tPt-Zn4PyP was then repeated starting from elemental 67Zn, which delivered complex tPt-67Zn4PyP (Fig. S5–S10, ESI†). The uptake study shows that only one quarter of all applied platinum atoms entered the HeLa cells, whereas three quarters of 67Zn atoms entered the cells (Fig. S13 and S14, ESI†). Subsequently, we studied the distribution of platinum and 67Zn in the different cell compartments (Table S4, ESI†). In the nucleus, the ratio of 67Zn to Pt was found to be 3:1 instead of 1:4. These results clearly demonstrate that the complex tPt-67Zn4PyP is disintegrated into the 67Zn containing porphyrin and the relatively non-toxic mono-activated transplatin.19 The analogous complex tPt-Zn4PyP with the natural Zn isotope distribution is expected to behave in the same way as tPt-67Zn4PyP. The transplatin moiety helps to improve solubility and transport the PS into the cell while not being toxic after the cleavage of the conjugate. Transplatin is known to be effluxed faster from the cell than cisplatin.20 This explains why there is less platinum found inside of the cells than 67Zn.
We also investigated the differences between the dark cytotoxcities of the transplatinated series of compounds, tPt-H24PyP, tPt-Zn4PyP and tPt-Cu4PyP and the cisplatinated series, cPt-H24PyP, cPt-Zn4PyP and cPt-Cu4PyP. The transplatinated series generates, as described before, the trans-diamminemonochloroplatinum(II) moiety21 after breakage of the N(pyridine)–platinum bond. This fragment has a much lower cytotoxcity than cis-diamminemonochloroplatinum(II), which is the first intermediate of cisplatin that must be generated in order to achieve any biological impact of cisplatin. Both, fragments of cis- and trans-diamminechloridoplatinum(II) in the conjugates c/tPt-H24PyP, c/tPt-Zn4PyP and c/tPt-Cu4PyP serve as a cancer-cell penetrating vector. However, the trans moiety has a much lower dark toxicity after hydrolysis. The cis-diamminemonochloroplatinum(II) fragment has been previously used as a chemotoxic part of a dual acting reagent.2b Our results imply that the trans-diamminemonochloroplatinum(II) conjugate would be even more interesting to study as well, maybe yielding a photosensitizer with an improved PI. Next, we examined whether the compounds induce damage to the DNA. It is established, that platinum drugs form drug-DNA adducts, leading to DNA damage, which triggers cell cycle arrest and DNA repair.22 The drug-induced DNA damage is followed by the phosphorylation of the histone H2AX, which is involved in the recruiting and localizing of the DNA damage repair proteins.
Therefore, γH2AX is considered an important marker to observe DNA damage, in particular double strand breaks.23 To determine the effectiveness of light-triggered DNA damage by the metal porphyrin conjugates, we treated HeLa cells with 500 nM tPt-H24PyP and tPt-Zn4PyP for 14 h and subsequent light irradiation. Cells were then stained with γH2AX-specific Alexafluor 594 antibody and visualized by confocal microscopy.
As shown in the immunofluorescent images (Fig. 3 and Fig. S15, ESI†), we could observe that tPt-H24PyP and tPt-Zn4PyP both induced severe DNA damage upon light irradiation, as observed by the increased accumulation of γH2AX. This means that tPt-H24PyP and tPt-Zn4PyP generate significantly more DNA damage after light irradiation compared with the experiments in the dark.
 | ||
| Fig. 3 DNA damage induced by light activated photosensitizer tPt-Zn4PyP. Immunofluorescent images of HeLa cells treated with 500 nM tPt-Zn4PyP for 14 h, then treated with light compared to the non-irradiated control. Cells were stained with γH2AX-Alexafluor 594 antibody (green) and DAPI. tPt-Zn4PyP: 458 nm ex., 630–750 nm em.; γH2AX-Alexafluor 594:594 nm ex., 610–630 em. | ||
To assess the intracellular localization of the photosensitizers, the compounds were co-stained with a marker for the Golgi organelle. tPt-Zn4PyP co-localized in the Golgi apparatus (Fig. 4). This experimental finding is pretty inspiring, as some reported drugs directed against the Golgi complex have been shown to be effective in both androgen-dependent and androgen-independent prostate cancer, through targeting abnormal glycosylation.24
 | ||
| Fig. 4 Co-localization of tPt-Zn4PyP with the Golgi apparatus. Representative images of live-cell imaging of HeLa cells treated with 50 μM tPt-Zn4PyP and stained with GOLGI-ID® Green dye (ENZO ENZ-51028). | ||
In summary, the insertion of a zinc cation into the phototoxic photosensitizer tPt-H24PyP yielded an even superior PS tPt-Zn4PyP with a phototoxic index of higher than 5880. This is likely due to the higher singlet oxygen quantum yield (ΦΔ) of the zinc porphyrin platinum complex tPt-Zn4PyP compared to the free-base porphyrin platinum complex tPt-H24PyP. The 1H-NMR experiments to follow the kinetics of the reaction of trans-[Pt(NH3)2(pyridine)Cl]NO3 (tPt-Py) with guanosine revealed that the monocation trans-[Pt(NH3)2(pyridine)Cl]+ reacts faster than the tetracation tPt-H24PyP. The tetracation has to overcome the barrier of generating an even five-fold charged state during the reaction with N7 of the guanine base. ICP-MS studies of the 67Zn labelled complex tPt-67Zn4PyP showed that the trans-diamminemonochloroplatinum(II) units of the complex tPt-Zn4PyP are dissociated from the conjugate inside the HeLa cells. For the first time, such a 67Zn labelled conjugate was employed to monitor the fate of the zinc conjugate within cells. The trans-diamminemonochloroplatinum(II) unit is thought to act as a cell-penetrating vector for the photosensitizer, which permits its activation inside the cancer cell by light. We could demonstrate that within the cell, the PS co-localizes with the Golgi apparatus and additionally, induces DNA damage in the nucleus. Unlike conjugates with a cisplatin, transplatin–photosensitizer conjugates have no increased dark toxicity. Therefore, we propose the further investigation of transplatin–drug conjugates for PDT and other oncological treatments.
Imaging was performed with support and equipment maintained by the Center for Microscopy and Image Analysis, University of Zurich. We thank the University of Zurich, the Swiss National Science Foundation (205321_159976) for financial support. W. W. was supported by a Swiss Government Excellence Scholarship and the Novartis Foundation for Medical-Biological Research. This work was financially supported by an ERC Consolidator Grant PhotoMedMet to G. G. (GA 681679) and has received support under the program “Investissements d’ Avenir” launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.). We thank Dr Thomas Fox for recording the 195Pt-NMR spectra, Martina Kalt and Dr Michal Shoshan for fruitful discussions and Beat Blattmann for setting up the under-oil crystallization trials.
Conflicts of interest
There are no conflicts to declare.References
- (a) T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS; (b) P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef; (c) P. M. Antoni, A. Naik, I. Albert, R. Rubbiani, S. Gupta, P. Ruiz-Sanchez, P. Munikorn, J. M. Mateos, V. Luginbuehl, P. Thamyongkit, U. Ziegler, G. Gasser, G. Jeschke and B. Spingler, Chem. – Eur. J., 2015, 21, 1179–1183 CrossRef CAS; (d) S. Mallidi, S. Anbil, A.-L. Bulin, G. Obaid, M. Ichikawa and T. Hasan, Theranostics, 2016, 6, 2458–2487 CrossRef CAS; (e) D. van Straten, V. Mashayekhi, H. S. de Bruijn, S. Oliveira and D. J. Robinson, Cancers, 2017, 9, 19 CrossRef.
- (a) C. Lottner, K.-C. Bart, G. Bernhardt and H. Brunner, J. Med. Chem., 2002, 45, 2064–2078 CrossRef CAS; (b) J. Mao, Y. Zhang, J. Zhu, C. Zhang and Z. Guo, Chem. Commun., 2009, 908–910 RSC; (c) G. Klein Couto, J. C. Rodrigues, B. S. Pacheco, L. Damé Simões, J. D. Paschoal, F. K. Seixas, T. V. Acunha, B. A. Iglesias and T. Collares, Photodiagn. Photodyn. Ther., 2020, 31, 101942 CrossRef CAS; (d) G. K. Couto, B. S. Pacheco, V. M. Borba, J. C. Rodrigues, T. L. Oliveira, N. V. Segatto, F. K. Seixas, T. V. Acunha, B. A. Iglesias and T. Collares, J. Photochem. Photobiol., B, 2020, 202, 111725 CrossRef CAS.
- V. Ramu, S. Gautam, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2019, 58, 9067–9075 CrossRef CAS.
- A. Naik, R. Rubbiani, G. Gasser and B. Spingler, Angew. Chem., Int. Ed., 2014, 53, 6938–6941 CrossRef CAS.
- J. Berlanda, T. Kiesslich, V. Engelhardt, B. Krammer and K. Plaetzer, J. Photochem. Photobiol., B, 2010, 100, 173–180 CrossRef CAS.
- (a) D. Praseuth, A. Gaudemer, J.-B. Verlhac, I. Kraljic, I. Sissoëff and E. Guillé, Photochem. Photobiol., 1986, 44, 717–724 CrossRef CAS; (b) K. Wang, C. T. Poon, C. Y. Choi, W.-K. Wong, D. W. J. Kwong, F. Q. Yu, H. Zhang and Z. Y. Li, J. Porphyrins Phthalocyanines, 2012, 16, 85–92 CrossRef CAS; (c) X. Hu, K. Ogawa, S. Li, T. Kiwada and A. Odani, Chem. Lett., 2017, 46, 764–766 CrossRef CAS; (d) P. J. Gonçalves, F. C. Bezzerra, A. V. Teles, L. B. Menezes, K. M. Alves, L. Alonso, A. Alonso, M. A. Andrade, I. E. Borissevitch, G. R. L. Souza and B. A. Iglesias, J. Photochem. Photobiol., A, 2020, 391, 112375 CrossRef.
- C. Pavani, A. F. Uchoa, C. S. Oliveira, Y. Iamamoto and M. S. Baptista, Photochem. Photobiol. Sci., 2009, 8, 233–240 RSC.
- J. M. Dabrowski, B. Pucelik, M. M. Pereira, L. G. Arnaut and G. Stochel, J. Coord. Chem., 2015, 68, 3116–3134 CrossRef CAS.
- (a) P. P. Nievergelt, M. Babor, J. Čejka and B. Spingler, Chem. Sci., 2018, 9, 3716–3722 RSC; (b) M. Babor, P. P. Nievergelt, J. Čejka, V. Zvoníček and B. Spingler, IUCrJ, 2019, 6, 145–151 CrossRef CAS; (c) R. Alvarez, P. P. Nievergelt, E. Slyshkina, P. Müller, R. Alberto and B. Spingler, Dalton Trans., 2020, 49, 9632–9640 RSC.
- (a) A. K. Bar, R. Chakrabarty, G. Mostafa and P. S. Mukherjee, Angew. Chem., Int. Ed., 2008, 47, 8455–8459 CrossRef CAS; (b) P. Lang, J. Habermehl, S. I. Troyanov, S. Rau and M. Schwalbe, Chem. – Eur. J., 2018, 24, 3225–3233 CrossRef CAS.
- L. Schneider, M. Larocca, W. Wu, V. Babu, R. Padrutt, E. Slyshkina, C. König, S. Ferrari and B. Spingler, Photochem. Photobiol. Sci., 2019, 18, 2792–2803 RSC.
- M. Leng, D. Locker, M.-J. Giraud-Panis, A. Schwartz, F. P. Intini, G. Natile, C. Pisano, A. Boccarelli, D. Giordano and M. Coluccia, Mol. Pharmacol., 2000, 58, 1525–1535 CrossRef CAS.
- J. M. Pascoe and J. J. Roberts, Biochem. Pharmacol., 1974, 23, 1345–1357 CrossRef CAS.
- M. D. Hall, K. A. Telma, K.-E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS.
- M. Huxley, C. Sanchez-Cano, M. J. Browning, C. Navarro-Ranninger, A. G. Quiroga, A. Rodger and M. J. Hannon, Dalton Trans., 2010, 39, 11353–11364 RSC.
- I. A. Riddell, T. C. Johnstone, G. Y. Park and S. J. Lippard, Chem. – Eur. J., 2016, 22, 7574–7581 CrossRef CAS.
- The maybe obvious alternative to 67Zn-4PyP would have been Pd-4PyP. We have tried many times to synthesize this compound, however the reactions products were always insoluble precipitates. Most likely, the metal coordinates to the exocyclic pyridyl nitrogen atoms, forming an infinite 2D-network.
- S. R. Hennigar and J. P. McClung, J. Nutr., 2016, 146, 2167–2173 CrossRef CAS.
- (a) G. Singh and J. Koropatnick, J. Biochem. Toxicol., 1988, 3, 223–233 CrossRef CAS; (b) K. S. Blisard, D. A. Harrington, D. A. Long and J. E. Jackson, J. Comp. Pathol., 1991, 105, 367–375 CrossRef CAS.
- X. Wu, S. Yuan, E. Wang, Y. Tong, G. Ma, K. Wei and Y. Liu, Metallomics, 2017, 9, 546–555 RSC.
- M. Coluccia and G. Natile, Anti-Cancer Agents Med. Chem., 2007, 7, 111–123 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS.
- L. J. Kuo and L.-X. Yang, In Vivo, 2008, 22, 305–309 CAS.
- T. Migita and S. Inoue, Int. J. Biochem. Cell Biol., 2012, 44, 1872–1876 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures to yield platinated porphyrins, Tables S1–S4 and Fig. S1–S15. CCDC 1977523. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05196a |
| ‡ These authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |